

Summary
The complex etiology of neurodegeneration continues to stifle efforts to develop effective therapeutics. New agents elucidating key pathways causing neurodegeneration might serve to increase our understanding and potentially lead to improved treatments. Here, we demonstrate that a water-soluble manganese(II) texaphyrin (MMn) is a suitable magnetic resonance imaging (MRI) contrast agent for detecting larger amyloid beta constructs. The imaging potential of MMn was inferred on the basis of in vitro studies and in vivo detection in Alzheimer’s disease C. elegans models via MRI and ICP-MS. In vitro antioxidant- and cellular-based assays provide support for the notion that this porphyrin analog shows promise as a therapeutic agent able to mitigate the oxidative and nitrative toxic effects considered causal in neurodegeneration. The present report marks the first elaboration of an MRI-active metalloantioxidant that confers diagnostic and therapeutic benefit in Alzheimer’s disease models without conjugation of a radioisotope, targeting moiety, or therapeutic payload.
Keywords: neurodegeneration, metalloantioxidant, MRI, Alzheimer’s disease, amyloid beta, expanded porphyrin, texaphyrin, amyloid aggregation, C. elegans AD models, amyloid beta modifications by ROS and RNS
Graphical Abstract
Highlights
-
•
Manganese texaphyrin binds to Aβ constructs (i.e., monomer, fibrils, and aggregates)
-
•
MRI studies demonstrate the utility of metallotexaphyrins for detecting Aβ aggregates
-
•
Antioxidant assays show potential utility of MMn for treating CNS neurodegeneration
The Bigger Picture
Neurodegenerative disease is correlated with aberrant oxidation and nitration of small-molecule neurotransmitters, proteins, DNA, and cellular membrane components. Cellular damage enhanced by these modifications propagates disease progression, leading to death. Effective tools that enable insight into the complex etiology of CNS diseases could allow researchers to find a cure. Part of this effort is focused on developing imaging modalities that can assist in understanding these diseases. Here, we demonstrate a new theranostic strategy that shows promise in addressing the oxidative and nitrative malfunctions considered causal in disease progression. The approach detailed in this report also allows the MRI of aggregated Aβ, which could lead to improvements in disease diagnosis. The present work is thus expected to set the stage for the mechanistic-based design of theranostics, useful in understanding and treating neurodegeneration.
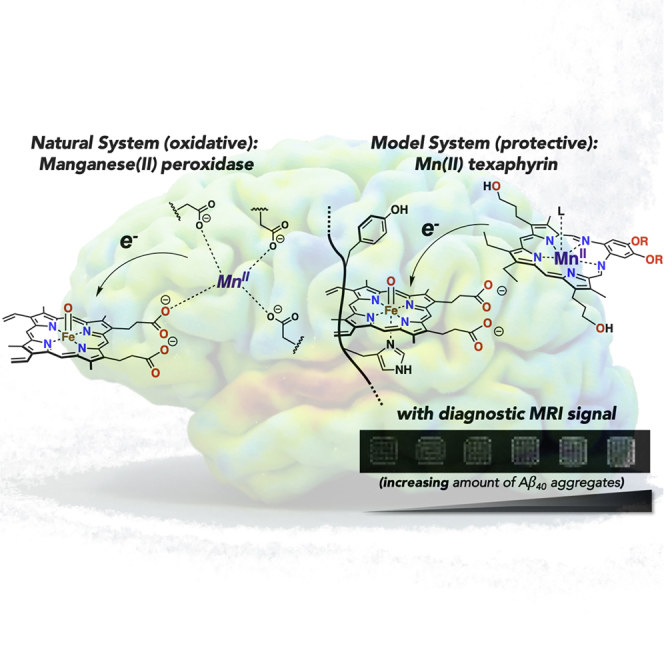
Sessler et al. have studied manganese(II) texaphyrin as a catalytic metalloantioxidant theranostic agent for the detection of Aβ aggregates. A series of in vitro and in vivo analyses allowed for the inference that metallotexaphyrins could be used for MRI-based detection of Aβ biomarkers. In vitro and cellular-based assays coupled with mechanistic analysis demonstrated an efficacious protective effect against oxidative and nitrative damage. The present results could thus guide future efforts in developing new theranostic modalities for use in neurodegenerative disease.
Introduction
Many seminal advances in physiology and medicine have been made by hewing to a reductionism-centered mantra wherein discrete biological functions are attributed to individual processes. However, an increased understanding of the complex interactions between cellular components (e.g., DNA, RNA, proteins, and small molecules) is leading to a more nuanced view.1 For instance, it is now recognized that a number of key biological effects arise from interaction webs involving multiple biochemical processes operating within a scale-free network.2 Such an appreciation is considered particularly relevant to the problem of understanding the central nervous system (CNS) and diseases that affect it.3
Among CNS disorders, neurodegeneration is particularly insidious. It defines a series of complex disease states that lead to death. Current therapies, at best, ameliorate the symptoms with limited long-term benefit to patients or relief to caregivers.4 The complex etiology of neurodegeneration derives from known and unknown initiators operating simultaneously within a feedback loop that is thought to exacerbate downstream toxicity.5 In particular, Alzheimer’s disease (AD) has been postulated to arise from abnormal protein deposits, excitotoxicity, disruption of metal-ion homeostasis, reduction in endogenous antioxidants, and neuroinflammation, among other hypotheses.6 Most, if not all, of these putative malfunctions are correlated with an increase in reactive oxygen species (ROS) and reactive nitrogen species (RNS).7 Both ROS and RNS are thought to disrupt standard cellular processes giving rise to dysfunction and neuronal cell death.8 Here, we report that a first-generation water-soluble manganese(II) texaphyrin (MMn) displays cell permeability and allows for the detection of larger order amyloid beta (Aβ) constructs (i.e., aggregates) by means of magnetic resonance imaging (MRI). Inductively coupled plasma-mass spectrometry (ICP-MS) and MRI analyses of in vivo (C. elegans) AD models provided further support for this notion. Based on in vitro mechanistic studies, this texaphyrin system also mitigates the oxidative and nitrative toxicity effects considered causal in CNS neurodegeneration. It thus shows promise as a new tool that may aid in understanding, imaging, and treating neurodegenerative disease.
Deciphering the precise biochemical deviations from a normal network that are associated with CNS neurodegeneration is considered key to finding a cure.9, 10, 11 Within this paradigm, significant efforts have focused on developing accurate biomarkers through cerebrospinal fluid (CSF) protein analysis and positron emission tomography (PET) imaging.12,13 These test methods have proven useful in furthering our understanding of disease progression; however, they can be invasive and expensive. MRI has emerged as a potential complement to CSF and PET analyses.14 Recent efforts have also focused on the use of theranostics, wherein a fluorophore, MRI-active metal ion, or radioisotope is conjugated with a therapeutic functionality.15 Many of these conjugates have relied on either Pittsburgh compound B (PiB)- or curcumin-based systems for imaging combined with an Aβ aggregation inhibitor to provide therapeutic efficacy. MR-based imaging systems for the study of neurodegeneration have generally relied on Gd-DOTA or related aminopolycarboxylate ligands conjugated to antibodies or other targeting moieties. Cell and blood-brain-barrier permeability remains a major issue in such agents and could represent a serious impediment to regulatory approval and ultimate clinical utility.16
Expanded porphyrins, porphyrin-like systems containing a larger internal cavity than natural tetrapyrrolic congeners, have received considerable attention in recent years.17 One such system is texaphyrin, a penta-aza Schiff base macrocycle.18 Compared with porphyrins, texaphyrins contain an approximately 20% larger core and display a unique electronic structure. These systems also exhibit efficacious biological activity with cell permeability and potential in human medicine.19 In fact, specific water-soluble gadolinium(III) and lutetium(III) texaphyrin derivatives, known as MGd and MLu, respectively, were explored early on as potential therapeutics; the pro-oxidative MGd was studied through phase III clinical trials as a radiosensitizer, and MLu was studied through phase I clinical trials for photoangioplasty.20, 21, 22 Notably, MGd demonstrated a high maximum tolerated dose (22.3 mg kg−1) and a median half-life of 7.4 h and could be detected within brain metastases even 14 h after administration, as determined by MRI. Recently, dual-modal MGd-platinum(IV) conjugates have shown promise as potential cancer drug leads.23
Mn(III) porphyrin and corrole metalloantioxidants have demonstrated an efficacious therapeutic effect that is thought to be mediated via decomposition of peroxynitrite (ONOO−) and, in some instances, superoxide and hydrogen peroxide (H2O2) (Figure 1).24 In particular, AEOL-10150, an octaethyl imidazolium Mn(III) porphyrin, has shown broad-spectrum superoxide dismutase (SOD)-like activity in animal models of amyotrophic lateral sclerosis (ALS), idiopathic pulmonary fibrosis, and nerve agent exposure (Figure 1).25 AEOL-10150 has also successfully completed a phase I clinical trial in ALS and healthy patients.26 Recent studies involving a water-soluble Fe(III)-corrole have also demonstrated utility in attenuating Aβ-Cu(II)-mediated ROS.23 However, the precise therapeutic mechanism remains recondite as Fe(III)- and Mn(III)-porphyrin and corrole complexes also catalyze oxidative transformations (e.g., peroxidase activity).27,28 Limited cellular uptake and acute toxicity in the case of polycationic pyridinium Mn porphyrins are also an issue.
Figure 1.

Chemical Structures of Representative Porphyrinoids
As discussed in the text proper, hemin catalyzes oxidative and nitrative damage. Manganese(II) texaphyrin (MMn) is the subject of this study. Manganese(III) tetramethylpyridinium porphyrin (MnTMPyP) is a manganese porphyrin metalloantioxidant. AEOL-10150 has successfully completed a phase I trial in ALS and healthy patients and has shown efficacy in animal models for radiation- and nerve-agent-induced toxicity. Axial ligands are omitted for clarity. See main text for further discussion of these agents.
