

Abstract
The lysyl oxidase (LOX) family of extracellular proteins plays a vital role in catalyzing the formation of cross-links in fibrillar elastin and collagens leading to extracellular matrix (ECM) stabilization. These enzymes have also been implicated in tumor progression and metastatic disease and have thus become an attractive therapeutic target for many types of invasive cancers. Following our recently published work on the discovery of aminomethylenethiophenes (AMTs) as potent, orally bioavailable LOX/LOXL2 inhibitors, we report herein the discovery of a series of dual LOX/LOXL2 inhibitors, as well as a subseries of LOXL2-selective inhibitors, bearing an aminomethylenethiazole (AMTz) scaffold. Incorporation of a thiazole core leads to improved potency toward LOXL2 inhibition via an irreversible binding mode of inhibition. SAR studies have enabled the discovery of a predictive 3DQSAR model. Lead AMTz inhibitors exhibit improved pharmacokinetic properties and excellent antitumor efficacy, with significantly reduced tumor growth in a spontaneous breast cancer genetically engineered mouse model.
Introduction
The lysyl oxidase (LOX) family of copper-dependent extracellular proteins comprises the founder member enzyme, LOX, and four LOX-like enzymes (LOXL1–4).1−6 While there is greater than 50% sequence identity between the isoforms, which includes a conserved C-terminal catalytic domain across the family containing the copper binding site and the lysine tyrosylquinone (LTQ) cofactor, the enzymes can be divided into two subgroups based on differences to the N-terminal structure. Indeed, LOX and LOXL1 contain a variable N-terminal propeptide that undergoes proteolytic cleavage to form the active enzyme extracellularly. LOXL2–4 differ in that they do not possess this propeptide region and instead contain four scavenger receptor cysteine-rich (SRCR) domains at the N-terminus, which are thought to mediate protein–protein interactions in the ECM. In the case of LOXL2, the protein undergoes proteolytic cleavage of the first two SRCR domains upon secretion; however, unlike LOX, processing is not required for catalytic activation.7,8
The most widely studied function of the LOX enzymes is their ability to form cross-links in fibrillar elastin and collagens through oxidative deamination of specific lysyl residues, thus stabilizing the ECM.3 However, recent reports suggest that these enzymes have a multitude of biological functions, which include cell proliferation and epithelial–mesenchymal transition (EMT).3,5,9 Consequently, the more widely studied LOX and LOXL2 isoforms have been implicated in tumor progression, where they are highly expressed and actively involved in remodeling the tumor microenvironment.10−16
The LOX family has thus become an attractive therapeutic target for the treatment of many types of invasive cancers, particularly those with poor patient outcomes. Targeting LOXs with small molecule inhibitors is very challenging owing to the lack of crystal structures useful for drug design for any of the isoforms (the only reported LOXL2 structure is a precursor state without cofactor formed)17 and difficulties associated with isolating several of the enzymes in an active form. Nevertheless, in recent years several LOXL2-selective inhibitors have been reported, including haloallylamine-based inhibitors PXS-S1A and the highly potent PXS-S2A (full structures not disclosed),18 as well as dual LOXL2/LOXL3 inhibitors PXS-5153A (1)19 and aminomethylenepyridine 2 (Figure 1).20 Our dual LOX/LOXL2 inhibitor CCT365623 (3a)21,22 is an orally efficacious aminomethylenethiophene (AMT) based inhibitor, which has been used to help elucidate mechanisms by which LOX drives tumor progression. These novel inhibitors offer significant advantages over the prototypical pan-LOX inhibitor β-aminopropionitrile (BAPN),23,24 whose lack of sites amenable for chemical modification precludes preclinical optimization.
Figure 1.

Small molecule inhibitors of LOX family enzymes.
We recently reported the discovery of AMT inhibitor CCT365623 (3a) following a significant medicinal chemistry campaign to elucidate the structure–activity relationship (SAR) of this class of compound with respect to LOX inhibition.22 Systematic modifications were made to a hit compound identified following a high-throughput screen (HTS), leading to development of submicromolar half maximal inhibitory concentration (IC50) inhibitors possessing desirable selectivity and pharmacokinetic (PK) properties.
During the course of these studies, the 2,5-substituted thiophene core was replaced with various other five-membered heterocyclic rings to ascertain the importance of this moiety on activity. Of those assessed, only a 2-aminomethylene-5-sulfonyl thiazole core retains activity, with naphthalenesulfonyl-substituted thiazole 6 showing comparable levels of LOX inhibition to the analogous thiophene compound 4 (Figure 2).22 By contrast, thiazole regioisomer 5 is relatively inactive. Interestingly, in the case of the active thiazole compound (6) we also observe a modest increase in potency toward LOXL2 inhibition, with 6 proving equipotent against both isoforms. These observations prompted parallel investigations into the development of 2-aminomethylene-5-sulfonylthiazoles (AMTz) as dual LOX/LOXL2 inhibitors, and given the commercial availability of purified LOXL2 enzyme, these studies were carried out using LOXL2 enzyme, both as a target and as a surrogate to assess LOX activity.
Figure 2.

LOX and LOXL2 activity of analogous AMT and AMTz inhibitors.
Results and Discussion
Time-Dependent Inhibition
Initial studies concerned the enzyme–inhibitor preincubation time in our biochemical assay, whereby we assessed whether our compounds inhibit enzyme activity in a time-dependent manner. Dual LOX/LOXL2 inhibitors 4 and 6, along with the less active 5-aminomethylene-2-sulfonyl thiazole regioisomer 5, were assessed using longer preincubation times of 1 and 3 h, and the activity was compared to previously obtained 20 min preincubation data (Table 1). The results of this study demonstrate that longer preincubation times result in increased levels of enzyme inhibition with the greatest difference in effect observed when the time is increased from 20 min to 1 h, upon which up to a 5-fold increase in potency is observed. Further increasing the time to 3 h has a smaller positive effect on activity while remaining within 2-fold of the 1 h data. On the basis of these findings, we decided to employ a 1 h preincubation time for the remainder of the studies described herein, which provides an optimal determination of enzyme inhibition at a physiologically relevant time point.
Table 1. Effect of Preincubation Time on LOXL2 Potency.

| entry | compd | X | Y | LOXL2, 20 min, IC50 (μM)a,b | LOXL2, 1 h, IC50 (μM)a,c | LOXL2, 3 h, IC50 (μM)a,d |
|---|---|---|---|---|---|---|
| 1 | 4 | CH | CH | 3.40 | 0.81 | 0.47 |
| 4.76 | 0.85 | 0.59 | ||||
| 2 | 5 | CH | N | 26.8 | 6.41 | 3.54 |
| 27.0 | 6.02 | 3.19 | ||||
| 3 | 6 | N | CH | 0.86 | 0.28 | 0.12 |
| 0.69 | [0.22, 0.36] | 0.12 |
Reported IC50 values were determined in at least two separate experiments (n ≥ 2). When n = 2, individual IC50 values are shown. When n > 2, the values are reported as the geometric mean with the corresponding 95% confidence interval in square brackets.
Compound was preincubated with enzyme for 20 min.
Compound was preincubated with enzyme for 1 h.
Compound was preincubated with enzyme for 3 h.
Thiophene vs Thiazole Core
Optimization studies began with our recently published AMT inhibitor (3a),22 whereby we modified the core to the analogous 1,3-thiazole compound (7a) and assessed LOXL2 activity inhibition (Table 2, compare entries 1 and 4). Pleasingly, this gives a modest increase in potency, with an IC50 of 0.086 μM. We then assessed whether this increase in activity, attributed to the presence of a nitrogen atom in the ring, could be mimicked through the incorporation of an electronegative halogen atom on the thiophene ring, which could also engender the formation of an intramolecular hydrogen bond to improve ligand binding:25 3-chlorothiophene (entry 2) was found to be less potent by >10-fold compared to the parent compound (entry 1), while a fluorine substituent was 4-fold less active (entry 3). A second matched pair was synthesized to confirm this trend, and again the potency achieved with the thiazole analogue was slightly greater than that of the thiophene compound (compare entries 6 and 7). This suggests that the presence of a nitrogen atom in the heterocyclic core is advantageous, providing additional stabilization to the protein–inhibitor complex through either resonance mechanisms or noncovalent interactions.
Table 2. Effects of Thiophene vs Thiazole Cores on LOXL2 Potency.

| entry | compd | X | R1 | R2 | SO2Me position | LOXL2, IC50 (μM)a |
|---|---|---|---|---|---|---|
| 1 | 3a | CH | H | H | 3 | 0.176 |
| [0.105, 0.295] | ||||||
| 2 | 3b | CCl | H | H | 3 | 2.488 |
| 2.164 | ||||||
| 3 | 3c | CF | H | H | 3 | 0.671 |
| 0.714 | ||||||
| 4 | 7a | N | H | H | 3 | 0.086 |
| [0.061, 0.119] | ||||||
| 5 | 7b | N | CH3 | H | 3 | 68.26 |
| [43.77, 106.5] | ||||||
| 6 | 8 | CH | H | CH3 | 4 | 0.686 |
| [0.456, 1.031] | ||||||
| 7 | 9 | N | H | CH3 | 4 | 0.423 |
| [0.302, 0.593] |
Reported IC50 values were determined using 1 h preincubation in at least two separate experiments (n ≥ 2). When n = 2, individual IC50 values are shown. When n > 2, the values are reported as the geometric mean with the corresponding 95% confidence interval in square brackets.
In order to confirm that the aminomethylene group was still essential for inhibition, we assessed the effect of an N-methyl substituent group (entry 5). As expected, this results in significant loss of activity, which is consistent with previous conclusions that the aminomethylene group is required to form a stable Schiff base between the inhibitor and the LTQ cofactor that is a feature of both LOX and LOXL2.
Modification of Thiazole C-5 Group
We next assessed the C-5 substituent group of the thiazole-based inhibitors to determine whether the same SAR trends were observed as in the thiophene series and whether the lead scaffold remained optimal. An aryl sulfonyl group is preferred to either an alkyl or cycloalkyl group (Table 3, compare entries 1–3). Focusing on the aryl substituents, we find that monosubstitution with either a methane sulfonyl (entry 4) or a phenyl group (entry 6) is tolerated since these are equipotent with the unsubstituted example (entry 3), but a disubstituted aryl group (entry 8) remains preferred for anti-LOXL2 activity.
Table 3. Effect of Thiazole C-5 Linker (L) and Substituent (R) Groups on LOXL2 Potency.

Reported IC50 values were determined using 1 h preincubation in at least two separate experiments (n ≥ 2). When n = 2, individual IC50 values are shown. When n > 2, the values are reported as the geometric mean with the corresponding 95% confidence interval in square brackets.
We assessed the effect of the oxidation level of the sulfonyl linker and find that use of a sulfoxide or sulfide linker results in less active compounds (compare entries 8–10). This is expected based on previous studies; however, it is noted that the impact resulting from a decrease in oxidation state is less significant in the AMTz series herein than it was in the AMT series.22 Removal of the sulfonyl linker is tolerated but results in partial loss of activity, with the most significant impact observed in the biaryl systems (compare entries 4 and 5; 6 and 7; 8 and 11). As explored later (Table 6), this increased tolerance to a range of linkers appears to be a feature of the thiazole compared to thiophene core, whereby the presence of the nitrogen atom in the ring increases the electron-withdrawing properties,26,27 thus mitigating the need for an electron-withdrawing linker.
Table 6. Activity of AMT and AMTz Inhibitors against LOX, LOXL2, and LOXL3.
| IC50 (μM)a |
|||
|---|---|---|---|
| compd | LOXL2 | LOX | LOXL3 |
| BAPN | 0.665 | 3.14 | 0.31 |
| [0.57, 0.77] | [1.79, 5.49] | [0.22, 0.45] | |
| 3a | 0.176 | 0.898 | 13.49 |
| [0.105, 0.295] | 0.728 | 18.54 | |
| 7a | 0.086 | 1.77 | 0.92 |
| [0.061, 0.12] | 1.49 | 0.86 | |
| 7d | 0.233 | 1.31 | 4.70 |
| [0.19, 0.28] | 1.13 | 3.85 | |
| 7e | 0.38 | 7.17 | 6.81 |
| [0.27, 0.54] | 6.91 | 6.78 | |
| 17 | 0.652 | 51.96 | 9.31 |
| 0.716 | 46.49 | 9.52 | |
| 21a | 0.109 | 3.42 | 1.22 |
| [0.066, 0.18] | 2.65 | 1.51 | |
| 21b | 0.151 | 3.3 | 1.25 |
| [0.12, 0.19] | 1.86 | 1.43 | |
| 22b | 0.302 | >100 | 3.86 |
| [0.22, 0.42] | >100 | 4.26 | |
| 22c | 0.37 | >100 | 7.56 |
| [0.23, 0.596] | >100 | 6.69 | |
| 22d | 0.425 | >100 | 24.24 |
| [0.35, 0.51] | >100 | 36.86 | |
Reported IC50 values were determined using 1 h preincubation in at least two separate experiments (n ≥ 2). When n = 2, individual IC50 values are shown. When n > 2, the values are reported as the geometric mean with the corresponding 95% confidence interval in square brackets.
Variation of the Phenylsulfonyl Ring Substituents
Incorporation of a bis-sulfonyl biaryl C-5 group is advantageous for activity and was previously found to improve oral in vivo PK exposure in the AMT series of LOX inhibitors.22 We subsequently looked to ascertain if there was scope to optimize the substituent effects further (Table 4). Varying the methanesulfonyl group (R2) to an ethyl or isopropyl group is tolerated, albeit resulting in a drop in potency; however, a larger tert-butyl group proves detrimental to activity (compare entries 1–4). A methoxy substituent is similarly well tolerated (entry 5), while a methylamino group appears to be less favorable (entry 6).
Table 4. Effects of 3,5-Disubstituted Phenylsulfonyl Groups on LOXL2 Potency.
![]()

Reported IC50 values were determined using 1 h preincubation in at least two separate experiments (n ≥ 2). When n = 2, individual IC50 values are shown. When n > 2, the values are reported as the geometric mean with the corresponding 95% confidence interval in square brackets.
Small modifications to the aryl substituent (R1) do not result in a significant change in potency. Inhibitors bearing either an electron-donating p-methyl group or an electron-withdrawing p-fluoro or p-trifluoromethyl substituent demonstrate comparable levels of activity to the parent compound (compare entries 7–9 to entry 1). Replacing the phenyl group with an N-methyl pyrazolyl group is found to be favorable in conjunction with an ethyl group (compare entries 2 and 11), though it does not appear to confer additional potency when R2 is a methanesulfonyl group (compare entries 1 and 10). Further modification of the ethyl group of 21b to an electron-withdrawing trifluoromethyl or chloro group has a slightly negative effect on activity, though a fluoro substituent is well tolerated (compare entries 11–14). Overall, from these results we conclude that the parent inhibitor 7a is well optimized while gaining further understanding of the SAR and identifying additional scaffolds for further study.
Activity of AMTz Regioisomers
Final SAR studies concerned the substitution pattern around the thiazole core, whereby we compared the anti-LOXL2 activity of selected 2,5-substituted AMTz inhibitors to that of their 2,4-substituted regioisomers (Table 5). Although there is a trend favoring 5-substitution vs 4-substitution, it is again less pronounced than that previously observed in the thiophene series against LOX22 (compare entries 1 and 2; 3 and 4; 5 and 6; 7 and 8).
Table 5. Effects of 2,4- vs 2,5-Substitution of Thiazole Core on LOXL2 Potency.

