

Abstract
Various species of non‐coding RNAs (ncRNAs) are enriched in specific subcellular compartments, but the mechanisms orchestrating their localization and their local functions remain largely unknown. We investigated both aspects using the elongating retinal ganglion cell axon and its tip, the growth cone, as models. We reveal that specific endogenous precursor microRNAs (pre‐miRNAs) are actively trafficked to distal axons by hitchhiking primarily on late endosomes/lysosomes. Upon exposure to the axon guidance cue semaphorin 3A (Sema3A), pre‐miRNAs are processed specifically within axons into newly generated miRNAs, one of which, in turn, silences the basal translation of tubulin beta 3 class III (TUBB3), but not amyloid beta precursor protein (APP). At the organismal level, these mature miRNAs are required for growth cone steering and a fully functional visual system. Overall, our results uncover a novel mode of ncRNA transport from one cytosolic compartment to another within polarized cells. They also reveal that newly generated miRNAs are critical components of a ncRNA‐based signaling pathway that transduces environmental signals into the structural remodeling of subcellular compartments.
Keywords: axon, neural circuit development, non‐coding RNAs, RNA localization, TUBB3
Subject Categories: Neuroscience, RNA Biology
Pre‐miRNAs transported to distal axons are locally processed upon exposure to Sema3A and are required for growth cone steering and a fully functional visual system.

Introduction
Most cells are polarized, with an intracellular milieu partitioned into various organelles, cytosolic and membrane microdomains that accomplish specialized biological and regulatory functions. Neurons are highly polarized, and their axons constitute a unique cellular outpost with distinctive autonomous functions. During development, axons grow and generate a complex network of interconnected neurons. To establish these connections, the tip of the growing axon, the growth cone, is guided with exquisite precision by attractant and repellent chemotropic cues en route to its target. Axons must sometimes navigate a significant distance before reaching their final destination. In large mammals, axons can even reach targets located meters away. The distance between growth cone and cell body poses a particular challenge to developing neurons, as growth cones must be able to rapidly and accurately transduce environmental information to ensure highly precise directional steering, without the immediate intervention from the soma. To overcome this challenge, growth cones store, locally produce, and fine‐tune the levels of their own proteins through local protein synthesis (LPS), from a rich repertoire of mRNAs that are selectively trafficked there (Cioni et al, 2018).
Overall, mRNA localization and its corollary, local translation, are key mechanisms to create and sustain polarity by conferring functional autonomy to a variety of subcellular compartments including axons (Chin & Lécuyer, 2017). Recent evidence suggests that not only mRNAs but also various additional RNA species such as the small non‐coding RNAs (ncRNAs) miRNAs (Kye et al, 2007; Lugli et al, 2008; Natera‐Naranjo et al, 2010), linear long ncRNAs (Cabili et al, 2015), and circular RNAs (You et al, 2015) are localized to and enriched within subcellular outposts. The mechanisms of ncRNA transport to these compartments and the biological functions of local ncRNAs have, however, remained elusive. Gaining such fundamental insight on axons is crucial, as derailed axonal transport is a common factor in several incurable neurodegenerative disorders (Liu et al, 2012).
Dicer is essential for the production of most ~22nt active, mature miRNAs from short hairpin precursor miRNAs (pre‐miRNAs) (Grishok et al, 2001; Hutvagner et al, 2001). Remarkably, several reports have revealed the presence of Dicer within growth cones (Hengst, 2006; Aschrafi et al, 2008; Zhang et al, 2013; Hancock et al, 2014; Kim et al, 2015; Vargas et al, 2016; Gershoni‐Emek et al, 2018). These findings raise the intriguing possibility that inactive pre‐miRNAs are trafficked along axons to the growth cone and locally activated to exert their function. Here, we thus explore the transport mechanisms of pre‐miRNAs within axons using a novel molecular beacon‐based approach. We discover that pre‐miRNAs exploit a vesicle‐based transport system to reach the axon tip, where they are subsequently stored. We then investigate the local function of pre‐miRNAs within growth cones. We uncover that upon repellent cue exposure, specific pre‐miRNAs are locally processed into active miRNAs that rapidly inhibit the basal translation of a selected transcript. This, in turn, induces directional steering ex vivo and in vivo, and promotes the development of a fully functional neuronal circuit. Collectively, these results reveal that miRNAs hitchhike on vesicles to distal axons in an inactive form. At the growth cone, they are activated, on demand, to acutely inhibit basal translation of specific transcripts ultimately promoting growth cone steering and the highly accurate assembly of neuronal circuits.
Results
Molecular beacons are new, specific tools to detect endogenous pre‐miRNAs in cells
To investigate pre‐miRNA transport mechanisms, we first established that these precursors are indeed located to axons. Analysis of our previously published dataset of small RNAs (Bellon et al, 2017) revealed the existence of sequences corresponding to the loop region of 42 pre‐miRNAs in axons and growth cones of Xenopus laevis retinal ganglion cells (RGCs), our model system (Appendix Fig S1A). We validated, by RT–PCR, the presence of the abundant pre‐miR‐182, pre‐miR‐181a‐1, and pre‐miR‐181a‐2 in eyes, and pure axonal RNA that we collected by laser capture microdissection (LCM; Fig 1A–C and Appendix Fig S1A). We also examined the relative expression levels of the two miR‐181a precursors by RT–qPCR, and found that a‐1 was 5.9× significantly less abundant than a‐2 in eyes (Appendix Fig S1B), yet 2.5× more abundant in isolated axons (Appendix Fig S1C). This indicates that pre‐miR‐181a‐1 may be preferentially targeted to axons and growth cones.
Figure 1. Molecular beacons are new, specific tools to detect endogenous pre‐miRNAs in cells.

-
ASchematic representation of the experimental protocol. LCM, laser capture microdissection.
-
B, CRT–PCR performed on RNA extracted from axons collected by LCM (Ax) or from eyes. (B) β‐Actin mRNA is present both in eyes and in axons, while MAP2 and H4 are present only in the eye sample, suggesting the absence of contamination from cell bodies or other cells in LCM axonal samples (Bellon et al, 2017). Ax, axonal sample; Eye, stage 37/38 eye; LCM, laser capture microdissection; –, PCR no template control; NT, RT no template control.
-
DSchematic of MB, pre‐miR‐181a‐1, and their hybridization complex. MB, molecular beacon.
-
ESchematic of thermal denaturation profile of the MB. MB, molecular beacon.
-
FThermal denaturation profiles of MB, in the absence (solid line) and presence (dashed line) of increasing target concentration. Each melting curve represents the average of three separate replicates. Yellow boxes indicate the range of working temperatures for ex vivo trafficking experiments. MB, molecular beacon; T m, MB melting temperature.
-
GSchematic representation of the experimental protocol. Concentrations used are as follows: 5 μM MB; 250 μM co‐MO; 250 μM pri‐miR‐MO. MB, molecular beacon; co‐MO, control morpholino; pri‐miR‐MO, morpholino blocking pre‐miR‐181a‐1 processing by targeting the Drosha cleavage site.
-
HQuantification of the expression levels of pre‐mir‐181a‐1 using the 2−ΔCt method and U6 as normalizer from small total RNA fraction (< ˜150 nt). Each data point corresponds to one independent experiment. n = 3 independent experiments. Values are mean ± SEM. ns, not significant.
-
ITotal number of MB puncta normalized to axon length (μm). Each data point corresponds to one axon. Total number of puncta and axons analyzed is as follows: 928 puncta and 61 axons (WT); 226 puncta and 15 axons (co‐MO); 208 puncta and 35 axons (MO). n = 4 independent experiments. Values are mean ± SEM. ns, not significant; co‐MO, control morpholino; pri‐miR‐MO, morpholino blocking pre‐miR‐181a‐1 processing by targeting the Drosha cleavage site.
-
JRepresentative axons. MB puncta are indicated (white arrows). Dashed white lines delineate axons. MB, molecular beacon. Scale bars: 5 μm.
-
KSchematic representation of the experimental protocol. Concentrations used are as follows: 5 μM MB; 200–250 ng/μl cy3‐pre‐miR‐181a‐1. MB, molecular beacon.
-
MSchematic representation of cy5‐labeled pre‐miR‐181a‐1.
-
NFrequency (in percentage) of puncta colocalization between MB and cy5‐pre‐miR‐181a‐1 (#MB+/pre‐miR+). Each data point corresponds to one axon. Total number of puncta and axons analyzed is as follows: 354 puncta and 32 axons (MB); 337 puncta and 32 axons (cy5‐pre‐miR‐181a‐1). n = 5 independent experiments. Values are mean ± SEM. MB, molecular beacon.
-
ORepresentative image of RGC axons. White, red, and blue arrows indicate, respectively, colocalized, single MB, and single pre‐miR‐181a‐1 puncta. Scale bars: 5 μm.
We next explored how pre‐miRNAs reach the growth cone. We specifically examined the transport dynamics of endogenous pre‐miR‐181a‐1, more abundant than pre‐miR‐181‐a2 in axons (Appendix Fig S1C), by adapting molecular beacon (MB) technology. MBs are single‐stranded oligonucleotide probes which fluoresce only when hybridized to their target (Santangelo et al, 2006) (Fig 1D). The MB backbone and sequence was carefully designed to maximize the probe's stability, signal‐to‐noise ratio, and specificity within the cell (see Methods for details). Thermal denaturation assay (Fig 1E) revealed increased fluorescence levels at low temperatures in the presence of in vitro transcribed pre‐miR‐181a‐1 (Fig 1F) but not in the presence of (i) pre‐miR‐187 which is absent from RGCs (this study and Ref. (Bellon et al, 2017)), (ii) a modified pre‐miR‐181a‐1 lacking the loop region, and (iii) short double‐stranded miR‐181 mimic (see Methods; Appendix Fig S1D–F). This confirms that our MB is able to specifically hybridize with and detect pre‐miR‐181a‐1 in vitro.
To test MB specificity ex vivo, we first examined change in MB signal when endogenous pre‐miR‐181a‐1 is knocked down (Fig 1G). Blocking Drosha cleavage with morpholino (pri‐miR‐MO) led to a significant decrease in pre‐miR‐181a‐1 levels by 56.3% in the eye (Fig 1H) and of MB puncta by 58.4% (Fig 1I and J). We did not observe significant differences between WT and co‐MO (Fig 1H–J). Second, we examined whether cy3‐labeled MB colocalized with exogenous cy5‐labeled pre‐miR‐181a‐1 (Fig 1L) or pre‐miR‐187 (Appendix Fig S1G) that we serially electroporated into eye primordia (Fig 1K and N, and Appendix Fig S1H–L). Within axons, 70.1–77.2 and 2.9% of MB puncta colocalized with pre‐miR‐181a‐1 and pre‐miR‐187 puncta, respectively (Fig 1M and Appendix Fig S1L). This suggests that the MB can specifically recognize pre‐miR‐181a‐1 ex vivo.
Endogenous and exogenous pre‐miRNAs are actively trafficked along axons
We then characterized endogenous pre‐miR‐181a‐1 trafficking dynamics in RGC axons by single axon live imaging and kymograph analysis following targeted electroporation of MBs into retinal cells (Fig 2A–C). MB‐labeled pre‐miR‐181a‐1 puncta were detected throughout the entire length of growing RGC axons and within the central domain of growth cones (Fig 2B). Equal frequencies of anterogradely and retrogradely moving puncta were detected (Fig 2D). Upon subdivision of the puncta population based on speed [stationary (< 0.2 μm/s), moving (0.2–0.5 μm/s), and fast‐moving puncta (> 0.5 μm/s) (Maday et al, 2014; Leung et al, 2018)], we observed that the fast anterogradely moving puncta were significantly faster on average than their retrograde counterpart (Fig 2E). These observations likely reflect the properties of molecular motors: Anterograde kinesins are highly processive motors, while retrograde dyneins make frequent back and side steps (Maday et al, 2014). No significant differences in puncta directionality (Fig 2D) and velocities (Fig 2E) between endogenous and exogenous pre‐miR‐181a‐1 were observed ([Link], [Link]).
Figure 2. Endogenous and exogenous pre‐miRNAs are actively trafficked along axon microtubules.
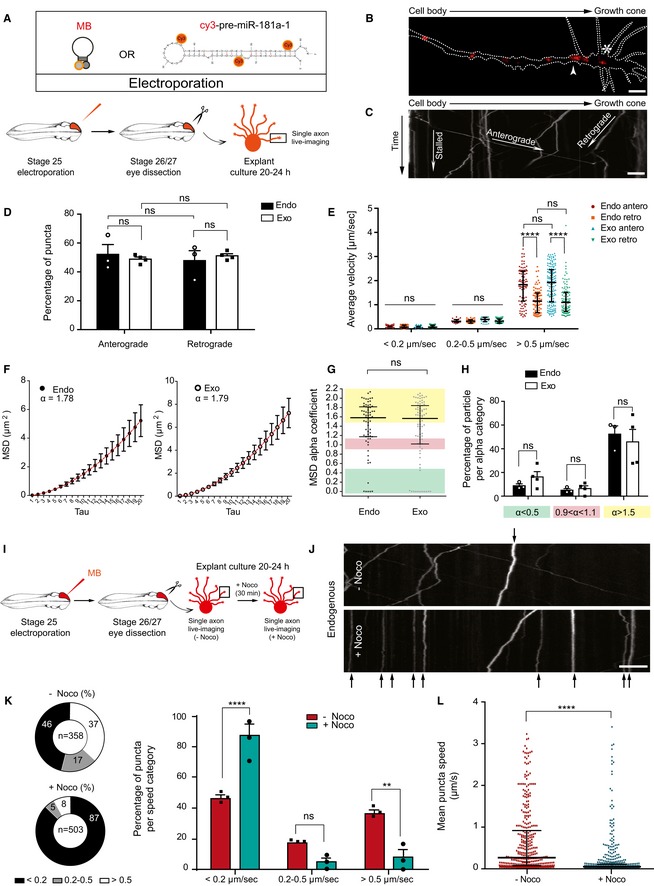
- Schematic representation of the experimental paradigm. Concentrations used are as follows: 5 μM MB; 200–250 ng/μl cy3‐pre‐miR‐181a‐1. MB, molecular beacon.
- Representative image of a single distal RGC axon from MB‐electroporated retina. Growth cone wrist and central domain are indicated with white arrow and star, respectively. Dashed white line delineates the axon. MB, molecular beacon. Scale bars: 5 μm.
- Illustrative kymograph. Scale bars: 5 μm.
- Frequency distribution (in percentage) of MB (endo) and cy3‐pre‐miR‐181a‐1 (exo) puncta along the RGC axon shaft. Each data point corresponds to one independent experiment. Total number of puncta and axons analyzed is as follows: 353 puncta and 20 axons (endo); 484 puncta and 29 axons (exo). n = 3 (endo) and n = 4 (exo) independent experiments. Values are mean ± SEM. MB, molecular beacon; ns, not significant.
- Average velocity of MB (endo) and cy3‐pre‐miR‐181a‐1 (exo) puncta. Each data point corresponds to one punctum. Total number of puncta and axons analyzed is as follows: 353 puncta and 20 axons (endo); 484 puncta and 29 axons (exo). n = 3 (endo) and n = 4 (exo) independent experiments. Values are median with interquartile range. MB, molecular beacon; ns, not significant; antero, anterograde movement; retro, retrograde movement.
- MSD data for MB (endo) and cy3‐pre‐miR‐181a‐1 (exo) tracked particles were fitted with an anomalous diffusion model and α thus calculated (red). Total number of particles and axons analyzed is as follows: 67 particles and 20 axons (endo); 82 particles and 29 axons (exo). n = 3 (endo) and n = 4 (exo) independent experiment. Values are mean ± SEM. MB, molecular beacon.
