

Abstract
Cytokines play crucial roles in orchestrating complex multicellular interactions between pancreatic β cells and immune cells in the development of type 1 diabetes (T1D) and are thus potential immunotherapeutic targets for this disorder. Cytokines that can induce regulatory functions—for example, IL‐10, TGF‐β and IL‐33—are thought to restore immune tolerance and prevent β‐cell damage. By contrast, cytokines such as IL‐6, IL‐17, IL‐21 and TNF, which promote the differentiation and function of diabetogenic immune cells, are thought to lead to T1D onset and progression. However, targeting these dysregulated cytokine networks does not always result in consistent effects because anti‐inflammatory or proinflammatory functions of cytokines, responsible for β‐cell destruction, are context dependent. In this review, we summarise the current knowledge on the involvement of well‐known cytokines in both the initiation and destruction phases of T1D and discuss advances in recently discovered roles of cytokines. Additionally, we emphasise the complexity and implications of cytokine modulation therapy and discuss the ways in which this strategy has been translated into clinical trials.
Keywords: cytokine, immunotherapy, type 1 diabetes, β‐cell destruction
Cytokines have been directly implicated in the pathogenesis of type 1 diabetes (T1D) via orchestrating complex multicellular interactions between pancreatic β cells and immune cells, and are thus potential immunotherapeutic targets for this disorder. In this review, we discuss the pleiotropic roles of cytokines, which determine whether a pathological or protective immune response occurs in both the initiation and destruction phases of T1D. We also discuss potential implications of cytokines in the deployment of immunotherapeutic strategies for T1D.

Introduction
Type 1 diabetes (T1D) is a chronic autoimmune disease characterised by the progressive and insidious destruction of β cells.1 This disease results from immune system breakdown, which is predominately mediated via T helper 1 (Th1) cells, accompanied by the activation and islet infiltration of immune cells, which collaborate to destroy pancreatic β cells and cause overt hyperglycaemia.2 Current T1D therapy mainly involves exogenous insulin replacement, highlighting the need for immunotherapies to limit the disease and improve clinical outcomes.
Genetic and immunopathogenic studies have directly implicated cytokines in the pathogenesis of T1D. In addition, cytokines are the major drivers of inflammation and play crucial roles in controlling ongoing β‐cell destruction.3 Studies in mouse models, particularly in nonobese diabetic (NOD) mice, a well‐established animal model of T1D, have shown that the modulation of cytokine function can be used for therapy and have identified several new cytokines as potential therapeutic targets to combat immune‐mediated β‐cell damage.4 The key roles of cytokines have also been highlighted by findings that blockade of tumor necrosis factor (TNF) results in the preservation of β‐cell function in children with new‐onset T1D5 and that IL‐2 treatment is able to increase the proportion of regulatory T cells (Tregs) without adverse effects in patients with established T1D.6, 7
However, the roles of cytokines in the pathophysiology of T1D are often unclear and complex, especially during inflammation and disease progression, when many more dysregulated cytokines are entangled into the dynamics of cytokine regulation (Figure 1 and Table 1). Very few cytokines have exclusively proinflammatory or anti‐inflammatory functions in the context of T1D. For purposes of this review, cytokines are classified into classical cytokines with anti‐inflammatory roles, such as IL‐10, TGF‐β and type‐2 cytokines; classical cytokines with proinflammatory roles, such as IL‐1, IL‐6 and TNF‐α; and cytokines with more recently described roles, such as the IL‐12 family, IL‐21 and IL‐33. We discuss the pleiotropic roles of cytokines, which determine whether a pathological or protective immune response occurs in both the initiation and destruction phases of T1D. We also discuss potential implications of cytokines in the deployment of immunotherapeutic strategies for T1D.
Figure 1.
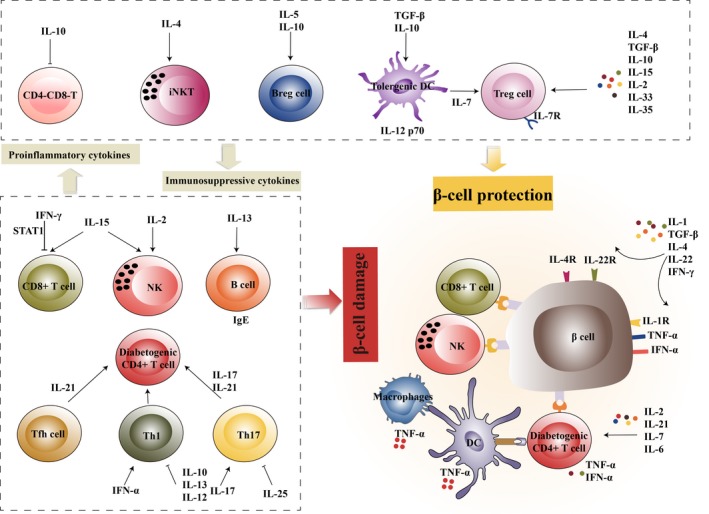
Cytokines involved in T1D. Cytokines produced by immune and pancreatic cells play diverse roles in T1D, which influences the development and progression of this disease. Cytokines such as IL‐10, TGF‐β, IL‐5, IL‐4, IL‐2, IL‐15, IL‐33 and IL‐35 can induce a regulatory phenotype in immune cells, which in turn release anti‐inflammatory cytokines such as IL‐10. In particular, IL‐7, released by regulatory DCs, is important for maintaining Tregs, which selectively express IL‐7Rα in T1D. Proinflammatory cytokines such as IL‐6, TNF‐α, IFN‐α, IL‐17 and IL‐21 amplify inflammation via proliferation and activation of diabetogenic immune cells, including Th1, Th17, CD8+ T cells and NK cells. However, because of the pleiotropic nature of cytokines, a given cytokine, such as IL‐2 or IL‐15, may trigger the activation of both diabetogenic and regulatory immune cells. In addition, β cells express high levels of cytokine receptors, such as IL‐1R, IL‐4R and IL‐22R, and exhibit increased sensitivity to cytokine‐induced apoptosis or regeneration. Thus, under in vivo conditions, the complexity of cytokine networks differentially contributes to the initiation and destruction phases of T1D.
Table 1.
Cytokines in T1D
| Cytokines | Pros | Cons | Outcomes of targeting cytokines in animals | Outcomes of targeting cytokines in humans |
|---|---|---|---|---|
| IL‐2 | Activating Tregs; shifting Th1 cytokine‐producing cells to Th2 and Th3 cytokine‐producing cells | Expanding effector T cells and NK cells; inducing IL‐17 | Low‐dose IL‐2 prevented disease development; IL‐2 combined with sirolimus induced disease remission | IL‐2 was well tolerated and increased the number of Tregs in patients with T1D |
| IL‐4 | Restoring IL‐12; activating and expanding iNKT and Tregs; activating PI3K and JAK/STAT pathways via IL‐4R in β cells | None | IL‐4 overexpression lowered the diabetes incidence, whereas complete elimination of IL‐4 did not accelerate or intensify insulitis | None |
| IL‐13 | Shifting a type 1 to a type 2 cytokine profile; increasing IgE production; promoting STAT6 and antiapoptotic gene expression in β cells | None | IL‐13 suppressed β‐cell destruction and prevented T1D development | None |
| IL‐10 | Inducing tolerogenic DCs, Tregs and Bregs; increasing Th2‐type cytokines and suppressing Th1‐type cytokines | Facilitating the apoptosis of CD4−CD8− T cells | IL‐10 prevented disease development, whereas local production of IL‐10 accelerated diabetes onset | None |
| TGF‐β | Proliferating Tregs; polarising islet antigen‐specific T‐cell responses towards a Th2 response | Promoting fibrosis and affecting pancreatic structure | TGF‐β inhibited the development of diabetes | None |
| IL‐1 | None | Triggering β‐cell apoptosis; inducing local inflammation | IL‐1R deficiency did not prevent disease progression | Anti‐IL‐1 antibodies (anakinra and canakinumab) did not prevent the decline in β‐cell function |
| IL‐6 | None | Promoting the migration and inflammatory responses of effector T cells | None | Therapeutic blockade of IL‐6 is being explored in clinical trials (NCT02293837) |
| TNF‐α | None | Inducing DC maturation; activating islet antigen‐specific T cells; accelerating β‐cell apoptosis | The protective effects of TNF‐α blockade on T1D were age‐dependent | Neutralisation of TNF‐α preserved β‐cell function in patients with recent‐onset T1D |
| IFN‐α | None | Augmenting Th1 responses; promoting the expression of HLA‐I molecules in β cells; mediating β‐cell endoplasmic reticulum stress and apoptosis | Blockade of IFN‐α signalling prevented disease development, whereas a certain dose of IFN‐α inhibited and prevented diabetes | Low‐dose IFN‐α had a beneficial effect in young patients with recent‐onset T1D |
| IFN‐γ | Inducing inhibitory STAT1 expression; suppressing diabetogenic CD8+ T cells and Th1‐type cytokines | Inducing aberrant expression of MHC‐I and MHC‐II in β cells | IFN‐γ had a dual role in T1D | None |
| IL‐15 | Enhancing Foxp3 expression in CD4+ Tregs; promoting Ly‐49+CD8+ Treg development | Proliferating and activating NK cells and CD8+ T cells | IL‐15 had a dual role in T1D | None |
| IL‐33 | Inducing Tregs; upregulating the expression of the ST2 molecule | None | IL‐33 prevented T1D development | None |
| IL‐35 | Inducing Tregs | None | IL‐35 prevented T1D development | None |
| IL‐12 | Suppressing proinflammatory cytokines; indirectly inhibiting Th17 cells | Increasing islet‐infiltrating CD4+ T cells | IL‐12 had a dual role in T1D | None |
| IL‐7 | Maintaining Tregs that selectively express IL‐7Rα | Promoting the differentiation of IFN‐γ‐producing cells; decreasing PD‐1 expression in diabetogenic T cells | Blockade of IL‐7 signalling reversed diseases | None |
| IL‐17 | None | Activating Th17 cells | Blockade of IL‐17 signalling prevented T1D development | None |
| IL‐21 | None | Promoting Th17 differentiation, DCs and Tfh migration | Blockade of IL‐21 signalling prevented T1D development. | Therapeutic blockade of IL‐21 is being explored in clinical trials (NCT02443155) |
| IL‐22 | Upregulating the expression of Bcl‐2, Bcl‐xl, Reg1 and Reg2 in β cells | None | Neither IL‐22 nor an anti‐IL‐22 antibody affected residual β‐cell function in a diabetic mouse model | None |
| IL‐25 | Inhibiting Th17 cells; inducing Th2 cytokine secretion | None | IL‐25 delayed diabetes progression | None |
Classical cytokines with anti‐inflammatory roles
Cytokines such as IL‐2, IL‐10, TGF‐β and type‐2 cytokines exert inhibitory or anti‐inflammatory effects in T1D, and their contribution to the disease progression is discussed below.
