

Abstract
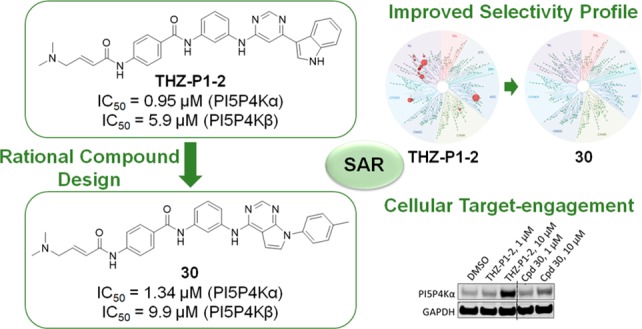
Phosphatidylinositol 5-phosphate 4-kinases (PI5P4Ks) are important molecular players in a variety of diseases, such as cancer. Currently available PI5P4K inhibitors are reversible small molecules, which may lack selectivity and sufficient cellular on-target activity. In this study, we present a new class of covalent pan-PI5P4K inhibitors with potent biochemical and cellular activity. Our designs are based on THZ-P1-2, a covalent PI5P4K inhibitor previously developed in our lab. Here, we report further structure-guided optimization and structure–activity relationship (SAR) study of this scaffold, resulting in compound 30, which retained biochemical and cellular potency, while demonstrating a significantly improved selectivity profile. Furthermore, we confirm that the inhibitors show efficient binding affinity in the context of HEK 293T cells using isothermal CETSA methods. Taken together, compound 30 represents a highly selective pan-PI5P4K covalent lead molecule.
Keywords: PI5P4K, lipid kinase inhibitors, structure−activity relationship, covalent inhibitors, phosphoinositide, drug discovery
The phosphatidylinositol 5-phosphate 4-kinases (PI5P4Ks), consisting of the three isoforms, PI5P4Kα, β, and γ, are lipid kinases that catalyze phosphorylation of phosphatidylinositol 5-phosphate (PI5P) on its 4-position to form phosphatidylinositol-4,5-bisphosphate (PI-4,5-P2).1 In the cellular membrane, PI-4,5-P2 is also produced by another signaling pathway in which phosphatidylinositol 4-phosphate (PI4P) is phosphorylated by phosphatidylinositol 4-phosphate 5-kinases (PI4P5Ks). Although the majority of PI-4,5-P2 is produced via the PI4P5K pathway, the PI5P4Ks have been recognized as key regulators of many cell functions including metabolism, stress response, autophagy, and immunological processes.2−8 Dysregulation of the PI5P4K signaling pathway has been further linked to diseases such as diabetes,3 neurodegenerative disorders,5 and cancers.9−11 These findings indicate that the inhibition of PI5P4K kinase activity with a small molecule inhibitor might have a therapeutic potential across various diseases.
To date, several scaffolds have been developed as PI5P4K inhibitors.5,12−15 For example, I-OMe Tyrphostin AG 538 was reported to inhibit PI5P4Kα with an IC50 of 1 μM,12 while NIH-12848 was found to be a PI5P4Kγ selective compound15 (Figure 1). However, though some of the reported compounds might seem able to covalently label kinases, such as a131, these inhibitors are reversible inhibitors with micromolar biochemical potency against PI5P4K, which limits their use in the investigation of PI5P4K function in the cellular context (Supporting Figure 6). In general, irreversible binding through a unique cysteine represents a viable strategy to improve potency and selectivity.16 We recently reported the discovery of THZ-P1-2 as a pan-PI5P4K covalent small molecule inhibitor by targeting conserved cysteines across all three PI5P4K isoforms that are situated outside of the ATP-binding pocket. Here we report the structure–activity relationship (SAR) for this series of compounds.
Figure 1.

Reported PI5P4K inhibitors.
