

Abstract

PROTACs-induced targeted protein degradation has emerged as a novel therapeutic strategy in drug development and attracted the favor of academic institutions, large pharmaceutical enterprises (e.g., AstraZeneca, Bayer, Novartis, Amgen, Pfizer, GlaxoSmithKline, Merck, and Boehringer Ingelheim, etc.), and biotechnology companies. PROTACs opened a new chapter for novel drug development. However, any new technology will face many new problems and challenges. Perspectives on the potential opportunities and challenges of PROTACs will contribute to the research and development of new protein degradation drugs and degrader tools.
Keywords: Proteolysis targeting chimeras, drug resistance, nonenzymatic function, undruggable target, reversible protein chemical knockdown
Proteolysis targeting chimeras (PROTACs) are heterobifunctional small molecules with three chemical elements: a ligand binding to a target protein, a ligand binding to E3 ubiquitin ligase, and a linker for conjugating these two ligands. PROTAC is a chemical knockdown strategy that degrades the target protein through the ubiquitin-proteasome system (Figure 1).1 Different from the competitive- and occupancy-driven process of traditional inhibitors, PROTACs are catalytic in their mode of action, which can promote target protein degradation at low exposures.2 PROTACs have the potential to degrade the target pathogenic proteins and regulate the related signaling pathways, which cannot be achieved by traditional therapy (inhibitor/activator). As a novel approach, PROTACs have gained great attention from academia and the pharmaceutical and biotechnology industry (e.g., Arvinas, C4 Therapeutics, Kymera Therapeutics, AstraZeneca, Bayer, Novartis, and Vertex, etc.).
Figure 1.
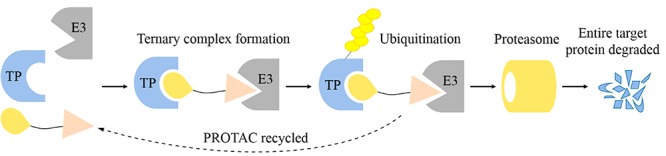
Overview steps of entire target protein degradation by PROTACs. TP, target protein.
At present, PROTACs have been successfully employed in the degradation of different types of target proteins related to various diseases, including cancer, viral infection, immune disorders, and neurodegenerative diseases. Some cases reported include PROTACs targeting B-cell lymphoma 6 (BCL6) from AstraZeneca,3 P300/CBP-associated factor and general control nonderepressible 5 (PCAF/GCN5) from GlaxoSmithKline (GSK),4 Bruton’s tyrosine kinase (BTK) from Pfizer,5 focal adhesion kinase (FAK) from Boehringer Ingelheim,6 and Interleukin-1 receptor-associated kinase 4 (IRAK4) from GSK.7 In addition, resistance caused by PROTACs was illustrated by researchers from Abbvie,8 and Promega reported the quantitative live-cell kinetic degradation and mechanistic profiling.9 Recently, ARV-110 (undisclosed structure)10 from Arvinas, Inc., an androgen (AR)-targeted PROTAC with high potency against both wild-type and mutants, exhibited satisfactory safety and tolerability in patients in a phase I clinical trial. ARV-471 (undisclosed structure),11 an estrogen (ER) degrader from Arvinas, Inc., is also in phase 1 studies in women with locally advanced or metastatic ER positive/HER2 negative breast cancer. PROTACs have opened a new chapter for the development of new drugs and novel chemical knockdown tools and brought unprecedented opportunities to the industry and academia, which are mainly reflected in the following aspects:
-
(1)
Overcoming drug resistance of cancer. In addition to traditional chemotherapy, kinase inhibitors have been developing rapidly in the past 20 years.12 Although kinase inhibitors are very effective in cancer therapy, patients often develop drug resistance and disease recurrence, consequently. PROTACs showed greater advantages in drug resistant cancers through degrading the whole target protein. For example, ARCC-4 targeting androgen receptor could overcome enzalutamide-resistant prostate cancer13 and L18I targeting BTK could overcome C481S mutation.14
-
(2)
Eliminating both the enzymatic and nonenzymatic functions of kinase. Traditional small molecule inhibitors usually inhibit the enzymatic activity of the target, while PROTACs affect not only the enzymatic activity of the protein but also nonenzymatic activity by degrading the entire protein. For example, FAK possesses the kinase dependent enzymatic functions and kinase independent scaffold functions, but regulating the kinase activity does not successfully inhibit all FAK function. In 2018, a highly effective and selective FAK PROTAC reported by Craig M. Crews’ group showed a far superior activity to clinical candidate drug in cell migration and invasion.15 Therefore, PROTAC can expand the druggable space of the existing targets and regulate proteins that are difficult to control by traditional small molecule inhibitors.
