

Abstract
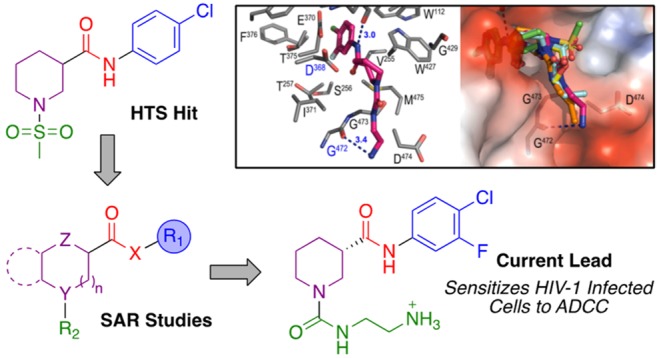
With approximately 37 million people living with HIV worldwide and an estimated 2 million new infections reported each year, the need to derive novel strategies aimed at eradicating HIV-1 infection remains a critical worldwide challenge. One potential strategy would involve eliminating infected cells via antibody-dependent cellular cytotoxicity (ADCC). HIV-1 has evolved sophisticated mechanisms to conceal epitopes located in its envelope glycoprotein (Env) that are recognized by ADCC-mediating antibodies present in sera from HIV-1 infected individuals. Our aim is to circumvent this evasion via the development of small molecules that expose relevant anti-Env epitopes and sensitize HIV-1 infected cells to ADCC. Rapid elaboration of an initial screening hit using parallel synthesis and structure-based optimization has led to the development of potent small molecules that elicit this humoral response. Efforts to increase the ADCC activity of this class of small molecules with the aim of increasing their therapeutic potential was based on our recent cocrystal structures with gp120 core.
Keywords: Antibody-dependent cellular cytotoxicity (ADCC), high throughput screen (HTS), structure−activity relationships (SARs), small molecule CD4 mimetic compounds (CD4mc)
The global acquired immunodeficiency syndrome (AIDS) pandemic caused by human immunodeficiency virus type 1 (HIV-1) represents one of the world’s leading health problems. Approximately 37.9 million people are living with HIV worldwide, with an additional 1.7 million new infections reported last year.1 Current clinical management is not curative and involves combinations of antiretrovirals (highly active antiretroviral therapies, HAART) targeting viral proteins involved in the intracellular replication or the viral entry process.2 Despite what currently appears to be a replete armamentarium of treatment for HIV, the emergence of multidrug resistance and issues of drug toxicity, intolerability, and lack of compliance limit effective therapies. Importantly, current therapies fail to deplete the latent HIV-1 viral reservoir persisting mainly in CD4+ T cells. Therefore, therapy interruption rapidly leads to the reemergence of viral replication and disease progression.2 This obstacle must be overcome to achieve a complete curative effect. Thus, developing novel strategies aimed at eradicating HIV-1 infection remains a critical worldwide challenge.
Entry of HIV-1 into host cells is initiated by attachment of the viral envelope glycoprotein (Env) trimer to the host cell CD4 receptor. The mature, unliganded Env (gp120/gp41)3 trimer adopts a “closed”, flexible, high energy conformation State 1.3,4 CD4 engagement leads to conformational rearrangements at the trimer apex, thus “opening” Env to form a prehairpin intermediate State 3 via a functional and obligate intermediate State 2. Binding of coreceptor (CCR5 or CXCR4) to State 3 then leads to viral entry.3−6 In addition to being expressed on the surface of viral particles, Env is the only virus-specific antigen exposed at the surface of HIV-1 infected cells, and thus represents a major cellular target for host antibodies (Ab).7 Importantly, conserved epitopes on Env that are recognized by Abs can elicit specific Fc-receptor-mediated effector functions that might be important for viral control, including antibody-dependent cellular cytotoxicity (ADCC).8,9 For example, the partially successful RV144 vaccine clinical trial in Thailand (31.2% protected) identified ADCC-mediating Abs, in the presence of low plasma immunoglobulin A (IgA) Env-specific Abs, as a correlate of protection.10 Conformational changes induced by Env:CD4 binding on the surface of infected cells leads to exposure of conserved CD4-induced (CD4i) epitopes on Env that are recognized by Abs able to mediate potent ADCC.8,11−15 To avoid exposure of the vulnerable CD4i epitopes, the virus has evolved sophisticated mechanisms to downregulate CD4 and prevent Env accumulation at the surface of infected cells, via Nef and Vpu accessory protein activities, as well as Vpu-mediated BST-2 antagonism.8,13,16 Evidence suggests that due to their size, small molecules might have an advantage over soluble CD4 (sCD4) to modulate Env conformation at the surface of cells infected with primary viruses, unmasking CD4i epitopes that are recognized by antibodies present in HIV+ sera.14,17 Specifically, sequential binding of small molecule CD4 mimetic compounds (CD4mc) and coreceptor binding site Abs (CoRBS, e.g. 17b) to Env opens the trimer and facilitates recognition by the ADCC-mediating anticluster A family of Abs. This results in the stabilization of the ADCC-vulnerable Env conformation State 2A.18−21
In search for small molecules that promote the ADCC immune response, we performed a high throughput screen (HTS) to identify molecules that open the trimeric Env on the surface of HIV-1 infected cells and expose anti-Env CD4i epitopes. We screened an internal collection (∼108,000 compounds) in a well-established cell-based ELISA (CBE) assay that detects the conformation of trimeric Env at the surface of transfected human osteosarcoma (HOS) cells, to evaluate the impact of different small molecules on HIV-1 Env conformation.22 More specifically, the CBE assay measures recognition (i.e., binding) of trimeric HIV-1 Env by monoclonal Abs, in this case anti-CoRBS Ab 17b, that only binds to “open” CD4i Env conformations. This is now automated and the simpler CBE assay is a useful surrogate for ADCC activity because anti-CoRBS Ab binding is a necessary prerequisite for ADCC mediated by antibodies present in most HIV-1-infected individuals. This effort identified a racemic hit (1, Figure 1), which could stabilize HIV-1 Env in “open” conformations.23 Further studies showed that while the activity of 1 is limited, it specifically binds to gp120 and sensitizes infected cells to modest HIV+ serum-mediated ADCC in a FACS-based assay (see Supporting Information).13,14,24 This assay specifically measures ADCC activity against productively infected cells and is not affected by the confounding effect of uninfected bystander cells.25,26 Encouraged by the modest but specific ADCC activity of 1, a series of parallel synthetic libraries aimed at increasing their capacity to expose the coreceptor binding site of Env was developed and compared by cell-based ELISA to the potent BNM-III-170, which binds within the same pocket18,21,27 and has been shown to protect from immunodeficiency virus infection in vivo.(28,29)
Figure 1.