IL‐2
IL‐2 was initially discovered as a T‐cell‐stimulatory cytokine that supported the proliferation and differentiation of effector T cells. Further studies have shown that low‐dose IL‐2 favors the development of Treg populations, thus controlling disease onset and progression in T1D.8, 9 Moreover, IL‐2 has been described to promote β‐cell proliferation in animal models.10 IL‐2, therefore, is a potential therapeutic target in T1D, as demonstrated in both preclinical and clinical studies (Table 1).
Indeed, IL‐2, via its receptor (IL‐2R), exerts pleiotropic effects on a range of immune cell populations, including natural killer (NK) cells and effector T cells, which play pathological roles in T1D onset. Mechanistic studies have shown that Tregs are distinct from other immune cells as they constitutively express high levels of IL‐2Rα (also referred to as CD25) and, therefore, exhibit a more sensitive response to IL‐2 through the activation of the trimeric IL‐2R. Furthermore, Tregs seem to be more dependent on the IL‐2/STAT5 pathway in response to active stimulation; by contrast, the MAPK, PI3K/AKT and STAT5 pathways are activated in effector T cells.11 These mechanistic studies have led to strategies for tipping the balance between Tregs and effector T cells in T1D towards Tregs via administration of low‐dose IL‐2.
Administration of a low dose of IL‐2 to NOD mice prevented the development of diabetes.8 In this context, IL‐2 is a selective Treg stimulator in the pancreas, facilitating the expression of cell‐surface markers such as CTLA‐4, ICOS and GITR, which promotes the generation and suppressive activity of Tregs.8 Combination therapy with IL‐2 and sirolimus, an immunosuppressant that inhibits IL‐2‐mediated T‐cell proliferation but not IL‐2‐induced T‐cell apoptosis, is effective in preventing autoimmune destruction of β cells in spontaneous and recurrent autoimmune diabetes.12 This protective effect is due to the inhibition of autoreactive Th1 cells, with a shift to Th2‐ and Th3‐type cytokine‐producing cells.12 Low doses of IL‐2 have also been reported to increase the proportion of Tregs in patients with T1D.6, 7 By contrast, high‐dose IL‐2 is less effective, resulting in a trend towards expansion of effector T cells and NK cells, as well as IL‐17 expression, although the effect on Tregs is more pronounced and prolonged than that of low‐dose IL‐27 (Figure 1). These data emphasise that low‐dose IL‐2 can specifically expand and activate Treg populations, reestablishing immune tolerance in T1D. Notably, residual β‐cell function is necessary for the therapeutic benefit following IL‐2 administration. IL‐2 therapy has not been reported in patients with new‐onset T1D, who have residual β‐cell function; thus, it is unclear whether this therapy is clinically effective.
Type‐2 cytokines
Type‐2 cytokines, including IL‐4, IL‐5, IL‐9 and IL‐13, which promote type‐2 immune responses, show promising effects in suppressing type 1‐driven autoimmune disease. Only limited studies have been reported to date, and thus, the roles of IL‐5 and IL‐9 are still poorly described in T1D. IL‐5 is important for the proliferation of CD19+IgM+ B cells in NOD mice and stimulates the secretion of IL‐10 by B cells, which exerts a robust suppressive effect on the pathogenesis of diabetes.13 IL‐9 expression is increased in the serum of patients with T1D; it is correlated with the size of Th9 effector subpopulations and predicts HbA1c values, but the role of IL‐9 in T1D is uncertain.14, 15 The IL‐4 and IL‐13 cytokines, encoded by adjacent genes, transduce signals via a partially shared receptor (IL‐4Rα), and their effects in T1D have been unveiled in a number of studies.
IL‐4 levels are decreased in both newly diagnosed T1D patients and in NOD mice,16, 17 but IL‐4 may play a dispensable role in controlling disease aggressiveness, a hypothesis supported by the finding that complete elimination of IL‐4 did not accelerate or intensify insulitis in NOD mice (Table 1).18 However, genetic overexpression of IL‐4 or systemic administration of the cytokine prevents the onset of insulitis and decreases the incidence of diabetes (Table 1).19, 20, 21 Similarly, local expression of IL‐4 in β cells or adoptive transfer of dendritic cells (DCs) expressing soluble IL‐4 delays the onset of diabetes in NOD mice.22, 23, 24 The protective effects of IL‐4 are in part due to the activation of the IL‐4R‐mediated PI3K and JAK/STAT pathways; these pathways promote the viability of β cells, in which IL‐4R is highly expressed and functionally competent.25, 26 Other mechanisms include the restoration of IL‐2 and the activation and expansion of iNKT cells and Tregs (Figure 1).24, 27
IL‐13, similar to IL‐4, suppresses the ongoing β‐cell destruction and prevents T1D development. IL‐13 production is decreased in patients with T1D, even in subjects carrying a genetic risk of T1D, compared with that in healthy control subjects.28, 29 Importantly, the defective IL‐13 production correlates with the reduced regulatory capacity of iNKT cells, implying that IL‐13 secretion is important for the regulatory role of iNKT cells in T1D.30 Consistent with these findings, treatment with a recombinant human IL‐13 markedly delays the progression of insulitis and diminishes the incidence of spontaneous diabetes.30 IL‐13 modifies the cytokine profile in the periphery from type 1 to type 2, with an increased level of IL‐4 and decreased levels of IFN and TNF (Figure 1 and Table 1).30 IL‐13 treatment also increases the blood levels of IgE in an adoptive transfer model, resulting in the prevention of diabetes development (Figure 1).30 In addition to affecting the immune response, IL‐13 has been proposed to protect β cells via the activation of STAT6 and upregulation of antiapoptotic genes in pancreatic β cells (Table 1).31, 32
Conversely, inhibiting the IL‐4/IL‐13 pathway via genetic deficiency of IL‐4Rα or the IL‐4Rα/IL‐13Rα1 heteroreceptor is effective in controlling the blood glucose levels and islet inflammation.33, 34 The proposed mechanism suggests that IL‐4Rα/IL‐13Rα1 receptor deficiency increases the frequency of mTGFβ+Foxp3int Tregs and the persistence of CD206+ macrophages in the pancreas.33 These results seem to fuel a debate about the controversial functions of these cytokines in the control of diabetogenic responses. IL‐4 and IL‐13 are examples of cytokines that can target the same part of a receptor and perform overlapping functions; thus, blocking the affected receptors may produce multiple effects on the inflammatory process, which are not completely clarified. Nevertheless, signalling via IL‐4 and IL‐13 receptors is likely to be an active participant in the pathogenesis of T1D.
IL‐10
IL‐10, a cytokine with anti‐inflammatory properties, plays a crucial role in preventing inflammatory and autoimmune pathologies. This role is mediated though IL‐10R and is associated with many immune cells, generating feedback regulation of diverse immune responses. Despite the immunosuppressive effects attributed to IL‐10, its ability to modulate disease progression remains controversial in T1D.
Several therapeutic interventions that activate IL‐10 have been reported to improve β‐cell function, inhibit insulitis progression, and prevent the occurrence of diabetes in animal models.35, 36 The protective effects of IL‐10 have also been reported in humans, as suggested by a strong correlation between increased IL‐10 expression and disease attenuation.37, 38 The mechanisms proposed for the regulation of T1D by IL‐10 involve increases in Treg frequencies and Th2‐type cytokine (IL‐4 and IL‐10) levels, as well as the suppression of Th1‐type cytokines (IL‐2 and IFN‐γ) (Figure 1 and Table 1).36, 39 IL‐10 is also associated with the tolerant state of immature DCs and IL‐10‐producing regulatory B cells (Bregs) in T1D.38, 40 Treatment of monocyte‐derived DCs with TGF‐β and IL‐10 induces tolerogenic properties, and this immature state has been shown to drive insulin‐specific tolerance in effector/memory T cells generated in patients with T1D.40 Similarly, both murine and human Bregs can significantly abrogate T‐cell‐mediated immune responses to an islet‐specific antigen, which is consistent with the reduction of this population in NOD mice and patients with T1D.38
However, IL‐10 also plays a pathogenic role at the initiation stage of the disease. As has already been reported, local production of IL‐10 accelerates the onset and increases the prevalence of diabetes.41, 42 These complicated and opposite biological effects have suggested that IL‐10 plays both immunosuppressive and stimulatory roles, which limits its translational potential. The exact mechanism of action is unclear, but it has been speculated that IL‐10 facilitates the apoptosis of double‐negative (CD4−CD8−) T cells, which exhibit prominent antigen‐specific immune tolerance properties (Figure 1 and Table 1)43 and whose reduction contributes to disease susceptibility in NOD mice. Furthermore, type 1 IFN signals, which drive prediabetic or susceptible individuals towards T1D, can inhibit IL‐10 signals via the JAK/STAT pathway in effector T cells and Tregs, subsequently altering the responses of these cells in T1D.44 Thus, because the effects of IL‐10 are dependent on the immune cell type and cytokine milieu, the physiological significance of this cytokine in T1D needs further clarification.