-
(3)
Degrade the “undruggable” protein target. At present, only 20–25% of the known protein targets (include kinases, G protein-coupled receptors (GPCRs), nuclear hormone receptors, and iron channels) can be targeted by using conventional drug discovery technologies.16,17 The proteins that lack catalytic activity and/or have catalytic independent functions are still regarded as “undruggable” targets. The involvement of Signal Transducer and Activator of Transcription 3 (STAT3) in the multiple signaling pathway makes it an attractive therapeutic target; however, the lack of an obviously druggable site on the surface of STAT3 limited the development of STAT3 inhibitors. Thus, there are still no effective drugs directly targeting STAT3 approved by the Food and Drug Administration (FDA). In November 2019, Shaomeng Wang’s group first reported a potent PROTAC targeting STAT3 with potent biological activities in vitro and in vivo.18 This successful case confirms the key potential of PROTAC technology, especially in the field of “undruggable” targets, such as K-Ras, a tricky tumor target activated by multiple mutations as G12A, G12C, G12D, G12S, G12 V, G13C, and G13D in the clinic.19
-
(4)
Fast and reversible chemical knockdown strategy in vivo. Traditional genetic protein knockout technologies, zinc-finger nuclease (ZFN), transcription activator-like effector nuclease (TALEN), or CRISPR-Cas9, usually have a long cycle, irreversible mode of action, and high cost, which brings a lot of inconvenience for research, especially in nonhuman primates. In addition, these genetic animal models sometimes produce phenotypic misunderstanding due to potential gene compensation or gene mutation. More importantly, the traditional genetic method cannot be used to study the function of embryonic-lethal genes in vivo. Unlike DNA-based protein knockout technology, PROTACs knock down target proteins directly, rather than acting at the genome level, and are suitable for the functional study of embryonic-lethal proteins in adult organisms. In addition, PROTACs provide exquisite temporal control, allowing the knockdown of a target protein at specific time points and enabling the recovery of the target protein after withdrawal of drug treatment. As a new, rapid and reversible chemical knockdown method, PROTAC can be used as an effective supplement to the existing genetic tools.20
Although PROTAC technology has a bright future in drug development, it also has many challenges as follows:
-
(1)
Until now, there is only one example of PROTAC reported for an “undruggable” target;18 more cases are needed to prove the advantages of PROTAC in “undruggable” targets in the future.
-
(2)
“Molecular glue”, existing in nature, represents the mechanism of stabilized protein–protein interactions through small molecule modulators of E3 ligases. For instance, auxin, the plant hormone, binds to the ligase SCF-TIR1 to drive recruitment of Aux/IAA proteins and subsequently triggers its degradation. In addition, some small molecules that induce targeted protein degradation through “molecular glue” mode of action have been reported.21,22 Furthermore, it has been recently reported that some PROTACs may actually achieve target protein degradation via a mechanism that includes “molecular glue” or via “molecular glue” alone.23 How to distinguish between these two mechanisms and how to combine them to work together is one of the challenges for future research.
-
(3)
Since PROTAC acts in a catalytic mode, traditional methods cannot accurately evaluate the pharmacokinetics (PK) and pharmacodynamics (PD) properties of PROTACs. Thus, more studies are urgently needed to establish PK and PD evaluation systems for PROTACs.
