

Abstract
A structure–activity relationship (SAR) for the oxadiazole class of antibacterials was evaluated by syntheses of 72 analogs and determination of the minimal-inhibitory concentrations (MICs) against the ESKAPE panel of bacteria. Selected compounds were further evaluated for in vitro toxicity, plasma protein binding, pharmacokinetics (PK), and a mouse model of methicillin-resistant Staphylococcus aureus (MRSA) infection. Oxadiazole 72c shows potent in vitro antibacterial activity, exhibits low clearance, a high volume of distribution, and 41% oral bioavailability, and shows efficacy in mouse models of MRSA infection.
Keywords: Antibacterials, oxadiazoles, penicillin-binding proteins, structure−activity relationship
Antibiotics have been called miracle drugs. Not only have they saved millions of lives in treatment of infections, they have also made possible other medical interventions such as surgery, dialysis, and organ transplantation. The availability of antibiotics has contributed to the increase in life expectancy in the United States from 59.7 years in 1930 to 78.8 years in 2010.1 Notwithstanding, emergence of resistance to any antibiotic is inevitable, threatening viability of any given class of such agents. Among Gram-positive bacteria, resistant Staphylococcus aureus and Enterococci pose the biggest threat.2S. aureus is a leading cause of health-related infections. Every year in the United States more than 80,000 severe methicillin-resistant S. aureus (MRSA) infections occur, resulting in 11,285 annual deaths.3 Enterococci cause infections, including endocarditis, urinary-tract infections, and wound infections. Over 66,000 Enterococcus infections are reported every year in the United States, of which 20,000 are vancomycin-resistant Enterococcus (VRE), resulting in 1,300 deaths.3 The limited pipeline of new antibiotics has exacerbated the antibiotic-resistance crisis.
The oxadiazoles are a newly discovered class of non-β-lactam antibiotics targeting cell-wall biosynthesis.4 These antibiotics show potent bactericidal activity against difficult-to-treat Gram-positive bacteria, including MRSA and VRE. They do not have Gram-negative antibacterial activity. Our initial studies into this class of antibiotics produced lead compounds 1–3, of which 2 and 3 exhibit 100% oral bioavailability and in vivo efficacy.4 Further studies on compound 2 suggested inhibition of the penicillin-binding proteins (PBPs) of S. aureus, which are responsible for bacterial cell-wall biosynthesis.4
With a view to explore the structural space of this class of antibiotics, we recently reported a structure–activity relationship (SAR) of the oxadiazoles against S. aureus(5) with a focus on modifications in ring A. We found that 4-phenol, 4-chloropyrazole, or 5-indole substitutions were generally favored at this position (1, 2, and 4–8; Figure 1). These compounds exhibit minimal-inhibitory concentration (MIC) values against S. aureus ranging from 0.5 to 4 μg/mL. Furthermore, antibiotic 8 gave excellent efficacy in MRSA-infected mice comparable to results obtained with linezolid, the gold standard, and exhibited oral bioavailability and low toxicity.5,6
Figure 1.

Structures of oxadiazoles active against S. aureus.
We report herein the synthesis and SAR results against S. aureus of 72 synthetic oxadiazoles with modifications in ring D (see Figure 1 for ring designations). To enable a comparison to our previously reported active compounds, we have limited our discussion in this report to compounds with the aforementioned ring A modifications. The SAR was established by initial screening of the synthetic derivatives against the ESKAPE panel of bacteria. This panel is comprised of Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter sp., and Escherichia coli. These bacteria account for the majority of problematic human infections.7 Selected compounds were further evaluated for in vitro toxicity, plasma protein binding, pharmacokinetics (PK), and in mouse models of MRSA infection.
The general synthetic routes to the oxadiazoles have been described previously.4,5 The same approach was used to access the analogs reported herein, except our focus was on modification of ring D, which necessitated diversification of the structures at the stage of the diphenyl ether construction. This was achieved either by direct nucleophilic substitution and Ullmann coupling reactions of nitriles 12–14 or by direct nucleophilic substitution and acylation of 4-hydroxybenzonitrile 15 (Figure 2). In many cases, ring D variations could be introduced by using commercially available substituted phenols (16–48), carboxylic acid derivatives (50, 51, 53, and 54), or halides (49 and 52). The substituents of several of these analogs could be used as synthetic handles for further elaboration, the reactions of which are described in the Supporting Information. In some cases where ring D was replaced by nonannular substituents, the requisite ethers were purchased (55–58).