TGF‐β
TGF‐β is a family of pleiotropic cytokines with potent immunosuppressive activities, which are exerted via Tregs against almost all types of immune cells. TGF‐β is generally accepted to play disease‐suppressive roles in autoimmune diseases. In the context of T1D, TGF‐β signalling is critical for the development of natural Tregs in the thymus and also induces Foxp3 expression and differentiation of peripheral Tregs.45 However, the molecular mechanisms that underlie the beneficial effects of TGF‐β exposure in T1D are more complicated than they might appear. Indeed, transgenic expression of TGF‐β in islets significantly inhibited the development of diabetes and was accompanied by the proliferation of Tregs,46 inhibition of antigen‐presenting cell function, and the polarisation of islet antigen‐specific T‐cell responses towards a Th2 response in local sets (Figure 1 and Table 1).47 Constitutive expression of TGF‐β in islets results in massive fibrosis and structural changes in the pancreas, which may compromise TGF‐β‐induced immunosuppression (Table 1)48. Similarly, TGF‐β, via its receptor, is strictly required for Treg‐mediated prevention of the disease, as supported by the finding that adoptively transferred Tregs could not control the islet‐reactive CD8+ T cells, which are deficient in TGF‐βR signalling.49
Notably, conventional CD4+ T cells but not Tregs are the main targets of TGF‐β in suppression of T1D. Ablation of TGF‐β signalling in Tregs did not result in the development of diabetes, whereas this ablation in peripheral effector CD4+ T cells and thymic T cells led to rapid development of T1D in BDC2.5 NOD mice.45 Although a reduced number of peripheral Tregs were also found, this effect was due to enhanced Th1‐cell responses.45 Additional support for the Treg‐independent regulation via TGF‐β signalling was provided using a Foxp3 deficiency diabetic model in which TGF‐β maintained peripheral T‐cell tolerance primarily via Foxp3‐independent regulation of autoreactive T cells.50 Thus, the beneficial role of TGF‐β in preventing T1D in mouse models encourages the development of new therapeutic strategies to target TGF‐β, although no drug has yet entered clinical trials for this application.
Classical cytokines with proinflammatory roles
Several classical cytokines that promote inflammatory responses, including IL‐1, IL‐6, TNF‐α and the IFN family, have been described in T1D, which leads to considerable interest in determining whether blocking these cytokines may be clinically beneficial.
IL‐1
IL‐1 is a prototypical mediator of innate immunity, with multiple biological actions.51 IL‐1 has long been known to cause β‐cell dysfunction and death.52 Islet β cells are more susceptible to IL‐1‐triggered apoptosis than other cell types, partly because of their higher IL‐1 receptor (IL‐1R) density and qualitative differences in NF‐κB and MAPK signalling (Figure 1).53 Thus, it has been hypothesised that IL‐1 blockade may have a synergistic effect on the preservation of β‐cell function by reducing local β‐cell inflammation and apoptosis.
However, these potential effects of IL‐1 blockade have not been confirmed in NOD mice and human patients with T1D. IL‐1R deficiency did not prevent the progression to diabetes in NOD mice, although islets were found to be protected from damaging effects of TNF and IFN‐γ in vitro (Table 1).54 Subsequently, two multicenter clinical trials were completed in 60 patients with recent‐onset T1D, who were treated with anti‐IL‐1 antibodies (anakinra and canakinumab) (Table 1).55 The effects of the treatments were not, however, what the investigators expected: single‐agent anti‐IL‐1 therapy did not prevent a decline in β‐cell function, as measured by the level of a stimulated C‐peptide.55 Notably, treatment with anakinra decreased systemic inflammation and improved insulin sensitivity in the insulin‐resistant patients with T1D, who had no residual β‐cell function, a result that was mirrored by improved glucose control and decreased insulin needs.56
Although these treatments were not effective, this approach may be attractive as a component of combination therapy. Consistent with this idea, a study indicated that the combination of IL‐1β blockade and an anti‐CD3 monoclonal antibody significantly enhanced clinical remission of diabetes, which was associated with an increase in Tregs and Th1‐to‐Th2 conversion.57 IL‐1 antibody treatment was shown to synergise with glutamic acid decarboxylase (GAD) immunisation to increase the number of Tregs and reduce the splenic cytotoxic T‐cell activity, as determined by the TNF and IFN‐γ levels; even more strikingly, IL‐1 antibody treatment reduced the numbers of islet CD11b+/high and cytotoxic T cells.58 These observations are consistent with an important role of IL‐1 inhibition in the prevention of local β‐cell inflammation and apoptosis.59, 60 Undoubtedly, IL‐1 signals facilitate β‐cell damage; however, their role in T1D must be further elucidated.
IL‐6
IL‐6 mediates the development and progression of autoimmune diseases; the pathological importance of this cytokine has been exemplified by the successful amelioration of a subset of autoimmune conditions by targeting the IL‐6/IL‐6 receptor (IL‐6R) axis.61 The efficacy of therapeutic blockade of IL‐6 in preserving β‐cell function in new‐onset T1D is being explored in an ongoing clinical trial (NCT02293837).
Although several reports have described a correlation between local IL‐6 production and the destructiveness of pancreatic proinflammatory infiltrates in NOD mice,62, 63 data on IL‐6 serum levels are inconsistent in patients with T1D.64 Overexpression of IL‐6 in local pancreatic β cells was shown to be associated with marked insulitis, accompanied by infiltration of B cells, macrophages and T cells.63 Other findings also support the disease relevance of the IL‐6 pathway, showing that STAT3 activation coordinates high expression of IL‐6R in CD4+ and CD8+ T cells from patients with T1D.65 However, activation of the IL‐6/STAT3 axis did not influence the frequencies or cytokine profiles (IL‐17 and IFN‐γ) of Helios− Teffs and Helios+ Tregs in the blood of patients with T1D.65 Despite these inconsistencies, dysregulated IL‐6 production and downstream receptor signalling are frequent events in T1D and are often associated with insulitis and β‐cell damage.
IL‐6 may regulate the migration and inflammatory responses of effector T cells in T1D (Figure 1 and Table 1). Whole‐transcriptome profiling revealed that IL‐6‐stimulated CD4+ T cells had a unique transcriptome and overexpressed genes implicated in T‐cell migration and activity (e.g. CXCR6 and CCR5).65 Notably, this effect might be regulated by ADAM17, as suggested by an inverse correlation between the expression of ADAM17 and the surface levels of IL‐6R in T cells.65 However, during exercise, contraction‐induced IL‐6 release is required for β‐cell viability, which lowers the expression of inducible nitric oxide synthase and reduces the level of cleaved caspase‐3.66 These studies demonstrate the context‐dependent nature of IL‐6 and suggest that it functions at several levels.
Tumor necrosis factor
Early studies described the presence of TNF‐α‐producing DCs and macrophages in pancreatic islet infiltrates and showed that these cells were the initial and major producers of TNF‐α.67 TNF‐α was found to upregulate MHC‐I molecules, thereby accelerating the apoptosis of β cells,68 and subsequently induce islet‐infiltrating DCs/macrophages to cross‐present an exogenous islet antigen to CD8+ T cells via a unique CD40/CD154‐independent pathway (Figure 1 and Table 1).69 Further studies demonstrated that TNF‐α upregulated the expression of DC maturation markers in the CD11b+CD11c+ subset, which were capable of activating islet‐specific T cells in pancreatic lymph nodes.70 These data suggest that TNF‐α plays an important role in the initiation of T1D. This hypothesis was confirmed by the finding that the protective effects of TNF‐α blockade were age dependent. When initiated at 4 weeks of age, anti‐TNF treatment was able to delay the disease onset but not completely prevent the disease in NOD mice.71
Despite these results, case reports have indicated that patients with rheumatoid arthritis can develop T1D when treated with a soluble TNF receptor (etanercept) or anti‐TNF therapy (adalimumab), but the cause of this adverse event is not understood.72, 73 These results suggest that this treatment strategy may be ineffective in preventing autoimmune diabetes in humans. Unexpectedly, neutralisation of TNF‐α in patients with recent‐onset T1D preserved β‐cell function, as measured by decreased A1C and increased endogenous insulin levels (Table 1).5 Furthermore, a small pilot trial (NCT02464033), which evaluated the influence of anti‐TNF‐α on the immune system, has been completed, but the results have not been published yet. Taken together, the results of these studies support a diabetogenic role for TNF‐α in the initiation of T1D and strengthen the targeting of TNF‐α as a new therapeutic strategy.
IFNs
IFN‐α and IFN‐γ, members of the IFN family, are implicated in the initiation and destruction phases of T1D, although the results are conflicting.
IFN‐α levels are increased in the blood of patients with T1D; the cytokine is also expressed by islet β cells and produced by plasmacytoid DCs (pDCs) from these patients.74, 75, 76 IFN‐α promotes the overexpression of HLA‐I molecules in human β cells and mediates their endoplasmic reticulum stress and apoptosis, which reflects the direct contribution of IFN‐α to β‐cell destruction (Figure 1).77, 78 Therapies targeting IFN‐α and its signalling have been shown to suppress β‐cell dysfunction and prevent the progression from prediabetes to T1D.79, 80, 81 IFN‐α produced by pDCs augments Th1 responses in patients with T1D, whereas blockade of IFN‐α signalling prevents the entry of autoreactive T cells into islets and induces an exhaustion signature in T cells, with upregulation of genes that encode negative immune regulators (Pdcd1, Lag3, Ctla4, Tigit and Btla), thereby limiting the diabetogenic ability of these cells (Figure 1).76, 79 Interestingly, low‐dose IFN‐α seems to have a beneficial effect on the preservation of β‐cell function in young patients with recent‐onset T1D82; this positive effect was also observed in NOD mice.83 However, the molecular mechanism underlying these controversial effects is not yet understood, and thus, the treatment of T1D by targeting IFN‐α presents a dilemma.