The interaction of hemin and other water-soluble phthalocyanines, corroles, and porphyrins with Aβ has been noted.29, 30, 31, 32 We thus envisaged that coupling the Aβ-recognition properties of metalloporphyrins with the biological activity of metallotexaphyrins would allow for MRI recognition of Aβ species with attendant utility as a potential metalloantioxidant therapeutic (Figure 2). As detailed further below, we have found that metallotexaphyrins, in particular MMn, interact with amyloid constructs, as inferred from ultraviolet-visible (UV-vis), fluorescence, and circular dichroism (CD) spectroscopies, as well as MRI studies. The protective effect of MMn against oxidative and nitrative damage was also demonstrated by means of in vitro studies with Aβ peptides and serotonin as model protein and small-molecule neurotransmitter substrates, respectively, and in cell culture with Neuro-2A cells. The protective and oxidative properties of Mn(III) tetramethylpyridinium porphyrin (MnTMPyP), a gold-standard Mn porphyrin metalloantioxidant, were also studied in vitro and in Neuro-2A cell cultures. These studies revealed rapid decomposition of MnTMPyP under oxidative conditions consistent with a sacrificial substrate-based antioxidant mechanism. Studies involving C. elegans models were also carried out with MMn and provide support for the notion that this first-generation metallotexaphyrin may be administered and detected in amyloid plaque-bearing AD models. It and related systems could thus have a role to play in the study and eventual management of neurodegenerative disease.
Figure 2.

The Aggregation State of Aβ Peptides as a Function of Incubation Time
The three Aβ40 constructs utilized in this study: (left) freshly dissolved peptide is predominantly monomeric in solution, (middle) soluble oligomeric species (partially aggregated) are produced after incubation in solution, and (right) fibrils and fully soluble and insoluble aggregates are formed after prolonged incubation. Aβ16 remains monomeric in solution.
Results and Discussion
Spectroscopic Analyses of Metallotexaphyrin-Aβ Interactions
Initial studies of the interactions between MMn and Aβ40 involved UV-vis spectroscopic analyses. This technique was chosen because changes in the intensity or wavelength (energy) of the Soret band (π-to-π∗ transition) of metalloporphyrinoids are often indicative of axial coordination.33 Reduction in the molar absorptivity can also arise from coordination within a peptide or protein tertiary structure.34 MMn, like other porphyrinoids, is intensely colored. It is dark green in most media in which it is soluble and characterized by strong Soret- and Q-like bands at λmax = 459 and 726 nm, respectively, in pH 7.4 phosphate-buffered saline (PBS). The addition of monomeric Aβ40 (5 μM, 1 equiv) into a PBS solution of MMn (5 μM) resulted in a decrease in spectral intensity for the Soret-like band (ε = 73,700 → 62,500 M−1cm−1; λmax = 459 nm) (Figure S4). Increasing the ratio of Aβ40 led to an enhancement in the molar absorptivity (ε = 72,450 M−1cm−1 at 20 equiv) with a slight spectral shift in the absorption maximum also being seen (λmax = 462 nm; Figure 3). UV-vis spectral analyses provided support for the conclusion that the Aβ40 peptide (ε = 1,480 M−1cm−1 at 280 nm) remains soluble upon addition to MMn. Specifically, no sign of precipitation was observed, even when tested at high concentrations of Aβ40 (i.e., 100 μM; Figure S5). A titration of MMn with human serum albumin (HSA) showed a similar reduction in absorbance intensity but yielded no shift in the Soret or Q bands (Figure S6). Considered in tandem, the changes to the UV-vis spectral features of MMn observed in the presence of monomeric Aβ40 are consistent with the coordination of one or more axial ligands to the texaphyrin metal center. However, the potential formation of a soluble, higher-order Aβ40 species (e.g., a dimeric-MMn complex) in solution cannot be excluded. Under these same conditions, a titration of MnTMPyP, a commercially available porphyrin considered as a gold-standard metalloantioxidant, with Aβ40 yielded no change in the observed spectrum (Figure S7). Similar spectral changes to the texaphyrin profile were, however, seen when Aβ40 was added to MGd (Figure S7). The marked differences between MMn (and MGd) and MnTMPyP when exposed to Aβ40 are believed to arise from differences in the structure and charge of the overall complex. MnTMPyP is decorated with four pyridinium functionalities positioned perpendicular to the porphyrin core, whereas MMn is formally monocationic, planar, and functionalized with flexible pendant substituents capable of hydrogen bonding.
Figure 3.

Interaction of MMn with Monomeric and Aggregated Aβ40
(A) Titration with MMn (5 μM) and monomeric Aβ40 with 0 (black), 1 (light blue), and 20 (purple) equiv and (B) photos of aggregated Aβ40 (100 μM; aged 2 months) in the absence and presence of MMn (100 μM) in PBS and after washing with PBS. The pellet shown in the right-most image was obtained by centrifugation and is in methanol.
Since Aβ16 remains monomeric in solution and conserves the relevant amino acids considered responsible for Aβ coordination chemistry, an analogous titration was carried out between MMn and Aβ16 (0–80 equiv) in an effort to differentiate between the effects of aggregation (hydrophobic interactions) and axial coordination on the observed UV-vis spectral changes. UV-vis spectra of MMn recorded as it was exposed to Aβ16 were characterized by an initial reduction in spectral intensity, just as was observed in the corresponding Aβ40 studies. This decrease was followed by a corresponding increase in spectral intensity. In analogy to what was seen in the case of monomeric Aβ40, approximately 17.5 equiv of Aβ16 (as opposed to 15 equiv for Aβ40) were required to reach an absorbance plateau. Subsequently, no further discernible increase or change in the Soret band (λmax) was observed (i.e., upon addition of >20 equiv Aβ16; cf. Figure S8). The spectral changes induced by Aβ16 and monomeric Aβ40 were thus attributed to axial ligand binding rather than sequestration by oligomeric or aggregated peptide species.
MMn was also titrated with Aβ40 (0–20 equiv) that was pre-incubated for 14 days in PBS at room temperature. This pre-incubated material is believed to contain higher-order oligomeric species (i.e., partial aggregates containing β sheets; cf. Figure 2). Treating MMn with oligomeric Aβ40 (1 equiv) engenders a decrease in the UV-vis spectral intensity (ε = 73,700 → ε = 45,600 M−1cm−1), a finding attributed to both axial ligand binding and incorporation of MMn within the aggregate structure. Upon adding additional oligomeric Aβ40 (up to 20 equiv), an increase in spectral intensity (ε = 50,000 M−1cm−1) and a bathochromic shift in the Soret band maximum (to λmax = 462 nm) were observed (Figure S9). Analyses of the relative spectral intensity of the Soret and Q bands (ratio of absorbance at 459 and 726 nm) revealed a minute change in this ratio for aggregated (3.39:1) and monomeric (3.43:1) forms of Aβ40. This was taken as further support for the suggestion that the UV-vis spectral changes seen for MMn upon exposure to Aβ40 originate from both axial coordination to the Mn(II) center and texaphyrin encapsulation within Aβ40 oligomers, fibrils, and aggregates.
To gain insights into the interactions between MMn and fully aggregated Aβ species, we mixed constructs composed of Aβ40 that had been aged for 2 months (a highly aggregated form) with MMn (100 μM, 1 equiv). After centrifugation, a green pellet was obtained. Even after washing and vortex mixing with deionized (DI) water (three times) and resuspension in methanol, the pellet remained green (Figure 3). A time-dependent UV-vis analysis of the spectral changes seen for MMn when exposed to this same aggregated Aβ40 species was carried out by following the decrease in the absorbance intensity of the Soret band (λ = 459 nm) as a function of time. Upon addition, an immediate decrease was observed. A further reduction in the absorbance intensity was then seen over the ensuing 30 min (Figure S10).
A competition experiment involving HSA and monomeric Aβ40 was also carried out in an effort to discriminate between selectivity for Aβ40 versus general biomolecule interactions. The first addition of Aβ40 (25 μM) to a preformed MMn (5 μM)-HSA (5.5 μM) complex induced a slight decrease in the absorption intensity of the Soret (5.8%) and Q (6.3%) bands. Subsequent addition of Aβ40 (25 μM) again resulted in a further decrease in the absorption intensity of the Soret (4.6%; 10.4% total) and Q (6.8%; 13.1% total) bands with concomitant bathochromic shifts in the Soret band from 459 to 460 nm and Q band from 726 to 728 nm (Figure S11). Increasing the concentration of Aβ40 (125 μM) and HSA (600 μM) yielded a green pellet by centrifugation in the presence and absence of HSA after the induction of Aβ fibrillization through storage at 4°C for 48 h (Figure S12).35 However, the MMn-Aβ40 pellet formed in the presence of both HSA and Aβ40 was smaller (ca. 0.2 cm) than that formed when MMn was treated with just Aβ40 (ca. 0.35 cm). No pellet was formed from MMn in the absence of Aβ40.
Further attempts to probe the interactions between MMn and oligomeric Aβ were made with an Aβ40 (aged 1 week) Thioflavin T (ThT) assay.36 The benzothiazole scaffold for ThT is a recognized binding agent for Aβ sequences conserved among many PET agents (e.g., the FDA-approved Vizamyl [flutemetamol (18F)]) and theranostics used in AD research.37 New small molecules with binding affinities similar to that of ThT could thus prove useful for clinical application.30 As important, the ThT assay can provide evidence of binding within Aβ40 because of the well-defined ThT-Aβ40 emission profile.36 Changes in the observed fluorescence correspond to ThT displacement or structural modification of the Aβ40 aggregate. To exploit this assay, we added MMn (1 to 10 equiv) in aliquots to Aβ40 (aged 1 week) containing ThT (ThT-Aβ40 constructs). These additions engendered a series of changes, including spectral shifts and an overall decrease in the intensity (Figure S13). Through-space quenching of bound or free MMn, as a result of the matched absorption spectrum of MMn and emission profile of ThT-Aβ40 constructs, accounts for some of the initial decrease (Figures S14 and S15). Continued modification of the fluorescence profile was seen upon the addition of each aliquot of MMn, a finding consistent with the presence of multiple binding sites within the Aβ40 aggregate. The observed modifications to the fluorescence profile are thus largely attributed MMn-induced structural reorganization that is triggered by binding to aggregated Aβ40.