Figure 2.

General synthetic routes to access the 1,2,4-oxadiazoles, and the starting materials used for variations within ring D. aNucleophilic substitution: NaH/K2CO3, DMSO/DMF, 60–100 °C, overnight; bUllmann coupling: CuI, Cs2CO3, N,N-dimethylglycine·HCl, 1,4-dioxane, 90 °C, 2–5 h ; cAcylation: anhydrous pyridine, 0 °C to rt, 4–6 h; dOutliers like dibenzo[b,d]furan and dibenzo[b,e][1,4]dioxine are not listed for simplicity of the graph. Compounds 55–58 are 4-cyanophenyl ethers purchased commercially; eAdditional modifications are listed in the Supporting Information.
We established the SAR for the oxadiazoles by screening them for antibacterial activity against the aforementioned ESKAPE panel of bacteria. The analogs reported herein show activity against Gram-positive bacteria. We specifically focused our attention on the analogs that exhibited activity against S. aureus (Figure 3).
Figure 3.

Antibacterial activities of the synthetic 1,2,4-oxadiazole derivatives with modifications of ring D. The MIC values (in μg/mL) measured for S. aureus ATCC 29213, with MIC of active compounds denoted in blue (MIC ≤ 8 μg/mL) and inactive ones in red (MIC > 8 μg/mL).
In general, hydrophobic substituents, and especially halogens, were well-tolerated in ring D. Addition of an extra fluorine (59b) or chlorine (69b) at the 3-position retained activity in the 4-chloropyrazole series, as did substitution by two chlorines at these same positions (60b). Replacement of the fluorine by an iodine, at either position 4 (61b, 75a), position 3 (74a), or position 2 (67a) also retained activity overall. The same trend was observed with introduction of a bromine atom at position 4 (63a) or position 3 (73a). Introduction of a trifluoromethoxy or methoxy group at position 4 of the ring (62a, 68a) also retained activity, as did the presence of a 3-trifluoromethyl ketone substituent (70a). An azide group at any position of the ring also maintained a favorable MIC against S. aureus (64a, 65a, 66a). Interestingly, a benzylic bromide at position 2 (76a) or position 3 (77a) of the ring did not result in a noticeable increase in MIC, but introduction of this substituent at position 4 (89a) completely abolished activity.
We also explored the effect of tethering rings C and D at their respective 2-positions to form a dibenzofuran, and we found that the resulting compounds where ring A was a phenol were inactive when ring D was unsubstituted (93a) or substituted with a 4-trifluoromethyl group (94a), 4-fluoro (90a), 4-methoxy (91a), or 4-trifluoromethoxy (92b). Likewise, expanding the furan portion with a heteroatom to form dibenzodioxine 105a resulted in loss of activity.
Replacing ring D with linear alkyl substituents resulted in only one active compound, the trifluoromethoxy derivative 78a, which contains a phenol for ring A. However, changing ring A to a 4-chloropyrazole (78b) or an indole (78c) resulted in loss of activity. The reasons for this are not clear at present. Other alkyl substituents introduced as surrogates for ring D, such as methyl (96a, 96b) and tert-butyl (97a), gave compounds with poor activity, as did sulfonates (98a, 99a), alkylcarbamates (100a), and amines (101a) and amides (102a, 103a, 104a).
