

Abstract
Objective
To identify the genetic cause of autosomal dominant ataxia complicated by behavioral abnormalities, cognitive decline, and autism in 2 families and to characterize brain neuropathologic signatures of dominant STUB1-related ataxia and investigate the effects of pathogenic variants on STUB1 localization.
Methods
Clinical and research-based exome sequencing was used to identify the causative variants for autosomal dominant ataxia in 2 families. Gross and microscopic neuropathologic evaluations were performed on the brains of 4 affected individuals in these families.
Results
Mutations in STUB1 have been primarily associated with childhood-onset autosomal recessive ataxia, but here we report heterozygous missense variants in STUB1 (p.Ile53Thr and p.The37Leu) confirming the recent reports of autosomal dominant inheritance. Cerebellar atrophy on imaging and cognitive deficits often preceded ataxia. Unique neuropathologic examination of the 4 brains showed the marked loss of Purkinje cells (PCs) without microscopic evidence of significant pathology outside the cerebellum. The normal pattern of polarized somatodendritic STUB1 protein expression in PCs was lost, resulting in aberrant STUB1 localization in the distal PC dendritic arbors.
Conclusions
This study confirms a dominant inheritance pattern in STUB1-ataxia in addition to a recessive one and documents its association with cognitive and behavioral disability, including autism. In the most extensive analysis of cerebellar pathology in this disease, we demonstrate disruption of STUB1 protein in PCs as part of the underlying pathogenesis.
There are more than 40 genetic types of autosomal dominant cerebellar ataxia referred to as the spinocerebellar ataxias (SCAs).1 In addition, there are more than 100 autosomal recessive disorders in which cerebellar ataxia is a prominent feature, variably denoted as spinocerebellar ataxia, recessive (SCAR) or ARCA (autosomal recessive cerebellar ataxia).1 One recessive variety, SCAR-16, is caused by homozygous or compound heterozygous mutations in the STUB1 (Stip1 homologous and Ubox-containing protein 1) gene.2,3 Most individuals with SCAR-16 have an onset of a cerebellar syndrome in childhood or adolescence sometimes associated with cognitive disability. The single brain autopsy reported showed severe loss of Purkinje cells (PCs) and neurons of the granular layer, accompanied by reactive Bergmann gliosis, but it lacked information about STUB1 staining in the cerebellum.4 Autosomal dominant inheritance of heterozygous STUB1 variants have been recently been reported but without neuropathologic evaluations.5,6
STUB1 protein, also called C terminus of HSP 70 interacting protein (CHIP), contains a tetratricopeptide repeat and a U-box. Among other functions, it is an E3 ubiquitin ligase/cochaperone that targets a broad range of chaperone protein substrates, including Hsp70, Hsc70, Hsp90, and DNA polymerase beta, for ubiquitin proteasome degradation.7
We report 2 families with autosomal dominant cerebellar ataxia, progressive cognitive decline, and, in one individual, childhood autism spectrum disorder associated with missense variants in STUB1. The availability of 4 brain autopsies from these families allowed a detailed evaluation of the cerebellar pathology and revealed aberrant intracellular location of STUB1.
Methods
Standard protocol approvals, registrations, and patient consents
Recruitment and sampling of the families was approved by the Institutional Review Board of the University of Washington (UW), and informed consent was provided by all patients.
Genetics studies
The pedigrees of the 2 families with familial cerebellar ataxia are shown in figure 1, A and B. Clinical testing for the common hereditary ataxias was performed for the probands, III-1 in family A and III-2 in family B, as described below in the clinical descriptions. Blood samples from the affected and unaffected patients (indicated in the pedigree) were collected, and genomic DNA was extracted by standard methods.
Figure 1. Four generation pedigrees of 2 families with hereditary cerebellar ataxia and cognitive disability.

If alive, current age in years is shown below the symbol. / = deceased; A = brain autopsy; ASD = autism spectrum disorder; black symbol = affected with ataxia; d = age at death. The variant or wild-type STUB1 allele is shown for individuals who were tested.
Exome sequencing was performed at the UW Center for Precision Diagnostics on affected patients A III-1 and A IV-1. Target enrichment by NimbleGen solution capture array and exome sequencing were performed and processed using Illumina HiSeq Analysis Software as previously described.8 The reads were aligned and compared with the Homo Sapiens GRCh37 reference genome. Sequences that failed Genome Analysis Toolkit quality filters were not further considered. Filtering of the variants was based on (1) coding region sequence effect—missense, nonsense, coding indels, and splice sites; (2) heterozygosity, given the autosomal dominant inheritance pattern of the disease in the family; (3) shared by both exomes or not read in one of the exomes (specified as “N” in the exome sequence data); (4) variant frequency—absence from the 1000 Genomes Project database and frequency threshold less than 0.0001 in gnomAD; (5) high evolutionary conservation, >3 by Genomic Evolutionary Rate Profiling (GERP)9; (6) a predicted damaging or probably damaging effect on protein structure and function by Sorting Intolerant From Tolerant10 and PolyPhen11,12; and (7) Combined Annotation Dependent Depletion (CADD) score13 >15. Variants that met these criteria were prioritized by gene expression, function in cerebellum, information from animal models, and relevance to neurologic diseases.
To confirm the exome variants and to investigate its cosegregation with the disease in family A, we performed PCR capillary sequencing using customized primers to amplify the fragment encompassing the candidate variants from genomic DNA using Hot Start Taq Polymerase (Qiagen, Hilden, Germany) as previously described.14
Neuropathologic studies
In both families, consent for autopsy was obtained from the legal next of kin according to the protocols approved by the UW Institutional Review Board. At the time of autopsy, the brain was removed in the usual fashion. In family A, for patients II-2 and III-1, the left halves were coronally sectioned and frozen for possible biochemical studies and the right halves were fixed in formalin. For patient A IV-1, the entire brain was fixed in formalin. After fixation, the cerebrum was sectioned coronally, the brainstem was sectioned axially, and the cerebellum sectioned sagittally. In family B, the brain was fixed in 10% neutral buffered formalin. After fixation, the cerebrum was sectioned coronally and the cerebellum and brainstem were sectioned axially.