Structures of HTS hit 1, the active and inactive enantiomers (2 and 3), and the strategy for the SAR study. See the Supporting Information for syntheses of 2 and 3.
Initially we focused on identifying the active conformer of racemic hit 1. Both enantiomers of 1 were synthesized from commercially available (R)- and (S)-4, via a two-step mesylation and EDC-mediated amide coupling procedure. The (S)-enantiomer 2 (MCG-II-153) proved to be the active conformer, while the (R)-enantiomer 3 (MCG-II-156) displayed no activity in the CBE assay. Having identified the eutomer of 1, we looked to modify different regions of the molecule through iterative parallel synthetic library development and to assess the effect on antibody binding in our CBE assay. Our strategy involved the design of libraries incorporating modifications of the following regions (Figure 1): (i) 4-chloro substituted aromatic ring: Region I; (ii) amide linker: Region II; (iii) piperidine core: Region III; and (iv) sulfonamide substituent: Region IV. At the time the work was conducted, no structural information on the compound’s specific binding site on gp120 was available. Therefore, we looked to augment the structure activity relationship (SAR) development via expedient syntheses that would produce libraries of compounds incorporating different diversity elements in Regions I to IV via late-stage diversification of common intermediates.
The initial library explored alternative functional groups attached to the benzene ring of Region I in place of the para-chlorine atom. This library was prepared by reaction of common acid chloride intermediate 5 (R: Me) with a variety of anilines (Scheme 1). The set chosen consisted of ortho-, meta-, para-, and disubstituted anilines in order to assess the tolerance for different functionality on the aromatic ring. While the 4-Br analog (6a) from the initial library displayed comparable activity to 2 (Table 1), the 4-chloro-3-fluoro-substituted compound (6l, MCG-III-027-D05) displayed enhanced 17b antibody binding. Based on this improved 17b binding activity of 6l, a library of 3,4-dihalo-substituted analogs was prepared that also included methyl-, ethyl-, and phenyl sulfonamides. The library was designed to test whether the 4-Cl-3-F-substitution could be replaced with other combinations of 3,4-dihalo-substituted analogs, as well as explore replacement of the methyl substitution on the sulfonamide of Region IV. To this end, common intermediates 5 (R: Me, Et, or Ph) were reacted with various 3,4-dihaloanilines to afford analogs 6 (Scheme 1).
Scheme 1. Synthesis of Region I Analogs.

Table 1. Structures and Activities of Select Analogs.
| entry | R | R′ | Z | n | ring | 17b |
|---|---|---|---|---|---|---|
| 2 | Me | 4-Cl | - | - | - | 0.31 |
| 3 | Me | 4-Cl | - | - | - | |
| 6a | Me | 4-Br | - | - | - | 0.36 |
| 6l | Me | 4-Cl-3-F | - | - | - | 0.58 |
| 6r | Et | 4-Cl-3-F | - | - | - | 0.35 |
| 6x | Ph | 4-Cl-3-F | - | - | - | 0.45 |
| 7a | Me | - | - | - | - | 0.08 |
| 8a | Me | - | - | - | - | 0.07 |
| 9 | - | - | - | - | 0.21 | |
| 11a | Me | - | CH2 | 0 | No | 0.36 |
| 11e | Me | - | O | 1 | No | 0.28 |
| 11l | Me | - | NH | 1 | No | 0.10 |
| 11p | Me | - | CH2 | 1 | Yes | 0.24 |
| 11t | Me | - | CH2 | 1 | No | 0.42 |
| 13 | - | - | - | - | - | 0.48 |
| (+)-13 | - | - | - | - | - | 0.58 |
| (−)-13 | - | - | - | - | - | 0.28 |
| 16a | Me | - | - | - | - | 0.50 |
| 16b | Et | - | - | - | - | 0.53 |
| 16d | nPr | - | - | - | - | 0.51 |
| 16e | iBu | - | - | - | - | 0.62 |
| 17d | nPr | H | - | - | - | 0.55 |
| 17f | (CH2)2NH3+ | H | - | - | - | 0.58 |
| 17g | (CH2)3NH3+ | H | - | - | - | 0.25 |
| 18b | nPr | - | - | - | - | 0.85 |
| 20 | (CH2)2NH3+ | - | - | - | - | 0.07 |
| 21a | nPr | - | - | - | - | 0.11 |
| 21b | (CH2)2NH3+ | - | - | - | - | 0.06 |
| 21c | (CH2)2NH3+ | - | - | - | - | 0.06 |
17b binding measured by CBE assay normalized to BNM-III-170 (17b binding in the presence of BNM-III-170 = 1 and in the absence of CD4mc is <0.05). See Supporting Information for assay details and an extended Table containing all analogs.