IFN‐γ, generally considered a proinflammatory cytokine, seems to be capable of exerting dual effects on the development of T1D. The ability of IFN‐γ to induce or promote T1D was found to be dependent on the dosage and time of interventions relative to the disease progression in NOD mice.84 The protective role of IFN‐γ is associated with the suppression of diabetogenic CD8+ T‐cell responses by inhibiting STAT1 expression85 and with a decrease in Th1 cell‐related cytokines, particularly IL‐12 and IL‐2, in spleen cells (Table 1).84 In some circumstances, IFN‐γ is a key participant in DC‐ or vaccine‐mediated peripheral tolerance in T1D.86, 87 However, this cytokine also mediates β‐cell destruction in local islets. IFN‐γ induces the aberrant expression of MHC‐I and MHC‐II in local pancreatic cells, through which inflammatory process could kill β cells (Figure 1, Table 1).88, 89 Moreover, mucosal‐associated invariant T (MAIT) cells, an innate‐like T‐cell subset, were activated and degranulated via incubation with IFN‐γ, IL‐1 and TNF‐α and thus became able to more efficiently kill a human β‐cell line.90 Together, the results of these studies support a complex role for IFN‐γ in both protection and destruction of β cells.
New players in the T1D cytokine milieu
Numerous studies have identified roles in T1D of more recently described cytokines, including members of the IL‐12 family (IL‐12, IL‐23, IL‐27 and IL‐35), Th17 cytokines (IL‐17, IL‐22 and IL‐25) and other cytokines, such as IL‐15, IL‐21, IL‐33 and IL‐7.
IL‐15
IL‐15, a member of the IL‐2 cytokine family, is expressed at high levels in mouse islets at the prediabetic stage.91 Double transgenic mice with pancreatic β‐cell expression of IL‐15 and IL‐15Rα developed hyperglycaemia, β‐cell destruction and anti‐insulin autoantibodies, mimicking early human T1D.91 By contrast, NOD mice treated with a monoclonal antibody against TMβ1, which blocks the transpresentation of IL‐15, or with the Jak2/3 inhibitor tofacitinib, which blocks IL‐15 signalling, displayed minimal infiltration of immune cells into pancreatic islets and reversion of hyperglycaemia.91 By promoting the proliferation and activation of NK cells and CD8+ T cells, IL‐15 plays a crucial role in the pathogenesis of T1D (Figure 1 and Table 1).91, 92
However, other data suggest a protective function of IL‐15 in T1D via inducing the production of Tregs. IL‐15 is required for the development of Ly49+CD8+ Tregs, whose deficiency is increased with ageing, as NOD mice progress towards diabetes (Figure 1 and Table 1).93 This finding led to the treatment of NOD mice with adoptively transferred IL‐15‐activated CD8+ Tregs, which resulted in delayed diabetes onset.93 Notably, IL‐15‐mediated Foxp3 expression in CD4+ Tregs could be abrogated by IL‐15‐activated NK cells.92 The results of these studies show that IL‐15 can accelerate T1D in vivo and, more importantly, also regulates diabetogenic effects via activation of Tregs, which, however, may be eliminated by simultaneous activation of diabetogenic T cells and NK cells.
IL‐33
IL‐33 is mainly expressed by cells of barrier tissues and is released as an alarmin to activate cells of both the innate and adaptive immune systems. A possible association of IL‐33 with T1D has been highlighted by the finding that soluble ST2, a decoy receptor for IL‐33, which sequesters circulating IL‐33, is increased in patients with T1D.94 The physiological function for soluble ST2 was suggested by the protective effects of IL‐33 administration in NOD mice and streptozotocin‐induced diabetic mice (Table 1). This function is mediated through the reduction in pancreas‐infiltrating cells and an increase in ST2+ Tregs in pancreatic lymph nodes, leading to a delay in disease onset.95, 96 Importantly, IL‐33 increases Treg frequencies and surface expression of ST2 molecules (Table 1 and Figure 1),94, 97 which fuels a positive feedback loop to enhance Treg differentiation. Given the important roles of Tregs in immunosuppression, these findings suggest a possible protective role of IL‐33 against T1D onset.
IL‐7
The IL‐7 cytokine is required for T‐cell development and homeostasis via interaction with the IL‐7 receptor (IL‐7R). Exposure of CD4+ or CD8+ naïve T cells to IL‐7 leads to the differentiation of IFN‐γ‐producing cells; in addition, IL‐7 was shown to decrease the expression of the inhibitory programmed death‐1 (PD‐1) receptor in diabetogenic T cells (Figure 1 and Table 1).98, 99 The results of these studies imply a pathogenic role of IL‐7 in T1D. However, for Treg‐mediated alleviation of T1D, IL‐7 is important for maintaining Tregs that selectively express IL‐7Rα (Figure 1 and Table 1).100 In addition, during suppression of T1D onset, the immunoregulatory function of DCs also relies on IL‐7.101 These observations have led to a concept that IL‐7 signalling is crucial for the balance between diabetogenic T cells and immunosuppressive cells in T1D.
Consistent with this hypothesis, IL‐7 accelerates the onset of diabetes in NOD mice,102 while blockade of IL‐7 signalling reverses the disease progression (Table 1).98, 99 Studies in mice have shown that blockade of IL‐7R reduces the population of IFN‐γ‐producing CD4+ and CD8+ T cells and increases the number of Tregs.98, 99 Importantly, an anti‐IL‐7Rα antibody enhanced the expression of the inhibitory PD‐1 receptor in effector T cells,98 suggesting a correlation between this critical inhibitory mechanism and therapeutic efficacy of the antibody. Together, these data indicate the potential of targeting the IL‐7/IL‐7R axis for the maintenance of β‐cell function and immune homeostasis in T1D, although the clinical benefit of this treatment has not been established.
IL‐21
The IL‐21 cytokine shows pleiotropic effects, which affect the differentiation and function of a broad range of immune cell types.103, 104 IL‐21 has been recognised for its ability to promote disease progression in T1D (Table 1). The main cellular source of IL‐21 in T1D is T follicular helper (Tfh) cells, which are enriched in the pancreas of NOD mice105 and peripheral blood of individuals with T1D.106 Analysis of autoantibody‐positive at‐risk children revealed that the elevation in activated Tfh cells, which produce IL‐21, could be detected even prior to the onset of diabetes.107 Abrogation of IL‐21 signalling protected mice from diabetes, while overexpression of IL‐21 was sufficient to induce diabetes.108, 109 It is currently evaluated in a phase II clinical trial (NCT02443155) whether blocking of IL‐21 would preserve β‐cell function in adult subjects with newly diagnosed T1D. These studies suggest that IL‐21‐producing Tfh cells could be used as a biomarker, and blocking of IL‐21 might be a promising approach to controlling T1D disease development.
Several ways may be postulated for IL‐21 mediation of immune responses associated with T1D (Figure 1). As discussed above, Th cells from inflamed tissues of NOD mice produced an abundance of IL‐21, and IL‐21‐producing Tfh cells played a major role in the disease development.105, 110 Coexpression of the chemokine receptor CCR9 was important for Tfh cell migration into pancreatic islets, which further promoted the proliferation and function of CD8+ T cells and subsequent destruction of islet β cells.105, 110 With respect to other immune cells, IL‐21R expression on DCs was required for their migration into draining lymph nodes, which induced diabetes in Il21r −/− knockout mice.111 IL‐21 acts on DCs to induce the production of PGE2 and IL‐6, which further promotes Th17 differentiation and, in turn, determines disease development.111 In addition, increased IL‐17 production was observed in the pancreas of β‐cell‐specific IL‐21‐expressing mice; by contrast, defective polarisation towards Th17 cells was observed in lymphocytes from Il21r −/− knockout NOD mice,108, 109 suggesting the potential role for IL‐21 in Th17 cells. Notably, IL‐21 blocking failed to change the number and function of Tregs in NOD mice,108 although IL‐21 has been described to inhibit the expansion of Tregs via reducing IL‐2 production112 or through transcription factor Bcl‐6‐mediated inhibition of CD25 expression.113 It is conceivable that Il21r −/− knockout mice are less susceptible to diabetes, which is independent of IL‐21 action on Tregs. Collectively, these results demonstrate that IL‐21 leads to a cascade of immune cell activation, with a clear role for a CD8+ T‐cell‐mediated attack on β cells. More importantly, the findings highlight that blocking of IL‐21 well controls the disease development, without a deleterious effect on regulatory immune cells.
IL‐12 family
The IL‐12 family currently includes IL‐12, IL‐23, IL‐27 and IL‐35. IL‐12 and IL‐23 are mainly proinflammatory cytokines, whereas IL‐27 and IL‐35 are immunoregulatory cytokines. Functional effects of IL‐23 and IL‐27 in T1D are a new area of research, and the importance of these two cytokines in T1D has not yet been established.
IL‐35, the most recently identified member of this family, is a potent inhibitory cytokine, produced by populations of immunosuppressive cells. An initial study showed that ectopic expression of IL‐35 in islets resulted in a substantial suppression of ongoing β‐cell destruction, accompanied by a reduction in the number of islet‐resident T cells, with limited cytokine alterations.114 Interestingly, although ectopic IL‐35 decreased the frequency of IFN‐γ‐expressing diabetogenic T cells, the number and proliferation of islet Foxp3+ Tregs were also reduced.115 These data suggest that the protective effect of islet IL‐35 is independent of the induction/expansion of pancreas‐resident immunosuppressive T cells. However, after systemic treatment of established diabetes with an IL‐35 recombinant protein, IL‐35 promoted a phenotypic shift of Tregs, which resulted in an increase in anti‐inflammatory (IL‐10, IL‐35 and TGF‐β) and a decrease in proinflammatory (IFN‐γ, IL‐2 and IL‐17) cytokines, counteracting the autoimmune attack in T1D.116 This effect is partly attributed to the induction of Eos expression and IL‐35 production in Tregs (Figure 1).116 Higher proportions of IL‐35‐producing Tregs and Bregs were also observed in the sera of long‐standing T1D patients with residual functional β cells than in those of patients without residual β cells.117 These findings in humans, together with the observation of hyperglycaemia‐reversing effects of systemic IL‐35 administration in established NOD mice (Table 1),116 suggest the application of IL‐35 as a treatment strategy in patients with either new‐onset or long‐standing T1D with residual β cells.