A microtome was used to cut 4 μm-thick tissue sections from formalin-fixed, paraffin-embedded tissue blocks. Hematoxylin and eosin (H&E), Luxol fast blue (LFB), and Bielschowsky silver stained slides were prepared. Using previously optimized conditions, immunohistochemistry was performed using a Leica Bond III Fully Automated IHC and ISH Staining System (Leica Biosystems, Wetzlar, Germany). The sections were immunostained with mouse monoclonal antibody against paired helical filament tau (AT8, 1:1,000 dilution) (Pierce Technology, Waltham, MA), mouse monoclonal against β-amyloid (6E10, 1:5,000) (Covance, Princeton, NJ), rat monoclonal against phosphorylated TDP-43 (ser409/ser410, 1:1,000) (Millipore, Burlington, MA), mouse monoclonal against α-synuclein (LB509, 1:500) (Invitrogen, Carlsbad, CA), rabbit polyclonal against glial fibrillar acidic protein (#Z033401-2, 1:2,000) (DAKO-Agilent, Santa Clara, CA), mouse monoclonal against calbindin protein (CB-955, 1:1,000) (Sigma-Aldrich, St. Louis, MO), mouse monoclonal against ubiquitin (ubi-1, 1:50,000) (Millipore), and mouse monoclonal against p62 (2C11, 1:4,000). Appropriate positive and negative controls were included with each antibody and each run.
STUB1 immunofluorescence microscopy
Paraffin-embedded brain sections were stained using reagents and protocols from a commercially available kit (Opal Manual IHC Kit; Akoya Bioscience, Menlo Park, CA). The primary antibodies used were: anti-calbindin D28k (1:1,000 dilution; EMD Millipore), anti-EAAT4 (1:200 dilution; Abcam, Cambridge, United Kingdom), and anti-STUB1 (1:1,000 dilution; Abcam). Briefly, the sections were deparaffinized in xylene and rehydrated through graded alcohol and fixed in neutral-buffered formalin. Heat-mediated antigen retrieval was performed in AR6 buffer for 15 minutes at 98°C. The sections were treated for 10 minutes in antibody diluent/block and incubated with primary antibody diluted in the same buffer. The slides were washed in tris-buffered saline and polysorbate 20, incubated with the peroxidase-conjugated antimouse/rabbit secondary antibody, and then washed and incubated with Opal fluorescent substrate diluted in amplification buffer (Opal-650, Opal-520, and Opal-570, presented as pseudocolored immunostaining in purple, green, and red, respectively). The sections were washed and heat-treated to remove the antibody and then blocked and incubated with the second primary antibody as followed by secondary and Opal substrate as before. The cycle was repeated once more with the third primary antibody. After the third cycle, the sections were incubated briefly with Hoechst 33,256, rinsed with distilled H2O, and mounted with ProLong Diamond Antifade Mountant (ThermoFisher, Waltham, MA). Confocal microscopy was performed using a Leica TCS SP5 II microscope. Confocal images were acquired with the Leica Application Suite and processed using identical data acquisition settings for the images in each specific figure. All confocal images are single z-plane scans. Postacquisition image processing and figure preparation were accomplished using Leica Application Suite and Photoshop software that were limited to linear contrast and brightness adjustments only and were applied identically to all images regarding each specific figure.
Data availability
Deidentified data included in this study are available from the corresponding author on reasonable request.
Results
Clinical descriptions
Family A: The index case (III-1 in figure 1A) is a 60-year-old man who developed cognitive problems and clumsiness in his early 40s. He had 2 years of college education and was employed as a mechanical engineer. Examination at 44 years of age showed delayed but accurate responses to cognitive questions, saccadic interruptions in smooth pursuit eye movements, and random brief jerking movements of his hands and legs. Repeat examination at 48 years of age showed a Mini-Mental Status Examination score of 26 of 30, mild gait ataxia, moderate dysarthria, irregular smooth pursuit and saccadic eye movements, and hyperactive tendon reflexes at the knees. A brain MRI revealed cerebellar atrophy that appeared out of proportion to the relatively mild physical findings. Formal neuropsychometric testing at 49 years of age revealed generally normal intelligence but abnormal scores for executive function and verbal memory (table 1). Repeat examination over the next 10 years showed slow and steady progression of his disease. His speech remained slurred, and his gait became more unsteady, although he was still ambulatory with a walker. He had a stooped posture and was bradykinetic but had no rigidity and no tremor. Repeat MRIs showed increased cerebellar atrophy (figure 2A). He had cognitive decline, became less verbal, and sometimes demonstrated agitation and belligerence. Psychometric testing documented deterioration with deficiencies in verbal and visual memory, general language ability, processing speed, executive functions, and abstract reasoning with many subtests in the 1st to 10th percentile. Medical comorbidities included adult-onset diabetes mellitus and central sleep apnea treated with nocturnal continuous positive airway pressure. Medications included olanzapine, lorazepam, Neurontin, and baclofen. Molecular testing for SCAs 1, 2, 3, 6, 7, 8, 14, and 12, and DRPLA were negative. He died at the age of 60 years in an assisted living facility, and his brain autopsy was obtained.
Table 1.