Our recently reported irreversible inhibitor of PI5P4Ks, THZ-P1-2, displays submicromolar biochemical potency against PI5P4Kα kinase activity.17 Although the THZ-P1-2/PI5P4Kα cocrystal structure (PDB-ID: 6OSP) did not resolve the loop that contains the covalently targeted cysteine (Cys293), the structure clearly revealed key noncovalent interactions, including two hydrogen bonds between the aminopyrimidine, as well as the benzamide carbonyl and Asn198, a hydrogen bond forming between the pyrimidine and Val199, and T-shaped π-stacking between the phenylenediamine group and Phe134 in the p-loop (Figure 2). In addition, Phe134 and Phe200 form a narrow passage encaging the phenylenediamine of THZ-P1-2 (Supporting Figure 7). Even though the electron-density for the warhead tail was insufficiently resolved, the acrylamide moiety seemed to be positioned toward the solvent exposed region, which allows proximity to the targetable cysteines for each isoform: Cys 293 (α), 307 or 318 (β), and 313 (γ) (Supporting Figure 8). Hydrophobic interactions between the indole and the hydrophobic pocket under the p-loop contribute to the overall affinity.
Figure 2.
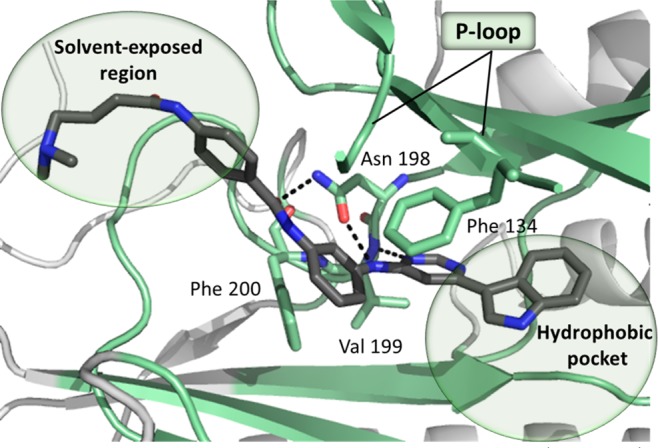
Cocrystal structure of PI5P4Kα and THZ-P1-2 (dark gray). The pocket bound by the inhibitor is highlighted in pale-green. H-bonds are shown as dashed bonds. For clarity, only Asn198, Val199, Phe134, and Phe200 are shown as capped sticks. PDB-ID: 6OSP.
To understand the SAR of THZ-P1-2 analogs, we employed biochemical assays for PI5P4Kα (ADP Glo) and PI5P4Kβ (Transcreener), which have produced reliable IC50 values for THZ-P1-2. The THZ-P1-2 analogs, which contain either a variety of different indole ring systems or bulkier mono- or bicyclic aromatic six-membered rings, were first designed to investigate the tolerability of the hydrophobic pocket that is occupied by the indole group in THZ-P1-2. The synthetic strategy for these analogs revolved around the 4,6-dichloropyrimidine, which underwent either a Suzuki coupling reaction with aromatic boronic acids/esters or an SN2 nucleophilic substitution reaction with aromatic amines. The second chloride group was then substituted with m-phenylenediamine, which was extended through an amide bond with 4-aminobenzoic acid, containing a free amino group for 4-dimethylaminocrotonamide transformation, as the final step (Scheme 1).
Scheme 1. Synthesis of THZ-P1-2 and Compounds 6–11.

Reagents and conditions: (a) (HO)2B-R or respective pinacol boronic acid esters, NaHCO3, Pd(PPh3)2Cl2, ACN/H2O, 90–100 °C, 2.5–19 h, 53–83% yield; (b) 4-methylaniline, TEA, BuOH, 140 °C, 1 h or 3/4-phenylaniline, TEA, EtOH, 120 °C, 3 h, 99%-quant. yield; (c) m-phenylenediamine, DIEA, NMP, 140–160 °C, 1.5 h to 2 d, 37–95% yield; (d) 4-nitrobenzoyl chloride, pyridine or TEA/DCM, rt, 5 h or 4-((tert-butoxycarbonyl)amino)benzoic acid, HATU, Hunig’s Base, DMF, rt, 1 h, then TFA/DCM, rt, 2 h, 37% yield; (e) SnCl2·2H2O, EtOAc/MeOH, 80 °C, 4.5 h to 2 d, or H2, 10% Pd/C, EtOAc/MeOH or MeOH, rt, 16 h, 4–87% yield over 2 steps; (f) 4-bromocrotonyl chloride, DIEA, DCM or ACN, 0 °C, then dimethylamine, THF, rt, 1 h, 24–71% yield; (g) 4-dimethylaminocrotonic acid, DIEA, HATU, DCM, or oxalylic chloride, DMF rt, 1–12 h, 6–30% yield; (h) 1.0 M NaOH/1,4-dioxane, rt, 4–6 h, 8–60% yield.
