

Abstract
Transcriptionally inactive genes are often positioned at the nuclear lamina (NL), as part of large lamina‐associated domains (LADs). Activation of such genes is often accompanied by repositioning toward the nuclear interior. How this process works and how it impacts flanking chromosomal regions are poorly understood. We addressed these questions by systematic activation or inactivation of individual genes, followed by detailed genome‐wide analysis of NL interactions, replication timing, and transcription patterns. Gene activation inside LADs typically causes NL detachment of the entire transcription unit, but rarely more than 50–100 kb of flanking DNA, even when multiple neighboring genes are activated. The degree of detachment depends on the expression level and the length of the activated gene. Loss of NL interactions coincides with a switch from late to early replication timing, but the latter can involve longer stretches of DNA. Inactivation of active genes can lead to increased NL contacts. These extensive datasets are a resource for the analysis of LAD rewiring by transcription and reveal a remarkable flexibility of interphase chromosomes.
Keywords: genome organization, lamina‐associated domains, nuclear lamina, replication timing, transcription
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics
Genomic maps acquired after transcriptional activation or inactivation of genes reveals how transcription affects their interaction with the nuclear lamina.
Introduction
In metazoan cell nuclei, large chromatin domains are associated with the nuclear lamina (NL) (Gonzalez‐Sandoval & Gasser, 2016; van Steensel & Belmont, 2017; de Leeuw et al, 2018; Kim et al, 2019; Lochs et al, 2019). Mammalian genomes have roughly one thousand of such lamina‐associated domains (LADs), which are typically hundreds of kb or even a few Mb in size. The NL contacts of some LADs are highly consistent between cell types, while other LADs interact in cell‐type‐specific (facultative) manners with the NL. How LAD‐NL contacts are regulated is poorly understood.
Most genes inside LADs have very low transcriptional activity (Guelen et al, 2008; Peric‐Hupkes et al, 2010; Leemans et al, 2019). When cells differentiate, detachment of genes from the NL often coincides with transcriptional activation, while increased NL interactions correlate with reduced transcription (Peric‐Hupkes et al, 2010; Lund et al, 2013; Robson et al, 2016, 2017). These observations raise the interesting possibility that the NL helps to establish a repressive environment. In support of this notion, depletion of lamins can lead to derepression of specific genes (primarily in Drosophila) (Shevelyov et al, 2009; Kohwi et al, 2013; Chen et al, 2014); transfer of human inactive promoters from LADs to a neutral chromatin environment can lead to activation of these promoters (Leemans et al, 2019); and artificial tethering of some genes to the NL can reduce their activity (Finlan et al, 2008; Kumaran & Spector, 2008; Reddy et al, 2008; Dialynas et al, 2010).
This, however, does not rule out that the contacts of genes with the NL are the consequence of a lack of transcriptional activity, and vice versa, that genes detach from the NL in response to their activation. This was initially suggested by experiments with fluorescently tagged lacO arrays that were integrated in a locus near the NL. Tethering of the transcriptional activator peptide VP16 to these arrays caused repositioning away from the NL (Tumbar & Belmont, 2001). Similar observations were made when VP64 (a tetramer of VP16) was tethered to promoters of three distinct genes in LADs in mouse embryonic stem (mES) cells (Therizols et al, 2014). Another study found that activation of the long non‐coding RNA gene ThymoD in mouse T‐cell progenitors contributed to the detachment of the neighboring gene Bcl11b from the NL (Isoda et al, 2017). The molecular signals that cause detachment of a locus from the NL are still poorly understood.
Analysis of NL detachment that follows forced activation of a gene has so far been limited to a handful of loci. It is thus unclear whether the observed detachment from the NL after transcription activation is universal, or limited to genes with particular features. For example, do the size of the gene and its level of transcription matter? Moreover, the previous studies of individual loci have only been based on microscopy‐based assays such as fluorescence in situ hybridization (FISH) or LacO tagging and have only visualized the targeted genes themselves, but not the flanking DNA sequences. It has therefore remained unclear what the impact of these repositioning events is on the surrounding chromosomal regions. One possible scenario is that activation of a single gene inside a LAD leads to movement of the whole surrounding LAD to the nuclear interior. Alternatively, detachment could be restricted to the target gene itself or only affect some of its flanking regions. Possibly, detachment of one locus from the NL could be compensated by increased NL contacts of another locus nearby. To investigate this, high‐resolution maps of NL interactions after manipulation of the activity of individual genes are needed.
Nuclear lamina interactions have also been associated with the timing of DNA replication during S‐phase. LADs typically coincide with late‐replicating domains, but the overlap is not complete, particularly at the edges of LADs (Guelen et al, 2008; Peric‐Hupkes et al, 2010; Pope et al, 2014). These local discrepancies are still poorly understood, but may provide important clues about the interplay between the mechanisms that establish LADs and late‐replicating domains. Above‐mentioned activation of genes in LADs with TALE‐VP64 was accompanied by a switch from late to early replication; however, it was not analyzed how far this switch extends across the locus and how well it tracks with the changes in NL contacts (Therizols et al, 2014).
To study these issues, we took three complementary approaches. First, we used two VP16‐tethering methods to activate a total of 14 different genes inside LADs, querying a variety of gene contexts. Second, we inactivated or truncated selected genes genetically to test whether they would re‐attach to the NL. Third, we integrated an active transgene driven by a strong promoter into multiple LADs and tested how this altered NL interactions of the integration sites and the flanking regions. In each instance, we used DamID to map NL interactions, enabling us to visualize the extent of NL detachment in detail along entire chromosomes. We also compared the changes in NL interactions to changes in replication timing.
Results
Detachment of genes from the NL upon activation by TALE‐VP64
We first employed a previously reported system in mouse embryonic stem (ES) cells, in which individual NL‐associated genes are upregulated by means of TALE‐VP64 fusion proteins that target the promoters (Therizols et al, 2014). In this system, relocation of the activated genes from the NL toward the nuclear interior was observed by FISH (Therizols et al, 2014). However, it is not known how much of the flanking DNA is involved in this detachment from the NL. We therefore repeated these experiments, but now we employed DamID mapping of lamin B1 interactions. This method has repeatedly been shown to correspond well with FISH microscopy (Guelen et al, 2008; Peric‐Hupkes et al, 2010; Harr et al, 2015; Kind et al, 2015; Robson et al, 2017), but it provides much more detailed maps of NL interactions.
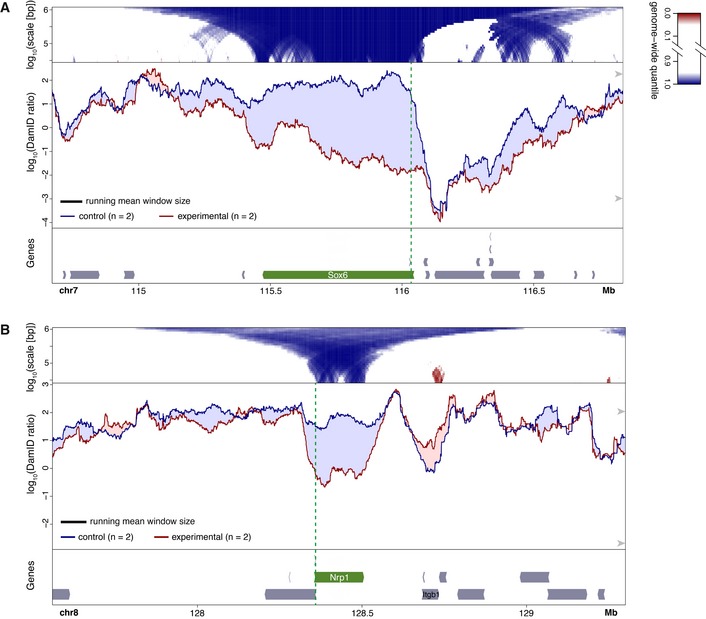
We focused on two previously studied genes, Sox6 and Nrp1 (Therizols et al, 2014). In line with the reported FISH results, we observed clear detachment of each gene from the NL, when activated by the corresponding TALE‐VP64 construct (Fig 1A and B middle panels). To assess the statistical significance of these changes, we compared their magnitude to those observed throughout the remainder of the genome. Because the size of the affected region is a priori not known, we calculated this comparison for various window sizes between about 30 kb and 1 Mb. This resulted in domainograms (de Wit et al, 2008; Tolhuis et al, 2011) that depict the genome‐wide ranking of displacement magnitudes as a function of window position as well as window size (Fig 1A and B top panels; see Appendix Fig S1 for an explanation of domainograms). We regard displacements that rank above the 95th percentile or below the 5th percentile (marked in shades of blue and red for decreased and increased NL interactions, respectively) and that occur locally near the targeted gene, to be highly likely due to direct effects. We note that some indirect displacements elsewhere in the genome may be expected, because the perturbations of Sox6 and Nrp1 may have secondary effects on gene expression.
Figure 1. Changes in NL interactions of Sox6 (A) and Nrp1 (B) in mES cells after activation by TALE‐VP64.
-
A, BBottom panels: gene annotation track (mm10); the activated gene is marked in green and the location of the TALE‐VP64 target sequence is shown by the vertical dashed green line. Middle panels: DamID tracks of NL interactions in control cells (“control”, blue line) and cells expressing TALE‐VP64 (“experimental”, red line). n indicates the number of independent biological replicates that were combined. Noise was suppressed by a running mean filter of indicated window size. Shading between the lines corresponds to the color of the sample with the highest value. Arrowheads on the right‐hand side mark the 5th and 95th percentiles of genome‐wide DamID values. Top panels: domainograms; for every window of indicated size (vertical axis) and centered on a genomic position (horizontal axis), the pixel shade indicates the ranking of the change in DamID score (experimental minus control) in this window compared to the genome‐wide changes in DamID scores across all possible windows of the same size. Blue: DamID score is highest in control samples; red: DamID score is highest in experimental samples (color key on the right of panel (A)). In (A) activation of Sox6 was the experimental perturbation, activation of Nrp1 (which is located on a different chromosome) served as control; in (B) activation of Nrp1 was the experimental condition and activation of Sox6 served as control.
The domainograms indicate that the displacements of the respective targeted genes were among the most extreme throughout the genome. For Sox6, the NL detachment included the entire gene, but it was more pronounced near the promoter than toward the 3′ end (Fig 1A). Upstream of the promoter the detachment extended over ~50–100 kb, up to the LAD border. Downstream of the gene, a modest detachment was observable that tapered off over ~300 kb. Interestingly, about 0.5 Mb upstream, across the LAD border, also some reduction in NL interactions is visible. For Nrp1, the detachment also involved the entire gene body but did not extend much beyond it (Fig 1B). We note that the loss of DamID signal along active transcription units cannot be attributed to the RNA polymerase machinery blocking access to Dam methylation, because all DamID data are normalized to a Dam‐only control that corrects for such accessibility biases (Greil et al, 2006), and the same experimental design has successfully detected interaction of specific proteins with actively transcribed regions (Filion et al, 2010). About 180 kb downstream of Nrp1, the gene Itgb1 showed a modest increase in NL interactions. This will be discussed below. Together, these data show that activation of two genes inside LADs of mES cells results in detachment from the NL along the entire gene body, possibly with some subtler involvement of flanking regions.