The 4-chloro-3-fluoro analogs (6l, 6r, and 6x, Table 1), not dissimilar to another structural class of small molecule CD4mc exemplified by BNM-III-170,27 proved to be the most effective in our CBE assay with alternate halogenation patterns proving less active. Notably, however, the phenyl derivatives all displayed poor aqueous solubility as evaluated by visual inspection. Efforts to improve the solubility and activity of 6x via the addition of polar groups to the benzene ring or replacement with heterocycles proved unsuccessful (see Supporting Information). Following the results of the initial compound libraries, our attention turned toward replacement of other regions of 2, while maintaining the 4-chloro-3-fluoro aromatic ring, to evaluate further the SAR of this class of molecules.
In an effort to understand the basic pharmacophore of this series of molecules, replacement of the amide linker in Region II with inverse amide (7) and ester (8) linkages was examined (Figure 2). Both replacements led to inactive analogs, as illustrated by 7a and 8a (R: Me, Table 1). Overall, this suggested that the original amide linker is important to expose the coreceptor binding site (17b binding); therefore, Region II modifications were deprioritized. In addition, the 4-position regioisomer 9 was also prepared and tested. This analog also exhibited lower 17b binding activity, thus validating the importance of the regiochemistry of the amide substituent.
Figure 2.

Structures of inverse amide (7) and ester (8) linked Region II analogs and regioisomeric analog 9. See the Supporting Information for syntheses.
Based on the modest improvements in 17b binding activity observed with the initial analogs (6–9), our attention turned to replacement of the piperidine core (Region III) with other nitrogen-based heterocycles (Scheme 2). Libraries of compounds in which the piperidine core was replaced with pyrrolidine, morpholine, piperazine, and tetrahydroquinoline were prepared (Scheme 2). The pyrrolidine, morpholine, and piperazine analogs all showed lower 17b binding activity (exemplified by 11a, 11e, and 11l, respectively, R:Me) to the racemic piperidine matched pair (e.g., 11t), with the tetrahydroquinoline analogs (e.g., 11p, R:Me) exhibiting poor aqueous solubility. Overall, replacement of the piperidine core with alternate N-heterocycles did not notably improve 17b binding activity.
Scheme 2. Synthesis of N-Heterocyclic Core Analogs.

In addition to the nitrogen-based heterocycles, the tetrahydropyran derivative (13, Scheme 3) was synthesized to investigate the importance of the piperidine nitrogen atom. Interestingly, racemic 13 exhibited promising biological activity. Importantly, we were able to identify chiral HPLC conditions to separate the enantiomers. Optical rotations were recorded, and the (+)-enantiomer proved to be more active.
Scheme 3. Synthesis of Tetrahydropyran Analog.

Having explored modifications to Regions I–III and various sulfonamide derivatives in Region IV with only minor improvements in 17b binding activity, our iterative synthetic library design strategy turned to introducing replacements of the sulfonamide substituent on the piperidine core to assess the impact on 17b antibody binding. The synthetic strategy focused on late-stage derivatization of common intermediate (+)-14, which was treated with various electrophiles to yield analogs with alternately N-substituted piperidine cores (Scheme 4). In particular, analogs in which the sulfonamide (15) was replaced with carbamate (16), urea (17), and guanidine (18) moieties were prepared. Gratifyingly, analogs 16–18 all exhibited improved activity. In particular, the carbamate analogs (16a–e) displayed marked improvement in 17b binding activity (Table 1) compared to their direct sulfonamide comparators, although the phenyl derivative (16c) exhibited poor aqueous solubility. The urea and guanidine analogs also displayed good 17b binding activity, particularly the n-propyl substituted analogs (17d and 18b, respectively).
Scheme 4. Synthesis of N-Substituted Piperidine Analogs.

Based on the literature precedent,30 a small library of analogs in which the 4-chloro substituent was replaced with a trifluoromethyl group was also prepared. From a common intermediate similar to 14 (CF3 in place of Cl), different N-substitutions were explored. The trifluoromethyl analogs displayed comparable, though slightly diminished, 17b binding activity to their chloro-substituted matched pairs (see Supporting Information). This suggests that, while the trifluoromethyl substituent is a reasonable replacement for the chlorine atom, the 4-chloro-3-fluoro substitution proved superior.
