

Abstract
Necroptosis has been implicated in a variety of disease states, and RIPK3 is one of the kinases identified to play a critical role in this signaling pathway. In an effort to identify RIPK3 kinase inhibitors with a novel profile, mechanistic studies were incorporated at the hit triage stage. Utilization of these assays enabled identification of a Type II DFG-out inhibitor for RIPK3, which was confirmed by protein crystallography. Structure-based drug design on the inhibitors targeting this previously unreported conformation enabled an enhancement in selectivity against key off-target kinases.
Keywords: RIPK3, type II kinase inhibitor, kinase, structure-based drug design (SBDD), high-throughput screening (HTS), necroptosis
Necroptosis is a programmed form of cell death and has been associated with a variety of diseases, including ischemia reperfusion injury, neurodegenerative disorders, pancreatic cancer, and autoimmune diseases such as inflammatory bowel disease (IBD).1−5 Upon stimulation of death receptors (such as the family of TNF receptors), signaling can initiate a necroptotic cell death process. This involves formation of a necrosome, which includes receptor interacting protein kinases 1 and 3 (RIPK1, RIPK3) in a cytosolic complex. RIPK3 phosphorylates the pseudokinase mixed-lineage kinase like (MLKL), which can subsequently induce cell death. Necroptotic cell death leads to the release of inflammatory damage-associated molecular patterns (DAMPs) into the extracellular environment and contributes to inflammation through activation of the Toll-like receptor pathway.3 Unlike RIPK3 and MLKL, RIPK1 can participate both in necroptosis and death receptor-mediated apoptosis. RIPK1, RIPK3, and pseudokinase MLKL have been the focus of drug discovery efforts in academia and industry.6−11
In 2013, the first X-ray crystal structures for RIPK3 were reported.12 As is common in kinases, RIPK3 was crystallized in its active form. To date, only Type I RIPK3 inhibitors have been reported13 (Figure 1), and they tend to suffer from poor RIPK family selectivity, notably with respect to RIPK2 activity.14,15 In contrast, the cocrystal structure of RIPK3 and MLKL with AMP-PNP indicated that RIPK3 was adopting an inactive conformation in the complex. Key findings included a shifting of the αC-helix along with partial unwinding of the helix. Significant movement and realignment in the hydrophobic spine was also noted. Based on these findings, we were intrigued by the potential opportunity to identify inhibitors of RIPK3, which target an inactive conformation of the kinase from a high-throughput screen (HTS). To assist in the identification of such inhibitors, biophysical screening was incorporated at the hit selection and triage phase.
Figure 1.

Reported RIPK3 inhibitors.
A common issue with pursuing kinase targets is kinome selectivity.16 Off-target kinase activities can lead to several challenges including off-target toxicology and opposing or confounding pharmacology to the target of interest. One potential way to mitigate issues related to kinome selectivity is to target the inactive form of the kinase. When targeting the inactive form of a kinase, both ATP competitive and noncompetitive binders can be found targeting Type II, III, or IV pockets.17−20 In addition to affecting kinome selectivity, kinase inhibitors targeting inactive conformations may differ from Type I inhibitors in other ways. Enhanced residence times are common in non-Type I inhibitors.21 The additional conformational changes required to access the pockets distal or allosteric to the ATP binding site is a likely contributing factor. Inhibitors targeting some inactive conformations (primarily Type III or Type IV) of the kinase may not be fully competitive with ATP leading to another source of differentiation.19,22
Numerous kinases that can adopt an inactive conformation have been identified to date.17 Several methods have been developed to further identify kinases, which may be able to adopt a Type II DFG-out conformation, including labeling approaches23,24 and predictive computational models.25 From an inhibitor standpoint, a characteristic pharmacophore for Type II kinase inhibitors has been established.17 Screening inhibitors designed on the pharmacophore has identified kinases capable of adopting a DFG-out inactive conformation.17,26
In this Letter, we disclose the use of high-throughput mechanistic assays in the hit triage stage to enable identification of non-Type I RIPK3 kinase inhibitors. For the follow-up mechanistic studies, we needed to consider what assays would be compatible with the HTS platform and automation. At the outset of the project, we considered several potential screening assays and deemed that a homogeneous time-resolved fluorescence assay (HTRF) would be most suitable for this kinase program.27 Although consideration of a RIPK3 construct was taken into account, we found that using the active, commercial full length enzyme was well-suited to the HTRF platform, demonstrating improved probe binding over the kinase domain construct (data not shown). After studying multiple preincubation times for the complex, the preincubation time for the HTS screen was increased to 60 min to accommodate any inhibitors with a slow on-rate. The complex was found to reach equilibrium within 45 min, with minimal changes beyond 60 min. Taking into account some of the reported attributes of non-Type I inhibitors, assays for time dependency (TD), and mode of inhibition (MOI) were deemed to be the most pertinent mechanistic studies to incorporate early in our screening paradigm. Use of the HTRF platform allowed for the option of utilizing probe displacement as opposed to ATP displacement as a tool for the studies.
