

Abstract
Supported Catalytically Active Liquid Metal Solutions (SCALMS) were recently described as a new class of heterogeneous catalysts, where the catalytic transformation takes place at the highly dynamic interface of a liquid alloy. Their application in alkane dehydrogenation has been claimed to be superior to classical heterogeneous catalysts, because the single atom nature of Rh dissolved in liquid Ga hinders the formation of significant amounts of coke, e. g. by oligomerisation of carbon fragments and excessive dehydrogenation. In the present study, we investigate the coking behaviour of Ga−Rh SCALMS during dehydrogenation of propane in detail by means of high‐resolution thermogravimetry. We report that the application of Ga−Rh SCALMS indeed limits the formation of coke when compared to the Ga‐free Rh catalyst, in particular when relating coke formation to the catalytic performance. Furthermore, the formed coke has been shown to be highly reactive during temperature programmed oxidation in 21 % O2/He with onset temperatures of approx. 150 °C enabling a regeneration of the Ga−Rh SCALMS system under mild conditions. The activation energy of the oxidation lies in the lower range of values reported for spent cracking catalysts. Monitoring the formation of coke and performance of SCALMS in situ via thermogravimetry coupled with mass spectrometry revealed the continuous formation of coke, which becomes the only process affecting the net weight change after a certain time on stream.
Keywords: Coke, Dehydrogenation, Gallium, SCALMS, TPO
High‐resolution thermogravimetric analysis coupled with mass spectrometry was employed to study the coke formation during propane dehydrogenation over Supported Catalytically Active Liquid Metal Solutions (SCALMS). Linking the coke analysis by means of temperature programmed oxidation with the catalytic performance data results in a decreased selectivity towards the formation of coke for SCALMS when compared to classical heterogeneous catalysts. In situ analysis of the formation of coke during catalysis provides insight into the evolution of coke during propane dehydrogenation.

Introduction
Thermocatalytical dehydrogenation of propane over classical heterogeneous Pt‐based catalysts under industrial conditions typically requires frequent regeneration of the catalysts under O2‐rich environment due to rapid deactivation via coke formation.1 Other side reactions, such as hydrogenolysis or cracking, also contribute to the observed severe loss in activity.2 Even though regeneration of the catalyst by combustion of coke may be realised within short time frames,1c an alternating operation with dehydrogenation‐regeneration cycles drastically reduces the process efficiency while increasing operational costs and required investments. Hence, a high resistance of the applied catalyst against coking is highly desired and a key success factor for technical processes applying alkanes as feedstock for alkene production by catalytic dehydrogenation. In the case of Pt‐based dehydrogenation catalysts, stabilisation of the Pt phase has been achieved via physical (entrapment in support) or chemical stabilisation (Ce doping of Al2O3 support material).3 Furthermore, Pt−Sn bimetallic catalysts have been reported to suppress rapid deactivation.1a, 1b Oxidative dehydrogenation represents another attractive alternative for producing alkenes without excessive coke formation due to the oxidative reaction environment. However, the undesired full oxidation of propane to CO2 is a relevant side reaction of oxidative dehydrogenation lowering the overall efficiency of the alkene production process.4
Supported Catalytically Active Liquid Metal Solutions (SCALMS) offer the potential for new and unusual catalytic reactivity in alkane dehydrogenation. SCALMS are composed of liquid alloy droplets of a catalytically active metal with a low melting point metal, such as Ga, on a porous support.5 In contrast to conventional supported liquid phase catalysis, the catalytic reaction in SCALMS occurs exclusively at the highly dynamic liquid metal/gas interface,5a, 5b, 5g, 6 as the liquid metal does not provide any relevant reactant solubility. Recently, the use of Ga‐rich supported bimetallic mixtures with Pd, Rh, or Pt (atomic Ga/Me ratio >10) has been reported for the highly endothermal dehydrogenation of n‐butane and propane.5a, 5c, 5g Remarkably, these SCALMS materials outperform, under some reaction conditions, commercial dehydrogenation catalysts (Pt/Al2O3, Cr2O3/Al2O3) without extensive material or process optimisation.5a Particularly noteworthy is the complete suppression of coking during n‐butane dehydrogenation over Ga−Pd SCALMS5a even though coking is the typical deactivation mechanism for high temperature reductive hydrocarbon transformation processes.2, 7 As explanation for this unexpected behaviour, angle‐resolved X‐ray photon spectroscopy (XPS) experiments and molecular dynamics simulations suggest depletion of the topmost layer in Ga‐rich alloys regarding the catalytically active metal, while the layer directly underneath is enriched.5a, 5g, 6 The atomic distribution at the liquid catalyst surface is strongly affected by substrates approaching the surface and coming into contact with the active metal atoms.5a, 5c, 5e–5g Dehydrogenation of propane is suggested to require only a single active metal atom.8 In turn, side reactions requiring a second active site are strongly reduced in the SCALMS approach due to the low probability of neighbouring active metal atoms at the surface of the liquid alloy5a, 5c, 5e–5g in combination with the generally low activation energy for desorption of propene.8 When compared to the dehydrogenation of butane, propane dehydrogenation (PDH) is expected to be rather vulnerable for the formation of coke. PDH has recently been studied using Ga‐rich Ga−Rh SCALMS.5g Analysis of the spent SCALMS by means of XPS suggested an increase of the C1s component in all samples when compared to the as prepared materials. Further, amorphous carbon was identified in the spent SCALMS after catalytic PDH at the highest studied temperature of 550 °C via Raman spectroscopy.5g
In this study we focus on nature and quantity of coke formed during propane dehydrogenation over said SCALMS system. In detail, we use temperature programmed oxidation (TPO), which is a widely applied technique for studying the reactivity/stability of carbon species.9 TPO describes the defined heating of a (partially) carbonaceous sample to high temperatures in the presence of an oxidising atmosphere, mostly diluted oxygen. Further, the activation energy can be determined using various techniques making use of the temperature dependency of the rate constant according to Arrhenius’ law. TPO of the spent SCALMS was conducted via high‐resolution thermogravimetric analysis coupled with mass spectrometry (HRTGA‐MS) in a XEMIS sorption analyser. Furthermore, we describe a novel approach to apply HRTGA‐MS for the in situ characterisation of coke formation during propane dehydrogenation over Ga−Rh SCALMS.