Administration of IL‐12 accelerates the onset of diabetes and increases the population of islet‐infiltrating Th1 cells (Figure 1 and Table 1).118 An antagonist of IL‐12 was shown to suppress the destruction of β cells in NOD mice at 3 or 4 weeks of age and lead to the skewing of islet‐infiltrating CD4+ T cells towards the Th2 phenotype.119, 120 When insulitis was established119 or treatment started at a very early age,121 an antagonist of IL‐12 failed to reverse the disease in NOD mice. These apparently inconsistent data regarding the function of IL‐12 in T1D reflect differences in the time of intervention. However, both wild‐type NOD mice and NOD mice with genetic ablation of IL‐12 were similar in the development of insulitis and diabetes, although IL‐12‐deficient NOD mice showed an impaired Th1 response to an islet autoantigen.122 Meanwhile, anti‐inflammatory effects of IL‐12 were also observed in NOD mice,123 with IL‐12 substantially suppressing the proinflammatory cytokines IL‐1β, IL‐6 and IL‐23 and indirectly inhibiting Th17 cells. IL‐12, therefore, is also able to play a protective role in the development of T1D (Figure 1 and Table 1).123 The above‐mentioned findings suggest that IL‐12 may play multifaceted roles in T1D, which have not been clearly addressed; thus, an IL‐12‐based approach may not be ideally suited for T1D therapy.
Th17 cytokines
Evidence indicates that Th17 cells play a pathological role in T1D onset. NOD mice treated with a selective inverse agonist of RORα/γ, a nuclear receptor that promotes Th17 cell development, displayed a reduced incidence of diabetes and insulitis.124 However, transfer of a pure population of Th17 cells into NOD/SCID mice failed to induce hyperglycaemia.125 More research is needed to understand the role of Th17 cells in T1D, as they may provide important new targets via their preferential production of diabetes‐related cytokines, including IL‐17A, IL‐21, IL‐22 and IL‐25, all of which play a role at the initiation or destruction phases of T1D.
IL‐17, a predominant product of Th17 cells, plays a pathogenic role in T1D (Table 1 and Figure 1). NOD and STZ‐induced diabetic mice exhibited significant increases in the percentages of IL‐17‐secreting T cells in the islet periphery following disease onset.126 Anti‐IL‐17 treatment, which antagonises Th17 cell differentiation, prevented diabetes development in the majority of NOD mice at 10 weeks of age, when effector destruction begins (Table 1).127 In this context, anti‐IL‐17 reduced the frequency of IL‐17+ cells and increased the proportion of Foxp3+ cells, surrounding the islets.127 However, anti‐IL‐17 did not alter the diabetes progression in NOD mice when treatment was initiated at 5 weeks of age, supporting a diabetogenic role for IL‐17 in the β‐cell destruction process but not in the initiation of this disease.127
IL‐22 is widely considered a Th17 cytokine because it is mostly coexpressed with IL‐17. Defective production of IL‐22 and IL‐17 by MAIT cells has been described to increase gut permeability in T1D, which suggests that these cytokines may play important roles in complex interactions between the immune system and intestinal microbiota.90 Although IL‐22 has been shown to be elevated in T1D,15 administration of either IL‐22 or an anti‐IL‐22 antibody failed to affect the onset of the disease and β‐cell residual function in a diabetic mouse model (Table 1),45, 128, 129 suggesting that IL‐22 is not required for the pathogenesis of T1D. Interestingly, after its administration, IL‐22 remains active and reaches the pancreas, where the highest level of IL‐22R expression is observed among human tissues (Figure 1).130 IL‐22 upregulates the expression of the antiapoptotic proteins Bcl‐2 and Bcl‐xl, as well as that of the regenerative proteins Reg1 and Reg2, in pancreatic β cells (Table 1).131, 132 The expression pattern of IL‐22/IL‐22R in the pancreas suggests that these molecules may play a vital but yet undefined role in β‐cell viability and regeneration.
IL‐25 is a member of the IL‐17 family of cytokines, but unlike IL‐17, IL‐25 inhibits Th17 cells and induces the secretion of Th2 cytokines (Figure 1). Studies investigating the involvement of IL‐25 in T1D have been limited. One study showed that IL‐25 treatment significantly reduced islet inflammation, prevented autoantibody formation, and delayed the progression to diabetes in prediabetic animals, which was associated with the alteration of the T‐cell repertoire (Table 1).127 The most intriguing finding is that IL‐25 could control diabetes after the disease was established and even delay the recurrence of autoimmunity after syngeneic islet transplantation,127 suggesting that IL‐25 treatment may be superior to IL‐17 neutralisation in regulating the effector autoimmune response during the destructive phase of T1D.
Conclusion and perspectives
As outlined above, cytokine networks play crucial roles in orchestrating inflammation during both the initiation and destruction phases of T1D. This realisation has provided remarkable insights into disease pathogenesis and potential therapeutic strategies. Consequently, approaches to targeting cytokines such as IL‐21, IL‐1β, IL‐2, IL‐6 and TNF‐α are currently evaluated in clinical trials. However, the contradictory functions of some cytokines (IL‐4, IL‐13 and IFN‐γ) have been supported by observations in murine models, indicating that these cytokines can either induce or prevent T1D, depending on the context. This duality may indicate that the roles of cytokines in the destruction of β cells are more complex than previously appreciated. The key feature of cytokine networks in T1D likely is that targeting a single cytokine axis may lead to the development of alternative, compensatory cytokine pathways or exert complex effects, because of partially shared receptors. Furthermore, some cytokine receptors, such as IL‐1βR and IL‐4R, are expressed at high levels not only by cells of the immune system but also by β cells, which seems to increase the sensitivity to cytokine‐induced direct β‐cell death or protection, apart from immune‐mediated effects.
Cytokine function can be affected by immune cell plasticity, which may contribute to the pathogenic or regulatory effects of cytokines within the different microenvironments of T1D. Thus, islet‐resident MAIT cells, which produce increased levels of IFN‐γ and granzyme B, directly participate in the process of β‐cell destruction. Conversely, IL‐17 and IL‐22 from MAIT cells of the gut mucosa may play a protective role against diabetes via preserving the integrity of the gut barrier.90 Therefore, the key issues that remain to be addressed are not identifying which cytokine is central to the disease but rather elucidating the interplay between changes in immune cell function during disease progression, as well as defining the different tissue microenvironments that participate in the disease development and the importance of cytokine networks involved in the pathogenesis of T1D.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgments
We apologise to all those authors whose work on this subject has not been cited owing to space constraints. We also gratefully acknowledge funding support from the National Natural Science Foundation of China (grant number 81603122).
References
- 1. Naushad N, Perdigoto AL, Rui J, Herold KC. Have we pushed the needle for treatment of Type 1 diabetes? Curr Opin Immunol 2017; 49: 44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. DiMeglio LA, Evans‐Molina C, Oram RA. Type 1 diabetes. Lancet 2018; 391: 2449–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Polychronakos C, Li Q. Understanding type 1 diabetes through genetics: advances and prospects. Nat Rev Genet 2011; 12: 781–792. [DOI] [PubMed] [Google Scholar]
- 4. Nepom GT, Ehlers M, Mandrup‐Poulsen T. Anti‐cytokine therapies in T1D: Concepts and strategies. Clin Immunol 2013; 149: 279–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mastrandrea L, Yu J, Behrens T et al Etanercept treatment in children with new‐onset type 1 diabetes: pilot randomized, placebo‐controlled, double‐blind study. Diabetes Care 2009; 32: 1244–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hartemann A, Bensimon G, Payan CA et al Low‐dose interleukin 2 in patients with type 1 diabetes: a phase 1/2 randomised, double‐blind, placebo‐controlled trial. Lancet Diabetes Endocrinol 2013; 1: 295–305. [DOI] [PubMed] [Google Scholar]
- 7. Rosenzwajg M, Churlaud G, Mallone R et al Low‐dose interleukin‐2 fosters a dose‐dependent regulatory T cell tuned milieu in T1D patients. J Autoimmun 2015; 58: 48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Grinberg‐Bleyer Y, Baeyens A, You S et al IL‐2 reverses established type 1 diabetes in NOD mice by a local effect on pancreatic regulatory T cells. J Exp Med 2010; 207: 1871–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tang Q, Adams JY, Penaranda C et al Central role of defective interleukin‐2 production in the triggering of islet autoimmune destruction. Immunity 2008; 28: 687–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Diaz‐de‐Durana Y, Lau J, Knee D et al IL‐2 immunotherapy reveals potential for innate beta cell regeneration in the non‐obese diabetic mouse model of autoimmune diabetes. PLoS One 2013; 8: e78483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Klatzmann D, Abbas AK. The promise of low‐dose interleukin‐2 therapy for autoimmune and inflammatory diseases. Nat Rev Immunol 2015; 15: 283–294. [DOI] [PubMed] [Google Scholar]