While time-dependent inhibition is not a defining characteristic for inhibitors targeting Type II conformations, it is found frequently enough to be suggestive of a Type II inhibitor.21 For the initial TD studies, several methods were evaluated based on probe or inhibitor displacement. Initially, probe displacement of the inhibitor at a single concentration wherein the output would be a percent shift in the displacement at 5 and 60 min was explored. This was based on forming an initial complex of the inhibitor, kinase, and antibody then treating with the probe at 30-fold the probe Kd. An alternative approach was to preform a complex consisting of the kinase, probe, and antibody. The inhibitor could then be added to displace the probe and diluted to determine an 11-point concentration response curve (CRC) at 5 and 60 min. Based on the results observed with a small test set of internal compounds, inhibitor displacement of the probe at 5 and 60 min in CRC format would be utilized for the time dependency studies.
Initial exploration of MOI assays to identify compounds that may bind in a pocket more removed from the ATP binding site proved challenging. Assays were attempted at both low ATP and high ATP concentrations and low probe and high probe concentrations in an effort to bin compounds as “competitive binders” or “mixed type binders” (uncompetitive or noncompetitive). The high hit rate in the initial study precluded the use of this assay in guiding a selection of chemotypes.
After running the HTS with an HTRF assay on the RIPK3 full length kinase, TD studies were run on a subset of compounds meeting potency requirements. With the establishment of a baseline expectation from the described validation runs, HTRF time dependency for RIPK3 hits at 5 and 60 min led to the identification of a small subset of compounds, which may be slow binding (Figure 2). Specifically 44 compounds out of 2571 tested showed a ≥5-fold shift in IC50 at 5 and 60 min.
Figure 2.
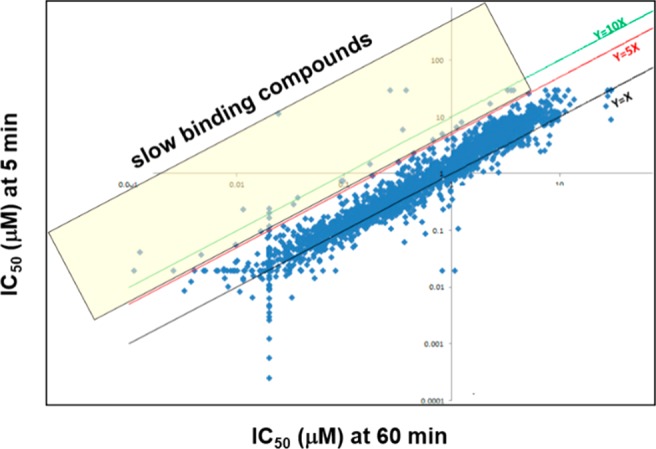
Analysis of high-throughput TD studies from the HTS. Compounds in the yellow box (44) had a shift of ≥5 in the RIPK3 IC50 at 5 and 60 min.
Multiple nontraditional kinase chemotypes were identified that represented interesting lead molecules. We were gratified to see that this screening methodology did identify a known Type II DFG-out kinase inhibitor, in addition to Type I kinase chemotypes (Figure 3).28,29 For example, compound 6 demonstrated exquisite binding potency for RIPK3 in the HTS assay along with superb selectivity over RIPK1 and RIPK2. As a key proof of concept to the incorporation of mechanistic tools at the hit triage stage, compound 6 demonstrated a greater than 6-fold shift in the TD assay. A shift of 2 in the TD assay was observed for compound 7, along with potent inhibition of RIPK2. Structurally similar compounds from the HTS shared similar profiles (data not shown). Two structurally related Type II DFG-out inhibitors, 8 and 9, were identified through internal deck-mining (Table 1). We were pleased to find that, while some binding potency was lost, the excellent RIPK family selectivity could be maintained.
Figure 3.