Neuropsychological testing
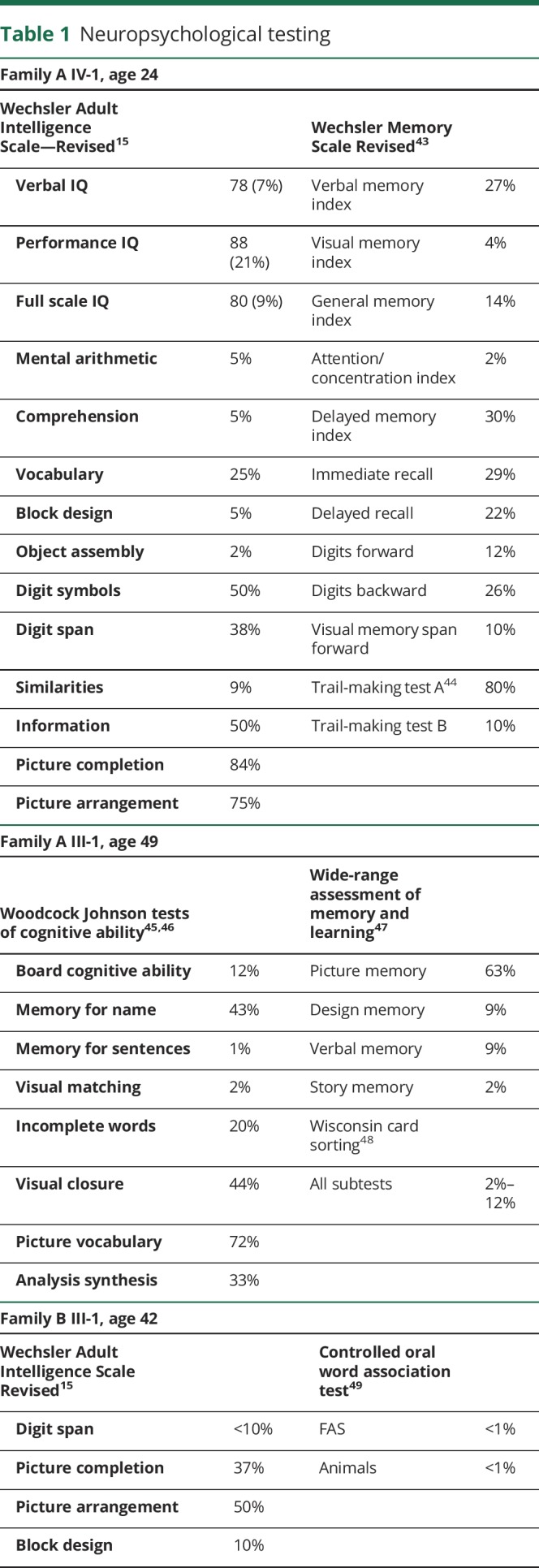

Figure 2. Neuroimaging.

(A) MRI of patient III-1 in family A at 53 years of age showing marked midline cerebellar atrophy, mildly enlarged lateral ventricle, and normal corpus callosum. (B) MRI of patient A IV-1 at 32 years of age showing marked midline cerebellar atrophy before the onset of ataxia but having cognitive disability and autism spectrum disorder. (C and D) MRI of patient III-2 in family B at 48 years of age showing midline (C) and lateral (D) cerebellar atrophy.
The father (II-2 in figure 1A) of the index patient had an onset of cognitive problems and poor coordination in his early 60s. He had been a pilot, mechanic, and homebuilder. A single neurologic examination at 68 years of age revealed mild gait ataxia, dysarthria, poor hand coordination, abnormal saccadic eye movements, hyperactive knee reflexes, and extensor plantar reflexes. He had intact short-term memory and could do simple arithmetic but exhibited paraphasias, inability to perform a three-step command or do a word problem in his head, and had concrete thinking and poor performance of the Luria hand sequence. His ataxia and cognitive problems progressed, and he started using a wheelchair. He died at the age of 79 years, and a brain autopsy was obtained.
The brother (III-3 in figure 1A) of the index patient had a junior college education. He had an onset of poor coordination in his late 20s. Examination at 34 years of age revealed marked gait ataxia, saccadic smooth pursuit eye movements with hypermetric saccades, dysarthria, dysmetria, and hyperactive tendon reflexes. Detailed mental status examination revealed moderate difficulty with memory and marked problems with visual spatial skills, calculations, insight, and judgment. Wechsler Adult Intelligence Scale—Revised (WAIS-R) testing15 revealed a verbal IQ of 74, performance IQ of 68, and full scale IQ of 71. He was labeled “demented.” He had a childlike affect with inappropriate and tangential responses. MRI revealed marked cerebellar atrophy. He experienced both alcohol and drug abuse and died at the age of 45 years without autopsy.
The daughter of the index patient (IV-1 in figure 1A) had an unremarkable labor and delivery and reached developmental motor milestones of childhood within the range of normal. She had average to below-average grades in school and was able to attend community college but dropped out of several courses. At 21 years of age, formal psychological testing showed a verbal IQ score of 84, performance score of 83, and full scale score of 83 (13th percentile; WAIS-R). Her highest score was in reading recognition (30th percentile), but other subscale scores were below the 20th percentile. Special weaknesses were in digit span, object assembly, arithmetic, and visual spatial analysis. She had great difficulty with social interactions and was labeled with “extreme shyness” and avoidant personality disorder. She seemed “disconnected from reality,” was frequently teased, and had a “nervous breakdown” at 13 years of age. A detailed analysis by a neuropsychologist at the age of 24 years demonstrated that she fell within the autism spectrum disorder. WAIS-R IQ and memory testing was similar to the age of 21 years, with mental arithmetic, comprehension, block design, object assembly, and visual memory at or below the fifth percentile (table 1). Neurologic examination at the age of 24 years was remarkable only for occasional saccadic interruptions in smooth pursuit and mild deep tendon hyper-reflexia. She had no nystagmus, no dysarthria, no intention tremor, and no gait ataxia. However, because of her family history of hereditary ataxia, she received a brain MRI that showed definite cerebellar atrophy. Over the next 10 years, there was no change in her neurologic examination and she did not develop ataxia. A repeat brain MRI at 32 years of age showed mild increase in the degree of cerebellar atrophy and mild generalized cerebral volume loss that was greater than expected for her age (figure 2B). She moved out of state and began to deteriorate in her late 30s and early 40s. She developed gait ataxia and started using a wheelchair. She developed severe cognitive decline with confusion, disorientation, poor judgment, and deficient problem solving. She developed bladder incontinence and became fully dependent on nursing care. She died at the age of 43 years, and her brain autopsy was obtained. Her brother (IV-2) had a normal examination and normal brain MRI at the age of 40 years.
Family B: The index patient (III-2 in figure 1B) was a woman who developed difficulty with coordination, mental confusion, and a personality change at approximately 40 years of age. Neurologic examination at the age of 42 years revealed gait ataxia, dysarthria, gaze-evoked horizontal nystagmus, and dysmetria. Tendon reflexes and sensory examination were unremarkable. Brain MRI at the age of 48 years showed moderate cerebellar atrophy (figure 2, C and D). The Mini-Mental Status Examination score was 28 of 30. Detailed formal neuropsychological testing revealed intact verbal memory but with numerous cognitive test scores at or below the fifth percentile, and she was noted to have marked general difficulties in the management and production of cognitive processes (table 1). She was a college graduate and had been a schoolteacher but was no longer able to perform her teaching duties. Multiple genetic tests for ataxia were all normal including SCAs 1, 2, 3, 5, 6, 7, 10, 14, 21, dentatorubral-pallidoluysian atrophy, Friedreich's ataxia, and senataxin.