Detachment span is linked to transcript length
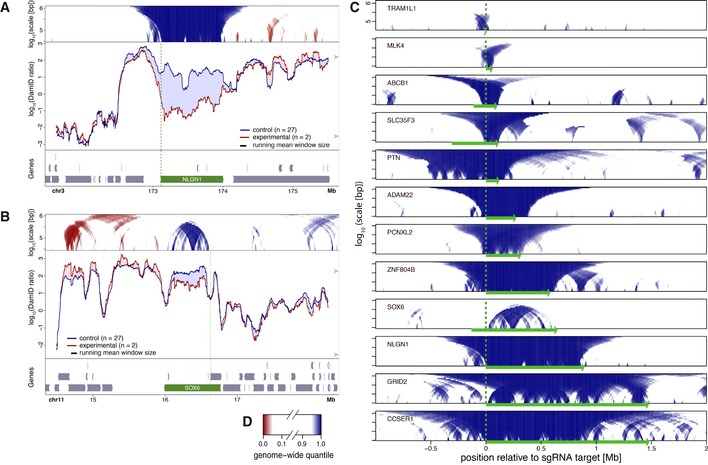
To extend this analysis to a larger number of genes, we switched to a more flexible gene activation system that does not require a custom‐made TALE for every promoter of interest. We chose a previously established human RPE‐1 cell line that stably expresses the SunCas‐CRISPRa system (Tanenbaum et al, 2014; Tame et al, 2017), in which multiple copies of VP64 can be targeted to a promoter of interest by a single sgRNA. We first used this system to activate NLGN1, a gene of 885 kb that is located in a LAD. Transfection with a sgRNA targeting the promoter caused ~80‐fold upregulation (Appendix Fig S2) and resulted in clear detachment from the NL (Fig 2A). Relocalization primarily affected the NLGN1 gene itself, with a mild 5′ to 3′ gradient inside the gene body and gradually decreasing along ~100 kb of flanking DNA. We also activated the SOX6 gene in RPE‐1 cells. Here, we activated one of the known alternative promoters located internally in the gene. Although the magnitude of the detachment was more modest, this gene also showed loss of NL interactions, but only downstream of the activated promoter (Fig 2B).
Figure 2. Local NL detachment caused by gene activation by CRISPRa in human RPE‐1 cells.
-
A, BPlots as in Fig 1, showing changes in lamin B1 DamID signals upon CRISPRa activation of NLGN1 (A) and SOX6 (B). Control cells were treated either without sgRNA or with one of various sgRNAs targeting promoters on different chromosomes. Vertical green dotted lines mark the position of the sgRNA target sequence.
-
CDomainograms showing regions with reduced NL interactions around 12 genes individually activated by CRISPRa. Genomic regions are aligned by the respective sgRNA target positions and oriented so that the activated genes are all transcribed from left to right. Corresponding DamID traces are shown in Figs 2A and B, and 4, 5, EV2, EV4.
-
DColor key of domainograms in (A–C). Increases in NL interactions (red) are not shown in (C).
We applied this analysis in RPE‐1 cells to 12 individual genes (Table 1) for which activation by the SunCas system could be achieved, as determined by RT–qPCR (Appendix Fig S2) or RNA‐seq (Fig EV1). We chose genes of a wide variety of lengths, from ~2 kb to ~1.5 Mb. In three cases (ABCB1, SLC35F3, and SOX6), we targeted a known alternative promoter located in the middle of the gene, instead of the promoter located most 5′. Strikingly, for all 12 activated genes we observed detachment of the entire region extending from the activated promoter to the 3′ end of the gene (Fig 2C). For most of the tested genes, detachment did not extend more than several tens of kb upstream of the activated promoter. A clear exception to this is PTN, which exhibited upstream detachment over nearly 0.5 Mb (see below). Likewise, for most activated genes the detachment did not extend more than 50–100 kb downstream of the 3′ end, although the precise range varied.
Table 1.
Genes and sequences targeted by CRISPRa in RPE‐1 cells
| Target gene | Target sequence | Target site (hg19) |
|---|---|---|
| ABCB1 | GGGCCGGGAGCAGTCATCTG | chr7:87230290‐87230310 |
| ABCB4a | TGCAACGGTAGGCGTTTCCC | chr7: 87105074‐87105093 |
| ADAM22 | CGGGCGACAAGAGCTCGGCA | chr7:87563472‐87563492 |
| CCSER1 | GTGCGCGGAGTGTGACTGTG | chr4:91048592‐91048612 |
| GRID2 | CAAAAGCATCCTGCAGCCTG | chr4:93225024‐93225044 |
| MLK4 | AGGGCGGAATGAACCTGGAG | chr1:233463313‐233463333 |
| NLGN1 | TGAAGGGTCAACCCTCCGCG | chr3:173115478‐173115498 |
| PCNX2 | TCCCTCCTTAGCCTTCGCTG | chr1:233431545‐233431565 |
| PTN | GAGCAGAGGAAAATCCAAAG | chr7:137028354‐137028374 |
| RUNDC3Ba | GCTGCTTTAAAAGGTCCGCG | chr7: 87257590‐87257609 |
| SLC35F3 | TAAAGGGCTTCTCAGAGAGG | chr1:234349808‐234349827 |
| SOX6 | GCTCCCCTCCCAGACAACAC | chr11:16629348‐16629368 |
| TRAM1L1 | AGAATTCAGGAGCATCTTGG | chr4:118006859‐118006879 |
| ZNF804B | AGGCGCGGGTACCCATCGTC | chr7:88388864‐88388884 |
ABCB4 and RUNDC3B were only targeted in combination with ADAM22 and ABCB1.
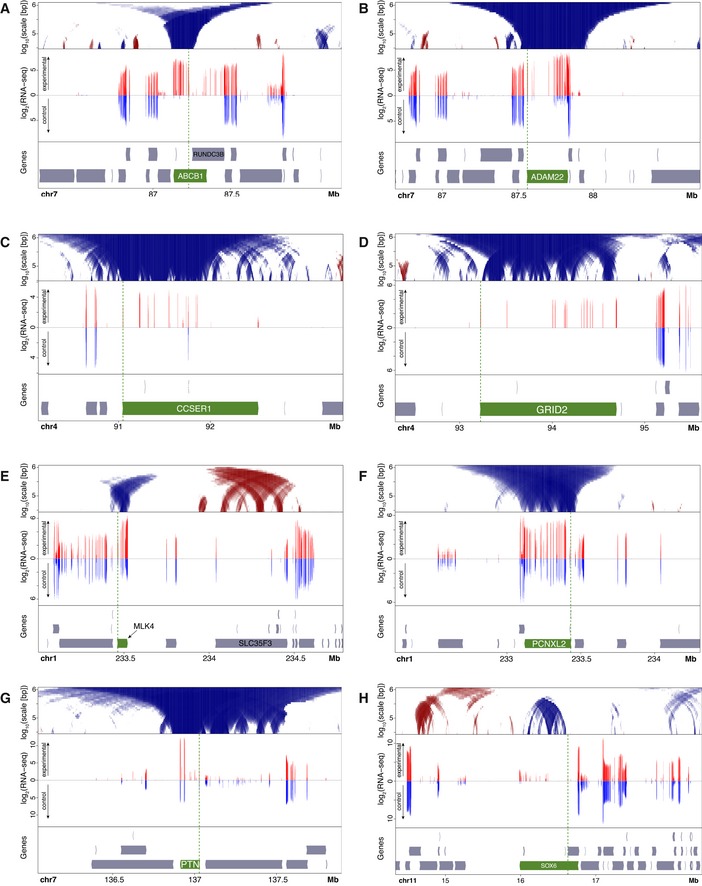
Figure EV1. Expression levels of CRISPRa‐targeted genes and neighboring genes in human RPE‐1 cells.
-
A–HTop panels: domainograms showing changes in NL interactions around genes activated by CRISPRa. Domainograms are same as in Fig 2C. Middle panels: log2 mRNA expression (normalized read counts in 50 bp bins) as determined by mRNA‐seq of CRISPRa‐activated cells (red, upward y‐axis) compared to untransfected control cells (blue, downward y‐axis). Note that only introns give detectable reads. Data are average of two independent replicate experiments each. Bottom panels: gene annotation track. Each CRISPRa‐targeted gene is highlighted in green; targeted promoters are marked by vertical green dotted line.
Quantitative link between gene expression level and NL detachment
We wondered whether the degree of NL detachment of a gene is quantitatively linked to the transcription level. To measure gene activity accurately, we performed RNA‐seq after activation of 8 of the 12 genes and in the untreated parental cell line (Fig EV1). Comparing RNA levels and average DamID scores across the SunCas‐activated genes before and after upregulation revealed a strong negative correlation (Fig 3). Thus, there is a remarkably quantitative inverse link between expression levels and NL interaction frequencies.
Figure 3. Inverse correlation between NL interaction and gene expression level in human RPE‐1 cells.
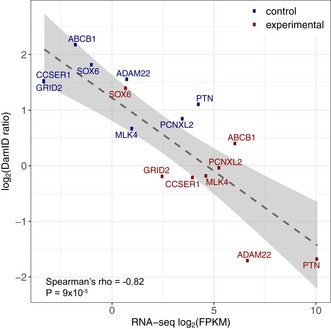
Average DamID values plotted against average expression levels of eight genes activated by CRISPRa (red; n = 2) or in control cells that were treated either without sgRNA or with one of various sgRNAs targeting promoters on different chromosomes (blue; n ranging from 19 to 27).
Neighboring genes of targeted genes do not generally show altered expression
We also queried our RNA‐seq data for neighboring genes of the activated genes. We examined genes within ~1 Mb of our targets and generally could not detect substantial up‐ or downregulation (Fig EV1A–H). A notable exception is the gene RUNDC3B that is partially overlapping and antisense to the activated gene ABCB1. RUNDC3B is co‐activated but does not show measurable detachment from the NL (Fig EV1A). Another, minor, exception is the gene STEAP4 nearby the activated ADAM22 gene (Fig EV1B). In this case, the absolute expression level of this co‐activated gene remained much lower than its CRISPRa‐targeted neighbor. Thus, strong SunCas‐induced upregulation is in most cases restricted to the targeted gene.
Some flanking genes co‐detached from the NL together with the activated gene, but showed no detectable change in expression. The most striking example is the gene DGKI that flanks PTN. Much of this ~0.5 Mb gene shows reduced NL interactions upon activation of PTN, but DGKI does not undergo a detectable upregulation (Fig EV1G). We conclude that CRISPRa activation and the ensuing changes in NL contacts generally do not have substantial effects on the expression of nearby genes.