We carried out high-performance liquid chromatography (HPLC) analyses in an effort to elucidate further the interactions between ThT (100 μM), MMn (100 μM), or MLu (100 μM) and 2-month-aged Aβ40 aggregates (100 μM) (Figure S16). After incubation at room temperature for 24 h, 77% ThT remained in the supernatant, as determined by integration against a ThT control (100 μM). Co-incubation with MMn or MLu (24 h at room temperature) effected little significant displacement given that 66.7% ThT and 7% MMn remained in the supernatant, as determined by HPLC analysis using ThT and MMn standards. A similar HPLC study using the diamagnetic MLu revealed that only 5.4% of the MLu remained in the supernatant (Figure S17). NMR spectroscopic experiments carried out with preformed ThT-Aβ40 samples (ThT [0.47 mM] and Aβ40 [0.40 mM]) in conjunction with MLu (0.49 mM) revealed signs of ThT displacement after incubation for 24 h at room temperature (Figures S18 and S19). Additional NMR spectroscopic analyses using monomeric Aβ40 (1.9 mM) and MLu (2.1 mM) in DMSO-d6 revealed changes to the proton signals ascribed to MLu and the Aβ40 peptide. The most notable difference after the addition of Aβ40 was a change in the chemical shift for the axial acetate anions originally bound to MLu; this is as would be expected under a scenario where the Aβ coordinates the metallotexaphyrin (Figure S20). In concert, these findings are taken as evidence that metallotexaphyrins bind well to aggregated Aβ, most likely through multiple binding sites, even if the specifics of the interaction are not fully elucidated.
Clinical trials aimed at the modification of higher-order oligomeric and aggregated Aβ have yet to yield viable therapeutics.11 An ability to produce and detect such modifications remains of interest in terms of assessing the potential impact of other treatment modalities. CD studies designed to assess the ability of MMn (20 μM) to interact with and modify the aggregate structure of Aβ (20 μM, aged 30 days) revealed no marked differences in the CD spectrum immediately after mixing. However, noticeable changes relative to the initial CD spectrum were observed 7 and 24 h after mixing (Figure 4). In particular, the intensity of the negative CD band at 220 nm seen for aggregated Aβ40 underwent a decrease in intensity of approximately 35% at 24 h with an attendant shift in the wavelength minimum to ca. 218 nm. These data are interpreted in terms of a reduction in the percentage of β sheet composition and modification of the overall aggregate structure.38 Control studies monitoring the progression over 24 h with aged Aβ40 in the absence of MMn revealed no significant change (Figure 4).
Figure 4.

Modification of Aggregated Aβ40 by MMn as Monitored by CD Spectroscopy and SEM Analyses
CD spectra (left) of aggregated (aged for 30 days) Aβ40 (40 μM) were recorded before and after the addition of MMn (20 μM) at t = 0, 7, and 24 h. SEM photographs (right) of (A) Aβ40 aggregate, (B) an Aβ40 aggregate incubated with MMn for 24 h, and (C) monomeric Aβ40 are shown.
Scanning electron microscopy (SEM) provided further support for the proposed modification of the Aβ40 aggregate structure (Figure 4). The Aβ40 aggregate (40 μM) utilized for CD experiments (aged 30 days plus 24 h) was characterized by fibril structures, observed at 5 μm (Figure 4A). In contrast, the Aβ40 aggregate (aged 30 days) incubated with MMn (20 μM) for 24 h displayed features at 3 μm more closely resembling those of an amorphous species (Figure 4B). Freshly prepared Aβ40 showed no discrete features even when magnified to 2 μm (Figure 4C).
In Vitro and In Vivo MRI
As a general rule, paramagnetic Gd(III) and high-spin Mn(II) ions enhance the relaxation rates of nearby proton nuclei. Not surprisingly, these cations have been incorporated into MRI contrast agents (CAs).39 Gd-based systems remain the current clinical standard. However, Mn-based CAs have recently been explored as surrogates for Gd systems and as new intramolecular MRI probes.40,41 Detailed MRI studies on MGd have previously been reported,42 including human trials where evidence of tumor-targeted imaging via enhanced drug uptake within tumor cells was obtained.18,19 However, the congeneric Mn(II) system MMn has yet to be the subject of extensive study in the context of MRI.43,44
MRI CAs are generally described by relaxivities, r1 and r2, in mM−1 s−1. These parameters are defined as the concentration-dependent enhancement of longitudinal (R1) and transverse (R2) water proton relaxation rates, each given in s−1, respectively. At the field used in this study (7 T), MMn displayed exceptional relaxivities in non-coordinating buffers (7 T, HEPES [pH 7.4], 25°C; MMn r1 = 4.59 ± 0.12 mM−1 s−1 and r2 = 32.2 ± 0.3 mM−1 s−1; MGd r1 = 16.0 ± 0.5 mM−1 s−1 and r2 = 23.5 ± 0.6 mM−1 s−1; Gd-DTPA r1 = 4.93 ± 0.06 mM−1 s−1 and r2 = 6.65 ± 0.02 mM−1 s−1) with well-behaved (i.e., linear) relaxation rates as a function of concentration. The longitudinal relaxivity of MMn in the coordinating buffer (i.e., PBS) was slightly reduced at r1 = 1.2 mM−1 s−1. Current CAs are known to be limited at these higher fields and previous examples of Aβ-binding MRI agents have, in general, been investigated at a lower magnetic-field strength.39,40,45 The ability of MMn to engender high relaxivities at a higher field could make such agents of clinical interest in the context of neurodegenerative disease. Studies with Aβ peptides were carried out as a first step toward exploring this potential.
Changes in the relaxation rates (R1 and R2) after addition of monomeric and aggregated Aβ40 (70 μM) to solutions of MMn (70 or 140 μM) at 25°C in PBS (pH 7.4) were monitored at 7 T with a multi-spin multi-echo scanning sequence. Bovine serum albumin (BSA, 70 μM) was used as a control biomolecule (Figure 5). All of the additives tested gave rise to an increase in the longitudinal relaxation rates of MMn. Control studies demonstrated that Aβ40 alone displayed no significant effect on the relaxation rate of the buffer (R1 = 0.400 ± 0.005 s−1 versus the PBS control R1 = 0.398 ± 0.005 s−1). Relative to the relaxation value generated by MMn alone, aggregated Aβ40 induced the largest increase in R1 (Figure 5A). The modest increase observed for monomeric Aβ40 and albumin coupled with the observed enhancement in the R1 parameter in the case of aggregated Aβ is taken as a further indication that such agents could find utility in the MRI-based detection of Aβ aggregate constructs. Interestingly, with the fully aggregated Aβ40, the R1 increase was substantially larger for 2 equiv of MMn (ca. 62% versus MMn alone) than for 1:1 MMn/Aβ40 (ca. 37% versus MMn alone) (Figure S21). Considered in light of the ThT-Aβ40 fluorescence assay results described above, these data are taken as a further indication that MMn binds to multiple sites within aggregated Aβ40. The MRI properties of Gd(III) texaphyrin (MGd, 70 μM) in the presence of aggregated Aβ28 (500 μM) and BSA (600 μM) were also monitored at 7 T with a multi-spin multi-echo scanning sequence at 25°C in PBS (pH 7.4). However, these studies failed to show selectivity or significant changes in either R1 or R2 in either case (Figure S22).
Figure 5.

In Vitro MRI Analyses of MMn with Various Aβ40 Constructs
(A) The longitudinal relaxation rate (R1) of MMn (140 μM) with monomeric (0.5 equiv, 70 μM), partial (1 week, 0.5 equiv, 70 μM), and aggregated (>2 months) Aβ40 (0.5 equiv, 70 μM) in comparison with MMn alone (140 μM) and in the presence of BSA (0.5 equiv, 70 μM). Dots represent individual samples; the mean (red line) and 95% confidence level for the mean (light red) are also displayed. Standard deviations are either indicated in blue or not shown if within the 95% confidence level.
(B) Phantom T1-weighted MRI images of 140 μM MMn were recorded in the presence of increasing concentrations of Aβ40 aggregate (0–70 μM) in PBS at 7 T and 25°C. Samples are normalized to MMn alone, shown in the left-most box.
Access to the intracellular intrinsically disordered proteins and peptides (IDPs) remains difficult for most MRI CAs and often comes at the cost of desirable physicochemical properties (e.g., LogP and solubility).16,40,41 Cell-uptake studies using MMn (30 μM), LogP 0.69, and the Neuro-2A cell line with a 9 h incubation period produced a green cell pellet with a 5.5-fold increase in the Mn content (2.2 ppb per million cells) versus the no-drug control (0.4 ppb per million cells), as quantified by ICP-MS analysis (Figure S23). MRI of the resultant cell pellet suspended in PBS provided additional support for the proposed increase in uptake given that both R1 (from 0.382 ± 0.004 to 0.412 ± 0.004 s−1) and R2 (from 2.43 ± 0.03 to 2.71 ± 0.03 s−1) were enhanced in comparison with the no-MMn control.
In order to probe the possible utility of MMn for in vivo MRI-based detection of neurodegenerative disease, we incubated an AD model of C. elegans (CL4176) expressing amyloid plaques in the presence of MMn (1 mL/100 μM).46 The resulting MMn content, measured by MRI and ICP-MS, was compared to what was seen in the case of the wild-type strain, N2. On the basis of reported drug absorption and retention within C. elegans, we envisaged that MMn would interact and be retained within the amyloid plaques, yielding higher MRI and ICP-MS signals than the control.47 We also used extended incubation times to allow for multiple uptake and excretion cycles. Control experiments using E. coli OP50 with MMn and worms without MMn were carried out concurrently with the experiments involving the N2 and CL4176 strains (Figure S24). MRI studies on the resulting pellets suspended in HEPES buffer (10 mM [pH 7.4], 1 mg pellet per 100 μL HEPES) showed an enhanced R1 signal in the CL4176 AD model (0.394 ± 0.012 s−1) when they were administered MMn versus the N2 treated with MMn in an equivalent way (0.360 ± 0.002 s−1), as well as no-MMn CL4176 (0.364 ± 0.003 s−1), N2 (0.344 ± 0.003 s−1), and E. coli with MMn control studies (0.333 ± 0.002 s−1). A similar trend was also seen in the case of the R2 with relevant values: CL4176 AD model with MMn (3.44 ± 0.20 s−1), N2 with MMn (2.82 ± 0.03 s−1), no-drug CL4176 (3.14 ± 0.02 s−1), no-drug N2 (2.59 ± 0.10 s−1), and E. coli with MMn control (2.450 ± 0.04 s−1) (Figure S25). A second batch displayed a similar trend (Figure S26).
As expected, both batches of C. elegans incubated with MMn showed higher Mn signal in the N2 and CL4176 strains than did the same strain without the texaphyrin (Figure 6). ICP-MS analyses of pellet suspensions (1 mg C. elegans per 100 μL HEPES) revealed Mn uptake values of 82 ± 11 μM (SD) for the CL4176 AD model and 48 ± 11 μM for the wild-type N2 (Figure S27). The increased metal uptake in the AD model is taken as evidence of the accumulation of MMn within the amyloid plaque over multiple uptake-drug-excretion cycles.