Her family history was positive for additional affected people. Her father had died at the age of 75 years with ataxia, and her paternal aunt died at the age of 73 years and had been using a wheelchair for 10 years with ataxia. Her paternal grandmother died at the age of 68 years after admission to a state mental institution with ataxia, cognitive decline, and psychotic features. The patient had younger siblings who were not symptomatic, one of whom (III-3) had a normal examination at the age of 56 years and remains unaffected at the age of 67 years.
The patient had a slowly progressive course, eventually requiring a wheelchair. Her personality change included episodes of agitation and belligerence treated with olanzapine. Later in life, she developed a large excursion movement disorder of her upper and lower limbs that was described as an unusual wing-beating tremor. She died at the age of 61 years, and her brain autopsy was obtained.
Genetic studies
Whole exome sequence data were obtained from the individuals III-1 and IV-1 in family A. The average allele depth of coverage was 100× with more than >99% of the exome covered at more than 20×. We first scanned the exome data for variants in genes for autosomal dominant SCAs; no potential pathogenic variants were present. Both exomes contained a small CTG repeat expansion (9CTG/13CTG) in ATXN3, but the same allele was carried by the unaffected patient (III-2) and is reported as a benign polymorphism in ClinVar. We then followed a stepwise filtering protocol to prioritize candidate causative variants. Based on the autosomal dominant inheritance pattern of the disease in the family and the population prevalence of 1–5/100,000 for combined SCAs,1,16 we selected heterozygous variants detected in both exomes with a conservative minor allele frequency threshold of <0.0001 to avoid overfiltering. Other criteria included high/moderate functional effect based on variant type (missense, nonsense, coding indels, and splice site) and prediction algorithms and high evolutionary conservation.
This process left 4 candidate variants (table e-1, links.lww.com/NXG/A222), of which the missense variant in STUB1 was the most compelling. The variant in exon 1, c.158T>C, p.Ile53Thr (RefSeq NM_005861) affects an evolutionarily conserved residue (GERP 3.66, phastCons 1) and is predicted to be deleterious by SIFT (0.001), PolyPhen2 (1.00), and CADD (24.1). The variant is not present in the gnomAD database. STUB1 has been associated with SCAR16, an autosomal recessive SCA.2,17 Sanger sequencing confirmed the variant and identified it in all 4 affected patients and not in the unaffected one. None of the other 3 prioritized candidate variants showed complete cosegregation with the disease in family A. In family B, clinical exome sequencing reported a heterozygous STUB1 variant of uncertain significance c.111C>G, p.The37Leu in the proband. The variant is located in a highly conserved residue (GERP 3.5; phastCons 1) and is predicted to be disease causing by SIFT (0.01), PolyPhen2 (1.00), and CADD (24.7). Furthermore, the variant is not present in the gnomAD database. This variant was not detected by targeted sequencing in the unaffected sibling. Other members of these 2 families were not available for testing.
We performed a literature search using PubMed and multiple public variant databases including ClinVar, MARRVEL,18 Varsome,19 and VarCards20 for variants reported in STUB1 (figure 3). Considering the phenotypes, evidence for cosegregation, and predicted or experimentally shown effect on protein function, we found 26 homozygous or compound heterozygous-likely pathogenic variants, all of which were reported in SCAR16 cases; none of these STUB1 variants were reported in autosomal dominant SCA. Most of these mutations are included in a recent report on SCAR16.21 While we were preparing this manuscript, one case with SCA and a frame-shift mutation p.L275Dfs*16 and 2 with missense mutations p.G33S and p.P228S in STUB1 were reported.5,6 This deletion and p.P228S are located in the U-box domain, whereas the 2 variants in families A and B are both in the N-terminal tetratricopeptide repeat domain (also p.G33S, figure 3).
Figure 3. Ataxia-related variants in STUB1.

(A) STUB1 gene showing reported variants associated with autosomal dominant and recessive ataxias and the corresponding locations in the protein. Colors indicate the TPR (turquoise), coiled-coil (purple), and E3 ubiquitin ligase (U-box) domains in STUB1. Bold = heterozygous autosomal dominant variants reported by Genis et al5 and De Michele et al6 and in this report (in red). Arrow = an intronic mutation at the donor splice site of exon 4. (B) Diagram depicts the location of the p.Ile53Thr (red amino acid) and p.Phe37Leu (yellow amino acid) missense variants within a 3-D model of a STUB1 homodimer (the functional isoform). STUB1 was modeled with PyMOL (pymol.org) using the data from the protein data bank (rcsb.org, PDB ID#2C2L). TPR = tetratricopeptide repeat.
Neuropathologic studies
Gross findings
Patient A II-2
The brain only showed atrophy of the superior vermis and dorsal region of the cerebellar hemisphere. No other significant pathologic findings were noted.
Patient A III-1
The 1,120 g (fresh) brain had mild cortical atrophy involving frontal lobes without significant temporal, parietal, or occipital cortical atrophy. On cut sections, the lateral ventricles were enlarged bilaterally and the hippocampus was moderately atrophic. The cerebellar vermis was small and firm, and the dentate nucleus appeared mildly atrophic on cut sections.
Patient A IV-1
The 939 g (fresh) brain had moderate cortical atrophy and severe cerebellar atrophy (figure e-1, links.lww.com/NXG/A221). The cerebellar atrophy globally included the cortex with marked thinning of the folia but the deep cerebellar nuclei appeared unremarkable (figure e-1).
Patient B III-2
The 1,040 g (fresh) brain had no significant cortical atrophy; however, the cerebellum was markedly atrophic in a diffuse global pattern. In addition, the basis pontis was mildly flattened.
Histopathologic findings
Each case in family A showed similar histologic findings in the cerebellum (figure 4). H&E/LFB-stained slides show thinning of the cerebellar folia with marked loss of PCs. Bielschowsky silver stain demonstrates empty baskets in association with the loss of PCs, and there is reduced calbindin immunohistochemistry. There is also increased astrogliosis and Bergmann gliosis highlighted on the GFAP immunostain. Scattered ubiquitinated inclusions are present, but no p62 inclusions are identified. Notably, there was no evidence of TDP-43 or alpha-synuclein-positive inclusions.