- 12. Rabinovitch A, Suarez‐Pinzon WL, Shapiro AM, Rajotte RV, Power R. Combination therapy with sirolimus and interleukin‐2 prevents spontaneous and recurrent autoimmune diabetes in NOD mice. Diabetes 2002; 51: 638–645. [DOI] [PubMed] [Google Scholar]
- 13. Vonberg AD, Acevedo‐Calado M, Cox AR et al CD19+IgM+ cells demonstrate enhanced therapeutic efficacy in type 1 diabetes mellitus. JCI Insight 2018; 3: e99860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shruthi S, Mohan V, Amutha A, Aravindhan V. Increased serum levels of novel T cell cytokines IL‐33, IL‐9 and IL‐17 in subjects with type‐1 diabetes. Cytokine 2016; 86: 6–9. [DOI] [PubMed] [Google Scholar]
- 15. Ryba‐Stanislawowska M, Werner P, Brandt A, Mysliwiec M, Mysliwska J. Th9 and Th22 immune response in young patients with type 1 diabetes. Immunol Res 2016; 64: 730–735. [DOI] [PubMed] [Google Scholar]
- 16. Kukreja A, Cost G, Marker J et al Multiple immuno‐regulatory defects in type‐1 diabetes. J Clin Invest 2002; 109: 131–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kent SC, Chen Y, Clemmings SM et al Loss of IL‐4 secretion from human type 1a diabetic pancreatic draining lymph node NKT cells. J Immunol 2005; 175: 4458–4464. [DOI] [PubMed] [Google Scholar]
- 18. Wang B, Gonzalez A, Hoglund P, Katz JD, Benoist C, Mathis D. Interleukin‐4 deficiency does not exacerbate disease in NOD mice. Diabetes 1998; 47: 1207–1211. [DOI] [PubMed] [Google Scholar]
- 19. Rapoport MJ, Jaramillo A, Zipris D et al Interleukin 4 reverses T cell proliferative unresponsiveness and prevents the onset of diabetes in nonobese diabetic mice. J Exp Med 1993; 178: 87–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cameron MJ, Arreaza GA, Zucker P et al IL‐4 prevents insulitis and insulin‐dependent diabetes mellitus in nonobese diabetic mice by potentiation of regulatory T helper‐2 cell function. J Immunol 1997; 159: 4686–4692. [PubMed] [Google Scholar]
- 21. Cameron MJ, Arreaza GA, Waldhauser L, Gauldie J, Delovitch TL. Immunotherapy of spontaneous type 1 diabetes in nonobese diabetic mice by systemic interleukin‐4 treatment employing adenovirus vector‐mediated gene transfer. Gene Ther 2000; 7: 1840–1846. [DOI] [PubMed] [Google Scholar]
- 22. Mueller R, Krahl T, Sarvetnick N. Pancreatic expression of interleukin‐4 abrogates insulitis and autoimmune diabetes in nonobese diabetic (NOD) mice. J Exp Med 1996; 184: 1093–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ruffner MA, Robbins PD. Dendritic cells transduced to express interleukin 4 reduce diabetes onset in both normoglycemic and prediabetic nonobese diabetic mice. PLoS One 2010; 5: e11848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rehman KK, Trucco M, Wang Z, Xiao X, Robbins PD. AAV8‐mediated gene transfer of interleukin‐4 to endogenous β‐cells prevents the onset of diabetes in NOD mice. Mol Ther 2008; 16: 1409–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kaminski A, Welters HJ, Kaminski ER, Morgan NG. Human and rodent pancreatic β‐cells express IL‐4 receptors and IL‐4 protects against β‐cell apoptosis by activation of the PI3K and JAK/STAT pathways. Biosci Rep 2009; 30: 169–175. [DOI] [PubMed] [Google Scholar]
- 26. Russell MA, Morgan NG. The impact of anti‐inflammatory cytokines on the pancreatic β‐cell. Islets 2014; 6: e950547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mi QS, Ly D, Zucker P, McGarry M, Delovitch TL. Interleukin‐4 but not interleukin‐10 protects against spontaneous and recurrent type 1 diabetes by activated CD1d‐restricted invariant natural killer T‐cells. Diabetes 2004; 53: 1303–1310. [DOI] [PubMed] [Google Scholar]
- 28. Ryden A, Faresjo M. Altered immune profile from pre‐diabetes to manifestation of type 1 diabetes. Diabetes Res Clin Pract 2013; 100: 74–84. [DOI] [PubMed] [Google Scholar]
- 29. Ryden A, Ludvigsson J, Fredrikson M, Faresjo M. General immune dampening is associated with disturbed metabolism at diagnosis of type 1 diabetes. Pediatr Res 2014; 75: 45–50. [DOI] [PubMed] [Google Scholar]
- 30. Zaccone P, Phillips J, Conget I et al Interleukin‐13 prevents autoimmune diabetes in NOD mice. Diabetes 1999; 48: 1522–1528. [DOI] [PubMed] [Google Scholar]
- 31. Rutti S, Howald C, Arous C, Dermitzakis E, Halban PA, Bouzakri K. IL‐13 improves β‐cell survival and protects against IL‐1β‐induced β‐cell death. Mol Metab 2016; 5: 122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Leslie KA, Russell MA, Taniguchi K, Richardson SJ, Morgan NG. The transcription factor STAT6 plays a critical role in promoting β cell viability and is depleted in islets of individuals with type 1 diabetes. Diabetologia 2019; 62: 87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ukah TK, Cattin‐Roy AN, Chen W, Miller MM, Barik S, Zaghouani H. On the role IL‐4/IL‐13 heteroreceptor plays in regulation of type 1 diabetes. J Immunol 2017; 199: 894–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Radu DL, Noben‐Trauth N, Hu‐Li J, Paul WE, Bona CA. A targeted mutation in the IL‐4α gene protects mice against autoimmune diabetes. Proc Natl Acad Sci USA 2000; 97: 12700–12704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li C, Zhang L, Chen Y, Lin X, Li T. Protective role of adenovirus vector‐mediated interleukin‐10 gene therapy on endogenous islet β‐cells in recent‐onset type 1 diabetes in NOD mice. Exp Ther Med 2016; 11: 1625–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Robert S, Gysemans C, Takiishi T et al Oral delivery of glutamic acid decarboxylase (GAD)‐65 and IL10 by Lactococcus lactis reverses diabetes in recent‐onset NOD mice. Diabetes 2014; 63: 2876–2887. [DOI] [PubMed] [Google Scholar]
- 37. Rapoport MJ, Mor A, Vardi P et al Decreased secretion of Th2 cytokines precedes up‐regulated and delayed secretion of Th1 cytokines in activated peripheral blood mononuclear cells from patients with insulin‐dependent diabetes mellitus. J Autoimmun 1998; 11: 635–642. [DOI] [PubMed] [Google Scholar]
- 38. Kleffel S, Vergani A, Tezza S et al Interleukin‐10+ regulatory B cells arise within antigen‐experienced CD40+ B cells to maintain tolerance to islet autoantigens. Diabetes 2015; 64: 158–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zheng XX, Steele AW, Hancock WW et al A noncytolytic IL‐10/Fc fusion protein prevents diabetes, blocks autoimmunity, and promotes suppressor phenomena in NOD mice. J Immunol 1997; 158: 4507–4513. [PubMed] [Google Scholar]
- 40. Torres‐Aguilar H, Sanchez‐Torres C, Jara LJ, Blank M, Shoenfeld Y. IL‐10/TGF‐β‐treated dendritic cells, pulsed with insulin, specifically reduce the response to insulin of CD4+ effector/memory T cells from type 1 diabetic individuals. J Clin Immunol 2010; 30: 659–668. [DOI] [PubMed] [Google Scholar]
- 41. Wogensen L, Lee MS, Sarvetnick N. Production of interleukin 10 by islet cells accelerates immune‐mediated destruction of β cells in nonobese diabetic mice. J Exp Med 1994; 179: 1379–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Moritani M, Yoshimoto K, Tashiro F et al Transgenic expression of IL‐10 in pancreatic islet A cells accelerates autoimmune insulitis and diabetes in non‐obese diabetic mice. Int Immunol 1994; 6: 1927–1936. [DOI] [PubMed] [Google Scholar]
- 43. Hillhouse EE, Beauchamp C, Chabot‐Roy G, Dugas V, Lesage S. Interleukin‐10 limits the expansion of immunoregulatory CD4−CD8− T cells in autoimmune‐prone non‐obese diabetic mice. Immunol Cell Biol 2010; 88: 771–780. [DOI] [PubMed] [Google Scholar]
- 44. Iglesias M, Arun A, Chicco M et al Type‐I interferons inhibit interleukin‐10 signaling and favor type 1 diabetes development in nonobese diabetic mice. Front Immunol 2018; 9: 1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ishigame H, Zenewicz LA, Sanjabi S et al Excessive Th1 responses due to the absence of TGF‐β signaling cause autoimmune diabetes and dysregulated Treg cell homeostasis. Proc Natl Acad Sci USA 2013; 110: 6961–6966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Peng Y, Laouar Y, Li MO, Green EA, Flavell RA. TGF‐β regulates in vivo expansion of Foxp3‐expressing CD4+CD25+ regulatory T cells responsible for protection against diabetes. Proc Natl Acad Sci USA 2004; 101: 4572–4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. King C, Davies J, Mueller R et al TGF‐β1 alters APC preference, polarizing islet antigen responses toward a Th2 phenotype. Immunity 1998; 8: 601–613. [DOI] [PubMed] [Google Scholar]
- 48. Grewal IS, Grewal KD, Wong FS et al Expression of transgene encoded TGF‐β in islets prevents autoimmune diabetes in NOD mice by a local mechanism. J Autoimmun 2002; 19: 9–22. [DOI] [PubMed] [Google Scholar]
- 49. Green EA, Gorelik L, McGregor CM, Tran EH, Flavell RA. CD4+CD25+ T regulatory cells control anti‐islet CD8+ T cells through TGF‐β‐TGF‐β receptor interactions in type 1 diabetes. Proc Natl Acad Sci USA 2003; 100: 10878–10883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Oh SA, Liu M, Nixon BG et al Foxp3‐independent mechanism by which TGF‐β controls peripheral T cell tolerance. Proc Natl Acad Sci USA 2017; 114: E7536–E7544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dinarello CA. Immunological and inflammatory functions of the interleukin‐1 family. Annu Rev Immunol 2009; 27: 519–550. [DOI] [PubMed] [Google Scholar]