THZ-P1-2 showed submicromolar potency (IC50 of 950 nM) against PI5P4Kα, as well as an IC50 of 5.9 μM against PI5P4Kβ. Pyrrolo[2,3-b]pyridine installed in compound 6 led to a slight decrease in potency on both isoforms, while a 2-methylindazole of compound 7 dampened the activity even further to 3.5 and 15.8 μM for PI5P4Kα and PI5P4Kβ, respectively (Table 1). Introducing an NH-linker, connecting the pyrimidine with a phenyl group carrying either a 4-methyl group or even a bulky 4-phenyl substituent (compounds 9 and 8, respectively) were well tolerated and maintained IC50 values of around 1.3 μM and 6.2–6.3 μM for the α- and β-isoform of the PI5P4Ks, respectively. In contrast, a 3-phenylaniline or direct conjugation of pyrimidine with a 3-phenylbenzene group resulted in a more than 10-fold loss in PI5P4Kα activity (compounds 10 and 11), underlining that these changes in the trajectory of binding seem to cause a steric clash with some residues in the hydrophobic pocket under the p-loop.
Table 1. IC50 Evaluation of THZ-P1-2 and Compounds 6–11.


Average ± SD of three experiments, each using an 11-point titration.
Next, to understand the SAR for the groups that bind the hinge region, as well as the solvent-exposed region, we designed a second batch of THZ-P1-2 analogs, shown in Table 2. To access these compounds, the second chloride moiety at the 6-position of the pyrimidine was substituted with different aromatic amines through SN2 reactions, which were then further conjugated with either 3- or 4-nitro benzoic acid, 4-aminocyclohexane-1-carboxylic acid, or 4-aminopiperidine through amide bond formation to yield compounds 14–19 and 25 (Schemes 2 and 4). We also synthesized compound 22 with a distinct 3-aminopiperidine group installed at the 6-position of the pyrimidine, followed by a urea functionality, which was utilized to conjugate with 3-aminobenzeneamine (Scheme 3). All these compounds contain a free amino group, which was eventually converted to a 4-dimethylaminocrotonamide as a cysteine reactive warhead.
Table 2. IC50 Evaluation of Compounds 14–19, 22, 25, and 30.

Average ± SD of three experiments, each using an 11-point titration.
Scheme 2. Synthesis of Compounds 14–19.

Reagents and conditions: (a) anilines, DIEA, NMP, 150 °C, 6–8 h, then TFA/DCM, rt, 1 h, 67–84% yield; (b) anilines, DIEA, NMP, 150 °C, 8 h to overnight, 46–60% yield; (c) nitroaniline, Cs2CO3, X-Phos, Pd2(dba)3, 1,4-dioxane, 104 °C, 3 h, then SnCl2, MeOH, 80 °C, 8 h, 61% yield; (d) carboxylic acid chlorides, pyridine, rt, overnight, then SnCl2·2H2O, EtOAc/MeOH, 80 °C, 1.5–2 d, 23–39% yield over 2 steps; (e) carboxylic acid chlorides, DIEA or TEA, DCM, 0 °C, 5 min or rt, 1.5 h, then H2, 10% Pd/C, MeOH or EtOAC/MeOH, rt, 12 h to overnight, 36–51% over 2 steps; (f) carboxylic acids, HATU, DIEA, DCM, rt, 0.5–3 h, then HCl, MeOH or EtOAc, rt, 0.5 h to overnight, 54–85% over 2 steps; (g) 4-bromocrotonyl chloride, DIEA, DCM or ACN, 0 °C, 5 min to 0.5 h, then dimethylamine (2.0 M in THF), rt, 0.5–2 h, 30–91% yield; (h) 4-dimethylaminocrotonic acid hydrochloride, DIEA, HATU, DCM, rt, 12 h, 30% yield; (i) 1.0 M NaOH/1,4-dioxane, 10 °C to rt, 3–6 h, 10–46% yield.
Scheme 4. Synthesis of Compound 25.

Reagents and conditions: (a) 2-(3-aminophenyl)acetic acid, DIEA, NMP, 140 °C, 12 h, 6% yield; (b) tert-butyl piperidin-4-ylcarbamate, HATU, DIEA, DMF, rt, 0.5 h, 51% yield; (c) HCl, EtOAc, rt, 0.5 h, 90% yield; (d) 4-dimethylaminocrotonic acid hydrochloride, DIEA, HATU, DCM, rt, 0.5 h, 50% yield; (e) 1.0 M NaOH/1,4-dioxane, rt, 4 h, 56% yield.