Results and Discussion
Coke analysis in spent Ga−Rh SCALMS
The reactivity of coke formed during PDH at 450 °C5g was first analysed by means of HRTGA‐MS during TPO (Figure S1) of spent Ga−Rh/Al2O3 SCALMS with atomic Ga/Rh ratios of 25 and 125, as well as of the benchmark cases of Ga/Al2O3 (Ga/Rh=∞) and Rh/Al2O3 (Ga/Rh=0). The overall weight loss between the exposure of the spent and dried catalysts to He at 100 °C and after exposure to 21 % O2/He at 500 °C for 12 h can be expected to mostly correspond to the amount of coke. However, a rapid initial weight increase is observed upon first exposure of the pre‐dried samples to the oxidising atmosphere (Figure 1a). Three processes may be at play.
Figure 1.
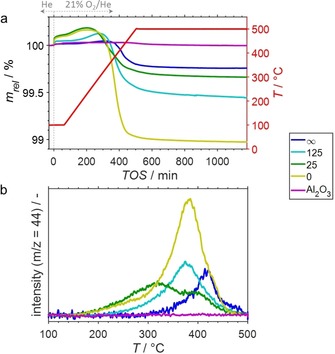
(a) Sample weight relative to the weight prior to exposure to 21 % O2/He at 100 °C and (b) formation of CO2 during temperature programmed oxidation (1 °C min−1) of spent SCALMS with atomic Ga/Rh ratios of 25 and 125, the spent Ga/Al2O3 (Ga/Rh=∞) and Rh/Al2O3 (Ga/Rh=0) reference catalysts, as well as the bare Al2O3 support material as monitored via high‐resolution thermogravimetry coupled with mass spectrometry. All catalysts were previously applied in propane dehydrogenation at 450 °C. Conditions of the experiment: He flow 100 mLN min−1 for TOS<0; He flow 79 mLN min−1 and O2 flow 21 mLN min−1 for TOS>0; GHSV 30000 mLN g−1 h−1.
Firstly, the formation of vacancies in the Al2O3 support material (due to desorption of chemisorbed water during the preceding drying step under inert atmosphere at up to 500 °C; Table S2) may facilitate a pronounced adsorption of O2.
Secondly, oxidic Ga species (GaOx) may form on the surface of the Ga‐rich phase forming a passivation layer due to the exposure to O2‐rich atmosphere at 100 °C.
Lastly, the initial weight increase may be originated in the presence of coke in the spent samples, which may allow for the formation of C(Ox) functionalities on the surface of coke (see Electronic Supporting Information (ESI) for detailed description).10
Only minor weight change is observed when analysing the bare Al2O3 support material in the same TPO sequence (Figure 1a). Hence, the observed weight increase is only to a small extent due to adsorption of O2 on the support material. The formation of GaOx species are indicated in an additional experiment exposing an as‐prepared SCALMS to two consecutive TPO sequences (Figure S2). While the first exposure to O2‐containing atmosphere resulted in a weight increase of 0.1 wt%, the second exposure after the first TPO sequence hardly affected the sample weight. Nevertheless, the relative weight increase correlates with the amount of coke present in each analysed spent SCALMS catalyst (Figure S3), in particular for samples with a high coke content. This points towards a significant contribution of the third hypothesised process to the initial weight increase. The oxygen‐containing functionalities on carbon are reported to be metastable at increased temperatures initiating the oxidation process of coke by O2 together with the formation of volatile CO or CO2.10a Noteworthy, minor amounts of CO2 have been detected in the off‐gas by means of MS indicating full oxidation of a small fraction of highly reactive coke in the spent catalysts during the first 60 min time on stream (TOS) at the initial isotherm at 100 °C (Figure S4).
After the initial isotherm, the temperature was increased inducing another weight increase of the spent catalysts (Figure 1a). This observation, once again, indicates the formation of (metastable) oxygen‐containing functionalities at the carbon surface or formation of GaOx species at the surface of the bimetallic Ga‐rich phase resulting in an increase of the sample weight. Volatisation of coke, that is decomposition of oxygen‐functionalised coke species in form of gaseous CO or CO2, becomes the dominating process at temperatures exceeding 200 °C and this results in a net decrease of weight. Nevertheless, the formation of CO2 is observed at temperatures as low as 150 °C (Figure 1b, Figures S5a), i. e. the combustion process initiates at even lower temperatures. The profile of CO2 formation during TPO indicates the presence of two classes of coke in the spent catalysts, denominated soft and hard coke (see ESI for further details on this classification), resulting in two overlapping peaks (Figure 1b). The soft coke is expected to have a lower degree of graphitisation and can be oxidised at temperatures as low as 150–200 °C (Figure 1b; Table S2). Seemingly, an increased fraction of reactive coke is formed when increasing the Rh content of the samples (Figures S6–S7). The coke in the Ga/Al2O3 reference sample displays a relatively low reactivity, i. e. mostly consists of hard coke with increased degree of graphitisation and an increased onset temperature of 390 °C (Table S2). However, it has to be emphasised that such temperatures are still relatively low for oxidation of carbonaceous deposits in spent catalysts.
The spent Ga−Rh SCALMS after PDH at 450 °C contain minor amounts of coke in the range of 0.3–0.6 wt% (Figure 1a). Contrary, the classical heterogeneous Rh/Al2O3 was shown to be more prone towards coking with a coke content of >1 wt% after PDH under identical conditions. In our previous contribution, the supported single‐phased Ga in the absence of Rh displayed a particularly special catalytic performance.5g The absence of Rh resulted in oxidation of Ga by oxygen containing impurities in the feed stream forming GaOx at the surface of the supported liquid Ga droplets. In turn, the Ga/Al2O3 sample activated over time, but describes a different catalytic system. The coke in this spent sample is relatively inactive requiring elevated temperatures when compared to the SCALMS samples. The overall amount of coke in all samples decreases in the order Rh/Al2O3>SCALMS>Ga/Al2O3 (Figure 1a), while a feasible correlation between the weight change and the integral formation of CO2 as monitored by HRTGA‐MS was obtained (Figure S8).