Type II and Type I kinase chemotypes from the HTS. HTRF assay data acquired at 60 min for all kinases shown.
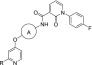
Table 1. Preliminary SARa,b.
![]()

HTRF assay protocol details are provided in the Supporting Information.
Assay results are the average of at least two replicates, and IC50 determination occurred after 60 min incubation.
Not tested in this assay.
Assay results are N of 1.
Full length construct used in assay instead of kinase domain.
While 8 and 9 are known Type II DFG-out inhibitors in c-Met kinase,30,31 it is possible for an inhibitor targeting an inactive conformation in one kinase to target the active conformation in a different kinase.17 As no inactive conformation inhibitors have been reported for RIPK3 previously, we were interested in building a model for a DFG-out conformation in RIPK3 to help guide structure design to improve kinase selectivity of our inhibitors; however, there were a number of challenges associated with building such a model. Based on the cocrystal structure with MLKL, RIPK3 is known to have a very short αC-helix, which is susceptible to unwinding, and would preclude the formation of a DFG-out conformation.12 In order to identify a conformation that may accommodate a DFG-out inhibitor, a model was constructed in which the activation loop from a RIPK2 DFG-out crystal structure32 was merged with the RIPK3 Type I/active conformation crystal structure. This provided a reasonable docking mode to use for evaluation of these compounds as putative RIPK3 Type II inhibitors. Ultimately this binding mode was confirmed by X-ray protein crystallography in a murine kinase domain construct bound with 9, demonstrating for the first time that RIPK3 can adopt this unreported conformation (Figure 4). The azaindole core engaged in hydrogen bond interactions with Met98 at the hinge. The DFG motif flipped outward, exposing the DFG backbone, which was able to form hydrogen bond interactions with the pyridone oxygen at Asp161. The pyridone oxygen also locked the conformation of the back pocket group through hydrogen bonding with the amide, further rigidifying the structure. The DFG flip revealed an available distal pocket that is filled by the pyridone and its substituent.
Figure 4.
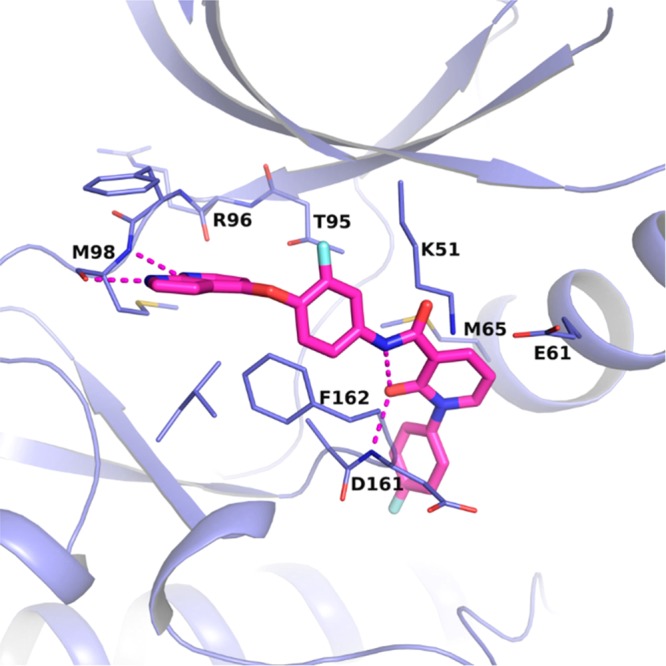
X-ray crystal structure of compound 9 bound to the catalytic domain of murine RIPK3. The carbons of 9 are colored in pink, and the carbons for RIPK3 are colored in purple. Oxygens are colored red, nitrogens blue, and fluorines light blue. Hydrogen bonds are indicated with dashed lines. PDB code 6OKO.
With Type II DFG-out inhibitors reported for both RIPK2 and RIPK1,6 our initial concern focused on RIP family selectivity. RIPK2 is part of the NOD signaling cascade, an innate immune pathway.14 The benefit of RIPK2 kinase inhibition is still somewhat controversial given that the RIPK2 kinase dead mutant (D146N) suggests that RIPK2 kinase activity and RIPK2 autophosphorylation were not linked to NOD2 signaling.33 RIPK1 is an important kinase in both necroptotic and death receptor-mediated apoptotic signaling.3 By targeting exclusive RIPK3 inhibition, we were seeking a profile devoid of the liabilities, which may be found with immunosuppression (RIPK2) or prevention of apoptotic cell death (RIPK1).