- MSD alpha‐coefficient distribution for each single MB (endo) and cy3‐pre‐miR‐181a‐1 (exo) tracked particle. Each data point corresponds to one particle. Total number of particles and axons analyzed is as followed: 67 particles and 20 axons (endo); 82 particles and 29 axons (exo). n = 3 (endo) and n = 4 (exo) independent experiments. Values are median with interquartile range. MB, molecular beacon; ns, not significant.
- Relative frequency distribution (percentage) of MB (endo) and cy3‐pre‐miR‐181a‐1 (exo) tracked particles. Each data point corresponds to one independent experiment. Total number of particles and axons analyzed is as follows: 67 particles and 20 axons (endo); 82 particles and 29 axons (exo). n = 3 (endo) and n = 4 (exo) independent experiments. Values are mean ± SEM. MB, molecular beacon; ns, not significant.
- Schematic representation of the experimental paradigm. Five micromolar MB was electroporated. MB, molecular beacon.
- Representative kymographs before nocodazole (top panel; −Noco) and 30 min after 2.4 μM Noco bath application (bottom panel; +Noco). Stationary puncta in both panels are indicated with black arrows. Scale bars: 5 μm.
- Frequency distribution (in percentage) of MB punctum speed. Each data point corresponds to one independent experiment. Total number of puncta and axons analyzed is as follows: 358 puncta and 25 axons (−Noco); 503 puncta and 34 axons (+Noco). n = 3 independent experiments. Values are mean ± SEM. MB, molecular beacon; ns, not significant; Noco, nocodazole.
- Frequency distribution of MB punctum speed. Each data point corresponds to one punctum. Total number of puncta and axons analyzed is as follows: 358 puncta and 25 axons (−Noco); 503 puncta and 34 axons (+Noco). n = 3 independent experiments. Values are median with interquartile range. MB, molecular beacon; ns, not significant; Noco, nocodazole.
To address whether moving (≥ 0.2 μm/s) endogenous pre‐miRNAs are driven by active transport or passive diffusion, we performed mean square displacement (MSD) analysis (see Methods for details). The MSD data were fitted with an anomalous diffusion model: MSD = Aτα + B (Eq1) (Otero et al, 2014). Trajectories were conservatively classified as actively driven (α > 1.5), diffusive (0.9 < α < 1.1), or confined (α < 0.5) (Otero et al, 2014). We obtained α = 1.78 (endogenous) for the combined trajectories of all particles (Fig 2F), suggesting that moving pre‐miRNAs were overall actively trafficked along axons. By analyzing the α distribution of individual moving particles, we detected that the majority assumed an active motion (Fig 2G and H). As above, exogenous and endogenous pre‐miRNAs appeared to behave similarly, since the computed α (Fig 2F) and α distribution (Fig 2G and H) were not significantly different. As expected, particle trajectory of each type of motion appeared to differ and most displayed a perfect or near perfect fitting reflecting a single‐mode behavior (Fig EV1A–C). Collectively, these data suggest that the majority of anterogradely displaced pre‐miRNAs are actively transported along the axon to reach the growth cone.
Figure EV1. Pre‐miRNAs are actively trafficked along microtubules.
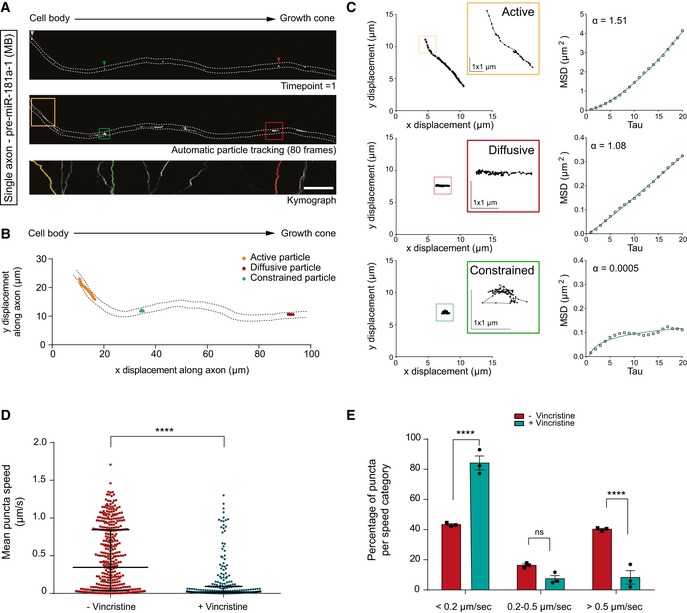
- Representative tracked particles. The dashed white lines delineate the axon. MB, molecular beacon. Scale bars: 10 μm.
- Representative tracked particles. The x‐y trajectory for each motion type is represented within the axon. Note the difference in trajectory length according to the particle type. The dashed black lines delineate the axon.
- High spatial resolution of x, y displacement for each motion type and corresponding MSD plot. Most trajectories analyzed displayed a perfect or near‐perfect fitting reflecting a single‐mode behavior. Note the striking difference in trajectories according to particle type. Only a small portion of the actively moving particle trajectory is shown in the yellow inset. Scale bars: 1 μmx1μm.
- Mean cy3‐pre‐miR‐181a‐1 puncta speed before and 30 min after vincristine bath application. Concentration used is as follows: 0.1 μM vincristine. Each data point corresponds to one punctum. Total number of puncta and axons analyzed is as follows: 397 puncta and 15 axons (−vincristine); 257 puncta and 15 axons (+vincristine). n = 3 independent experiments. Values are median with interquartile range.
- Frequency distribution (in percentage) of cy3‐pre‐miR‐181a‐1 puncta per speed categories before and 30 min after vincristine bath application. Concentration used is as follows: 0.1 μM vincristine. Each data point corresponds to one independent experiment. Total number of puncta and axons analyzed is as follows: 397 puncta and 15 axons (−vincristine); 257 puncta and 15 axons (+vincristine). n = 3 independent experiments. Values are mean ± SEM. ns, not significant.
Pre‐miRNAs are trafficked along microtubules, associated primarily with late endosomes/lysosomes
Next, we dissected the mechanisms mediating pre‐miRNA axonal transport. As axonal‐directed trafficking relies on microtubules (MTs) (Maday et al, 2014), we explored whether pre‐miR‐181a‐1 is also transported via this cytoskeletal component using the MT‐destabilizing drug nocodazole (Noco; Fig 2I). Kymograph analysis of time‐lapse movies before (−) or after (+) a 30‐min Noco incubation revealed a strong and significant increase in the proportion of stationary (< 0.2 μm/s) puncta (Fig 2J and K) and an overall 91.5% decrease in puncta's average velocities (from 0.2357 to 0.0199 μm/s; Fig 2L) after drug application. Bath application of vincristine, another MT‐destabilizing agent (Jordan et al, 1985), led to similar results (Fig EV1D and E). This indicates that pre‐miRNA transport along the axon shaft is mediated via microtubules.
In neurons, mRNAs are trafficked along axons packaged within ribonucleoparticles (RNPs) (Bauer et al, 2017). Pre‐miRNAs may also be dynamically trafficked within RNPs, as recent data on dendrites suggest (Bicker et al, 2013). However, mature miRNAs, miRNA‐repressible mRNAs, and components of the miRNA processing machinery associate with late endosomes/multivesicular bodies and lysosomes (LE/Ly) in non‐neuronal cells (Gibbings et al, 2009; Lee et al, 2009) and LE/Ly are detected in axons and growth cones (Falk et al, 2014; Konopacki et al, 2016). It is thus possible that pre‐miRNAs adopt a non‐canonical transport mode associated with LE/Ly within the axon shaft. Using CD63, a transmembrane protein enriched in a LE/Ly (Pols & Klumperman, 2009), as a marker, we examined whether CD63‐eGFP‐labeled vesicles and MB‐labeled pre‐miR‐181a‐1 puncta were co‐trafficked within axons (Fig 3A). 71.4% of MB‐positive moving puncta were co‐transported with CD63‐eGFP‐labeled vesicle‐like focal puncta, and an equal percentage of these moved anterogradely and retrogradely (Fig 3B–D, F and G, and [Link], [Link]). This bidirectional transport is consistent with previously published studies on LE/Ly trafficking in embryonic neurons (Farías et al, 2017). Similar results were obtained when growth cones were cultured from older embryos (Appendix Fig S2A–G and Movie EV5). These percentages are likely an underestimation, as endogenous unlabeled vesicle present in these axons may mask the extent of this co‐traffic. These results indicate that CD63‐positive vesicles contribute, to a large extent, to pre‐miRNA axonal transport. Furthermore, numerous co‐trafficked puncta reached the growth cones and appeared to stall and accumulate within the organelle‐rich central domain (Dent & Gertler, 2003) (star, Figs 2B and 3E and H–L, and Appendix Fig S2E and H–I), and in the perinuclear region (Fig 3H–L). Taken together, these results suggest that pre‐miRNAs hitchhike on vesicles to reach the growth cone central domain where they are stored.
Figure 3. Pre‐miRNAs are trafficked along microtubules associated with vesicles.
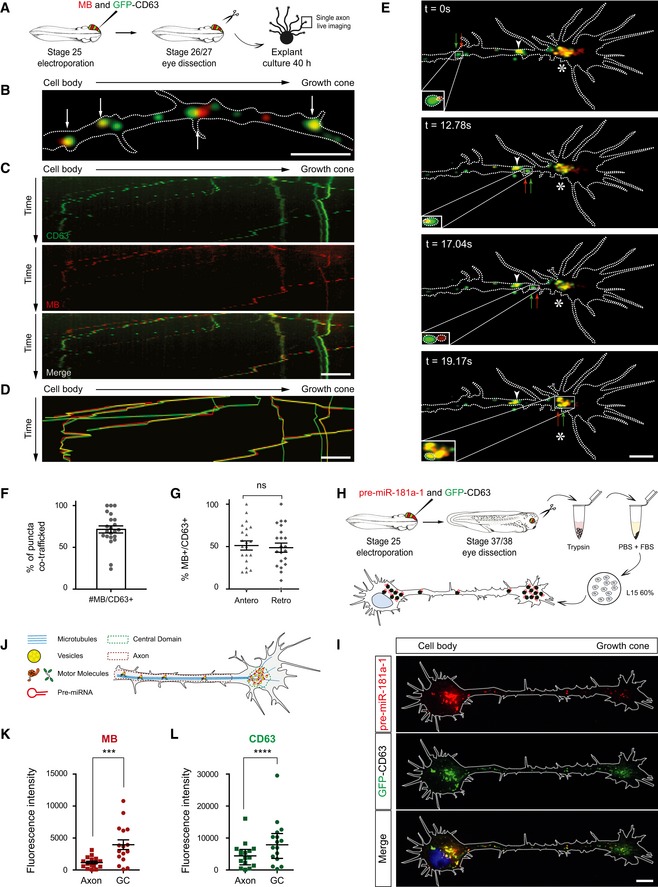
-
ASchematic of the experimental paradigm. Concentrations used are as follows: 5 μM MB; 0.5 μg/μl pCS2‐CD63‐eGFP. CD63, CD63‐eGFP; MB, molecular beacon.
-
BRepresentative axon where MB‐labeled pre‐miR‐181a‐1 (red) and CD63‐eGFP‐labeled vesicles (green) are co‐trafficked (white arrows). MB, molecular beacon. Scale bars: 5 μm.
-
CRepresentative kymographs. CD63, CD63‐eGFP; MB, molecular beacon. Scale bars: 5 μm.
-
DComposite kymograph shown in (C) where the individual traces were drawn and color‐coded. Yellow trajectories represent co‐trafficking MB‐labeled pre‐miRNA (red) and CD63‐eGFP‐labeled vesicle (green). MB, molecular beacon. Scale bars: 5 μm.
-
ERepresentative time‐lapse depicting MB‐labeled pre‐miR‐181a‐1 (red arrow) and CD63‐eGFP‐positive vesicle (green arrow) co‐trafficked along the axon shaft to the growth cone (delineated with dashed white lines) wrist (white arrowhead) and central domain (white star). Scale bars: 5 μm.
-
FFrequency (in percentage) of MB‐labeled pre‐miR‐181a‐1 co‐trafficked with CD63‐eGFP‐positive vesicles. Each data point corresponds to one axon. Total number of puncta and axons analyzed is as follows: 253 puncta and 22 axons (MB+); 306 puncta and 22 axons (CD63+). n = 5 independent experiments. Values are mean ± SEM. CD63, CD63‐eGFP; MB, molecular beacon.
-
GFrequency distribution (in percentage) of MB‐labeled pre‐miR‐181a‐1 co‐trafficked with CD63‐eGFP‐positive vesicles. Each data point corresponds to one axon. Total number of puncta and axons analyzed is as follows: 92 puncta and 22 axons (anterograde); 78 puncta and 22 axons (retrograde). n = 5 independent experiments. Values are mean ± SEM. CD63, CD63‐eGFP; MB, molecular beacon.
-
HSchematic of the experimental paradigm. Concentrations used are as follows: 250 ng/μl cy3‐pre‐miR‐181a‐1; 0.5 μg/μl pCS2‐CD63‐eGFP. CD63, CD63‐eGFP.
-
IRepresentative image of an electroporated and dissociated RGC. CD63, CD63‐eGFP. Scale bars: 5 μm.
-
JSchematic of the analysis. Pre‐miRNA, pre‐miRNA‐181a‐1.
-
K, LFluorescence intensity of MB (K) or CD63‐eGFP (L) measured in axons and growth cone central domains. Each data point corresponds to one axon or a growth cone central domain. Total number of axons analyzed is as follows: 16. n = 5 independent experiments. Values are mean ± SEM. (K) and median ± interquartile range (L). CD63, CD63‐eGFP; MB, molecular beacon; GC, growth cone.
To further characterize the vesicles on which pre‐miR‐181a‐1 hitchhikes, we use a panel of exogenous markers, linked to fluorescent reporters, enriched in early (Rab5a) and late (Rab7a) endosomes and lysosomes (Lamp1; Fig 4A and B) (Saftig & Klumperman, 2009; Langemeyer et al, 2018). We detected that CD63 colocalized with Rab7a (81%) and Lamp1 (81%) but much less with Rab5a (39%) (Fig 4C and D and Appendix Fig S3A and B). We then examined whether these vesicles have degradative properties and found that CD63 colocalized with LysoTracker (73%), a marker of acidic organelles (Fig 4D and Appendix Fig S3C–F). This suggests that the CD63‐eGFP that we have used to study pre‐miRNA co‐traffic is primarily expressed in acidic late endosomes/lysosomes in Xenopus RGC axons in culture. Subsequently, we investigated whether pre‐miR‐181a‐1 similarly associates with these markers and detected a high colocalization with Rab7a (90%), Lamp1 (91%), and LysoTracker (90%), similar to that found with CD63 (80%), but a significantly lower colocalization with Rab5a (50%) (Fig 4E and F). Furthermore, pre‐miRNAs colocalized with CD63/LysoTracker (80%) and CD63/Rab7a (68.5%) double‐marked vesicles (Fig 4G and H). The distribution of the colocalization frequency of pre‐miR‐181‐a1 with LysoTracker or CD63/LysoTracker (Fig 4F–H) indicates that this RNA does not always associate with degradative, acidic vesicles. Collectively, these data suggest that pre‐miRNAs are primarily transported by late endosomes/lysosomes.
Figure 4. pre‐miR‐181a‐1 is trafficked docked to late endosomes/lysosomes.