Figure 4. Microscopic neuropathology.

Microscopic sections of the patients from both families showing marked cerebellar atrophy with thinning of the folia (H&E/LFB low magnification) and Purkinje cell (PC) loss (H&E/LFB high magnification). The loss of PCs is also highlighted by the empty baskets noted on Bielschowsky silver stain and by the severe reduction in calbindin-positive fibers (calbindin and GFAP immunostaining available on request). H&E = hematoxylin and eosin.
In patient A II-2, other neuropathologic findings including neurofibrillary tangles present within several regions of the hippocampus, including CA2 and subiculum, in addition to the transentorhinal cortex (Braak III) without evidence of beta-amyloid pathology (Thal 0, Consortium to Establish a Registry for Alzheimer's Disease [CERDAD] score absent). In patient A III-1, scattered neurofibrillary tangles were limited to the pre-alpha cells of the transentorhinal cortex (Braak I) with patchy, diffuse neocortical amyloid plaques (Thal 1), and no neuritic plaques were identified (CERAD absent). In patient A IV-1, there are rare neurofibrillary tangles limited to the pre-alpha cells of the transentorhinal cortex without the evidence of beta-amyloid pathology (Braak I, Thal 0, CERAD absent). There were also patchy white matter lesions located in the subcortical white matter, corpus callosum, anterior commissure, and the internal capsule characterized by a loss of myelinated axons, most consistent with acute microinfarcts. No pathologic findings were noted in the basal ganglia in any of the cases in this family.
In patient B III-2, H&E-stained slides show diffuse, bilateral PC loss and cerebellar cortical atrophy. Bielschowsky stains confirm PC loss highlighted by diffuse empty baskets and rare torpedo axons. Calbindin immunostain showed marked loss of fibers, and GFAP immunostain highlighted Bergman and astrogliosis. Scattered ubiquitin-positive inclusions are noted, but no p62- TDP-43-positive or alpha-synuclein-positive inclusions are identified. Other neuropathologic changes include sparse neuritic plaques in the neocortex (CERAD sparse) and diffuse amyloid plaques present in isocortex, hippocampus, and striatum (Thal 3). Neurofibrillary tangles are limited to the pre-alpha cells of the transentorhinal cortex (Braak I). No pathologic changes were noted in the basal ganglia.
STUB1 confocal microscopy findings
There are at least 29 previously reported pathogenic variants in STUB1 (figure 3), 3 dominant and 26 recessive, some of which destabilize the protein in vitro,3 but currently, there are no reported data regarding the effects of mutant STUB1 protein expression in the cerebellum of an individual harboring an ataxia-related STUB1 variant. To address this question, we carried out confocal microscopy using the rare neuropathologic cerebellum specimens from our 4 autopsied cases (II-2, III-1, and IV-1 in family A and III-2 in family B).
Figure 5A shows results from 2 normal controls (a–h), where the STUB1 expression in the cerebellar molecular layer was expressed prominently in PCs. Additional information about the localization of STUB1 was revealed by coimmunostaining calbindin and the neuronal glutamate transporter EAAT4, which is known to be expressed virtually exclusively in PC plasma membranes.22,23 EAAT4 immunostaining was carried out to augment the calbindin staining because EAAT4 more comprehensively labels distal dendritic compartments of the PCs (compare b and c in figure 5A). In a normal control, STUB1 expression was polarized in PCs, with the most prominent expression confined to cell bodies and proximal dendrites (somatodendritic) and much less in the distal dendritic compartments that project prominently into the cerebellar molecular layer. Note that although EAAT4 immunostaining (figure 5A.b) confirmed that the image field included extensive normal-appearing PC dendritic arbors, STUB1 expression was restricted mostly to the PC cell bodies and proximal dendrites (figure 5A.a). In marked contrast to this, STUB1 immunostaining from patient III-1 in family A with Ile53Thr-STUB1 (figure 5A.i–l) revealed an apparent loss of normal STUB1 polarization that was evidenced by expression throughout the distal PC arbors in addition to the somatodendritic compartments. As with patient A III-1, STUB1 immunoreactivity was aberrantly expressed in distal PC dendritic processes of patient A IV-1 (figure 5B.e–h) and A II-2 (figure 5B.i–l). Inspection of the cerebellum from family B patient III-2 with Phe37Leu-STUB1 (figure 5C.e–h) also revealed the same pattern of aberrant distal PC arbor STUB1 expression.
Figure 5. Loss of polarized STUB1 expression in Purkinje cells in patients with STUB1-cerebellar ataxia.

Immunostaining was performed with antibodies to STUB1 (pseudocolored purple), EAAT4 (green), and calbindin (red), and all scanning acquisition and postprocessing parameters were carried out identically among the cases about the controls in each panel. In 3 different normal control individuals (A.a–A.d, B.a–B.d, C.a–C.d), STUB1 immunoreactivity was uniformly localized primarily in PC cell bodies and proximal dendrites, with little expression in distal PC dendritic arbors. (A and B) Family A with STUB1-Ile53Thr. Loss of STUB1 polarization results in aberrant expression in distal PC dendritic arbors and cell bodies in patients III-1 (A.e–A.h), IV-1 (B.e–B.h), and II-2 (B.i–B.l). Scale bars = 100 μm. (C) Family B with STUB1-Phe37Leu. In individual III-2, STUB1 was aberrantly expressed in PC distal arbors (C.e–C.h). Scale bars = 50 μm.
To address the possibility that loss of polarized PC STUB1 expression could be due to nonspecific neurodegenerative processes, we also examined STUB1 expression in SCA5, SCA3, and SCA-7. In SCA5 (figure 6, E–H) and SCA3 (figure 6, I–L), STUB1 immunostaining was not different from the control (figure 6, A–D). These findings suggest that aberrant STUB1 expression in PCs is not mediated solely by nonspecific PC degeneration. However, in the SCA7 case (figure 6, M–P), STUB1 appeared mislocalized in distal dendritic arbors. Thus, it is unlikely that PC STUB1 mislocalization is mediated selectively by these 2 dominantly inherited STUB1 variants.