Aside from CO2, small amounts of water were released during TPO in the temperature range of 350–500 °C for all catalysts containing Rh (Figure S5b). However, the low signal‐to‐noise ratio indicates an extremely low H‐content of the hard coke. In general, full conversion of coke was achieved at moderate temperatures for all catalyst systems allowing for full regeneration at temperatures as low as 500 °C. Noteworthy. full combustion of coke formed during commercial fluid catalytic cracking requires increased temperatures of approx. 900 °C,10c while the initiation of coke oxidation is reported at 200–250 °C. Hence, both classes of coke in the herein analysed samples are highly reactive.
At 450 °C, the highly diluted SCALMS with an atomic Ga/Rh ratio of 125 is expected to form a fully liquid supported alloy in accordance with the concept of SCALMS (Figure S9).5g, 11 Intermetallic solid compounds may form when increasing the Rh content. Hence, the different CO2 formation profile during TPO of the SCALMS with a reduced Ga/Rh ratio of 25 may be originated in the presence of such intermetallic solids. Hence, the coke analysis was extended to samples after PDH at an increased temperature of 550 °C allowing for a higher solubility of Rh in the liquid Ga phase at reaction conditions, i. e. atomic ratios of Ga/Rh exceeding 50 form fully liquid supported alloys. The catalytic performance of SCALMS samples with atomic Ga/Rh ratios of 125, 82 and 34 was previously evaluated during PDH at 550 °C.5g Decreasing the Ga/Rh ratio from 82 to 34 implicates a crossing of the liquidus line (Figure S9). Hence, the SCALMS with the lowest ratio is expected to represent mostly liquid supported alloys with a minor fraction of intermetallic solids, which may affect the catalytic performance and coking behaviour alike, e. g. the productivity based on the overall amount of Rh in the samples increases with dilution of Rh in Ga.5g Indeed, analysis of the coke in the spent samples by means of HRTGA‐MS once again suggests a pronounced formation of coke with increased Ga/Rh ratio (Figures S10–S12). The fraction of soft coke is increased for the fully liquid SCALMS with an atomic Ga/Rh ratio of 125, while said fraction decreases when lowering the ratio (Figure S13).
Previously, PDH was also conducted using the SCALMS with the highest atomic Ga/Rh ratio of 125 at intermediate temperatures of 480 and 500 °C. When comparing the productivity per Rh atom, this SCALMS sample outperformed both, classical heterogeneous Rh catalysts and other Ga−Rh SCALMS systems with lower atomic Ga/Rh ratios.5g Analysis of this SCALMS after application in PDH by means of HRTGA‐MS demonstrates a strong correlation between the amount of coke and the reaction temperature (Figure 2a; Figure S14). This behaviour is expected as increased temperatures typically accelerate coking. In addition, the conversion of propane increases with temperature providing increased concentrations of alkenes and other precursors of coke, i. e. an enhanced formation of coke is expected at increased conversion levels.12 In the analysed SCALMS, the amount of identified coke increases exponentially with dehydrogenation temperature (Figure 3). Accordingly, the required temperature for conversion of 50 % coke during the temperature ramp of the TPO increases with dehydrogenation temperature (Figure 2b; Table S2).
Figure 2.
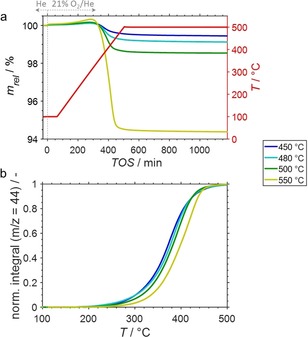
(a) Sample weight relative to the weight prior to exposure to 21 % O2/He at 100 °C and (b) normalised cumulative formation of CO2 during temperature programmed oxidation (1 °C min−1) of spent SCALMS with an atomic Ga/Rh ratio of 125 as monitored via high‐resolution thermogravimetry coupled with mass spectrometry. The catalysts were previously applied in propane dehydrogenation at 450, 480, 500, and 550 °C. Conditions of the experiment: He flow 100 mLN min−1 for TOS<0; He flow 79 mLN min−1 and O2 flow 21 mLN min−1 for TOS>0; GHSV 30000 mLN g−1 h−1.
Figure 3.
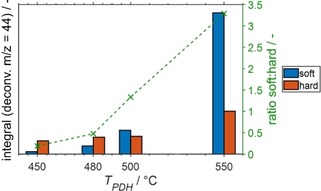
Integral CO2 formation from temperature programmed oxidation of soft and hard coke in spent SCALMS with an atomic Ga/Rh ratio of 125 after previous application in propane dehydrogenation at various temperatures as monitored via high‐resolution thermogravimetry coupled with mass spectrometry.
The normalised integrals of the CO2 formation shift to increased temperature with increased PDH temperature, which is particularly pronounced for the SCALMS after PDH at 550 °C (Figure 2b). This observation suggests a change in the composition of the coke in the spent SCALMS. Indeed, a clear trend to increased amounts of soft coke with increased PDH temperatures is observed (Figure 3; Figure S15). Further, the temperatures required for 50 % conversion of coke increase with PDH reaction temperature from 331 to 376 °C for the soft coke and from 378 to 417 °C for the hard coke (Figure S16; Table S2). Note, that the soft coke after PDH at 550 °C displays a reactivity similar to the hard coke formed during PDH at 450 °C. Nevertheless, the distinct shift may be an indicator for a pronounced contribution of the Al2O3 support to coking at increased temperatures (Figure 3). An increased formation of soft coke at increased temperatures may be caused by the support, while the amount of hard coke remains almost constant in the temperature range of 450–500 °C and hence may be a result of catalytic activity. Consequentially, the identified ratio of soft/hard coke increases drastically to 3.5, while the ratio is below 1.4 after operating at reduced temperatures (Figure 3).
The presented results strongly demand for a correlation of the conducted coke analyses with the performance data from our previous catalytic study. In order to draw valid conclusions, the actual conditions during PDH have to be taken into account, e. g. the actual conversion level represents an additional parameter for the formation of coke. Calculation of the carbon‐based selectivities of the SCALMS towards coke (S C,coke) using the obtained fractions of coke as obtained by means of HRTGA‐MS [Eq. (1)] demonstrates the great benefits of the SCALMS concept.5a
| (1) |
Where mcoke is the amount of coke identified by means of HRTGA‐MS and extrapolated to the loading of catalyst in our previous catalytic studies,5g m C3H8,feed(t) is the amount of propane in the feed stream at each time interval, XC3H8(t) is the conversion of propane at each time interval, and MC and MC3H8 are the molar masses of carbon and propane, respectively.