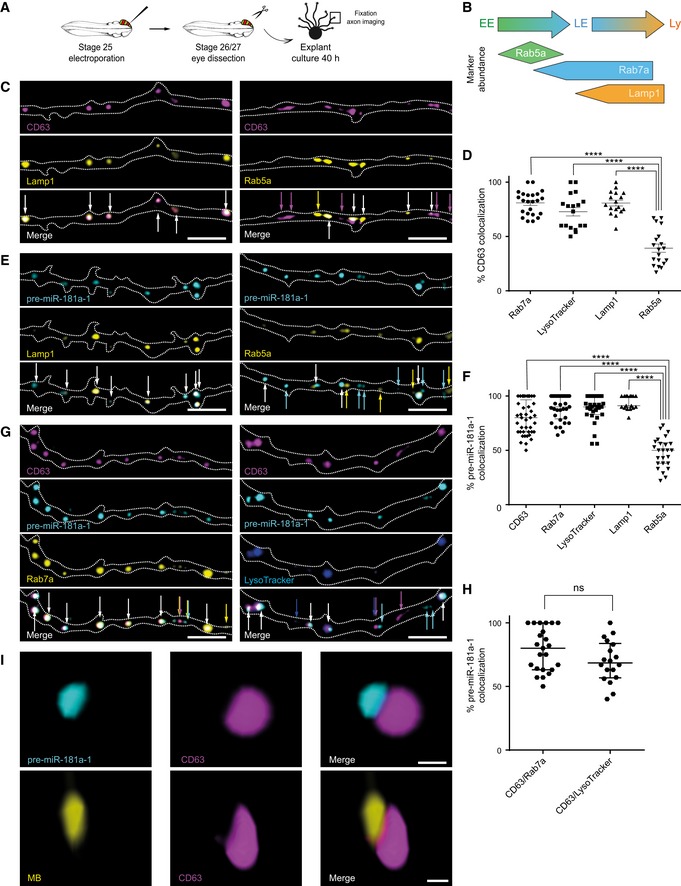
- Schematic representation of the experimental paradigm. Plasmid concentrations used are as follows: 0.5 μg/μl pCS2‐CD63‐eGFP, pCS2‐CD63‐mRFP, pCS2‐Rab7a‐mRFP, or pCS2‐Lamp1‐eGFP; 0.4 μg/μl pCS2‐Rab7a‐eGFP or pCS2‐Rab5a‐mRFP; 0.3 μg/μl pCS2‐Rab5a‐eGFP; 50 nM LysoTracker; 200 ng/μl cy3‐pre‐miR‐181a‐1 or cy5‐pre‐miR‐181a‐1.
- Overview of the markers used. Figure adapted from Ref. (Yap et al, 2018). EE, early endosome; LE, late endosome; Ly, lysosome.
- Representative images of co‐electroporated axons. Dashed white lines delineate axons. White arrows indicate colocalized puncta. Colored arrows represent non‐colocalized signal as follows: magenta for CD63 and yellow for Lamp1 or Rab5a. CD63, CD63‐eGFP or CD63‐mRFP; Lamp1, Lamp1‐eGFP; Rab5a, Rab5a‐eGFP or Rab5a‐mRFP. Scale bars: 5 μm.
- Frequency (in percentage) of CD63 colocalization with specific organelle markers. Each data point corresponds to one axon. Total number of puncta, axons, and independent experiments analyzed is as follows: 540 puncta, 23 axons, and n = 4 (CD63/Rab7a); 416 puncta, 18 axons, and n = 3 (CD63/LysoTracker); 584 puncta, 17 axons, and n = 2 (CD63/Lamp1); 649 puncta, 19 axons, and n = 3 (CD63/Rab5a). Values are mean ± SEM. CD63, CD63‐eGFP or CD63‐mRFP; Lamp1, Lamp1‐eGFP; Rab5a, Rab5a‐eGFP or Rab5a‐mRFP; Rab7a, Rab7a‐eGFP or Rab7a‐mRFP.
- Representative images of co‐electroporated axons. Dashed white lines delineate axons. White arrows indicate colocalized puncta. Colored arrows represent non‐colocalized signal as follows: cyan for pre‐miR‐181a‐1 and yellow for Lamp1 or Rab5a. Lamp1, Lamp1‐eGFP; Rab5a, Rab5a‐eGFP or Rab5a‐mRFP; pre‐miR‐181a‐1, cy3‐pre‐miR‐181a‐1 or cy5‐pre‐miR‐181a‐1. Scale bars: 5 μm.
- Frequency (in percentage) of pre‐miR‐181a‐1 colocalization with specific organelle markers. Each data point corresponds to one axon. Total number of puncta, axons, and independent experiments analyzed is as follows: 825 puncta, 41 axons, and n = 8 (pre‐miR‐181a‐1/CD63); 732 puncta, 33 axons, and n = 7 (pre‐miR‐181a‐1/Rab7a); 681 puncta, 29 axons, and n = 5 (pre‐miR‐181a‐1/LysoTracker); 501 puncta, 17 axons, and n = 2 (pre‐miR‐181a‐1/Lamp1); 791 puncta, 25 axons, and n = 3 (pre‐miR‐181a‐1/Rab5a). Values are median with interquartile range. CD63, CD63‐eGFP or CD63‐mRFP; Rab7a, Rab7a‐eGFP or Rab7a‐mRFP; Lamp1, Lamp1‐eGFP; Rab5a, Rab5a‐eGFP or Rab5a‐mRFP; pre‐miR‐181a‐1, cy3‐pre‐miR‐181a‐1 or cy5‐pre‐miR‐181a‐1.
- Representative images of co‐electroporated axons. Dashed white lines delineate axons. White arrows indicate colocalized puncta. Colored arrows represent non‐colocalized signal as follows: magenta for CD63, cyan for pre‐miR‐181a‐1, yellow for Rab7a, and blue for LysoTracker. CD63, CD63‐eGFP or CD63‐mRFP; Rab7a, Rab7a‐eGFP or Rab7a‐mRFP; pre‐miR‐181a‐1, cy3‐pre‐miR‐181a‐1 or cy5‐pre‐miR‐181a‐1. Scale bar: 5 μm.
- Frequency (in percentage) of pre‐miR‐181a‐1 colocalization with vesicles double‐positive for organelle markers. Each data point corresponds to one axon. Total number of puncta, axons, and independent experiments analyzed is as follows: 745 puncta, 23 axons, and n = 5 (CD63/pre‐miR‐181a‐1/Rab7a); and 577 puncta, 18 axons, and n = 3 (CD63/pre‐miR‐181a‐1/LysoTracker). Values are median with interquartile range. ns, not significant; CD63, CD63‐eGFP or CD63‐mRFP; Rab7a, Rab7a‐eGFP or Rab7a‐mRFP; pre‐miR‐181a‐1, cy3‐pre‐miR‐181a‐1 or cy5‐pre‐miR‐181a‐1.
- Representative 3D‐STED super‐resolution image. Total number of puncta, axons, and growth cones analyzed is as follows : 99 puncta, 16 axons, and 10 growth cones (MB); 291 puncta, 36 axons, and 15 growth cones (cy3‐pre‐miR‐181a‐1). n = 2 (MB) and n = 3 (cy3‐pre‐miR‐181a‐1) independent experiments. CD63, CD63‐eGFP; pre‐miR‐181a‐1, cy3‐pre‐miR‐181a‐1; MB; molecular beacon. Scale bars: 200 nm.
Finally, using 3D‐STED super‐resolution imaging analysis, we detected that pre‐miR‐181a‐1 molecules were associated with the outer surface of CD63‐eGFP‐marked vesicles in both axons and growth cones (Fig 4I, [Link], [Link]). Occasionally, we noticed that two pre‐miRNAs were carried in polar opposite orientation (Appendix Fig S3G). Collectively, our data suggest that pre‐miRNAs are not incorporated inside intraluminal vesicles of late endosomes/lysosomes for degradation or exocytosis. Rather, they are docked to endosomes for transport, readily available for cytoplasmic function.
Pre‐miRNAs are processed locally in response to cues
Our data thus far indicate that pre‐miRNAs are actively delivered to growth cones in their inactive precursor form. We next wondered whether they are locally processed into active mature miRNAs in this compartment. To address this, we first examined in Xenopus the distribution of Dicer, an endoribonuclease that cleaves pre‐miRNA. We observed a clear punctate immunoreactive signal in growth cones not only for Dicer but also for Ago2, a component of the miRNA‐induced silencing complex miRISC (Fig 5A and B). To confirm the bona fide axonal localization of Dicer, we used an endogenous epitope‐tagged (FLAG‐HA2)‐Dicer knock‐in mouse allele (Much et al, 2016) (Fig EV2A and B) and anti‐HA immunofluorescence (Fig EV2C). At E13.5, when RGC axons reach the chiasm (Bovolenta & Mason, 1987), Dicer was not detected in RGC axons marked with anti‐neurofilament antibody but instead appeared in cells of the optic nerve (Fig 5C, arrowheads). However, by P0, when axons start innervating their target centers (Godement et al, 1984), Dicer was detected within RGC axons where HA‐associated and axonal neurofilament‐associated signals clearly overlapped (Figs 5D and EV2D). This strongly suggests that Dicer may process pre‐miRNA into miRNAs within RGC axons during the developmental time window of axon targeting. It further points to the possibility that such processing might be triggered by cues expressed by the target region itself.
Figure 5. Dicer and Ago2 are present within axons.
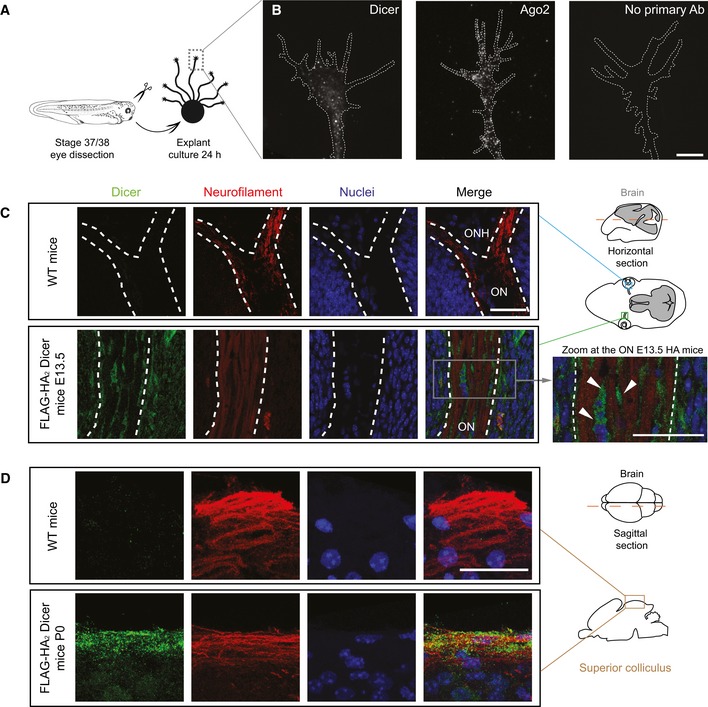
- Schematic representation of the experimental protocol.
- Representative Xenopus growth cones stained with anti‐Dicer and anti‐Ago2. White dashed lines delineate growth cones. Scale bars: 5 μm.
- Representative E13.5 mice brain cross‐section (dashed red line in the schematic) comprising the optic nerve (ON) stained with anti‐neurofilament and anti‐HA antibodies to detect RGC axons and (FLAG‐HA2)‐Dicer, respectively. Note the absence of HA signal in wild‐type (WT) mice. The white dashed line delineates the ON. Zoom of the triple‐stained ON (right panel): Dicer signal is detected inside ON cells surrounding axon bundles but not in axons per se (arrowheads). n = 3 independent experiments. WT, wild type; HA, (FLAG‐HA2)‐Dicer; E13.5, embryonic day 13.5; ONH, optic nerve head; ON, optic nerve. Scale bars: 30 μm.
- Representative P0 superior colliculus stratum griseum superficiale (SGS; sagittal sectioning of P0 brains along the dashed red line in the schematic, maximal projection of 5 μm depth). Note the presence of Dicer within the SGS marked with neurofilament. n = 3 independent experiments. WT, wild type; P0, postnatal day 0. Scale bars: 30 μm.
Figure EV2. Dicer is present in RGC axonal compartment.

- Schematic of FLAG‐HA2‐Dicer. Red arrows indicate primers used for the genotyping spanning the FLAG‐HA2 region. WT, wild type; HA, (FLAG‐HA2)‐Dicer.
- Genotyping PCR. WT, wild type; HA, (FLAG‐HA2)‐Dicer; P0, postnatal day 0; E13.5, embryonic day 13.5.
- Representative E13.5 retina stained for anti‐HA antibody. Dashed light blue box indicates the region zoomed. WT, wild type; E13.5, embryonic day 13.5. Scale bars: 50 μm.
- Representative P0 superior colliculus stained with anti‐neurofilament and anti‐HA antibodies to detect RGC axons in the stratum griseum superficiale of the superior colliculus and (FLAG‐HA2)‐Dicer, respectively. Dashed white box indicates the region zoomed. Zoom of the triple‐stained P0 superior colliculus in a nucleus‐free region. WT, wild type; P0, postnatal day 0. Scale bars: 30 μm.
Source data are available online for this figure.
We therefore next asked whether axonal pre‐miRNAs are locally processed into active mature miRNAs upon exposure to cues regulating axon targeting. To address this, we used isolated axons, which yield pure axons and growth cones responsive to cues (Appendix Fig S4A and B). We exposed isolated axons to the well‐described repellent cues, Sema3A and Slit2, at concentrations that generate protein synthesis‐dependent growth cone responses (Bellon et al, 2017) (Appendix Fig S4C). These cues are expressed within the optic tectum, a midbrain structure targeted by Xenopus RGC axons, and act as chemorepellent for these axons (Campbell et al, 2001; Piper et al, 2006). We then assessed by RT–PCR whether any of these cues triggered pre‐miRNA processing (Fig 6A). Pre‐miR‐181a‐1 and pre‐miR‐181a‐2 are derived from two distinct primary transcripts and both give rise to miR‐181a‐5p and, respectively, to miR‐181a‐1‐3p and miR‐181a‐2‐3p (Appendix Fig S5A and B). Upon Sema3A bath application of isolated axons, we detected a significant increase in mature miR‐181a‐5p, miR‐181a‐1‐3p, and miR‐181a‐2‐3p concomitant with a significant decrease in pre‐miR‐181a‐1 and pre‐miR‐181a‐2, but unchanged pre‐miR‐182, miR‐182‐5p, and miR‐182‐3p levels (Fig 6B and Appendix Fig S4D and E). Additionally, Slit2 exposure did not significantly alter the levels of any of the tested miRNAs (Fig 6C) or pre‐miRNAs. Interestingly, miR‐181a‐5p levels were far higher than miR‐181a‐1‐3p and miR‐181a‐2‐3p following Sema3A exposure (Appendix Fig S5C), suggesting that the 3p forms are rapidly degraded and unlikely to be functional. Taken together, these results suggest that Sema3A but not Slit2 triggers the processing of pre‐miR‐181a‐1 and to some extent of pre‐miR‐181a‐2 into mature miR‐181a‐5p, miR‐181a‐1‐3p, and miR‐181a‐2‐3p. Since pre‐miR‐182 expression levels are unaltered, our results further indicate that specific cues induce the maturation of specific axonal pre‐miRNAs.
Figure 6. Pre‐miRNAs are processed locally in response to cues and newly generated miRNAs are important for growth cone steering ex vivo .

-
ASchematic representation of the experimental paradigm. Concentrations used are as follows: 200 ng/ml Sema3A and Slit2.
-
B, CQuantification of miRNA and pre‐miRNA expression levels using the 2−ΔCt method and U6 as normalizer, upon Sema3A (B) or Slit2 (C) stimulation. Data are normalized to PBS control. Each data point represents a single RT–qPCR. n = 3 (B) and n = 4 (C) independent experiments. Values are mean ± SEM. ns, not significant.
-
DSchematic representation of the experimental paradigm. co‐MO, control morpholino.
-
E, FFrequency (in percentage) of collapsed growth cones from stage 37/38 embryos, following a 10 min Sema3A bath application. Concentration used is as follows: 200 ng/ml (Sema3A), 2 μM (MOs‐5p), (E) or 2 μM (MOs‐3p) (F). Total number of counted growth cones is reported in the column. Each data point represents one independent experiment. n = 3 independent experiments. Values are mean ± SEM. ns, not significant; co‐MO, control morpholino.