Figure 6. STUB1 expression pattern in Purkinje cells in a number of non-STUB1-associated ataxias and cerebellar degeneration.

In a normal individual (A–D), STUB1 immunoreactivity was expressed mostly in PC cell bodies and proximal dendrites (pseudocolored purple). Calbindin (red) and EAAT4 (green) immunostained PC bodies and dendrites. STUB1 localization appeared normal in PCs from SCA5 (E–H) and SCA3 (I–L). In an SCA7 case, STUB1 expression was aberrantly localized in distal PC dendritic arbors (M–P). Scale bars = 100 μm.
Discussion
STUB1 was previously associated with SCAR16, an autosomal recessive ataxia.3 Recently, 3 families with ataxia segregating heterozygous frameshift (p.L275Dfs*16) or a missense (p.Gly33Ser or p.Pro228Ser) variant in STUB1 have been reported, but neuropathology was not included.5,6 The 2 new families we describe herein confirm that heterozygous variants in STUB1 can cause autosomal dominant hereditary cerebellar ataxia, expand the associated pathogenic missense variants, broaden the age at onset range, and neurodevelopmental manifestations to include childhood autism spectrum disorder, document that the cerebellum is the prominent target of the pathologic process, and provide evidence that there is a change in cellular distribution of STUB1 in PCs. Taken together, these 5 families document that STUB1-related dominant ataxia is commonly associated with cognitive and behavioral disturbances that may precede the ataxia by years.
With advances in sequencing and improvements in variant interpretation techniques, the tremendous heterogeneity of neurogenetic disorders is more evident and it is increasingly apparent that genes once associated only with one form of inheritance are in fact capable of causing both recessive and dominant forms.24–27 The nomenclature that assigns a sequential number to each gene in a disease category provides no clarifying information and has limited clinical or research utility. As proposed, regarding the nomenclature of recessive cerebellar ataxia28 and other genetic movement disorders,29 we suggest ATX-STUB1 and SCA-STUB1 to replace SCAR16 and SCA48.
The features of the recessive and dominant forms of SCA-STUB1 ataxia overlap, including progressive cognitive impairment. However, in the recessive form, the behavioral deficit is usually mild and the onset of ataxia is usually in early childhood to the mid-20s.2,30,31 Additional features of the recessive form may include spasticity of the lower limbs, mild peripheral sensory neuropathy, and hypogonadotropic hypogonadism.30,32,33 As with other dominant disorders, clinical manifestations and age at onset are variable even within families. The mild parkinsonian features noted in III-1 (family A) and the wing beating-like tremor seen late in the disease in III-2 (family B) are apparently additional features that may occur in the dominant form of this syndrome.
A role for the cerebellum in autism has been previously suggested, and both decreased cerebellar vermis volume and PC density have been observed.34–36 The single case of autism spectrum disorder in the present family does not prove an association with STUB1 mutations but is noteworthy and continued search for additional examples in other families. It is of interest that this family member's MRI showed cerebellar atrophy at the time of her diagnosis of autism but before the onset of ataxia and cognitive decline.
The neuropathologic findings in the 4 brains from the 2 families reported here are remarkably similar in the prominent cerebellar atrophy with the loss of PCs. Our cases support the direct effect of cerebellar pathology on the cognitive/behavioral features of SCA-STUB1 because cerebellar atrophy was found on imaging when the cognitive behavioral abnormality was evident before the clinical signs of ataxia were displayed and because no significant brain pathology outside the cerebellum was identified on autopsy. This is consistent with the hypothesis that the cerebellum participates in cognitive processing and emotional control.37,38 The only previous neuropathologic description associated with SCA-STUB1 disease is in an autosomal recessive case that also showed marked PC loss.4 CHIP (STUB1) immunostaining labeled primarily cytoplasm in the neurons of the paraventricular nuclei of the hypothalamus and nuclear staining of pyramidal neurons of the medial-entorhinal cortex. No CHIP staining of the cerebellum was reported. The authors speculated that the involvement of frontal and temporal cortex was likely related to the dementia that developed in the later stages of the disease. The remarkably low total brain weight in our patient A IV-1 suggests there may be abnormalities in the cerebral cortex beyond what we were able to detect microscopically and may be related to the hypometabolism on fluorodeoxyglucose-PET studies of the cerebellum, striatum, and cerebral cortex reported.6
The CHIP ubiquitin ligase encoded by STUB1 functions as a homodimer.39 A number of variants responsible for the recessive form of ATX-STUB1 have been shown to reduce STUB1 protein stability in vitro, suggesting that these mutations may lead to lower steady-state STUB1 expression levels,3 yet the carriers remain unaffected. Therefore, haploinsufficiency and dominant negative mechanisms seem unlikely for the dominant presentation. The autosomal dominant missense and frameshift variants may have a toxic gain of function effect.5,6
In support of a toxic gain of function, we found that the intracellular location of STUB1 protein in PCs is altered by the missense variants. In PCs, STUB1 is normally polarized, with the most prominent expression in somatodendritic compartments and lower expression in the distal dendritic arbors that extend into the cerebellar molecular layer. The molecular layer is filled with a dense network of EAAT4-positive “knotted lace-like” PC arbors that is quite large compared with the PC somatodendritic compartments, hence further emphasizing the highly polarized nature of normal STUB1 protein expression in PCs. In the p.Ile53Thr and p.Phe37Leu cases in this study, STUB1 appeared to lose somatodendritic polarization, allowing STUB1 to localize in distal PC arbors and cell bodies. The faint cytoplasmic staining of STUB1 and extension into dendrites and axons were also observed in the cortex of the patient with recessive ATX-STUB14; however, the cerebellum was not evaluated for STUB1 in that study. Additional functional studies of the pathogenic variants responsible for these ataxias may clarify the biochemical consequences of this location shift.