Scheme 3. Synthesis of Compound 22.

Reagents and conditions: (a) tert-butyl 3-aminopiperidine-1-carboxylate, DIEA, NMP, 110 °C, overnight, 50% yield; (b) TFA/DCM, rt, overnight, quant. yield; (c) 3-nitroaniline, triphosgene, TEA, DCM, 0 °C, 2 h, then rt, overnight, 83% yield; (d) SnCl2, EtOAc/MeOH, reflux, overnight, 74% yield; (e) 4-bromocrotonyl chloride, DIEA, DCM, 0 °C, 5 min, then dimethylamine (2.0 M in THF), rt, 1 h, 68% yield; (f) 1.0 M NaOH/1,4-dioxane, 10 °C to rt, 4 h, 30% yield.
We first found that introducing a methyl group to either carbon-4 (compound 15) or carbon-6 (compound 16) led to up to 5-fold higher IC50s compared to THZ-P1-2 and compound 6. An explanation could be that the conformational changes in these compounds are potentially causing steric clashes with the relatively small passage (Supporting Figure 7) or are jeopardizing the π-stacking interaction with Phe134. Going even further, using a bulky, nonaromatic piperidine ring resulted in the overall inactive compound 22.
A scan of different crotonamide warhead-containing linkers pointing toward the solvent-exposed area, including piperidine, cyclohexane, or phenyl rings with different substitution patterns, demonstrated high tolerability to both the composition and the conformation of the linker with respect to activity against PI5P4Kα. For example, both piperidine and cyclohexane containing compounds (compounds 25 and 19, respectively) showed almost identical activity with IC50 values of 2.0–2.5 μM on PI5P4Kα. Interestingly, PI5P4Kβ seems to be more sensitive to changes in this motif since the IC50 of compound 19 drops by 5-fold, compared to THZ-P1-2. Similarly, the regiochemical variation of compound 6 to compound 17, attaching the crotonamide residue in either the 3- or 4-position of the benzamide, or to compound 18, carrying either a m- or p-phenylenediamine, displayed again only minor effects on the inhibitory activity on PI5P4Kα. While changing the warhead attachment-site on the benzamide did not have any effect on PI5P4Kβ activity either (compounds 6 and 17), changes in the phenylenediamine moiety led to a 3.5-fold higher IC50 (compound 18), underlining a higher sensitivity of the β-isoform to alterations in this region. However, an extended linker with an insertion of a methylene group, shifting the benzamide carbonyl further away from the hinge region, in compound 14 became more resistant to both, PI5P4Kα and PI5P4Kβ.
Inspired by the SAR information we obtained above, as well as the cocrystal structure of our inhibitor THZ-P1-2 with PI5P4Kα,17 we cyclized the pyrimidine ring from carbon-4 and -5 to yield a bicyclic pyrrolo[2,3-d]pyrimidine and introduced a 4-methylphenyl, a favored residue (e.g., compound 9), attached to the N atom of the pyrrolo ring system. We hypothesized that this could potentially improve the overall selectivity of the scaffold while maintaining on-target activity. The compound was synthesized through the coupling of 4-chloro-7H-pyrrolo[2,3-d]pyrimidine with 4-iodotoluene in a copper-catalyzed nucleophilic substitution reaction. The free chloride was then coupled with m-phenylenediamine, extended with benzoic acid via an amide bond, and the free amino group was finally converted to a crotonamide as already described in Scheme 1 (Scheme 5), leading to active compound 30 with IC50 values of 1.3 and 9.9 μM for PI5P4Kα and PI5P4Kβ, respectively.
Scheme 5. Synthesis of Compound 30.

Reagents and conditions: (a) 4-iodotoluidine, trans-1,2-cyclohexanediamine, K3PO4, CuI, toluene, 110 °C, 1 d, 48% yield; (b) m-phenylenediamine, DIEA, NMP, 150 °C, 4 d, quant. yield; (c) 4-nitrobenzoyl chloride, pyridine, rt, overnight; (d) SnCl2·2H2O, EtOAc/MeOH, 80 °C, 1 d, 40% yield over 2 steps; (e) 4-bromocrotonyl chloride, DIEA, ACN, 0 °C; (f) dimethylamine (2.0 M in THF), rt, 1 h, 4% yield over 2 steps.