Isothermal titration calorimetry (ITC) was next used to evaluate the binding affinities of select analogs with monomeric YU-2 gp120 (Figure 3). Compound 2, the (S)-enantiomer of the original HTS hit, exhibited poor binding affinity with an estimated Kd value of >10 μM. The addition of the 3-fluoro substituent in analog 6l improved the binding affinity with a measured Kd value of 2.8 μM. The carbamate analogs 16a and 16b also displayed higher binding affinities with Kd values of 1.8 and 0.77 μM, respectively. Importantly, the improved binding affinity observed correlated with higher 17b Ab binding.
Figure 3.
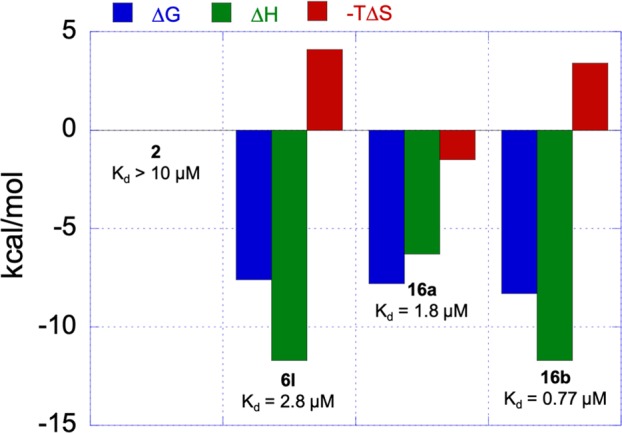
Binding thermodynamics as determined by ITC employing YU-2 gp120.
Our approach to develop active compounds capable of triggering the HIV Env trimer to expose ADCC vulnerable epitopes was then coupled with structural analyses of our more active molecules with gp120 antigen. The structure-based design was guided by the X-ray crystal structures of complexes formed between compounds A, 6l; B, 16a; C, 16b; D, 17d and LM/HT gp120CRF01_AE core (Figure 4, for data collection and refinement statistics for the structures, see ref (23)). As shown in Figure 4 the small molecules bind in the previously disclosed cavity of gp120 that binds the Phe43 of the CD4 receptor.27,31 This result is perhaps unsurprising due to the similarity of the 4-chloro-3-fluoro substituted aromatic ring attached to an amide (or oxalamide) linker observed in both families of small molecule CD4mc (vide supra).27
Figure 4.
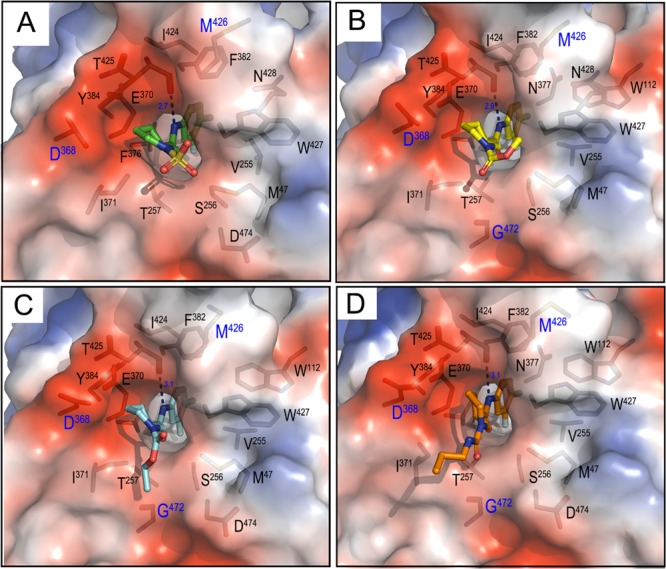
Protein–analog structures with LM/HT gp120CRF01_AE core: A, 6l (PDB code: 6ONV); B, 16a (PDB code: 6ONE); C, 16b (PDB code: 6ONF); D, 17d (PDB code: 6ONVH). Structures were aligned based on gp120, and the electrostatic potential is displayed over the gp120 molecular surface colored red for negative, blue for positive, and white for apolar. Analogs are shown in a ball–stick representation, and residues lining the analogs binding cavities (buried at the interface as calculated by PISA software) are shown as sticks. There is one direct H-bond at the contact interface of each analog formed between Thr425 O of gp120 and amide N of the compound. Key conserved residues in the Phe43 pocket: Asp368, Gly472, and Met426 are labeled in blue.
Structural analyses indicated that in addition to hydrophobic contacts of the 4-chloro-3-fluoro substituted aromatic ring observed deeply within the Phe43 cavity, the compounds interact with highly conserved gp120 residues of the Phe43 cavity rim, including Asp368, Gly472, and Met426. These interactions are mediated mostly through residues of the piperidine ring (Figures 4 and 5). Based on these analyses, we hypothesized that the addition of an amino group at the terminus of the carbamate or urea alkyl chain and/or at the 3-position of the piperidine core could establish additional hydrogen bonding interactions with Asp368, Gly472, and/or Met426, key conserved residues in the Phe43 pocket rim.
Figure 5.