We examined whether Sema3A alters the trafficking dynamics of pre‐miR‐181a‐1 (Fig EV3A). We found that Sema3A but not Netrin‐1 nor Slit2 elicited an increase in the proportion of static puncta and a decrease in the proportion of fast‐moving puncta 7 min following cue exposure (Fig EV3B–D), suggesting that fewer puncta would reach and leave the growth cone. Puncta speed and directionality were unaffected (Fig EV3E and F). These data indicate that Sema3A‐induced processing and trafficking might be coupled.
Figure EV3. The fast transport of pre‐miR‐181a‐1 is decreased upon Sema3A exposure.
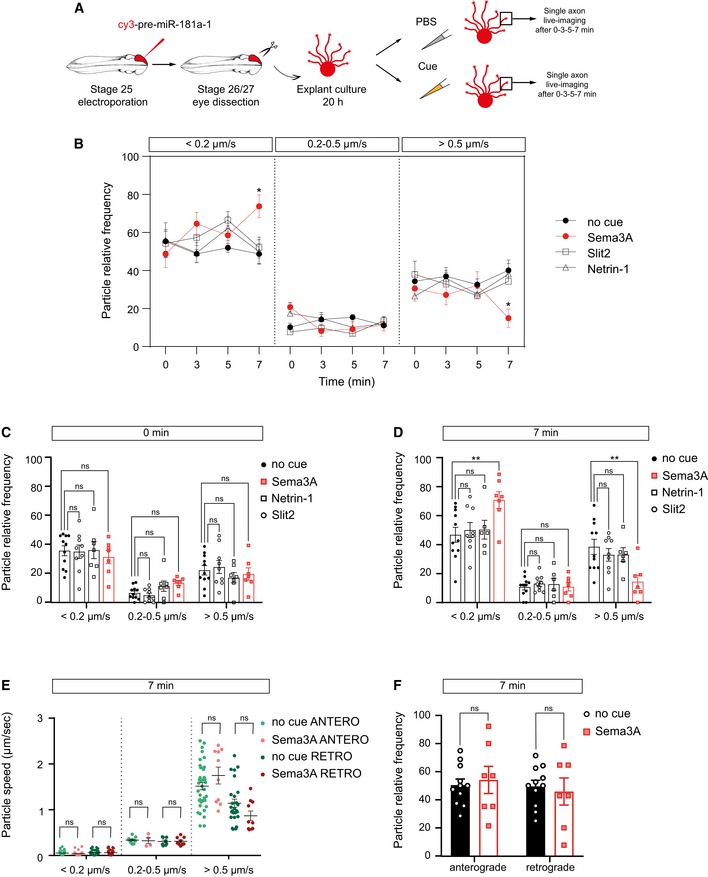
-
ASchematic representation of the experimental protocol. Two hundred to two hundred fifty nanogram per microliter cy3‐pre‐miRNA‐181a‐1, 100 ng/ml Netrin‐1, 200 ng/ml Sema3A, and 200 ng/ml Slit2 or no cue control (1× PBS) were bath applied to the cultures for up to 7 min. Zero minute in the graph corresponds to time‐lapse movies acquired before treatment. The same axon was analyzed for each condition at 0, 3, 5, and 7 min.
-
B–DFrequency distribution of tracked puncta. Each data point corresponds to one axon. Total number of puncta analyzed is as follows: 244, 221, 211, and 158 (no cue at 0, 3, 5, and 7 min); 231, 245, 203, and 178 (Slit2 at 0, 3, 5, and 7 min); 133, 160, 119, and 85 (Netrin‐1 at 0, 3, 5, and 7 min); 125, 134, 106, and 111 (Sema at 0, 3, 5, and 7 min). Total number of axons analyzed is as follows: 11 (no cue), 9 axons (Slit2), 7 axons (Netrin‐1), and 7 axons (Sema3A). n = 6 (no cue), n = 3 (Slit, Netrin), and n = 4 (Sema) independent experiments. Values are mean ± SEM. ns, not significant.
-
EAverage velocity of puncta. Each data point corresponds to one axon. Total number of puncta and axons analyzed is as follows: 150 puncta and 11 axons (no cue); 99 puncta and 7 axons (Sema3A). n = 4 (Sema3A) and n = 6 (no cue) independent experiments. Values are mean ± SEM. ns, not significant.
-
FFrequency distribution of tracked puncta. Each data point corresponds to one axon. Total number of puncta and axons analyzed is as follows: 150 puncta and 11 axons (no cue); 99 puncta and 7 axons (Sema). n = 4 (Sema) and n = 6 (no cue) independent experiments. Values are mean ± SEM. ns, not significant.
Newly generated miRNAs are important for growth cone steering ex vivo
We next explored whether newly generated miRNAs (NGmiRNAs) are important players in modulating growth cone behavior using a loss‐of‐function approach. We blocked miRNA biogenesis by preventing Dicer‐mediated cleavage using two separate mixes of MOs (MOs‐5p and MOs‐3p; Fig EV4A and Appendix Fig S5D and E). As expected, MOs‐5p and MOs‐3p both led to the strong reduction in mature miR‐181a isoforms (Fig EV4B and C), confirming that these MOs block miRNA biogenesis. Next, we assessed whether these MOs prevent Sema3A‐induced pre‐miR‐181a‐1 and pre‐miR‐181a‐2 processing from pure isolated axons ex vivo (Fig EV4D). While Sema3A induced the expected increase in axonal miR‐181a levels, MO‐transfected axons did not show such changes (Fig EV4E and F). This indicates that these MOs block Dicer‐induced pre‐miRNA cleavage in axons and also confirms the specificity of Sema3A‐induced pre‐miR‐181a‐1 and pre‐miR‐181a‐2 processing, described above (Fig 6B).
Figure EV4. Morpholino mix validation.
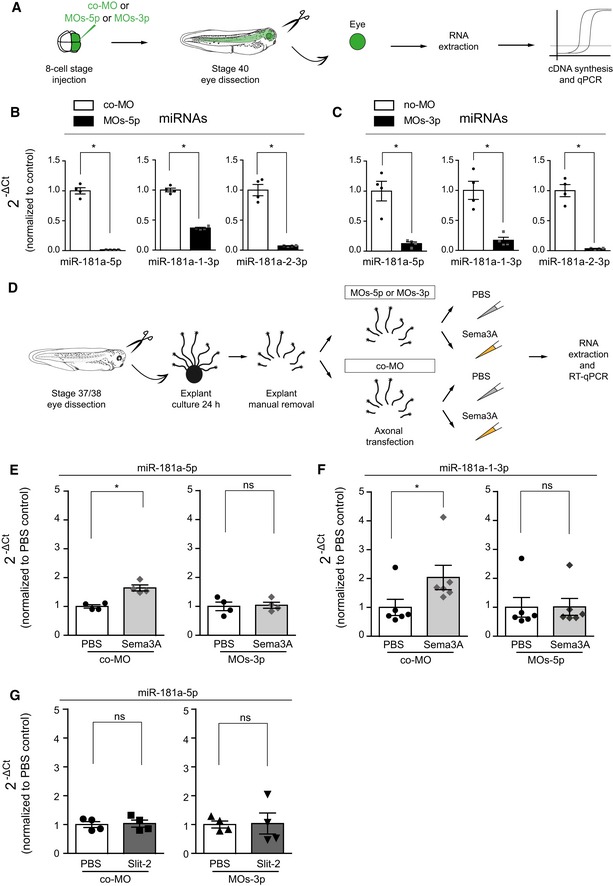
-
ASchematic representation of the experimental paradigm. co‐MO, control morpholino.
-
B, CQuantification of the expression levels of miRNAs using the 2−ΔCt method and U6 as normalizer. Concentrations used are as follows: 150–200 μM co‐MO or MOs‐5p or MOs‐3p. Data are presented normalized to co‐MO. Each data point corresponds to one independent experiment. n = 4 independent experiments. Values are mean ± SEM. co‐MO, control morpholino.
-
DSchematic representation of the experimental paradigm. co‐MO, control morpholino.
-
E–GQuantification of the expression levels of miR‐181a‐5p (E, G) and miR‐181a‐1‐3p (F) and using the 2−ΔCt method and U6. Concentrations used are as follows: 2 μM MOs‐5p or MOs‐3p or co‐MO, 200 ng/ml Sema3A or Slit‐2. Data are presented normalized to PBS control. Each data point represents a single RT–qPCR. n = 4 (E, G) and n = 6 (F) independent experiments. Values are mean ± SEM. ns, not significant
We then examined whether blocking pre‐miRNA processing, as above, impairs growth cone responsiveness to Sema3A (Fig 6D). We observed a significant reduction in Sema3A‐induced collapse in axons transfected with either MO mixes compared with co‐MO (Fig 6E and F). No such effect was observed when Slit2 was used instead (Fig EV4G). These results reveal that pre‐miR‐181a‐1 and pre‐miR‐181a‐2 processing is required for growth cone responsiveness to Sema3A. They further suggest that NGmiRNAs impinge on the Sema3A signaling pathway by targeting transcripts important for Sema3A‐mediated growth cone turning.
Mature miRNAs are important for axon targeting in vivo and for a fully functional visual system
As NGmiRNAs are essential for growth cone behavior ex vivo, we assessed whether they are also important for growth cone turning in vivo using a loss‐of‐function approach. For this, we electroporated a miR‐181‐MO cocktail blocking mature miRNA formation and function directly into stage 26 developing eyes, bypassing all earlier developmental stages, and examined axon projections at stage 40, when pioneer axons have reached the optic tectum (Holt & Harris, 1983) (Fig 7A). While long‐range guidance was unaffected, a significant subset of axons strayed within the optic tectum following aberrant trajectories (Fig 7B, arrows and C). Serial electroporation of miR‐181‐MO and of miRNA mimics into stage 26 embryos rescued the phenotype (Fig 7D and E, and Appendix Fig S6A–C), confirming the specificity of the miR‐181‐MO. Taken together, these results suggest that mature miRNAs are important for axon targeting in vivo. Since the onset of Dicer temporal expression occurs when axons are reaching their target (Fig 5D), these results overall suggest a restricted role of NGmiRNAs during this critical decision period.
Figure 7. Mature miRNAs are important for axon targeting in vivo and for a fully functional visual system.
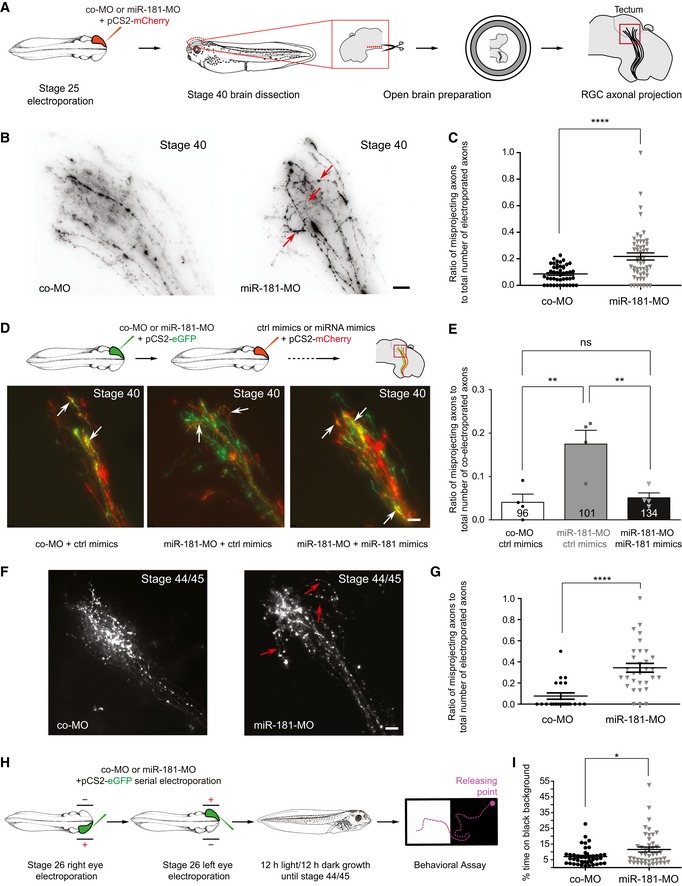
- Schematic representation of the experimental paradigm. Concentrations used are as follows: 0.5 μg/μl pCS2‐mCherry plasmid; 250 μM miR‐181‐MO or control MO cocktail. co‐MO, control morpholino.
- Representative images of RGC axons within the optic tectum. A subset of aberrantly projecting axons are indicated (red arrows). Note that a few straying axons are always observed within the wild‐type tectum. co‐MO, control morpholino. Scale bars: 20 μm.
- Quantification of misprojecting axons. Each data point corresponds to one brain. Total number of brains analyzed is as follows: 45 brains (co‐MO) and 52 brains (miR‐181‐MO). n = 4 independent experiments. Values are mean ± SEM. co‐MO, control morpholino.
- Schematic representation of the experimental paradigm. Concentrations used are as follows: 0.5 μg/μl pCS2‐mCherry or pCS2‐eGFP plasmids; 250 μM miR‐181‐MO or control MO cocktail; 50 μM miR‐181 or control mimics (top). Representative images of RGC axons within the optic tectum. White arrows indicate axons targeted both by MO (green) and by miRNA mimics (red; bottom). co‐MO, control morpholino. Scale bars: 20 μm.
- Quantification of misprojecting axons. The number reported on the bars is the total number of co‐electroporated axons. Note how miR‐181 mimics rescued aberrant misprojection of morphant axons in vivo. Each data point corresponds to one independent experiment. Total number of axons and brains analyzed is as follows: 96 axons and 4 brains (co‐MO + ctrl mimics); 101 axons and 4 brains (miR‐181‐MO + ctrl mimics), 134 axons and 4 brains (miR‐181‐MO + miR‐181 mimics). n = 4 independent experiments. Values are mean ± SEM. co‐MO, control morpholino; ns, not significant.
- Representative images of RGC axons within the optic tectum. A subset of aberrantly projecting axons are indicated (red arrows). Note that a few straying axons are always observed within the wild‐type tectum. co‐MO, control morpholino. Scale bars: 20 μm.
- Quantification of misprojecting axons. Each data point corresponds to one brain. Total number of brains analyzed is as follows: 21 brains (co‐MO) and 31 brains (miR‐181‐MO). n = 3 independent experiments. Values are mean ± SEM. co‐MO, control morpholino.
- Schematic representation of the experimental paradigm. Concentrations used are as follows: 0.5 μg/μl pCS2‐eGFP plasmid; 250 μM miR‐181‐MO or control MO cocktail. co‐MO, control morpholino.
- Frequency (in percentage) of the amount of time embryos spent on black background. Each data point corresponds to one embryo. Total number of embryos analyzed is as follows: 43 embryos (co‐MO) and 45 embryos (miR‐181‐MO). n = 4 independent experiments. Values are mean ± SEM. co‐MO, control morpholino.
By stage 44/45, a similar subset of miR‐181‐MO electroporated axons projected aberrantly within the tectum (Fig 7F, arrows and G) suggesting that this phenotype is not transient. Finally, we evaluated whether these straying axons impact vision by performing a behavioral assay which measures visual discrimination between white and black backgrounds (Viczian & Zuber, 2014) (Fig 7H). Contrary to blind embryos, WT embryos normally seek a light‐colored environment, therefore lingering little on a dark background (Appendix Fig S6D). Following serial miR‐181‐MO electroporation into right and left retinas at stage 26, stage 44/45 embryos displayed a significant decrease in the preference for the white background compared with co‐MO controls (Fig 7I). The miR‐181 family is therefore important to ensure accurate targeting of RGC axons within the tectum and subsequently for fully functional vision.