As previously reported, the selectivity profiling of THZ-P1-217 with KINOMEscan has shown that this compound exhibits cross activity with the type I lipid kinase PIKFYVE, as well as several protein kinases including BRK, TYK2, and Abl. We next questioned whether the bicyclic pyrrolo[2,3-d]pyrimidine scaffold on compound 30 could dial out these off-targets, making it a more selective PI5P4K inhibitor. Therefore, another KINOMEscan profiling assay for compound 30 was undertaken. At 1 μM, compound 30 lost almost all off-targets seen with THZ-P1-2, including the PI5P4K-related PIKFYVE kinase (Figure 3). An ADP-Glo biochemical assay (commercially available from Carna Biosciences, Inc.) done with PIKFYVE confirmed these findings as an IC50 value of 40.1 nM for THZ-P1-2, whereas compound 30 did not show any activity in this assay format, which was also true for other off-targets (Supporting Table 2). Since the two compounds solely differ in their headgroup moieties, facing the hydrophobic pocket in the binding site, potential reasons for the selectivity improvement could be the slightly bulkier headgroup in compound 30, as well as the missing hydrogen bond offering NH group of the indole found in THZ-P1-2. It should be noted that PI5P4Kα is not assessed in the KINOMEscan panel. Although PI5P4Kβ and PI5P4Kγ were included in the kinases tested, we could not see significant binding of both compounds tested, despite their biochemical potency and cellular target engagement. Hence, the kinome-wide profiling data (Figure 3A) was used to investigate potential off-targets.
Figure 3.

(A) KINOMEscan results for THZ-P1-2 and compound 30. Selectivity profile was determined for 469 kinases as percent DMSO control at 1 μM inhibitor concentration (excluding mutant, atypical, and pathogen). (B) Table comparing percent DMSO control for top five off-targets of THZ-P1-2 to compound 30.
To assess the cellular activity of the THZ-P1-2 analogs, we used a cellular thermal shift assay (CETSA) in which we treated HEK 293T cells with our most potent pan-PI5P4K inhibitors. We first determined the optimal incubation temperature for the assay, by comparing melting curves of cells treated with either DMSO or 10 μM THZ-P1-2 at temperatures ranging from 40 to 67 °C (Figure 4A). While we observed loss of soluble PI5P4Kα and PI5P4Kβ between 49 and 52 °C with the DMSO control, THZ-P1-2 stabilized the enzymes to temperatures up to 58 to 61 °C, showing that THZ-P1-2 exhibits good pan-PI5P4K binding in the cellular context. We determined 52 °C to be the optimal assay temperature to investigate cellular target engagement of our compounds since incubation at this temperature resulted in still optimal stabilization of soluble PI5P4Kα/β with THZ-P1-2, while the DMSO-treated cells show already an immense loss of soluble protein. This temperature was used for further CETSA experiments. Next, an isothermal CETSA was performed to compare cellular target engagement of our top inhibitors, showing good target binding activity of all selected compounds in HEK 293T cells (Figure 4B,C). Dose–response curves were obtained using the same isothermal CETSA conditions. Surprisingly, compounds 8 and 30 generally showed slightly less cellular activity compared to THZ-P1-2, while compound 9 seems to be slightly more active in cells (Figure 4D,E and Supporting Figure 5A,B).
Figure 4.

(A) Melting-curves (Tm) of PI5P4Kα and PI5P4Kβ. Treatment with THZ-P1-2 at 10 μM for 1 h, in comparison to DMSO. (B) Representative Western blot of isothermal CETSA at 52 °C (HEK 293T cells) with selected PI5P4Kα/β inhibitors at 1 or 10 μM. (C) Quantified relative band intensity (%) of isothermal CETSA experiment (Figure 4B). Mean of three independent experiments. (D) Dose–response curve of isothermal CETSA for PI5P4Kα at 52 °C (HEK 293T cells) with selected PI5P4Kα/β inhibitors. Mean of three independent experiments (representative Western blot, see Supporting Figure 5A). (E) Dose–response curve of isothermal CETSA for PI5P4Kβ at 52 °C (HEK 293T cells) with selected PI5P4Kα/β inhibitors. Mean of three independent experiments (representative Western blot, see Supporting Figure 5B).