The supported Rh catalyst was the least active catalyst, but formed the highest amount of coke at 450 °C (1.0 wt%; Figure 1) demonstrating the high vulnerability of conventional heterogeneous catalysts to coking during dehydrogenation, even at low temperatures (Figure S17). Both SCALMS catalyst outperformed the Rh/Al2O3 catalyst with a carbon‐based, undesired selectivity towards coke of ≤1.5 % (Rh/Al2O3 : 10.5 %) after PDH at 450 °C. As for the productivity per Rh atom,5g the liquidus line of the Ga−Rh phase diagram may be traced by the coke selectivity (Figure S18). Remarkably, the coke selectivity of the SCALMS with an atomic Ga/Rh ratio of 125 after dehydrogenation at 450–500 °C decreases with increasing reaction temperature, which is a remarkable finding. As discussed earlier on, increased temperatures are typically associated with an increased formation of coke. The actual conversion level dictates the potential for an extended coke formation by creating an atmosphere that is more prone for coking. Hence, the coke selectivity is related to the average conversion of propane during the particular experiments in order to consider this important parameter (Figure 4). This results in the lowest value for the SCALMS with an atomic Ga/Rh ratio of 125 after PDH at 550 °C, which is significantly lower than both SCALMS tested at 450 °C exhibiting the beneficial properties of fully liquid supported alloys according to the SCALMS concept.5a Further, normalising the coke selectivities to the average conversion of propane results in a lower value for the SCALMS with an atomic Ga/Rh ratio of 125 at 550 °C compared to PDH using the same SCALMS at 450 °C (Figure 4). As previously suggested, the Al2O3 support material may result in pronounced coking at 550 °C increasing the coke selectivity from 1.0 to 4.0 % when compared to PDH at 500 °C (Figure S18).
Figure 4.
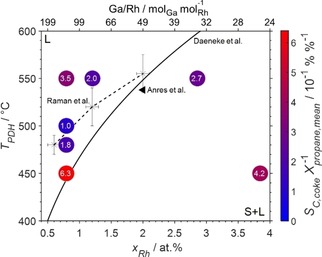
Integral carbon based selectivity towards coke relative to the average conversion of propane over time on stream (circles) of the various Ga−Rh SCALMS compositions at different temperatures superimposed on the liquidus line of the Ga−Rh phase diagram (solid) according to Daeneke et al.,11b an experimental data point for the solubility of Rh in Ga (filled triangle) by Anres et al.,11a as well as an experimentally obtained solubility data from XPS measurements (crosses connected by dashed line to guide the eye) by Raman et al.5g
Activation energy of coke in spent Ga−Rh SCALMS
The activation energy (Ea) of coke formed during PDH at the reaction temperature of 500 °C with the SCALMS catalyst of an atomic ratio of Ga/Rh of 125 was exemplarily determined via an isoconversional method according to Starink (see ESI for further details) using Equation (2).13
| (2) |
Where β is the heating rate, T α is the temperature at which the particular fractions of coke are oxidised, and R is the natural gas constant.
The heating ramp during TPO of the spent samples was varied from 0.5 to 5 °C min−1 (Figure S19) and the conversion of coke (α) was compared using the observed weight loss or the integrated CO2 formation peak from the analysis of the off gas by means of mass spectrometry (Figure 5). At least 95 % conversion of coke were achieved during the ramp to 500 °C (Figure 5) allowing for the comparison of a broad range of conversion levels. A linear dependency is obtained when analysing the weight change for conversion levels from 10–70 % in the Arrhenius plot (Figure S20), while the integral CO2 formation allows for comparison of 30–90 % and generally provides superior linear trends (Figure 6). Hence, observed adsorption/desorption effects, as well as the formation of carbon complexes (Figure 1a) seemingly affect the measurement of the weight change and can be expected to blur the determination of the activation energy by this isoconversional method. An activation energy of 121.4 kJ mol−1 (Figure S21) was obtained for 50 % conversion based on CO2 formation (132.3 kJ mol−1 based on weight change), which is in the lower range when compared to activation energies for coke formed on catalysts during other catalytic processes such as fluid catalytic cracking (110‐167 kJ mol−1).10c–10e, 14 Lastly, the pre‐exponential factor (A=1.900 ⋅ 104 s−1) and the rate constant of oxidation of coke by O2 (k=8.005 ⋅ 10−4 s−1 at 500 °C) were calculated according to Arrhenius’ law (Equations S6–S9) based on the maximum formation of CO2. The corresponding conversions of coke during TPO with varying heating rate are within 60–65 % and the calculations based on the formation rate of CO2 is expected to be more reliable than the calculation based on the observed weight change.
Figure 5.
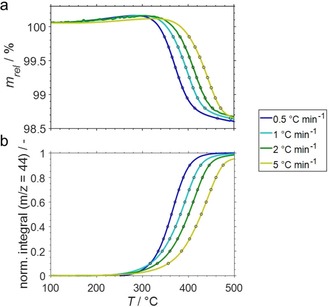
(a) Sample weight relative to the weight prior to exposure to 21 % O2/He at 100 °C (solid) and (b) normalised cumulative formation of CO2 (solid) together with selected data points for isoconversional analysis (10–90 %, open circles) during temperature programmed oxidation of spent SCALMS with an atomic Ga/Rh ratio of 125 as monitored via high‐resolution thermogravimetry coupled with mass spectrometry. The catalyst was previously applied in propane dehydrogenation at 500 °C. Conditions of the experiment: He flow 100 mLN min−1 for TOS<0; He flow 79 mLN min−1 and O2 flow 21 mLN min−1 for TOS>0; GHSV 30000 mLN g−1 h−1.
Figure 6.