NGmiRNAs silence the locally translated transcript TUBB3
To explore the molecular mechanisms through which NGmiRNAs act, we reasoned that NGmiRNAs could either (i) silence translationally active transcripts acting as a switch, or (ii) prevent translational onset of axonal mRNAs as a fail‐safe mechanism to avoid spurious translation. We thus investigated the effect of NGmiRNAs on local translation.
We first identified putative direct targets of NGmiRNA miR‐181a‐5p, the most abundant miR‐181a variants in axon (Appendix Fig S5C), among axonal transcripts profiled by total RNA‐seq (see Methods for details, Table EV1). Since blocking NGmiRNA biogenesis impaired Sema3A‐induced collapse (Fig 6D and E), we inferred that under normal conditions, the NGmiRNA‐induced silencing of candidate mRNAs would assist the collapse response. It could do so by supporting mechanisms involved in repulsive turning such as by impairing cytoskeleton polymerization, impairing cell adhesion to laminin, and/or enabling Sema3A signaling. We selected one representative miR‐181 target candidate reflecting each of these three possibilities: tubulin beta 3 class III (TUBB3), a microtubule beta isotype needed for proper axon targeting (Poirier et al, 2010; Tischfield et al, 2010), thrombospondin 1 (THBS1), an adhesive glycoprotein mediating the interaction between cells and the extracellular matrix (Resovi et al, 2014), and amyloid beta precursor protein (APP) known to prevent Sema3A‐induced collapse (Magdesian et al, 2011).
We assessed whether local translation of these three candidate mRNAs could be regulated by NGmiRNAs using fluorescence recovery after photobleaching (FRAP) of a fast‐folding and fast‐bleaching translational reporter, Venus, carrying the 3′UTR of genes of interest (Ströhl et al, 2017; Wong et al, 2017) (Figs 8A and EV5A). After confirming that FRAP can be successfully used to measure LPS of a well‐established axonal mRNA, β‐actin (ACTB; Fig EV5B and C), we examined the de novo synthesis of TUBB3. Venus‐TUBB3‐3′UTR expressing growth cones exhibited rapid fluorescence recovery which was suppressed upon Sema3A exposure to recovery levels similar to Venus alone (Fig 8B and H). These data suggest that TUBB3 is rapidly synthesized in growth cones and that Sema3A prevents local translation of TUBB3. To test the role of mature miRNAs in mediating Sema3A‐induced repression of TUBB3, we mutated the two miR‐181a‐5p miRNA responsive elements (MREs) within the TUBB3 3′UTR to decouple Sema3A‐induced miRNA maturation from mRNA binding and silencing (Fig EV5A). Upon Sema3A exposure, fluorescence recovery of Venus‐TUBB3‐3′UTR(mut) was significantly higher than Venus‐TUBB3‐3′UTR(WT), similar to Venus‐TUBB3‐3′UTR(mut) without Sema3A, and abolished by CHX (Fig 8B and H). This indicates that Sema3A elicits mature miRNA activity, which in turn represses TUBB3 translation. Similar results were obtained when isolated axons were used (Fig 8C and I) or when pre‐miRNA processing was blocked with MOs‐3p prior to FRAP (Figs 8D and EV5D). Taken together, these results reveal that repression of TUBB3 translation is mediated by Sema3A‐activated NGmiRNAs locally within axons.
Figure 8. Newly generated miRNAs silence the locally translated transcript TUBB3.
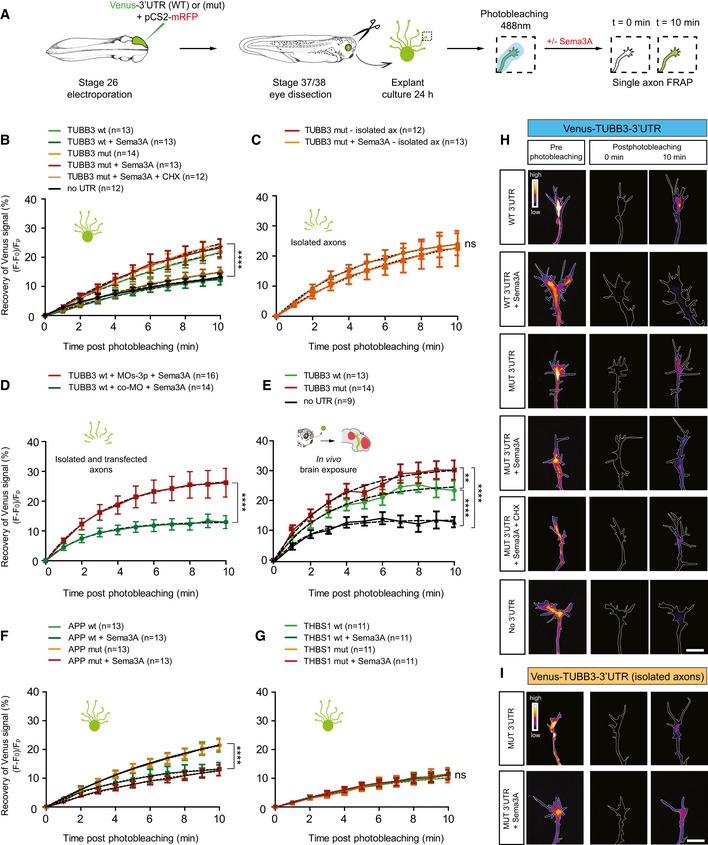
-
ASchematic representation of the experimental paradigm. Plasmid concentrations used are as follows: 1 μg/μl pCS2‐mRFP; 0.7 μg/μl pCS2 + Venus‐no 3′UTR or pCS2 + Venus‐3′UTR of interest. wt, wild type; mut, miR‐181‐5p responsive elements mutated.
-
B–GQuantification (in percentage) of the axonal fluorescence recovery after photobleaching (FRAP) of different Venus‐3′UTR constructs ex vivo and in vivo. Concentrations used are as follows: 200 ng/ml Sema3A; 100 μM cycloheximide; 2 μM co‐MO or MOs‐3p. Growth cones expressing Venus alone (no 3′UTR) displayed a minimal amount of recovery after photobleaching corresponding to Venus diffusion from adjacent, non‐bleached regions to the bleached growth cone (Wong et al, 2017). Ex vivo, whole explants (B, F, G), isolated axons (C), or isolated transfected axons (D) were used for FRAP analysis. In vivo (E), whole embryos with exposed brain were used. The electroporated eye was removed prior to mounting the embryo to eliminate somatic contribution. (B) 200 ng/ml Sema3A was bath applied with or without 100 μM cycloheximide (CHX, a translational blocker). Mutating TUBB3 3′UTR did not affect fluorescence recovery in basal conditions compared with WT indicating that mature miRNAs do not regulate the constitutive TUBB3 expression in distal axons. (E) The red area on the brain schematic indicates Sema3A expressing territories. Numbers of axons analyzed are reported between brackets. n = 5 (B), n = 4 (F), and n = 3 (C, D, E, G) independent experiments. Values are mean ± SEM. ns, not significant; wt, wild type; mut, miR‐181‐5p responsive elements mutated; CHX, cycloheximide; TUBB3, tubulin beta 3 class III; APP, amyloid beta precursor protein; THBS1, thrombospondin 1; co‐MO, control morpholino.
-
H, IRepresentative growth cones depicting Venus fluorescence intensity as a heatmap. Dashed white lines delineate the growth cones. wt, wild type; mut, miR‐181‐5p responsive elements mutated; CHX, cycloheximide; TUBB3, tubulin beta 3 class III. Scale bars: 10 μm.
Figure EV5. FRAP construct and experimental paradigm.
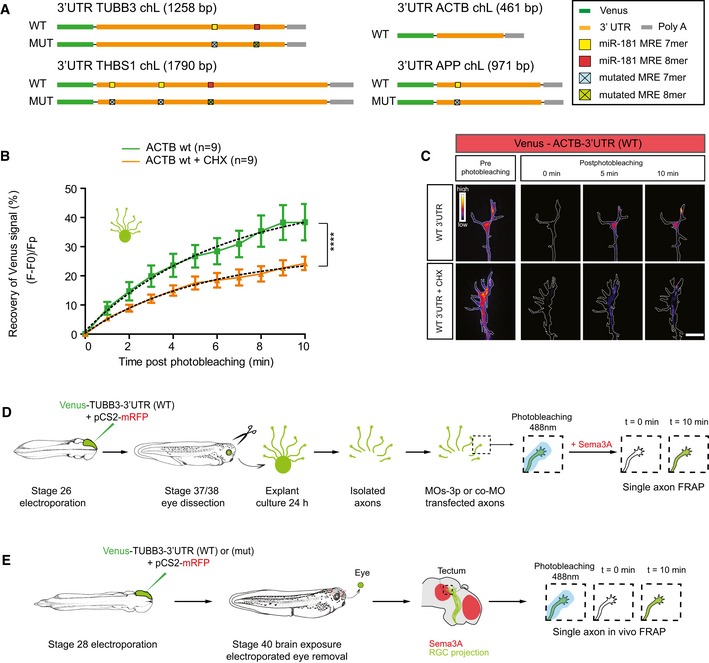
- Schematic of Venus‐3′UTR constructs used as reporters of axonal translation in FRAP experiments. ACTB, β‐actin; bp, base pair; TUBB3, tubulin beta 3 class III; APP, amyloid precursor protein; THBS1, thrombospondin 1; wt, wild type.
- Quantification (in percentage) of the axonal fluorescence recovery after photobleaching (FRAP) of pCS2‐Venus‐ACTB‐3′UTR construct ex vivo using whole explants. Concentration used is as follows: 0.7 μg/μl. Total number of axons analyzed is reported in the legend. n = 3 independent experiments. Values are mean ± SEM. ACTB, β‐actin; CHX, cycloheximide.
- Representative growth cones depicting Venus fluorescence intensity as a heatmap. Dashed white line delineates the growth cones. ACTB, β‐actin; CHX, cycloheximide; wt, wild type. Scale bars: 10 μm.
- Schematic representations of the experimental paradigm.
- Schematic representations of the experimental paradigm. Axons in the vicinity of tectal Sema3A expression territories were selected for FRAP analysis.
We next performed in vivo FRAP on somaless RGC distal axons within the tectum in the vicinity of Sema3A expressing territories (Figs 8E and EV5E). Venus‐TUBB3‐3′UTR(WT) displayed a rapid fluorescence recovery, significantly higher than Venus alone, but lower than Venus‐TUBB3‐3′UTR(mut). This indicates that mature miR‐181a‐5p normally represses and fine‐tunes TUBB3 local translation within RGC growth cones near sources of tectal Sema3A in vivo.
Finally, we assessed whether Sema3A‐induced NGmiRNAs modulate the translation of APP and THBS1 (Fig EV5A). Fluorescence from Venus‐APP‐3′UTR expressing axons recovered following photobleaching, and this recovery was significantly impaired when growth cones were exposed to Sema3A (Fig 8F). Recovery of Venus‐APP‐3′UTR(mut) and (WT) constructs did not, however, differ. This indicates that APP is locally translated in growth cones and that APP LPS is repressed by Sema3A independently from NGmiRNAs (Figs 8F and EV5A). Since APP interferes with Sema3A‐induced growth cone collapse (Magdesian et al, 2011), Sema3A may increase growth cone sensitivity to itself by controlling APP expression level. Furthermore, Venus‐THBS1‐3′UTR(WT) constructs showed similar recovery irrespective of Sema3A exposure and/or MRE mutation status, indicating that THBS1 is not locally translated in axons (Fig 8G). Collectively, this indicates that not all predicted miR‐181 targets are regulated by mature miRNAs within RGC axons.
Taken together, these results demonstrate that Sema3A inhibits basal translation of key molecules. They further reveal that NGmiRNAs are a major component of the Sema3A signaling pathway that is required for the repression of specific, translationally active transcripts within growth cones.
Endogenous TUBB3 is a critical target of NGmiRNAs in growth cone steering
Since the results obtained above employed a reporter system as an indirect readout of translational regulation of specific transcripts, we further investigated whether endogenous TUBB3 is similarly regulated. To address this, we used puro‐PLA, a technique that enables the detection of newly synthesized proteins in situ (tom Dieck et al, 2015) (Fig 9A). We detected that Sema3A exposure induced a decrease in TUBB3‐associated signal in growth cones of isolated axon culture compared with PBS control (Fig 9B–E) but not when pre‐miR‐181a‐1 processing was blocked with MOs‐3p (Fig 9D and E). Together, these data suggest that endogenous TUBB3 is silenced by the Sema3A‐NGmiRNA axis ex vivo specifically in axons.
Figure 9. Endogenous TUBB3 is a key mediator of NGmiRNA‐mediated Sema3A signaling.
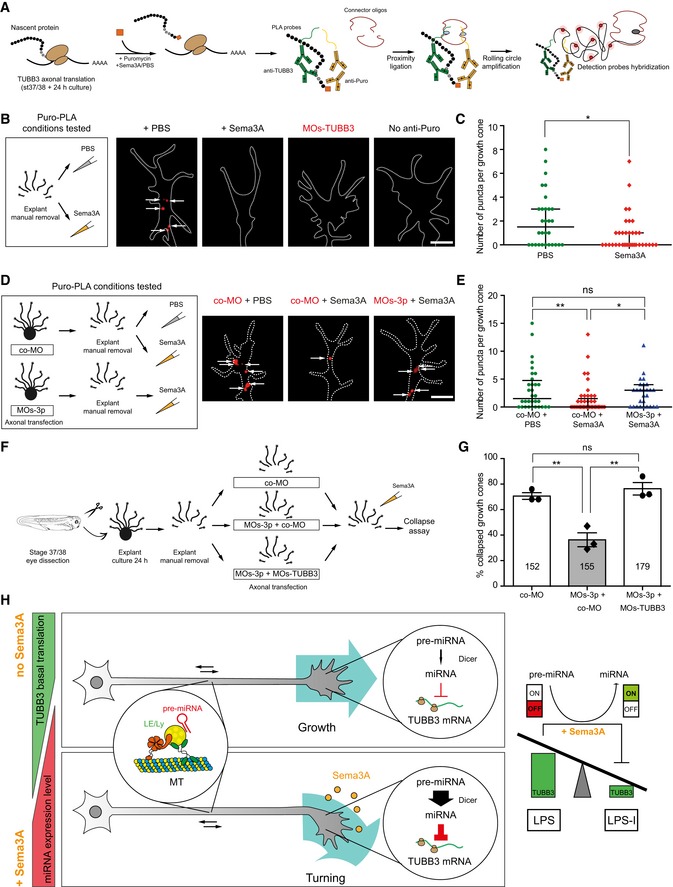
- Schematic representation of puromycin‐proximity ligation assay. Concentration used is as follows: 2 ng/μl (puromycin). TUBB3, tubulin beta 3 class III; PLA probes, proximity ligation assay probes.
- Schematic representation of the experimental paradigm. Representative images of local translation events of TUBB3 in the growth cone. Dashed white line delineates the growth cone. Each white arrow points to a puro‐PLA puncta. TUBB3, tubulin beta 3 class III; Puro, puromycin. Scale bars: 5 μm.
- Quantification of number of puncta per growth cone. Each data point corresponds to one growth cone. Total number of growth cones analyzed is as follows: 30 (PBS) and 36 (Sema3A). n = 2 independent experiments. Values are median with interquartile range.
- Schematic representation of the experimental paradigm. Concentrations used are as follows: 2 μM co‐MO; 2 μM MOs‐3p; 200 ng/ml Sema3A. Representative images of local translation events of TUBB3 in the growth cone of transfected axons. Dashed white line delineates the growth cone. White arrows represent number of puro‐PLA puncta per growth cone. co‐MO, control morpholino. Scale bars: 5 μm.
- Quantification of number of puncta per growth cone. Each data point corresponds to one growth cone. Total number of growth cones analyzed is as follows: 32 (co‐MO + PBS), 45 (co‐MO + Sema3A), 27 (MOs‐3p + Sema3A). n = 2 independent experiments. Values are median with interquartile range. co‐MO, control morpholino; ns, not significant.