Ultimately, comparing our lead compounds, THZ-P1-2 and compound 30, with their reversible analogs (THZ-P1-2-R and compound 30-R, respectively) led to an overall decrease in biochemical activity of 3–4-fold, except for compound 30 and its PI5P4Kα activity, which did not change significantly (Supporting Figure 9A). The same trend was seen in an isothermal CETSA experiment, although compound 30 now seems to show similar potency changes on both, PI5P4Kα and -β (Supporting Figure 9B,C). However, the observed potency changes between covalent and noncovalent analogs were again more pronounced with THZ-P1-2. To make sure that compound 30 is still a covalent PI5P4K inhibitor, an intact mass spectrometry experiment was undertaken, which showed indeed covalent labeling of PI5P4Kα, -β, and -γ by compound 30 (Supporting Figure 10). These results indicate that compound 30 might be a slower covalent binder than THZ-P1-2, which could explain not only the biochemical results but also the weaker cellular activity of compound 30.
In conclusion, the discovery of THZ-P1-2 and its analogs present a novel class of covalent inhibitors for PI5P4Ks. The SAR study reported here provides a rationale for further development, by systematically exploring the tolerability of each binding region for chemical modification. The selectivity profiling indicated a highly selective PI5P4K inhibitor is achievable through the modification of the core 4,6-pyrimidine scaffold, shown by compound 30. Furthermore, investigating the cellular target engagement, using isothermal CETSA methods, we were able to confirm that the inhibitors show efficient binding affinity in the context of HEK 293T cells. These findings suggest that compound 30 is a selective, covalent pan-PI5P4K inhibitor, which may overcome limitations of cellular activity of reported PI5P4K inhibitors. Further efforts to optimize cellular potency of compound 30 and its pharmacokinetics for in vivo studies are ongoing.
Acknowledgments
The authors would like to thank M. Kostic for her help and expertise with writing the manuscript.
Glossary
ABBREVIATIONS
- ACN
acetonitrile
- BRK
breast tumor-related kinase
- CETSA
cellular thermal shift assay
- Cpd
compound
- DCM
dichloromethane
- DIEA
N,N-diisopropylethylamine
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate
- NMP
N-methyl-2-pyrrolidone
- Pd2(dba)3
tris(dibenzylideneacetone)dipalladium(0)
- PI-4,5-P2
phosphatidylinositol-4,5-bisphosphate
- PI4P
phosphatidylinositol 4-phosphate
- PI4P5K
phosphatidylinositol 4-phosphate 5-kinase
- PI5P
phosphatidylinositol 5-phosphate
- PI5P4K
phosphatidylinositol 5-phosphate 4-kinase
- PIKFYVE
phosphatidylinositol 3-phosphate 5-kinase
- rt
room temperature
- SAR
structure–activity relationship
- SD
standard deviation
- TEA
triethylamine
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- TYK2
tyrosine kinase 2
- X-Phos
2-dicyclohexylphosphino-2′,4′,6′-triisoropylbiphenyl
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00402.
Experimental and characterization data for all new compounds; additional biological data and protocols (PDF)
Author Contributions
T.D.M., T.Z., F.M.F., and N.S.G. conceived the project and designed the research strategy. T.D.M., T.Z., and F.M.F. designed and synthesized the compounds. S.C.S. performed compound testing and helped with planning experimental strategies. M.B.B. synthesized additional compounds for the biochemical assay validation. A.Y., M.I.D., A.S., and M.D.H. developed and validated the biochemical assays. A.Y. performed the biochemical assays under supervision of M.D.H. and M.S. H.-S.S. and S.D.-P. generated the cocrystal structure. A.T.S. provided reagents and methods for the biochemical assays. S.C.S., J.D.C., S.B.F., and J.A.M. performed the intact mass spectrometry analysis. T.D.M. planned and performed the cellular assays. H.S. and L.C.C. were involved in the planning and provided expertise and feedback for the project. The manuscript was written by T.D.M. and contributions of all authors. All authors have given approval to the final version of the manuscript. T.Z., F.M.F., and N.S.G. supervised the research.