Figure 6.

ICP-MS and In Vivo MRI Analyses of MMn in C. elegans AD Models
Mn concentrations (A), as determined by ICP-MS, and (B) MRI analyses of MMn for pellets of wild-type (N2) and AD model (CL4176) C. elegans with and without MMn. Pellets were standardized by dissolving every 1 mg C. elegans in 100 μL HEPES and digesting in HNO3. n = 5–8 per sample, 2 replicates of each for ICP-MS measurements and n = 2–5 for MRI measurements. Dots represent individual samples; the mean (red line) and 95% confidence level for the mean (light red) are shown. Standard deviation (blue) is shown where larger than the 95% confidence level. *Two-sample t test on ICP-MS variance between MMn-wild-type (WT) and -AD models; p = 0.00018.
In Vitro ROS and RNS Reactivity Studies
Nitrative and oxidative damage is prevalent in AD brains.48 Oxidized and nitrated neurotransmitters, DNA bases, proteins, metalloenzymes, and cellular membrane components are also correlated with disease progression. These chemical changes are thought to alter up- and downstream biochemical processes via inactivation, hyperactivation, and off-target effects.49,50 Recent studies have thus led to the suggestion that oxidation and nitration of the amyloid peptide and tau proteins leads to enhanced cytotoxicity with attendant neuronal cell death.51,52 MMn is known to display distinct redox behavior in comparison with its related Mn(III)porphyrin and corrole congeners.53 Previous work on MMn has also illustrated the possibility of deactivating ONOO− catalytically in the presence of sodium ascorbate (NaAsc) with attendant application in preventing formation of nitrotyrosine in ALS mice models.54,55 The propensity of metallotexaphyrins to interact with Aβ constructs coupled with the unique redox capabilities of MMn led us to explore whether this specific metallotexaphyrin could be used to mitigate the effects of hemin-mediated oxidation and nitration and copper-mediated oxidation of small-molecule neurotransmitters and amyloid peptides. The pro-oxidative and protective (antioxidant) activity of MnTMPyP has also previously been reported.24,56 It was thus studied for comparative purposes.
Serotonin was used as the first test substrate. When hemin is activated in the presence of hydrogen peroxide, serotonin undergoes an oxidative transformation to yield the neurotoxic 4,4′-serotonin dimer (5,5′-dihydroxy-4,4′-bitryptamine [DHBT]). The antioxidant (protective) effect of MMn was studied with serotonin (1 mM) with hemin (15 μM) and an oxidative mixture consisting of hydrogen peroxide (1 mM) and NaAsc (40 μM) in PBS (pH 7.4) at 37°C for 2 h. By comparing the changes observed in the corresponding 1H NMR spectra in D2O, we could compare the antioxidant effect of MMn against that of MnTMPyP, a well-studied Mn(III)-porphyrin metalloantioxidant, and the pro-oxidative hemin control (Figure 7). Under these conditions, MMn proved the most protective, leaving behind 63% of unmodified serotonin. In contrast, the hemin/H2O2/ascorbate and hemin/H2O2/ascorbate/MnTMPyP mixtures showed a marked increase in serotonin oxidation with 36% and 4% remaining serotonin, respectively. An analogous experiment using hemin (15 μM) and H2O2 (1 mM) without NaAsc revealed that MMn was protective, yielding 70% of unmodified serotonin versus 60% and 40% remaining for hemin/H2O2 and hemin/H2O2/MnTMPyP, respectively (Figure S28). Additional NMR spectroscopic experiments designed to evaluate the protective effect of MMn against Cu(II), another metal ion considered relevant to oxidation in CNS disorders, were also carried out (Figure S28). Qualitative 1H NMR spectroscopic analyses using Cu(II)Cl2 (100 μM), serotonin (1 mM), and H2O2 (1 mM) without and with MMn (120 μM) revealed that prolonged incubation times and high concentrations of the metal salt were necessary for producing low levels of oxidation. Shorter reaction times and lower concentrations of Cu(II) led to no detectable oxidation, as inferred from 1H NMR spectral studies.
Figure 7.

1H NMR Spectroscopic Study of the Hemin-Mediated Oxidation of Serotonin
NMR yields were calculated by comparing the spectra of (A) serotonin (1 mM) in pH 7.4 PBS at 37°C in the presence of (B) hemin (15 μM) and an oxidative mixture containing H2O2 (1 mM) and sodium ascorbate (40 μM), (C) an H2O2-ascorbate oxidative mixture plus MnTMPyP (120 μM), and (D) an H2O2-ascorbate oxidative mixture plus MMn (120 μM) recorded after a reaction time of 120 min in all cases.
MMn also proved efficient at deactivating the oxidative modification of histidine (H13-H14), nitration of tyrosine (Y10), and oxidative dimerization of tyrosine (Y10) in Aβ16 by the more reactive hemin, as determined by high-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) analysis. The Aβ16 sequence serves as a soluble surrogate for larger amyloid species or other proteins that could suffer degradation when treated with the oxidative mixture of hemin-hydrogen peroxide (15 μM-1 mM) or copper(II) (15, 60, or 150 μM) with hydrogen peroxide (1 mM) or nitrative mixtures of hemin-hydrogen peroxide-sodium nitrite (15 μM-1 mM-1 mM) and hemin-ONOO− (15 μM-1 mM) at 37°C in phosphate buffer (pH 7.4, 5 mM). A range of reaction variables—such as the use of acidic pH thought relevant to neurodegeneration,57 solution ionic strength (5 versus 50 mM), reaction time (2–24 h), and the presence or absence of MnTMPyP, MGd, and curcumin—were also studied in the context of hemin-mediated oxidation and nitration of Aβ16 (Figures S29–S32). Within the range of a normal physiological pH of 7.4 in low-ionic-strength 5 mM phosphate buffer, only 25.1% ± 2.1% (SD) of the Aβ16 remained in the case of the additive-free control, composed of Aβ16 (300 μM), hemin (15 μM), H2O2 (1 mM), and NaNO2 (1 mM). MMn was found to militate against oxidation and nitration of Aβ16 with an IC50 = 4.22 μM and a maximal inhibition of 97.4% ± 0.4% at 60 μM MMn under nitrative conditions (Figure 8A). A similar protective effect by MMn was also seen under oxidative conditions of Aβ16 (300 μM), hemin (15 μM), H2O2 (1 mM), and MMn (30 μM), yielding 98% ± 0.4% of unchanged Aβ16. Control experiments without MMn, with MGd, and with the sacrificial antioxidant curcumin gave rise to 83.5% ± 5% (Δ = −13.9%), 77.5% ± 5.5% (Δ = −19.9%), and 84.5% ± 2% (Δ = −12.9%) of unchanged Aβ16, respectively. At a more acidic pH of 6.3 and the higher ionic strength (i.e., 50 mM phosphate buffer), under nitrative conditions, the amount of remaining Aβ16 (300 μM) decreased from 63.5% ± 1.4% at pH 7.4 to 33.3% at pH 6.3 (heme-H2O2-NaNO2; 10 μM-1 mM-1 mM). However, the protective effect of MMn (30 μM) was enhanced such that 98.8% ± 0.6% of the Aβ16 remained intact. This increased benefit is believed to arise from deactivation rates that are accelerated in more acidic media. The results of the Cu(II)-H2O2 experiments proved difficult to quantify. This difficulty reflects the minimal oxidative reactivity of Cu(II) at levels appropriate for HPLC-MS analysis (Figure S33). Under conditions analogous to those tested in the case of hemin, Cu(II) (15 μM) with H2O2 (1 mM) incubated at 37°C for 2 h in phosphate buffer (pH 7.4, 5 mM) returned 96.9% ± 1.7% of unchanged Aβ16. The addition of MMn (30 μM) to Aβ16 (300 μM), Cu(II) (15 μM), and H2O2 (1 mM) yielded 98.7% ± 0.4% unchanged Aβ16.
Figure 8.

HPLC-MS Analyses of the Oxidative and Nitrative Modification of Aβ16
(A) Aβ16 incubated under hemin-nitration conditions using heme-H2O2-NaNO2 with varying concentrations of MMn. Percent composition of Aβ16 is shown in black and, as expected, increases with the [MMn].
(B) The protective effect of MMn was also tested against heme-peroxynitrite-mediated modification of Aβ16 in comparison with MnTMPyP. Sodium ascorbate (NaAsc) was added to regenerate the MMn catalyst under the reaction conditions. Reported values are averages with the corresponding standard deviation as obtained from two independent measurements.
It is worth stressing that although it promotes the oxidation of serotonin, the gold-standard control system, MnTMPyP, was found to be protective against the oxidation and nitration of Aβ16. In particular, MnTMPyP showed an excellent protective effect against oxidative modification by yielding 97.4% ± 0.6% unmodified Aβ16. MnTMPyP also proved protective against nitrative modification by yielding 88.7% ± 0.6% unmodified Aβ16 (Figure S34). These are both similar to the values observed for MMn (98% ± 0.4% for oxidation and 91% ± 0.1% for nitration). The observed protective effect of MnTMPyP is thought to arise from rapid oxidative degradation of hemin and MnTMPyP via the iron- and Mn-oxo porphyrin intermediates. This loss in complex arising from MnTMPyP serving as a sacrificial antioxidant would, in turn, we propose, preclude extensive oxidation or nitration of this amyloid peptide (i.e., Aβ16) during the time course of the 2 h reactivity study. UV-vis analyses provided further support for the notion that MnTMPyP (5 μM) serves as a sacrificial substrate in the presence of hemin (15 μM) and H2O2 (1 mM) given that only 13% MnTMPyP and 60% hemin remain after incubation for 10 min at 37°C in phosphate buffer (pH 7.4) (Figure S35). A control experiment with hemin alone under the same conditions yielded 56% unmodified hemin.
Reactivity studies designed to test the protective effect against ONOO−-mediated damage of Aβ16 were also carried out. Here, a mixture of hemin (15 μM), Aβ16 (300 μM), and ONOO− (1 mM) were incubated at 37°C for 30 min in phosphate buffer (5 mM, pH 7.4) and yielded 0.2% ± 0.1% unmodified Aβ16. In the presence of MMn (30 μM) with NaAsc (500 μM) as the reductant, 82.6% ± 3.7% of the original Aβ16 remained. Notably, in the presence of MnTMPyP and NaAsc, only 0.4% Aβ16 was observed intact under otherwise identical conditions (Figure 8B). We attribute this lack of protection for MnTMPyP to the rapid oxidative action of ONOO− and formation of an oxidative high-valent Mn- or Fe-oxo species that could oxidize Aβ16 prior to decomposition.58 According to early mechanistic studies, such oxo species are not thought to be produced when MMn is used to deactivate ONOO− in the presence of NaAsc.54 Control experiments with NaAsc (500 μM), MMn (30 μM) alone (not containing a reductant to regenerate the active Mn(II) species), and MGd as a texaphyrin control containing a non-redox-active metal conferred little protection, such that 20.6% ± 20.4%, 1.6% ± 1.2%, and 1.6% ± 0.2% Aβ16 remained intact, respectively (Figure S32).