- Schematic representation of the experimental paradigm. Concentrations used are as follows: 4 μM co‐MO; 2 μM MOs‐3p + 2 μM co‐MO; 2 μM MOs‐3p + 2 μM MOs‐TUBB3; 200 ng/ml Sema3A. co‐MO, control morpholino; TUBB3, tubulin beta 3 class III.
- Percentage of collapsed growth cones in axons transfected with co‐MO, MOs‐3p + co‐MO, and MOs‐3p + MOs‐TUBB3. Each data point corresponds to one independent experiment. Total number of growth cones counted is as follows: 152 (co‐MO), 155 (MOs‐3p + co‐MO), 179 (MOs‐3p + MOs‐TUBB3). n = 3 independent experiments. Values are mean ± SEM. ns, not significant; co‐MO, control morpholino; TUBB3, tubulin beta 3 class III.
- Working model: Pre‐miR‐181a‐1 is transported along RGC axons tethered to CD63‐positive late endosomes/lysosomes. Under non‐stimulated conditions, TUBB3 undergoes basal translation in the axonal compartment. Upon Sema3A exposure, pre‐miR‐181a‐1 is locally processed and the expression level of the concomitant newly generated miRNAs increases locally within the growth cone. miR‐181a‐5p, the predominant mature miRNA generated from pre‐miR‐181a‐1, targets TUBB3, thereby silencing protein synthesis through LPS inhibition (LPS‐I). ns, not significant; LE/Ly, late endosomes/lysosomes; LPS, local protein synthesis; LPS‐I, LPS inhibition; TUBB3, tubulin beta 3 class III.
Finally, we assessed whether endogenous TUBB3 is responsible for NGmiRNA‐mediated growth cone responsiveness to Sema3A. For this, we knocked down TUBB3 in axons transfected with MOs‐3p prior to Sema3A stimulation (Fig 9F). This double knockdown restored the responsiveness of growth cones to Sema3A (Fig 9G), suggesting that endogenous TUBB3 is a key mediator of NGmiRNAs in the Sema3A signaling pathway.
Discussion
In this study, we show that pre‐miR‐181a‐1 is actively transported, tethered to CD63‐positive vesicles along axonal microtubules to the growth cone central domain using a novel approach based on MB. Sema3A bath application of isolated axons leads to the processing of pre‐miR‐181a‐1 and pre‐miR‐181a‐2 into mature miRNAs in a cue and pre‐miRNA‐specific manner. These mature miRNAs are important for growth cone collapse ex vivo, axon guidance in vivo and necessary for a normally functioning visual system. Mechanistically, cue‐generated NGmiRNAs silence the basal translation of a specific transcript, TUBB3, at the growth cone ex vivo and in vivo. Collectively, our findings are consistent with a model in which pre‐miRNAs are delocalized to and stored within growth cones in an inactive form. Upon cue exposure, they are rapidly processed into active miRNAs to inhibit the basal local translation of transcripts, thereby ensuring accurate axon trajectories.
miRNAs are transported to the growth cone as inactive precursors via vesicles
Transport of mRNA as a means to delocalize genetic material is well‐described. While emerging evidence strongly suggests that ncRNAs and in particular miRNAs are differentially distributed in compartments (Kye et al, 2007; Lugli et al, 2008; Natera‐Naranjo et al, 2010; Iyer et al, 2014), mechanisms leading to the compartmentalization of ncRNAs are largely unexplored. It is technically very challenging to study transport of endogenous miRNAs due to their short length, making classic tools such as the long MS2 tagging ineffective. To investigate mechanisms of miRNA dynamics, we have adapted MB, a technique already employed by others to track miRNA localization, although not their transport dynamics [e.g., (Földes‐Papp et al, 2009)], and mRNA trafficking in living cells including in Xenopus RGC axons (Turner‐Bridger et al, 2018). The series of data we provide here collectively suggest that the MB we designed specifically detects pre‐miRNAs in axons. First, in RGC axons the MB is complementary only to pre‐miR‐181a‐1 as shown through combined Xenopus genome blast and axonal RNA‐seq analysis (see Methods and Table EV2). Second, the MB recognizes and binds to exogenous pre‐miRNA in vitro as revealed by the thermal denaturation assay and not to RNA lacking the MB target sequence. Third, at least 71% of MB colocalizes with its exogenous pre‐miRNA target within axons ex vivo and only very little (3%) to an unrelated pre‐miRNA. This level of colocalization appears particularly high considering that MB and pre‐miRNA may not be delivered to axons with similar copy numbers when using serial electroporation and that the MB likely also detects endogenous pre‐miRNAs besides the exogenous ones. Fourth, pre‐miRNA abundance detected by qPCR and number of MB‐positive puncta in axons change to a virtually equal extent (56 and 58% decrease, respectively) in response to impairment of primary miRNA processing (Fig 1H and I). Fifth, MB and pre‐miRNA trafficking dynamics are highly similar. MBs are thus a powerful new tool to specifically track pre‐miRNA transport.
Recent studies have provided great insight into axonal mRNA trafficking (Alami et al, 2014; Leung et al, 2018; Turner‐Bridger et al, 2018). Many components of pre‐miRNA dynamics that we report here resemble those of mRNA in axons. We document similar bidirectional trafficking (Leung et al, 2018; Turner‐Bridger et al, 2018) and faster anterograde than retrograde transport (Turner‐Bridger et al, 2018). Maximal velocities and average speed of active puncta are also comparable (Leung et al, 2018; Turner‐Bridger et al, 2018). In contrast to our study, the dominant mRNA trajectories previously measured (Turner‐Bridger et al, 2018) were confined and diffusive, with only a small proportion being directed, while the majority of puncta we detected adopted an overall active and directed trajectory. This disparity in motion type frequencies may be due to technical disparities, or reflect actual biological differences. In axons, mRNA can be reused for multiple rounds of translation, while pre‐miRNA cannot be reutilized after they have been processed. It is thus tempting to speculate that a constant supply of fresh pre‐miRNAs may be required to replenish the growth cone pre‐miRNA storage. We calculated (see Methods for details) that pre‐miRNA diffusion to the growth cone would take 20 days on average, while it takes merely 2 days for X. laevis RGC axons to navigate to their main target (Holt & Harris, 1983). Thus, rapid active transport would be required to match the ever‐changing demands of fast‐elongating axons.
We show that Sema3A increases the proportion of static pre‐miR‐181a‐1 puncta in axons at the expense of fast‐moving ones. As a consequence, Sema3A would reduce the number of pre‐miRNA molecules reaching and leaving the growth cones. This effect appears to be specific to Sema3A as neither Netrin‐1 nor Slit2 perturbs the axonal trafficking of this miRNA. Since Slit2 does not lead to pre‐miR‐181a‐1 processing, it is tempting to speculate that Sema3A regulation of miRNA precursor processing and trafficking is coupled. Sema3A‐induced NGmiRNA production would cease when the local supply of precursor runs out and is not replenished due to decreased trafficking. This would result in a discrete, self‐limiting burst of miRNA production, ensuring that only a specific number of NGmiRNAs are generated in response to cue to induce appropriate mRNA silencing.
Intriguingly, we find that pre‐miRNAs are retrogradely transported. We speculate that the retrograde transport of a pool of pre‐miRNAs could support various physiological mechanisms. Similarly to the sushi‐belt model of mRNA localization proposed in dendrites (Doyle & Kiebler, 2011), pre‐miRNAs may be scanning the axons until they are required for mRNA silencing at the growth cone. It is thus interesting that we observe an increase in static pre‐miRNA puncta upon Sema3A stimulation. Retrograde transport could also be a means to preserve the required copy number of pre‐miRNAs within the growth cone and ship back any extra molecules, thereby ensuring that miRNA:mRNA pair stoichiometry is maintained for adequate silencing. Finally, retrogradely transported pre‐miRNAs could act as a long‐range intracellular messenger and inform the soma of events occurring at the periphery. Such mechanisms have been documented for locally synthesized proteins to promote neuron survival and regeneration responses (Panayotis et al, 2015).
We reveal that axonal pre‐miRNAs hitchhike onto LE/Ly. Although we do not know how pre‐miRNA docks to LE/Ly, it is possible that it employs a similar lysosome molecular tether as that recently described for RNA granules (Liao et al, 2019). Remarkably, such non‐canonical hitchhiking onto membrane‐bound vesicles to achieve subcellular motility has been described for a small subset of cargoes (Salogiannis & Reck‐Peterson, 2017). In particular, mRNAs in fungi were found to translocate docked to cytoplasmic surface of early endosomes (Baumann et al, 2012, 2014). This suggests that the pre‐miRNA trafficking mode belongs to an ancient and evolutionary conserved transport system that spans across species and subcellular compartments. Intriguingly, mRNAs were recently shown to associate with early and late endosomes in developing Xenopus RGC axons (Cioni et al, 2019). Contrary to pre‐miRNAs, mRNAs appear to utilize these organelles as a docking platform for translation rather than for axonal trafficking per se. Why are pre‐miRNAs transported by vesicles in vertebrate axons while mRNAs employ a canonical ribonucleoprotein particle‐based transport? It is possible that vesicles are linked to inactive regulators, such as pre‐miRNAs, so that translation can be modulated rapidly, locally, and on demand once these regulatory molecules are activated. Future work will be required to unravel whether NGmiRNAs indeed silence vesicle‐associated mRNAs.
Basal local protein translation of TUBB3 is silenced by Sema3A‐induced NGmiRNAs
A widely accepted view posits that local translation in axons is triggered by stimuli, by either chemotropic and maturation cues during development or under injury conditions in adults (Cioni et al, 2018). For instance, Sema3A induces the synthesis of proteins that elicit cytoskeletal remodeling and steering (Campbell et al, 2001; Wu et al, 2005). Here, however, we report the basal translation of APP, TUBB3, and β‐actin in individual axons elongating ex vivo on laminin substrate in the absence of chemotropic and trophic cues. This is in agreement with a few studies that have documented the basal translation of specific transcripts (Eng et al, 1999; Taylor et al, 2013; Preitner et al, 2014; Batista et al, 2017). A recent report furthermore revealed wide‐scale protein synthesis occurring in isolated unstimulated X. laevis axons within minutes (Cagnetta et al, 2018). These newly synthesized proteins represent one‐third of the total axonal proteome, suggesting the existence of an unsuspected rich and complex basal translatome.
While the induction of global translation by chemotrophic cues is well‐established, very little is known about the fate of the basal translatome upon cue exposure. Here, using single axon FRAP of Venus translational reporter constructs, we reveal that Sema3A rapidly suppresses the basal translation of TUBB3 and APP ex vivo. This is in line with two other studies which also measured cue‐induced decreases in the translation of specific molecules in distal axons (Yao et al, 2006; Cagnetta et al, 2018). Overall, it is conceivable that two cue‐activated pathways may co‐exist in parallel to regulate the expression of two separate sets of proteins: a dominant pathway eliciting a burst of LPS and a secondary pathway inducing a trough of LPS or “LPS inhibition” (LPS‐I). Both cue‐induced LPS and LPS‐I may ultimately lead to cytoskeleton remodeling and changes in growth cone behavior. LPS‐I may be used as an alternative to proteasome degradation, which is not systematically employed for cue‐mediated growth cone response (Campbell et al, 2001).
One key unresolved question is how cues inhibit basal LPS in axons. Here, we provide a series of evidence demonstrating that cue‐induced LPS‐I can be mediated by NGmiRNAs. Using single axon FRAP of Venus‐TUBB3‐3′UTR constructs, we show that a cue‐induced miRNA silences TUBB3 translation locally and that this effect is not due to a generic activation of miRNAs but to the cue‐induced local biogenesis of specific miRNAs. Overall, our data suggest that an RNA‐based signaling pathway exists, composed of mRNA and NGmiRNAs as two serially connected components of the same regulatory circuit. In response to cue, NGmiRNAs impinge on basal LPS of mRNAs to induce LPS‐I. LPS and LPS‐I are thereby coupled and coordinately regulated by NGmiRNAs acting on 3′UTR regulatory motifs. This coupling may generate a crucial leverage point for repellent cues to quickly and accurately adjust the desired level of individual proteins, including tubulin isotypes, within growth cones. Since Sema3A‐induced growth cone turning does not depend on proteome degradation, contrary to other cues such as Netrin‐1 (Campbell et al, 2001), this RNA‐based mechanism may be crucial to regulate rapid changes in protein expression in response to proteasome‐independent cues. To a large extent, this type of RNA‐based signaling would allow to tightly control the rate and type of protein production for cytoskeletal remodeling, and thereby confer a higher order of regulatory potential to ensure the exquisite precision required for brain wiring.
We speculate that Sema3A‐induced ERK1/2 signaling, involved in triggering LPS (Campbell & Holt, 2003), may also elicit LPS‐I. It may do so by generating specific NGmiRNAs via two mechanisms. First, NGmiR‐181 might be produced following the phosphorylation/activation of a Dicer cofactor. Indeed, activation of given cofactors, such as TRBP, bestows specificity to the Dicer complex (Pullagura et al, 2018) (Venigalla & Turner, 2012; Darnell, 2013). Furthermore, in neuronal progenitor cells, TRBP becomes phosphorylated upon ERK activation and promotes miR‐181a maturation (Xu et al, 2015). It is therefore possible that Sema3A activates ERK pathway leading in turn to the phosphorylation/activation of TRBP and pre‐miR‐181a‐1 processing. Second, NGmiR‐181 might be generated following the phosphorylation/release of a different, as yet unknown RNA‐binding protein (RBP). The binding affinity of specific RBPs for their target RNAs can change depending on their phosphorylation status (Venigalla & Turner, 2012; Darnell, 2013). Furthermore, RBPs can compete with Dicer to hinder pre‐miRNA processing (Bicker et al, 2013). It is thus conceivable that pre‐miR‐181a‐1 is bound by a specific RBP, preventing Dicer from accessing its cleavage site. Upon activation of the Sema3A‐induced ERK signaling, such hypothetical RBP would be phosphorylated and release its target. Pre‐miRNA would thereby be freely accessible for Dicer cleavage. Further investigation will be necessary to help uncover the signaling pathway that confers specificity to the pre‐miRNA processing.
What is the biological implication of the NGmiRNA‐triggered LPS‐to‐LPS‐I switch for axon development? We uncover that this switch mediates cue‐induced growth cone steering. More specifically, we reveal that Sema3A triggers growth cone collapse response ex vivo and axon targeting in vivo through NGmiRNAs. NGmiRNAs, in turn, silence TUBB3 mRNA translation upon Sema3A exposure ex vivo and in the vicinity of Sema3A expressing territories in vivo. Taken together, these data suggest that Sema3A‐induced NGmiRNAs lead to the silencing of MT tubulin isotype TUBB3, MT depolymerization, and ultimately, growth cone steering. Collectively, our data thus support a model whereby Sema3A‐produced NGmiRNAs elicit a rapid shift in axon behavior from axon elongation supported by basal TUBB3 LPS, to growth cone collapse prompted by TUBB3 LPS‐I (Fig 9H). According to this model, blocking NGmiRNA production prevents Sema3A‐induced LPS‐I of TUBB3 leading to the persistent production of TUBB3 and the maintenance of MT throughout the growth cone. As a consequence, growth cones fail to collapse ex vivo and aberrantly continue to grow and meander in vivo, consistently with the observed phenotype.
In conclusion, our results reveal that inactive pre‐miRNAs are actively transported to local sites for miRNA biogenesis and function, similarly to the subcellular translocation and subsequent local translation of silent mRNAs into functional proteins. At the growth cone, cue‐induced NGmiRNAs impinge on local protein production by inhibiting basal LPS of their target mRNAs, thereby contributing to changing growth cone direction. This type of ncRNA‐based signaling pathway constitutes an additional regulatory layer in brain wiring. Many drug targets are detected within specific subcellular compartments. However, drug design does not often incorporate strategies for subcellular delivery (Rajendran et al, 2010). Since miRNA‐based therapy using miRNA mimics and anti‐miRs is emerging as promising new therapeutics (Rupaimoole & Slack, 2017), our results lay important ground for the design of new clinical tools based on the targeted delivery and local activation of miRNAs.