We observed that introducing partially or fully saturated systems at ring D gave compounds with good activity in a few cases. Introduction of a nitrogen atom to give 3-pyrrolidines 113a and 114b, or of an oxygen atom to give 3-tetrahydrofuran 115a, decreased or abolished the activity. As a general trend, introduction of hydrogen-bond-donating substituents at ring D resulted in compounds with decreased antimicrobial activity against S. aureus. Thus, the 4-phenol derivative 79a was only modestly active (MIC = 16 μg/mL), and 2-benzyl alcohol 88a exhibited no activity. Similarly, benzoic acid derivatives 80a, 81a, and 82a were either poorly active or inactive, and 2-carboxamide 83a and 2-aniline 84a were inactive. Introduction of polar substituents onto an aliphatic system also resulted in compounds with no activity, as observed with 4-aminocyclohexane derivatives 111a and 112a. Electron-withdrawing substituents introduced onto ring D also resulted in compounds with no activity, as evidenced by 4-benzonitriles 85a and 85b, as well as 4-methylsulfonate 86b and 4-acetate 87b. One exception is the 2-nitro derivative 71a, which had an MIC of 4 μg/mL against S aureus.
Replacement of the phenyl ring at position D with heteroaromatic systems was also detrimental to activity. Thus, the benzothiazole derivative 106b was modestly active (MIC = 16 μg/mL), while the pyridine derivative 107a was inactive (MIC > 128 μg/mL). We observed a similar trend with heteroaliphatic ring systems, such as 4-piperidines (108a, 108b, 109b, 110a), 3-piperidines (117a, 117b, 118a, 118b), 3-azetidines (119a), as well as the pyrrolidines and tetrahydrofuran discussed above. Finally, we introduced more complex aliphatic amine-containing ring systems, giving quinuclidine derivatives 120a and 120b and tropine derivatives 121a and 121b; however, these exhibited poor activity against S. aureus (MIC ≥ 32 μg/mL).
Compounds 59b, 60b, 61b, 62a, 69b, 78a, and 72c were selected and further evaluated for in vitro toxicity using the XTT assay with HepG2 cells and hemolysis of red blood cells (Table 1). Compound 78a, in which ring D was replaced with trifluoromethoxy, caused 16% hemolysis at a concentration of 64 μg/mL; no further studies were done with this compound. Plasma protein binding was between 90% to 99%. Although high, it is within the range for 43% of the 1500 most frequently prescribed drugs,8 including many antibiotics on the market such as daptomycin, oxacillin, teicoplanin, rifampicin, and clindamycin.9−12 Compounds 59b, 60b, 61b, 62a, and 69b showed > 1% hemolysis or < 35 for the ratio of XTT/MIC and were not studied further.
Table 1. In Vitro Evaluation of Oxadiazoles.
| MIC (μg/mL) | Human plasma protein binding (%) | XTT IC50(μg/mL) | Hemolysis (%) | |
|---|---|---|---|---|
| 8a | 2 | 98.2 ± 3.2 | 75.7 ± 7.3 | <1 |
| 59b | 0.5 | 98.9 ± 1.6 | 17.7 ± 1.0 | 1.3 |
| 60b | 1 | 97.8 ± 0.3 | 17.2 ± 1.0 | 1.8 |
| 61b | 1 | 90.5 ± 0.3 | 30.7 ± 0.8 | 3.1 |
| 62a | 2 | 99.8 ± 0.1 | 16.9 ± 0.9 | 2.1 |
| 69b | 2 | 91.2 ± 0.6 | 21.0 ± 0.9 | 2.9 |
| 78a | 1 | ND | 84.9 ± 0.9 | 16 |
| 72c | 1 | 98.8 ± 0.7 | 37.0 ± 0.9 | <1 |
Data from Spink et al., reproduced for comparison.
Compound 72c was evaluated in a broader panel of Gram-positive bacteria and compared to the previous lead oxadiazole 8 and the standards vancomycin and linezolid (Table 2). Oxadiazole 72c showed better activity against MRSA strains NRS70, VRS1, and VRS2, and S. hemolyticus, B. cereus, B. licheniformis, and E. faecalis ATCC 29212 than oxadiazole 8.