UV-vis spectroscopic studies revealed that upon addition of MMn (1 equiv, 10 μM) to the six-coordinate 2:1 low-spin Aβ16-heme (200-10 μM) complex, formed by the addition of an excess of Aβ16 to hemin, a hypsochromic shift in the absorption of the hemin-derived features from λmax = 412 to 409 nm occurred (Figure S36). Such changes can be ascribed to two possible mechanisms. First, upon addition of MMn, the reversion from 412 to 409 nm in the Soret band could reflect deconstruction of the 2:1 heme-Aβ16 construct to yield a mixed low- (2:1) and high-spin (1:1) Aβ16-heme species via MMn displacement of the heme bound to Aβ16.59 A separate mechanism could also be operable wherein MMn co-binds within the 2:1 Aβ16-heme construct to distrupt the Aβ axial coordination to hemin or modification of the secondary coordination environment responsible for the enhanced peroxidase activity. To such an extent that this latter rationale is correct, it would support a mechanism where MMn-mediated protection results from a proximity effect, as well as an attenuation of the heme peroxidase activity that is normally enhanced upon coordination of hemin to amyloid peptides.59 Both proposed mechanisms would be expected to induce changes to the UV-vis spectrum of the 2:1 hemin-Aβ16 complex.
A proposed mechanism for the deactivation of iron-oxo-mediated oxidation and nitration is shown in Scheme 1. In briefl, the hemin-Aβ complex is activated by H2O2 or ONOO− to yield a highly oxidative porphyrin π-radical cation/iron(IV)-oxo species (compound I in Scheme 1A).60 The resultant species readily oxidizes hydrogen peroxide or nitrite as well as tyrosine, histidine, or methionine residues in Aβ (producing compound II in Scheme 1). MMn could confer a protective effect by reducing compound I and/or compound II, resulting in the formation of an oxidized Mn(III) texaphyrin. In contrast to Mn peroxidase and certain model Mn complexes, such as Mn(III)TMPyP (vide infra), the resulting oxidized Mn(III) texaphyrin fails to carry out additional oxidative chemistry on the peptide or small redox-active molecules. The oxidized Mn(III) texaphyrin can then be reduced by ascorbate or oxidative decomposition of H2O2 to O2 and 2 H+ (Figure S37).54 Support for this H2O2-decomposition mechanism comes from UV-vis analyses. Complete reduction of Mn(III) texaphyrin (generated with ONOO−) was observed after the addition of 10 equiv H2O2 within 3 min at room temperature in PBS (Figure S38). A control experiment that involved monitoring the reduction of Mn(III) texaphyrin to Mn(II) texaphyrin without H2O2 yielded approximately 7% reduced Mn(III) after 20 min when left open to air at room temperature. Under the reaction conditions containing a large excess of H2O2 (1 mM) versus MMn (30 μM), it is expected that the H2O2 oxidation mechanism will dominate the catalytic cycle to regenerate Mn(II) texaphyrin. No oxidation of Mn(II) texaphyrin to the corresponding Mn(III) species was observed with H2O2 alone (Figure S39).
Scheme 1.

Proposed Mechanism for the MMn-Based Deactivation of Hemin- and Peroxynitrite (ONOO−)-Mediated Oxidation and Nitration, as well as Oxidative Decomposition of Hydrogen Peroxide (H2O2)
Redox cycle of Mn(II/III) texaphyrin. Compounds I and II are generated by the reaction of hemin with H2O2. The Aβ peptide and axial ligands are omitted for clarity.
As implied above, these proposed MMn-relevant mechanisms stand in contrast to what is thought to occur with Mn(III)TMPyP. Under oxidizing conditions, such as those employed in this study, the Mn(III) center of MnTMPyP is oxidized to give a Mn(IV)-oxo species that promotes self-decomposition and substrate oxidation. The mixed protective (antioxidant) and oxidative properties of MnTMPyP is thus attributed to variations in the respective redox profile of MnTMPyP versus various biological substrates.61 Indeed, in instances where the substrate is more prone to oxidative modification (e.g., serotonin or methionine), a pro-oxidative-type mechanism dominates.55 In cases where the substrate is less labile under oxidative conditions (e.g., histidine and tyrosine) and generation of the oxidative species is slow (i.e., hemin-hydrogen peroxide), MnTMPyP instead acts as an antioxidant engendering a protective effect as a sacrificial substrate. Under more strongly oxidizing conditions, such as hemin-ONOO−, MnTMPyP again fails to convey any protection. The observed toxic effect for MnTMPyP in in vitro cell culture could also reflect the release of free Mn as a result of the rapid oxidative decomposition of this particular porphyrin complex seen upon treatment with hemin and H2O2.61
Cellular Assays and Stability Studies
In vitro toxicity experiments with MMn using Neuro-2A revealed that MMn displays an IC50 of 95 μM after an 18 h incubation time at 37°C, as inferred from a standard MTT assay (Figure S40). MMn has been previously determined to be well tolerated in vivo, as deduced from studies involving 2.5 mg kg−1 daily injections in ALS G93A mice.55 Notably, in these same studies, HPLC analysis of Mn texaphyrin-treated mice (end of study) revealed lower levels of nitrotyrosine in the spinal cord than in untreated controls. MMn-based nanoparticles have also been evaluated in vivo by subjecting healthy female BABL/c mice (n = 5) to high-dose injections (10 mg kg−1).42 Again, good tolerability was seen.
In vitro stability studies in the presence of ascorbate, albumin, zinc, copper, and citric acid revealed good stability at 37°C over 24 h (Figure S41). In the presence of the oxidative mixture composed of hemin and H2O2, MMn displayed increased stability versus hemin and MnTMPyP when incubated at 37°C (Figure S42). MMn, MnTMPyP, and hemin all displayed moderate stability when incubated with ONOO− (2 equiv) at 37°C for 0.5 h with 80%, 84%, and 71% of these complexes, respectively, after the addition of excess NaAsc (Figure S43).
The cellular protective effect of MMn was demonstrated via MTT assays with Neuro-2A cells. Previous studies have shown that the cytotoxic effects of Aβ are dependent upon the aggregate structure (i.e., fibril, oligomer, and aggregate).62,63 Oxidative and nitrative modifications of the peptide alters its aggregation properties and can enhance cytotoxic pathways.64, 65, 66, 67 We thus sought to determine how partial oxidation and nitration of Aβ40, coupled with modification of the aggregated Aβ40 architecture, affects cell viability. The Aβ40 peptide was chosen since, together with Aβ42, it is one of the predominant Aβ species found within the AD brain. Aβ40 also displays a slower aggregation rate and lower toxicity than Aβ42, allowing for enhanced control in cell experiments. We assessed the protective effects of MMn and MnTMPyP by incubating Aβ40 for 3 h at 37°C in the presence of a nitration mixture containing hemin, H2O2, and NaNO2 in PBS. MGd was utilized as a negative control because of its ability to propagate oxidative damage.19,23 We then stored the reaction mixture at 4°C overnight to allow the formation of additional toxic fibrils35 before transferring it to wells containing Neuro-2A cells and subjecting it to incubation for an additional 24 h. In comparison to the unmodified Aβ40 not subjected to the nitration conditions (90.4% ± 2%), the mixed oxidized-nitrated Aβ40 was more toxic, resulting in only 64.1% ± 0.1% cell viability. In the presence of MMn (30 μM), the neuroblastoma Neuro-2A cell viability was increased to 85.5% ± 1.5%. MGd (30 μM) displayed cytotoxicity similar to that of the mixed oxidized-nitrated Aβ40 system, where only 58.25% ± 13% cell viability was observed (Figure 9). MnTMPyP (30 μM) also produced a lower level of cell viability (44.62% ± 10%). On the basis of these experiments, we conclude that MMn provides a protective effect against the oxidative and nitrative modifications that are thought to yield the neurotoxic fibril and oligomeric species that lead to cell death. In the event, the incubation with MMn translates into improved cell viability.
Figure 9.

Cellular (Neuro-2A) MTT Assay
Neuro-2A cell viability upon exposure to Aβ40 and mixed oxidized-nitrated Aβ40 (hemin-H2O2-NaNO2) with and without MMn, MGd, and MnTMPyP. Reported values are the average with standard deviation of three independent assays each tested four times. The Aβ40 “NO2” mix is an average of two independent assays each tested four times. *Two-sample t test on percent cell viability (MTT assay) between the Aβ40 “NO2” mix with and without MMn; p = 0.00016.
Qualitative HPLC-MS analyses were carried out in an effort to correlate further the level of Aβ40 oxidation and the observed survival effects seen in Neuro-2A (Figure S44). A mixture of Aβ40 (100 μM), hemin (15 μM), and H2O2 (1 mM) was incubated with or without MMn (30 μM), MnTMPyP (30 μM), or MGd (30 μM) in PBS at 37°C for 3 h. MMn conferred the greatest protective effect, yielding an approximately 1:1 ratio of oxidized (Aβ40-[O] to native Aβ40). This stands in contrast to the sample containing MGd, where the resultant HPLC trace showed a 1.7:1 ratio of Aβ40-[O]/Aβ40. Finally, MnTMPyP induced a high level of oxidative modification with an approximately 3:1 ratio of Aβ40-[O] to Aβ40 being seen. Tandem mass spectrometry (MS/MS) analysis proved consistent with oxidation occurring primarily at His13-14 and Met35. When considered in conjunction with the N2 cell data, the observed toxicity data are found to correlate with the levels of oxidative and nitrative modification in the case of the Aβ40 peptide. Previous studies have demonstrated that variations in cell toxicity are correlated with the Aβ40 aggregation state, meaning that they could be subject to potential modulation (i.e., enhanced or retarded toxicity) by porphyrinoid additives.30, 31, 32 MMn appears particularly effective in this regard.