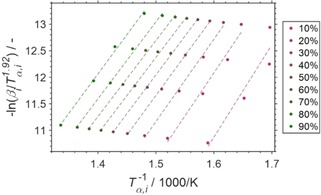
Isoconversional analysis in the Arrhenius plot according to Starink13 of various conversion levels of coke (α, filled circles) with linear trendlines (dashed) as monitored by the formation of CO2 using mass spectrometry during temperature programmed oxidation in 21 % O2/He at different heating rates (β) for a spent SCALMS with an atomic Ga/Rh ratio of 125. The catalyst was previously applied in propane dehydrogenation at 500 °C.
In situ thermogravimetry during propane dehydrogenation with cyclic regeneration
The performance during PDH, the coking behaviour and the regeneration of the most promising SCALMS catalyst in terms of efficient use of precious Rh (Ga/Rh=125) with limited selectivity towards coke (1.0 %) was studied in five consecutive dehydrogenation‐regeneration cycles at 500 °C by means of in situ HRTGA‐MS in the XEMIS sorption analyser. An initial sharp increase of the weight of the pre‐dried sample by ∼0.5 wt% (Figure 7a) may be explained by the adsorption of propane, which strongly interacts with Rh atoms of the alloy at the liquid‐gas interface.5a, 5g The weight increases further by ∼0.2 wt% and peaks at 60 min TOS indicating potential continuous formation of small amounts of coke. However, a subsequent first decrease in weight indicates a second simultaneous processes with opposing effects regarding the sample weight. While the expected formation of coke results in a weight increase, an expected reduction of GaxO species by the in situ formed H2 leads to abstraction of oxygen atoms and water formation and hence decreases the weight. Oxidic gallium species are present in the as‐prepared SCALMS5g as metallic Ga forms an oxide passivation layer when exposed to air. Further, the catalysts were also exposed to various atmospheres and solvents during preparation including oxidative H2O. The reductive dehydrogenation environment is expected to initiate the oxygen abstraction from GaxO species, in particular in the presence of Rh atoms promoting H2 adsorption and dissociation.5g, 15 The formation of H2O as the product of such a reduction with H2 is also identified via mass spectrometry for the first 180 min TOS (Figure S22c). Hence, adsorption and coke formation are seemingly dominating the net weight change in the first 60 min TOS, while the reduction of oxidic gallium overcomes this (most likely) ongoing mass increase after 90 min TOS.
Figure 7.
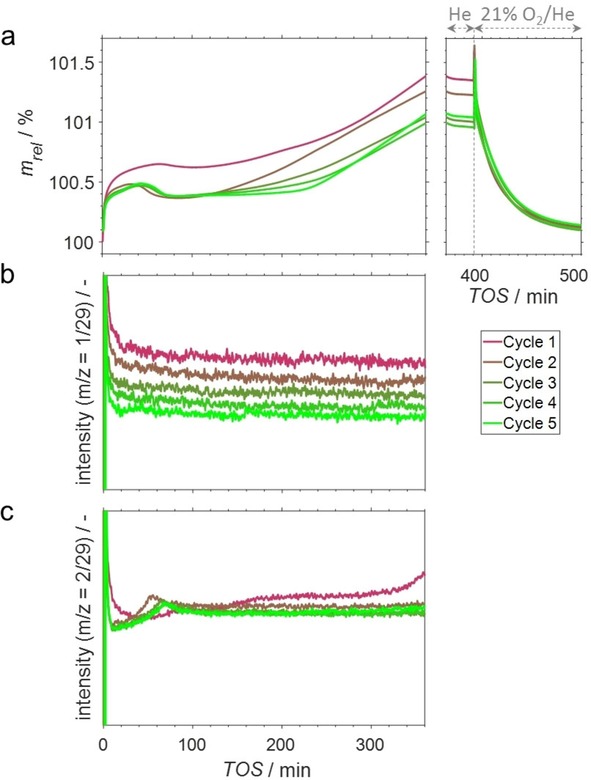
(a) Sample weight relative to the weight prior to propane dehydrogenation during 5 consecutive propane dehydrogenation‐regeneration cycles at 500 °C using a SCALMS with an atomic Ga/Rh ratio of 125 and (b‐c) mass‐to‐charge ratios of 1 (almost exclusively propene) and 2 (almost exclusively H2) relative to the mass‐to‐charge ratio of 29 (exclusively propane) during the dehydrogenation cycles as monitored via high‐resolution thermogravimetry coupled with mass spectrometry. Conditions of the experiment: He flow 80 mLN min−1 and C3H8 flow 20 mLN min−1 for 0<TOS<360 min; He flow 100 mLN min−1 for 360<TOS<390 min; He flow 79 mLN min−1 and O2 flow 21 mLN min−1 for TOS>390 min; GHSV 30000 mLN g−1 h−1.
The weight increases again after ∼120 min TOS (Figure 7a). A constant weight increase is observed during the last 60 min TOS, which indicates a continuous build‐up of coke. Hence, other processes affecting the sample weight are seemingly completed, i. e. the catalyst performs at steady‐state with a constant build‐up of coke. In line with the present findings, such a continuous build‐up of coke has been suggested in literature for alkane dehydrogenation over Pt‐based catalysts.16 After an initial formation of highly dispersed carbonaceous species on the active metal with consequential deactivation of the catalyst, the subsequent migration of these species onto the support material resulted in a continuous build‐up of a more graphitic coke with a less profound effect on the catalyst's activity.
The conversion of propane to propene is qualitatively monitored via mass spectrometry for a mass‐to‐charge ratio of 1 (Figure S23). It is noted, that the fragmentation pattern of propene is almost fully covered by propene, i. e. also the parent ion peak (m/z=41) is strongly affected by the presence of propane. Only a mass‐to‐charge ratio of 1 is exclusively due to the formation of propene with an expected less pronounced contribution of the second dehydrogenation product H2 to this ratio (Figure S23). In order to circumvent initial unsteady flow patterns within the analyser, the intensity ratio of propene (m/z=1)/propane (m/z=29) is analysed (Figure 7b). The latter ratio is the parent ion peak for propane (Figure S23). A continuously decreasing ratio resembles the trend in propane conversion during dehydrogenation in a lab‐scale fixed‐bed reactor.5g However, deactivation of the SCALMS is mostly observed within the first 25 min TOS (Figure 7b; Figure S24) followed by a subsequent robust performance of SCALMS in alkane dehydrogenation,5a, 5g most likely due to an increased resistance against coke formation when compared to traditional heterogeneous dehydrogenation catalysts. Nevertheless, the hypothesised continuous formation of coke results in slow deactivation of the catalyst, most likely via pore blockage due to migration of coke species onto the support material.16 However, the exact location of coke cannot be determined at this stage and will be identified in systematic studies in the future.