Reported IC50 values were determined using 1 h preincubation in at least two separate experiments (n ≥ 2). When n = 2, individual IC50 values are shown. When n > 2, the values are reported as the geometric mean with the corresponding 95% confidence interval in square brackets.
Selectivity Studies
We assessed the selectivity profiles of our AMTz inhibitors against LOX and LOXL3 isoforms (Table 6) and against common amine oxidases and the potassium ion channel hERG (Table 7).
Table 7. Potency and Selectivity of AMTz Inhibitors over Common Amine Oxidases and hERG.
| IC50 (μM)a |
||||||
|---|---|---|---|---|---|---|
| compd | LOXL2 | MAO-A | MAO-B | DAO | SSAO | hERG |
| 6 | 0.28 | >100 | >100 | >100 | 47.2 | 51 |
| [0.22, 0.36] | >100 | >100 | >100 | 47.1 | ||
| 7a | 0.086 | 63.8 | >100 | >100 | 3.2 | 10 |
| [0.061, 0.12] | 59.3 | >100 | >100 | 1.2 | ||
| 7e | 0.38 | 29.2 | 64.3 | >100 | 5.2 | 36 |
| [0.27, 0.54] | 31.4 | 46.2 | >100 | 3.6 | ||
| 18 | 0.103 | 100 | 16.4 | >100 | 2.7 | 3.2 |
| [0.083, 0.13] | 88.3 | 20 | >100 | 1.3 | ||
| 21a | 0.109 | >100 | >100 | >100 | 13.3 | nd |
| [0.066, 0.18] | >100 | >100 | >100 | 17.2 | ||
| 21b | 0.151 | >100 | >100 | >100 | 28.6 | 47 |
| [0.12, 0.19] | >100 | >100 | >100 | 19.6 | ||
| 21e | 0.143 | 61.7 | >100 | 21.3 | 36.3 | 66 |
| [0.057, 0.36] | 66.8 | >100 | 22.5 | 40.3 | ||
| 22b | 0.302 | >100 | >100 | >100 | >100 | nd |
| [0.22, 0.42] | >100 | >100 | >100 | >100 | ||
| 22c | 0.37 | >100 | >100 | >100 | >100 | nd |
| [0.23, 0.596] | >100 | >100 | >100 | >100 | ||
| 22d | 0.425 | >100 | >100 | >100 | 102.2 | 29 |
| [0.35, 0.51] | >100 | >100 | >100 | 74.3 | ||
Reported IC50 values were determined using 1 h preincubation in at least two separate experiments (n ≥ 2). When n = 2, individual IC50 values are shown. When n > 2, the values are reported as the geometric mean with the corresponding 95% confidence interval in square brackets. nd: not determined.
Activity Profiles against LOX and LOXL3 Isoforms
While use of LOXL2 as a surrogate for LOX was a practical and informative approach, it was necessary to assess the activity of a range of AMTz inhibitors against LOX to confirm our belief that these are in fact dual LOX/LOXL2 inhibitors. A diverse set of compounds were selected, encompassing 2-aminomethylene-5-sulfonyl- (7a, 7e, 21a, and 21b) and 2-aminomethylene-4-sulfonyl- (22b, 22c, and 22d) AMTz regioisomers, sulfide-linked 7d, and 17 with no linker, as well as thiophene 3a and BAPN. From this study we were able to confirm that 2-aminomethylene-5-sulfonyl thiazoles 7a, 21a, and 21b, thiophene 3a, and BAPN exhibit good anti-LOX activity that is in accordance with previously obtained data.21,22,28 A slight decrease in potency toward LOX inhibition is observed with compound 7e, which is consistent with LOXL2 activity. Removal of the sulfonyl linker (17) is detrimental to activity; however, a sulfide linker (7d) is well tolerated which, as discussed previously, can be attributed to the presence of a nitrogen atom in the thiazole core which presumably increases the ability of these compounds to form a stable covalent bond upon binding. Interestingly, 2,4-AMTz regioisomers (22b, 22c, and 22d) are found to be inactive against LOX, which follows a similar trend to the thiophene series in which 2,4-regioisomers were demonstrated to be 15-fold less potent.22 As seen previously, all AMTz inhibitors demonstrate increased potency toward LOXL2 inhibition versus LOX.
As discussed previously, literature compounds 1 and 2 are in fact dual LOXL2/LOXL3 inhibitors,19,20 while selectivity data concerning LOXL isoforms have not been reported for other literature inhibitors. As a member of the LOX family, LOXL3 is known to modulate the ECM,29−31 though there is tissue expression variance compared to other LOX proteins, and it has been shown to play a significant role in muscular, skeletal, and lung development in mice.32−34 More recently, studies have demonstrated an involvement of LOXL3 in cancer and metastasis, suggesting it to be a potential therapeutic target for malignant disease.35,36
With regards to LOXL3 inhibition, it is interesting to observe that our AMT and AMTz compounds exhibit moderate to high selectivity toward LOXL2 in all cases (Table 6), unlike the nonselective LOX-family inhibitor BAPN and reported literature compounds 1 and 2 (activity within 3-fold for LOXL2 and LOXL3 in all cases).19,20 2,5-AMTz inhibitors 7a, 7e, 21a, and 21b demonstrate ≥10-fold selectivity toward LOXL2, with IC50 values of approximately 1 μM; compound 7e is consistently less potent against all isoforms. Modification of the sulfonyl linker to a sulfide (7d) or direct aryl-linked compound (17) results in a decrease in potency, comparable to that observed against LOXL2. Increased selectivity is observed with 2,4-AMTz regioisomers (22b–d), in particular compound 22d which does not possess a bis-sulfonyl group. AMT inhibitor 3a is found to be a weak inhibitor of LOXL3, providing selectivity in excess of 100-fold. This study demonstrates that 2-aminomethylene-5-sulfonyl thiazoles are potent inhibitors of three LOX isoforms, while selectivity toward LOXL2, particularly 2,4-AMTz regioisomers, supports their use as valuable tool compounds to study the biology and functions of LOXL2. Owing to the lack of availability and difficulties involved in obtaining other LOXL enzymes in an active form,20 we have been unable to assess selectivity of our compounds over other LOX-family members.
Selectivity over Common Amine Oxidases and hERG
A selection of compounds including our most potent AMTz inhibitors were assessed for their selectivity over the flavin-containing monoamine oxidases (MAO) A and B, copper-containing diamine oxidase (DAO) and semicarbazide-sensitive amine oxidase (SSAO), and the hERG channel (Table 7). In general, our inhibitors show excellent selectivity over MAO-A and -B and DAO. Bis-sulfonyl compounds 7a, 18, and 21a, along with biphenyl compound 7e, demonstrate moderate SSAO inhibition. N-Methyl pyrazolyl inhibitors 21b and 21e show improved selectivity, and further studies carried out with 21b indicate that it is not a substrate of SSAO, unlike 3a.22 Bis-sulfonyl compounds 7a and 18 are also found to be moderate inhibitors of the hERG channel, while others compounds assessed show good selectivity. On the basis of the in vitro activity and selectivity profiles, 2-aminomethylene-5-sulfonylthiazole inhibitors 6, 7e, 21b, and 21e, along with 7a for direct comparison with thiophene 3a and 22d as an exemplar of the 2,4-AMTz subseries, were advanced to metabolic stability assessment and in vivo mouse PK studies.
Pharmacokinetic Evaluation
All compounds assessed in vivo demonstrate good metabolic stability against mouse liver microsomes (MLM) and inhibitor exposure following oral administration in mouse PK studies (Table 8). Naphthalenesulfonyl compound 6 shows low to moderate plasma exposure (AUC = 1–10 μM), as does 3-ethyl-5-phenyl inhibitor 7e and 21e bearing N-methyl pyrazolyl and fluoro substituents. Pleasingly, a number of compounds assessed demonstrate desirable PK profiles, achieving greater plasma exposure levels than that of our previously published inhibitor (3a),22 and excellent oral bioavailability. Indeed, compounds 7a, 21b, and 22d have AUCs above 18 μM·h, achieving Cmax concentrations of up to 32 μM. These compounds also exhibit good permeability in the Caco-2 assay, used to model adsorption of orally administered drugs in the small intestine, and present lower efflux levels than those seen previously for thiophene 3a. Given our observations that these compounds demonstrate time-dependent inhibition (see Tables 1 and 9), it is likely that efficacy would be driven by Cmax.
Table 8. In Vitro Mouse Liver Microsome (MLM) Stability, in Vivo PK Properties, and Caco-2 Permeability of AMTz Inhibitors.
| Caco-2 Pappe,f (10–6 cm/s) |
|||||||
|---|---|---|---|---|---|---|---|
| compd | MLM stability (%)a | Cmax(PO) (μM)c | AUC(PO) (μM·h)d | t1/2(PO) (h) | F (%)e | A → B | B → A |
| 6 | 89 | 6.35 | 4.20 | 0.8 | nd | nd | nd |
| 21e | 90 | 9.75 | 4.48 | 2.0 | nd | nd | nd |
| 7e | 70 | 10.16 | 9.97 | 0.6 | nd | nd | nd |
| 3a | |||||||
| (i) mouse | (i) 64 | (i) 16.83 | (i) 14.89 | (i) 0.45 | (i) 45 | 8.5 | 35 |
| (ii) rat | (ii) 35b | (ii) 0.34 | (ii) 0.22 | (ii) 0.5 | (ii) 0.2 | ||
| 21b | |||||||
| (i) mouse | (i) 76 | (i) 26.71 | (i) 18.5 | (i) 1.12 | (i) 98 | 31 | 29 |
| (ii) rat | (ii) 82b | (ii) 6.5 | (ii) 18.5 | (ii) 1.3 | (ii) 68 | ||
| 22d | 97 | 27.22 | 20.98 | 1.0 | 68 | 23 | 51 |
| 7a | 100 | 32.03 | 21.61 | 0.89 | nd | 23 | 33 |
Mouse liver microsome (MLM) stability values represent the percentage of compound remaining after 30 min.
Rat liver microsome (RLM) stability values represent the percentage of compound remaining after 30 min. Mouse plasma PK parameters were determined following a single po dose at 50 mg/kg or iv dose at 10 mg/kg. Rat plasma PK parameters were determined following a single po dose at 20 mg/kg or iv dose at 4 mg/kg.
Cmax: maximum concentration.
AUC: area under curve.
nd: not determined.
Papp: permeability coefficient.
Table 9. Time-Dependent Activity of Lead AMTz Inhibitors.
| compd | LOXL2, 20 min, IC50 (μM)a,b | LOXL2, 1 h, IC50 (μM)a,c | LOXL2, 3 h, IC50 (μM)a,d |
|---|---|---|---|
| BAPN | 4.26 | 0.665 | 0.372 |
| [3.63, 5.00] | [0.57, 0.77] | [0.31, 0.45] | |
| 3a | 0.439 | 0.176 | 0.157 |
| 0.46 | [0.105, 0.295] | 0.126 | |
| 7a | 0.587 | 0.086 | 0.069 |
| 0.648 | [0.061, 0.12] | 0.075 | |
| 21a | 0.544 | 0.109 | 0.072 |
| 0.331 | [0.066, 0.18] | 0.069 | |
| 21b | 0.71 | 0.151 | 0.078 |
| 0.687 | [0.12, 0.19] | 0.079 | |
| 22b | 1.252 | 0.302 | 0.09 |
| 0.53 | [0.22, 0.42] | 0.085 | |
| 22c | 0.795 | 0.37 | 0.146 |
| 0.787 | [0.23, 0.596] | 0.157 | |
| 22d | 1.238 | 0.425 | 0.199 |
| 1.373 | [0.35, 0.51] | 0.176 |
Reported IC50 values were determined in at least two separate experiments (n ≥ 2). When n = 2, individual IC50 values are shown. When n > 2, the values are reported as the geometric mean with the corresponding 95% confidence interval in square brackets.
Compound was preincubated with enzyme for 20 min.
Compound was preincubated with enzyme for 1 h.
Compound was preincubated with enzyme for 3 h.
Further PK studies were carried out using AMT compound 3a and AMTz 21b in a rat model. Metabolic stability against rat liver microsomes (RLM) is moderate in 3a and good in 21b; however, a significant difference in oral bioavailability is observed between the compounds, with 3a demonstrating very poor levels of plasma exposure. In contrast, 21b exhibits comparable AUCs of 18 μM·h between the species and maintains a good Cmax concentration (6.5 μM) and oral bioavailability (68%). On the basis of the balance of potency, selectivity profile, PK, and permeability, compound 21b is determined to have the best profile overall and thus was chosen for further in vivo antitumor efficacy evaluation.
Evaluation of Anti-Tumor Efficacy
In vivo efficacy studies involving compound 21b were carried out using a genetically engineered mouse model (GEMM) that functions as a LOX-driven spontaneous breast cancer model.21 Mice were dosed daily via oral gavage (70 mg/kg) from around 60 days after birth, once primary tumors become palpable. Inhibitor 21b was very well tolerated, with no observed body weight loss.
Pleasingly, we observe a delay in primary tumor development and a significant reduction in tumor growth rate in the 21b-treated group compared to that of the controls (Figure 3a), with no deaths due to tumor volume reaching ethical size limits necessitated in the inhibitor-treated group during the course of this study (Figure 3b).
Figure 3.