RIPK2 selectivity, a particular issue for most of the reported RIPK3 Type I inhibitors, was not a concern for the RIPK3 Type II inhibitors. Mono- and bidentate hinge binders surveyed showed a moderate degree of RIPK family selectivity (Table 1). Notably, 9 and 10 demonstrated 64- and 38-fold selectivity over RIPK1. Over 180-fold selectivity for RIPK2 was observed in all of the examples. In an effort to understand the importance of the hydrogen bonding interaction of the pyridone oxygen with the DFG backbone in the crystal structure and the internal amide, biphenyl analog 12 was prepared. The inability of 12 to inhibit RIPK3 suggests a strong dependence on the presence of the hydrogen bond network of the pyridone exists in this class of inhibitors.
As can be seen in Table 1, the selected compounds showed potent binding to c-Met kinase in addition to RIPK3. As the crystal structure for 9 in c-Met kinase had already been obtained,31 we were able to compare the protein crystal structures of c-Met (human) and RIPK3 (murine) and define spaces in which optimization targeting improved kinome selectivity could be realized (Figure 5). In both kinases, the inhibitor binds to an inactive conformation. The azaindole moiety is engaged in hydrogen bonding at the hinge, and the amide and pyridone form a hydrogen bond network with the protein backbone of the kinases at the DFG motif of the activation loop. The 4-fluorophenyl moiety occupies the distal pocket created by the DFG flip. Of note, the smaller gatekeeper residue present in RIPK3 (Thr95 vs Leu1157 in c-Met kinase) could provide an opportunity to increase bulk around the phenyl linker moiety and thereby improve selectivity over c-Met kinase.
Figure 5.
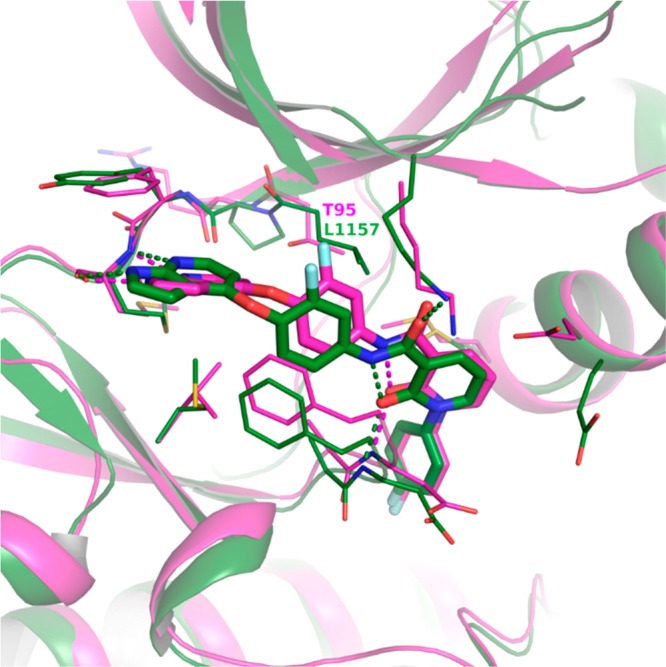
X-ray crystal structure of 9 in murine RIPK3 (magenta, PDB code 6OKO) overlaid with 9 in human c-Met kinase (green, PDB code 3CE3). Crystal structures of compound 9 are bound to the kinase catalytic domains. Oxygens are colored red, nitrogens blue, and fluorines light blue. Hydrogen bonds are indicated with dashed lines.
Initial work focused on improving selectivity of 11 versus c-Met kinase via modification of the A-ring that binds near the gatekeeper residue (Table 2). Incorporation of methyl groups ortho to the oxygen yielded 13. While only a small improvement in RIPK3 potency was observed, a significant decrease in c-Met kinase potency indicated that changes to the linker phenyl group were having the desired effect. Placing the methyl groups on the same side of the phenyl linker (14) further improved selectivity against c-Met kinase (>10-fold). Further increasing the bulk to naphthyl (15) was effective in both improving RIPK3 potency and increasing selectivity over c-Met kinase by >150-fold; however, 15 also demonstrated potent binding to p38α kinase (data not shown).
Table 2. Hinge Binder and Phenyl Linker SARa,b.

HTRF assay protocol details are provided in the Supporting Information.
Assay results are the average of at least two replicates, and IC50 determination occurred after 60 min incubation.
Assay results are N of 1.