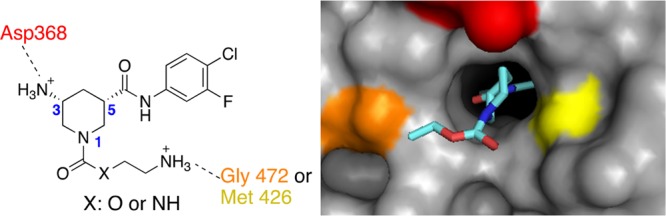
Design of new analogs with additional amino groups.
Synthetic efforts thus turned toward identifying an enantioselective synthesis of our designed analogs (Figure 6) containing an additional amino group directly attached to the piperidine core. Unfortunately, all of the analogs containing a 3-amino substituent proved inactive in our CBE assays (20a and 21a–c, Table 1). Furthermore, crystallographic analysis of 21a with gp120 monomer revealed that the compound adopted a different conformation in the solvent exposed vestibule of the Phe43 pocket than predicted computationally (see Supporting Information for gp120 cocrystal structure). Thus, the desired hydrogen bonding interaction with Asp368 and the 3-amino substituent was not realized.
Figure 6.

Designed analogs with additional amino group. See the Supporting Information for syntheses.
Preparation of compounds with an amino group at the terminus of the alkyl carbamate or urea was next achieved in a similar fashion to that of the alkyl congeners (Scheme 4). Excitingly, the amino-substituted compounds revealed improved 17b binding activity compared to the direct alkyl analogs. Compound 17f (MCG-IV-210, Figure 7) is therefore currently the lead compound for this structural class of small molecules that sensitize HIV-1 infected cells to ADCC (Figure 8). Structural analyses of 17f in complex with LM/HT gp120CRF01_AE core reveal that 17f establishes an additional H-bond with a main chain atom of Gly472 (Figure 7) and has the highest buried surface area (BSA) at the interface, 692 Å2 (as compared with other compounds with BSA in a range of 610–683 Å2).
Figure 7.
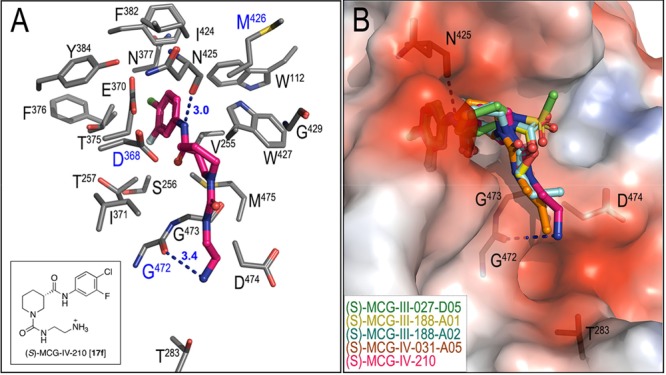
Structure of lead compound 17f in complex with LM/HT gp120CRF01_AE core (PDB code: 6P9N). (A) The 17f binding interface. LM/HT gp120CRF01_AE core residues contributing to 17f compound binding are shown as sticks. H-bonds are shown as blue dashes. (B) Comparison of bound conformations of 17f to other compounds. Structures were aligned based on gp120 and the electrostatic potential displayed over the gp120 molecular surface of the 17f-LM/HT gp120CRF01_AE core complex. Compounds are shown as sticks and H-bonds formed by 17f shown as blue dashes. Compound 17f is the only compound that establishes an H-bond with the main chain atoms of Gly472.
Figure 8.

CD4mc sensitize HIV-1 infected cells to ADCC. Primary CD4 T cells isolated from PBMC were infected with HIV-1CH58TF for 48 h. For cell surface staining, 5 μg/mL 17b (A) or 1:1000 diluted HIV+ plasma (n = 5) (B) were used in the presence of 50 μM of the different MCG analogs, (+)-BNM-III-170, or with equivalent volume of vehicle (DMSO), and an Alexa Fluor 647-conjugated antihuman IgG secondary Ab was then used for fluorescent labeling. For ADCC (C), infected cells were used as target cells in a FACS-based ADCC assay that measures the killing of infected (p24+ cells) to determine their susceptibility to ADCC mediated by a 1:1000 dilution of plasma from 5 HIV-1-infected individuals in the presence of 50 μM different MCG analogs, (+)-BNM-III-170, or with equivalent volume of vehicle (DMSO). Data shown are the mean ± SD of three independent experiments. Statistical significance was evaluated using paired t test (*, P < 0.05; **, P < 0.01; ***, p < 0.001; ns, not significant).
To determine if there was a correlation between anti-CoRBS Ab 17b binding and ADCC activity for this structural class of small molecule CD4mc, we evaluated the ability of select active analogs to sensitize primary CD4+ T cells infected with a clinically relevant transmitted/founder virus (CH58TF) to HIV+ serum-mediated ADCC in our previously described flow cytometry-based assay14 (Figure 8). Analogs 6l (MCG-III-027-D05), 16a (MCG-III-188-A01), 16b (MCG-III-188-A02), 17d (MCG-IV-031-A05), and 17f (MCG-IV-210) all enhanced the recognition of infected cells by the anti-CoRBS 17b antibody (Figure 8A) and by sera from HIV-1 infected individuals (Figure 8B). Importantly, these analogs displayed improved ADCC activity (Figure 8C), thus revealing a correlation between enhanced 17b/HIV+ sera Ab binding and ADCC activity.