- 52. Maedler K, Schumann DM, Sauter N et al Low concentration of interleukin‐1β induces FLICE‐inhibitory protein‐mediated β‐cell proliferation in human pancreatic islets. Diabetes 2006; 55: 2713–2722. [DOI] [PubMed] [Google Scholar]
- 53. Boni‐Schnetzler M, Boller S, Debray S et al Free fatty acids induce a proinflammatory response in islets via the abundantly expressed interleukin‐1 receptor I. Endocrinology 2009; 150: 5218–5229. [DOI] [PubMed] [Google Scholar]
- 54. Thomas HE, Irawaty W, Darwiche R et al IL‐1 receptor deficiency slows progression to diabetes in the NOD mouse. Diabetes 2004; 53: 113–121. [DOI] [PubMed] [Google Scholar]
- 55. Moran A, Bundy B, Becker DJ et al Interleukin‐1 antagonism in type 1 diabetes of recent onset: two multicentre, randomised, double‐blind, placebo‐controlled trials. Lancet 2013; 381: 1905–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. van Asseldonk EJ, van Poppel PC, Ballak DB, Stienstra R, Netea MG, Tack CJ. One week treatment with the IL‐1 receptor antagonist anakinra leads to a sustained improvement in insulin sensitivity in insulin resistant patients with type 1 diabetes mellitus. Clin Immunol 2015; 160: 155–162. [DOI] [PubMed] [Google Scholar]
- 57. Ablamunits V, Henegariu O, Hansen JB et al Synergistic reversal of type 1 diabetes in NOD mice with anti‐CD3 and interleukin‐1 blockade: evidence of improved immune regulation. Diabetes 2012; 61: 145–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pagni PP, Bresson D, Rodriguez‐Calvo T et al Combination therapy with an anti‐IL‐1β antibody and GAD65 DNA vaccine can reverse recent‐onset diabetes in the RIP‐GP mouse model. Diabetes 2014; 63: 2015–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Maedler K, Sergeev P, Ris F et al Glucose‐induced β cell production of IL‐1β contributes to glucotoxicity in human pancreatic islets. J Clin Invest 2002; 110: 851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yamada K, Takane‐Gyotoku N, Yuan X, Ichikawa F, Inada C, Nonaka K. Mouse islet cell lysis mediated by interleukin‐1‐induced Fas. Diabetologia 1996; 39: 1306–1312. [DOI] [PubMed] [Google Scholar]
- 61. Jones SA, Jenkins BJ. Recent insights into targeting the IL‐6 cytokine family in inflammatory diseases and cancer. Nat Rev Immunol 2018; 18: 773–789. [DOI] [PubMed] [Google Scholar]
- 62. Teros T, Hakala R, Ylinen L et al Cytokine balance and lipid antigen presentation in the NOD mouse pancreas during development of insulitis. Pancreas 2000; 20: 191–196. [DOI] [PubMed] [Google Scholar]
- 63. Campbell IL, Hobbs MV, Dockter J, Oldstone MB, Allison J. Islet inflammation and hyperplasia induced by the pancreatic islet‐specific overexpression of interleukin‐6 in transgenic mice. Am J Pathol 1994; 145: 157–166. [PMC free article] [PubMed] [Google Scholar]
- 64. Chen YL, Qiao YC, Pan YH et al Correlation between serum interleukin‐6 level and type 1 diabetes mellitus: a systematic review and meta‐analysis. Cytokine 2017; 94: 14–20. [DOI] [PubMed] [Google Scholar]
- 65. Hundhausen C, Roth A, Whalen E et al Enhanced T cell responses to IL‐6 in type 1 diabetes are associated with early clinical disease and increased IL‐6 receptor expression. Sci Transl Med 2016; 8: 356ra119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Paula FM, Leite NC, Vanzela EC et al Exercise increases pancreatic β‐cell viability in a model of type 1 diabetes through IL‐6 signaling. FASEB J 2015; 29: 1805–1816. [DOI] [PubMed] [Google Scholar]
- 67. Dahlen E, Dawe K, Ohlsson L, Hedlund G. Dendritic cells and macrophages are the first and major producers of TNF‐α in pancreatic islets in the nonobese diabetic mouse. J Immunol 1998; 160: 3585–3593. [PubMed] [Google Scholar]
- 68. Green EA, Eynon EE, Flavell RA. Local expression of TNFα in neonatal NOD mice promotes diabetes by enhancing presentation of islet antigens. Immunity 1998; 9: 733–743. [DOI] [PubMed] [Google Scholar]
- 69. Green EA, Wong FS, Eshima K, Mora C, Flavell RA. Neonatal tumor necrosis factor α promotes diabetes in nonobese diabetic mice by CD154‐independent antigen presentation to CD8+ T cells. J Exp Med 2000; 191: 225–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lee LF, Xu B, Michie SA et al The role of TNF‐α in the pathogenesis of type 1 diabetes in the nonobese diabetic mouse: analysis of dendritic cell maturation. Proc Natl Acad Sci USA 2005; 102: 15995–16000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yang XD, Tisch R, Singer SM et al Effect of tumor necrosis factor α on insulin‐dependent diabetes mellitus in NOD mice. I. The early development of autoimmunity and the diabetogenic process. J Exp Med 1994; 180: 995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tack CJ, Kleijwegt FS, Van Riel PL, Roep BO. Development of type 1 diabetes in a patient treated with anti‐TNF‐α therapy for active rheumatoid arthritis. Diabetologia 2009; 52: 1442–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bloom BJ. Development of diabetes mellitus during etanercept therapy in a child with systemic‐onset juvenile rheumatoid arthritis. Arthritis Rheum 2000; 43: 2606–2608. [DOI] [PubMed] [Google Scholar]
- 74. Morgan NG, Leete P, Foulis AK, Richardson SJ. Islet inflammation in human type 1 diabetes mellitus. IUBMB Life 2014; 66: 723–734. [DOI] [PubMed] [Google Scholar]
- 75. Chehadeh W, Weill J, Vantyghem MC et al Increased level of interferon‐α in blood of patients with insulin‐dependent diabetes mellitus: relationship with coxsackievirus B infection. J Infect Dis 2000; 181: 1929–1939. [DOI] [PubMed] [Google Scholar]
- 76. Xia CQ, Peng R, Chernatynskaya AV et al Increased IFN‐α‐producing plasmacytoid dendritic cells (pDCs) in human Th1‐mediated type 1 diabetes: pDCs augment Th1 responses through IFN‐α production. J Immunol 2014; 193: 1024–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lombardi A, Tomer Y. Interferon α impairs insulin production in human β cells via endoplasmic reticulum stress. J Autoimmun 2017; 80: 48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Marroqui L, Dos Santos RS, Op de Beeck A et al Interferon‐α mediates human β cell HLA class I overexpression, endoplasmic reticulum stress and apoptosis, three hallmarks of early human type 1 diabetes. Diabetologia 2017; 60: 656–667. [DOI] [PubMed] [Google Scholar]
- 79. Marro BS, Ware BC, Zak J, de la Torre JC, Rosen H, Oldstone MB. Progression of type 1 diabetes from the prediabetic stage is controlled by interferon‐α signaling. Proc Natl Acad Sci USA 2017; 114: 3708–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Stewart TA, Hultgren B, Huang X, Pitts‐Meek S, Hully J, MacLachlan NJ. Induction of type I diabetes by interferon‐α in transgenic mice. Science 1993; 260: 1942–1946. [DOI] [PubMed] [Google Scholar]
- 81. Li Q, Xu B, Michie SA, Rubins KH, Schreriber RD, McDevitt HO. Interferon‐α initiates type 1 diabetes in nonobese diabetic mice. Proc Natl Acad Sci USA 2008; 105: 12439–12444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Rother KI, Brown RJ, Morales MM et al Effect of ingested interferon‐α on β‐cell function in children with new‐onset type 1 diabetes. Diabetes Care 2009; 32: 1250–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Sobel DO, Ahvazi B. Alpha‐interferon inhibits the development of diabetes in NOD mice. Diabetes 1998; 47: 1867–1872. [DOI] [PubMed] [Google Scholar]
- 84. Sobel DO, Han J, Williams J, Yoon JW, Jun HS, Ahvazi B. Gamma interferon paradoxically inhibits the development of diabetes in the NOD mouse. J Autoimmun 2002; 19: 129–137. [DOI] [PubMed] [Google Scholar]
- 85. Driver JP, Racine JJ, Ye C et al Interferon‐γ limits diabetogenic CD8+ T‐cell effector responses in type 1 diabetes. Diabetes 2017; 66: 710–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Shinomiya M, Fazle Akbar SM, Shinomiya H, Onji M. Transfer of dendritic cells (DC) ex vivo stimulated with interferon‐gamma (IFN‐γ) down‐modulates autoimmune diabetes in non‐obese diabetic (NOD) mice. Clin Exp Immunol 1999; 117: 38–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Mori Y, Kodaka T, Kato T, Kanagawa EM, Kanagawa O. Critical role of IFN‐γ in CFA‐mediated protection of NOD mice from diabetes development. Int Immunol 2009; 21: 1291–1299. [DOI] [PubMed] [Google Scholar]
- 88. Campbell IL, Wong GH, Schrader JW, Harrison LC. Interferon‐γ enhances the expression of the major histocompatibility class I antigens on mouse pancreatic β cells. Diabetes 1985; 34: 1205–1209. [DOI] [PubMed] [Google Scholar]
- 89. Sarvetnick N, Liggitt D, Pitts SL, Hansen SE, Stewart TA. Insulin‐dependent diabetes mellitus induced in transgenic mice by ectopic expression of class II MHC and interferon‐γ. Cell 1988; 52: 773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Rouxel O, Da Silva J, Beaudoin L et al Cytotoxic and regulatory roles of mucosal‐associated invariant T cells in type 1 diabetes. Nat Immunol 2017; 18: 1321–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Chen J, Feigenbaum L, Awasthi P et al. Insulin‐dependent diabetes induced by pancreatic β cell expression of IL‐15 and IL‐15α. Proc Natl Acad Sci USA 2013; 110: 13534–13539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Xia J, Liu W, Hu B, Tian Z, Yang Y. IL‐15 promotes regulatory T cell function and protects against diabetes development in NK‐depleted NOD mice. Clin Immunol 2010; 134: 130–139. [DOI] [PubMed] [Google Scholar]