Conclusions
In conclusion, we have demonstrated that metallotexaphyrins interact with Aβ peptides and aggregates and could hold promise for the MRI-based recognition of amyloid aggregates. The first-generation water-soluble MMn that is the subject of the present study also protects against oxidative and nitrative damage of small molecules and peptides and reduces Aβ-mediated cell death. MMn was also shown to permeate the cellular membrane and be taken up within C. elegans animal models of AD. The present results thus serve to highlight the potential of MRI-active metalloantioxidants as potential treatment- and diagnostic-related modalities for use in the diagnosis and management of neurodegenerative disease.
Experimental Procedures
Synthesis of Aβ16 and Aβ40
Aβ peptides were prepared via Fmoc amino solid-phase peptide synthesis with a Liberty Blue microwave peptide synthesizer. Preparative reverse-phase high-performance liquid chromatography (RP-HPLC) purification of peptides was performed with an Agilent Zorbax SB-C18 Prep HT column 21.2 × 250 mm. Analytical RP-HPLC characterization of peptides was performed with an Agilent Zorbax column 4.6 × 250 mm. An Agilent Technologies 6530 Accurate Mass QT of LC/MS was used for high-resolution mass spectra of purified peptides. Solvents used were HPLC grade.
Synthesis of MGd, MMn, and MLu
Motexafin gadolinium (gadolinium(III) texaphyrin [MGd]) was a gift from Pharmacyclics Inc., recently acquired by Abbvie. MLu and MMn were synthesized according to literature protocols.53,68
Cell-Uptake Studies
Neuro-2A cells were grown until confluent in T-75 flasks. MMn was added (100 μM) and cells were incubated for 9 h, upon which cells were washed twice with PBS, incubated with trypsin (1×) for 2 min, diluted with supplemented Eagle's minimal essential medium (EMEM), counted, and pelleted by centrifugation (3 min at 2,000 rpm). After supernatant removal, the pellet was suspended and washed one more time with PBS (5 mL), and the pellet was reformed by centrifugation (3 min at 2,000 rpm). The cell pellet was digested in concentrated nitric acid (100 μL) for 15 h at 60°C and diluted with miliQ water to a final concentration of 2% HNO3 before being analyzed by ICP-MS.
C. elegans Culture
The C. elegans strains N2 and CL4176 were provided by CGC (nematodes shipped on 35 mm diameter plates), which is funded by the NIH Office of Research Infrastructure Programs (P40-OD010440). C. elegans were grown and maintained as described in “WormBook: The Online Review of C. elegans Biology” by Theresa Stiernagle (https://www.ncbi.nlm.nih.gov/books/NBK19649/).
C. elegans MMn Uptake
After the C. elegans animals, N2 and CL4176, were received from the CGC (University of Minnesota) on petri dishes, agar matrix was cut in small chunks (0.5 × 0.5 cm) with a razor blade and transferred on nematode growth medium (NGM) agar plates previously seeded with E. coli strain OP50 as a food source. NGM plates were then placed at 16°C in order to increase the population of C. elegans before drug exposure and prevent significant plaque formation in the CL4176 AD model. After 7 days, MMn was added to plates (1 mL per plate of a solution at 100 μM), which were incubated at 25°C for 48 h. The animals were then transferred to Eppendorf tubes (previously weighted empty on an analytical balance) with 1.5 mL of water. The tubes were put on ice for 5 min and centrifuged (14,000 rpm for 1 min), and the supernatant was removed. The plate was washed again with 1.5 mL of water and centrifuged again. The resulting pellets were washed twice with 1 mL of DI water. C. elegans pellets were lyophilized over a period of 15 h before being weighed (dry pellet weights obtained by lyophilization were used to normalize the MRI and ICP-MS results).
Preparation of Peroxynitrite
Three separate solutions of NaNO2 (0.6 M), HCl (0.68 M) with H2O2 (0.72 M), and NaOH (3.6 M) were prepared and stored in the freezer at −20°C for 30 min. The solutions were transferred to an ice bath and allowed to sit for 30 min. The NaNO2 solution was added to a 100 mL beaker placed in an ice bath and stirred at >500 rpm. The HCl/H2O2 solution and NaOH solutions were added sequentially such that the NaOH came immediately after the HCl/H2O2 solution. These solutions must be added in sequence (i.e., one then the other) with as little time between the two additions as possible. The best results were obtained by adding the two solutions with a very small offset. The mixture turned yellow and was stirred in the ice bath for 5 min. MnO2 was added portion-wise until gas evolution ceased. The mixture was split into aliquots and centrifuged, and the peroxynitrite solution was decanted with a glass pipette. The concentration could be calculated with λmax = 302 nm ε = 1,670 M−1cm−1. The best results were obtained by centrifugation in a cold room at 4°C. [peroxynitrite] = 72–86 mM.
Serotonin-Hemin Reactivity Studies
Serotonin (1 mM), H2O2 (1 mM), hemin (15 μM), and sodium ascorbate (40 μM) without and with MMn (120 μM) or MnTMPyP (120 μM) were incubated in PBS (pH 7.4, 1.5 mL total volume) at 37°C for 2 h in the dark. The resulting mixture was passed through a reverse-phase tC18 SPE (Waters Sep-Pak, Waters) column containing 10 g of the C-18 substrate. The column was washed with 25 mL of 5% acetonitrile: H2O to separate the serotonin and 4,4′-serotonin dimer from paramagnetic species. The resulting solution was concentrated with a rotary evaporator. The mixture was taken up in D2O (0.7 mL) and analyzed via 1H NMR spectroscopy. NMR spectroscopic yields were calculated by comparing the relative concentration of serotonin to the 4,4′-serotonin dimer. Note that additional washing of the column failed to yield additional serotonin or the 4,4′-serotonin dimer.
Aβ16 and Aβ40 Reactivity Studies
Hemin Oxidative Reactivity Conditions
Hemin (15 μM) and H2O2 (1 mM) and Aβ16 (300 μM) or Aβ40 (100 and 200 μM) in PBS (200 μL total volume) were incubated together at 37°C. The resulting mixtures were immediately subjected to HPLC-MS analysis. Vials were cooled to 4°C inside the HPLC-MS queue to prevent additional reactivity.
Hemin Nitrative Reactivity Conditions
Hemin (15 μM), NaNO2 (1 mM), H2O2 (1 mM), and Aβ16 (300 μM) or Aβ40 (100 and 200 μM) in PBS (200 μL total volume) were incubated together at 37°C. The resulting mixtures were immediately subjected to HPLC-MS analysis. Vials were cooled to 4°C inside the HPLC-MS queue to prevent additional reactivity.
Hemin-Peroxynitrite Nitrative Reactivity Conditions
Hemin (15 μM), peroxynitrite (1 mM), and Aβ16 (300 μM) in PBS (200 μL total volume) were incubated together at 37°C. All compounds were added and then vortexed to provide a homogenous mixture. Immediately after peroxynitrite was added, the mixture was vortexed again and allowed to incubate at 37°C for 30 min. The resulting mixtures were immediately subjected to HPLC-MS analysis. Vials were cooled to 4°C inside the HPLC-MS queue to prevent additional reactivity.
Cell Culture
The neuroblastoma cell line Neuro-2A was bought from ATCC. Cells were grown in EMEM containing 10% heat-inactivated fetal bovine serum (FBS) and antibiotics (100 mg/mL streptomycin and 100 U/mL penicillin) at 37°C under 5% CO2 atmosphere. The cells were sub-cultured once every 4 to 5 days.
Neuro-2A MTT Assay
Aβ40 (90 μM) was incubated in PBS alone or in the presence of H2O2 (1 mM), hemin (15 μM), and NaNO2 (1 mM) (referred to as nitration mixture) for 3 h at 37°C followed by 24 h at 4°C. 250 μL of each of the solutions was transferred into each well containing about 50,000 Neuro-2A cells (plated 24 h earlier) in 250 μL of supplemented EMEM without phenol red. After 24 h, the medium was removed and replaced with 500 μL of fresh EMEM not containing FBS or phenol red. To this was added 200 μL of an MTT solution at 3 mg/mL in EMEM not containing phenol red or FBS. After 4 h at 37°C, the supernatant was removed and 200 μL of DMSO was added to each well to dissolve the formazan purple crystals. Aliquots (50 μL) of each well were transferred into a 96-well plate well, and the absorbance at 570 nm was read with a plate reader (see description above).
Acknowledgments
The work in Austin was supported by the National Institutes of Health (grant RO1 CA68682 to J.L.S.) and the Robert A. Welch Foundation (F-0018 to J.L.S.). The work at MIT was supported by NIH grant UF1 NS107712 to A.J. and P.H. In addition, P.H. would like to thank a Wellcome Trust-MIT Postdoctoral Research Fellowship (105932/Z/14/Z) and a Nottingham Research Fellowship at the University of Nottingham for support. S.D. and L.C. would like to thank the Italian Ministry of Education, University, and Research (MIUR) for a Research Project of National Interest (PRIN) 2015, Prot. 2015T778JW. NMR probes at the University of Texas at Austin (UT Austin) were purchased with the shared NIH grant 1-S10-OD021508-01. The C. elegans strains N2 and CL4176 were provided by CGC (nematodes shipped on 35-mm-diameter plates), which is funded by the NIH Office of Research Infrastructure Programs (P40-OD010440). J.T.B. would like to thank the UT Austin for a Global Research Fellowship at the University of Pavia, Italy, and a Scientist in Residence Fellowship. H.D.R. would like to thank the UT Austin for a Provost’s Graduate Excellence Fellowship and a Scientist in Residence Fellowship. J.T.B. and G.D.T. would like to thank Dr. Ian Riddington and Andrew Kalamarides (UT Austin) for assistance with MS analyses, Professor Jonathan Pierce and Momo Sae-Lee (UT Austin) for helpful discussions on C. elegans work, Dr. Timothy Johnstone and Professor Kelvin Gee (UC Irvine) for helpful discussions on CNS disorders, and Dr. Steve Sorey (UT Austin) for assistance with NMR analyses.