-
(4)
How to quickly and effectively screen for target protein ligands that can be used in PROTACs, especially those targeting protein–protein interactions, is another challenge.
-
(5)
How to understand the degradation activity, selectivity, and possible off-target effects (based on different targets, different cell lines, and different animal models) and how to rationally design PROTACs etc. are still unclear.
-
(6)
The human genome encodes more than 600 E3 ubiquitin ligases. However, there are only very few E3 ligases (VHL, CRBN, cIAPs, and MDM2) used in the design of PROTACs. How to expand E3 ubiquitin ligase scope is another challenge faced in this area.
PROTAC technology is rapidly developing, and with the joint efforts of the vast number of scientists in both academia and industry, these problems shall be solved in the near future.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (#81573277, 81622042, 81773567) and the National Major Scientific and Technological Special Project for “Significant New Drugs Development” (#SQ2017ZX095003, 2018ZX09711001).
Glossary
Abbreviations
- PROTACs
proteolysis targeting chimeras
- BCL6
B-cell lymphoma 6
- PCAF/GCN5
P300/CBP-associated factor and general control nonderepressible 5
- GSK
GlaxoSmithKline
- BTK
Bruton’s tyrosine kinase
- FAK
focal adhesion kinase
- IRAK4
interleukin-1 receptor-associated kinase 4
- AR
androgen
- ER
estrogen
- GPCRs
G-protein-coupled receptors
- STAT3
signal transducer and activator of transcription 3
- FDA
Food and Drug Administration
- ZFN
zinc-finger nuclease
- TALEN
transcription activator-like effector nuclease
- PK
pharmacokinetics
- PD
pharmacodynamics
Views expressed in this editorial are those of the authors and not necessarily the views of the ACS.
The authors declare no competing financial interest.
References
- Gadd M. S.; Testa A.; Lucas X.; Chan K. H.; Chen W.; Lamont D. J.; Zengerle M.; Ciulli A. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 2017, 13, 514–521. 10.1038/nchembio.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson D. P.; Mares A.; Smith I. E.; Ko E.; Campos S.; Miah A. H.; Mulholland K. E.; Routly N.; Buckley D. L.; Gustafson J. L.; Zinn N.; Grandi P.; Shimamura S.; Bergamini G.; Faelth-Savitski M.; Bantscheff M.; Cox C.; Gordon D. A.; Willard R. R.; Flanagan J. J.; Casillas L. N.; Votta B. J.; den Besten W.; Famm K.; Kruidenier L.; Carter P. S.; Harling J. D.; Churcher I.; Crews C. M. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–7. 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoull W.; Cheung T.; Anderson E.; Barton P.; Burgess J.; Byth K.; Cao Q.; Castaldi M. P.; Chen H.; Chiarparin E.; Carbajo R. J.; Code E.; Cowan S.; Davey P. R.; Ferguson A. D.; Fillery S.; Fuller N. O.; Gao N.; Hargreaves D.; Howard M. R.; Hu J.; Kawatkar A.; Kemmitt P. D.; Leo E.; Molina D. M.; O’Connell N.; Petteruti P.; Rasmusson T.; Raubo P.; Rawlins P. B.; Ricchiuto P.; Robb G. R.; Schenone M.; Waring M. J.; Zinda M.; Fawell S.; Wilson D. M. Development of a Novel B-Cell Lymphoma 6 (BCL6) PROTAC To Provide Insight into Small Molecule Targeting of BCL6. ACS Chem. Biol. 2018, 13, 3131–3141. 10.1021/acschembio.8b00698. [DOI] [PubMed] [Google Scholar]