An equimolar formation of propene and H2 is expected for the dehydrogenation of propane. However, the formation of H2 (m/z=2) differs drastically from the one of propene (Figure 7c). A minimal net formation rate is observed after 40–50 min TOS due to the reduction of GaxO species, which is strongly suggested by a decrease of the sample weight after 60 min TOS (Figure 7a). The net H2 formation rate suggests a rather slow reduction of such species over 180 min TOS, which is also the time frame for the formation of significant amounts of H2O (Figure S22c). These observations strongly support the previously suggested simultaneous formation of coke and reduction of GaxO species with opposing effects on the catalyst weight.
After flushing the reactor for 30 min with He, the SCALMS was exposed to 21 % O2/He. A pronounced spike of the weight, once again, indicates adsorption of O2 to re‐form Ga oxide species (Figure 7c). Metastable carbon oxide functionalities are expected to volatise immediately at given temperature forming CO2 (Figure S25). The final mass is only increased by 0.1 wt% when compared to the dry sample prior to dehydrogenation indicating full oxidation of coke within only 120 min TOS during regeneration at 500 °C. The marginal weight increase may be caused by minor carbonaceous residuals or a more severe oxidation of the Ga phase after exposure to the oxidising 21 % O2/He atmosphere at 500 °C when compared to the as prepared SCALMS.
The observed weight changes in the subsequent dehydrogenation cycles become repeatable after the second cycle for the first 120 min TOS (Figure 7a). The maximum sample weight shifts to 40 min TOS, after which a distinct weight decrease is observed. Subsequently, the weight is rather constant for increasing TOS with each cycle (constant weight for 140 min TOS during PDH cycle 5) suggesting less coke formation with each dehydrogenation‐regeneration cycle. However, a constant formation of coke is observed during the last 100 min TOS (Figure 7a). Comparable amounts of coke were formed in the last three cycles (∼1 wt%), while larger fractions were accumulated in the first two cycles (1st cycle: 1.39 wt%; 2nd cycle: 1.24 wt%; Figure 7b). Nevertheless, the activity over time decreases with every cycle. Contrarily, the generation of H2 is rather constant in the last three cycles (Figure 7c), which may be explained by additional generation of H2 as a product from excessive abstraction of H from hydrocarbons during coke formation. Hence, this observation is in line with the comparable weight increase of the catalyst in the last three cycles showing a constant formation of coke and H2, while the actual dehydrogenation activity is further decreasing with each cycle.
Conclusions
This detailed analysis of coke in Supported Catalytically Active Liquid Metal Solutions (SCALMS) after catalytic application sheds more light on the effect of liquid alloy catalysis on coke formation. Moreover, our study may serve as blueprint for an elaborate characterisation of coke in spent catalysts by means of temperature programmed oxidation (TPO) and high‐resolution thermogravimetric analysis coupled with mass spectrometry (HRTGA‐MS). Making use of a XEMIS sorption analyser with a superior resolution of the sample weight, the formation of oxygen‐functionalised coke and the formation of GaOx passivation layers was monitored during HRTGA‐MS upon first exposure of the spent SCALMS to oxygen containing atmospheres at 100 °C. Functionalised coke species are typically metastable and rapidly decompose to CO or CO2 at elevated temperatures. However, the low starting temperature of 100 °C allowed for coke functionalisation while preventing major decomposition. Coke was identified in all spent samples and only tiny amounts of H2O were formed during TPO. The overall amount of coke was shown to strongly depend on the reaction temperature and catalyst design, that is classical solid heterogeneous catalysts and SCALMS. Only small amounts of coke (coke selectivity <1.5 %) were identified in Ga−Rh SCALMS after propane dehydrogenation (PDH) at 450 °C. In contrast, twice the amount of coke was identified in a classical heterogeneous Rh/Al2O3 catalyst (coke selectivity>10 %). Remarkably, the selectivity towards coke of SCALMS with an atomic Ga/Rh ratio of 125 decreased with increasing PDH temperature in the temperature range 450–500 °C. This is most likely due to a crossing of the liquidus line resulting in fully liquid supported alloy system. An increased coke formation after PDH at 550 °C may indicate the pronounced contribution of the Al2O3 support material to the coking process that generates reactive “soft” coke. Lastly, in situ HRTGA‐MS of dehydrogenation‐regeneration cycles using Ga−Rh SCALMS at 500 °C provided insight into the evolution of coke during catalysis. Coke is seemingly formed continuously during PDH with the Ga−Rh SCALMS system. In addition, HRTGA‐MS provided valuable information on the regeneration of SCALMS via oxidation of coke. The regeneration was successful, however, a certain degree of deactivation was observed for the first three regeneration cycles. We take the finding that the subsequent regeneration cycles gave almost identical results as strong indication that recyclability of the investigated Ga−Rh SCALMS by oxidative treatment is feasible.