NL detachment partially overlaps with changes in replication timing
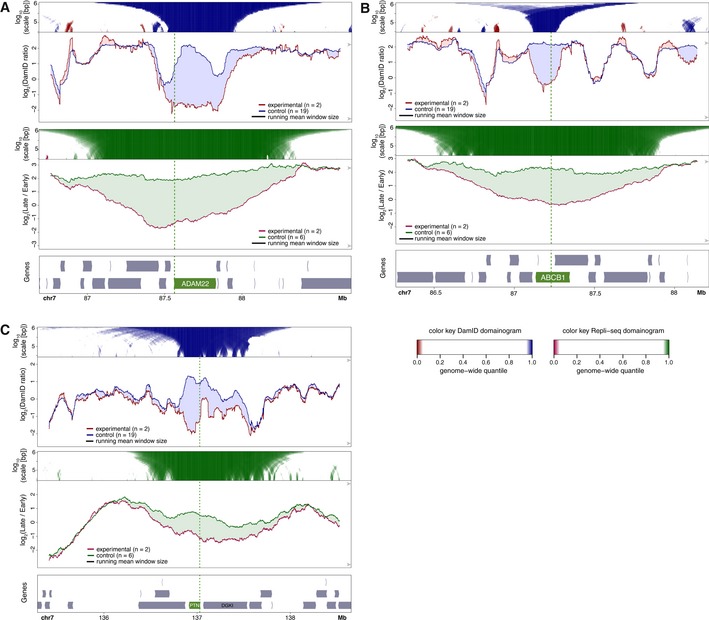
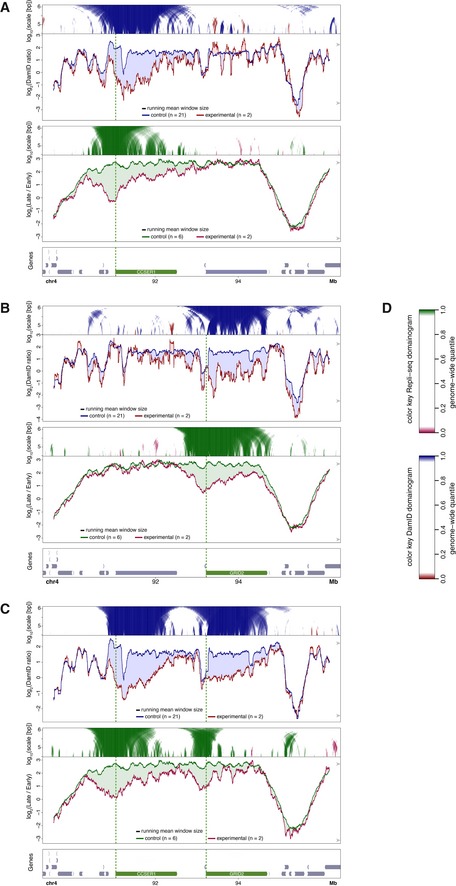
Next, we investigated the link between changes in NL interactions and replication timing. We applied Repli‐seq (Marchal et al, 2018) to visualize replication timing, revisiting five genes that exhibited NL detachment in RPE‐1 cells upon activation by CRISPRa. For all five activated genes, we observed a clear shift toward earlier replication. When the activated genes were relatively small (ADAM22, ABCB1, PTN), this shift was more or less symmetrical around the activated promoter and extended about 0.4–0.8 Mb on each side, i.e., well beyond the activated transcription units and also beyond the changes in NL interactions (Fig 4A–C). With longer activated genes (CCSER1, GRID2, both about 1.5 Mb long), again the shift in replication timing was strongest around the targeted promoter and extended about 0.6 Mb upstream (Fig EV2). Downstream of these promoters, the shift declined gradually toward the end of the gene, similar to the detachment from the NL.
Figure 4. NL interactions and replication timing around activated genes.
-
A–CCRISPRa activation of ADAM22 (A), ABCB1 (B), and PTN (C) in human RPE‐1 cells. Top panels visualize DamID data similar to Fig 2A and B. Middle panels show maps of replication timing at the same resolution and in the same plotting style as panels, except that different colors are used as indicated. Bottom panels show gene track, with activated gene highlighted in green.
Figure EV2. Changes in NL interactions and replication timing in the CCSER1/GRID2 locus.
-
A–DEffects of CRISPRa activation of CCSER1 (A), GRID2 (B) or both genes (C) in human RPE‐1 cells. Top panels show DamID data similar to Fig 2A and B. Middle panels show maps of replication timing at the same resolution and in the same plotting style as for DamID, except that different colors are used as indicated in (D). Bottom panels show gene annotation; activated gene(s) highlighted in green; targeted promoters are marked by vertical green dotted line. DamID data are the same as in Figs 2C and 6A.
We investigated whether the changes in NL interactions and replication timing were linked to topologically associated domains (TADs) as detected by the Hi‐C technology (Dixon et al, 2012; Nora et al, 2012). For this, we analyzed previously reported Hi‐C data from wild‐type RPE‐1 cells (Darrow et al, 2016; Data ref: Darrow et al, 2016). We focused on the PTN locus, which showed the most striking difference between the changes in NL contacts and Repli‐seq patterns after CRISPRa. Interestingly, the change in Repli‐seq pattern appeared not visibly linked to the (pre‐existing) TAD pattern, while the changes in NL interactions showed a partial correlation with the TAD organization (Fig EV3). Together, these results reveal that changes in replication timing only partially overlap with changes in NL interactions (see Discussion).
Figure EV3. Comparison of DamID and replication timing patterns around the PTN gene to Hi‐C data.
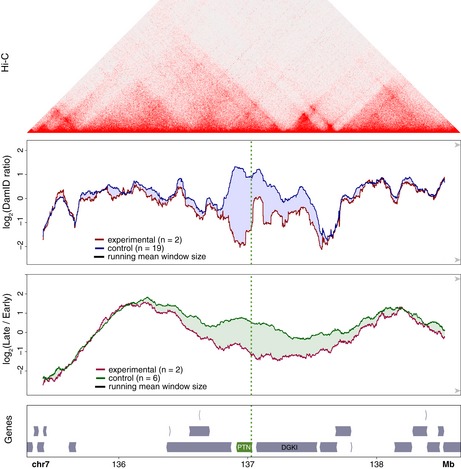
DamID data and gene annotation track of Fig 4C (three bottom panels) aligned to Hi‐C data from untreated human RPE‐1 cells (top panel). Hi‐C data are from (Darrow et al, 2016) (Data ref: Darrow et al, 2016).
Possible compensatory movements
In a few instances, we observed that the loss of NL interactions of the activated gene was accompanied by a gain of NL interactions of a nearby region. This was particularly notable for a region ~0.6 Mb downstream of the activated MLK4 gene (Fig 5). This region coincides approximately with gene SLC35F3. The expression of SLC35F3 is reduced by ~30% (P = 0.02, DESeq2 analysis) when MLK4 was activated (Fig EV1E). Possibly, detachment of MLK4 leads to compensatory movement of SLC35F3 toward the NL, which in turn may contribute to slightly stronger repression of SLC35F3. Forced activation of SLC35F3 caused its own NL detachment as expected (Fig 2C), but it did not alter the NL interactions of MLK4 (Fig EV4A). This suggests that the putative compensatory relationship is not reciprocal, but we note that this latter experiment was done only once and should therefore be interpreted with caution.
Figure 5. Putative compensatory movement near the activated MLK4 gene.
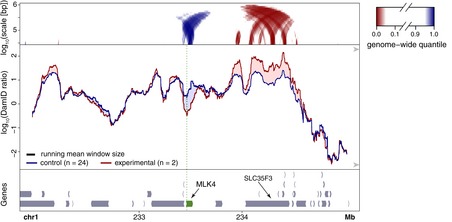
Changes in NL interactions after CRISPRa activation of MLK4 in human RPE‐1 cells. Visualization of DamID data as in Fig 2A and B.
Figure EV4. Changes in NL interactions of genes upregulated by CRISPRa.
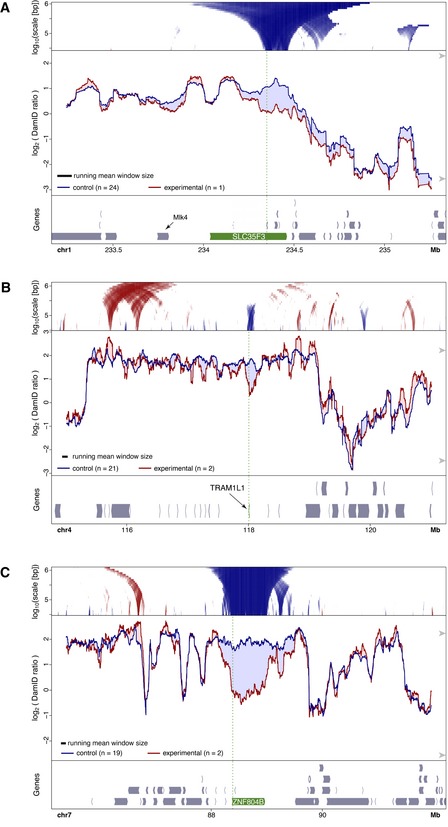
-
A–CDamID data obtained after CRISPRa in human RPE‐1 cells of SLC35F3 (A), TRAM1L1 (B), ZNF804B (C). Domainograms are the same as in Fig 2C, but additionally show increased NL contacts in red.
We also observed moderately enhanced NL interactions of a region ~1.3 Mb downstream of the activated SOX6 gene (Fig 2B). This region coincided with the promoters of two divergent genes that were not significantly up‐ or downregulated (Fig EV1H). Likewise, in case of Nrp1 activated by TALE‐VP64, the gene Itgb1 (about 180 kb downstream of Nrp1) showed a modest increase in NL interactions (Fig 1B), but its expression was not found to be detectably altered by TALE‐VP64 targeting of Nrp1 (Therizols et al, 2014). We found also minor local increases in NL interactions within ~2 Mb of the activated genes NLGN1 (Fig 2A), TRAM1L1 and ZNF804 (Fig EV4B and C). However, because of their modest magnitude, we did not further investigate these movements. In summary, possible compensatory changes in NL interactions around activated genes are relatively modest and may only anecdotally affect gene expression, at least in the cell systems we studied. These increases in NL interactions may reflect compensatory movement to fill up space at the NL vacated by the activated genes, but other secondary mechanisms cannot be ruled out.
Detachment remains local even if multiple genes are activated
We next explored whether it is possible to detach a whole LAD by activating multiple genes in the domain. We first tested this for two neighboring large genes, CCSER1 (1,475 kb) and GRID2 (1,468 kb). Activation of each gene individually caused clear detachment from the NL (Figs 6A and EV2A and B). When both genes were activated simultaneously (by co‐transfection of the respective sgRNAs), both genes detached, but the intervening ~700 kb region showed no significant reduction in NL association (Fig 6A). Under this double‐activating condition, the intervening region also continued to be replicated a bit later in S‐phase than the two activated genes (Fig EV2C).
Figure 6. Effects of activation of multiple neighboring genes on NL interactions.