- Bassi Z. I.; Fillmore M. C.; Miah A. H.; Chapman T. D.; Maller C.; Roberts E. J.; Davis L. C.; Lewis D. E.; Galwey N. W.; Waddington K. E.; Parravicini V.; Macmillan-Jones A. L.; Gongora C.; Humphreys P. G.; Churcher I.; Prinjha R. K.; Tough D. F. Modulating PCAF/GCN5 Immune Cell Function through a PROTAC Approach. ACS Chem. Biol. 2018, 13, 2862–2867. 10.1021/acschembio.8b00705. [DOI] [PubMed] [Google Scholar]
- Zorba A.; Nguyen C.; Xu Y.; Starr J.; Borzilleri K.; Smith J.; Zhu H.; Farley K. A.; Ding W.; Schiemer J.; Feng X.; Chang J. S.; Uccello D. P.; Young J. A.; Garcia-Irrizary C. N.; Czabaniuk L.; Schuff B.; Oliver R.; Montgomery J.; Hayward M. M.; Coe J.; Chen J.; Niosi M.; Luthra S.; Shah J. C.; El-Kattan A.; Qiu X.; West G. M.; Noe M. C.; Shanmugasundaram V.; Gilbert A. M.; Brown M. F.; Calabrese M. F. Delineating the role of cooperativity in the design of potent PROTACs for BTK. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E7285–E7292. 10.1073/pnas.1803662115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popow J.; Arnhof H.; Bader G.; Berger H.; Ciulli A.; Covini D.; Dank C.; Gmaschitz T.; Greb P.; Karolyi-Ozguer J.; Koegl M.; McConnell D. B.; Pearson M.; Rieger M.; Rinnenthal J.; Roessler V.; Schrenk A.; Spina M.; Steurer S.; Trainor N.; Traxler E.; Wieshofer C.; Zoephel A.; Ettmayer P. Highly Selective PTK2 Proteolysis Targeting Chimeras to Probe Focal Adhesion Kinase Scaffolding Functions. J. Med. Chem. 2019, 62, 2508–2520. 10.1021/acs.jmedchem.8b01826. [DOI] [PubMed] [Google Scholar]
- Nunes J.; McGonagle G. A.; Eden J.; Kiritharan G.; Touzet M.; Lewell X.; Emery J.; Eidam H.; Harling J. D.; Anderson N. A. Targeting IRAK4 for Degradation with PROTACs. ACS Med. Chem. Lett. 2019, 10, 1081–1085. 10.1021/acsmedchemlett.9b00219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Riley-Gillis B.; Vijay P.; Shen Y. Acquired Resistance to BET-PROTACs (Proteolysis-Targeting Chimeras) Caused by Genomic Alterations in Core Components of E3 Ligase Complexes. Mol. Cancer Ther. 2019, 18, 1302–1311. 10.1158/1535-7163.MCT-18-1129. [DOI] [PubMed] [Google Scholar]
- Riching K. M.; Mahan S.; Corona C. R.; McDougall M.; Vasta J. D.; Robers M. B.; Urh M.; Daniels D. L. Quantitative Live-Cell Kinetic Degradation and Mechanistic Profiling of PROTAC Mode of Action. ACS Chem. Biol. 2018, 13, 2758–2770. 10.1021/acschembio.8b00692. [DOI] [PubMed] [Google Scholar]
- Neklesa T.; Snyder L. B.; Willard R. R.; Vitale N.; Pizzano J.; Gordon D. A.; Bookbinder M.; Macaluso J.; Dong H.; Ferraro C.; Wang G.; Wang J.; Crews C. M.; Houston J.; Crew A. P.; Taylor I.. ARV-110: an oral androgen receptor PROTAC degrader for prostate cancer; ASCO-GU: San Francisco, California, USA, 2019, February 14–16. [Google Scholar]
- Flanagan J. J.; Qian Y.; Gough S. M.; Andreoli M.; Bookbinder M.; Cadelina G.; Bradley J.; Rousseau E.; Chandler J.; Willard R.; Pizzano J.; Crews C. M.; Crew A. P.; Taylor I.; Houston J.. ARV-471, an oral estrogen receptor PROTAC protein degrader for breast cancer; SABCS: San Antonio, Texas, USA, 2018, December 4–8. [Google Scholar]
- Wu P.; Nielsen T. E.; Clausen M. H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–39. 10.1016/j.tips.2015.04.005. [DOI] [PubMed] [Google Scholar]