(a) Antitumor efficacy of compound 21b in MMTV-PyMT GEMM model: control animals (n = 5; gray) or treated with compound 21b at 70 mg/kg q.d. (n = 3; green); day 91 *p = 0.0367, Welch’s t test. (b) Kaplan–Meier survival analysis.
Potency and Mode of Inhibition (MOI) of AMT and AMTz Inhibitors
We assessed the time-dependent inhibitory activity of select AMT and AMTz compounds (Table 9) and investigated the mode of enzyme inhibition for key series examples (Figure 4).
Figure 4.

LOXL2 jump dilution assay to assess MOI of key LOXL2 inhibitors.
Time-Dependent Inhibition of Lead Compounds
We previously demonstrated that series exemplars of AMT and AMTz compounds inhibit LOXL2 in a time-dependent manner (Table 1); we now sought to ascertain if this trend remained true for the series in general. As such, a range of AMTz inhibitors including potent 2-aminomethylene-5-sulfonyl compounds 7a, 21a, and 21b, 2,4-regioisomers 22b, 22c, and 22d, along with AMT compound 3a and BAPN, were assessed for anti-LOXL2 activity following different enzyme preincubation times (Table 9). All compounds were found to exhibit time-dependent inhibition, with the most significant effect again observed on increasing the time from 20 min to 1 h, whereupon up to a 7-fold increase in activity is observed; further increasing the preincubation time to 3 h provides a small additional increase in activity, with IC50 < 0.1 μM attained for all 2-aminomethylene-5-sulfonylthiazoles (7a, 21a, and 21b) assessed.
MOI Studies
Given the time-dependent inhibition observed with these compounds, we wanted to clarify the mechanism of LOXL2 inhibition; as such, we set up a jump dilution assay, which readily distinguishes between reversible and irreversible modes of enzyme inhibition (Figure 4).37 Leading AMT and AMTz inhibitors 3a and 21b, respectively, along with BAPN were assessed, whereby the enzyme was preincubated for 1 h with 10 × IC50 of these compounds, at which concentration we see complete inhibition of the enzyme in all cases. The enzyme/inhibitor mixture was then diluted 100-fold into a solution containing all enzyme reaction components, and the activity of the enzyme was assessed.
The resulting curve obtained for 3a displays about 80% recovery of activity following dilution, as compared to the DMSO control, which suggests that this compound behaves as a reversible inhibitor under these assay conditions, unlike BAPN which can be characterized as an irreversible inhibitor. In the case of 21b a more modest regain in activity of around 30% is observed following the jump dilution. This is suggestive of either a slowly reversible compound or an irreversible inhibitor whereby a residual amount of enzyme has not committed to forming a stable covalent bond during the two-step inhibition process. The results obtained from this study suggest that the electronic nature of the heterocyclic core affects the binding mechanism of these inhibitors, with the AMTz inhibitors better able to form stable, irreversible Schiff base intermediates following initial reversible binding to the LTQ cofactor.20,38 One possible explanation for this is that the initial Schiff base formed is more susceptible to hydrolysis in the case of 3a compared with 21b, which is better able to rearrange to form a more stable intermediate as a result of either increased resonance stabilization or noncovalent electrostatic interactions. Alternatively, this could also be the result of differences in enzyme–inhibitor kinetics, with initial rates of binding affecting the ability of these inhibitors to form covalent bonds.
Pharmacophore and QSAR Modeling
Binding Mode/Pharmacophore Hypothesis Generation
With the dearth of protein–ligand crystal structures for LOXL2 in its active form, we decided to embark upon a ligand-based approach to propose a legitimate binding conformation for SAR analysis and modeling. FieldTemplater39,40 was used to derive a pharmacophore model by comparing conformational ensembles of molecules using their electrostatic and hydrophobic characters to identify common motifs. The field point pattern for a conformer of 7a is shown in Figure 5.
Figure 5.
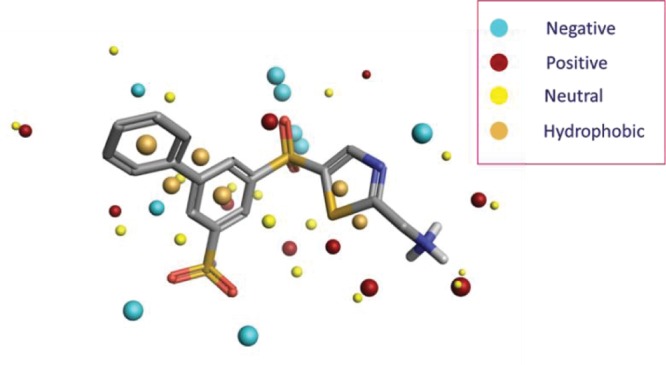
Sample conformation of 7a showing electrostatic fields and 3D field point pattern, which provides a concise 3D pharmacophore.
The field-based alignment can be independent of chemical structure, allowing alignment of molecules from different series. The compounds reported in this article are thus augmented with a set of LOXL2 inhibitors obtained from the literature,20 along with in-house inhibitors not explicitly described in this article, with only those compounds with pIC50 > 5 included (see Supporting Information). The collection of 54 molecules (with IC50 data) were visually inspected, with the four most active, most structurally diverse compounds selected for pharmacophore modeling (7a, pIC50 = 7.07; 21e, pIC50 = 6.84; JMC2017-31,20 pIC50 = 6.55; and JMC2017-33,20 pIC50 = 6.51).
The FieldTemplater experiment was run using Normal (large molecules) conformation hunt settings, with a group constraint placed on the cationic NH3+ groups to force their alignment in the templated result, since these compounds are all believed to follow the same mechanism of inhibition via covalent binding of the aminomethylene group to the LTQ cofactor. The best scoring template consisting of all four compounds was taken forward to Forge,40,41 and the 7a and 21e conformations were used as references for the field-based alignment of the other molecules in the data set.
Structure–Activity Relationship Modeling
We attempted to calculate statistically relevant mathematical models to predict activities of new compounds. Pleasingly, we were rewarded with two predictive, complementary models as described below.
Field-Based 3DQSAR40,41
Cresset’s approach to 3DQSAR is similar to traditional CoMFA;42,43 however, there are some striking differences around how sampling points are selected and the use of irregular grids, such that calculation speed is greatly improved. The 54 compound data set was randomly partitioned to put 15% in the test set (8 molecules), leaving 46 in the training set, upon which the model was built; tested with 50 y-scrambles, followed by leave-one-out (LOO) cross-validation. The resulting three-component model features an r2 of 0.856, q2 of 0.665, RMSE of 0.147, and a Kendall’s tau44 value of 0.760, a model we believe to be statistically relevant45 and able to predict rank order of activity.
Forge can be used to visualize the field/steric contributions to predicted activities, as demonstrated in Figure 6c; these plots were helpful in rationalizing the SAR observed for the reported compounds. The details of this field-based 3DQSAR model are shown in Figure 6a and Figure 6b, with the model details included in the Supporting Information.
Figure 6.

(a) Plot of predicted vs actual activity for training, training cross-validation, and test sets. (b) 7a with the electrostatic and steric coefficient positions/values displayed. (c) Example of the field/steric contributions to predicted activity for 7a.
As a follow-up and to build confidence in the model and binding mode hypothesis, an alternative set of machine-learning algorithms, k-nearest neighbor (kNN), random forest (RF), support vector machine (SVM), and relevance vector machine (RVM) regression models were used to model the data.41 The statistics for RVM for the full model on the training set were r2 = 0.852, RMSE = 0.150, and Kendall’s tau = 0.757. For the cross-validation, the model statistics were q2 = 0.649, RMSE = 0.230, Kendall’s tau = 0.638. This model is similarly predictive to the field-based 3DQSAR model described earlier, and the calculation of multiple predictive models from a set of molecules aligned to a common binding mode hypothesis lends support to our having derived a sensible binding mode in the absence of protein–ligand crystallographic information. While not reported here, it is noteworthy that the other machine learning models also have statistics that suggest that they are predictive.
Activity Atlas Analysis: Activity Cliff Summary
In tandem with the predictive 3DQSAR model, Activity Atlas/Activity Miner40,41 was used to conduct a qualitative assessment of the SAR for the data set. The summary plots of the calculated electrostatic and steric activity cliffs are shown in Figure 7 and compare favorably to the 3DQSAR model visualizations. This information is built up from doing a pairwise analysis of all molecules in their aligned conformation and automatically examining activity cliffs, highly similar pairs where there is a large change in activity. The most potent and least potent compounds are shown in the context of the activity cliff summary.
Figure 7.

Activity Atlas/activity cliff summary plots for LOXL2 activity shown with (a) 7a around thiophene/thiazole core, (b) 7a, 7e, and 7g to illustrate steric and electrostatic contributions (using field points, as described above) that rationalize the observed SAR. All isosurfaces are shown at ≥2.0 confidence level.
The predicted SAR around the thiazole moiety (Figure 7a) is consistent with the observed decrease in potency when the thiazole nitrogen is replaced by the larger CX groups (3b, 3c), suggesting that this position is sterically restricted and requires negative electrostatics. In addition, the requirement for negative electrostatics is consistent with the moderate decrease in potency of the thiophene analogues (3a) and the 2,4 substituted thiazoles (22b). Examination of the SAR around the thiazole C-5 substituent using Activity Atlas suggests that the meta-position of the phenyl group (R1) is predicted to favor large groups; in 21b, replacement of phenyl with N-methyl pyrazolyl substituent meets both the desired steric and electrostatic conditions near the 5-position. With regard to the 3-position, the activity cliff summary (Figure 7b) can be used to rationalize the drop in potency on varying the methanesulfonyl group of 7a to an ethyl (7e) or tert-butyl (7g) group, which increases the number of cliff violations (i.e., a mismatching of field points with activity cliff summary): In 7a, the 3-sulfonyl group has favorable electrostatics and is able to thread the needle of a sterically unfavorable region; a larger tert-butyl group has unfavorable electrostatics and multiple steric clashes that result in diminished activity. Further, in the context of the model, the central phenyl group is predicted to have a less electron-rich π-cloud, in line with the observed improvement in potency when small electron-withdrawing groups are present in the 3-position.
We have developed predictive field-based 3DQSAR and visual qualitative models of activity based on the LOXL2 inhibition data acquired during the development of the aminomethylenethiazole inhibitors. This will be a useful tool to aid further development of the series as well as to design new chemical inhibitors in the future.
Synthetic Chemistry
Sulfone-linked AMTz analogues were synthesized from N-Boc-protected (5-bromothiazol-2-yl)methanamine 23 and the appropriate thiol, using palladium-catalyzed coupling methods, as shown in Scheme 1. In the case of aryl thiols, tris(dibenzylideneacetone)dipalladium catalyst and XantPhos ligand were used in conjunction with sodium tert-butoxide base in a 4:1 solvent mixture of toluene/tert-butanol. In the case of alkyl thiols, DIPEA was found to be preferable as a base and toluene was used as the solvent. Oxidation of the sulfide was achieved using m-CPBA to afford the desired sulfone, and subsequent Boc-removal using 4 M HCl in dioxane gave rise to the target AMTz inhibitors.
Scheme 1. General Route to Sulfur-Linked 2,5-AMTz Analogues.

Reagents and conditions: (a) Pd2dba3, XantPhos, NaOtBu, PhMe, tBuOH, 110 °C; (b) Pd2dba3, XantPhos, DIPEA, PhMe, 110 °C; (c) m-CPBA, DCM, rt; (d) 4 M HCl in dioxane, rt.
Substituted aryl thiols that were not commercially available were synthesized according to the method shown in Scheme 2.46 Palladium-catalyzed coupling of an aryl halide with 2-ethylhexyl 3-mercaptopropanoate afforded a thiol surrogate that also functions as a thiol protecting group for further chemical modification. The thiol surrogate could then be used directly in a palladium-catalyzed coupling reaction, whereby deprotection was achieved in situ, or the protecting group can be removed using sodium ethoxide to afford the desired aryl thiol.
Scheme 2. General Synthesis of Noncommercially Available Thiols.

Reagents and conditions: (a) 2-ethylhexyl 3-mercaptopropanoate, Pd2dba3, XantPhos, DIPEA, PhMe, 110 °C; (b) NaOEt, PhMe, EtOH, rt; then acid; (c) KOtBu, THF, rt; then acid.
AMTz analogues bearing aryl groups directly attached to the thiazole ring were synthesized according to the method shown in Scheme 3. Suzuki reaction of N-Boc-protected (5-bromothiazol-2-yl)methanamine (23) with a boronic acid or ester, followed by acid-mediated deprotection, afforded the desired phenyl-linked target compounds.
Scheme 3. General Route to Phenyl-Linked AMTz Analogues.