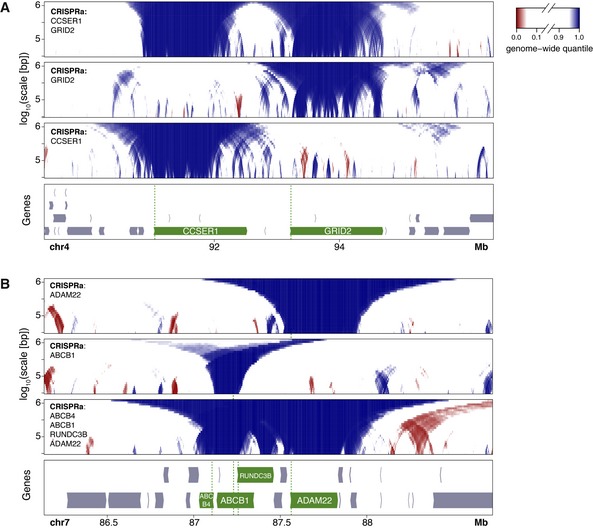
- DamID domainograms of NL interactions after activation of CCSER1, GRID2, or both.
- DamID domainograms after activation of ADAM22, ABCB1, or the genes ABCB4, ABCB1, ADAM22, and RUNC3B simultaneously.
We also applied CRISPRa simultaneously to the much more closely spaced genes ABCB4, ABCB1, ADAM22, and RUNDC3B by co‐transfection of four sgRNAs. All four genes were induced to varying levels (Fig EV5A and B). However, the activity of both ABCB4 and RUNDC3B remained rather low under this quadruple activation condition, compared to their levels after CRISPRa of ABCB1 alone (cf. Fig EV1A). This may be due to competition of the multiple sgRNAs for the available SunCas. We compared the resulting DamID maps to those obtained after activation of ABCB1 or ADAM22 alone (Figs 6B and EV5C–E). While the single gene activations resulted in selective detachment of the respective genes, the quadruple activation caused detachment of each gene, with the degree of detachment roughly corresponding to the activity of each gene after activation (Fig EV5A and E). There was no indication that the observed detachment of the four genes involved a more extensive region than a simple combination of their independent detachments. However, it is noteworthy that the very modest activity of ABCB4 and RUNDC3B after quadruple CRISPRa is sufficient to partially detach these two genes from the NL. This suggests that the detachment of these genes is facilitated by the detachment of the nearby ABCB1 and ADAM22 genes. Together, these results indicate that co‐activation of multiple neighboring genes may lead to more efficient detachment of moderately active genes, but not to a broader detachment of flanking regions.
Figure EV5. Expression and NL interactions of ABCB4,ABCB1,RUNDC3B, and ADAM22 after combined activation.
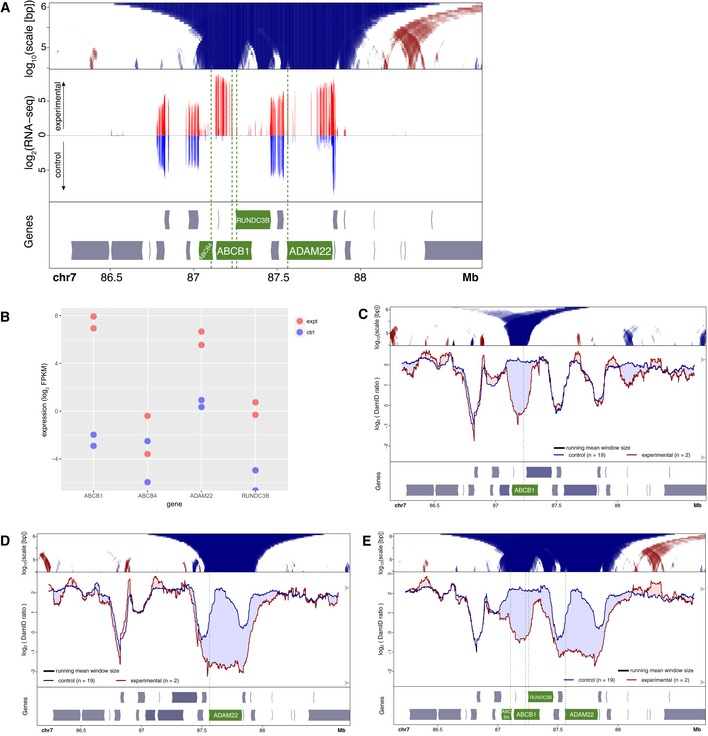
- Visualization of mRNA expression (middle panel), together with DamID domainogram (top panel) and gene annotation track (bottom panel) in human RPE‐1 cells. CRISPRa was done using four sgRNAs simultaneously, one for each promoter. Middle panel shows log2 mRNA expression (normalized read counts in 50 bp bins) as determined by mRNA‐seq of CRISPRa‐activated cells (red, upward y‐axis) compared to untransfected control cells (blue, downward y‐axis). Note that only introns give detectable reads. Data are average of two independent replicate experiments each.
- Quantification of mRNA expression of the four genes, same mRNA‐seq data as in (A).
- DamID profiles after activation of ABCB1 (C), ADAM22 (D), or ABCB4, ABCB1, ADAM22, and RUNDC3B simultaneously (E). Data visualization as in Fig 2A and B.
Inactivation of genes can promote NL interactions
Our observations so far strongly suggested that the act of transcription is a driving force that localizes genes to the nuclear interior. To test this further, we set out to block transcription by two complementary genetic strategies.
In the first strategy, we aimed to disrupt all transcription in an inter‐LAD region (iLAD), to test whether this would lead to increased NL interactions of the entire region. We focused on the mouse genes Dppa2, Dppa4, Morc1, and Morc (a shorter form of Morc1 that initiates from an alternative transcription start site). In ES cells, these genes are localized in an approximately 500 kb‐sized iLAD. However, this region is NL‐associated in mouse neural precursor cells (Peric‐Hupkes et al, 2010), and therefore, it has the potential to become a LAD. We used recently reported (Sima et al, 2019) F1 hybrid Cast/129Sv mES cell clones (named E2 and A6) with a heterozygous triple deletion of the promoters of DppA2, Morc1, and Morc on the 129Sv‐derived chr16. These deletions also stop transcription of the Dppa4 gene and therefore essentially abolish transcriptional activity in the whole iLAD (Sima et al, 2019). Owing to the high density of SNPs that differ between the 129Sv and Cast genomes, we could generate allele‐specific DamID maps, enabling us to compare the mutated and wild‐type chromosomes.
DamID profiles of the locus revealed that Morc1 and Morc on the mutated chromosome had moved toward the NL in the mutant cells, when compared to control cells carrying an unrelated mutation on a different chromosome (Fig 7A). This effect was not observed for the wild‐type Cast chr16 in the same cells (Fig 7B). Interestingly, the region containing Dppa2 and Dppa4 was unaffected and clearly remained detached from the NL. This suggests that a transcription‐independent detachment mechanism may exist in addition to a transcription‐linked mechanism. To determine whether ablation of the most prominent transcript would be sufficient to induce attachment, we also tested a single deletion of the Morc promoter (clones A12 and B11), which reduces transcription of Morc1 by ~2‐fold and presumably ablates expression of Morc, but does not alter expression of Dppa2 and Dppa4 (Sima et al, 2019). In the mutated 129Sv‐derived locus, this perturbation resulted in a more restricted increase in NL interactions of Morc while the 5′ end of Morc1 was much less affected (Fig 7C). Again, in the wild‐type Cast‐derived locus only minor changes were observed between mutated and control clones (Fig 7D). These data show that inactivation of one or more genes in a facultative iLAD can lead in cis to locally increased NL interactions of the inactivated genes.
Figure 7. Increased NL interactions upon allele‐specific transcription inactivation in F1 hybrid mES cells.
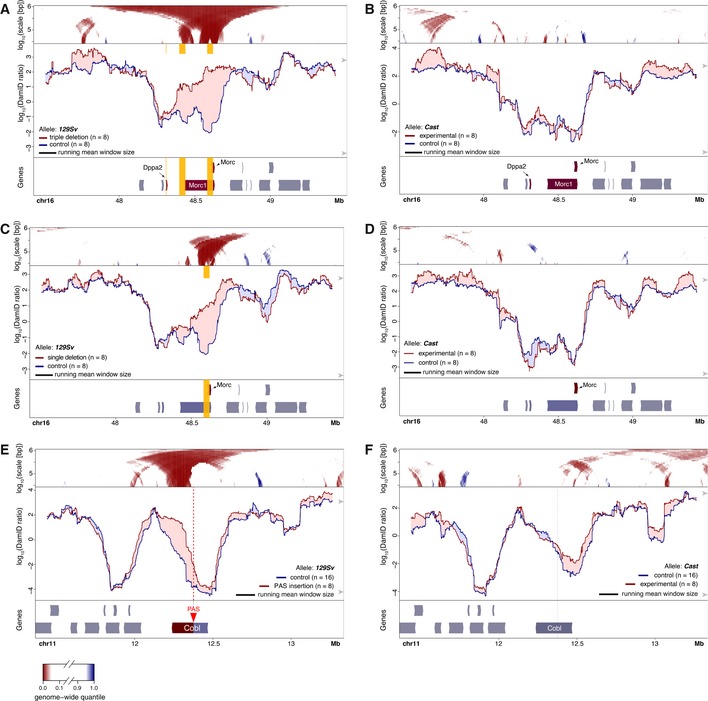
-
ADamID profiles of the 129Sv allele of the Morc1 locus with deletions of the promoters of Morc, Morc1, and DppA2 (deletions marked by yellow vertical boxes) and in control cells.
-
BSame as (A), but for the non‐mutated Cast allele.
-
C, DSame design as (A, B), but with only a single mono‐allelic deletion of the Morc promoter (vertical yellow box).
-
E, FEffect of PAS insertion on NL interactions of Cobl gene locus. Same design as (A, B) but with insertion of a PAS (located at red triangle and vertical dotted line) that truncates the 129Sv allele of the Cobl transcription unit. 129Sv allele is shown in (E), non‐mutated Cast allele in (F).
In the second genetic approach, we aimed to truncate a single transcript, to test directly whether transcription elongation is required for detachment from the NL. We chose the 228 kb Cobl gene, which is active and locally detached from the NL in mES cells but inactive and NL‐associated in neuronal precursor cells (NPCs), indicating that its detached state is facultative and linked to transcription. We created a heterozygous truncation of the Cobl transcription unit in F1 hybrid Cast/129 mES cells by insertion of a polyadenylation sequence (PAS) in the 129 allele of Cobl, 89 kb downstream of the TSS. Analysis of Cobl allelic sequence variants in mRNA‐seq data confirmed the premature termination of transcription at the 129Sv allele (Appendix Fig S3). Allele‐specific DamID profiles show increased NL interactions of the 129Sv allele of Cobl, particularly downstream of the PAS integration (Fig 7E). This did not occur at the unmodified CAST allele, although some modest changes in NL interactions were detected in the surrounding region (Fig 7F). We conclude that blocking of Cobl transcription elongation causes local increases in NL interactions.