Author Contributions
J.T.B. and G.D.T. contributed equally. J.T.B. and G.D.T. conceived the project. J.F.R. synthesized the Aβ16 and Aβ40 peptides. J.T.B. and G.D.T. designed and performed the spectroscopic analyses and serotonin reactivity studies. G.D.T., J.T.B., and H.Z. carried out UV-vis studies. J.T.B. and G.D.T. designed and performed the Neuro-2A reactivity studies. P.H. designed and performed all of the MRI experiments. J.T.B., G.D.T., and P.H. analyzed the NMR, fluorescence, CD, reactivity studies, MRI, C. elegans uptake data, and ICP-MS analyses. G.D.T., H.Z., and J.W. carried out the MTT assay and cell-uptake studies. H.D.R. performed and analyzed the SEM studies. J.T.B., G.D.T., S.D., E.M., and L.C. designed and analyzed the in vitro HPLC-MS reactivity studies. J.T.B and S.D. performed the HPLC-MS reactivity studies. J.T.B., G.D.T., H.Z., R.M.J., and P.M. designed and performed the C. elegans studies. J.T.B., G.D.T, P.H., S.D., L.C., and J.L.S. prepared the manuscript. All authors approved the final version of the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: January 9, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.chempr.2019.12.016.
Contributor Information
Peter Harvey, Email: peter.harvey@nottingham.ac.uk.
Simone Dell’Acqua, Email: simone.dellacqua@unipv.it.
Luigi Casella, Email: luigi.casella@unipv.it.
Jonathan L. Sessler, Email: sessler@cm.utexas.edu.
Supplemental Information
References
- 1.Hopkins A.L. Network pharmacology: the next paradigm in drug discovery. Nat. Chem. Biol. 2008;4:682–690. doi: 10.1038/nchembio.118. [DOI] [PubMed] [Google Scholar]
- 2.Albert R. Scale-free networks in chemical biology. Journal of Cell Science. 2005;118:4847–4857. doi: 10.1242/jcs.02714. [DOI] [PubMed] [Google Scholar]
- 3.Gitler A.D., Dhillon P., Shorter J. Neurodegenerative disease: models, mechanisms, and a new hope. Dis. Models Mech. 2017;10:499–502. doi: 10.1242/dmm.030205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wyss-Coray T. Ageing, neurodegeneration, and brain rejuvenation. Nature. 2016;539:180–186. doi: 10.1038/nature20411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prezedborski S., Vila M., Jackson-Lewis V. Neurodegeneration: what is it and where are we? J. Clin. Invest. 2003;1:3–10. doi: 10.1172/JCI17522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Monzani E., Nicolis S., Dell’Acqua S., Capucciati A., Bacchella C., Zucca F.A., Mosharov E.V., Sulzer D., Zecca L., Casella L. Dopamine, oxidative stress and protein-quinone modifications in Parkinson’s and other neurodegenerative diseases. Angew. Chem. Int. Ed. Engl. 2019;58:6512–6527. doi: 10.1002/anie.201811122. [DOI] [PubMed] [Google Scholar]
- 7.Dos Santos Picanco L.C., Ozela P.F., de Fatima de Brito Brito M., Pinheiro A.A., Padilha E.C., Braga F.S., de Paula da Silva C.H.T., Dos Santos C.B.R., Rosa J.M.C., da Silva Hage-Melim L.I. Alzheimer’s disease: a review from the pathophysiology to diagnosis, new perspectives for pharmacological treatment. Curr. Med. Chem. 2018;25:3141–3159. doi: 10.2174/0929867323666161213101126. [DOI] [PubMed] [Google Scholar]
- 8.Kumar A., Singh A., Ekavali A review on Alzheimer’s disease pathophysiology and its management: an update. Pharmacol. Rep. 2015;67:195–203. doi: 10.1016/j.pharep.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 9.Parikshak N.N., Gandal M.J., Geschwind D.H. Systems biology and gene networks in neurodevelopmental and neurodegenerative disorders. Nat. Rev. Genet. 2015;16:441–458. doi: 10.1038/nrg3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arneson D., Zhang Y., Yang X., Narayanan M. Shared mechanisms among neurodegenerative diseases: From genetic factors to gene networks. J. Genet. 2018;97:795–806. [PMC free article] [PubMed] [Google Scholar]
- 11.Cummings J. Lessons learned from Alzheimer disease: clinical trials with negative outcomes. Clin. Transl. Sci. 2018;11:147–152. doi: 10.1111/cts.12491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang Z.L., Zhang L.J. PET/MRI of central nervous system: current status and future perspective. Eur. Radiol. 2016;26:3534–3541. doi: 10.1007/s00330-015-4202-5. [DOI] [PubMed] [Google Scholar]
- 13.Rosén C., Hansson O., Blennow K., Zetterberg H. Fluid biomarkers in Alzheimer’s disease – current concepts. Mol. Neurodegener. 2013;8:20. doi: 10.1186/1750-1326-8-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frisoni G.B., Fox N.C., Jack C.R., Scheltens P., Thompson P.M. The clinical use of structural MRI in Alzheimer disease. Nat. Rev. Neurol. 2010;6:67–77. doi: 10.1038/nrneurol.2009.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bolognesi M.L., Gandini A., Prati F., Uliassi E. From companion diagnostics to theranostics: a new avenue for Alzheimer’s disease? J. Med. Chem. 2016;59:7759–7770. doi: 10.1021/acs.jmedchem.6b00151. [DOI] [PubMed] [Google Scholar]
- 16.Wang X., Wang X., Guo Z. Metal-involved theranostics: an emerging strategy for fighting Alzheimer’s disease. Coord. Chem. Rev. 2018;362:72–84. [Google Scholar]
- 17.Sessler J.L., Wegnorn S.J. Elsevier; 1997. Expanded, Contracted & Isomeric Porphyrins. [Google Scholar]
- 18.Sessler J.L., Hemmi G., Mody T.D., Murai T., Burrell A., Young S.W. Texaphyrins: synthesis and applications. Acc. Chem. Res. 1994;27:43–50. [Google Scholar]
- 19.Preihs C., Arambula J.F., Magda D., Jeong H., Yoo D., Cheon J., Siddik Z.H., Sessler J.L. Recent developments in texaphyrin chemistry and drug discovery. Inorg. Chem. 2013;52:12184–12192. doi: 10.1021/ic400226g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mehta M.P., Shapiro W.R., Phan S.C., Gervais R., Carrie C., Chabot P., Patchell R.A., Glantz M.J., Recht L., Sur R.K. Motexafin gadolinium combined with prompt whole brain radiotherapy prolongs time to neurologic progression in non-small-cell lung cancer patients with brain metastases: results of a phase III trial. Int. J. Radiat. Oncol. Biol. Phys. 2009;73:1069–1076. doi: 10.1016/j.ijrobp.2008.05.068. [DOI] [PubMed] [Google Scholar]
- 21.Rosenthal D.I., Nurenberg P., Becerra C.R., Frenkel E.P., Carbone D.P., Lum B.L., Miller R., Engel J., Young S., Miles D. A phase I single-dose trial of gadolinium texaphyrin (Gd-Tex), a tumor selective radiation sensitizer detectable by magnetic resonance imaging. Clin. Cancer Res. 1999;5:739–745. [PubMed] [Google Scholar]
- 22.Kereiakes D.J., Szyniszewski A.M., Wahr D., Herrmann H.C., Simon D.I., Rogers C., Kramer P., Shear W., Yeung A.C., Shunk K.A. Phase I drug and light dose-escalation trial of motexafin lutetium and far red light activation (phototherapy) in subjects with coronary artery disease undergoing percutaneous coronary intervention and stent deployment: procedural and long-term results. Circulation. 2003;108:1310–1315. doi: 10.1161/01.CIR.0000087602.91755.19. [DOI] [PubMed] [Google Scholar]
- 23.Thiabaud G., McCall R., He G., Arambula J.F., Siddik Z.H., Sessler J.L. Activation of platinum(IV) prodrugs by motexafin gadolinium as a redox mediator. Angew. Chem. Int. Ed. Engl. 2016;55:12626–12631. doi: 10.1002/anie.201604236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Batinic-Haberle I., Tovmasyan A., Spasojevic I. Mn porphyrin-based redox-active drugs- Differential effects as cancer therapeutics and protectors of normal tissue against oxidative injury. Antioxid. Redox Signal. 2018;29:1691–1724. doi: 10.1089/ars.2017.7453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang X.R., Zhou W.X., Zhang Y.X. Improvements in SOD mimic AEOL-10150, a potent broad-spectrum antioxidant. Mil. Med. Res. 2018;5:30. doi: 10.1186/s40779-018-0176-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.WCG’s FDAnews Aeolus announces optimistic AEOL 10150 Phase I trial interim results. 2005. https://www.fdanews.com/articles/70464-aeolus-announces-optimistic-aeol-10150-phase-i-trial-interim-results March 30, 2005.