Table 2. Activity of Oxadiazoles against Gram-Positive Organismsa.
| Organism | 72c | 8 | vancomycin | linezolid |
|---|---|---|---|---|
| S. aureus ATCC 29213b | 1–2 | 4 | 1 | 2 |
| S. aureus ATCC 27660c | 2–4 | 4 | 2 | 1 |
| S. aureus N315 (NRS70)c | 1 | 2 | 1 | 1 |
| S. aureus NRS100 (COL)c | 2 | 2 | 2 | 1 |
| S. aureus NRS119d | 1–2 | 2 | 1 | 32 |
| S. aureus NRS120d | 2 | 2 | 1 | 32 |
| S. aureus VRS1e | 1 | 2 | 64 | 1 |
| S. aureus VRS2f | 1 | 2 | 32 | 1 |
| S. epidermidis ATCC 35547 | 1 | 2 | 4 | 0.5 |
| S. hemolyticus ATCC 29970 | 2 | 4–8 | 4 | 1 |
| B. cereus ATCC 13061 | 4 | 16 | 1 | 0.5 |
| B. licheniformis ATCC 12759 | 2 | 8 | 0.5 | 0.5 |
| E. faecalis ATCC 29212b | 4 | 8 | 2 | 1 |
| E. faecalis 201 (Van S)g | 4 | 4 | 1 | 1 |
| E. faecalis 99 (Van R)h | 4 | 4 | 128 | 1 |
| E. faecium 119–39A (Van S)g | 1 | 1 | 0.5 | 1 |
| E. faecium 106 (Van R)h | 1 | 2 | 128 | 1 |
| E. faecium NCTC 7171 | 1 | 2 | 0.5 | 1 |
MIC in μg/mL.
A quality-control strain to monitor accuracy of MIC testing.
mecA positive, resistant to methicillin, oxacillin, and tetracycline; susceptible to vancomycin and linezolid.
mecA positive, resistant to ciprofloxacin, gentamicin, oxacillin, penicillin, and linezolid.
Vancomycin-resistant MRSA (vanA) clinical isolate from Michigan.
Vancomycin-resistant MRSA (vanA) clinical isolate from Pennsylvania.
Vancomycin-susceptible clinical isolate.
Vancomycin-resistant clinical isolate.
A PK study was conducted with 72c after intravenous (iv) and oral (po) administration (Figure 4). Oxadiazole 72c had low clearance of 7.8 mL/min/kg, a large volume of distribution of 14.2 L/kg, and a long terminal half-life of 21.1 h after iv administration (Table 3). Maximum concentration of 1.0 μg/mL was achieved at 2.5 h, and oral bioavailability was 41%. Systemic exposure and oral bioavailability for 72c were generally lower than the corresponding parameters for oxadiazole 8. In addition, the half-life for distribution after iv administration for oxadiazole 8 was shorter than that for 72c.
Figure 4.
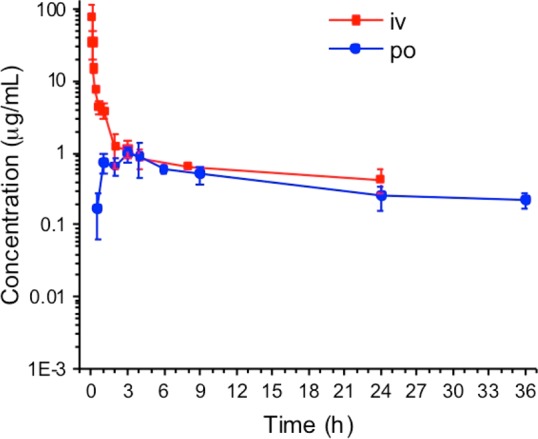
Pharmacokinetics of 72c after single iv and po administration at 20 mg/kg to mice (n = 3 mice per time point).
Table 3. Pharmacokinetic Parameters of 72c in Mice.