The involvement of STUB1 in disease extends beyond its causative role in STUB1-ataxia. The CHIP and ataxin-1 proteins directly interact, and CHIP promotes ubiquitination of expanded ataxin-1.40 Ataxin-3 is involved in the degradation of misfolded chaperone substrates through its interaction with STUB1/CHIP.41 Ataxin-3 is recruited to monoubiquitinated STUB1/CHIP where it prevents further ubiquitin chain extension. These reports led us to examine STUB1 mislocalization in other type of SCAs. The shift in subcellular localization in our patients was not seen in SCA3, SCA5, or a nongenetic case with chronic ischemic cerebellar injury (data not shown). Taken together, these findings argue against the possibility that STUB1 mislocalization is mediated nonspecifically by degenerating PCs. However, we also found evidence of disturbed STUB1 polarization in the SCA7 case. Therefore, it is unlikely that STUB1 mislocalization is restricted to these dominantly expressed STUB1 mutations. Detailed study of more cases and more subtypes of SCA may shed light on the possible role of STUB1 in a common pathway with relevance to SCA development.
It is also noteworthy that the 4 brains from the 2 families we report demonstrated no tau pathology other than low Braak stages consistent with the patients' ages. This is in contrast to a report of substantial tau pathology in CHIP/STUB1 knockout mice and Caenorhabditis elegans consistent with this protein being involved in the ubiquitination of tau.42 This contrast is additional evidence that the missense mutations reported here are unlikely to cause a loss of function.
In conclusion, a variety of mutations in STUB1 can cause either autosomal recessive or autosomal dominant cerebellar ataxia often associated with a prominent cognitive affective syndrome that may include the autism spectrum disorder. The dominant form presents with cognitive impairment at an earlier stage and ataxia at a later stage, in comparison to the recessive form. The brunt of the pathologic process affects PCs where there is a mislocalization of the STUB1 protein from the cytoplasm to the dendritic arbor. Further resolution of this pathologic process will help explain the extensive variability of the hereditary ataxias.
Acknowledgment
The authors are grateful to the families whose participation made this work possible and to John Wolff, Allison Beller, Lisa Keene, Kim Howard, and Emily Trittschuh for their excellent technical assistance.
Glossary
- CADD
Combined Annotation Dependent Depletion
- CHIP
C terminus of HSP 70 interacting protein
- GERP
Genomic Evolutionary Rate Profiling
- H&E
hematoxylin and eosin
- PC
Purkinje cell
- SCA
Spinocerebellar Ataxia
- SCAR
Spinocerebellar Ataxia, Recessive
- UW
University of Washington
- WAIS-R
Wechsler Adult Intelligence Scale—Revised
Appendix. Authors
Study funding
Supported by NIH (1R01NS069719, U01AG005136, and 5T32GM007454) and the United States (U.S.) Department of Veterans Affairs (Merit Review Award Number 101 CX001702 from the Clinical Sciences R&D [CSRD] Service and RDIS# 0005 from the Office of Research and Development Medical Research Service).
Disclosure
Disclosures available: Neurology.org/NG.
References
- 1.Bird T. Hereditary ataxia overview. In: GeneReviews at GeneTests: Medical Genetics Information Resource [Database Online] 1993-2019. Seattle: University of Washington; 2019. [Google Scholar]
- 2.Depondt C, Donatello S, Simonis N, et al. . Autosomal recessive cerebellar ataxia of adult onset due to STUB1 mutations. Neurology 2014;82:1749–1750. [DOI] [PubMed] [Google Scholar]
- 3.Kanack AJ, Newsom OJ, Scaglione KM. Most mutations that cause spinocerebellar ataxia autosomal recessive type 16 (SCAR16) destabilize the protein quality-control E3 ligase CHIP. J Biol Chem 2018;293:2735–2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bettencourt C, de Yebenes JG, Lopez-Sendon JL, et al. . Clinical and neuropathological features of spastic ataxia in a Spanish family with novel compound heterozygous mutations in STUB1. Cerebellum 2015;14:378–381. [DOI] [PubMed] [Google Scholar]
- 5.Genis D, Ortega-Cubero S, San Nicolas H, et al. . Heterozygous STUB1 mutation causes familial ataxia with cognitive affective syndrome (SCA48). Neurology 2018;91:e1988–e1998. [DOI] [PubMed] [Google Scholar]
- 6.De Michele G, Lieto M, Galatolo D, et al. . Spinocerebellar ataxia 48 presenting with ataxia associated with cognitive, psychiatric, and extrapyramidal features: a report of two Italian families. Parkinsonism Relat Disord 2019;65:91–96. [DOI] [PubMed] [Google Scholar]
- 7.McDonough H, Patterson C. CHIP: a link between the chaperone and proteasome systems. Cell Stress Chaperones 2003;8:303–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen D-H, Below JE, Shimamura A, et al. . Ataxia-pancytopenia syndrome is caused by missense mutations in SAMD9L. Am J Hum Genet 2016;98:1146–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput Biol 2010;6:e1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res 2003;31:3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sunyaev S, Ramensky V, Koch I, Lathe W III, Kondrashov AS, Bork P. Prediction of deleterious human alleles. Hum Mol Genet 2001;10:591–597. [DOI] [PubMed] [Google Scholar]
- 12.Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res 2002;30:3894–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014;46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen YZ, Matsushita MM, Robertson P, et al. . Autosomal dominant familial dyskinesia and facial myokymia: single exome sequencing identifies a mutation in adenylyl cyclase 5. Arch Neurol 2012;69:630–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wechsler D. Wechsler Adult Intelligence Scale—Revised (WAIS-R). San Antonio: The Psychological Corporation; 1981. [Google Scholar]
- 16.Ruano L, Melo C, Silva MC, Coutinho P. The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies. Neuroepidemiology 2014;42:174–183. [DOI] [PubMed] [Google Scholar]