Reagents and conditions: (a) Pd(PPh3)4, K2CO3, 1,2-DME, H2O, 95 °C; (b) 4 M HCl in dioxane, rt.
Synthesis of the thiol surrogate used in the synthesis of 7a and close analogues was achieved starting from 1,3-dibromo-5-(methylsulfonyl)benzene, Scheme 4. Palladium cross-coupling with 2-ethylhexyl 3-mercaptopropanoate installed the protected thiol group and Suzuki coupling with phenyl boronic acid at the remaining bromo-position completed the synthesis of the required thiol reactant. In situ deprotection followed by coupling with a 5-bromo-2-AMTz intermediate (23 or 24) resulted in the corresponding sulfide-linked compound. Direct N-Boc-deprotection provided the sulfide-linked final compound 7d, or oxidation using either 1 or 2 equiv of m-CPBA prior to deprotection resulted in the sulfoxide or sulfone-linked inhibitors, 7c and 7a or 7b, respectively.
Scheme 4. AMTz Sulfide, Sulfoxide, and Aminomethylene Modifications.

Reagents and conditions: (a) 2-ethylhexyl 3-mercaptopropanoate, Pd2dba3, XantPhos, DIPEA, PhMe, 110 °C; (b) PhB(OH)2, Pd(PPh3)4, K2CO3, 1,2-DME, H2O, 95 °C; (c) Pd2dba3, XantPhos, NaOtBu, PhMe, tBuOH, 110 °C; (d) 4 M HCl in dioxane, rt; (e) m-CPBA (1 equiv), DCM, 0 °C; (f) m-CPBA (2.2 equiv), DCM, rt.
Thiophene analogues 3b and 3c, bearing a halogen atom in the 3-position, were synthesized from the corresponding 3-halo-5-bromo-AMT precursors (25 and 26), which were synthesized according to literature procedures,47,48Scheme 5. Palladium coupling was then carried out with the thiol surrogate, and m-CPBA oxidation of the resulting bis-sulfide followed by treatment with HCl afforded the desired halogenated thiophene analogues.
Scheme 5. General Route to Sulfur-Linked AMT Analogues.

Reagents and conditions: (a) NaOEt, PhMe, EtOH, rt; then acid; (b) KOtBu, THF, rt; then acid; (c) Pd2dba3, XantPhos, NaOtBu, PhMe, tBuOH, 110 °C; (d) m-CPBA, DCM, rt; (e) 4 M HCl in dioxane, rt.
Synthesis of the 2,4-thiazole regioisomers was achieved in the same manner as described previously, starting from N-Boc-protected (4-bromothiazol-2-yl)methanamine (27),49Scheme 6. Palladium-catalyzed coupling with the respective thiol followed by subsequent oxidation using m-CPBA and Boc-deprotection using HCl afforded the desired regioisomers.
Scheme 6. General Route to Sulfur-Linked 2,4-AMTz Regioisomer Analogues.