| 72c | 8a | |
|---|---|---|
| Dose 20 mg/kg iv | ||
| AUC0–last (μM·min/mL) | 1790 | 3140 |
| AUC0–∞ (μM·min/mL) | 2570 | 3520 |
| CL (mL/min/kg) | 7.8 | 5.7 |
| Vd (L/kg) | 14.2 | 4.7 |
| t1/2 dist (h) | 0.4 | 0.009 |
| t1/2 elim (h) | 21.1 | 9.6 |
| Dose 20 mg/kg po | ||
| AUC0–last (μM·min/mL) | 718 | 3060 |
| AUC0–∞ (μM·min/mL) | 1040 | 4060 |
| Cmax (μg/mL) | 1.0 | 2.3 |
| Tmax (h) | 2.5 | 6 |
| t1/2 abs (h) | 1.3 | 0.8 |
| t1/2 dist (h) | 6.2 | 15.9 |
| t1/2 elim (h) | 17.0 | 18.6 |
| F (%) | 41 | 97 |
Data from Spink et al., reproduced for comparison
Oxadiazole 72c was evaluated in the mouse peritonitis MRSA model of infection. The compound showed efficacy, with half of the mice surviving after two 20 mg/kg doses of the compound given iv at 30 min and 7.5 h after infection. Compound 72c was further evaluated in a mouse neutropenic thigh MRSA infection model using linezolid-sensitive (NRS70) and linezolid-resistant (NRS119) MRSA strains (Figure 5). The efficacy of 72c after a single oral dose at 40 mg/kg was comparable to that of compound 8, with 1.1-log and 1.5-log reduction in bacterial count compared to vehicle in the NRS70 and NRS119 strains, respectively. In the NRS119 study, bacterial counts after treatment with 72c and 8 were significantly lower (p < 0.05) than in the linezolid group. Drug concentrations at 48 h after a single oral 40 mg/kg dose of compound 8 were 7.6 ± 2.3 μg/g and 6.0 ± 1.2 μg/g in the NRS70 and NRS119 uninfected thighs, respectively (Table S5), which corresponded to 3- to 4-fold above MIC. Likewise, levels of compound 72c were 5.8 ± 1.4 μg/g and 6.4 ± 1.3 μg/g in the NRS70 and NRS119 uninfected thighs, respectively (Table S5), 6-fold above MIC. The levels of linezolid had dropped significantly and could not be quantified in the uninfected thighs. In summary, 72c showed potent in vitro activity against MRSA, oral bioavailability, and efficacy in mouse models of MRSA infection.
Figure 5.
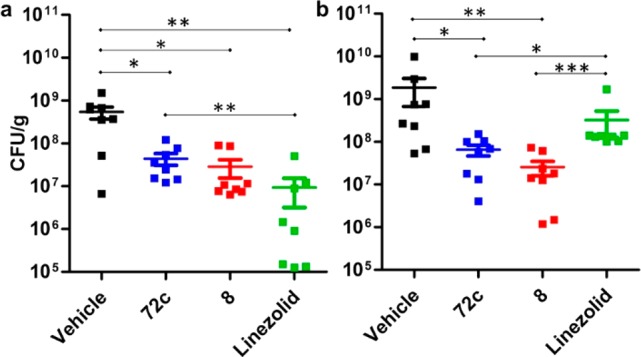
Efficacy of 72c in a mouse neutropenic thigh MRSA infection model using (a) NRS70 (linezolid-sensitive) and (b) NRS119 (linezolid-resistant). Mice (n = 8 per group) were made neutropenic by administration of cyclophosphamide. Bacteria (105 cfu) were injected intramuscularly into the right thigh. A single 40 mg/kg oral dose of vehicle, 72c, 8, or linezolid was given 1 h after infection. The thighs were harvested 48 h after infection, homogenized, and plated for colony counts. Mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001 by Mann–Whitney U test two-paired.
Acknowledgments
We thank Pablo Cabrera for assistance with the syntheses of two analogs.
Glossary
Abbreviations
- AUC
area-under-the curve
- iv
intravenous
- MIC
minimal-inhibitory concentration
- MRSA
methicillin resistant Staphylococcus aureus
- PK
pharmacokinetics
- po
per os
- SAR
structure–activity relationship
- VRE
vancomycin-resistant Enterococci
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00379.