Full length construct used in assay instead of kinase domain.
To improve c-Met kinase selectivity in the bidentate hinge binder, the direct replacement of the A-ring fluorine in 10 with a methyl group (16) was explored. While this modification failed to improve c-Met kinase selectivity, incorporation of dimethyl substitution (17 and 18) yielded more promising results. Placement of the dimethyl groups on the same side of the phenyl ring (18) maintained the majority of RIPK3 potency while increasing selectivity over c-Met kinase to 100-fold. Consistent with other results in this series, especially the disubstitued analogs, 18 was exquisitely selective among the RIPK family members. More extensive screening across the kinome revealed that 18 inhibited roughly 40 kinases (out of 246 tested) with an IC50 < 1 μM (data in Supporting Information), which may impact its ability to serve as a useful tool to probe RIPK3 pharmacology.
The synthesis of 8 and 9 has been previously described.30,31 Modifications to the phenyl linker and the hinge binder are detailed in Scheme 1. Reaction of p-nitrophenols 20 with 4-halopyridine or 2,4-dichloropyridine (19) provided the nitro intermediate 21. While most analogs were made using nitrophenols, use of the aniline was also tolerated. Reduction of the nitro group and coupling with previously reported 1-(4-fluorophenyl)-2-oxo-1,2-dihydropyridine-3-carboxylic acid31 led to final monodentate hinge binding compounds exemplified by 23. Palladium-mediated couplings with cyclopropanecarboxamide yielded bidentate hinge binding compounds exemplified by 24.
Scheme 1. Synthesis of Hinge Binder and Linker Analogs.

Reagents and conditions: (a) iPr2NEt, NMP, microwave, 210 °C, 33–94%; (b) SnCl2, EtOAc, 80 °C, 24–33%; (c) 1-(4-fluorophenyl)-2-oxo-1,2-dihydropyridine-3-carboxylic acid, BOP, Et3N, DMF, 22–87%; (d) cyclopropanecarboxamide, Pd2(dba)3, Xantphos, Cs2CO3, dioxane, 100 °C, 7–20%; (e) H2(g), Pd/C, MeOH, HCl, 6–61%.
In conclusion, the use of mechanistic screening to assess HTS hits can assist in the triaging of chemotypes with alternate binding modes. Time dependency studies on the HTS hits led to the identification of a putative Type II kinase inhibitor for RIPK3. An X-ray structure cocrystal of compound 9 with RIPK3 demonstrated that the kinase can adopt a previously unreported DFG-out conformation. Structure-based drug design considerations aided the optimization effort to identify compounds with improved selectivity over c-Met kinase (e.g., 18). Importantly, RIPK3 Type II kinase inhibitors were found to have excellent RIPK family selectivity, although broader kinome selectivity needs further optimization.
Acknowledgments
Special thanks to Randi Brown, Sarah Malmstrom, and Alan Futran for assay support.
Glossary
ABBREVIATIONS
- DAMP
damage-associated molecular patterns
- IBD
inflammatory bowel disease
- RIPK
receptor interacting protein kinase
- MLKL
mixed-lineage kinase like
- AMP-PNP
adenylyl-imidodiphosphate
- ATP
adenosine triphosphate
- DFG
aspartic acid, phenylalanine, glycine
- HTS
high-throughput screen
- HTRF
homogeneous time-resolved fluorescence
- CRC
concentration response curve
- TD
time dependency
- MOI
mode of inhibition
- Met98
methionine 98
- Asp161
aspartic acid 161
- Thr95
threonine 95
- Leu1157
leucine 1157
- c-Met
a receptor tyrosine kinase
- TNF
tumor necrosis factor
- NOD
nucleotide binding oligomerization domain
- iPr2NEt
diisopropylethylamine
- NMP
N-methyl-2-pyrrolidinone
- EtOAc
ethyl acetate
- BOP
(benzotriazol-1-yloxy)tris(dimethylamine)phosphonium hexafluorophosphate
- Et3N
triethylamine
- DMF
dimethylformamide
- dba
dibenzylideneacetone
- Xantphos
4,5-bis(diphenylphosphino)-9,9-dimethylxanthene
- MeOH
methanol
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00065.
Full experimental details for key compounds, biological protocols, and screening protocols (PDF)
Author Present Address
† Agios, 88 Sidney Street, Cambridge, Massachusetts 02139, United States.
Author Present Address
‡ EMD Serono, 45A Middlesex Turnpike, Billerica, Massachusetts 01821, United States.