Reagents and conditions: (a) Pd2dba3, XantPhos, NaOtBu, PhMe, tBuOH, 110 °C; (b) m-CPBA, DCM, rt; (c) 4 M HCl in dioxane, rt.
Conclusion
Following our recent discovery of a potent, selective, and orally bioavailable LOX inhibitor we observed that replacement of the thiophene core with a 2-aminomethylene-5-sulfonyl thiazole core leads to potent, irreversible LOXL2 inhibitors. SAR investigations revealed similar trends as seen in the analogous AMT series, resulting in potencies of <0.1 μM achieved, and enabled development of a predictive LOXL2 3DQSAR model. Selectivity studies concerning LOX and LOXL3 isoforms revealed a modest selectivity toward LOXL2 in our main series inhibitors, while 2-aminomethylene-4-sulfonyl thiazole regioisomers exhibit excellent selectivity for LOXL2 and thus have the potential to be used as probe compounds. Further selectivity and ADME assessment leads to the discovery of 21b, which has an improved PK profile and demonstrates excellent antitumor efficacy in a LOX-driven GEMM breast cancer model.
Experimental Section
Synthesis of Inhibitors
All chemicals, reagents, and solvents were purchased from commercial sources and were used as received. Flash chromatography was performed on a Biotage Isolera or Combiflash Rf+ UV–vis flash purification system using prepacked silica gel cartridges (Biotage) with HPLC grade solvents. Thin layer chromatography (TLC) analysis was performed using silica gel 60 F-254 thin layer plates and visualized using UV light (254 nm) and/or developed with vanillin stain. LCMS and HRMS analyses of chemical compounds were performed on an Agilent 1200 series HPLC and diode array detector coupled to a 6210 time-of-flight mass spectrometer with a multimode ESI source; or a Waters Acquity UPLC or I-class UPLC with a diode array detector coupled to a Waters G2 QToF, SQD, or QDa mass spectrometer fitted with a multimode ESI/APCI source. 1H, 19F, and 13C NMR spectra were recorded using a Bruker Avance 500, 400, or 300 MHz spectrometer using an internal deuterium lock. Chemical shifts are expressed in parts per million (ppm), and splitting patterns are indicated as follows: br, broad; s, singlet; d, doublet; t, triplet; q, quartet; p, pentet; h, hextet; m, multiplet. All coupling constants (J) are reported in hertz (Hz). All final inhibitors submitted for biological evaluation were at least 95% pure by HPLC–MS. Synthesis of inhibitors 3a, 4, 5, 6, and 8 has previously been described in the literature.22 Below are a representative synthesis of compound 7a and analytical data for all final inhibitors. All tested inhibitors have purity of >95% (LCMS/UV).
(3-Chloro-5-((5-(methylsulfonyl)-[1,1′-biphenyl]-3-yl)sulfonyl)thiophen-2-yl)methanamine Hydrochloride (3b)
Pale yellow solid (>99% purity). 1H NMR (CDCl3/methanol-d4, 500 MHz) δ 8.52–8.45 (m, 3H), 7.94 (s, 1H), 7.76–7.70 (m, 2H), 7.58–7.53 (m, 2H), 7.52–7.47 (m, 1H), 4.37 (s, 2H), 3.27 (s, 3H) ppm; 13C NMR (CDCl3/methanol-d4, 125 MHz) δ 146.03, 144.53, 144.28, 143.73, 138.08, 137.97, 135.13, 131.81, 131.23, 130.39, 130.35, 129.33, 128.23, 125.55, 44.17, 36.07 ppm; LCMS m/z 424.9734 found (M-NH2)+, 424.9737 calculated for C18H14S3O4Cl.
(3-Fluoro-5-((5-(methylsulfonyl)-[1,1′-biphenyl]-3-yl)sulfonyl)thiophen-2-yl)methanamine Hydrochloride (3c)
White solid (>99% purity). 1H NMR (DMSO-d6, 500 MHz) δ 8.65 (br s, 3H), 8.59–8.49 (m, 2H), 8.39 (s, 1H), 8.27 (s, 1H), 7.99–7.81 (m, 2H), 7.68–7.45 (m, 3H), 4.20 (s, 2H), 3.46 (s, 3H) ppm; 13C NMR (DMSO-d6, 125 MHz) δ 155.78 (d, J = 265.9 Hz), 144.21, 143.82, 142.92, 139.39 (d, J = 7.6 Hz), 137.00, 131.30, 130.08, 129.88, 129.81, 128.00, 124.96 (d, J = 17.6 Hz), 124.57 (d, J = 25 Hz), 124.28, 43.51, 32.77 ppm; 19F NMR (DMSO-d6, 471 MHz) δ −122.29 ppm; LCMS m/z 409.0026 found (M – NH2)+, 409.0033 calculated for C18H14S3O4F.
(5-((5-(Methylsulfonyl)-[1,1′-biphenyl]-3-yl)sulfonyl)thiazol-2-yl)methanamine Hydrochloride (7a)
2-Ethylhexyl 3-((3-Bromo-5-(methylsulfonyl)phenyl)thio)propanoate
A solution of 1,3-dibromo-5-(methylsulfonyl)benzene (500 mg, 1.59 mmol) and DIPEA (0.55 mL, 3.18 mmol) in toluene (10 mL) was degassed with nitrogen for 5 min. Pd2dba3 (36 mg, 2.5 mol %), XantPhos (46 mg, 5 mol %), and 2-ethylhexyl-3-mercaptopropionate (0.36 mL, 1.59 mmol) were then added with stirring, and the solution was bubbled with nitrogen for a further 5 min before sealing the flask and heating to 110 °C, with stirring for 18 h. After cooling to room temperature, the reaction mixture was diluted with ethyl acetate (60 mL) and washed with water (50 mL) and brine (50 mL), dried over anhydrous magnesium sulfate, filtered, and concentrated to give the crude product, which was purified using flash column chromatography (5–100% EtOAc in PE) to give the title compound (664 mg, 72% purity) as a clear pale yellow oil, which is used in the subsequent transformation as an impure mixture. 1H NMR (CDCl3, 300 MHz) δ 7.84 (t, 1H, J = 1.6 Hz), 7.76 (t, 1H, J = 1.6 Hz), 7.66 (t, 1H, J = 1.7 Hz), 4.04–4.01 (m, 2H), 3.26 (t, 2H, J = 7.2 Hz), 3.06 (s, 3H), 2.67 (t, 2H, J = 7.2 Hz), 1.61–1.51 (m, 1H), 1.39–1.25 (m, 8H), 0.91–0.85 (m, 6H) ppm; LCMS, did not ionize.
2-Ethylhexyl 3-((5-(Methylsulfonyl)-[1,1′-biphenyl]-3-yl)thio)propanoate
2-Ethylhexyl 3-((3-bromo-5-(methylsulfonyl)phenyl)thio)propanoate (1.80 g, 72% purity, 2.88 mmol) and phenylboronic acid (422 mg, 3.46 mmol) were dissolved in 1,2-DME/H2O (5:1, 24 mL), and the solution was bubbled with nitrogen for 5 min. Pd(PPh3)4 (333 mg, 10 mol %) and K2CO3 (796 mg, 5.76 mmol) were then added with stirring, and the mixture was bubbled with nitrogen for a further 5 min before sealing the flask and heating at 100 °C with stirring for 18 h. After cooling to room temperature the reaction mixture was diluted with ethyl acetate (60 mL) and washed with brine (50 mL), dried over anhydrous magnesium sulfate, filtered, and concentrated to give the crude product, which was purified using flash column chromatography (5–80% EtOAc in PE) to give the title compound (998 mg, 72% purity) as a clear pale yellow oil, which is used in the subsequent transformation as an impure mixture. 1H NMR (CDCl3, 500 MHz) δ 7.94 (t, 1H, J = 1.6 Hz), 7.84 (t, 1H, J = 1.7 Hz), 7.77 (t, 1H, J = 1.7 Hz), 7.60–7.58 (m, 2H), 7.48 (t, 2H, J = 7.4 Hz), 7.43 (t, 1H, J = 7.3 Hz), 4.06–3.99 (m, 2H), 3.31 (t, 2H, J = 7.2 Hz), 3.10 (s, 3H), 2.70 (t, 2H, J = 7.2 Hz), 1.59–1.55 (m, 1H), 1.37–1.31 (m, 2H), 1.30–1.25 (m, 6H), 0.88 (t, 6H, J = 7.5 Hz) ppm. LCMS, did not ionize.
tert-Butyl ((5-((5-(Methylsulfonyl)-[1,1′-biphenyl]-3-yl)thio)thiazol-2-yl)methyl)carbamate
A solution of 2-ethylhexyl 3-((5-(methylsulfonyl)-[1,1′-biphenyl]-3-yl)thio)propanoate (830 mg, 72% purity, 1.34 mmol), tert-butyl ((5-bromothiazol-2-yl)methyl)carbamate (431 mg, 1.47 mmol) in toluene/tBuOH (4:1, 15 mL) was degassed with nitrogen for 5 min. Pd2dba3 (128 mg, 10 mol %), XantPhos (162 mg, 20 mol %), and NaOtBu (283 mg, 2.94 mmol) were then added with stirring, and the solution was bubbled with nitrogen for a further 5 min before sealing the flask and heating to 110 °C, with stirring for 18 h. After cooling to room temperature, the reaction mixture was diluted with ethyl acetate (60 mL) and washed with water (50 mL) and brine (50 mL), dried over anhydrous magnesium sulfate, filtered, and concentrated to give the crude product, which was purified using flash column chromatography (5–100% EtOAc in PE) followed by reversed-phase chromatography (C18 silica, 5–95% CH3CN in water) to give the title compound (108 mg, 17%) as a yellow oil. 1H NMR (CDCl3, 500 MHz) δ 7.94 (t, 1H, J = 1.6 Hz), 7.88 (s, 1H), 7.73 (t, 1H, J = 1.7 Hz), 7.63 (t, 1H, J = 1.6 Hz), 7.53–7.51 (m, 2H), 7.48–7.45 (m, 2H), 7.44–7.41 (m, 1H), 5.29 (br s, 1H), 4.62 (d, 2H, J = 6.0 Hz), 3.07 (s, 3H), 1.45 (s, 9H) ppm; LCMS m/z 421.0358 found (M – tBu + H)+, 421.0345 calculated for C18H17N2O4S3.
tert-Butyl ((5-((5-(Methylsulfonyl)-[1,1′-biphenyl]-3-yl)sulfonyl)thiazol-2-yl)methyl)carbamate
m-CPBA (77%, 41 mg, 0.184 mmol) was added over 2 min to a solution of tert-butyl ((5-((5-(methylsulfonyl)-[1,1′-biphenyl]-3-yl)thio)thiazol-2-yl)methyl)carbamate (35 mg, 0.073 mmol) in dichloromethane (3 mL), with stirring at room temperature under air for 18 h. The reaction mixture was then quenched with aq NaHCO3 solution and extracted with dichloromethane (2 × 20 mL). The combined organic extracts were washed with brine (30 mL), dried over anhydrous sodium sulfate, filtered, and concentrated to give the crude product, which was purified using flash column chromatography (25–75% EtOAc in cyclohexane) to give the title compound (27 mg, 72% yield) as a clear pale yellow oil. 1H NMR (CDCl3, 500 MHz) δ 8.48 (t, 1H, J = 1.7 Hz), 8.41 (t, 1H, J = 1.7 Hz), 8.36 (t, 1H, J = 1.6 Hz), 8.28 (s, 1H), 7.63–7.61 (m, 2H), 7.55–7.49 (m, 3H), 5.27 (br s, 1H), 4.60 (d, 2H, J = 6.0 Hz), 3.14 (s, 3H), 1.44 (s, 9H) ppm; LCMS m/z 453.0214 found (M – tBu + H)+, 453.0243 calculated for C18H17N2O6S3.
(5-((5-(Methylsulfonyl)-[1,1′-biphenyl]-3-yl)sulfonyl)thiazol-2-yl)methanamine Hydrochloride (7a)
tert-Butyl ((5-((5-(methylsulfonyl)-[1,1′-biphenyl]-3-yl)sulfonyl)thiazol-2-yl)methyl)carbamate (25 mg, 0.185 mmol) was dissolved in 4 M HCl in dioxane (0.25 mL, 0.98 mmol) and the reaction mixture was stirred at room temperature under nitrogen for 4 h, before concentrating to dryness. Dichloromethane (1 mL) was added and the mixture was concentrated again, and the resulting precipitate was washed with diethyl ether and dried under vacuum to give the title compound (20 mg, 91%) as a white solid that requires no further purification. 1H NMR (DMSO-d6, 500 MHz) δ 8.83 (s, 1H), 8.72 (br s, 3H), 8.58 (t, 1H, J = 1.7 Hz), 8.53 (t, 1H, J = 1.6 Hz), 8.43 (t, 1H, J = 1.7 Hz), 7.87 (d, 2H, J = 7.0 Hz), 7.58 (t, 2H, J = 7.3 Hz), 7.53 (t, 1H, J = 7.3 Hz), 4.50 (br s, 2H), 3.45 (s, 3H) ppm; 13C NMR (DMSO-d6, 125 MHz) δ 171.0, 148.8, 143.8, 143.4, 142.6, 138.9, 136.5, 130.9, 129.5, 129.4, 129.35, 127.5, 123.8, 43.0, 39.8 ppm; LCMS m/z 409.0328 found (M + H)+, 409.0345 calculated for C17H17N2S3O4; LCMS/UV analysis >99% purity.
N-Methyl-1-(5-((5-(methylsulfonyl)-[1,1′-biphenyl]-3-yl)sulfonyl)thiazol-2-yl)methanamine Hydrochloride (7b)
Off-white solid (96% purity). 1H NMR (DMSO-d6, 500 MHz) δ 9.51 (br s, 2H), 8.85 (s, 1H), 8.59 (t, 1H, J = 1.7 Hz), 8.54 (t, 1H, J = 1.6 Hz), 8.44 (t, 1H, J = 1.7 Hz), 7.87 (dd, 2H, J = 8.1, 1.5 Hz), 7.61–7.50 (m, 3H), 4.60 (s, 2H), 3.45 (s, 3H), 2.62 (s, 3H) ppm; 13C NMR (DMSO-d6, 125 MHz) δ 169.2, 149.0, 143.8, 143.4, 142.6, 139.2, 136.5, 130.9 129.6, 129.4, 129.3, 127.6, 123.9, 47.8, 43.0, 32.7 ppm; LCMS m/z 423.1 found (M + H)+, 423.0501 calculated for C18H19N2S3O4.
(5-((5-(Methylsulfonyl)-[1,1′-biphenyl]-3-yl)sulfinyl)thiazol-2-yl)methanamine Hydrochloride (7c)
Off-white solid (96% purity). 1H NMR (methanol-d4, 500 MHz) δ 8.51 (s, 1H), 8.37 (t, 1H, J = 1.6 Hz), 8.31–8.29 (m, 2H), 7.75–7.72 (m, 2H), 7.57–7.48 (m, 3H), 4.51 (s, 2H), 3.25 (s, 3H) ppm; 13C NMR (DMSO-d6, 125 MHz) δ 170.0, 149.0, 147.4, 146.9, 145.7, 144.8, 138.9, 130.5, 130.4, 129.8, 128.3, 128.0, 122.4, 44.1, 41.3 ppm; LCMS m/z 393.2 found (M + H)+, 393.0396 calculated for C17H17N2O3S3.
(5-((5-(Methylsulfonyl)-[1,1′-biphenyl]-3-yl)thio)thiazol-2-yl)methanamine Hydrochloride (7d)
Pale yellow solid (95% purity). 1H NMR (DMSO-d6, 500 MHz) δ 8.73 (br s, 3H), 8.28 (s, 1H), 8.06 (t, 1H, J = 1.6 Hz), 7.90 (t, 1H, J = 1.7 Hz), 7.76–7.72 (m, 3H), 7.55–7.44 (m, 3H), 4.48 (s, 2H), 3.33 (s, 3H) ppm; 13C NMR (DMSO-d6, 125 MHz) δ 168.2, 150.0, 142.8, 142.7, 138.7, 137.5, 130.3, 129.2, 128.9, 127.2, 127.0, 124.1, 123.9, 43.2 (1 × C missing; under DMSO peak) ppm; LCMS m/z 377.0 found (M + H)+, 377.0447 calculated for C17H17N2S3O2.
(5-((5-Ethyl-[1,1′-biphenyl]-3-yl)sulfonyl)thiazol-2-yl)methanamine Hydrochloride (7e)
Pale yellow solid (>99% purity). 1H NMR (DMSO-d6, 500 MHz) δ 8.86 (br s, 3H), 8.68 (s, 1H), 8.04 (s, 1H), 7.91 (s, 1H), 7.88 (s, 1H), 7.74 (d, 2H, J = 7.2 Hz), 7.52 (t, 2H, J = 7.5 Hz), 7.45 (t, 1H, J = 7.3 Hz), 4.47 (s, 2H), 2.80 (q, 2H, J = 7.6 Hz), 1.25 (t, 3H, J = 7.6 Hz) ppm; 13C NMR (DMSO-d6, 125 MHz) δ 170.2, 147.6, 147.1, 142.1, 141.6, 140.2, 138.2, 132.4, 129.2, 128.5, 127.1, 124.9, 122.4, 39.7, 27.9, 15.3 ppm; LCMS m/z 359.0882 found (M + H)+, 359.0882 calculated for C18H19N2S2O2.