- Salami J.; Alabi S.; Willard R. R.; Vitale N. J.; Wang J.; Dong H.; Jin M.; McDonnell D. P.; Crew A. P.; Neklesa T. K.; Crews C. M. Androgen receptor degradation by the proteolysis-targeting chimera ARCC-4 outperforms enzalutamide in cellular models of prostate cancer drug resistance. Commun. Biol. 2018, 1, 100. 10.1038/s42003-018-0105-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y.; Ding N.; Song Y.; Yang Z.; Liu W.; Zhu J.; Rao Y. Degradation of Bruton’s tyrosine kinase mutants by PROTACs for potential treatment of ibrutinib-resistant non-Hodgkin lymphomas. Leukemia 2019, 33, 2105–2110. 10.1038/s41375-019-0440-x. [DOI] [PubMed] [Google Scholar]
- Cromm P. M.; Samarasinghe K. T. G.; Hines J.; Crews C. M. Addressing Kinase-Independent Functions of Fak via PROTAC-Mediated Degradation. J. Am. Chem. Soc. 2018, 140, 17019–17026. 10.1021/jacs.8b08008. [DOI] [PubMed] [Google Scholar]
- Wells J. A.; McClendon C. L. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 2007, 450, 1001–9. 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- Kim J.; Kim H.; Park S. B. Privileged structures: efficient chemical ″navigators″ toward unexplored biologically relevant chemical spaces. J. Am. Chem. Soc. 2014, 136, 14629–38. 10.1021/ja508343a. [DOI] [PubMed] [Google Scholar]
- Bai L.; Zhou H.; Xu R.; Zhao Y.; Chinnaswamy K.; McEachern D.; Chen J.; Yang C. Y.; Liu Z.; Wang M.; Liu L.; Jiang H.; Wen B.; Kumar P.; Meagher J. L.; Sun D.; Stuckey J. A.; Wang S. A Potent and Selective Small-Molecule Degrader of STAT3 Achieves Complete Tumor Regression In Vivo. Cancer Cell 2019, 36, 498–511. 10.1016/j.ccell.2019.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; e17.
- Jinesh G. G.; Sambandam V.; Vijayaraghavan S.; Balaji K.; Mukherjee S. Molecular genetics and cellular events of K-Ras-driven tumorigenesis. Oncogene 2018, 37, 839–846. 10.1038/onc.2017.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X.; Wang J.; Yao X.; Zheng W.; Mao Y.; Lan T.; Wang L.; Sun Y.; Zhang X.; Zhao Q.; Zhao J.; Xiao R. P.; Zhang X.; Ji G.; Rao Y. A chemical approach for global protein knockdown from mice to non-human primates. Cell Discov 2019, 5, 10. 10.1038/s41421-018-0079-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel P.; Kaiser M.; Ottmann C. Small-molecule stabilization of protein-protein interactions: an underestimated concept in drug discovery?. Angew. Chem., Int. Ed. 2012, 51, 2012–8. 10.1002/anie.201107616. [DOI] [PubMed] [Google Scholar]
- Chamberlain P. P.; D’Agostino L. A.; Ellis J. M.; Hansen J. D.; Matyskiela M. E.; McDonald J. J.; Riggs J. R.; Hamann L. G. Evolution of Cereblon-Mediated Protein Degradation as a Therapeutic Modalit. ACS Med. Chem. Lett. 2019, 10, 1592–1602. 10.1021/acsmedchemlett.9b00425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Li Y.; Aguilar A.; Liu Z.; Yang C. Y.; Wang S. Simple Structural Modifications Converting a Bona fide MDM2 PROTAC Degrader into a Molecular Glue Molecule: A Cautionary Tale in the Design of PROTAC Degraders. J. Med. Chem. 2019, 62, 9471–9487. 10.1021/acs.jmedchem.9b00846. [DOI] [PMC free article] [PubMed] [Google Scholar]