In summary, we have identified a new class of small molecule CD4mc that “open-up” Env, thus allowing enhanced recognition and binding by antibodies resulting in the sensitization of HIV-1 infected cells to ADCC. Use of parallel synthesis and purification enabled rapid access to libraries of analogs of the original HTS hit. Addition of a fluorine atom ortho to the chlorine atom of the original hit improved binding, while other modifications to the aromatic ring were poorly tolerated. Replacement of the piperidine core did not improve 17b binding activity. The most substantial improvement in Env opening and 17b Ab binding was observed upon replacing the sulfonamide substituent attached to the piperidine nitrogen with urea, carbamate, and guanidine substituents. Addition of a terminal amino group on the alkyl urea (e.g., 17f, MCG-IV-210) further improved the capacity of the compound to expose the CoRBS. Importantly, the most active analogs in our CBE assay displayed improved ADCC activity over the HTS hit and were recently shown to have some viral neutralization activity.23 With the specific binding site on gp120 identified, current efforts are focused on using computational modeling and structural data to augment the rational design of more potent analogs.
Acknowledgments
The authors thank the CRCHUM BSL3 and Flow Cytometry Platforms, Jean Duchaine and Dominic Salois from the IRIC HTS platform, the FRQS AIDS and Infectious Diseases network, and Mario Legault for cohort coordination and clinical samples. Dr. Charles W. Ross, III and Drs. Simon Berritt and Charles E. Hendrick (University of Pennsylvania) are also acknowledged for their assistance obtaining mass spectra and utilizing the mass-directed purification system, respectively.
Glossary
ABBREVIATIONS
- AIDS
acquired immunodeficiency syndrome
- HIV-1
human immunodeficiency virus type 1
- HAART
highly active antiretroviral therapies
- Env
envelope glycoprotein
- Ab
antibodies
- ADCC
antibody-dependent cellular cytotoxicity
- CD4i
CD4-induced
- CD4mc
CD4 mimetic compounds
- CoRBS
coreceptor binding site
- HTS
high throughput screen
- CBE
cell-based ELISA
- HOS
human osteosarcoma
- DMF
N,N-dimethylformamide
- DCM
dichloromethane
- TFA
trifluoroacetic acid
- HATU
hexafluorophosphate azabenzotriazole tetramethyl uronium
- DIPEA
diisopropylethylamine
- EDC
1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide
- ITC
isothermal titration calorimetry
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00445.
Synthesis, experimental methods, cell-based ELISA results, computational modeling, and crystallographic data (PDF)
Author Contributions
∇ Equal contribution. A.F. designed the HTS screening. M.C.G. and A.B.S designed and synthesized analogs. W.D.T., R.S., and M.P. performed structural studies of analogs. S.D., D.V., and J.P.C. performed CBE and ADCC experiments. A.S. performed ITC experiments. M.C.G., A.F., and A.B.S. wrote the manuscript. All authors have read, edited, and approved the final manuscript.
Funding was provided by Program Project (P01 GM 56550) to M.C.G., A.F., and A.B.S.; this work was also supported by an internal CRCHUM grant (for the HTS screening) awarded to A.F., by a CIHR foundation grant #352417 to A.F., and by a NIH R01 to A.F. and M.P. (AI129769) and to M.P. (AI116274). A.F. is the recipient of a Canada Research Chair on Retroviral Entry # RCHS0235. S.D. is the recipient of a FRSQ postdoctoral fellowship award.
The views expressed in this presentation are those of the authors and do not reflect the official policy or position of the Uniformed Services University, US Army, the Department of Defense, or the US Government.
The authors declare no competing financial interest.
Supplementary Material
References
- UNAIDS . Global HIV & AIDS statistics – 2019 fact sheet. http://www.unaids.org/en/resources/fact-sheet (accessed October 24, 2019).