(5-((5-Isopropyl-[1,1′-biphenyl]-3-yl)sulfonyl)thiazol-2-yl)methanamine Hydrochloride (7f)
Off-white solid (>99% purity). 1H NMR (DMSO-d6, 500 MHz) δ 8.83 (br s, 3H), 8.70 (s, 1H), 8.04 (t, 1H, J = 1.7 Hz), 7.92 (t, 1H, J = 1.4 Hz), 7.88 (t, 1H, J = 1.5 Hz), 7.74 (d, 2H, J = 7.1 Hz), 7.52 (t, 2H, J = 7.5 Hz), 7.45 (t, 1H, J = 7.3 Hz), 4.47 (s, 2H), 3.13 (p, 1H, J = 6.9 Hz), 1.28 (d, 6H, J = 6.9 Hz) ppm; 13C NMR (DMSO-d6, 125 MHz) δ 170.2, 151.7, 147.7, 142.2, 141.6, 140.2, 138.2, 131.0, 129.2, 128.5, 127.2, 123.5, 122.7, 39.7, 33.4, 23.5 ppm; LCMS m/z 373.1030 found (M + H)+, 373.1039 calculated for C19H21N2S2O2.
(5-((5-(tert-Butyl)-[1,1′-biphenyl]-3-yl)sulfonyl)thiazol-2-yl)methanamine Hydrochloride (7g)
Off-white solid (>99% purity). 1H NMR (DMSO-d6, 500 MHz) δ 8.75 (br s, 3H), 8.72 (s, 1H), 8.04 (t, 1H, J = 1.7 Hz), 8.01 (t, 1H, J = 1.7 Hz), 7.97 (t, 1H, J = 1.7 Hz), 7.74 (d, 2H, J = 7.1 Hz), 7.52 (t, 2H, J = 7.5 Hz), 7.46 (t, 1H, J = 7.3 Hz), 4.47 (s, 2H), 1.38 (s, 9H) ppm; 13C NMR (DMSO-d6, 125 MHz) δ 170.2, 153.9, 147.7, 142.1, 141.5, 140.2, 138.5, 130.0, 129.2, 128.5, 127.3, 122.5, 122.2, 39.7, 35.2, 30.8 ppm; LCMS m/z 409.1007 found (M + Na)+, 409.1015 calculated for C20H22N2S2O2Na.
(5-((5-Methoxy-[1,1′-biphenyl]-3-yl)sulfonyl)thiazol-2-yl)methanamine Hydrochloride (7h)
White solid (>99% purity). 1H NMR (DMSO-d6, 500 MHz) δ 8.79 (br s, 3H), 8.71 (s, 1H), 7.80 (t, 1H, J = 1.5 Hz), 7.76–7.74 (m, 2H), 7.57–7.56 (m, 1H), 7.53–7.49 (m, 3H), 7.46 (t, 1H, J = 7.3 Hz), 4.47 (s, 2H), 3.93 (s, 3H) ppm; 13C NMR (DMSO-d6, 125 MHz) δ 170.3, 160.5, 147.9, 143.7, 142.7, 139.9, 137.9, 129.1, 128.7, 127.2, 118.3, 117.1, 111.0, 56.1, 39.7 ppm; LCMS m/z 361.00677 found (M + H)+, 361.0675 calculated for C17H17N2S2O3.
5-((2-(Aminomethyl)thiazol-5-yl)sulfonyl)-N-methyl-[1,1′-biphenyl]-3-amine Dihydrochloride (7i)
White solid (>99% purity). 1H NMR (methanol-d4, 500 MHz) δ 8.53 (s, 1H), 8.23 (s, 1H), 8.11 (s, 1H), 8.03 (s, 1H), 7.71 (d, 2H, J = 7.5 Hz), 7.52 (t, 2H, J = 7.4 Hz), 7.47 (t, 1H, J = 7.3 Hz), 4.60 (s, 2H), 3.17 (s, 3H) ppm; 13C NMR (methanol-d4, 125 MHz) δ 170.95, 149.41, 146.60, 145.10, 141.90, 141.63, 138.69, 130.44, 130.36, 128.32, 126.18, 125.77, 119.59, 41.34, 37.04 ppm; LCMS m/z 360.0848 found (M + H)+, 360.0835 calculated for C17H18N3S2O2.
(5-((4′-Methyl-6-(methylsulfonyl)-[1,1′-biphenyl]-3-yl)sulfonyl)thiazol-2-yl)methanamine Hydrochloride (9)
Yellow solid (>99% purity). 1H NMR (methanol-d4, 500 MHz) δ 8.50 (s, 1H), 8.41 (d, 1H, J = 8.4 Hz), 8.26 (dd, 1H, J = 8.4, 2.0 Hz), 7.97 (d, 1H, J = 1.9 Hz), 7.39–7.30 (m, 4H), 4.56 (s, 2H), 2.71 (s, 3H), 2.44 (s, 3H) ppm; 13C NMR (methanol-d4, 125 MHz) δ 171.23, 149.67, 146.55, 145.92, 144.88, 141.50, 140.66, 135.27, 132.37, 131.10, 131.05, 129.95, 128.01, 43.41, 41.24, 21.31 ppm; LCMS m/z 423.0507 found (M + H)+, 423.0501 calculated for C18H19N2S3O4.
(5-(Propylsulfonyl)thiazol-2-yl)methanamine Hydrochloride (10)
White solid (>99% purity). 1H NMR (DMSO-d6, 500 MHz) δ 8.81 (br s, 3H), 8.47 (s, 1H), 4.54 (s, 2H), 3.54–3.48 (m, 2H), 1.65 (s, 2H, J = 7.5 Hz), 0.96 (t, 3H, J = 7.4 Hz) ppm; 13C NMR (DMSO-d6, 125 MHz) δ 169.5, 147.6, 137.9, 57.9, 39.7, 16.5, 12.4 ppm; LCMS m/z 221.1 found (M + H)+, 221.0413 calculated for C7H13N2S2O2.
(5-(Cyclohexylsulfonyl)thiazol-2-yl)methanamine Hydrochloride (11)
White solid (>99% purity). 1H NMR (DMSO-d6, 500 MHz) δ 8.86 (br s, 3H), 8.42 (s, 1H), 4.53 (d, 2H, J = 5.4 Hz), 3.43–3.38 (m, 1H), 2.0–1.95 (m, 2H), 1.83–1.78 (m, 2H), 1.62–1.60 (m, 1H), 1.35–1.23 (m, 4H), 1.13–1.05 (m, 1H) ppm; 13C NMR (DMSO-d6, 125 MHz) δ 170.0, 148.4, 135.5, 63.1, 39.7, 25.2, 24.6 24.1 ppm; LCMS m/z 261.0723 found (M + H)+, 261.0726 calculated for C10H17N2S2O2.
(5-(Phenylsulfonyl)thiazol-2-yl)methanamine Hydrochloride (12)
Off-white solid (>99% purity). 1H NMR (DMSO-d6, 500 MHz) δ 8.78 (br s, 3H), 8.60 (s, 1H), 8.04 (d, 2H, J = 7.2 Hz), 7.78 (t, 1H, J = 7.4 Hz), 7.69 (at, 2H, J = 7.7 Hz), 4.47 (s, 2H) ppm; 13C NMR (DMSO-d6, 125 MHz) δ 170.2, 147.4, 140.7, 140.1, 134.6, 130.1, 127.1, 39.7 ppm; LCMS m/z 255.0255 found (M + H)+, 255.0256 calculated for C10H11N2O2S2.
(5-((3-(Methylsulfonyl)phenyl)sulfonyl)thiazol-2-yl)methanamine Hydrochloride (13)
Off-white solid (97% purity). 1H NMR (DMSO-d6, 500 MHz) δ 8.72 (s, 1H), 8.61 (br s, 3H), 8.48 (t, 1H, J = 1.7 Hz), 8.43–8.39 (m, 1H), 8.34–8.31 (m, 1H), 7.99 (t, 1H, J = 7.9 Hz), 4.47 (s, 2H), 3.37 (s, 3H) ppm; 13C NMR (DMSO-d6, 125 MHz) δ 171.7, 148.5, 142.6, 141.8, 138.9, 133.0, 132.2, 131.9, 125.4, 43.1, 41.6 ppm; LCMS m/z 333.1 found (M + H)+, 333.0032 calculated for C11H13N2S3O4.
(5-(3-(Methylsulfonyl)phenyl)thiazol-2-yl)methanamine Hydrochloride (14)
White solid (>99% purity). 1H NMR (DMSO-d6, 500 MHz) δ 8.77 (s, 3H), 8.47 (s, 1H), 8.18 (t, 1H, J = 1.6 Hz), 8.04 (d, 1H, J = 7.8 Hz), 7.94 (d, 1H, J = 7.8 Hz), 7.76 (t, 1H, J = 7.8 Hz), 4.49 (s, 2H), 3.32 (s, 3H) ppm; 13C NMR (DMSO-d6, 125 MHz) δ 162.6, 142.5, 140.3, 139.0, 132.1, 131.9, 131.1, 127.2, 124.9, 45.7, 43.8 ppm; LCMS m/z 269.0415 found (M + H)+, 269.0413 calculated for C11H13N2O2S2.
(5-([1,1′-Biphenyl]-3-ylsulfonyl)thiazol-2-yl)methanamine Hydrochloride (15)
White solid (>99% purity). 1H NMR (DMSO-d6, 500 MHz) δ 8.74 (br s, 3H), 8.70 (s, 1H), 8.23 (t, 1H, J = 1.8 Hz), 8.07 (ddd, 1H, J = 7.8, 1.8, 1.0 Hz), 8.04 (ddd, 1H, J = 7.9, 1.9, 1.0 Hz), 7.79 (t, 1H, J = 7.9 Hz), 7.76–7.73 (m, 2H), 7.53 (t, 2H, J = 7.5 Hz), 7.46 (t, 1H, J = 7.3 Hz), 4.48 (s, 2H) ppm; 13C NMR (DMSO-d6, 125 MHz) δ 170.3, 147.7, 142.0, 141.5, 140.0, 138.0, 132.8, 130.9, 129.2, 128.6, 127.1, 126.0, 124.8, 39.7 ppm; LCMS m/z 331.0554 found (M + H)+, 331.0569 calculated for C16H15N2S2O2.
(5-([1,1′-Biphenyl]-3-yl)thiazol-2-yl)methanamine Hydrochloride (16)
Off-white solid (>99% purity). 1H NMR (DMSO-d6, 500 MHz) δ 8.75 (br s, 3H), 8.42 (s, 1H), 7.95 (t, 1H, J = 1.6 Hz), 7.75 (dd, 2H, J = 8.3, 1.3 Hz), 7.71–7.63 (m, 2H), 7.59–7.48 (m, 3H), 7.42 (t, 1H, J = 7.3 Hz), 4.46 (q, 2H, J = 5.7 Hz) ppm; 13C NMR (DMSO-d6, 125 MHz) δ 161.0, 141.3, 140.2, 139.4, 138.9, 131.1, 130.1, 129.0, 127.9, 127.1, 127.0, 125.8, 124.6 (1 × C missing; under DMSO peak) ppm; LCMS m/z 267.3 found (M + H)+, 267.0950 calculated for C16H15N2S.
(5-(5-(Methylsulfonyl)-[1,1′-biphenyl]-3-yl)thiazol-2-yl)methanamine Hydrochloride (17)
White solid (>99% purity). 1H NMR (DMSO-d6, 500 MHz) δ 8.82 (br s, 3H), 8.60 (s, 1H), 8.29 (t, 1H, J = 1.7 Hz), 8.15 (t, 1H, J = 1.6 Hz), 8.09 (t, 1H, J = 1.6 Hz), 7.89–7.85 (m, 2H), 7.59–7.46 (m, 3H), 4.50 (s, 2H), 3.39 (s, 3H) ppm; 13C NMR (methanol-d4, 125 MHz) δ 162.6, 145.3, 144.1, 141.3, 140.8, 139.8, 134.1, 131.0, 130.4, 129.9, 128.4, 126.7, 124.9, 44.2, 41.0 ppm; LCMS m/z 345.2 found (M + H)+, 345.0726 calculated for C17H17N2O2S2.
(5-((4′-Methyl-5-(methylsulfonyl)-[1,1′-biphenyl]-3-yl)sulfonyl)thiazol-2-yl)methanamine Hydrochloride (18)
Off-white solid (>99% purity). 1H NMR (DMSO-d6, 500 MHz) δ 8.82 (s, 1H), 8.77 (br s, 3H), 8.55 (t, 1H, J = 1.7 Hz), 8.51 (t, 1H, J = 1.6 Hz), 8.40 (t, 1H, J = 1.6 Hz), 7.78 (d, 2H, J = 8.2 Hz), 7.38 (d, 2H, J = 7.9 Hz), 4.49 (s, 2H), 3.45 (s, 3H), 2.39 (s, 3H) ppm; 13C NMR (methanol-d4, 125 MHz) δ 171.15, 149.57, 146.12, 144.89, 144.70, 141.58, 140.98, 135.43, 131.70, 131.17, 130.85, 128.22, 125.25, 44.00, 41.18, 21.19 ppm; LCMS m/z 423.0488 found (M + H)+, 423.0501 calculated for C18H19N2S3O4.
(5-((5-(Methylsulfonyl)-4′-(trifluoromethyl)-[1,1′-biphenyl]-3-yl)sulfonyl)thiazol-2-yl)methanamine Hydrochloride (19)
Off-white solid (>99% purity). 1H NMR (methanol-d4, 500 MHz) δ 8.64–8.55 (m, 4H), 7.98 (d, 2H, J = 8.0 Hz), 7.86 (d, 2H, J = 7.8 Hz), 4.59 (s, 2H), 3.30 (s, 3H) ppm; 13C NMR (methanol-d4, 125 MHz) δ 171.30, 149.78, 145.16, 144.98, 144.53, 142.17, 141.36, 132.60, 132.22 (q, J = 32.8 Hz), 131.63, 129.33, 127.63 (q, J = 272.2 Hz), 127.34 (q, J = 3.8 Hz), 126.63, 44.01, 41.23 ppm; 19F NMR (methanol-d4, 471 MHz) δ −61.78 ppm; LCMS m/z 477.0230 found (M + H)+, 477.0219 calculated for C18H16N2S3O4F3.
(5-((4′-Fluoro-5-(methylsulfonyl)-[1,1′-biphenyl]-3-yl)sulfonyl)thiazol-2-yl)methanamine Hydrochloride (20)
White solid (>99% purity). 1H NMR (methanol-d4, 500 MHz) δ 8.79–8.32 (m, 4H), 7.79 (br, 2H), 7.28 (br, 2H), 4.58 (s, 2H), 3.28 (s, 3H) ppm; 13C NMR (methanol-d4, 125 MHz) δ 173.79, 167.59 (d, J = 249.5 Hz), 152.53, 147.74, 147.61, 147.40, 144.20, 137.27, 134.82, 133.97, 133.55 (d, J = 7.6 Hz), 128.42, 120.05 (d, J = 22.7 Hz), 47.48, 45.02 ppm; LCMS m/z 427.0242 found (M + H)+, 427.0251 calculated for C17H16FN2S3O4.
(5-((3-(1-Methyl-1H-pyrazol-4-yl)-5-(methylsulfonyl)phenyl)sulfonyl)thiazol-2-yl)methanamine Hydrochloride (21a)
Off-white solid (95% purity). 1H NMR (DMSO-d6, 500 MHz) δ 8.78 (s, 1H), 8.70 (br s, 3H), 8.57 (s, 1H), 8.50 (t, 1H, J = 1.7 Hz), 8.45 (t, 1H, J = 1.6 Hz), 8.20 (t, 1H, J = 1.7 Hz), 8.19 (d, 1H, J = 0.7 Hz), 4.50 (br d, 2H, J = 5.2 Hz), 3.90 (s, 3H), 3.41 (s, 3H) ppm; 13C NMR (DMSO-d6, 125 MHz) δ 171.0, 148.5, 143.4, 142.6, 139.0, 137.1, 136.7, 129.9, 128.3, 127.1, 121.6, 118.8, 42.9, 38.9, 38.8 ppm; LCMS m/z 413.1 found (M + H)+, 413.0406 calculated for C15H17N4S3O4.
(5-((3-Ethyl-5-(1-methyl-1H-pyrazol-4-yl)phenyl)sulfonyl)thiazol-2-yl)methanamine Hydrochloride (21b)
White solid (>99% purity). 1H NMR (DMSO-d6, 500 MHz) δ 8.87 (br s, 3H), 8.63 (s, 1H), 8.37 (s, 1H), 8.01 (d, 1H, J = 0.7 Hz), 7.97 (t, 1H, J = 1.7 Hz), 7.81 (t, 1H, J = 1.5 Hz), 7.66 (t, 1H, J = 1.5 Hz), 4.46 (q, 2H, J = 5.6 Hz), 3.87 (s, 3H), 2.72 (q, 2H, J = 7.6 Hz), 1.22 (t, 3H, J = 7.6 Hz) ppm; 13C NMR (DMSO-d6, 125 MHz) δ 170.1, 147.4, 146.8, 141.5, 140.3, 136.5, 134.7, 130.1, 129.0, 123.3, 120.4, 120.1, 39.7, 38.8, 27.9, 15.3 ppm; LCMS m/z 363.0940 found (M + H)+, 363.0944 calculated for C16H19N4S2O2.
(5-((3-(1-Methyl-1H-pyrazol-4-yl)-5-(trifluoromethyl)phenyl)sulfonyl)thiazol-2-yl)methanamine Hydrochloride (21c)
Pale yellow solid (>99% purity). 1H NMR (methanol-d4, 500 MHz) δ 8.56 (s, 1H), 8.45 (s, 1H), 8.41 (s, 1H), 8.23–8.21 (m, 2H), 8.11 (s, 1H), 4.57 (s, 2H), 4.01 (s, 3H) ppm; 13C NMR (methanol-d4, 125 MHz) δ 171.1, 149.6, 144.8, 141.7, 137.3, 137.2, 134.0 (q, J = 34.0 Hz), 131.7, 128.34, 128.31, 124.5 (q, J = 272.2 Hz), 122.6 (q, J = 3.8 Hz), 121.4, 41.3, 39.3 ppm; 19F NMR (methanol-d4, 471 MHz) δ −64.40 (s) ppm; LCMS m/z 403.0517 found (M + H)+, 403.0505 calculated for C15H14F3N4S2O2.
(5-((3-Chloro-5-(1-methyl-1H-pyrazol-4-yl)phenyl)sulfonyl)thiazol-2-yl)methanamine Hydrochloride (21d)
Yellow solid (97% purity). 1H NMR (DMSO-d6, 500 MHz) δ 8.91 (br s, 3H), 8.72 (s, 1H), 8.48 (s, 1H), 8.13 (t, 1H, J = 1.6 Hz), 8.11 (s, 1H), 8.07 (t, 1H, J = 1.7 Hz), 7.84 (t, 1H, J = 1.8 Hz), 4.47 (q, 2H, J = 5.4 Hz), 3.87 (s, 3H) ppm; 13C NMR (DMSO-d6, 125 MHz) δ 170.8, 148.3, 143.3, 139.3, 137.0, 136.9, 135.3, 129.9, 129.7, 123.4, 121.4, 118.9, 39.8, 38.9 ppm; LCMS m/z 369.0234 found (M + H)+, 369.0241 calculated for C14H14N4S2O2Cl.
(5-((3-Fluoro-5-(1-methyl-1H-pyrazol-4-yl)phenyl)sulfonyl)thiazol-2-yl)methanamine Hydrochloride (21e)
Yellow solid (>99% purity). 1H NMR (methanol-d4, 500 MHz) δ 8.40 (s, 1H), 8.17 (br, 1H), 8.01–7.90 (m, 2H), 7.65–7.48 (m, 2H), 4.53 (s, 2H), 3.98 (s, 3H) ppm; 13C NMR (methanol-d4, 125 MHz) δ 169.90, 163.89 (d, J = 252 Hz), 148.66, 144.34 (d, J = 7.6 Hz), 141.33, 137.62 (d, J = 10.1 Hz), 136.99, 130.68, 121.23, 120.57, 118.51 (d, J = 23 Hz), 112.79 (d, J = 25 Hz), 40.99, 39.30 ppm; 19F NMR (methanol-d4, 471 MHz) δ −109.29 ppm; LCMS m/z 353.0541 found (M + H)+, 353.0537 calculated for C14H14N4S2O2F.
(4-(Naphthalen-2-ylsulfonyl)thiazol-2-yl)methanamine Hydrochloride (22a)
Off-white solid (97% purity). 1H NMR (DMSO-d6, 500 MHz) δ 8.83 (s, 1H), 8.72 (d, 1H, J = 1.5 Hz), 8.66 (br s, 3H), 8.26 (d, 1H, J = 8.1 Hz), 8.18 (d, 1H, J = 8.8 Hz), 8.08 (d, 1H, J = 8.2 Hz), 7.95 (dd, 1H, J = 8.7, 1.9 Hz), 7.77 (t, 1H, J = 6.9 Hz), 7.72 (t, 1H, J = 6.9 Hz), 4.42 (s, 2H) ppm; 13C NMR (DMSO-d6, 125 MHz) δ 166.1, 152.3, 136.3, 134.9, 131.7, 130.5, 129.9, 129.7, 129.6, 129.4, 127.9 (2 x C), 122.7, 39.3 ppm; LCMS m/z 305.0403 found (M + H)+, 305.0413 calculated for C14H13N2O2S2.
(4-((5-(Methylsulfonyl)-[1,1′-biphenyl]-3-yl)sulfonyl)thiazol-2-yl)methanamine Hydrochloride (22b)