Author Present Address
§ Sanofi, 153 2nd Avenue, Waltham, Massachusetts 02451, United States.
Author Contributions
The manuscript was written through contributions of all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Grootjans S.; Vanden Berghe T.; Vandenabeele P. Initiation and execution mechanisms of necroptosis: an overview. Cell Death Differ. 2017, 24, 1184–1195. 10.1038/cdd.2017.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad M.; Angeli J. P. F.; Vandenabeele P.; Stockwell B. R. Regulated necrosis: disease relevance and therapeutic opportunities. Nat. Rev. Drug Discovery 2016, 15, 348–366. 10.1038/nrd.2015.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silke J.; Rickard J. A.; Gerlic M. The diverse role of RIP kinases in necroptosis and inflammation. Nat. Immunol. 2015, 16, 689–697. 10.1038/ni.3206. [DOI] [PubMed] [Google Scholar]
- Wang W.; Marinis J. M.; Beal A. M.; Savadkar S.; Wu Y.; Khan M.; Taunk P. S.; Wu N.; Su W.; Ahson A.; Kurz E.; Chen T.; Yaboh I.; Li F.; Gutierrez J.; Diskin B.; Hundeyin M.; Reilly M.; Lich J. D.; Harris P. A.; Mahajan M. K.; Thorpe J. H.; Massau P.; Mosley J. E.; Leinwand J.; Kochen Rossi J. A.; Mishra A.; Aykut B.; Glacken M.; Ochi A.; Verma N.; Kim J. I.; Vasudevaraja V.; Adeegbe D.; Almonte C.; Bagdatlioglu E.; Cohen D. J.; Wong K.-K.; Berton J.; Miller G. RIP1 Kinase drives macrophage-mediated adaptive immune tolerance in pancreatic cancer. Cancer Cell. 2018, 34, 757–774. 10.1016/j.ccell.2018.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werba G.; Seifert L.; Miller G. Necroptotic cell death – an unexpected driver on pancreatic oncogenesis. Cell Cycle 2016, 15, 2095–2096. 10.1080/15384101.2016.1191251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan M. C.; Rajapaksa N. S. Kinase inhibitors for the treatment of immunological disorders: recent advances. J. Med. Chem. 2018, 61, 9030. 10.1021/acs.jmedchem.8b00667. [DOI] [PubMed] [Google Scholar]
- Yoshikawa M.; Saitoh M.; Katoh T.; Seki T.; Bigi S. V.; Shimizu Y.; Ishii T.; Okai T.; Kuno M.; Hattori H.; Watanabe E.; Saikatendu K. S.; Zou H.; Nakakariya M.; Tatamiya T.; Nakada Y.; Yogo T. Discovery of 7-oxo-2,4,5,7-tetrahydro-6H-pyrazolo[3,4-c]pyridine derivatives as potent, orally available, and brain penetrant receptor interacting protein 1 (RIP1) kinase inhibitors: analysis of structure-kinetic relationships. J. Med. Chem. 2018, 61, 2384–2409. 10.1021/acs.jmedchem.7b01647. [DOI] [PubMed] [Google Scholar]
- Patel S.; Hamilton G.. Inhibitors of RIP1 and methods of use thereof. PCT Int. Appl. 2018WO A12018/109097 A1.
- Delehouzé C.; Leverrier-Penna S.; Le Cann F.; Comte A.; Jacquard-Fevai M.; Delalande O.; Desban N.; Baratte B.; Gallais I.; Faurez F.; Bonnet M. C.; Hauteville M.; Goekjian P. G.; Thuillier R.; Favreau F.; Vandenabeele P.; Hauet T.; Dimanchi-Boitrel M. T.; Bach S. 6E11, a highly selective inhibitor of receptor-interacting protein kinase 1, protects cells against cold hypoxia-reoxygenation injury. Sci. Rep. 2017, 7, 12931. 10.1038/s41598-017-12788-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan B.; Liu L.; Huang S.; Ren Y.; Wang H.; Yao Z.; Lin L.; Chen S.; Wang X.; Zhang Z. Discovery of a new class of highly potent necroptosis inhibitors targeting the mixed lineage kinase domain-like protein. Chem. Commun. 2017, 53, 3637–3640. 10.1039/C7CC00667E. [DOI] [PubMed] [Google Scholar]
- Lessene G. L.; Wilks A. F.; Murphy J. M.; Garnier J.-M.; Czabotar P. E.; Hildebrand J. M.; Lucet I.; Silke J. H.; Feutrill J. T.; Cuzzupe A. N.; Sharma P.. Methods for inhibiting necroptosis. PCT Int. Appl. 2015WO 2015/172203 A1.