Insertion of a small active gene causes moderate detachment from the NL
Finally, complementary to the activation and inactivation of genes in their native context, we tested whether insertion of a highly active transgene into a LAD is sufficient to cause local detachment from the NL. For this purpose, we designed an expression cassette consisting of a transcription unit encoding enhanced green fluorescent protein (eGFP) driven by the strong human PGK promoter, cloned into a PiggyBac transposable element vector (Fig 8A). We integrated this cassette randomly in the genome of F1 hybrid mouse ES cells by co‐transfection with PiggyBac transposase. We then isolated clonal cell lines and focused on two with a large number of integrations, reasoning that by random chance several integrations would occur inside LADs. Indeed, by inverse PCR and Tn5 mapping (see Materials and Methods) we found 17 uniquely mappable integrations to be inserted inside LADs, out of a total of 80 in the two cell clones combined (Fig 8B). In comparison with the corresponding wild‐type alleles in the same cells, a roughly twofold reduction in average DamID signal was detected around the integration sites, spanning approximately 20 kb on each side (Fig 8C). We conclude that the integrated transcription units tend to detach the directly flanking DNA from the NL, but only partially and within a range of roughly 20 kb.
Figure 8. Effects of a highly active integrated transgene on NL interactions in mES cells.
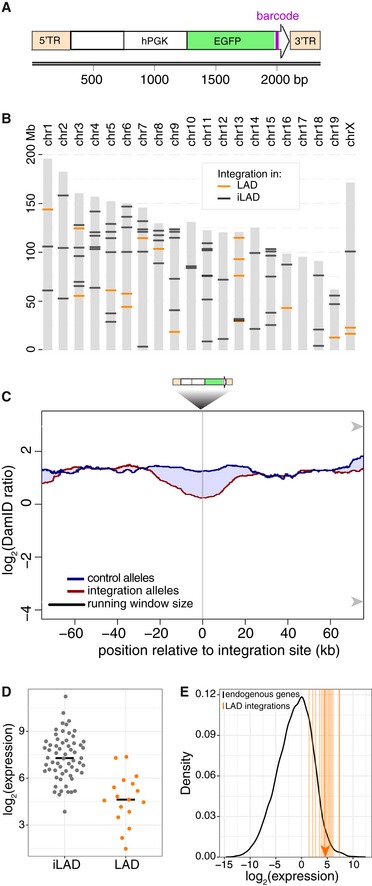
- Design of the transgene construct, consisting of an enhanced green fluorescent protein (EGFP) transcript, marked at its 3′ end by a random barcode (purple bar) and driven by the human PGK promoter. The construct is flanked by the terminal repeats (5′TR and 3′TR) of the Piggybac transposon that are used for random integration in the genome.
- Summary of the mapped locations of integrations in the genomes of two F1 hybrid mES cell clonal cell lines. LAD and iLAD integrations are shown in orange and black, respectively.
- Average DamID profiles across 17 transgene integration sites inside LADs. Blue curve shows alleles without integrations; red curve shows the corresponding alleles with integrations. Shading between the lines shows which curve has the highest value.
- Relative expression levels of individual barcoded transgenes in LADs and iLADs.
- Estimated expression levels of integrated transgenes in LADs (vertical orange lines; median value indicated by arrowhead), compared to the distribution of expression levels of all active endogenous genes (black curve).
We considered the possibility that our expression cassette was not strong enough to cause more pronounced or extended detachments from the NL. To determine the expression level relative to endogenous genes, we performed RNA‐seq and used the barcodes to estimate the expression levels of individual integrations (see Materials and Methods). Transcriptional activity was readily detectable for LAD integrations, although their median expression was about eightfold lower compared to the iLAD integrations (Fig 8D). However, the median expression level of the integrated transgenes inside LADs still ranks approximately in the upper 97th percentile of all active endogenous genes (Fig 8E). Thus, even within LADs, most of the integrated transgenes are expressed at very high levels. These expression levels can be sufficient to reduce NL interactions, but only moderately and locally.
Discussion
Evidence that components of the transcription machinery can affect the spatial organization of the genome is accumulating, but the underlying processes are still poorly understood (van Steensel & Furlong, 2019; Vermunt et al, 2019). The data presented here consistently show that activation of genes in LADs leads to detachment from the NL and conversely that inactivation can lead to increased NL contacts. Moreover, the results point to a remarkable flexibility of the chromatin fiber, allowing for the repositioning of individual genes without much effect on flanking DNA.
Several of our results point to a role for transcription elongation in counteracting NL interactions. First, activity‐induced detachment from the NL generally extends across the entire activated transcription unit, from the activated promoter until the 3′ end of the gene. We observed this for a wide range of gene sizes. A role for elongation is also strongly supported by premature termination of the active Cobl gene by insertion of a PAS, which primarily caused an increase of NL interactions downstream but not upstream of the new termination site. These results are consistent with a study of the ThymoD non‐coding RNA gene in mouse T‐cell progenitors, where insertion of a PAS prevented detachment from the NL as observed by FISH (Isoda et al, 2017). Conversely, read‐through transcription into heterochromatin, elicited by influenza virus NS1 protein, was found to cause relocation from the heterochromatic compartment “B” to the euchromatic compartment “A” (Heinz et al, 2018), which largely correspond to LADs and iLADs, respectively (van Steensel & Belmont, 2017). How transcription elongation may prevent NL interactions remains to be elucidated. It could be a physical effect, for example when a transcribed gene is tethered to a structure in the nuclear interior. It may also be a biochemical effect, such as the removal of particular NL‐interacting chromatin proteins by the elongating RNA polymerase complex. We note that our dataset is skewed toward long genes (median length: 366 kb, n = 14) compared to the average genome‐wide gene length (which is about 10–15 kb); we cannot rule out that smaller genes often behave differently, although our results for TRAM1L1 and the transgene insertions (both ~2 kb) suggest that transcription‐induced detachment of smaller genes from the NL is also mostly limited to a region of several tens of kb around the transcription unit.
Earlier work found that VP16‐induced movement of a LacO repeat toward the nuclear interior could not be blocked by elongation inhibitors (Chuang et al, 2006). This is not necessarily contradictory to our evidence that supports a role for elongation; it is possible that a transcription activator like VP16 also promotes detachment from the NL independently of transcription elongation. In support of such an additional mechanism, some of the genes that we studied (e.g., Figs 1A and 2A, and 6A) showed the strongest loss of NL interactions near their 5′ end. Similarly, a class of naturally active genes inside LADs exhibits more prominent detachment of their TSS compared to the downstream transcription units (preprint: Luperchio et al, 2017; Wu & Yao, 2017; Leemans et al, 2019). Furthermore, global tethering of VP64 across all LADs caused virtually no changes in transcription yet triggered loosening of LAD‐NL interactions (Kind et al, 2013), underscoring that VP16 can counteract NL interactions without activating transcription.
That detachment from the NL can involve non‐transcribed regions is also suggested by our study of the PTN gene. CRISPRa of this gene causes detachment that extends several hundred kb into the neighboring DGKI gene, even though the latter gene is not detectably activated. Furthermore, the transcriptionally inactivated Dppa2/4 genes remained mostly dissociated from the NL (Fig 7A). In two earlier reports, relocation from the NL to the nuclear interior was also achieved by tethering of an artificial peptide that induces chromatin decondensation without detectable recruitment of RNA polymerase II (Chuang et al, 2006; Therizols et al, 2014). It is not understood how this peptide (which is not derived from a naturally occurring protein) exerts this effect, but it suggests that a transcription‐independent mechanism of relocation exists in addition to transcription‐linked mechanisms. Recent evidence suggests that active chromatin marks such as H3K27ac deposited by p300 may counteract NL interactions (Cabianca et al, 2019).
In most cases, the transcriptionally inactive regions adjacent to our activated genes remain relatively unaffected in their NL contacts. Conversely, inhibition of transcription (either by deleting promoters or by insertion of a PAS) leads to increased NL interactions in a very local manner. The latter results were obtained in genomic regions that are facultative LADs, i.e., they may have an intrinsic ability to interact with the NL in the absence of transcription. Genes in constitutive iLADs may lack this ability, either due to spatial constraints or because they lack certain sequence features or chromatin characteristics. It will be of interest to further dissect the molecular mechanisms that underlie the apparent competition between forces that tether chromatin to the NL and forces (such as transcription elongation) that counteract these interactions.
These and previously reported data (Gonzalez‐Sandoval & Gasser, 2016; van Steensel & Belmont, 2017; de Leeuw et al, 2018; Kim et al, 2019; Lochs et al, 2019) together suggest a balancing act between transcription and LADs: For many genes, LADs pose a repressive environment (Leemans et al, 2019). This, however, may be overcome by strong transcription activators. Once transcription is active, it causes detachment of the gene from the NL. Possibly this helps to reinforce the active state. It was previously found that a transiently activated gene can remain detached from the NL for several days after the activation signal has subsided (Therizols et al, 2014).
We observed changes from late to early replication timing for all five upregulated genes that we assayed by Repli‐Seq. The regions that exhibit shifts in replication timing are roughly 1–2 Mb long, matching in size with the usual span of replication domains observed in vivo (Hiratani et al, 2008; Pope et al, 2014). This suggests that there may be a fundamental minimal size of such domains. This is different from the regions that change NL interactions, which can be smaller. Indeed, the overlap between changes in NL interactions and changes in replication timing, while substantial, was imperfect in most instances. The shifts in replication timing tended to involve a larger region than the shift in NL interactions and were centered around the targeted promoters rather than the entire transcription unit. An exception to this is the PTN locus, where changes in NL interactions and replication timing roughly coincide; this may be due to the putative transcription‐independent mechanism discussed above. Together, these results suggest that both NL interactions and replication timing can be modulated by the transcription machinery, but elongation appears to play a more prominent role in counteracting NL interactions, while a signal emanating from activated promoters may evoke a change in replication timing. These distinct but closely linked mechanisms may explain why LADs and late‐replicating domains overlap strongly but imperfectly. Together, the large datasets presented here provide a wealth of information on the spatial rewiring of chromosomes in response to transcription activation or inactivation.
Materials and Methods
Cell culture
The RPE‐1 cell line stably expressing SunTag‐CRISPRa (Tame et al, 2017) was kindly provided by the R. Medema lab (Netherlands Cancer Institute, Amsterdam) and cultured in DMEM‐F12 supplemented with 10% FCS. F121‐9 mES cells were kindly provided by J. Gribnau (Erasmus Medical Center, Rotterdam, the Netherlands) and cultured in feeder‐free 2i medium according to the 4D Nucleome protocol (https://data.4dnucleome.org/protocols/cb03c0c6-4ba6-4bbe-9210-c430ee4fdb2c/).
Primers
Primer sequences are listed in Tables 2 and 3.
Table 2.
Miscellaneous primers used
| Oligonucleotide ID | Sequence (5′–3′) |
|---|---|
| lb877 | GACATGGTGCTTGTTGTCCTC |
| lb982 | CTGAGAACGCAGAAGGCTGT |
| lb991 | CTGTTGTCCCACGCATACAG |
| lb1010 | CTGGACCCACCAACTTTGTGG |
| EB66 | CGACAACCACTACCTGAGCA |
| EB38 | CGAACTCCAGCAGGACCATGT |
| JOYC231 | CTCCACTTCCCTCCACCTCT |
| JOYC232 | GAGAGCTTGAACGAAAAACCA |
| lb563_enrichm_R | CATTGACAAGCACGCCTCAC |
| lb564_enrichm_F | TAAACCTCGATATACAGACC |
| lb565_Badapter_ME_5TR_r | GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGCAATTTTACGCAGACTATCTTTCTAG |
| lb566_Badapter_ME_3TR_f | GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGTACGTCACAATATGATTATCTTTCTAG |
Table 3.