- 27.Abu-Omar M.M. High-valent iron and manganese complexes of corrole and porphyrin in atom transfer and dioxygen evolving catalysis. Dalton Trans. 2011;40:3435–3444. doi: 10.1039/c0dt01341b. [DOI] [PubMed] [Google Scholar]
- 28.Gross Z. High-valent corrole metal complexes. J. Biol. Inorg. Chem. 2001;6:733–738. doi: 10.1007/s007750100273. [DOI] [PubMed] [Google Scholar]
- 29.Ghosh C., Seal M., Mukherjee S., Ghosh Dey S.G. Alzheimer’s disease: A heme-Aβ perspective. Acc. Chem. Res. 2015;48:2556–2564. doi: 10.1021/acs.accounts.5b00102. [DOI] [PubMed] [Google Scholar]
- 30.Valiente-Gabioud A.A., Riedel D., Outeiro T.F., Menacho-Márquez M.A., Griesinger C., Fernández C.O. Binding modes of phthalocyanines to amyloid β peptide and their effects on amyloid fibril formation. Biophys. J. 2018;114:1036–1045. doi: 10.1016/j.bpj.2018.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fan Y., Wu D., Yi X., Tang H., Wu L., Xia Y., Wang Z., Liu Q., Zhou Z., Wang J. TMPyP inhibits amyloid-β aggregation and alleviates amyloid-induced cytotoxicity. ACS Omega. 2017;2:4188–4195. doi: 10.1021/acsomega.7b00877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gomes L.M.F., Mahammed A., Prosser K.E., Smith J.R., Silverman M.A., Walsby C.J., Gross Z., Storr T. A catalytic antioxidant for limiting amyloid-beta peptide aggregation and reactive oxygen species generation. Chem. Sci. 2019;10:1634–1643. doi: 10.1039/c8sc04660c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brothers P.J. Organoelement chemistry of main-group porphyrin complexes. Adv. Organomet. Chem. 2001;48:289–342. [Google Scholar]
- 34.Nienhaus K., Nienhaus G.U. Probing heme protein-ligand interactions by UV/visible absorption spectroscopy. Methods Mol. Biol. 2005;305:215–242. [Google Scholar]
- 35.Stine W.B., Jungbauer L., Yu C., LaDu M.J. Humana Press; 2010. Preparing Synthetic Aβ in Different Aggregation States. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xue C., Lin T.Y., Chang D., Guo Z. Thioflavin T as an amyloid dye: fibril quantification, optimal concentration and effect on aggregation. R. Soc. Open Sci. 2017;4:160696. doi: 10.1098/rsos.160696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alavez S., Vantipalli M.C., Zucker D.J.S., Klang I.M., Lithgow G.J. Amyloid-binding compounds maintain protein homeostasis during ageing and extend lifespan. Nature. 2011;472:226–229. doi: 10.1038/nature09873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cukalevski R., Yang X., Meisl G., Weininger U., Bernfur K., Frohm B., Knowles T.P.J., Linse S. The Aβ40 and Aβ42 peptides self-assemble into separate homomolecular fibrils in binary mixtures but cross-react during primary nucleation. Chem. Sci. 2015;6:4215–4233. doi: 10.1039/c4sc02517b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wahsner J., Gale E.M., Rodríguez-Rodríguez A., Caravan P. Chemistry of MRI contrast agents: current challenges and new frontiers. Chem. Rev. 2019;119:957–1057. doi: 10.1021/acs.chemrev.8b00363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gale E.M., Atanasova I.P., Blasi F., Ay I., Caravan P. A Manganese alternative to gadolinium for MRI contrast. J. Am. Chem. Soc. 2015;137:15548–15557. doi: 10.1021/jacs.5b10748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barandov A., Bartelle B.B., Gonzalez B.A., White W.L., Lippard S.J., Jasanoff A. Membrane-permeable Mn(III) complexes for molecular magnetic resonance imaging of intracellular targets. J. Am. Chem. Soc. 2016;138:5483–5486. doi: 10.1021/jacs.5b13337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sessler J.L., Mody T.D., Hemmi G.W., Lynch V., Young S.W., Miller R.A. Gadolinium(III) texaphyrin: a novel MRI contrast agent. J. Am. Chem. Soc. 1993;115:10368–10369. [Google Scholar]
- 43.Keca J.M., Chen J., Overchuk M., Muhanna N., MacLaughlin C.M., Jin C.S., Foltz W.D., Irish J.C., Zheng G. Nanotexaphyrin: one-pot synthesis of a manganese texaphyrin-phospholipid nanoparticle for magnetic resonance imaging. Angew. Chem. Int. Ed. Engl. 2016;55:6187–6191. doi: 10.1002/anie.201600234. [DOI] [PubMed] [Google Scholar]
- 44.Keca J.M., Valic M.S., Cheng M.H.Y., Jiang W., Overchuk M., Chen J., Zheng G. Mixed and matched metallo-nanotexaphyrin for customizable biomedical imaging. Adv. Healthc. Mater. 2019;8:e1800857. doi: 10.1002/adhm.201800857. [DOI] [PubMed] [Google Scholar]
- 45.Martins A.F., Morfin J.-F., Geraldes C.F.C., Toth E. Gd3+ complexes conjugated to Pittsburgh compound B: potential MRI markers of β-amyloid plaques. J Biol Inorg Chem. 2014;19:281–295. doi: 10.1007/s00775-013-1055-8. [DOI] [PubMed] [Google Scholar]
- 46.Dostal V., Link C.D. Assaying β-amyloid toxicity using a transgenic C. elegans model. J. Vis. Exp. 2010;44:e2252. doi: 10.3791/2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zheng S.Q., Ding A.J., Li G.P., Wu G.S., Luo H.R. Drug absorption efficiency in Caenorhbditis elegans delivered by different methods. PLoS One. 2013;8:e56877. doi: 10.1371/journal.pone.0056877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mangialasche F., Polidori M.C., Monastero R., Ercolani S., Camarda C., Cecchetti R., Mecocci P. Biomarkers of oxidative and nitrosative damage in Alzheimer’s disease and mild cognitive impairment. Ageing Res. Rev. 2009;8:285–305. doi: 10.1016/j.arr.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 49.Radi R. Protein tyrosine nitration: biochemical mechanisms and structural basis of functional effects. Acc. Chem. Res. 2013;46:550–559. doi: 10.1021/ar300234c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fiedorowicz M., Grieb P. Nitrooxidative stress and neurodegeneration. In: Gonzalez-Quevedo A., editor. Brain Damage – Bridging Between Basic Research and Clinics. InTech Open; 2012. pp. 130–162. [Google Scholar]
- 51.Zhang Y.J., Xu Y.F., Chen X.Q., Wang X.C., Wang J.Z. Nitration and oligomerization of tau induced by peroxynitrite inhibit its microtubule-binding activity. FEBS Lett. 2005;579:2421–2427. doi: 10.1016/j.febslet.2005.03.041. [DOI] [PubMed] [Google Scholar]
- 52.Butterfield D.A., Swomley A.M., Sultana R. Amyloid β-Peptide (1-42)-induced oxidative stress in Alzheimer disease: importance in disease pathogenesis and progression. Antioxid. Redox Signal. 2013;19:823–835. doi: 10.1089/ars.2012.5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hannah S., Lynch V., Guldi D.M., Gerasimchuk N., MacDonald C.L.B., Magda D., Sessler J.L. Late first-row transition-metal complexes of texaphyrin. J. Am. Chem. Soc. 2002;124:8416–8427. doi: 10.1021/ja012747a. [DOI] [PubMed] [Google Scholar]
- 54.Shimanovich R., Hannah S., Lynch V., Gerasimchuk N., Mody T.D., Magda D., Sessler J.L., Groves J.T. Mn(II)-texaphyrin as a catalyst for the decomposition of peroxynitrite. J. Am. Chem. Soc. 2001;123:3613–3614. doi: 10.1021/ja005856i. [DOI] [PubMed] [Google Scholar]
- 55.Crow J.P. Administration of Mn porphyrin and Mn texaphyrin at symptom onset extends survival of ALS mice. In: ACS Symp. S., Sessler J.L., Doctrow S.R., McMurry J.T., Lippard S.J., editors. Medicinal inorganic chemistry. 2005. pp. 295–318. [Google Scholar]
- 56.Groves J.T., Marla S.S. Peroxynitrite-induced DNA strand scission mediated by a manganese porphyrin. J. Am. Chem. Soc. 1995;117:9578–9579. [Google Scholar]
- 57.Chesler M. Regulation and modulation of pH in the brain. Physiol. Rev. 2003;83:1183–1221. doi: 10.1152/physrev.00010.2003. [DOI] [PubMed] [Google Scholar]
- 58.Arasasingham R.D., He G.X., Bruice T.C. Mechanism of manganese porphyrin-catalyzed oxidation of alkenes. Role of manganese(IV)-oxo species. J. Am. Chem. Soc. 1993;115:7985–7991. [Google Scholar]
- 59.Thiabaud G., Pizzocaro S., Garcia-Serres R., Latour J.M., Monzani E., Casella L. Heme binding induces dimerization and nitration of truncated β-amyloid peptide Aβ16 under oxidative stress. Angew. Chem. Int. Ed. Engl. 2013;52:8041–8044. doi: 10.1002/anie.201302989. [DOI] [PubMed] [Google Scholar]
- 60.Hiner A.N.P., Raven E.L., Thorneley R.N.F., García-Cánovas F., Rodríguez-López J.N. Mechanisms of compound I formation in heme peroxidases. J. Inorg. Biochem. 2002;91:27–34. doi: 10.1016/s0162-0134(02)00390-2. [DOI] [PubMed] [Google Scholar]
- 61.Batinić-Haberle I., Benov L., Spasojević I., Fridovich I. The Ortho effect makes manganese(III)meso-tetrakis(N-Methylpyridinium-2-yl)porphyrin a powerful and potentially useful superoxide dismutase mimic. J. Biol. Chem. 1998;273:24521–24528. doi: 10.1074/jbc.273.38.24521. [DOI] [PubMed] [Google Scholar]
- 62.Davies M.J. Protein oxidation and peroxidation. Biochem. J. 2016;473:805–825. doi: 10.1042/BJ20151227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Irie K., Murakami K., Masuda Y., Morimoto A., Ohigashi H., Ohashi R., Takegoshi K., Nagao M., Shimizu T., Shirasawa T. Structure of β-amyloid fibrils and its relevance to their neurotoxicity: implications for the pathogenesis of Alzheimer’s disease. J. Biosci. Bioeng. 2005;99:437–447. doi: 10.1263/jbb.99.437. [DOI] [PubMed] [Google Scholar]
- 64.Kummer M.P., Hermes M., Delekarte A., Hammerschmidt T., Kumar S., Terwel D., Walter J., Pape H.C., König S., Roeber S. Nitration of tyrosine 10 critically enhances amyloid β aggregation and plaque formation. Neuron. 2011;71:833–844. doi: 10.1016/j.neuron.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 65.Yuan C., Yi L., Yang Z., Deng Q., Huang Y., Li H., Gao Z. Amyloid beta-heme peroxidase promoted protein nitrotyrosination: relevance to widespread protein nitration in Alzheimer’s disease. J. Biol. Inorg. Chem. 2012;17:197–207. doi: 10.1007/s00775-011-0842-3. [DOI] [PubMed] [Google Scholar]
- 66.Hou L., Shao H., Zhang Y., Li H., Menon M.K., Neuhaus E.B., Brewer J.M., Byeon J.M., Ray D.G., Vitek M.P. Solution NMR studies of the Aβ(1–40) and Aβ(1–42) peptides establish that the Met35 oxidation state affects the mechanism of amyloid formation. J. Am. Chem. Soc. 2004;126:1992–2005. doi: 10.1021/ja036813f. [DOI] [PubMed] [Google Scholar]
- 67.Guivernau B., Bonet J., Valls-Comamala V., Bosch-Morató M., Godoy J.A., Inestrosa N.C., Perálvarez-Marín A., Fernández-Busquets X., Andreu D., Oliva B. Amyloid-β peptide nitrotyrosination stabilizes oligomers and enhances NMDAR-mediated toxicity. J. Neurosci. 2016;36:11693–11703. doi: 10.1523/JNEUROSCI.1081-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sessler J.L., Mody T.D., Hemmi G.W., Lynch V. Synthesis and structural characterization of lanthanide(III) texaphyrins. Inorg. Chem. 1993;32:3175–3187. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.