Methods
Xenopus laevis
Xenopus laevis embryos were obtained by in vitro fertilization, raised in 0.1× MMR, pH 7.5, at 14–22°C, and staged according to Nieuwkoop and Faber (1994). All animal experiments were approved by the Italian “Ministero della Salute” with the authorization no. 1159/2016‐PR and no. 546/2017‐PR according to art.31 of D.lgs. 26/2014.
Mus musculus
Flag‐HA2‐Dicer C57BL6 mice (Comazzetto et al, 2014) were kindly donated by Dónal O'Carroll. Mice were housed and maintained, in accordance with the Decreto Legislativo 4 marzo 2014, no. 26. All animal experiments were approved by the Italian “Ministero della Salute” with the authorization no. 1159/2016‐PR according to art.31 of D.lgs. 26/2014.
Xenopus retinal organoculture
Glass coverslips (Bellco) or glass‐bottom dishes (MatTek) were subsequently coated with poly‐l‐lysine (Sigma, 10 μg/ml) and with laminin (Sigma, 10 μg/ml). Eyes were dissected from anesthetized embryos and cultured at 20°C for 20–24 h in 60% L‐15 and 1% antibiotic‐antimycotic (Thermo Fisher).
Electroporation
Stage 26 embryos (or stage 28 for the in vivo FRAP experiment, Figs 8E and EV5E) were anesthetized in 0.3 mg/ml MS222 (Sigma) in 1× MBS. The nucleic acid mixture was injected in retinal primordium using a 1.0 mm outer diameter (OD) × 0.5 mm or 0.78 mm internal diameter (ID) glass capillary (Harvard Apparatus). After injection, the mixture was delivered at 18V, by applying eight electric pulses of 50‐ms duration at 1000‐ms intervals, except for in vivo FRAP experiment, where two pulses were applied instead of eight.
Collapse assay
Explant cultures were bathed in 200 ng/ml human recombinant Sema3A‐FC (R&D System), 200 ng/ml Slit2 (R&D System), or 1× PBS (for control) for 10 min and then fixed. Postfixation, coverslips were washed three times with 1 ml 1× PBS (Gibco), mounted in ImmunoHistoMount (Sigma), and collapsed growth cones (central part of the growth cone shrunk and less than two filopodia present) were counted blind. For isolated axon preparations, axons closer than 100 μm to the cut were excluded from the count, and collapsed axons counted in PBS wash without mounting it.
Drug bath application
Two hundred nanogram per milliliter human recombinant Sema3A‐FC (R&D System), 200 ng/ml Slit2 (R&D System), and 100 ng/ml Netrin‐1 or PBS (for control) were bathed to explant culture for 10 min and then fixed. 0.1 μM vincristine (Alexis Biochemicals, kind gift from HTS facility, CIBIO) or 2.4 μM of nocodazole diluted in 60% L‐15 and 1% antibiotic–antimycotic (Thermo Fisher) was added to stage 26/27 cultured explants at a concentration sufficient to rapidly disrupt MT in Xenopus RGC axons (Leung et al, 2018). Live imaging was performed before and 30 min after nocodazole or vincristine treatment.
Isolated axon
Axons were severed from the explants at the stereomicroscope by manually cut them with 0.20‐mm pins and gently removing the dislodged entire eye explant from the plate with a p10 pipette. The complete experimental procedure was concluded within 1.5 h after the first cut.
Axonal transfection
NeuroMag Transfection Reagent (OZ Biosciences) was mixed and incubated for 20 min with MOs in a 1:100 proportion, added to the culture, and incubated for 15 min on the magnetic base (OZ Biosciences). After transfection, the plates were incubated for 30 min at 20°C, followed by three consecutive washes of 200 μl culture medium.
RT–qPCR
Pre‐miRNA expression levels were investigated using Power SYBR Green PCR Master Mix (×2) (Applied Biosystem) and 0.25 μl of each 10 μM primer, while TaqMan™ Universal Master Mix II (Thermo Fisher) was used for miRNA amplification with the following TaqMan™ MicroRNA Assay (Table EV4). For RT–qPCR quantitative analysis, cycle threshold (Ct) was defined with CFX96 Bio‐Rad software v3.1, as mean of three technical replicates per sample. All technical replicates have a standard deviation smaller than 0.35. All Ct values are smaller than 35. Amplification efficiency of the new designed primers (pre‐miR‐181a‐1, pre‐miR‐181a‐2, and pre‐miR‐182) was investigated with standard curves independently from the actual experiments. To calculate miRNA or pre‐miRNA differential expression, ΔCt method (Schmittgen et al, 2008) was applied as follows: .
miR‐181 target prediction and candidate selection
Xenopus laevis miR‐181a targets were predicted using custom scripts and Perl scripts downloaded from the TargetScan website (TargetScan 6 (Garcia et al, 2011)), with the X. laevis 3′UTR sequences. All predicted human and mouse miR‐181a targets, including those with poorly conserved target sites but removing non‐canonical ones, were downloaded from TargetScanHuman 7.1 and TargetScanMouse 7.1 (Garcia et al, 2011), respectively.
Axonally present predicted miR‐181a‐5p X. laevis targets with an annotated 3′UTR of at least 50 bp length were selected, and, using their human ortholog's NCBI Entrez ID, screened against the following Reactome (Fabregat et al, 2016) pathways of interest: axon guidance (R‐HSA‐422475) and integrin cell surface interactions (R‐HSA‐216083). In addition, we filtered the obtained predicted miR‐181a‐5p targets for those that are within the top 20% of predicted targets, according to TargetScan ContextScore, and are also predicted to be targeted by miR‐181‐5p in human or mouse (Table EV1). This filtering resulted in 526 predicted targets (Table EV1).
Pre‐miRNA candidate selection
Axonal pre‐miRNA candidates were selected from the published data [GEO: GSE86883 (Bellon et al, 2017)]. The two libraries yielded 7.8 and 10.8 million reads. Candidate pre‐miRNAs were identified by reads spanning at least the entire loop sequence of the pre‐miRNA. The sequencing data were mapped to the available Xenopus tropicalis, X. laevis, and Danio rerio pre‐miRNA sequences present in miRBase v21 (Bellon et al, 2017). For the pre‐miRNA candidate selection, all sequences were required to perfectly match to X. laevis, to check these sequences were blasted against the J‐strain 9.2 Genome in Xenbase (using the default settings E value 0.1, BLOSUM62 matrix).
Vesicles characterization and pre‐miRNA colocalization
Xenopus laevis electroporated eyes were dissected at stage 26/27 and cultured for 40 h at 20°C. Images were acquired after fixation (Fig 4C, E, and G) or after a 30‐min incubation with 50 nM LysoTracker Red (Thermo Fisher, kind gift from Paola Bellosta) with subsequent phenol red‐free 60% L15 (Gibco) washes (Fig 4G and Appendix Fig S3E and F). Images were performed with an inverted Nikon Ti2 microscope equipped with a Plan Apochromatic 60x/1.4 Oil PH3 objective and a Andor Zyla 4.2 PLUS camera (4.2 Megapixel). The acquisition mode was set at 12 bit with gain 4 and 540 MHz. Fluorescent images were taken using Lumencor Spectra X LED light source with excitation spectra 470/24 nm, 550/15 nm, and 640/30 nm for eGFP, mRFP/Cy3, and Cy5, respectively. The concentration and mixture used in each experiment are specified in the figure legends.
Colocalization analysis was performed using ImageJ. An axon length of 50–100 μm measured from the growth cone wrist was analyzed. Within this area, the number of positive vesicles for each fluorophore and for multiple fluorophores (colocalized vesicles) was counted. The percentage of colocalization was measured for each axon. Isolated electroporated axons were selected strictly avoiding bundles, and the ones containing < 5 puncta in any fluorophore were excluded to avoid biasing the data. Colocalization percentages were calculated for each axon.
Single axon analysis
Axons were defined as single when their distal end was not fasciculated with other axons and were not touching any neighboring cell body, axons, or growth cones.
Live imaging of pre‐miR trafficking
Time lapses for pre‐miR‐181a‐1 trafficking analysis consisted of 150–300 consecutive frames recorded continuously, for a total time of 17–44 s, respectively, with a 0.144‐s delay between two consecutive frames, while it consisted of 130 frames per channel for co‐trafficking with CD63. The distal end of single axons was chosen by the phase for imaging, strictly avoiding bundles, and had to comprise a stereotypical growth cone.
The axon segment was selected strictly avoiding bundles, 50–100 μm to the growth cone and at least 100 μm far from the soma.
Movies were mapped back onto kymographs of the live pre‐miR‐181a‐1, MB, or CD63 movement using ImageJ. The “KymoReslicedWide” FIJI/ImageJ plugin was used to generate kymographs from time‐lapse movies, and when necessary, a “Bleach Correction” FIJI/ImageJ plugin was used (author: Kota Miura + Jens Rietdorf EMBL Heidelberg). For pre‐miR‐181a‐1 trafficking studies, the specific macro‐tsp050706.txt for FIJI/ImageJ software (author: J. Rietdorf FMI Basel + A. Seitz EMBL Heidelberg) was used to extrapolate from the tracked trace information about velocity and spatial directionality of the puncta. The plugin was custom‐modified to also obtain puncta directionality. Puncta's velocity was calculated by considering the average speed of its segmental components.
For co‐trafficking analysis, the two kymographs (MB and CD63‐eGFP puncta) were merged and artificial color assigned using FIJI. Overlapping trace was considered as co‐transported puncta when CD63 trace overlapped over the entire trajectory of the MB trace. Puncta were considered moving when the spatial displacement was greater than zero. The two kymographs (MB and CD63‐eGFP puncta) were used to extrapolate puncta directionality.
For live imaging upon treatment (Fig EV3), movies were 16.5 s long (115 frames in total). Movies were acquired on the same axon before treatment (0 min) or at 3, 5, or 7 min after treatment with a given cue at 100‐ms exposure using LasX software (Leica). Movies were subsequently mapped back onto kymographs of the live pre‐miR‐181a‐1 movement using ImageJ. Static puncta were not included in directionality analysis. Only single axons were analyzed.
FRAP analysis
The mean intensity of the Venus signal normalized per growth cone area was measured with Volocity 64× software (Figs 8B, C and E‐G, and EV5B) or ImageJ (Fig 8D), by manually tracing the terminal of RGC axons on the sum of the z‐stack for that specific time point using mRFP signal. From the background‐corrected fluorescence intensity at each time point (F), the fluorescence signal after photobleaching (F 0) was subtracted and normalized to the fluorescence signal pre‐photobleaching: FRAP = (F −F 0)/F p.
Fluorescence recovery after photobleaching data were reported as curves of the recovered fluorescence signal in a 10‐min timeframe. The different curves were described by fitting the data with a non‐linear model (“one‐phase decay” option in Prism), and differences between conditions were analyzed for statistical significance with an extra sum‐of‐square F‐test. Each axon acquired belonged to a different eye explant. To avoid technical biases, the order of processing of the stimulation conditions with Sema3A, MO and co‐MO samples, or WT and mutated was randomized both in terms of organoculture and acquisition. Only single axons were analyzed.
Statistic analysis
All data were analyzed with Prism (GraphPad 6 or GraphPad 7). For all tests, the significance level was α = 0.05. Exact number of replicates, tests used, qPCR fold changes, and statistics are reported in Table EV3.
Data and software availability
The GEO entry for the RNA sequencing data reported in this paper for the miR‐181a‐5p target candidate selection (Table EV1) is GSE124168.
Author contributions
Conceptualization: M‐LB; Methodology: AG, MR; Software: SS, CA‐G; Formal analysis: all authors; Investigation: EC, IDC, AG, AI, MR, TAO, EO, SB, DV, GSR; Writing—original draft: EC, M‐LB; Writing—review & editing: all authors; Visualization: EC, IDC, AG, MR, AI, TAO, EO, SB, SS, DV; Supervision: EC, MR, GS, CA‐G, M‐LB; Project administration and funding acquisition, M‐LB.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7
Source Data for Expanded View
Review Process File
Acknowledgements
The authors would like to thank Giovanna Berto for help with IF on transgenic mice; Charlotte Hardion and Sara Longhi for help with plasmid design; Emanuela Kerschbamer for help with python script; Benita Turner‐Bridger for discussions on MB technology; Daniele Arosio, Angela Re, and Silvia Pizzini for help with MSD analysis; Julie Lin and Hovy Ho‐Wai Wong for help with ex vivo and in vivo single axon FRAP; Joshua Fortriede (Xenbase) for help with MO off‐target analysis; Marco Canossa's and Marino Zerial's group for help with super‐resolution pilot experiments; and C.E. Holt for granting access to HH Lab Spinning disk confocal microscope, Institute for biomedicine EURAC imaging, CIBIO Imaging, and MOF facilities. We also would like to thank Nick Ingoglia for stimulating discussions; and Paolo Macchi, Giovanni Stefani, Dario Bonanomi, and Jean‐Michel Cioni for critically reading the manuscript. This study was supported by Erasmus+ Travel grant (E.C.), University of Trento PhD studentship (to A.I.), Fondazione AIRC (IG grants #21315 to G.S.; #20366 to D.V.), Marie Curie Career Integration (618969 GUIDANCE‐miR), G. Armenise‐Harvard Foundation Career, and MIUR SIR (RBSI144NZ4) grants (to M.‐L.B.).