Off-white solid (97% purity). 1H NMR (DMSO-d6, 500 MHz) δ 8.97 (s, 1H), 8.65 (br s, 3H), 8.54 (t, 1H, J = 1.6 Hz), 8.49 (t, 1H, J = 1.7 Hz), 8.41 (t, 1H, J = 1.6 Hz), 7.85 (dd, 2H, J = 8.1, 1.4 Hz), 7.61–7.50 (m, 3H), 4.46 (s, 2H), 3.45 (s, 3H) ppm; 13C NMR (DMSO-d6, 125 MHz) δ 167.0, 151.5, 143.9, 143.6, 141.8, 137.1, 132.3, 131.3, 130.8, 129.9, 129.7, 128.0, 125.0, 43.6 (1 × C missing; under DMSO peak) ppm; LCMS m/z 409.2 found (M + H)+, 409.0345 calculated for C17H17N2O4S3.
(4-((3-(1-Methyl-1H-pyrazol-4-yl)-5-(methylsulfonyl)phenyl)sulfonyl)thiazol-2-yl)methanamine Hydrochloride (22c)
Off-white solid (>99% purity). 1H NMR (methanol-d4, 500 MHz) δ 8.80 (s, 1H), 8.45 (t, 1H, J = 1.7 Hz), 8.41 (t, 1H, J = 1.7 Hz), 8.36 (t, 1H, J = 1.6 Hz), 8.26 (s, 1H), 8.03 (d, 1H, J = 0.8 Hz), 4.52 (s, 2H), 3.96 (s, 3H), 3.25 (s, 3H) ppm; 13C NMR (DMSO-d6, 125 MHz) δ 166.9, 154.2, 144.6, 143.2, 138.1, 137.8, 131.5, 130.8, 130.0, 129.8, 125.1, 121.1, 44.0, 40.8, 39.3 ppm; LCMS m/z 413.0 found (M + H)+, 413.0406 calculated for C15H17N4O4S3.
(4-((3-Ethyl-5-(1-methyl-1H-pyrazol-4-yl)phenyl)sulfonyl)thiazol-2-yl)methanamine Hydrochloride (22d)
White solid (>99% purity). 1H NMR (DMSO-d6, 500 MHz) δ 8.78 (s, 1H), 8.75 (br s, 3H), 8.33 (s, 1H), 7.97 (d, 1H, J = 0.7 Hz), 7.90 (t, 1H, J = 1.7 Hz), 7.79 (t, 1H, J = 1.5 Hz), 7.62 (t, 1H, J = 1.6 Hz), 4.44 (q, 2H, J = 5.7 Hz), 3.87 (s, 3H), 2.72 (q, 2H, J = 7.6 Hz), 1.22 (t, 3H, J = 7.6 Hz) ppm; 13C NMR (methanol-d4, 125 MHz) δ 166.38, 155.19, 148.39, 141.75, 136.26, 134.47, 131.72, 131.62, 130.54, 126.83, 123.52, 123.14, 40.85, 39.11, 29.58, 15.76 ppm; LCMS m/z 363.0959 found (M + H)+, 363.0944 calculated for C16H19N4O2S2.
In Silico Modeling
All compounds were imported to Forge and subjected to a field-based alignment to the reference structure (the FieldTemplater model described in the article), using maximum common substructure to guide the alignment; a similarity score was calculated as the average of the field and shape similarity scores.40 The calculated alignments were visually inspected to ensure best alignment and adjusted as required.
PAINS Assessment
To identify reactive compounds that might exhibit interference in biochemical assays, the PAINS filters as described by Baell and Holloway50 were curated as SMARTS and scripted as a flagging protocol deployed in Vortex (version 2018.09.76561.53-s, 2018, https://www.dotmatics.com/products/vortex) and Pipeline Pilot (Dassault Systèmes BIOVIA, BIOVIA Pipeline Pilot, release 2018, San Diego, Dassault Systèmes, 2018). The 480 patterns were used to recognize structures that may result in nonspecific binding to multiple biological targets by virtue of comprising one or more fragments established to be of concern. No LOXL2 inhibitor in this study showed any potential PAINS liability when screened against this PAINS filter.
LOX Protein Preparation and Enzyme Assays
LOX enzyme was extracted from pig skin by the method of Shackleton and Hulmes.51 LOXL2 and LOXL3 were purchased from R&D Systems. LOX, LOXL2, and LOXL3 catalytic activity were determined using the Promega ROS-Glo assay kit with cadaverine dihydrochloride as substrate, BAPN as the reference inhibitor control, a preincubation time of 20 min, 1 h, or 3 h, with nine dilutions from a top concentration of 10 μM or 100 μM.
LOXL2 Jump Dilution Assay
LOXL2 catalytic activity was determined using the Amplex Red hydrogen peroxide assay kit with cadaverine dihydrochloride as substrate. LOXL2 at 100-fold final assay concentration and compound at 10 × IC50 were preincubated for 1 h. The enzyme/inhibitor mixture was then diluted 100-fold into a solution containing substrate and detection reagents and read kinetically every 5 min.
Amine Oxidase Assays
All amine oxidase assays were performed with concentrations as above. MAO-A and MAO-B enzymes were purchased from Sigma. The catalytic activity of MAO-A and MAO-B was determined using the Promega MAO-Glo assay kit (substrate included), with clorgyline and deprenyl as reference inhibitor controls, respectively. DAO was purchased from Sigma, and the catalytic activity was determined using the Promega ROS-Glo assay kit, with aminoguanidine as the reference inhibitor control. SSAO was purchased from Sigma. SSAO catalytic activity was determined using the Promega MAO-Glo assay kit, with mofegiline as the reference inhibitor control.
Assessment of Compound 21b as a Substrate for Amine Oxidases
The catalytic activities of MAO-A, MAO-B, and SSAO with compound 21b as a substrate were determined using the respective enzymes described above, and the hydrogen peroxide produced was quantified using an Amplex red monoamine oxidase assay kit. p-Tyramine was used as the positive substrate control for MAO-A and MAO-B, and benzylamine was used for SSAO.
MLM Stability Assay
Mouse liver microsomes (CD1 female; M1500) and rat liver microsomes (Sprague Dawley female; R1500) were purchased from Tebu-bio, and the assay was performed by methods previously described.21 Inhibitors at 10 μM concentration incubated with the microsomes were assessed at 0, 15, and 30 min. Control samples containing no microsomes and no cofactors were also assessed at 0 and 30 min. Samples were extracted by protein precipitation, and centrifugation for 20 min in a refrigerated centrifuge (4 °C) at 3700 rpm. The supernatant was analyzed by LC–MS/MS for % metabolized over time.
Animal Procedures
All procedures involving animals were performed under licenses PPL-70/7635, 70/7701, and PE3DF1A5B and National Home Office regulations under the Animals (Scientific Procedures) Act 1986. Procedures were approved by the Animal Welfare and Ethical Review Bodies (AWERB) of the CRUK Manchester Institute and the Institute of Cancer Research and reported in accordance with ARRIVE guidelines. All mice and rats were maintained in pathogen-free, ventilated cages in the Biological Resources Unit at Cancer Research UK Manchester Institute and the Biological Services Unit at The Institute of Cancer Research. All mice and rats were allowed free access to irradiated food and autoclaved water in a 12 h light/dark cycle with room temperature at 21 ± 2 °C. All cages contained wood shavings, bedding, and a cardboard tube for environmental enrichment. PyMT-driven breast cancer model mice (FVB background) were bred in a specific pathogen-free facility at The University of Manchester (U.K.) under a Home Office approved license.
In Vivo PK
Female Balb/C or CD1 nude mice (Charles River Laboratories) at 6 weeks of age were used for the mouse PK analyses. The mice were dosed orally by gavage (50 mg kg–1 in DMSO/water 1:19 v:v; n = 21) or intravenously in the tail vein (10 mg kg–1 in DMSO/Tween20/saline 10:1:89 v:v:v; n = 24). Blood samples were taken at seven (po) or eight (iv) time-points between 5 min and 24 h, after one single dose of the inhibitor. Three mice were used per time-point per route; average values are reported. They were placed under halothane or isoflurane anesthesia, and blood for plasma preparation was taken by terminal cardiac puncture into heparinized syringes. Female Sprague Dawley (Charles River Laboratories) weighing between 170 g and 200 g were used for the rat PK analyses. The rats were dosed orally by gavage (20 mg kg–1 in DMSO/water 1:19 v:v; n = 21) or intravenously in the tail vein (4 mg kg–1 in DMSO/Tween20/saline 10:1:89 v:v:v; n = 24). Blood samples were taken at five (po and (iv) time-points between 5 min and 8 h, after 1 single dose of the inhibitor. One rat was used per route with serial bleeds taken through the time points. They were placed in a heated box for 10 min prior to sampling to increase vasodilation, and blood for plasma preparation was taken by tail vain bleed into heparinized tubes. Plasma samples, obtained after blood spun at 1300 rpm for 3 min, were pipetted into cryovials and immediately snap frozen in liquid nitrogen and then stored at −80 °C prior to analysis.
In Vivo Antitumor Efficacy
LOX inhibitor treatment was carried out in a genetically engineered MMTV-PyMT driven mouse breast cancer model where female mice were randomized as described previously.21 Mice were treated daily by oral gavage with 70 mg/kg compound 21b (n = 3) in vehicle (5% DMSO/2.5% Tween20 in water), and controls (n = 5) received vehicle alone or were untreated. Oral administration of 21b was initiated at day 57 (n = 1) and day 61 (n = 2) with all treatments continuing for 34 days. The spontaneous breast tumors arising in the model were measured twice weekly. All of the controls bar one were culled due to large tumor size by day 95. All of the treated were culled at the end of treatment at day 91 (n = 1) and day 95 (n = 2); none were culled due to reaching license limit tumor volumes. Statistical significance was calculated using Welch’s t test on day 91 (*p = 0.0367), utilizing the final measured tumor volume of the culled control mice and linear interpolation for the two remaining control mice (between measurements made on days 89 and 92). All animals allocated to the study were used.
Commercial ADME-T Services
hERG inhibition was determined using the “hERG human potassium ion channel cell based antagonist Qpatch assay” by Eurofins Ltd. Cell permeability was determined using the “Caco-2 permeability assay” by Cyprotex Ltd.
Acknowledgments
This work was supported by the CRUK Manchester Institute (Grants C5759/A12328, C5759/A27412), CRUK Manchester Institute Drug Discovery Unit (Grant C480/A17098), the Division of Cancer Therapeutics at The Institute of Cancer Research (Grants C309/A11566, C309/A8274, C107/A10433), and the Wellcome Trust (Grants 1003X, 103021/Z/13/Z). We thank Dr. Maggie Liu, Dr. Amin Mirza, Meirion Richards, Dr. Ali Raoof, and Dr. Christopher Kershaw for assistance with NMR and MS instrumentation.
Glossary
Abbreviations Used
- AMT
aminomethylenethiophene
- AMTz
aminomethylenethiazole
- BAPN
3-aminopropionitrile
- DAO
diamine oxidase
- dba
dibenzylideneacetone
- Cl
clearance
- Cmax
maximal concentration
- DIPEA
N,N-diisopropylethylamine
- DPPA
diphenylphosphoryl azide
- dppf
(diphenylphosphino)ferrocene
- EtOAc
ethyl acetate
- GEMM
genetically engineered mouse model
- LOX
lysyl oxidase
- LOXL
lysyl oxidase-like
- LTQ
lysine tyrosylquinone
- MAO
monoamine oxidase
- MLM
mouse liver microsome
- MMTV
mouse mammary tumor virus
- MOI
mode of inhibition
- PAINS
pan assay interference compounds
- PE
petroleum ether
- PyMT
polyomavirus middle T-antigen
- ROS
reactive oxygen species
- SSAO
semicarbazide-sensitive amine oxidase
- p-Tol
para-toluene
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.9b01112.
Experimental procedures and characterization data for all synthetic intermediates; NMR and LCMS/UV spectra for lead inhibitors (PDF)
Compound alignment data set used to derive pharmacophore/binding mode hypothesis; calculated field-based 3DQSAR and RVM model details (ZIP)
Molecular formula strings and some data for all final inhibitors (CSV)
The authors declare the following competing financial interest(s): C.S., R.Ma., D.N.-D., L.M.H.L., D.A.S., and H.T. have filed a patent application which includes the therapeutic use of aminomethylenethiophene and aminomethylenethiazole inhibitors such as CCT365623 (3a), 7a, 21b, and 22d. All authors D.A.S., L.M.H.L., M.C., R.L., H.T., D.N.-D., S.P.P., R.Mc., F.L., M.A., M.B., L.J., G.T., R.Ma., and C.S. could benefit financially if any of the described inhibitors such as CCT365623 (3a), 7a, 21b, and 22d are licenced, as part of a reward scheme for employees at ICR and The University of Manchester.
Supplementary Material
References
- Kagan H. M.; Ryvkin F.. Lysyl oxidase and lysyl oxidase-like enzymes. In The Extracellular Matrix: An Overview, 1st ed.; Mecham R. P., Ed.; Springer, 2011; pp 303–335. [Google Scholar]
- Finney J.; Moon H.-J.; Ronnebaum T.; Lantz M.; Mure M. Human copper-dependent amine oxidases. Arch. Biochem. Biophys. 2014, 546, 19–32. 10.1016/j.abb.2013.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucero H. A.; Kagan H. M. Lysyl oxidase: an oxidative enzyme and effector of cell function. Cell. Mol. Life Sci. 2006, 63 (19–20), 2304–2316. 10.1007/s00018-006-6149-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan H. M.; Li W. Lysyl oxidase: Properties, specificity, and biological roles inside and outside of the cell. J. Cell. Biochem. 2003, 88 (4), 660–672. 10.1002/jcb.10413. [DOI] [PubMed] [Google Scholar]