- 17.Pakdaman Y, Sanchez-Guixe M, Kleppe R, et al. . In vitro characterization of six STUB1 variants in spinocerebellar ataxia 16 reveals altered structural properties for the encoded CHIP proteins. Biosci Rep 2017;37:BSR20170251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang J, Al-Ouran R, Hu Y, et al. . MARRVEL: integration of human and model organism genetic resources to facilitate functional annotation of the human genome. Am J Hum Genet 2017;100:843–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kopanos C, Tsiolkas V, Kouris A, et al. . VarSome: the human genomic variant search engine. Bioinformatics 2018;35:1978–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li J, Shi L, Zhang K, et al. . VarCards: an integrated genetic and clinical database for coding variants in the human genome. Nucleic Acids Res 2018;46:D1039–D1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Turkgenc B, Sanlidag B, Eker A, et al. . STUB1 polyadenylation signal variant AACAAA does not affect polyadenylation but decreases STUB1 translation causing SCAR16. Hum Mutat 2018;39:1344–1348. [DOI] [PubMed] [Google Scholar]
- 22.Bar-Peled O, Ben-Hur H, Biegon A, et al. . Distribution of glutamate transporter subtypes during human brain development. J Neurochem 1997;69:2571–2580. [DOI] [PubMed] [Google Scholar]
- 23.Dehnes Y, Chaudhry FA, Ullensvang K, Lehre KP, Storm-Mathisen J, Danbolt NC. The glutamate transporter EAAT4 in rat cerebellar Purkinje cells: a glutamate-gated chloride channel concentrated near the synapse in parts of the dendritic membrane facing astroglia. J Neurosci 1998;18:3606–3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen DH, Meneret A, Friedman JR, et al. . ADCY5-related dyskinesia: broader spectrum and genotype-phenotype correlations. Neurology 2015;85:2026–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barrett MJ, Williams ES, Chambers C, Dhamija R. Autosomal recessive inheritance of ADCY5-related generalized dystonia and myoclonus. Neurol Genet 2017;3:193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lise S, Clarkson Y, Perkins E, et al. . Recessive mutations in SPTBN2 implicate beta-III spectrin in both cognitive and motor development. PLoS Genet 2012;8:e1003074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morimoto Y, Yoshida S, Kinoshita A, et al. . Nonsense mutation in CFAP43 causes normal-pressure hydrocephalus with ciliary abnormalities. Neurology 2019;92:e2364–e2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rossi M, Anheim M, Durr A, et al. . The genetic nomenclature of recessive cerebellar ataxias. Mov Disord 2018;33:1056–1076. [DOI] [PubMed] [Google Scholar]
- 29.Marras C, Lang A, van de Warrenburg BP, et al. . Nomenclature of genetic movement disorders: recommendations of the international Parkinson and movement disorder society task force. Mov Disord 2016;31:436–457. [DOI] [PubMed] [Google Scholar]
- 30.Shi Y, Wang J, Li JD, et al. . Identification of CHIP as a novel causative gene for autosomal recessive cerebellar ataxia. PLoS One 2013;8:e81884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heimdal K, Sanchez-Guixe M, Aukrust I, et al. . STUB1 mutations in autosomal recessive ataxias—evidence for mutation-specific clinical heterogeneity. Orphanet J Rare Dis 2014;9:146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shi CH, Schisler JC, Rubel CE, et al. . Ataxia and hypogonadism caused by the loss of ubiquitin ligase activity of the U box protein CHIP. Hum Mol Genet 2014;23:1013–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Synofzik M, Schule R, Schulze M, et al. . Phenotype and frequency of STUB1 mutations: next-generation screenings in Caucasian ataxia and spastic paraplegia cohorts. Orphanet J Rare Dis 2014;9:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Webb SJ, Sparks BF, Friedman SD, et al. . Cerebellar vermal volumes and behavioral correlates in children with autism spectrum disorder. Psychiatry Res 2009;172:61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Skefos J, Cummings C, Enzer K, et al. . Regional alterations in purkinje cell density in patients with autism. PLoS One 2014;9:e81255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang SS, Kloth AD, Badura A. The cerebellum, sensitive periods, and autism. Neuron 2014;83:518–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmahmann JD. The cerebellum and cognition. Neurosci Lett 2019;688:62–75. [DOI] [PubMed] [Google Scholar]
- 38.Liang KJ, Carlson ES. Resistance, vulnerability and resilience: a review of the cognitive cerebellum in aging and neurodegenerative diseases. Neurobiol Learn Mem 2019. doi: 10.1016/j.nlm.2019.01.004. Epub 2019 Jan 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nikolay R, Wiederkehr T, Rist W, Kramer G, Mayer MP, Bukau B. Dimerization of the human E3 ligase CHIP via a coiled-coil domain is essential for its activity. J Biol Chem 2004;279:2673–2678. [DOI] [PubMed] [Google Scholar]
- 40.Al-Ramahi I, Lam YC, Chen HK, et al. . CHIP protects from the neurotoxicity of expanded and wild-type ataxin-1 and promotes their ubiquitination and degradation. J Biol Chem 2006;281:26714–26724. [DOI] [PubMed] [Google Scholar]
- 41.Scaglione KM, Zavodszky E, Todi SV, et al. . Ube2w and ataxin-3 coordinately regulate the ubiquitin ligase CHIP. Mol Cell 2011;43:599–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dickey CA, Yue M, Lin WL, et al. . Deletion of the ubiquitin ligase CHIP leads to the accumulation, but not the aggregation, of both endogenous phospho- and caspase-3-cleaved tau species. J Neurosci 2006;26:6985–6996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wechsler D. WMS-R. Wechsler Memory Scale—Revised. San Antonio: Psychological Corp: Harcourt Brace Jovanovich; 1987. [Google Scholar]
- 44.King FW, Bird TD. Trail making test: norms for college males and relationship to scholastic aptitude. Percept Mot Skills 1965;21:199–206. [DOI] [PubMed] [Google Scholar]
- 45.Woodcock RW, Johnson B. Woodcock-Johnson Psycho-Educational Battery–Revised. Rolling Meadows, IL: Riverside; 1989. [Google Scholar]
- 46.Woodcock R, Johnson B. Woodcock-Johnson Psychoeducational Battery-Revised Tests of Achievement. Chicago: Riverside Publishing; 1990. [Google Scholar]
- 47.Sheslow D, Adams W. Wide Range Assessment of Memory and Learning (WRAML). Wilmington, DE: Jastak; 1990. [Google Scholar]
- 48.Grant DA, Berg E. A behavioral analysis of degree of reinforcement and ease of shifting to new responses in Weigl-type card-sorting problem. J Exp Psychol 1948;38:404–411. [DOI] [PubMed] [Google Scholar]
- 49.Hooper H. Hooper Visual Organization Test (VOT). Los Angeles: Western Psychological Services; 1983. [Google Scholar]
- 50.Wiens AN, Tindall AG, Crossen JR. California verbal learning test: a normative data study. J Clin Neuropsychologist 1994;8:75–90. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Deidentified data included in this study are available from the corresponding author on reasonable request.