- Delannoy A.; Poirier M.; Bell B. Cat and Mouse: HIV Transcription in Latency, Immune Evasion and Cure/Remission Strategies. Viruses 2019, 11, 269. 10.3390/v11030269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herschhorn A.; Gu C.; Castillo-Menendez L.; Sodroski J. G.; Ma X.; Ventura J. D.; Mothes W.; Melillo B. N.; Smith A. B. III; Terry D. S.; et al. Release of Gp120 Restraints Leads to an Entry-Competent Intermediate State of the HIV-1 Envelope Glycoproteins. mBio 2016, 7 (5), e01598 10.1128/mBio.01598-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro J. B.; Gorman J.; Ma X.; Zhou Z.; Arthos J.; Burton D. R.; Koff W. C.; Courter J. R.; Smith A. B. III; Kwong P. D.; et al. Conformational Dynamics of Single HIV-1 Envelope Trimers on the Surface of Native Virions. Science 2014, 346 (6210), 759–763. 10.1126/science.1254426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herschhorn A.; Gu C.; Moraca F.; Ma X.; Farrell M.; Smith A. B. III; Pancera M.; Kwong P. D.; Schön A.; Freire E.; et al. The Β20−Β21 of Gp120 Is a Regulatory Switch for HIV-1 Env Conformational Transitions. Nat. Commun. 2017, 8 (1), 1049. 10.1038/s41467-017-01119-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro J. B.; Mothes W. Structure and Dynamics of the Native HIV-1 Env Trimer. J. Virol. 2015, 89 (11), 5752–5755. 10.1128/JVI.03187-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J.; Prévost J.; Alsahafi N.; Ding S.; Finzi A. Impact of HIV-1 Envelope Conformation on ADCC Responses. Trends Microbiol. 2018, 26 (4), 253–265. 10.1016/j.tim.2017.10.007. [DOI] [PubMed] [Google Scholar]
- Veillette M.; Coutu M.; Richard J.; Batraville L.-A.; Dagher O.; Bernard N.; Tremblay C.; Kaufmann D. E.; Roger M.; Finzi A. The HIV-1 Gp120 CD4-Bound Conformation Is Preferentially Targeted by Antibody-Dependent Cellular Cytotoxicity-Mediating Antibodies in Sera from HIV-1-Infected Individuals. J. Virol. 2015, 89 (1), 545–551. 10.1128/JVI.02868-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forthal D. N.; Finzi A. Antibody-Dependent Cellular Cytotoxicity (ADCC) in HIV Infection. AIDS 2018, 32 (17), 1. 10.1097/QAD.0000000000002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes B. F.; Gilbert P. B.; McElrath M. J.; Zolla-Pazner S.; Tomaras G. D.; Alam S. M.; Evans D. T.; Montefiori D. C.; Karnasuta C.; Sutthent R.; et al. Immune-Correlates Analysis of an HIV-1 Vaccine Efficacy Trial. N. Engl. J. Med. 2012, 366 (14), 1275–1286. 10.1056/NEJMoa1113425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding S.; Tolbert W. D.; Prévost J.; Pacheco B.; Coutu M.; Debbeche O.; Xiang S.-H.; Pazgier M.; Finzi A. A Highly Conserved Gp120 Inner Domain Residue Modulates Env Conformation and Trimer Stability. J. Virol. 2016, 90 (19), 8395–8409. 10.1128/JVI.01068-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prévost J.; Richard J.; Ding S.; Pacheco B.; Charlebois R.; Hahn B. H.; Kaufmann D. E.; Finzi A. Envelope Glycoproteins Sampling States 2/3 Are Susceptible to ADCC by Sera from HIV-1-Infected Individuals. Virology 2018, 515, 38–45. 10.1016/j.virol.2017.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veillette M.; Désormeaux A.; Medjahed H.; Gharsallah N.-E.; Coutu M.; Baalwa J.; Guan Y.; Lewis G.; Ferrari G.; Hahn B. H.; et al. Interaction with Cellular CD4 Exposes HIV-1 Envelope Epitopes Targeted by Antibody-Dependent Cell-Mediated Cytotoxicity. J. Virol. 2014, 88 (5), 2633–2644. 10.1128/JVI.03230-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J.; Veillette M.; Brassard N.; Iyer S. S.; Roger M.; Martin L.; Pazgier M.; Schön A.; Freire E.; Routy J.-P.; et al. CD4Mimetics Sensitize HIV-1-Infected Cells to ADCC. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (20), e2687–94. 10.1073/pnas.1506755112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y.; Pazgier M.; Sajadi M. M.; Kamin-Lewis R.; Al-Darmarki S.; Flinko R.; Lovo E.; Wu X.; Robinson J. E.; Seaman M. S.; et al. Diverse Specificity and Effector Function among Human Antibodies to HIV-1 Envelope Glycoprotein Epitopes Exposed by CD4 Binding. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (1), e69–78. 10.1073/pnas.1217609110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias J. F.; Heyer L. N.; von Bredow B.; Weisgrau K. L.; Moldt B.; Burton D. R.; Rakasz E. G.; Evans D. T. Tetherin Antagonism by Vpu Protects HIV-Infected Cells from Antibody-Dependent Cell-Mediated Cytotoxicity. Proc. Natl. Acad. Sci. U. S. A. 2014, 111 (17), 6425–6430. 10.1073/pnas.1321507111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J.; Prevost J.; Ding S.; Brassard N.; Medjahed H.; Coutu M.; Kaufmann D. E.; Finzi A.; von B. B.; Evans D. T. BST-2 Expression Modulates Small CD4-Mimetic Sensitization of HIV-1-Infected Cells to Antibody-Dependent Cellular Cytotoxicity. J. Virol. 2017, 91 (11), e00219-17. 