The authors would like to thank the National Institutes of Health for their generous financial support through grants R01 CA197329 (to N.S.G., subcontracted to S.D.P.), R01 NS089815 and R03 MH096575 (to A.T.S.), R01 CA222218 and R03 CA250020 (to J.A.M.), as well as R35 CA197588, R01 GM041890, and U54 CA210184 (to L.C.C.). This work was further supported by the Breast Cancer Research Foundation (to L.C.C.) and the NIH intramural research program (NCATS).
The authors declare the following competing financial interest(s): N.S.G. is a founder, SAB member, and equity holder in Gatekeeper, Syros, Petra, C4, B2S, and Soltego. The Gray lab receives or has received research funding from Novartis, Takeda, Astellas, Taiho, Janssen, Kinogen, Voronoi, Her2llc, Deerfield, and Sanofi. N.S.G. and T.Z. are inventors on a patent application covering chemical matter in this publication, owned by Dana-Farber Cancer Institute. L.C.C. is a founder and member of the Board of Directors (BOD) of Agios Pharmaceuticals and is a founder and receives research support from Petra Pharmaceuticals. These companies are developing novel therapies for cancer. J.A.M. serves on the SAB of 908 Devices.
Supplementary Material
References
- Rameh L. E.; Tolias K. F.; Duckworth B. C.; Cantley L. C. A new pathway for synthesis of phosphatidylinositol-4,5-bisphosphate. Nature 1997, 390 (6656), 192–196. 10.1038/36621. [DOI] [PubMed] [Google Scholar]
- Hu A.; Zhao X.-T.; Xiao T.; Fu T.; Wang Y.; Liu Y.; Shi X.-J.; Luo J.; Song B.-L. PIP4K2A regulates intracellular cholesterol transport through modulating PI(4,5)2 homeostasis. J. Lipid Res. 2018, 59, 507–514. 10.1194/jlr.M082149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamia K. A.; Peroni O. D.; Kim Y.-B.; Rameh L. E.; Kahn B. B.; Cantley L. C. Increased Insulin Sensitivity and Reduced Adiposity in Phosphatidylinositol 5-Phosphate 4-Kinase β–/– Mice. Mol. Cell. Biol. 2004, 24 (11), 5080–5087. 10.1128/MCB.24.11.5080-5087.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim H.; Wu C.; Ramsamooj S.; Bosch K. N.; Chen Z.; Emerling B. M.; Yun J.; Liu H.; Choo-Wing R.; Yang Z.; Wulf G. M.; Kuchroo V. K.; Cantley L. C. Deletion of the gene Pip4k2c, a novel phosphatidylinositol kinase, results in hyperactivation of the immune system. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (27), 7596–7601. 10.1073/pnas.1600934113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Ramahi I.; Giridharan S. S. P.; Chen Y.-C.; Patnaik S.; Safren N.; Hasegawa J.; de Haro M.; Wagner Gee A. K.; Titus S. A.; Jeong H.; Clarke J.; Krainc D.; Zheng W.; Irvine R. F.; Barmada S.; Ferrer M.; Southall N.; Weisman L. S.; Botas J.; Marugan J. J. Inhibition of PIP4Kγ ameliorates the pathological effects of mutant huntingtin protein. eLife 2017, 6, e29123 10.7554/eLife.29123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundquist M. R.; Goncalves M. D.; Loughran R. M.; Possik E.; Vijayaraghavan T.; Yang A.; Pauli C.; Ravi A.; Verma A.; Yang Z.; Johnson J. L.; Wong J. C. Y.; Ma Y.; Hwang K. S.-K.; Weinkove D.; Divecha N.; Asara J. M.; Elemento O.; Rubin M. A.; Kimmelman A. C.; Pause A.; Cantley L. C.; Emerling B. M. Phosphatidylinositol-5-Phosphate 4-Kinases Regulate Cellular Lipid Metabolism by Facilitating Autophagy. Mol. Cell 2018, 70 (3), 531–543. 10.1016/j.molcel.2018.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulley S. J.; Droubi A.; Clarke J. H.; Anderson K. E.; Stephens L. R.; Hawkins P. T.; Irvine R. F. In B cells, phosphatidylinositol 5-phosphate 4-kinase-α synthesizes PI(4,5)P2 to impact mTORC2 and Akt signaling. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (38), 10571–10576. 10.1073/pnas.1522478113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keune W.-J.; Jones D. R.; Divecha N. PtdIns5P and Pin1 in oxidative stress signaling. Advances in Biological Regulation 2013, 53 (2), 179–189. 10.1016/j.jbior.2013.02.001. [DOI] [PubMed] [Google Scholar]