Primers for RT–qPCR
| Target gene | Oligonucleotide ID | Sequence (5′–3′) |
|---|---|---|
| ABCB1 | lb667_ABCB1_hs_qPCR_f | CAGTTGAGTGGTGGGCAGAA |
| lb668_ABCB1_hs_qPCR_r | GCCTTATCCAGAGCCACCTG | |
| ZNF804B | lb669_ZNF804B_hs_qPCR_f | GCAATCTGAATGTGTTTCTGGA |
| lb670_ZNF804B_hs_qPCR_r | ATTCCTTGCTGGAGTTGCT | |
| PTN | lb671_PTN_hs_qPCR_f | CCCAAACCTCAAGAGAAGG |
| lb672_PTN_hs_qPCR_r | ACCATCTTCTCAAACTCTCC | |
| SOX6 | lb673_SOX6_hs_qPCR_f | TACCAACACTTGTCAGTACCA |
| lb674_SOX6_hs_qPCR_r | TCTCTGATTCCATTCTTTGCTG | |
| TRAM1L1 | lb675_TRAM1L1_hs_qPCR_f | TCACTGTTGGGTTTCACCT |
| lb676_TRAM1L1_hs_qPCR_r | TTTCCAGTAAGGGCATCAG | |
| NLGN1 | lb817_qPCR_NLGN1_hs_f | GGTTTCTTGAGTACAGGCG |
| lb818_qPCR_NLGN1_hs_r | TGTATGAGATCAAGGAGTCCA | |
| MLK4 | lb821_qPCR_KIAA1804_hs_f | GAGGAAGGGCAAGTTTAAGAG |
| lb822_qPCR_KIAA1804_hs_r | TTGTGCTGGAAATCTGAAGG | |
| SLC35F3 | lb823_qPCR_SLC35F3_hs_f | TTGCCGTTACATATCCCAC |
| lb824_qPCR_SLC35F3_hs_r | TGGTGTAGTGATCAATCACTG | |
| ADAM22 | lb829_qPCR_ADAM22_hs_f | GTTACTACCAGGGCCATATCC |
| lb830_qPCR_ADAM22_hs_r | AGAACATCCCATGAAGTCCG |
TALE‐VP64 experiments
TALE‐VP64 constructs (Therizols et al, 2014) with Puro resistance marker were kindly provided by Pierre Therizols. The Sox6 TALE target coordinate is chr7: 116034554–116034570, and the Nrp1 TALE target coordinate is chr8:128358929–128358945 (mm10 coordinates).
F121‐9 cells were transfected with TALE‐VP64 constructs targeting Nrp1 or Sox2 by electroporation using Lonza Mouse Embryonic Stem Cell Nucleofector™ Kit (VPH‐1001) according to the manufacturer's instructions. Cells were selected with Puromycin (1 μg/μl) for 1 week to obtain stable polyclonal cell pools.
CRISPRa experiments
sgRNAs were cloned into LentiGuide‐Puro vector (Addgene #52963) using restriction enzyme BsmBI, and lentivirus was prepared. RPE‐1 cells stably expressing SunTag‐CRISPRa were infected with LentiGuide virus and selected with 10 μg/μl puromycin for 1 week to obtain stable polyclonal cell pools.
PAS integration
A PAS was inserted by in‐frame integration of a blasticidin resistance (BlastR) cassette followed by the PAS into the Cobl gene. For this purpose, sgRNA sequence AGTCATCTGTGCGAAGTGTG was cloned into Blast‐TIA vector (Lackner et al, 2015) (kindly supplied by the Brummelkamp lab, Netherlands Cancer Institute) via BbsI restriction digestion. Cells were transfected with the resulting Blast‐TIA vector co‐transfected with Cas9 expression vector pX330 (Addgene #42230) by nucleofection and subjected to selection by culturing in the presence of 10 μg/μl blasticidin for 1 week. Clones were picked and screened for correct integration of the BlastR cassette by PCR with primers lb877 and lb982. Heterozygosity of the integration was confirmed by PCR using primers lb982 and lb991, and the 129Sv allele was identified as the targeted allele by PCR using primers lb877 and lb1010, followed by Sanger sequencing with the same primers.
Repli‐seq
Repli‐seq was performed as described (Marchal et al, 2018). Sequencing was done on a NovaSeq 6000 system (Illumina), 50‐bp read length.
mRNA‐seq
As previously described (Gogola et al, 2018), mRNA‐seq was performed as follows. Quality and quantity of the total RNA were assessed by the 2100 Bioanalyzer using a Nano chip (Agilent, Santa Clara, CA). Total RNA samples having RIN>8 were subjected to library generation. Strand‐specific libraries were generated using the TruSeq Stranded mRNA sample preparation kit (Illumina Inc., San Diego, RS‐122‐2101/2) according to the manufacturer's instructions (Illumina, Part # 15031047 Rev. E). Briefly, polyadenylated RNA from intact total RNA was purified using oligo‐dT beads. Following purification, the RNA was fragmented, random‐primed, and reverse‐transcribed using SuperScript II Reverse Transcriptase (Invitrogen, part # 18064‐014) with the addition of actinomycin D. Second‐strand synthesis was performed using polymerase I and RNaseH with replacement of dTTP for dUTP. The generated cDNA fragments were 3′ end adenylated and ligated to Illumina paired‐end sequencing adapters and subsequently amplified by 12 cycles of PCR. The libraries were analyzed on a 2100 Bioanalyzer using a 7500 chip (Agilent, Santa Clara, CA), diluted and pooled equimolar into a multiplex sequencing pool, and stored at −20°C. The libraries were sequenced with 65 base single reads on a HiSeq2500 using V4 chemistry (Illumina Inc., San Diego). Reads were aligned to hg19 or mm10 using TopHat version 2.1, with Ensembl genome build 75. For Cobl PAS integration experiments in the F1 hybrid ES cells, mRNA‐seq reads were aligned to mm10 using STAR (Dobin et al, 2013).
RT–qPCR
Cells were collected in TRIsure and total RNA was extracted using PureLink RNA Mini Kit (Thermo Fisher Scientific) according to the manufacturer's instructions. RNA was reverse‐transcribed using Tetro Reverse Transcriptase (Bioline) with Oligo(dT)20 primers (Thermo Fisher Scientific) according to the manufacturer's instructions. qPCR was performed using SensiFast no‐ROX mix (Bioline) in a 10 μl reaction. Primers are listed in Table 3.
Generation and mapping of random integrations
The hPGK‐EGFP cassette was derived from TRIP vector pPTK‐Gal4‐mPGK‐Puro‐IRES‐eGFP‐sNRP‐pA (Akhtar et al, 2014) by replacing mPGK‐Puro‐IRES with the human PGK promoter using restriction enzyme cloning with SalI and NcoI. Generation of a barcoded plasmid pool and integration into F121‐9 mES cells was performed as described (Akhtar et al, 2014). Clones with high EGFP expression were sorted by FACS and screened for high integration copy number by qPCR with EGFP‐specific primers EB66 and EB38, using Lbr‐specific primers (JOYC231 and JOYC232) for normalization.
Mapping of integrations without linking to barcodes was done by Tagmentation as described (preprint: Stern, 2017) with minor modifications: Before PCR for Tn5 adapters, linear amplification of PiggyBac integrations was performed using primers lb565 or lb566 for mapping in reverse or forward orientation respectively. Linear amplification was performed using 0.5 U Phusion polymerase (Bioline) in a 20 μl reaction with Phusion GC‐rich buffer, 1 mM dNTPs, 50 nM primer. Reaction was incubated at 98°C for 30 s, then 45 cycles of 98°C for 8 s, 60°C for 5 s and 72°C for 30 s followed by a final step at 72°C for 20 s. For PCR amplification, PiggyBac‐specific primers lb565 or 566 were used for mapping in reverse or forward orientation, respectively.
To process the tagmentation mapping reads, the Tn5 adaptor sequence and PiggyBac primer sequence at the ends of the paired‐end reads were removed using an adaptation of cutadapt v1.11. The genomic part of the sequence was mapped to strain‐specific versions of GRCm38 release 68 from Ensembl using bowtie v2.3.4.1 with mapping set to “very‐sensitive”. To create these strain‐specific genomes, SNP information was downloaded from the Mouse Genomes Project (Keane et al, 2011; http://www.sanger.ac.uk/science/data/mouse-genomes-project) as VCF files “CAST_EiJ.mgp.v5.snps.dbSNP142.vcf.gz” and “129S1_SvImJ.mgp.v5.snps.dbSNP142.vcf.gz” (version 1 May 2015). Bcftools was used to incorporate all SNPs into the GRCm38 reference genome. After mapping to strain‐specific genomes, bam files were compared, and for each read, the alignment with the highest alignment score (AS) was used. When the AS was identical, a random choice was made. Read‐pairs aligning in opposite orientation and <1,500 bp apart were converted to genomic regions using the bamToBed from bedtools and awk, covering both reads as well as the region in between. Genomecov from bedtools and awk was used to combine regions and calculate coverage. Integration sites were called by combining regions from PCRs from both transposon arms using closest from bedtools and awk. Regions on opposite strands that were at most 5 bp apart were regarded to represent an integration. Next, the allele of the integration was determined by using mpileup from Samtools (Li et al, 2009) v1.5 with a maximum depth of 50 to count the number of mismatch positions over the complete region compared to both of the strain‐specific GRCm38 modifications. Each position with the allele of the strain‐specific genome occurring in a ratio < 0.5 was considered a mismatch position. The allele with the lowest number of mismatch positions was then considered the allele of integration. In case of equal number of mismatch positions, the integration allele was classified as ambiguous. We selected only putative integration sites with at least 1 read having a mapping quality > 10 and > 500 reads mapped on both sides of it.
In addition, allele‐specific mapping of the integrations and linking to their barcodes was performed by inverse PCR as described (Akhtar et al, 2014), except that the mapping of reads was confined to regions that were initially found by tagmentation mapping. Tagmentation alone identified 50 integrations for clone CM1407 and 56 for clone CM1420, of which 37 and 43 were linked to a single, unique barcode, respectively.
Expression analysis of ES cell clones with random integrations
Clones CM1407 and CM1420 were subjected to mRNA‐seq as above. For comparison between eGFP and endogenous mRNA expression, a fasta entry for eGFP was added to the mouse genome version mm10 chromosomes 1‐19, X, Y, and M without alternative contigs. The annotation of eGFP transcript was also added to gencode version M18. STAR (Dobin et al, 2013) version 2.6.0c was used to align the cDNA reads to this modified reference genome, and for each transcript, reads were counted. DESeq2 (Love et al, 2014) was used to calculate fragments per kilobase million (FPKM) values for each gene, including eGFP.