10.1128/JVI.00219-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J.; Pacheco B.; Gohain N.; Veillette M.; Ding S.; Alsahafi N.; Tolbert W. D.; Prévost J.; Chapleau J.-P.; Coutu M.; et al. Co-Receptor Binding Site Antibodies Enable CD4-Mimetics to Expose Conserved Anti-Cluster A ADCC Epitopes on HIV-1 Envelope Glycoproteins. EBioMedicine 2016, 12, 208–218. 10.1016/j.ebiom.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolbert W. D.; Gohain N.; Veillette M.; Chapleau J.-P.; Orlandi C.; Visciano M. L.; Ebadi M.; DeVico A. L.; Fouts T. R.; Finzi A.; et al. Paring Down HIV Env: Design and Crystal Structure of a Stabilized Inner Domain of HIV-1 Gp120 Displaying a Major ADCC Target of the A32 Region. Structure 2016, 24 (5), 697–709. 10.1016/j.str.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand S. P.; Prevost J.; Baril S.; Richard J.; Medjahed H.; Chapleau J.-P.; Finzi A.; Tolbert W. D.; Pazgier M.; Kirk S.; et al. Two Families of Env Antibodies Efficiently Engage Fc-Gamma Receptors and Eliminate HIV-1-Infected Cells. J. Virol. 2019, 93 (3), e01823 10.1128/JVI.01823-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsahafi N.; Bakouche N.; Kazemi M.; Richard J.; Ding S.; Bhattacharyya S.; Das D.; Anand S. P.; Prévost J.; Tolbert W. D.; et al. An Asymmetric Opening of HIV-1 Envelope Mediates Antibody-Dependent Cellular Cytotoxicity. Cell Host Microbe 2019, 25 (4), 578–587. 10.1016/j.chom.2019.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veillette M.; Coutu M.; Richard J.; Batraville L.-A.; Désormeaux A.; Roger M.; Finzi A. Conformational Evaluation of HIV-1 Trimeric Envelope Glycoproteins Using a Cell-Based ELISA Assay. J. Visualized Exp. 2014, 91, 51995. 10.3791/51995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding S.; Grenier M. C.; Tolbert W. D.; Vézina D.; Sherburn R.; Richard J.; Prévost J.; Chapleau J.-P.; Gendron-Lepage G.; Medjahed H.; et al. A New Family of Small-Molecule CD4-Mimetic Compounds Contact the Highly Conserved Aspartic Acid 368 of HIV-1 Gp120 and Mediates ADCC. J. Virol. 2019, JVI, 01325-19. 10.1128/JVI.01325-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J.; Veillette M.; Batraville L.-A.; Coutu M.; Chapleau J.-P.; Bonsignori M.; Bernard N.; Tremblay C.; Roger M.; Kaufmann D. E.; et al. Flow Cytometry-Based Assay to Study HIV-1 Gp120 Specific Antibody-Dependent Cellular Cytotoxicity Responses. J. Virol. Methods 2014, 208, 107–114. 10.1016/j.jviromet.2014.08.003. [DOI] [PubMed] [Google Scholar]
- Richard J.; Prevost J.; Baxter A. E.; Ding S.; Medjahed H.; Delgado G. G.; Brassard N.; Kaufmann D. E.; Finzi A.; Richard J.; et al. Uninfected Bystander Cells Impact the Measurement of HIV-Specific Antibody-Dependent Cellular Cytotoxicity Responses. mBio 2018, 9 (2), e00358 10.1128/mBio.00358-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J.; Veillette M.; Ding S.; Zoubchenok D.; Alsahafi N.; Coutu M.; Brassard N.; Park J.; Courter J. R.; Melillo B. N.; et al. Small CD4Mimetics Prevent HIV-1 Uninfected Bystander CD4 + T Cell Killing Mediated by Antibody-Dependent Cell-Mediated Cytotoxicity. EBioMedicine 2016, 3, 122–134. 10.1016/j.ebiom.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melillo B. N.; Liang S.; Park J.; Schön A.; Courter J. R.; LaLonde J. M.; Wendler D. J.; Princiotto A. M.; Seaman M. S.; Freire E.; et al. Small-Molecule CD4-Mimics: Structure-Based Optimization of HIV-1 Entry Inhibition. ACS Med. Chem. Lett. 2016, 7 (3), 330–334. 10.1021/acsmedchemlett.5b00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madani N.; Princiotto A. M.; Mach L.; Ding S.; Prevost J.; Richard J.; Hora B.; Sutherland L. L.; Zhao C. A.; Conn B. P.; et al. A CD4-Mimetic Compound Enhances Vaccine Efficacy against Stringent Immunodeficiency Virus Challenge. Nat. Commun. 2018, 9 (1), 2363. 10.1038/s41467-018-04758-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Princiotto A. M.; Sodroski J. G.; Madani N.; Vrbanac V. D.; Tager A. M.; Melillo B. N.; Park J.; Smith A. B. III A Small-Molecule CD4-Mimetic Compound Protects Bone Marrow-Liver-Thymus Humanized Mice From HIV-1 Infection. J. Infect. Dis. 2018, 218 (3), 471–475. 10.1093/infdis/jiy174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curreli F.; Belov D. S.; Ahmed S.; Ramesh R. R.; Kurkin A. V.; Altieri A.; Debnath A. K. Synthesis, Antiviral Activity, and Structure-Activity Relationship of 1,3-Benzodioxolyl Pyrrole-Based Entry Inhibitors Targeting the Phe43 Cavity in HIV-1 Gp120. ChemMedChem 2018, 13 (21), 2332–2348. 10.1002/cmdc.201800534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courter J. R.; Madani N.; Sodroski J. G.; Schön A.; Freire E.; Kwong P. D.; Hendrickson W. A.; Chaiken I. M.; LaLonde J. M.; Smith A. B. III Structure-Based Design, Synthesis and Validation of CD4-Mimetic Small Molecule Inhibitors of HIV-1 Entry: Conversion of a Viral Entry Agonist to an Antagonist. Acc. Chem. Res. 2014, 47 (4), 1228–1237. 10.1021/ar4002735. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.