The EMBO Journal (2020) 39: e102513
References
- Alami NH, Smith RB, Carrasco MA, Williams LA, Winborn CS, Han SSW, Kiskinis E, Winborn B, Freibaum BD, Kanagaraj A et al (2014) Axonal transport of TDP‐43 mRNA Granules Is Impaired by ALS‐Causing Mutations. Neuron 81: 536–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschrafi A, Schwechter AD, Mameza MG, Natera‐Naranjo O, Gioio AE, Kaplan BB (2008) MicroRNA‐338 regulates local cytochrome c oxidase IV mRNA levels and oxidative phosphorylation in the axons of sympathetic neurons. J Neurosci 28: 12581–12590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista AFR, Martínez JC, Hengst U (2017) Intra‐axonal synthesis of SNAP25 is required for the formation of presynaptic terminals. Cell Rep 20: 3085–3098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer KE, Kiebler MA, Segura I (2017) Visualizing RNA granule transport and translation in living neurons. Methods 126: 177–185 [DOI] [PubMed] [Google Scholar]
- Baumann S, Pohlmann T, Jungbluth M, Brachmann A, Feldbrügge M (2012) Kinesin‐3 and dynein mediate microtubule‐dependent co‐transport of mRNPs and endosomes. J Cell Sci 125: 2740–2752 [DOI] [PubMed] [Google Scholar]
- Baumann S, König J, Koepke J, Feldbrügge M (2014) Endosomal transport of septin mRNA and protein indicates local translation on endosomes and is required for correct septin filamentation. EMBO Rep 15: 94–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellon A, Iyer A, Bridi S, Lee FCY, Ovando‐Vázquez C, Corradi E, Longhi S, Roccuzzo M, Strohbuecker S, Naik S et al (2017) miR‐182 regulates Slit2‐mediated axon guidance by modulating the local translation of a specific mRNA. Cell Rep 18: 1171–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bicker S, Khudayberdiev S, Weiss K, Zocher K, Baumeister S, Schratt G (2013) The DEAH‐box helicase DHX36 mediates dendritic localization of the neuronal precursor‐microRNA‐134. Genes Dev 27: 991–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bovolenta P, Mason C (1987) Growth cone morphology varies with position in the developing mouse visual pathway from retina to first targets. J Neurosci 7: 1447–1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabili MN, Dunagin MC, McClanahan PD, Biaesch A, Padovan‐Merhar O, Regev A, Rinn JL, Raj A (2015) Localization and abundance analysis of human lncRNAs at single‐cell and single‐molecule resolution. Genome Biol 16: 20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagnetta R, Frese CK, Shigeoka T, Krijgsveld J, Holt CE (2018) Rapid cue‐specific remodeling of the nascent axonal proteome. Neuron 99: 29–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DS, Regan AG, Lopez JS, Tannahill D, Harris WA, Holt CE (2001) Semaphorin 3A elicits stage‐dependent collapse, turning, and branching in Xenopus retinal growth cones. J Neurosci 21: 8538–8547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DS, Holt CE (2003) Apoptotic pathway and MAPKs differentially regulate chemotropic responses of retinal growth cones. Neuron 37: 939–952 [DOI] [PubMed] [Google Scholar]
- Chin A, Lécuyer E (2017) RNA localization: making its way to the center stage. Biochim Biophys Acta 1861: 2956–2970 [DOI] [PubMed] [Google Scholar]
- Cioni J‐M, Koppers M, Holt CE (2018) Molecular control of local translation in axon development and maintenance. Curr Opin Neurobiol 51: 86–94 [DOI] [PubMed] [Google Scholar]
- Cioni J‐M, Lin JQ, Holtermann AV, Koppers M, Jakobs MAH, Azizi A, Turner‐Bridger B, Shigeoka T, Franze K, Harris WA et al (2019) Late endosomes act as mRNA translation platforms and sustain mitochondria in axons. Cell 176: 56–72.e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comazzetto S, Di Giacomo M, Rasmussen KD, Much C, Azzi C, Perlas E, Morgan M, O'Carroll D (2014) Oligoasthenoteratozoospermia and infertility in mice deficient for miR‐34b/c and miR‐449 loci. PLoS Genet 10: e1004597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell RB (2013) RNA protein interaction in neurons. Annu Rev Neurosci 36: 243–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dent EW, Gertler FB (2003) Cytoskeletal dynamics and transport in growth cone motility and axon guidance. Neuron 40: 209–227 [DOI] [PubMed] [Google Scholar]
- tom Dieck S, Kochen L, Hanus C, Heumüller M, Bartnik I, Nassim‐Assir B, Merk K, Mosler T, Garg S, Bunse S et al (2015) Direct visualization of newly synthesized target proteins in situ . Nat Methods 12: 411–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle M, Kiebler MA (2011) Mechanisms of dendritic mRNA transport and its role in synaptic tagging: mechanisms of dendritic mRNA transport. EMBO J 30: 3540–3552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng H, Lund K, Campenot RB (1999) Synthesis of beta‐tubulin, actin, and other proteins in axons of sympathetic neurons in compartmented cultures. J Neurosci 19: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabregat A, Sidiropoulos K, Garapati P, Gillespie M, Hausmann K, Haw R, Jassal B, Jupe S, Korninger F, McKay S et al (2016) The reactome pathway knowledgebase. Nucleic Acids Res 44: D481–D487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk J, Konopacki FA, Zivraj KH, Holt CE (2014) Rab5 and Rab4 regulate axon elongation in the Xenopus visual system. J Neurosci 34: 373–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farías GG, Guardia CM, Pace RD, Britt DJ, Bonifacino JS (2017) BORC/kinesin‐1 ensemble drives polarized transport of lysosomes into the axon. PNAS 114: E2955–E2964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Földes‐Papp Z, König K, Studier H, Bückle R, Breunig HG, Uchugonova A, Kostner GM (2009) Trafficking of mature miRNA‐122 into the nucleus of live liver cells. Curr Pharm Biotechnol 10: 569–578 [DOI] [PubMed] [Google Scholar]
- Garcia DM, Baek D, Shin C, Bell GW, Grimson A, Bartel DP (2011) Weak seed‐pairing stability and high target‐site abundance decrease the proficiency of lsy‐6 and other microRNAs. Nat Struct Mol Biol 18: 1139–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershoni‐Emek N, Altman T, Ionescu A, Costa CJ, Gradus‐Pery T, Willis DE, Perlson E (2018) Localization of RNAi machinery to axonal branch points and growth cones is facilitated by mitochondria and is disrupted in ALS. Front Mol Neurosci 11: 311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbings DJ, Ciaudo C, Erhardt M, Voinnet O (2009) Multivesicular bodies associate with components of miRNA effector complexes and modulate miRNA activity. Nat Cell Biol 11: 1143–1149 [DOI] [PubMed] [Google Scholar]
- Godement P, Salaün J, Imbert M (1984) Prenatal and postnatal development of retinogeniculate and retinocollicular projections in the mouse. J Comp Neurol 230: 552–575 [DOI] [PubMed] [Google Scholar]
- Grishok A, Pasquinelli AE, Conte D, Li N, Parrish S, Ha I, Baillie DL, Fire A, Ruvkun G, Mello CC (2001) Genes and mechanisms related to RNA interference regulate expression of the small temporal RNAs that control C. elegans developmental timing. Cell 106: 23–34 [DOI] [PubMed] [Google Scholar]
- Hancock ML, Preitner N, Quan J, Flanagan JG (2014) MicroRNA‐132 is enriched in developing axons, locally regulates Rasa1 mRNA, and promotes axon extension. J Neurosci 34: 66–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengst U (2006) Functional and selective RNA interference in developing axons and growth cones. J Neurosci 26: 5727–5732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt CE, Harris WA (1983) Order in the initial retinotectal map in Xenopus: a new technique for labelling growing nerve fibres. Nature 301: 150–152 [DOI] [PubMed] [Google Scholar]
- Hutvagner G, McLachlan J, Pasquinelli AE, Bálint E, Tuschl T, Zamore PD (2001) A cellular function for the RNA‐interference enzyme dicer in the maturation of the let‐7 small temporal RNA. Science 293: 834–838 [DOI] [PubMed] [Google Scholar]
- Iyer AN, Bellon A, Baudet M‐L (2014) microRNAs in axon guidance. Front Cell Neurosci 8: 78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan MA, Hirnes RH, Wilson L (1985) Comparison of the effects of vinblastine, vincristine, vindesine, and vinepidine on microtubule dynamics and cell proliferation in vitro . Cell Prolif 45: 8 [PubMed] [Google Scholar]
- Kim HH, Kim P, Phay M, Yoo S (2015) Identification of precursor microRNAs within distal axons of sensory neuron. J Neurochem 134: 193–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopacki FA, Wong HH‐W, Dwivedy A, Bellon A, Blower MD, Holt CE (2016) ESCRT‐II controls retinal axon growth by regulating DCC receptor levels and local protein synthesis. Open Biol 6: 150218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kye M‐J, Liu T, Levy SF, Xu NL, Groves BB, Bonneau R, Lao K, Kosik KS (2007) Somatodendritic microRNAs identified by laser capture and multiplex RT‐PCR. RNA 13: 1224–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langemeyer L, Fröhlich F, Ungermann C (2018) Rab GTPase function in endosome and lysosome biogenesis. Trends Cell Biol 28: 957–970 [DOI] [PubMed] [Google Scholar]
- Lee YS, Pressman S, Andress AP, Kim K, White JL, Cassidy JJ, Li X, Lubell K, Lim DH, Cho IS et al (2009) Silencing by small RNAs is linked to endosomal trafficking. Nat Cell Biol 11: 1150–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung K‐M, Lu B, Wong HH‐W, Lin JQ, Turner‐Bridger B, Holt CE (2018) Cue‐polarized transport of β‐actin mRNA depends on 3′UTR and microtubules in live growth cones. Front Cell Neurosci 12: 300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y‐C, Fernandopulle MS, Wang G, Choi H, Hao L, Drerup CM, Patel R, Qamar S, Nixon‐Abell J, Shen Y et al (2019) RNA granules hitchhike on lysosomes for long‐distance transport, using annexin A11 as a molecular tether. Cell 179: 147–164.e20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X‐A, Rizzo V, Puthanveettil SV (2012) Pathologies of axonal transport in neurodegenerative diseases. Transl Neurosci 3: 355–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugli G, Torvik VI, Larson J, Smalheiser NR (2008) Expression of microRNAs and their precursors in synaptic fractions of adult mouse forebrain. J Neurochem 106: 650–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maday S, Twelvetrees AE, Moughamian AJ, Holzbaur ELF (2014) Axonal transport: cargo‐specific mechanisms of motility and regulation. Neuron 84: 292–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magdesian MH, Gralle M, Guerreiro LH, Beltrão PJI, Carvalho MMVF, Santos LE, de Mello FG, Reis RAM, Ferreira ST (2011) Secreted human amyloid precursor protein binds semaphorin 3a and prevents semaphorin‐induced growth cone collapse. PLoS ONE 6: e22857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Much C, Auchynnikava T, Pavlinic D, Buness A, Rappsilber J, Benes V, Allshire R, O'Carroll D (2016) Endogenous mouse dicer is an exclusively cytoplasmic protein. PLoS Genet 12: e1006095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natera‐Naranjo O, Aschrafi A, Gioio AE, Kaplan BB (2010) Identification and quantitative analyses of microRNAs located in the distal axons of sympathetic neurons. RNA 16: 1516–1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwkoop PD (Pieter D), Faber J (1994) Normal Table of Xenopus laevis (Daudin): A Systematical and Chronological Survey of the Development from the Fertilized Egg Till the End of Metamorphosis. New York, NY: Garland Pub; [Google Scholar]
- Otero MG, Alloatti M, Cromberg LE, Almenar‐Queralt A, Encalada SE, Pozo Devoto VM, Bruno L, Goldstein LSB, Falzone TL (2014) Fast axonal transport of the proteasome complex depends on membrane interaction and molecular motor function. J Cell Sci 127: 1537–1549 [DOI] [PubMed] [Google Scholar]
- Panayotis N, Karpova A, Kreutz MR, Fainzilber M (2015) Macromolecular transport in synapse to nucleus communication. Trends Neurosci 38: 108–116 [DOI] [PubMed] [Google Scholar]
- Piper M, Anderson R, Dwivedy A, Weinl C, van Horck F, Leung KM, Cogill E, Holt C (2006) Signaling mechanisms underlying Slit2‐induced collapse of Xenopus retinal growth cones. Neuron 49: 215–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier K, Saillour Y, Bahi‐Buisson N, Jaglin XH, Fallet‐Bianco C, Nabbout R, Castelnau‐Ptakhine L, Roubertie A, Attie‐Bitach T, Desguerre I et al (2010) Mutations in the neuronal β‐tubulin subunit TUBB3 result in malformation of cortical development and neuronal migration defects. Hum Mol Genet 19: 4462–4473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pols MS, Klumperman J (2009) Trafficking and function of the tetraspanin CD63. Exp Cell Res 315: 1584–1592 [DOI] [PubMed] [Google Scholar]
- Preitner N, Quan J, Nowakowski DW, Hancock ML, Shi J, Tcherkezian J, Young‐Pearse TL, Flanagan JG (2014) APC is an RNA‐binding protein, and its interactome provides a link to neural development and microtubule assembly. Cell 158: 368–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullagura SRN, Buaas B, Gray N, Krening LC, Srivastava A, Braun RE (2018) Functional redundancy of DICER co‐factors TARBP2 and PRKRA during murine embryogenesis does not involve miRNA biogenesis. Genetics 208: 1513–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran L, Knölker H‐J, Simons K (2010) Subcellular targeting strategies for drug design and delivery. Nat Rev Drug Discovery 9: 29–42 [DOI] [PubMed] [Google Scholar]
- Resovi A, Pinessi D, Chiorino G, Taraboletti G (2014) Current understanding of the thrombospondin‐1 interactome. Matrix Biol 37: 83–91 [DOI] [PubMed] [Google Scholar]
- Rupaimoole R, Slack FJ (2017) MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discovery 16: 203–222 [DOI] [PubMed] [Google Scholar]
- Saftig P, Klumperman J (2009) Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol 10: 623–635 [DOI] [PubMed] [Google Scholar]
- Salogiannis J, Reck‐Peterson SL (2017) Hitchhiking: a non‐canonical mode of microtubule‐based transport. Trends Cell Biol 27: 141–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santangelo P, Nitin N, LaConte L, Woolums A, Bao G (2006) Live‐cell characterization and analysis of a clinical isolate of bovine respiratory syncytial virus, using molecular beacons. J Virol 80: 682–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Lee EJ, Jiang J, Sarkar A, Yang L, Elton TS, Chen C (2008) Real‐time PCR quantification of precursor and mature microRNA. Methods 44: 31–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ströhl F, Lin JQ, Laine RF, Wong HH‐W, Urbančič V, Cagnetta R, Holt CE, Kaminski CF (2017) Single molecule translation imaging visualizes the dynamics of local β‐actin synthesis in retinal axons. Sci Rep 7: 709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, Wu J, Tai H‐C, Schuman EM (2013) Axonal translation of β‐catenin regulates synaptic vesicle dynamics. J Neurosci 33: 5584–5589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischfield MA, Baris HN, Wu C, Rudolph G, Van Maldergem L, He W, Chan W‐M, Andrews C, Demer JL, Robertson RL et al (2010) Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell 140: 74–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner‐Bridger B, Jakobs M, Muresan L, Wong HH‐W, Franze K, Harris WA, Holt CE (2018) Single‐molecule analysis of endogenous β‐actin mRNA trafficking reveals a mechanism for compartmentalized mRNA localization in axons. Proc Natl Acad Sci USA 115: E9697–E9706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas JNS, Kar AN, Kowalak JA, Gale JR, Aschrafi A, Chen C‐Y, Gioio AE, Kaplan BB (2016) Axonal localization and mitochondrial association of precursor microRNA 338. Cell Mol Life Sci 73: 4327–4340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venigalla RKC, Turner M (2012) RNA‐binding proteins as a point of convergence of the PI3K and p38 MAPK pathways. Front Immunol 3: 398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viczian AS, Zuber ME (2014) A simple behavioral assay for testing visual function in Xenopus laevis . J Vis Exp 88: e51726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong HH‐W, Lin JQ, Ströhl F, Roque CG, Cioni J‐M, Cagnetta R, Turner‐Bridger B, Laine RF, Harris WA, Kaminski CF et al (2017) RNA docking and local translation regulate site‐specific axon remodeling in vivo. Neuron 95: 852–868.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu KY, Hengst U, Cox LJ, Macosko EZ, Jeromin A, Urquhart ER, Jaffrey SR (2005) Local translation of RhoA regulates growth cone collapse. Nature 436: 1020–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Zheng H, Loh HH, Law P‐Y (2015) Morphine promotes astrocyte‐preferential differentiation of mouse hippocampal progenitor cells via PKCε‐dependent ERK activation and TRBP phosphorylation. Stem Cells 33: 2762–2772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Sasaki Y, Wen Z, Bassell GJ, Zheng JQ (2006) An essential role for beta‐actin mRNA localization and translation in Ca2+‐dependent growth cone guidance. Nat Neurosci 9: 1265–1273 [DOI] [PubMed] [Google Scholar]
- Yap CC, Digilio L, McMahon LP, Garcia ADR, Winckler B (2018) Degradation of dendritic cargos requires Rab7‐dependent transport to somatic lysosomes. J Cell Biol 217: 3141–3159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- You X, Vlatkovic I, Babic A, Will T, Epstein I, Tushev G, Akbalik G, Wang M, Glock C, Quedenau C et al (2015) Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat Neurosci 18: 603–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Ueno Y, Liu XS, Buller B, Wang X, Chopp M, Zhang ZG (2013) The MicroRNA‐17‐92 cluster enhances axonal outgrowth in embryonic cortical neurons. J Neurosci 33: 6885–6894 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7
Source Data for Expanded View
Review Process File
Data Availability Statement
The GEO entry for the RNA sequencing data reported in this paper for the miR‐181a‐5p target candidate selection (Table EV1) is GSE124168.