In addition, barcode‐specific expression was determined using sequencing of the barcodes in cDNA similar to the standard TRIP protocol (Akhtar et al, 2014). To discriminate between genuine barcodes and sequencing errors of these barcodes, starcode (Zorita et al, 2015) was used. Unlike the standard TRIP protocol, reads were not normalized by gDNA counts. Finally, the eGFP FPKM was scaled by the number of integrations for each clone in order to determine the average eGFP expression per integrated reporter. These numbers were 52 for clone CM1417 and 55 for clone CM1420; barcodes were counted 2 times when found at 2 integration sites.
Lamina‐associated domains coordinates in mouse ES cells were obtained from Peric‐Hupkes et al (2010) and adjusted to mm10 using the LiftOver tool (Hinrichs et al, 2006). To estimate FPKM's for integrations in LADs and iLADs separately, barcodes were used that had a unique location according to a combination of iPCR and tagmentation mapping. In total, 17 barcodes could be confidently linked to LAD locations. To determine LAD and iLAD‐specific expression levels, the average eGFP FPKM per integration was scaled by the median LAD and iLAD expression. Finally, the percentile of eGFP FPKM relative to endogenous active genes was calculated by counting the number of genes with higher FPKM than the eGFP estimation, divided by the total number of active genes (defined as genes with FPKM > 0).
DamID‐seq
DamID‐seq was performed as described (Brueckner et al, 2016) with minor modifications. Dam fused to human LMNB1 protein (Dam‐LMNB1) or unfused Dam were expressed in cells by lentiviral transduction (Vogel et al, 2007). Three days after infection, cells were collected for genomic DNA (gDNA) isolation. gDNA was pre‐treated with SAP (10 U, New England Biolabs #M0371S) in CutSmart buffer in a total volume of 10 μl at 37°C for 1 h, followed by heat inactivation at 65°C for 20 min to suppress signal from apoptotic fragments. This gDNA was then digested with DpnI (10 U, New England Biolabs #R0176L) in CutSmart buffer in a total volume of 10 μl at 37°C for 8 h followed by heat inactivation at 80°C for 20 min. Fragments were ligated to 12.5 pmol DamID adapters using T4 ligase (2.5 U, Roche #10799009001) in T4 ligase buffer in a total volume of 20 μl incubated at 16°C for 16 h. The reaction was heat‐inactivated for 10 min at 65°C. Products were then digested with DpnII to destroy partially methylated fragments. DpnII buffer and DpnII (10 U, New England Biolabs #R0543L) were added in a total volume of 50 μl and incubated at 37°C for 1 h. Next, 8 μl of DpnII‐digested products was amplified by PCR with MyTaq Red Mix (Bioline #BIO‐25044) and 1.25 μM primers Adr‐PCR‐Rand1 in a total volume of 40 μl. PCR settings were 8 min at 72°C (1×) followed by 20 s at 94°C, 30 s at 58°C, 20 s at 72°C (24× for Dam, 28× for Dam‐LMNB1 samples) and 2 min at 72°C (1×). Remaining steps were performed as previously described. Samples were sequenced on an Illumina HiSeq2500.
Processing of RPE‐1 and ES cell DamID data
First, the constant DamID adapter was trimmed from the 65‐bp single‐end reads using cutadapt (Martin, 2011) version 1.11 and custom scripts. The remaining sequence starting with GATC was mapped to hg19 with bowtie2 (Langmead & Salzberg, 2012) version 2.2.6. Uniquely mapped reads (filtered for bowtie's XS‐tag) were then assigned to individual gDNA sequences between two GATC motifs (referred to as GATC fragments), which are the units of further data processing and analysis because Dam‐only methylates GATC motifs. Further processing and analysis were done in R (R Core Team, 2017) versions 3.4–3.6 using Bioconductor (Huber et al, 2015), in particular the packages GenomicRanges (Lawrence et al, 2013) and Sushi (Phanstiel et al, 2014).
Replicate experiments were combined by summing the reads for each GATC fragment. Hence, experiments with more reads were weighed proportionally stronger than experiments with fewer reads. Extremely high read counts of individual GATC fragments (> 100 times the genome‐wide average) were assumed to be due to PCR artifacts; these read counts were replaced with the genome‐wide average read count. Next, smoothing was applied by summing read counts over a running window of 201 consecutive GATC fragments. A pseudocount of 30 was added to each window. The ratio Dam‐lamin B1/Dam‐only was calculated for each window and log2‐transformed. Finally, the log2 ratios were normalized by subtracting the genome‐wide average log2 ratio.
When comparing experimental and control DamID log ratios in genome‐wide scatterplots, we noticed modest systematic biases and skews (visible as point clouds that were somewhat banana‐shaped rather than cigar‐shaped) or differences in the dynamic ranges of the DamID values that are likely to be of technical nature (under the assumption that CRISRPa activation of a single gene is unlikely to cause a genome‐wide systematic effect). We estimated such skews empirically by applying a lowess fit (span = 0.5) to the experimental ~ control comparison of a random selection of 50,000 GATC fragments and then used this fit to correct the genome‐wide comparison. This effectively removed genome‐wide biases, thereby enhancing the sensitivity to detect local changes in NL contacts around the targeted genes.
Processing of F1 hybrid mouse ES cell DamID data
For DamID on F1 hybrid 129/Cast mouse ES cells, 150 or 200 nt single reads were trimmed to remove the DamID adaptor sequence and then strain‐specifically mapped to mm10 with WASP (van de Geijn et al, 2015) using bowtie2 and VCF files from the mouse genomes project (Keane et al, 2011; https://www.sanger.ac.uk/science/data/mouse-genomes-project; version 5). Data were further processed as described for RPE‐1 cells, except that a smoothing window size of 301 instead of 201 GATC fragments was applied.
Domainograms
The domainograms in this study are related to those reported previously (de Wit et al, 2008; Tolhuis et al, 2011), but do not show estimated P‐values, which are not easily calculated for our experimental design. Rather, for a given window size, they show the ranking of changes in DamID log ratios (experimental minus control) relative to windows of the same size genome‐wide. Briefly, in a window of w neighboring GATC fragments, the difference in mean DamID log‐ratio is calculated between the experimental and control samples. This is done for all possible windows of size w genome‐wide. Next, windows in which both experimental and control sample showed only baseline DamID signals (i.e., both log ratios are in the respective lower 0.3 quantiles genome‐wide) are discarded. This is done because baseline fluctuations can appear strong on a logarithmic scale but are generally of minor amplitude on a linear scale and therefore unlikely to be of biological relevance. The remaining windows are ranked by their log‐ratio differences; ranks < 5% or > 95% are visualized by blue or red color scales, respectively. This is repeated for 28 different window sizes w that are logarithmically ranging from 67 and 2,917 GATC fragments, i.e., from ~30 kb to ~1 Mb.
Data analysis of Repli‐seq samples
Repi‐seq reads from early and late‐replicating fractions were mapped and processed in the same way as DamID reads, using the same smoothing window size. Instead of Dam‐lamin B1/Dam‐only, the ratio late/early replication was calculated.
Hi‐C data analysis
Hi‐C data from wild‐type RPE‐1 cells are from Darrow et al (2016) (Data ref: Darrow et al, 2016) and visualized using Juicebox 1.8.8 (Durand et al, 2016).
Author contributions
LB: conceived and designed study, conducted majority of experiments, initial data analysis, and wrote manuscript. PAZ: performed Repli‐seq experiments and initial Repli‐seq data analysis. DP‐H: performed experiments and data analysis. TS: processing of DamID data. CL: performed data analysis. JS: Morc1 locus deletions and initial data analysis. DMG: supervised Repli‐seq experiments, generation of Morc1 locus deletions and initial data analysis. BS: designed study, performed coding and data analysis, wrote manuscript, and supervised project.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgments
We thank the NKI Genomics, Flow Cytometry, and RHPC core facilities, as well as Tom Rieuwerts and Ludo Pagie for technical assistance and Lorenzo Bombardelli for Tn5 protein. We thank Andrew Belmont, Jian Ma, and other members of the 4DN Center for Nuclear Cytomics for helpful discussions. Supported by NIH Common Fund “4D Nucleome” Program grant U54DK107965 (BvS and DMG) and European Research Council Advanced Grant 694466 (BvS). The Oncode Institute is partly supported by KWF Dutch Cancer Society.
The EMBO Journal (2020) 39: e103159
Data availability
The datasets (and computer code) produced in this study are available in the following databases:
Sequencing reads and processed data of DamID, Repli‐seq and RNA‐seq experiments: Gene Expression Omnibus GSE133275 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE133275).
Data analysis code: https://github.com/vansteensellab/LAD_rewiring.