- Molnar J.; Fong K. S. K.; He Q. P.; Hayashi K.; Kim Y.; Fong S. F. T.; Fogelgren B.; Molnarne Szauter K.; Mink M.; Csiszar K. Structural and functional diversity of lysyl oxidase and the LOX-like proteins. Biochim. Biophys. Acta, Proteins Proteomics 2003, 1647 (1–2), 220–224. 10.1016/S1570-9639(03)00053-0. [DOI] [PubMed] [Google Scholar]
- Csiszar K. Lysyl oxidases: A novel multifunctional amine oxidase family. Prog. Nucleic Acid Res. Mol. Biol. 2001, 70, 1–32. 10.1016/S0079-6603(01)70012-8. [DOI] [PubMed] [Google Scholar]
- Okada K.; Moon H.-J.; Finney J.; Meier A.; Mure M. Extracellular processing of lysyl oxidase-like 2 and its effect on amine oxidase activity. Biochemistry 2018, 57 (51), 6973–6983. 10.1021/acs.biochem.8b01008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Jimenez A. J.; Basak T.; Vanacore R. M. Proteolytic processing of lysyl oxidase-like-2 in the extracellular matrix is required for crosslinking of basement membrane collagen IV. J. Biol. Chem. 2017, 292 (41), 16970–16982. 10.1074/jbc.M117.798603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith-Mungo L. I.; Kagan H. M. Lysyl oxidase: properties, regulation and multiple functions in biology. Matrix Biol. 1998, 16 (7), 387–398. 10.1016/S0945-053X(98)90012-9. [DOI] [PubMed] [Google Scholar]
- Johnston K. A.; Lopez K. M. Lysyl oxidase in cancer inhibition and metastasis. Cancer Lett. (N. Y., NY, U. S.) 2018, 417, 174–181. 10.1016/j.canlet.2018.01.006. [DOI] [PubMed] [Google Scholar]
- Wang T.-H.; Hsia S.-M.; Shieh T.-M. Lysyl oxidase and the tumor microenvironment. Int. J. Mol. Sci. 2017, 18 (1), 62–73. 10.3390/ijms18010062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox T. R.; Gartland A.; Erler J. T. Lysyl oxidase, a targetable secreted molecule involved in cancer metastasis. Cancer Res. 2016, 76 (2), 188–192. 10.1158/0008-5472.CAN-15-2306. [DOI] [PubMed] [Google Scholar]
- Trackman P. C. Lysyl oxidase isoforms and potential therapeutic opportunities for fibrosis and cancer. Expert Opin. Ther. Targets 2016, 20 (8), 935–945. 10.1517/14728222.2016.1151003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L.; Zhu Y. The function and mechanisms of action of LOXL2 in cancer (Review). Int. J. Mol. Med. 2015, 36 (5), 1200–1204. 10.3892/ijmm.2015.2337. [DOI] [PubMed] [Google Scholar]
- Nishioka T.; Eustace A.; West C. Lysyl oxidase: from basic science to future cancer treatment. Cell Struct. Funct. 2012, 37 (1–2), 75–80. 10.1247/csf.11015. [DOI] [PubMed] [Google Scholar]
- Xiao Q.; Ge G. Lysyl oxidase, extracellular matrix remodeling and cancer metastasis. Cancer Microenviron. 2012, 5 (3), 261–273. 10.1007/s12307-012-0105-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Wang Q.; Wu J.; Wang J.; Shi Y.; Liu M. Crystal structure of human lysyl oxidase-like 2 (hLOXL2) in a precursor state. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (15), 3828–3833. 10.1073/pnas.1720859115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J.; Lucas M. C.; Leonte L. E.; Garcia-Montolio M.; Singh L. B.; Findlay A. D.; Deodhar M.; Foot J. S.; Jarolimek W.; Timpson P.; Erler J. T.; Cox T. R. Pre-clinical evaluation of small molecule LOXL2 inhibitors in breast cancer. Oncotarget 2017, 8 (16), 26066–26078. 10.18632/oncotarget.15257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilter H.; Findlay A. D.; Perryman L.; Yow T. T.; Moses J.; Zahoor A.; Turner C. I.; Deodhar M.; Foot J. S.; Zhou W.; Greco A.; Joshi A.; Rayner B.; Townsend S.; Buson A.; Jarolimek W. The lysyl oxidase like 2/3 enzymatic inhibitor, PXS-5153A, reduces crosslinks and ameliorates fibrosis. J. Cell. Mol. Med. 2019, 23 (3), 1759–1770. 10.1111/jcmm.14074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowbottom M. W.; Bain G.; Calderon I.; Lasof T.; Lonergan D.; Lai A.; Huang F.; Darlington J.; Prodanovich P.; Santini A. M.; King C. D.; Goulet L.; Shannon K. E.; Ma G. L.; Nguyen K.; MacKenna D. A.; Evans J. F.; Hutchinson J. H. Identification of 4-(aminomethyl)-6-(trifluoromethyl)-2-(phenoxy)pyridine derivatives as potent, selective, and orally efficacious inhibitors of the copper-dependent amine oxidase, lysyl oxidase-like 2 (LOXL2). J. Med. Chem. 2017, 60 (10), 4403–4423. 10.1021/acs.jmedchem.7b00345. [DOI] [PubMed] [Google Scholar]
- Tang H. R.; Leung L.; Saturno G.; Viros A.; Smith D.; Di Leva G.; Morrison E.; Niculescu-Duvaz D.; Lopes F.; Johnson L.; Dhomen N.; Springer C.; Marais R. Lysyl oxidase drives tumour progression by trapping EGF receptors at the cell surface. Nat. Commun. 2017, 8, 14909–14922. 10.1038/ncomms14909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung L.; Niculescu-Duvaz D.; Smithen D.; Lopes F.; Callens C.; McLeary R.; Saturno G.; Davies L.; Aljarah M.; Brown M.; Johnson L.; Zambon A.; Chambers T.; Menard D.; Bayliss N.; Knight R.; Fish L.; Lawrence R.; Challinor M.; Tang H.; Marais R.; Springer C. Anti-metastatic inhibitors of lysyl oxidase (LOX): design and structure-activity relationships. J. Med. Chem. 2019, 62 (12), 5863–5884. 10.1021/acs.jmedchem.9b00335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S. S.; Trackman P. C.; Kagan H. M. Reaction of aortic lysyl oxidase with β-aminopropionitrile. J. Biol. Chem. 1983, 258 (7), 4331–4338. [PubMed] [Google Scholar]
- Pinnell S. R.; Martin G. R. The cross-linking of collagen and elastin: enzymatic conversion of lysine in peptide linkage to alpha-aminoadipic-delta-semialdehyde (allysine) by an extract from bone. Proc. Natl. Acad. Sci. U. S. A. 1968, 61 (2), 708–716. 10.1073/pnas.61.2.708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riel A. M. S.; Decato D. A.; Sun J.; Massena C. J.; Jessop M. J.; Berryman O. B. The intramolecular hydrogen bonded-halogen bond: a new strategy for preorganization and enhanced binding. Chem. Sci. 2018, 9 (26), 5828–5836. 10.1039/C8SC01973H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung I. H.; Jung Y. K.; Lee J.; Park J.-H.; Woo H. Y.; Lee J.-I.; Chu H. Y.; Shim H.-K. Synthesis and electroluminescent properties of fluorene-based copolymers containing electron-withdrawing thiazole derivatives. J. Polym. Sci., Part A: Polym. Chem. 2008, 46 (21), 7148–7161. 10.1002/pola.23019. [DOI] [Google Scholar]
- Lin Y.; Fan H.; Li Y.; Zhan X. Thiazole-based organic semiconductors for organic electronics. Adv. Mater. (Weinheim, Ger.) 2012, 24 (23), 3087–3106. 10.1002/adma.201200721. [DOI] [PubMed] [Google Scholar]
- Springer C.; Marais R.; Niculescu-Duvaz D.; Leung L.; Smithen D.; Callens C.; Tang H.. Heteroarylmethylamine Derivatives as Lysyl Oxidase Inhibitors for the Treatment of Cancer and Their Preparation. Patent WO2017141049A1, 2017.
- Huang Y.; Dai J.; Tang R.; Zhao W.; Zhou Z.; Wang W.; Ying K.; Xie Y.; Mao Y. Cloning and characterization of a human lysyl oxidase-like 3 gene (hLOXL3). Matrix Biol. 2001, 20 (2), 153–157. 10.1016/S0945-053X(01)00124-X. [DOI] [PubMed] [Google Scholar]
- Maki J. M.; Kivirikko K. I. Cloning and characterization of a fourth human lysyl oxidase isoenzyme. Biochem. J. 2001, 355 (2), 381–387. 10.1042/bj3550381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.-E.; Kim Y. A tissue-specific variant of the human lysyl oxidase-like protein 3 (LOXL3) functions as an amine oxidase with substrate specificity. J. Biol. Chem. 2006, 281 (49), 37282–37290. 10.1074/jbc.M600977200. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Liu Z.; Zhang T.; Lin Z.; Li Z.; Zhang A.; Sun X.; Gao J. Loss of lysyl oxidase-like 3 attenuates embryonic lung development in mice. Sci. Rep. 2016, 6, 33856. 10.1038/srep33856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft-Sheleg O.; Zaffryar-Eilot S.; Genin O.; Yaseen W.; Soueid-Baumgarten S.; Kessler O.; Smolkin T.; Akiri G.; Neufeld G.; Cinnamon Y.; Hasson P. Localized LOXL3-dependent fibronectin oxidation regulates myofiber stretch and integrin-mediated adhesion. Dev. Cell 2016, 36 (5), 550–561. 10.1016/j.devcel.2016.02.009. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Yang R.; Liu Z.; Hou C.; Zong W.; Zhang A.; Sun X.; Gao J. Loss of lysyl oxidase-like 3 causes cleft palate and spinal deformity in mice. Hum. Mol. Genet. 2015, 24 (21), 6174–6185. 10.1093/hmg/ddv333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santamaria P. G.; Floristan A.; Fontanals-Cirera B.; Vazquez-Naharro A.; Santos V.; Morales S.; Yuste L.; Peinado H.; Garcia-Gomez A.; Portillo F.; Hernando E.; Cano A. Lysyl oxidase-like 3 is required for melanoma cell survival by maintaining genomic stability. Cell Death Differ. 2018, 25 (5), 935–950. 10.1038/s41418-017-0030-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasashima H.; Yashiro M.; Okuno T.; Miki Y.; Kitayama K.; Masuda G.; Kinoshita H.; Morisaki T.; Fukuoka T.; Hasegawa T.; Sakurai K.; Toyokawa T.; Kubo N.; Tanaka H.; Muguruma K.; Hirakawa K.; Ohira M. Significance of the lysyl oxidase members lysyl oxidase like 1, 3, and 4 in gastric cancer. Digestion 2018, 98 (4), 238–248. 10.1159/000489558. [DOI] [PubMed] [Google Scholar]
- Copeland R. A.; Basavapathruni A.; Moyer M.; Scott M. P. Impact of enzyme concentration and residence time on apparent activity recovery in jump dilution analysis. Anal. Biochem. 2011, 416 (2), 206–210. 10.1016/j.ab.2011.05.029. [DOI] [PubMed] [Google Scholar]
- Moon H.-J.; Finney J.; Ronnebaum T.; Mure M. Human lysyl oxidase-like 2. Bioorg. Chem. 2014, 57, 231–241. 10.1016/j.bioorg.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FieldTemplater, Forge, version 10.6.0; Cresset: Litlington, Cambridgeshire, U.K.; http://www.cresset-group.com/forge/.
- Cheeseright T.; Mackey M.; Rose S.; Vinter A. Molecular field extrema as descriptors of biological activity: definition and validation. J. Chem. Inf. Model. 2006, 46 (2), 665–676. 10.1021/ci050357s. [DOI] [PubMed] [Google Scholar]
- Forge, version 10.6.0; Cresset: Litlington, Cambridgeshire, .K.; http://www.cresset-group.com/forge/.
- Cramer R. D. III; Patterson D. E.; Bunce J. D. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988, 110 (18), 5959–5967. 10.1021/ja00226a005. [DOI] [PubMed] [Google Scholar]
- Clark M.; Cramer R. D. III; Jones D. M.; Patterson D. E.; Simeroth P. E. Comparative molecular field analysis (CoMFA). 2. Toward its use with 3D-structural databases. Tetrahedron Comput. Methodol. 1990, 3 (1), 47–59. 10.1016/0898-5529(90)90120-W. [DOI] [Google Scholar]
- McLellan M. R.; Ryan M. D.; Breneman C. M. Rank order entropy: why one metric is not enough. J. Chem. Inf. Model. 2011, 51 (9), 2302–2319. 10.1021/ci200170k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golbraikh A.; Tropsha A. Beware of q2!. J. Mol. Graphics Modell. 2002, 20 (4), 269–276. 10.1016/S1093-3263(01)00123-1. [DOI] [PubMed] [Google Scholar]
- Itoh T.; Mase T. Practical thiol surrogates and protective groups for arylthiols for suzuki-miyaura conditions. J. Org. Chem. 2006, 71 (5), 2203–2206. 10.1021/jo052624z. [DOI] [PubMed] [Google Scholar]
- Crowley V. M.; Khandelwal A.; Mishra S.; Stothert A. R.; Huard D. J. E.; Zhao J.; Muth A.; Duerfeldt A. S.; Kizziah J. L.; Lieberman R. L.; Dickey C. A.; Blagg B. S. J. Development of glucose regulated protein 94-selective inhibitors based on the BnIm and radamide scaffold. J. Med. Chem. 2016, 59 (7), 3471–3488. 10.1021/acs.jmedchem.6b00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black C.; Beaulieu C.. Preparation of Dipeptide Nitriles as Papain Family Cysteine Protease Inhibitors for the Treatment of Parasitic Diseases. Patent WO2010148488A1, 2010.
- Siddiqui M. A.; Mansoor U. F.; Reddy P. A. P.; Madison V. S.. Preparation of Tartaric Acid Functional Compounds for the Treatment of Disorders Mediated by MMPs, Aggrecanase, ADMP, LpxC, ADAMs, TACE and TNF-α. Patent US20070167426A1, 2007.
- Baell J. B.; Holloway G. A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53 (7), 2719–2740. 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Shackleton D. R.; Hulmes D. J. S. Purification of lysyl oxidase from piglet skin by selective interaction with Sephacryl S-200. Biochem. J. 1990, 266 (3), 917–919. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.