- Xie T.; Peng W.; Yan C.; Wu J.; Gong X.; Shi Y. Structural insights into RIP3-mediated necroptotic signaling. Cell Rep. 2013, 5, 70–78. 10.1016/j.celrep.2013.08.044. [DOI] [PubMed] [Google Scholar]
- Hart A. C.; Shaw P. J.. Targeting Necroptosis for Therapeutic Intervention in Inflammatory Diseases. In Medicinal Chemistry Reviews; Desai M. J., Ed.; ACS: Washington, DC, 2016; Vol. 51, pp 101–114. [Google Scholar]
- Haile P. A.; Votta B. J.; Marquis R. W.; Bury M. J.; Mehlmann J. F.; Singhaus R. Jr.; Charnley A. K.; Lakdawala A. S.; Convery M. A.; Lipshutz D. B.; Desai B.; Swift B.; Capriotti C. A.; Berger S. B.; Mahajan M. K.; Reilly M. A.; Rivera E. J.; Sun H. H.; Nagilla R.; Beal A. M.; Finger J. N.; Cook M. N.; King B. W.; Ouellette M. T.; Totritis R. D.; Pierdomenico M.; Negroni A.; Stronati L.; Cucchiara S.; Ziólkowski B.; Vossenkämper A.; MacDonald T. T.; Gough P. J.; Bertin J.; Casillas L. N. The identification and pharmacological characterization of 6-(tert-butylsulfonyl)-N-(5-fluoro-1H-indazol-3-yl)quinolin-4-amine (GSK583), a highly potent and selective inhibitor of RIP2 kinase. J. Med. Chem. 2016, 59, 4867–4880. 10.1021/acs.jmedchem.6b00211. [DOI] [PubMed] [Google Scholar]
- http://lincs.hms.harvard.edu/db/datasets/20131/main.
- Davis M. I.; Hunt J. P.; Herrgard S.; Ciceri P.; Wodicka L. M.; Pallares G.; Hocker M.; Treiber D. K.; Zarrinkar P. P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- Zhao Z.; Wu H.; Wang L.; Liu Y.; Knapp S.; Liu Q.; Gray N. S. Exploration of Type II binding mode: a privileged approach for kinase inhibitor focused drug discovery?. ACS Chem. Biol. 2014, 9, 1230–1241. 10.1021/cb500129t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Z.; Grütter C.; Rauh D. Strategies for the selective regulation of kinases with allosteric modulators: exploiting exclusive structural features. ACS Chem. Biol. 2013, 8, 58–70. 10.1021/cb300663j. [DOI] [PubMed] [Google Scholar]
- Garuti L.; Roberti M.; Bottegoni G. Non-ATP competitive protein kinase inhibitors. Curr. Med. Chem. 2010, 17, 2804–2821. 10.2174/092986710791859333. [DOI] [PubMed] [Google Scholar]
- Alton G. R.; Lunney E. A. Targeting the unactivated conformations of protein kinases for small molecule drug discovery. Expert Opin. Drug Discovery 2008, 3, 595–605. 10.1517/17460441.3.6.595. [DOI] [PubMed] [Google Scholar]
- Roskoski R. Jr. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 2016, 103, 26–48. 10.1016/j.phrs.2015.10.021. [DOI] [PubMed] [Google Scholar]
- Palmieri L.; Rastelli G. P-C helix displacement as a general approach for allosteric modulation of protein kinases. Drug Discovery Today 2013, 18, 407–414. 10.1016/j.drudis.2012.11.009. [DOI] [PubMed] [Google Scholar]
- Simard J. R.; Rauh D. FliK: a direct-binding assay for the identification and kinetic characterization of stabilizers of inactive kinase conformations. Methods Enzymol. 2014, 548, 147–171. 10.1016/B978-0-12-397918-6.00006-9. [DOI] [PubMed] [Google Scholar]
- Ranjitkar P.; Perera B. G. K.; Swaney D. L.; Hari S. B.; Larson E. T.; Krishnamurty R.; Merritt E. A.; Villén J.; Maly D. J. Affinity-based probes based on Type II kinase inhibitors. J. Am. Chem. Soc. 2012, 134, 19017–19025. 10.1021/ja306035v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ung P.M.-U.; Schlessinger A. DFGModel: predicting protein kinase structures in inactive states for structure-based discovery of Type-II inhibitors. ACS Chem. Biol. 2015, 10, 269–278. 10.1021/cb500696t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich J.; Hulme C.; Hurley L. H. The design, synthesis and evaluation of 8 hybrid DFG-out allosteric kinase inhibitors: a structural analysis of the binding interactions of Gleevec®, Nexavar®, and BIRB796. Bioorg. Med. Chem. 2010, 18, 5738–5748. 10.1016/j.bmc.2010.05.063. [DOI] [PubMed] [Google Scholar]
- Jia Y. Current status of HTRF(®) technology in kinase assays. Expert Opin. Drug Discovery 2008, 3, 1461–1474. 10.1517/17460440802518171. [DOI] [PubMed] [Google Scholar]
- Borzilleri R. M.; Cornelius L. A. M.; Schmidt R. J.; Schroeder G. M.; Kim K. S. Monocyclic heterocycles as kinase inhibitors. U.S. Patent 2008, 7459562 B2.