References
- Akhtar W, Pindyurin AV, de Jong J, Pagie L, Ten Hoeve J, Berns A, Wessels LF, van Steensel B, van Lohuizen M (2014) Using TRIP for genome‐wide position effect analysis in cultured cells. Nat Protoc 9: 1255–1281 [DOI] [PubMed] [Google Scholar]
- Brueckner L, van Arensbergen J, Akhtar W, Pagie L, van Steensel B (2016) High‐throughput assessment of context‐dependent effects of chromatin proteins. Epigenetics Chromatin 9: 43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabianca DS, Munoz‐Jimenez C, Kalck V, Gaidatzis D, Padeken J, Seeber A, Askjaer P, Gasser SM (2019) Active chromatin marks drive spatial sequestration of heterochromatin in C. elegans nuclei. Nature 569: 734–739 [DOI] [PubMed] [Google Scholar]
- Chen H, Zheng X, Zheng Y (2014) Age‐associated loss of lamin‐B leads to systemic inflammation and gut hyperplasia. Cell 159: 829–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang CH, Carpenter AE, Fuchsova B, Johnson T, de Lanerolle P, Belmont AS (2006) Long‐range directional movement of an interphase chromosome site. Curr Biol 16: 825–831 [DOI] [PubMed] [Google Scholar]
- Darrow EM, Huntley MH, Dudchenko O, Stamenova EK, Durand NC, Sun Z, Huang SC, Sanborn AL, Machol I, Shamim M et al (2016) Deletion of DXZ4 on the human inactive X chromosome alters higher‐order genome architecture. Proc Natl Acad Sci USA 113: E4504–E4512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darrow EM, Huntley MH, Lieberman Aiden E (2016) Gene Expression Omnibus GSE71831 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE71831). [DATASET]
- Dialynas G, Speese S, Budnik V, Geyer PK, Wallrath LL (2010) The role of Drosophila Lamin C in muscle function and gene expression. Development 137: 3067–3077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B (2012) Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485: 376–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES, Aiden EL (2016) Juicebox provides a visualization system for Hi‐C contact maps with unlimited zoom. Cell Syst 3: 99–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filion GJ, van Bemmel JG, Braunschweig U, Talhout W, Kind J, Ward LD, Brugman W, de Castro IJ, Kerkhoven RM, Bussemaker HJ et al (2010) Systematic protein location mapping reveals five principal chromatin types in Drosophila cells. Cell 143: 212–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlan LE, Sproul D, Thomson I, Boyle S, Kerr E, Perry P, Ylstra B, Chubb JR, Bickmore WA (2008) Recruitment to the nuclear periphery can alter expression of genes in human cells. PLoS Genet 4: e1000039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Geijn B, McVicker G, Gilad Y, Pritchard JK (2015) WASP: allele‐specific software for robust molecular quantitative trait locus discovery. Nat Methods 12: 1061–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogola E, Duarte AA, de Ruiter JR, Wiegant WW, Schmid JA, de Bruijn R, James DI, Guerrero Llobet S, Vis DJ, Annunziato S et al (2018) Selective loss of PARG restores PARylation and counteracts PARP inhibitor‐mediated synthetic lethality. Cancer Cell 33: 1078–1093.e1012 [DOI] [PubMed] [Google Scholar]
- Gonzalez‐Sandoval A, Gasser SM (2016) On TADs and LADs: spatial control over gene expression. Trends Genet 32: 485–495 [DOI] [PubMed] [Google Scholar]
- Greil F, Moorman C, van Steensel B (2006) DamID: mapping of in vivo protein‐genome interactions using tethered DNA adenine methyltransferase. Methods Enzymol 410: 342–359 [DOI] [PubMed] [Google Scholar]
- Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W et al (2008) Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 453: 948–951 [DOI] [PubMed] [Google Scholar]
- Harr JC, Luperchio TR, Wong X, Cohen E, Wheelan SJ, Reddy KL (2015) Directed targeting of chromatin to the nuclear lamina is mediated by chromatin state and A‐type lamins. J Cell Biol 208: 33–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Texari L, Hayes MGB, Urbanowski M, Chang MW, Givarkes N, Rialdi A, White KM, Albrecht RA, Pache L et al (2018) Transcription elongation can affect genome 3D structure. Cell 174: 1522–1536.e1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinrichs AS, Karolchik D, Baertsch R, Barber GP, Bejerano G, Clawson H, Diekhans M, Furey TS, Harte RA, Hsu F et al (2006) The UCSC genome browser database: update 2006. Nucleic Acids Res 34: D590–D598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiratani I, Ryba T, Itoh M, Yokochi T, Schwaiger M, Chang CW, Lyou Y, Townes TM, Schubeler D, Gilbert DM (2008) Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol 6: e245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T et al (2015) Orchestrating high‐throughput genomic analysis with Bioconductor. Nat Methods 12: 115–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isoda T, Moore AJ, He Z, Chandra V, Aida M, Denholtz M, Piet van Hamburg J, Fisch KM, Chang AN, Fahl SP et al (2017) Non‐coding transcription instructs chromatin folding and compartmentalization to dictate enhancer‐promoter communication and T cell fate. Cell 171: 103–119.e118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, Heger A, Agam A, Slater G, Goodson M et al (2011) Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477: 289–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Zheng X, Zheng Y (2019) Role of lamins in 3D genome organization and global gene expression. Nucleus 10: 33–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kind J, Pagie L, Ortabozkoyun H, Boyle S, de Vries SS, Janssen H, Amendola M, Nolen LD, Bickmore WA, van Steensel B (2013) Single‐cell dynamics of genome‐nuclear lamina interactions. Cell 153: 178–192 [DOI] [PubMed] [Google Scholar]
- Kind J, Pagie L, de Vries SS, Nahidiazar L, Dey SS, Bienko M, Zhan Y, Lajoie B, de Graaf CA, Amendola M et al (2015) Genome‐wide maps of nuclear lamina interactions in single human cells. Cell 163: 134–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohwi M, Lupton JR, Lai SL, Miller MR, Doe CQ (2013) Developmentally regulated subnuclear genome reorganization restricts neural progenitor competence in Drosophila . Cell 152: 97–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaran RI, Spector DL (2008) A genetic locus targeted to the nuclear periphery in living cells maintains its transcriptional competence. J Cell Biol 180: 51–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackner DH, Carre A, Guzzardo PM, Banning C, Mangena R, Henley T, Oberndorfer S, Gapp BV, Nijman SMB, Brummelkamp TR et al (2015) A generic strategy for CRISPR‐Cas9‐mediated gene tagging. Nat Commun 6: 10237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pages H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, Carey VJ (2013) Software for computing and annotating genomic ranges. PLoS Comput Biol 9: e1003118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leemans C, van der Zwalm MCH, Brueckner L, Comoglio F, van Schaik T, Pagie L, van Arensbergen J, van Steensel B (2019) Promoter‐intrinsic and local chromatin features determine gene repression in LADs. Cell 177: 852–864.e814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leeuw R, Gruenbaum Y, Medalia O (2018) Nuclear lamins: thin filaments with major functions. Trends Cell Biol 28: 34–45 [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing S (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochs SJA, Kefalopoulou S, Kind J (2019) Lamina associated domains and gene regulation in development and cancer. Cells 8: E271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund E, Oldenburg AR, Delbarre E, Freberg CT, Duband‐Goulet I, Eskeland R, Buendia B, Collas P (2013) Lamin A/C‐promoter interactions specify chromatin state‐dependent transcription outcomes. Genome Res 23: 1580–1589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luperchio TR, Sauria MEG, Wong X, Gaillard MC, Tsang P, Pekrun K, Ach RA, Yamada NA, Taylor J, Reddy KL (2017) Chromosome conformation paints reveal the role of lamina association in genome organization and regulation. bioRxiv 10.1101/122226 [PREPRINT] [DOI] [Google Scholar]
- Marchal C, Sasaki T, Vera D, Wilson K, Sima J, Rivera‐Mulia JC, Trevilla‐Garcia C, Nogues C, Nafie E, Gilbert DM (2018) Genome‐wide analysis of replication timing by next‐generation sequencing with E/L Repli‐seq. Nat Protoc 13: 819–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011) Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnetjournal 17: 10–12 [Google Scholar]
- Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J et al (2012) Spatial partitioning of the regulatory landscape of the X‐inactivation centre. Nature 485: 381–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peric‐Hupkes D, Meuleman W, Pagie L, Bruggeman SW, Solovei I, Brugman W, Graf S, Flicek P, Kerkhoven RM, van Lohuizen M et al (2010) Molecular maps of the reorganization of genome‐nuclear lamina interactions during differentiation. Mol Cell 38: 603–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phanstiel DH, Boyle AP, Araya CL, Snyder MP (2014) Sushi.R: flexible, quantitative and integrative genomic visualizations for publication‐quality multi‐panel figures. Bioinformatics 30: 2808–2810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope BD, Ryba T, Dileep V, Yue F, Wu W, Denas O, Vera DL, Wang Y, Hansen RS, Canfield TK et al (2014) Topologically associating domains are stable units of replication‐timing regulation. Nature 515: 402–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2017) R: a language and environment for statistical computing. R Core Team; https://www.R-projectorg [Google Scholar]
- Reddy KL, Zullo JM, Bertolino E, Singh H (2008) Transcriptional repression mediated by repositioning of genes to the nuclear lamina. Nature 452: 243–247 [DOI] [PubMed] [Google Scholar]
- Robson MI, de Las Heras JI, Czapiewski R, Le Thanh P, Booth DG, Kelly DA, Webb S, Kerr AR, Schirmer EC (2016) Tissue‐specific gene repositioning by muscle nuclear membrane proteins enhances repression of critical developmental genes during myogenesis. Mol Cell 62: 834–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson MI, de Las Heras JI, Czapiewski R, Sivakumar A, Kerr ARW, Schirmer EC (2017) Constrained release of lamina‐associated enhancers and genes from the nuclear envelope during T‐cell activation facilitates their association in chromosome compartments. Genome Res 27: 1126–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevelyov YY, Lavrov SA, Mikhaylova LM, Nurminsky ID, Kulathinal RJ, Egorova KS, Rozovsky YM, Nurminsky DI (2009) The B‐type lamin is required for somatic repression of testis‐specific gene clusters. Proc Natl Acad Sci USA 106: 3282–3287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sima J, Chakraborty A, Dileep V, Michalski M, Klein KN, Holcomb NP, Turner JL, Paulsen MT, Rivera‐Mulia JC, Trevilla‐Garcia C et al (2019) Identifying cis elements for spatiotemporal control of mammalian DNA replication. Cell 176: 816–830.e818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B, Belmont AS (2017) Lamina‐associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell 169: 780–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B, Furlong EEM (2019) The role of transcription in shaping the spatial organization of the genome. Nat Rev Mol Cell Biol 20: 327–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern DL (2017) Tagmentation‐based mapping (TagMap) of mobile DNA genomic insertion sites. bioRxiv 10.1101/037762 [PREPRINT] [DOI] [Google Scholar]
- Tame MA, Manjon AG, Belokhvostova D, Raaijmakers JA, Medema RH (2017) TUBB3 overexpression has a negligible effect on the sensitivity to taxol in cultured cell lines. Oncotarget 8: 71536–71547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanenbaum ME, Gilbert LA, Qi LS, Weissman JS, Vale RD (2014) A protein‐tagging system for signal amplification in gene expression and fluorescence imaging. Cell 159: 635–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therizols P, Illingworth RS, Courilleau C, Boyle S, Wood AJ, Bickmore WA (2014) Chromatin decondensation is sufficient to alter nuclear organization in embryonic stem cells. Science 346: 1238–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolhuis B, Blom M, Kerkhoven RM, Pagie L, Teunissen H, Nieuwland M, Simonis M, de Laat W, van Lohuizen M, van Steensel B (2011) Interactions among Polycomb domains are guided by chromosome architecture. PLoS Genet 7: e1001343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumbar T, Belmont AS (2001) Interphase movements of a DNA chromosome region modulated by VP16 transcriptional activator. Nat Cell Biol 3: 134–139 [DOI] [PubMed] [Google Scholar]
- Vermunt MW, Zhang D, Blobel GA (2019) The interdependence of gene‐regulatory elements and the 3D genome. J Cell Biol 218: 12–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel MJ, Peric‐Hupkes D, van Steensel B (2007) Detection of in vivo protein‐DNA interactions using DamID in mammalian cells. Nat Protoc 2: 1467–1478 [DOI] [PubMed] [Google Scholar]
- de Wit E, Braunschweig U, Greil F, Bussemaker HJ, van Steensel B (2008) Global chromatin domain organization of the Drosophila genome. PLoS Genet 4: e1000045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F, Yao J (2017) Identifying novel transcriptional and epigenetic features of nuclear lamina‐associated genes. Sci Rep 7: 100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorita E, Cusco P, Filion GJ (2015) Starcode: sequence clustering based on all‐pairs search. Bioinformatics 31: 1913–1919 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Darrow EM, Huntley MH, Lieberman Aiden E (2016) Gene Expression Omnibus GSE71831 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE71831). [DATASET]
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Data Availability Statement
The datasets (and computer code) produced in this study are available in the following databases:
Sequencing reads and processed data of DamID, Repli‐seq and RNA‐seq experiments: Gene Expression Omnibus GSE133275 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE133275).
Data analysis code: https://github.com/vansteensellab/LAD_rewiring.