- 93. Stocks BT, Wilson CS, Marshall AF, Hoopes EM, Moore DJ. Regulation of diabetogenic immunity by IL‐15‐activated regulatory CD8 T cells in type 1 diabetes. J Immunol 2019; 203: 158–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Ryba‐Stanislawowska M, Werner P, Skrzypkowska M, Brandt A, Mysliwska J. IL‐33 effect on quantitative changes of CD4+CD25highFOXP3+ regulatory T cells in children with type 1 diabetes. Mediators Inflamm 2016; 2016: 9429760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Lu J, Liang Y, Zhao J, Meng H, Zhang X. Interleukin‐33 prevents the development of autoimmune diabetes in NOD mice. Int Immunopharmacol 2019; 70: 9–15. [DOI] [PubMed] [Google Scholar]
- 96. Pavlovic S, Petrovic I, Jovicic N et al IL‐33 prevents MLD‐STZ induction of diabetes and attenuate insulitis in prediabetic NOD mice. Front Immunol 2018; 9: 2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Ryba‐Stanislawowska M, Buksa L, Brandt A, Juhas U, Mysliwiec M. IL‐33 improves the suppressive potential of regulatory T cells in patients with type 1 diabetes. Diabetes Res Clin Pract 2017; 128: 67–73. [DOI] [PubMed] [Google Scholar]
- 98. Lee LF, Logronio K, Tu GH et al. Anti‐IL‐7 receptor‐α reverses established type 1 diabetes in nonobese diabetic mice by modulating effector T‐cell function. Proc Natl Acad Sci USA 2012; 109: 12674–12679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Penaranda C, Kuswanto W, Hofmann J et al. IL‐7 receptor blockade reverses autoimmune diabetes by promoting inhibition of effector/memory T cells. Proc Natl Acad Sci USA 2012; 109: 12668–12673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Li CR, Deiro MF, Godebu E, Bradley LM. IL‐7 uniquely maintains FoxP3+ adaptive Treg cells that reverse diabetes in NOD mice via integrin‐β7‐dependent localization. J Autoimmun 2011; 37: 217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Harnaha J, Machen J, Wright M et al Interleukin‐7 is a survival factor for CD4+ CD25+ T‐cells and is expressed by diabetes‐suppressive dendritic cells. Diabetes 2006; 55: 158–170. [PubMed] [Google Scholar]
- 102. Calzascia T, Pellegrini M, Lin A et al CD4 T cells, lymphopenia, and IL‐7 in a multistep pathway to autoimmunity. Proc Natl Acad Sci USA 2008; 105: 2999–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Chtanova T, Tangye SG, Newton R et al T follicular helper cells express a distinctive transcriptional profile, reflecting their role as non‐Th1/Th2 effector cells that provide help for B cells. J Immunol 2004; 173: 68–78. [DOI] [PubMed] [Google Scholar]
- 104. Vogelzang A, McGuire HM, Yu D, Sprent J, Mackay CR, King C. A fundamental role for interleukin‐21 in the generation of T follicular helper cells. Immunity 2008; 29: 127–137. [DOI] [PubMed] [Google Scholar]
- 105. McGuire HM, Vogelzang A, Ma CS et al A subset of interleukin‐21+ chemokine receptor CCR9+ T helper cells target accessory organs of the digestive system in autoimmunity. Immunity 2011; 34: 602–615. [DOI] [PubMed] [Google Scholar]
- 106. Ferreira RC, Simons HZ, Thompson WS et al IL‐21 production by CD4+ effector T cells and frequency of circulating follicular helper T cells are increased in type 1 diabetes patients. Diabetologia 2015; 58: 781–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Viisanen T, Ihantola EL, Nanto‐Salonen K et al Circulating CXCR5+PD‐1+ICOS+ follicular T helper cells are increased close to the diagnosis of type 1 diabetes in children with multiple autoantibodies. Diabetes 2017; 66: 437–447. [DOI] [PubMed] [Google Scholar]
- 108. Spolski R, Kashyap M, Robinson C, Yu Z, Leonard WJ. IL‐21 signaling is critical for the development of type I diabetes in the NOD mouse. Proc Natl Acad Sci USA 2008; 105: 14028–14033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Sutherland AP, Van Belle T, Wurster AL et al Interleukin‐21 is required for the development of type 1 diabetes in NOD mice. Diabetes 2009; 58: 1144–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Van Belle TL, Nierkens S, Arens R, von Herrath MG. Interleukin‐21 receptor‐mediated signals control autoreactive T cell infiltration in pancreatic islets. Immunity 2012; 36: 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Liu SM, Lee DH, Sullivan JM et al Differential IL‐21 signaling in APCs leads to disparate Th17 differentiation in diabetes‐susceptible NOD and diabetes‐resistant NOD.Idd3 mice. J Clin Invest 2011; 121: 4303–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Attridge K, Wang CJ, Wardzinski L et al IL‐21 inhibits T cell IL‐2 production and impairs Treg homeostasis. Blood 2012; 119: 4656–4664. [DOI] [PubMed] [Google Scholar]
- 113. Jandl C, Liu SM, Canete PF et al IL‐21 restricts T follicular regulatory T cell proliferation through Bcl‐6 mediated inhibition of responsiveness to IL‐2. Nat Commun 2017; 8: 14647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Bettini M, Castellaw AH, Lennon GP, Burton AR, Vignali DA. Prevention of autoimmune diabetes by ectopic pancreatic β‐cell expression of interleukin‐35. Diabetes 2012; 61: 1519–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Manzoor F, Johnson MC, Li C, Samulski RJ, Wang B, Tisch R. β‐cell‐specific IL‐35 therapy suppresses ongoing autoimmune diabetes in NOD mice. Eur J Immunol 2017; 47: 144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Singh K, Kadesjo E, Lindroos J et al Interleukin‐35 administration counteracts established murine type 1 diabetes–possible involvement of regulatory T cells. Sci Rep 2015; 5: 12633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Espes D, Singh K, Sandler S, Carlsson PO. Increased interleukin‐35 levels in patients with type 1 diabetes with remaining C‐peptide. Diabetes Care 2017; 40: 1090–1095. [DOI] [PubMed] [Google Scholar]
- 118. Trembleau S, Penna G, Gregori S, Giarratana N, Adorini L. IL‐12 administration accelerates autoimmune diabetes in both wild‐type and IFN‐γ‐deficient nonobese diabetic mice, revealing pathogenic and protective effects of IL‐12‐induced IFN‐γ. J Immunol 2003; 170: 5491–5501. [DOI] [PubMed] [Google Scholar]
- 119. Trembleau S, Penna G, Gregori S, Gately MK, Adorini L. Deviation of pancreas‐infiltrating cells to Th2 by interleukin‐12 antagonist administration inhibits autoimmune diabetes. Eur J Immunol 1997; 27: 2330–2339. [DOI] [PubMed] [Google Scholar]
- 120. Nicoletti F, Di Marco R, Zaccone P et al Endogenous interleukin‐12 only plays a key pathogenetic role in non‐obese diabetic mouse diabetes during the very early stages of the disease. Immunology 1999; 97: 367–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Fujihira K, Nagata M, Moriyama H et al Suppression and acceleration of autoimmune diabetes by neutralization of endogenous interleukin‐12 in NOD mice. Diabetes 2000; 49: 1998–2006. [DOI] [PubMed] [Google Scholar]
- 122. Trembleau S, Penna G, Gregori S et al Pancreas‐infiltrating Th1 cells and diabetes develop in IL‐12‐deficient nonobese diabetic mice. J Immunol 1999; 163: 2960–2968. [PubMed] [Google Scholar]
- 123. Zhang J, Huang Z, Sun R, Tian Z, Wei H. IFN‐γ induced by IL‐12 administration prevents diabetes by inhibiting pathogenic IL‐17 production in NOD mice. J Autoimmun 2012; 38: 20–28. [DOI] [PubMed] [Google Scholar]
- 124. Solt LA, Banerjee S, Campbell S, Kamenecka TM, Burris TP. ROR inverse agonist suppresses insulitis and prevents hyperglycemia in a mouse model of type 1 diabetes. Endocrinology 2015; 156: 869–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Bending D, De la Pena H, Veldhoen M et al Highly purified Th17 cells from BDC2.5NOD mice convert into Th1‐like cells in NOD/SCID recipient mice. J Clin Invest 2009; 119: 565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Li CR, Mueller EE, Bradley LM. Islet antigen‐specific Th17 cells can induce TNF‐α‐dependent autoimmune diabetes. J Immunol 2014; 192: 1425–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Emamaullee JA, Davis J, Merani S et al Inhibition of Th17 cells regulates autoimmune diabetes in NOD mice. Diabetes 2009; 58: 1302–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Borg DJ, Wang R, Murray L et al The effect of interleukin‐22 treatment on autoimmune diabetes in the NOD mouse. Diabetologia 2017; 60: 2256–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Bellemore SM, Nikoopour E, Krougly O, Lee‐Chan E, Fouser LA, Singh B. Pathogenic T helper type 17 cells contribute to type 1 diabetes independently of interleukin‐22. Clin Exp Immunol 2016; 183: 380–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Shioya M, Andoh A, Kakinoki S, Nishida A, Fujiyama Y. Interleukin 22 receptor 1 expression in pancreas islets. Pancreas 2008; 36: 197–199. [DOI] [PubMed] [Google Scholar]
- 131. Hill T, Krougly O, Nikoopour E et al The involvement of interleukin‐22 in the expression of pancreatic β cell regenerative Reg genes. Cell Regen (Lond) 2013; 2: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Feng D, Park O, Radaeva S et al Interleukin‐22 ameliorates cerulein‐induced pancreatitis in mice by inhibiting the autophagic pathway. Int J Biol Sci 2012; 8: 249–257. [DOI] [PMC free article] [PubMed] [Google Scholar]