- For advancement in the Type I kinase chemotype, see: Hart A. C.; Pitts W. J.; Mastalerz H.; Guo J.; Brown G. D.. Preparation of substituted dihydro-1H-pyrrolo[3,2-c]pyridin-4(5H)-ones as RIPK3 inhibitors. PCT Int. Appl. 2016, WO 2016100166.
- Schroeder G. M.; An Y.; Cay Z.-W.; Chen X.-T.; Clark C.; Cornelius L. A. M.; Dai J.; Gullo-Brown J.; Gupta A.; Henley B.; Hunt J. T.; Jeyaseelan R.; Kamath A.; Kim K.; Lippy J.; Lombardo L. J.; Manne V.; Oppenheimer S.; Sack J. S.; Schmidt R. J.; Shen G.; Stefanski K.; Tokarski J. S.; Trainor G. L.; Wautlet B. S.; Wei D.; Williams D. K.; Zhang Y.; Zhang Y.; Fargnoli J.; Borzilleri R. M. Discovery of N-(4-(2-amino-3-chloropyridin-4-yloxy)-3-fluorophenyl)-4-ethoxy-1-(4-fluorophenyl)-2-oxo-1,2-dihydropyridine-3-carboxamide (BMS-777607), a selective and orally efficacious inhibitor of the Met kinase superfamily. J. Med. Chem. 2009, 52, 1251–1254. 10.1021/jm801586s. [DOI] [PubMed] [Google Scholar]
- Kim K. S.; Zhang L.; Schmidt R.; Cai Z.-W.; Wei D.; Williams D. K.; Lombardo L. J.; Trainor G. L.; Xie D.; Zhang Y.; An Y.; Sack J. S.; Tokarski J. S.; Darienzo C.; Kamath A.; Marathe P.; Zhang Y.; Lippy J.; Jeyaseelan R. Sr.; Wautlet B.; Henley B.; Gullo-Brown J.; Manne V.; Hunt J. T.; Fargnoli J.; Borzilleri R. M. Discovery of pyrrolopyridine-pyridone based inhibitors of Met kinase: synthesis, X-ray crystallographic analysis, and biological activities. J. Med. Chem. 2008, 51, 5330–5341. 10.1021/jm800476q. [DOI] [PubMed] [Google Scholar]
- Canning P.; Ruan Q.; Schwerd T.; Hrdinka M.; Maki J. L.; Saleh D.; Suebsuwong C.; Ray S.; Brennan P. E.; Cuny G. D.; Uhlig H. H.; Gyrd-Hansen M.; Degterev A.; Bullock A. N. Inflammatory signaling by NOD-RIPK2 is inhibited by clinically relevant Type II kinase inhibitors. Chem. Biol. 2015, 22, 1174–1184. 10.1016/j.chembiol.2015.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncharov T.; Hedayati S.; Mulvihill M. M.; Izrael-Tomasevic A.; Zobel K.; Jeet S.; Gedoroba A. V.; Eidenschenk C.; DeVoss J.; Yu K.; Shar A. S.; Kirkpatrick D. S.; Fairbrother W. J.; Deshayes K.; Vucic D. Disruption of XIAP-RIPK2 association blocks NOD2-mediated inflammatory signaling. Mol. Cell 2018, 69, 551–565. 10.1016/j.molcel.2018.01.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.