Synthetic and experimental procedures (PDF)
Author Contributions
MAB, DD, ES, TY, SAT, PIO, and EL synthesized compounds. YQ conducted some of the MS and NMR work and helped in preparing the manuscript. JEM, ZP, and WS conducted plasma protein binding and PK. JEM determined drug concentrations in plasma and thighs in the mouse neutropenic thigh infection study. JJ conducted the mouse peritonitis and mouse neutropenic thigh infection studies and MIC against Gram-positive organisms. E. Lastochkin conducted MIC against the ESKAPE organisms and in vitro cytotoxicity. VAS, WRW, and MAS conducted the in-life portion of mouse peritonitis and PK studies. SM and MC designed the studies and wrote the manuscript, with contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by grants AI090818 (to MC and SM) and by AI104987 (to SM) from the National Institutes of Health. YQ is a Ruth L. Kirschstein National Research Service Award Fellow of the Chemistry-Biochemistry-Biology Interface Program at the University of Notre Dame, supported by training grant T32 GM075762 from the National Institutes of Health.
The authors declare no competing financial interest.
Supplementary Material
References
- National Center for Health Statistics . National Vital Statistics Report. http://www.infoplease.com/ipa/A0005148.html.
- Rossolini G. M.; Arena F.; Pecile P.; Pollini S. Update on the antibiotic resistance crisis. Curr. Opin. Pharmacol. 2014, 18, 56–60. 10.1016/j.coph.2014.09.006. [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention . Antibiotic Resistance Threats in the United States, 2013; 2013.
- O’Daniel P. I.; Peng Z.; Pi H.; Testero S. A.; Ding D.; Spink E.; Leemans E.; Boudreau M. A.; Yamaguchi T.; Schroeder V. A.; Wolter W. R.; Llarrull L. I.; Song W.; Lastochkin E.; Kumarasiri M.; Antunes N. T.; Espahbodi M.; Lichtenwalter K.; Suckow M. A.; Vakulenko S.; Mobashery S.; Chang M. Discovery of a new class of non-beta-lactam inhibitors of penicillin-binding proteins with Gram-positive antibacterial activity. J. Am. Chem. Soc. 2014, 136 (9), 3664–3672. 10.1021/ja500053x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spink E.; Ding D.; Peng Z.; Boudreau M. A.; Leemans E.; Lastochkin E.; Song W.; Lichtenwalter K.; O’Daniel P. I.; Testero S. A.; Pi H.; Schroeder V. A.; Wolter W. R.; Antunes N. T.; Suckow M. A.; Vakulenko S.; Chang M.; Mobashery S. Structure-activity relationship for the oxadiazole class of antibiotics. J. Med. Chem. 2015, 58 (3), 1380–1389. 10.1021/jm501661f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janardhanan J.; Meisel J. E.; Ding D.; Schroeder V. A.; Wolter W. R.; Mobashery S.; Chang M. In Vitro and In Vivo Synergy of the Oxadiazole Class of Antibacterials with beta-Lactams. Antimicrob. Agents Chemother. 2016, 60, 5581–5588. 10.1128/AAC.00787-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher H. W.; Talbot G. H.; Bradley J. S.; Edwards J. E.; Gilbert D.; Rice L. B.; Scheld M.; Spellberg B.; Bartlett J. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48 (1), 1–12. 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- Kratochwil N. A.; Huber W.; Muller F.; Kansy M.; Gerber P. R. Predicting plasma protein binding of drugs: a new approach. Biochem. Pharmacol. 2002, 64 (9), 1355–1374. 10.1016/S0006-2952(02)01074-2. [DOI] [PubMed] [Google Scholar]
- Dykhuizen R. S.; Harvey G.; Stephenson N.; Nathwani D.; Gould I. M. Protein binding and serum bactericidal activities of vancomycin and teicoplanin. Antimicrob. Agents Chemother. 1995, 39 (8), 1842–1847. 10.1128/AAC.39.8.1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochansky C. J.; McMasters D. R.; Lu P.; Koeplinger K. A.; Kerr H. H.; Shou M.; Korzekwa K. R. Impact of pH on plasma protein binding in equilibrium dialysis. Mol. Pharmaceutics 2008, 5 (3), 438–448. 10.1021/mp800004s. [DOI] [PubMed] [Google Scholar]
- Lee B. L.; Sachdeva M.; Chambers H. F. Effect of protein binding of daptomycin on MIC and antibacterial activity. Antimicrob. Agents Chemother. 1991, 35 (12), 2505–2508. 10.1128/AAC.35.12.2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargent P.Oxacillin for injection USP. Oxacillin package insert. 2013.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.