- Jude J. G.; Spencer G. J.; Somerville T. D. D.; Jones D. R.; Divecha N.; Somervaille T. C. P. A targeted knockdown screen of genes coding for phosphoinositide modulators identifies PIP4K2A as required for acute myeloid leukemia cell proliferation and survival. Oncogene 2015, 34 (10), 1253–1262. 10.1038/onc.2014.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luoh S.-W.; Venkatesan N.; Tripathi R. Overexpression of the amplified Pip4k2β gene from 17q11–12 in breast cancer cells confers proliferation advantage. Oncogene 2004, 23, 1354–1363. 10.1038/sj.onc.1207251. [DOI] [PubMed] [Google Scholar]
- Emerling B. M.; Hurov J. B.; Poulogiannis G.; Tsukazawa K. S.; Choo-Wing R.; Wulf G. M.; Bell E. L.; Shim H.-S.; Lamia K. A.; Rameh L. E.; Bellinger G.; Sasaki A. T.; Asara J. M.; Yuan X.; Bullock A.; DeNicola G. M.; Song J.; Brown V.; Signoretti S.; Cantley L. C. Depletion of a Putatively Druggable Class of Phosphatidylinositol Kinases Inhibits Growth of p53-Null Tumors. Cell 2013, 155 (4), 844–857. 10.1016/j.cell.2013.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M. I.; Sasaki A. T.; Shen M.; Emerling B. M.; Thorne N.; Michael S.; Pragani R.; Boxer M.; Sumita K.; Takeuchi K.; Auld D. S.; Li Z.; Cantley L. C.; Simeonov A. A Homogeneous, High-Throughput Assay for Phosphatidylinositol 5-Phosphate 4-Kinase with a Novel, Rapid Substrate Preparation. PLoS One 2013, 8 (1), e54127 10.1371/journal.pone.0054127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa M.; Liao P.-J.; Lee K. H.; Wong J.; Shang S. C.; Minami N.; Sampetrean O.; Saya H.; Lingyun D.; Prabhu N.; Diam G. K.; Sobota R.; Larsson A.; Nordlung P.; McCormick F.; Ghosh S.; Epstein D. M.; Dymock B. W.; Lee S. H. Dual blockade of the lipid kinase PIP4Ks and mitotic pathways leads to cancer-selective lethality. Nat. Commun. 2017, 8 (1), 2200. 10.1038/s41467-017-02287-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss M. D.; Czechtizky W.; Li Z.; Rudolph C.; Petry S.; Brummerhop H.; Langer T.; Schiffer A.; Schaefer H.-L. Discovery and pharmacological characterization of a novel small molecule inhibitor of phosphatidylinositol-5-phosphate 4-kinase, type II, beta. Biochem. Biophys. Res. Commun. 2014, 449 (3), 327–331. 10.1016/j.bbrc.2014.05.024. [DOI] [PubMed] [Google Scholar]
- Clarke J. H.; Giudici M.-L.; Burke J. E.; Williams R. L.; Maloney D. J.; Marugan J.; Irvine R. F. The function of phosphatidylinositol 5-phosphate 4-kinase γ (PI5P4Kγ) explored using a specific inhibitor that targets the PI5P-binding site. Biochem. J. 2015, 466 (2), 359–367. 10.1042/BJ20141333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q.; Sabnis Y.; Zhao Z.; Zhang T.; Buhrlage S. J.; Jones L. H.; Gray N. S. Developing Irreversible Inhibitors of the Protein Kinase Cysteinome. Chem. Biol. 2013, 20 (2), 146–159. 10.1016/j.chembiol.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivakumaren S. C.; Shim H.; Zhang T.; Ferguson F. M.; Lundquist M. R.; Browne C. M.; Seo H.-S.; Paddock M. N.; Manz T. D.; Jiang B.; Hao M.-F.; Krishnan P.; Wang D. G.; Yang J.; Kwiatkowski N. P.; Ficarro S. B.; Cunningham J. M.; Marto J. A.; Dhe-Paganon S.; Cantley L. C.; Gray N. S.. Targeting the PI5P4K lipid kinase family in cancer using novel covalent inhibitors. bioRxiv 2019; 10.1101/819961. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.