Experimental Section
Preparation of Supported Catalytically Active Liquid Metal Solutions (SCALMS)
At first, (Et3N)GaH3 was synthesised according to the published procedure,17 while triethylammonium chloride (Sigma‐Aldrich) was used instead of trimethylammonium chloride. The compound was not isolated and used as ethereal solution. The Ga content of this solution was determined as follow: 1 mL of ethereal solution was quenched in 10 mL of HCl (∼2 M) and the resulting mixture shortly boiled to evaporate the diethyl ether. The homogeneous solution was diluted to 500 mL and the Ga content determined by inductively coupled plasma atomic emission spectroscopy (ICP−AES). Given the low stability of gallane complexes, the (Et3N)GaH3 solution was analysed immediately after its preparation. The second part of the synthesis procedure includes the decoration of the support material with Ga according to a previously published procedure.5a, 5g A total of 100 g of Al2O3 (Sigma‐Aldrich; grade: Brockmann I, activated, standard; particle size: 0.05–0.15 mm; BET surface area: 155 m2 g−1; pH: 7.0±0.5) was heated under vacuum (1 mbar) at 300 °C overnight in a Schlenck flask. After cool‐down, the carrier material was suspended in 200 mL dry diethyl ether under an inert argon atmosphere. The required amount of an ethereal solution of (Et3N)GaH3 was added to this suspension in order to obtain the targeted loading of Ga (ca. 6–7 % w/w of Ga) with respect to the support. After complete removal of diethyl ether under vacuum at ca. −30 °C, the flask was heated up to 300 °C (ca. 10 °C min−1) until no gaseous products were observed indicating the termination of the gallane decomposition. The resulting grey solid was continuously heated to 300 °C under vacuum (1 mbar) overnight and stored under argon after cool‐down to room temperature. In the last step of the synthesis of Ga−Rh SCALMS, 5 g of Ga decorated Al2O3 were suspended in 50 mL of distilled water under vigorous stirring at 50 °C.5a, 5g The required volume of a stock solution of RhCl3 ⋅ 3H2O (with a nominal Rh concentration of 1 mg ml−1) in distilled water was added to obtain the targeted atomic Ga/Rh ratio taking the loss of Ga due to galvanic replacement18 into account. The resulting suspension was stirred for 15 min, filtered, and thoroughly washed with distilled water (500 ml). The solid obtained was dried in an oven at 130 °C overnight.
Elemental analysis
The Ga and Rh loading and the corresponding Ga/Rh ratios of the SCALMS were determined by ICP−AES (Table S1) using a Ciros CCD (Spectro Analytical Instruments GmbH). The solid samples were digested in 3 : 1 : 1 volumetric ratio of concentrated HCl:HNO3:HF using microwave heating up to 220 °C for 40 min. The instrument was calibrated for Rh (343.489 nm) and Ga (417.206 nm) with standard solutions of the particular elements prior to the analyses.
Propane dehydrogenation (PDH)
Catalytic testing was previously conducted in a fixed‐bed tubular quartz reactor placed inside a tubular split furnace at atmospheric pressure generating the herein analysed spent catalysts (for details on the laboratory set‐up and analysis of the catalytic performance please refer to Raman et al.).5g In short, a total of 1.2 g of catalyst was placed in the fixed‐bed reactor and heated to the desired temperature (450, 480, 500, or 550 °C) at 10 °C/min under a flow of Ar. After 15 min at the final temperature, 9.1 % propane (Linde, purity 2.5) in He (Linde, purity 4.6) were introduced to the feed stream at a total gas hourly space velocity (GHSV) of 4900 mLSTP mLcat −1 h−1 (residence time τ=0.7 s at standard conditions) for ∼20 h.
High‐resolution thermogravimetric analysis coupled with mass spectrometry (HRTGA−MS)
Spent samples were analysed via temperature programmed oxidation (TPO) in 21 % O2/He in order to determine the amount of coke formed during PDH. The weight change was monitored by means of high‐resolution thermogravimetric analysis using a XEMIS sorption analyser (Hiden Isochema)19 equipped with a mass spectrometer (Hiden Analytical). The XEMIS sorption analyser has a high resolution of ±0.1 μg and can be operated at low vacuum or pressures up to 200 bar, while temperatures up to 500 °C can be realised. A total of ∼200 mg of the sample was placed in cylindrical stainless steel mesh sample holder, heated to 120 °C (5 °C min−1) under an inert He atmosphere for 6 h to remove physisorbed H2O in the Al2O3 support material. A subsequent heating to 500 °C (5 °C min−1) for 12 h can be expected to remove chemisorbed H2O. After cool‐down to 100 °C under a continuous He flow, 21 % O2/He was introduced for 1 h at 100 °C. Subsequently, the temperature was increased with a heating rate of 0.5, 1, 2, or 5 °C min−1 up to the maximum temperature of 500 °C for 12 h. The overall flow rate was 100 mL min−1 throughout TPO. The baselines of the particular MS signals were deducted in order to fit the peaks in MATLAB20 with the peakfit.m algorithm of the file exchange database.21 The reproducibility of the experiments in the XEMIS sorption analyser was conducted in three repetitions using the same spent SCALMS sample (Figure S26). The mean confidence interval for the identified sample weight was ±0.0054 wt % (selected confidence level: 95 %) with a corresponding effective resolution of ±11 μg. The mean confidence interval of the mass spectrometer was exemplarily calculated for m/z=44 (CO2) and amounts to ±0.55 % of the maximum ratio detected in the reproducibility studies, which are representative for all samples studied.
In addition to TPO, an in situ HRTGA‐MS experiment was conducted in order to monitor the weight change and off‐gas during 5 consecutive PDH‐regeneration cycles in the XEMIS sorption analyser. A total of 200 mg of as prepared SCALMS with an atomic Ga/Rh ratio of 125 was loaded. At first, the sample was dried in 100 mL min−1 He at 500 °C for 6 h (5 °C min−1). PDH was conducted at 500 °C for 6 h using a feed gas composition of 20 % C3H8/He and followed regeneration of the catalyst in 21 % O2/He for 2 h, i. e. formed coke was oxidised after each dehydrogenation step. The reactor was flushed with 100 mL min−1 He for 0.5 h after each dehydrogenation and regeneration step.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support by the European Research Council is gratefully acknowledged (Project 786475: Engineering of Supported Catalytically Active Liquid Metal Solutions).
M. Wolf, N. Raman, N. Taccardi, M. Haumann, P. Wasserscheid, ChemCatChem 2020, 12, 1085.
References
- 1.
- 1a. Iglesias-Juez A., Beale A. M., Maaijen K., Weng T. C., Glatzel P., Weckhuysen B. M., J. Catal. 2010, 276, 268–279; [Google Scholar]
- 1b. Pham H. N., Sattler J. J. H. B., Weckhuysen B. M., Datye A. K., ACS Catal. 2016, 6, 2257–2264; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Otroshchenko T., Sokolov S., Stoyanova M., Kondratenko V. A., Rodemerck U., Linke D., Kondratenko E. V., Angew. Chem. Int. Ed. 2015, 54, 15880–15883; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 16107–16111. [Google Scholar]
- 2. Sattler J. J. H. B., Ruiz-Martinez J., Santillan-Jimenez E., Weckhuysen B. M., Chem. Rev. 2014, 114, 10613–10653. [DOI] [PubMed] [Google Scholar]
- 3. Im J., Choi M., ACS Catal. 2016, 6, 2819–2826. [Google Scholar]
- 4. Rostom S., de Lasa H., Ind. Eng. Chem. Res. 2018, 57, 10251–10260. [Google Scholar]
- 5.
- 5a. Taccardi N., Grabau M., Debuschewitz J., Distaso M., Brandl M., Hock R., Maier F., Papp C., Erhard J., Neiss C., Peukert W., Görling A., Steinrück H. P., Wasserscheid P., Nat. Chem. 2017, 9, 862–867; [DOI] [PubMed] [Google Scholar]
- 5b. Rupprechter G., Nat. Chem. 2017, 9, 833–834; [DOI] [PubMed] [Google Scholar]
- 5c. Bauer T., Maisel S., Blaumeiser D., Vecchietti J., Taccardi N., Wasserscheid P., Bonivardi A., Görling A., Libuda J., ACS Catal. 2019, 2842–2853; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5d. Esposito D., Nat. Can. 2019, 2, 179–179; [Google Scholar]
- 5e. Hohner C., Kettner M., Stumm C., Schuschke C., Schwarz M., Libuda J., Top. Catal. 2019; [Google Scholar]
- 5f. Kettner M., Maisel S., Stumm C., Schwarz M., Schuschke C., Görling A., Libuda J., J. Catal. 2019, 369, 33–46; [Google Scholar]
- 5g. Raman N., Maisel S., Grabau M., Taccardi N., Debuschewitz J., Wolf M., Wittkämper H., Bauer T., Wu M., Haumann M., Papp C., Görling G., Spiecker E., Libuda J., Steinrück H.-P., Wasserscheid P., ACS Catal. 2019, 9, 9499–9507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Grabau M., Krick Calderón S., Rietzler F., Niedermaier I., Taccardi N., Wasserscheid P., Maier F., Steinrück H.-P., Papp C., Surf. Sci. 2016, 651, 16–21; [Google Scholar]
- 6b. Grabau M., Erhard J., Taccardi N., Calderon S. K., Wasserscheid P., Gorling A., Steinruck H. P., Papp C., Chem. Eur. J. 2017, 23, 17701–17706. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Rostrup-Nielsen J. R., J. Catal. 1984, 85, 31–43; [Google Scholar]
- 7b. Kuznetsov V. L., Usol′tseva A. N., Butenko Y. V., Kinet. Catal. 2003, 44, 726–734. [Google Scholar]
- 8. Biloen P., Dautzenberg F. M., Sachtler W. M. H., J. Catal. 1977, 50, 77–86. [Google Scholar]
- 9. Mahamulkar S., Yin K., Agrawal P. K., Davis R. J., Jones C. W., Malek A., Shibata H., Ind. Eng. Chem. Res. 2016, 55, 9760–9818. [Google Scholar]
- 10.
- 10a. Lear A. E., Brown T. C., Haynes B. S., Symp. Int. Combust. 1991, 23, 1191–1197; [Google Scholar]
- 10b. Le Minh C., Jones R. A., Craven I. E., Brown T. C., Energ. Fuel. 1997, 11, 463–469; [Google Scholar]
- 10c. Le Minh C., Li C., Brown T. C., Catalyst Deactivation 1997, Vol. 111 (Eds.: C. H. Bartholomew, G. A. Fuentes), 1997, pp. 383–390; [Google Scholar]
- 10d. Li C., Le Minh C., Brown T. C., J. Catal. 1998, 178, 275–283; [Google Scholar]
- 10e. Li C., Brown T. C., Carbon 2001, 39, 725–732. [Google Scholar]
- 11.
- 11a. Anres P., Gaune-Escard M., Bros J. P., J. Alloys Compd. 1998, 265, 201–208; [Google Scholar]
- 11b. Daeneke T., Khoshmanesh K., Mahmood N., De Castro I. A., Esrafilzadeh D., Barrow S. J., Dickey M. D., Kalantar-Zadeh K., Chem. Soc. Rev. 2018, 47, 4073–4111. [DOI] [PubMed] [Google Scholar]
- 12. Towfighi J., Sadrameli M., Niaei A., J. Chem. Eng. Jpn. 2002, 35, 923–937. [Google Scholar]
- 13. Starink M. J., Thermochim. Acta 2003, 404, 163–176. [Google Scholar]
- 14.
- 14a. Querini C. A., Fung S. C., Appl. Catal. A 1994, 117, 53–74; [Google Scholar]
- 14b. Kanervo J. M., Krause A. O. I., Aittamaa J. R., Hagelberg P. H., Lipiäinen K. J. T., Eilos I. H., Hiltunen J. S., Niemi V. M., Chem. Eng. Sci. 2001, 56, 1221–1227; [Google Scholar]
- 14c. Ochoa A., Ibarra Á., Bilbao J., Arandes J. M., Castaño P., Chem. Eng. Sci. 2017, 171, 459–470; [Google Scholar]
- 14d. Müller N., Moos R., Jess A., Chem. Eng. Technol. 2010, 33, 103–112. [Google Scholar]
- 15.
- 15a. Redekop E. A., Galvita V. V., Poelman H., Bliznuk V., Detavernier C., Marin G. B., ACS Catal. 2014, 4, 1812–1824; [Google Scholar]
- 15b. Föttinger K., Catal. Today 2013, 208, 106–112. [Google Scholar]
- 16. Liwu L., Tao Z., Jingling Z., Zhusheng X., Appl. Catal. 1990, 67, 11–23. [Google Scholar]
- 17. Shriver D. F., Shirk A. E., Dilts J. A., Inorg. Synth. 1977, 17, 42–45. [Google Scholar]
- 18. Alia S. M., Yan Y. S., Pivovar B. S., Catal. Sci. Technol. 2014, 4, 3589–3600. [Google Scholar]
- 19. Minnick D. L., Turnaoglu T., Rocha M. A., Shiflett M. B., J. Vac. Sci. Technol. A 2018, 36, 050801. [Google Scholar]
- 20.The MathWorks Inc., MATLAB 2018b, Natick, USA, 2018.
- 21. O′Haver T., peakfit.m, Version 9.0, MATLAB File Exchange, 2018. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary