

Abstract
CD8+ T-cell exhaustion is a dysfunctional state that is regulated through the expression of inhibitory checkpoint receptor genes including the cytotoxic T-lymphocyte–associated antigen 4, programmed death 1, and DNA methylation of effector genes interferon-γ, perforin, and granzyme B. Different strategies have been used to reverse T-cell exhaustion, which is an adverse event of checkpoint inhibitor blockade. Here, we present the mechanisms by which DNA methyltransferase inhibitors and Simian virus 40 large T antigen through viral mimicry can promote the reversion of exhausted CD8+ T cells. We examine how these pharmacological strategies can work together to improve the clinical efficacy of immunotherapies.
Keywords: CD8+ T cells, cell immortalization, checkpoint inhibitors, epigenetics, methylation, methyltransferase inhibitors, oncovirus, SV40-T antigen
Introduction
Our understanding of the signaling pathways that control naïve CD8+ T-cell differentiation into effector and memory cells upon tissue damage and acute and chronic pathogen infections has been providing new avenues for improving immune therapies based on cells, antibodies, or cytokines.1–4 However, one concern in the checkpoint blockade therapy in cancer is heterogeneity of response rate, which can vary from 10% to 40% in those patients with immunologically hot tumors.1–4 The CD8+ T-cell activation and differentiation into memory and effector cells depends on instructive genomic, epigenomic, metabolic, and mitotic division programs triggered by cytokines, chemokines, and cell–cell interactions as well as an intricate balance of downstream signaling pathways.5–8 The terminal effector CD8+ T-cell populations exhibit various dysfunctional or hyporesponsive states induced by persistent antigen exposure, costimulatory and coinhibitory receptor expression, telomere crisis, and senescence.9,10 These states are distinct from tolerance and anergy, which are acquired at early T-cell receptor (TCR) negative selection and via central and peripheral immune tolerance mechanisms.9,10 Endogenous and exogenous factors may contribute to T-cell dysfunctions, including defects in cytotoxicity, proliferation and secretion of cytokines interleukin (IL)-2, IL-7, IL-15, tumor necrosis factor (TNFα and β), and interferons. As highlighted in recent studies, the blockade of the immune checkpoint receptors on the surface of T cells, such as cytotoxic T lymphocyte antigen 4 (CTLA-4), lymphocyte activation gene-3 (LAG-3), T-cell immunoglobulin mucin domain 3 (TIM-3), B- and T-lymphocyte attenuator (BTLA), T -ell immunoglobulin and T-cell immunoreceptor tyrosine-based inhibitory motif (ITIM) domain, and programmed cell death 1 (PD-1) can rescue the cytotoxic function of CD8+ T cells and brings benefits to patients undergoing combined chemoimmunotherapies.1–4 Recent studies revealed that these T-cell dysfunctional states could be rescued by epigenetic reprogramming therapy.11,12 Thus, erasing epigenomic signatures of exhausted T cells appears to be one of the critical steps toward developing therapeutic strategies for overcoming T-cell dysfunction. First, we briefly describe molecular mechanisms by which immune-checkpoint blockade modulates T-cell activation and exhaustion. Second, we provide an update on cellular epigenetic programs that control naïve T cell differentiation into effector and memory cell subsets and vice versa. Third, we describe viral mimicry as the mechanism by which demethylating agents enhance tumor immunogenicity as well as the survival and functionality of CD8+ T cells. At the end, we introduce SV40 T antigen protein as a biological tool for restoration of senescence and exhaustion of CD8+ T cells.
T-cell development and CD4 and CD8 T lineage differentiation
T-cell development is characterized by expression and rearrangement of the TCR genes coding for γ, δ, α, and β chains during passage of lymphoid progenitors into the thymus.13–15 This stochastic process leads to V(D)J somatic recombination of TCR genes to give rise to either γ and δ or α and β progenitors at the CD4 and CD8 double-negative stage. CD8+ cytotoxic and CD4+ helper/inducer T cells develop from common thymocyte precursors that are TCR+, CD3+, CD4+, and CD8+. Hematopoietic dendritic cells (DCs) and thymic epithelial cells express major histocompatibility (MHC), class I and II, which process and present self- and non-self-antigens to TCRs. The selection of TCR+, CD3+, and CD4+ T cells depends on class II expression, whereas TCR+,CD3+, and CD8+ T cells depend on class I. Thus, when TCRs on the T-cell membrane recognize self-antigens with high affinity using class II molecules, the CD4+ T-cell population is eliminated (negative selection) and only the TCR+, CD3+, and CD8+ T-cell population remains alive. Conversely, if TCRs recognize self-antigens with high affinity using class I, CD8+ T cells are eliminated (negative selection), and only the TCR+, CD3+, and CD4+ T-cell population remains alive. It has been shown recently that the zinc finger transcription factor Th-POK (T-cell-inducing POS/Kruppel-like factor), the core-binding factor β (RUNX/CBFβ) transcription factor complexes, and the thymocyte selection associated high mobility group box protein (TOX) are the master regulators of CD4/CD8 lineage commitment.14,15 Th-POK gene is only detected in thymocytes after positive selection. Expression of RUNX complexes and TOX occur in mature CD4−CD8+ single-positive thymocytes and deficiency of the TOX gene in mice blocked the production of all CD4 T lineage cells, however, CD8+ T cells developed normally.14
Naïve CD4+ and CD8+ T cells display a diverse repertoire of TCR specificity that recognize, in a specific manner, self and non-self molecular motifs via MHC class I or class II.16 The self-reactivity of T cells is regulated through negative factors that, in principle, prevent or tolerate inappropriate T-cell activation.9 Not tolerated, mutated self-proteins (neo-antigens) or foreigner protein epitopes, which arise from degradation of cell proteins, are recognized either through cross-presentation by professional antigen presenting cells (APCs) or directly when presented by tumor or infected cells. Following a positive stimulation, naïve CD4+/CD8+ T cells initiate a burst of tyrosine phosphorylation of various transducing and transcription factors, including nuclear factor activated T cells, (NAFT1), mammalian target of rapamycin (mTOR), protein-kinase B (AKT), and nuclear factor-κB (NF-κB), which orchestrate downstream biochemical events.5,6 The phosphoinositide 3-kinase (PI3K)/AKT/mTOR pathway is well established in regulating cell survival, proliferation, and metabolism of immune cells throughout mitochondrial bioenergetics pathways.17,18 These events are depicted in Figure 1.
Figure 1.
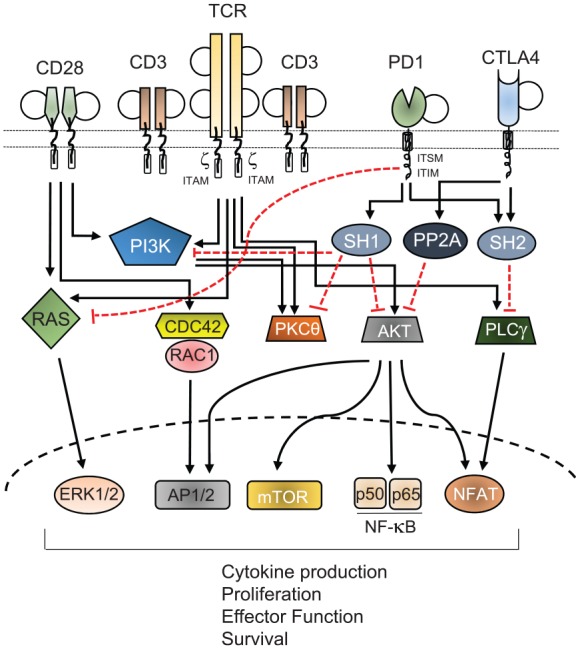
TCR/CD3 complex and CD28 downstream signaling pathways leading to activation of transcription of genes for cytokines, chemokines, cell division, activation of effector function, and survival. The coinhibitory receptors programmed cell death 1 (PD-1) and cytotoxic T lymphocyte antigen 4 (CTLA-4) suppress T-cell activation and function through the recruitment of the phosphatases SH2 domain-containing tyrosine phosphatase 1 (SHP1), SHP2 and serine/threonine protein phosphatase 2A (PP2A) via their ITAM, ITIM, or ITSM motif. These phosphatases dephosphorylate critical serine/threonine protein kinases PI3K, AKT, and PLCθ that play roles in multiple cellular processes for stimulation of T cells. PD-1 inhibits the RAS-extracellular signal-regulated kinase (ERK) pathway and CTLA-4 inhibits PLCγ and thereby the NFAT transcriptional activity. NFAT activation and its nuclear translocation requires cooperation of calmodulin, a well-known calcium sensor protein, which activates the serine/threonine phosphatase calcineurin. Engagement of PD-1 receptor with PD-L1 or PD-L2 recruits SHP2 phosphatase to its cytoplasmic domain, which functions to inhibit TCR signaling pathway by preventing ZAP70 phosphorylation and its association with CD3ζ at TCR complex.
ζ, TCR homodimeric domain; AKT, protein-kinase B; AP, activator protein; CDC42, cell division control protein 42 homolog; FOXO1, forkhead box protein O1; ITAM, T-cell immunoreceptor tyrosine-based activation motif; ITIM, T-cell immunoreceptor tyrosine-based inhibitory motif; ITSM, immunoreceptor tyrosine-based switch motif; mTOR, mammalian target of rapamycin; NF-κB, nuclear factor-κB; NFAT, nuclear factor of activated T cells; PKCθ, protein kinase Cθ; PLCγ, phospholipase Cγ; RAC1, Ras-related C3 botulinum toxin substrate 1; TCR, T-cell receptor.
T-cell exhaustion and checkpoint inhibitor receptor expression
T-cell exhaustion refers to functional unresponsiveness due to antigen overstimulation commonly observed alongside chronic viral infection and cancer overgrowth.19,20 Various studies have been performed to characterize the cellular and molecular features associated with the unresponsiveness of human tumor-associated T cells (TSTs) and tumor infiltrating lymphocytes.21–26 CD8+ T-cell effector activities are constrained by various inhibitory signaling pathways from an extrinsic immunosuppressive tumor environment, including those mediated by myeloid-derived suppressor cells, CD4+CD25+ T regulatory cells, IL-10, transforming growth factor-β (TGF-β), reactive oxygen and nitrogen species, hypoxia, and low pH, which impede their proliferative and metabolic demands.21–26 Dysfunctional CD8+ T cells display a higher expression of surface markers CD101 and CD38, costimulatory receptors and inhibitory receptors CTLA-4, PD-1, LAG-3, BTLA, and TIM-3.21–26 The exhausted T cells display an impaired production of effector cytokines and cytotoxic molecules, including TNFα, interferon γ (IFNγ), perforin, granzymes, and granulysin.21–26
The primary objective of the checkpoint therapies is to reactivate cytotoxic activity of dysfunctional CD8+ T cells in peripheral lymphoid tissues and inflammatory tissue enviroments.2 The over-activation of TCRs is inhibited through competition by costimulatory and coinhibitory families and subfamilies of the transmembrane receptors, which share extracellular and intracellular domains for recognition and activation of downstream signaling upon ligand binding.5,6 Their interaction and molecular mechanisms are quite complex and not well understood. The activating and inhibitory receptors containing ITIMs and immunoreceptor tyrosine-based activation motif molecules, which serve as intracellular docking sites for members of the spleen tyrosine kinase and ζ-chain-associated protein kinase 70 kDa (ZAP-70) family. When inhibitory phosphorylated receptors interact via ITIM molecules with cytosolic SHP-1 and SHP-2 tyrosine phosphatases, the downstream TCR (signal 1) and CD28-mediated costimulation (signal 2) signaling pathway are inhibited.2,5,6 CTLA-4 is expressed following T-cell activation and is necessary to effectively attenuate T-cell activation through competition with the costimulatory molecule CD28 for the B7 ligands B7-1 (CD80) and B7-2 (CD86), for which CTLA-4 has higher avidity and affinity.27,28 These interactions take place at the immune synapse between T cells and APCs. This results in the activation of a number of signaling molecules including the p85 subunit of PI3 kinase, the Src family kinase Lck, IL-2 inducible family kinase Itk, protein kinase C theta, and adaptor proteins Grb2.27,28 Through this mechanism, CTLA-4 also attenuates downstream signaling mediated by phosphoinositide 3-kinase (PI3K) and AKT during TCR signaling. These events are illustrated in Figure 1. CTLA-4 blockade therefore increases CD28 costimulation and, thus, reactivates TCR signaling and T-cell expansion. CTLA-4 is typically found in intracellular vesicles and degraded in lysosomal compartments or recycled to the plasma membrane.28 This is the mechanism for depleting the CD80/86 from APC and avoiding overstimulation.28 Accordingly, CTLA-4-deficiency leads to a hyperactivation of the immune system that causes lymphoproliferative disease, tissue damage, and ultimately death within days of age in transgenic mice.29
PD-1 inhibitory receptor is an immunoglobulin family member related to the CD28 and CTLA-4 family.30 It is expressed on T cells, B cells, monocytes, and natural killer (NK) cells and is induced on peripheral CD4+ and CD8+ cells to regulate T-cell activation. Their ligands, PD-L1 and PD-L2, are expressed on some tissues, including the endothelium, placenta, macrophages, and DCs, and mainly in tumor cells. PD-1-deficient mice develop autoimmune diseases, such as lupus-like diseases or dilated cardiomyopathy. Interestingly, PD-1 deficiency causes an increased proliferation as compared with normal T cells. Like CTLA-4, PD-1 expression is induced on T cells following activation and is linked to exhaustion events.28,30
Given that engagement with both PD-1 and CTLA-4 attenuate T-cell activity, mono or multiple combinations of monoclonal antibodies, such as anti-CTLA-4, anti-PD-1, or PD-L1, have been used to increase cell survival and proliferative lifecycles of T cells and attenuate or overcome exhaustion.1–4 PD-1 inhibits cell proliferation by attenuating TCR-mediated activation and synthesis of IL-2 and its receptor, which are important steps for the activation of downstream PI3K-AKT and ERK pathways and cell cycle progression through the G1 phase to mitosis. Moreover, ligation of PD-1 modulates cellular metabolism by inhibiting PI3K and the mTOR pathway that causes shifts of glycolysis toward oxidative phosphorylation.17,18,22 It is interesting that pharmacological inhibition of phosphoinositide 3-kinase δ (PI3Kδ) with small molecules antagonized the antitumor effects of CD4+ and CD8+ T cells and checkpoint blockade therapies.31
The activity of many transcription factors including T-bet, eomesodermin homolog (Eomes), PR domain zinc finger protein 1 (PRDM1), B-lymphocyte-induced maturation protein-1 (Blimp-1), early gene response 1 (Egr1), and Egr2 are reduced or altered in dysfunctional CD8+ T cells.7,8,32 TOX is highly expressed in dysfunctional TSTs and in exhausted T cells in chronic viral infection.33 In agreement, comparative gene expression and chromatin accessibility profiles from exhausted versus control CD8+ T cells between wild-type and TOX-deficient transgenic mice showed decreased expression of multiple inhibitory receptors and the reversal of the exhausted phenotype.34 These studies confirmed that the expression of TOX and TOX2 as well as NR4A, a nuclear receptor of the calcium/calcineurin-regulated transcription factor NFAT family act as major inducers of the exhausted state observed in CD8+ CAR+ PD-1high TIM3high tumor-infiltrating lymphocytes.34 CD8+ T cells with ‘stem-like’ and pluripotency properties have a greater ability to self-renew and reinvigorate after PD-1 blockade.35,36 Therefore, further manipulation (with drugs, cytokines, or genetic engineering) in this complex framework of cytoplasmic and nuclear signaling pathways may be required for achieving long-term effects and successful adoptive immunotherapy.
Epigenetics mechanisms
Epigenetic transmission of information plays a critical role in a number of important cellular reprogramming events during embryonic and adult life.37 Specific epigenetic modifications in a DNA molecule determine accessibility of genetic loci to transcriptional machinery and the sequential heritable gene expressions. A double strand DNA molecule is wrapped around a core complex of four histone proteins, which bind to DNA and form a nucleosome. Methylation marks on lysine or arginine on histones H3 and H4, typically, acetylation of H3/H4 in their lysine residue and methylation of H3 at its lysine 4 (H3K4me and H3K4me3) residue mark for transcriptionally active chromatin whereas methylation of H3 at its Lysine 9 and 27 (H3K9me2 and H3K27me) mark for repressive chromatin in genomic loci.38 These histone epigenetic modifications are combinatorial with DNA methylation, the covalent addition of a methyl group to the C5 of the cytosine base, present exclusively in CpG sites that form 5-methylcytosine. The methyltransferases DNMT1, 3A, and 3B, using methyl donor S-adenosyl-methionine, promote inheritance DNA methylation that allows the preservation of methylation marks on the new DNA strand of daughter cells during cellular division.37 Methylated DNA provides a platform for the binding of methyl-CpG-binding domain proteins, which prevent gene expression by recruiting chromatin remodeling corepressor complexes, containing histone deacetylases (HDACs) and histone methyltransferases (HMTs). Together with the chromatin remodelers, ten–eleven translocation proteins (TET1–3) and repressive histone-modifying enzymes of the polycomb repressive complex 1 (PRC1) and PRC2, through a HMT called enhancer of zeste homologue 2 (EZH2), exert their repressive function. These enzymes act as writers (methyltransferases) and erasers (demethylases) that directly regulate the epigenetic patterns on DNA and histone complexes.38 Post-translational modification of histones contributes to a histone code, which regulates the accessibility of DNA to transcriptional machinery, including RNA polymerase II and transcription factors. Noncoding RNAs and micro RNAs, which act by altering histone–DNA interactions and chromatin state, can impose another epigenetic layer at global and loci-specific levels.37,38
The standardization and significant evolution of the assays chromatin immunoprecipitation with DNA sequencing (ChIP-seq) and transposase accessible chromatin with high-throughput DNA sequencing (ATAC-Seq) have allowed the systematic evaluation of epigenetic patterns in promoters, enhancers, and insulators in various DNA regions involved in gene transcriptional regulation.39,40 Large-scale deep DNA sequencing and mapping of differentially accessible regions in immune cells at distinct states have revealed major locus regions under epigenetic regulation during innate and adaptive responses to tissue damage, infections, and autoimmune diseases.41,42 These studies have given further support to the direct interplay of heritable genetic and epigenetic mechanisms in the regulation of effector T-cell activation, survival, and functions, and self-renewing of memory T cells. The reversible and dynamic nature of the epigenetic changes create potential opportunities for cellular reprogramming therapy.
Epigenetic reprogramming of memory and effector T cells after virus infection
Previous studies have identified the molecular basis for epigenetic changes tied to transition of T-cell effector and memory function in mice during acute lymphocytic choriomeningitis virus (LCMV) infection and in humans after yellow fever (YFV) virus vaccination.43 More precisely, Youngblood and colleagues followed up de novo DNA methylation by DNMT3A in an isolated population of naïve, memory, and effector CD8+ T cells at various times during virus infection. They evaluated the activation and binding of the transcription factors T-bet (also known as Tbx21), Eomes, and Blimp-1 and their the epigenetic states using ChIP-seq and ATAC-Seq assays.44,45 These transcription factors bound exactly on a DNA locus that encompasses the promoters for L-selectin/Sell, B-cell lymphoma 2 (BCL-2), granzymes, perforin 1, and CD127 genes.44,45 The Sell promoter was completely unmethylated in naïve cells, whereas the Sell promoter was significantly methylated in LCMV-specific effector CD8+ T cells. The methylation profiling imprinted in terminal effector CD8+ T-cell populations (95%) marked them for cell death, whereas the methylation of the Sell promoter in a minority (10%) of memory precursor (MP) CD8+ T-cell subsets (stem cell memory T and central memory T) increased their survival. Next, the authors longitudinally tracked MP CD8+ T cells and noted that these cells re-expressed naïve associated genes thorough a cellular dedifferentiation process as they developed into memory cells. More importantly, they confirmed these results after the immunization of mice and humans with LCMV and YFV, respectively. Longitudinal phenotypic and epigenetic studies done 1–2 years later demonstrated that memory CD8+ T cells could reactive the epigenetic program and rapidly become again effector CD8+ T cells. They concluded that memory T cells arise from effector T-cell subsets.44,45
High-dimensional cellular phenotyping (CyTOF analysis) combined with bioinformatic analysis revealed that upregulation of chemokines and low or absence of production of IFNγ, TNF, and IL-2 as specific features of exhausted CD8+ T cells with high viral load.46 The regulatory region of the Pdcd1 locus is completely demethylated in exhausted CD8+ T cells, thus these cells display sustained PD-1 expression after virus infection.47,48 CD8+ T cells displaying stem-like properties have a very distinct epigenetic signature.35,36 The expression of CXCR5, a chemokine receptor, in a specific CD8+ T-cell population (CXCR5+ and CD8+ T cells) appears to be responsible for the regulation of long-term memory CD8+ T cells that selectively proliferate after PD-1 blockade.36,49
DNA methyltransferase inhibitors and induction of viral mimicry
Previous studies identified molecular basis for phenotype and epigenetic changes tied to dysfunctional CD8+ T cells isolated from tissue microenvironment of tumor xenografts in cancer mouse models.25,26 Remarkably, among the intrinsic mechanisms identified are those associated with the epigenetic reprograming and chromatin remodeling.25,26 Importantly, these studies revealed that these T-cell dysfunctional states could be rescued by pharmacological approaches.12,33
The DNA demethylating inhibitors (DDMTi) 5-aza-2’-deoxycytidine (decitabine) and 5-azacytidine are cytosine analogs that, when incorporated into DNA molecules stop methyltransferase activity and, thereby, proper DNA methylation reaction.50 Recent studies have demonstrated that treatment with DDMTi in vitro and in vivo can restore not only the immunogenicity of cancer cells but also the cytotoxic activity of cytolytic T cells and their chemoattraction into the tumor tissue.51,52 DNA methylation by inactivating viral gene transcription and replication within the host cells promotes an antiviral defense. Most endogenous retroviruses and retrotransposons in the human genome are inactivated by DNA hypermethylation.53 DDMTi treatment induces a viral mimicry effect on several cancer cells.50,51 The viral mimicry refers to global epigenetic modification in DNA molecules that leads to reactivation of genes encoding for ancient retroviruses including endogenous retroviruses (ERVs) and non-LTR retrotransposons, such as long interspersed nuclear elements (LINEs) and short interspersed nuclear elements.53 These endogenous double-strand RNAs (dsRNAs) are recognized by the mitochondrial cytosolic pattern recognition receptor melanoma differentiation-associated protein, IFIH1 (MDA5), retinoic acid-inducible gene I protein, DDX58 (RIG-I), and cGAMP synthase.54 MDA5 and RIG-I contain two N-terminal CARD domains and a central DEAD box helicase/ATPase domain. These viral RNA cytosolic sensors interact with the mitochondrial adaptor protein – mitochondrial antiviral-signaling protein (MAVS).54 MAVS has a CARD domain-containing protein that interacts with NLRP3 protein and stimulator of interferon genes (STING) at the mitochondrion membrane. MAVS antiviral signaling propagated through assembly of a MAVS signalosome, which contains TRAF3, TRAF6, TRAF family member-associated NF-κB activator, and TNFR1-associated death domain protein.54 Toll-like receptor 3 (TLR3), TLR7, and TLR9 are cytoplasmic endosomal receptors that also detect free short hairpin RNA (shRNA).55 Together, these dsRNA cytosolic sensors ultimately promote phosphorylation and nuclear translocation of the interferon regulatory factor 1 (IRF1) and IRF7. IRFs promote the transcriptional induction of type I and III IFNs. IFNs, through the JAK-STAT pathway, lead to the transcription of hundreds of IFN-stimulated genes (ISGs) coding for many antiviral proteins.12,54,55 DDMTi treatment also increases the expression of MHC class I and antigen peptide transporter 1 (TAP) and latent cancer-testis antigens (CTAs) only expressed in testis, placenta, and in special embryonic stem cells and tumor cells.51,52 These events simultaneously increase CTAs presentation onto class I MHC molecules that boost T- and B-cell immunoreactivity, thus increasing patient immune response against cancer cells.12 In an ongoing study, Yau and collaborators demonstrated that DDMTi treatment also promotes the activation of the dsRNA-mediated viral mimicry in CD8+ T cells infiltrated in tumor tissue.56 DDMTi induces double-stranded human endogenous retrovirus (dsHERV) expression, in particular, HERV9 of LTR12 family.12,53,56 This is followed by MAV aggregation and activation of IRF7, which upregulate the expression of IFN genes.56 It can be assumed that these biological events are similar to what is observed in cancer cells. T-cell recognition of HERV families is characterized in few T-cell epitopes in patient samples from a variety of cancers.53,57 Therefore, it will be important that these experiments can be replicated in future studies.
Consistent with the results described above, Sheng and colleagues discovered that the ablation of the histone H3K4 demethylase LSD1 stimulates antitumor immunity by improving the checkpoint blockade response to melanoma cells in a mouse model.56 LSD1 mediated effects were driven through upregulation of ERV transcripts, type I and III IFNs, and consequently ISG expression.58 Additional support for the viral mimicry pathway also appeared in the studies describing the function of RNA editing enzyme adenosine deaminase acting on RNA (ADAR1) in a transgenic mouse model.57 ADAR1 catalyzes the deamination of adenosine to inosine in dsRNA in viral and cellular RNAs, which is referred to as A-to-I RNA editing.59 The ablation of the ADAR1 gene is embryonically lethal. Mice die from fatal immune disease owing to suppression of MAVS cytosolic RNA sensor signaling that mediate phosphorylation of IRF7 and transcription of ISGs.59 Finally, another study describing the pharmacological effects of the small molecule cyclin-dependent kinases 4 and 6 (CDK4/6) inhibitors, yielded similar results.60 It was observed that the CDK4/6 inhibitor abemaciclib, while decreasing cell proliferation, enhances shRNA levels (viral mimicry) in tumor cells, and consequently the production of type III IFNs and IFN-induced ISGs in an autocrine fashion.57 Accordingly, treatment with abemaciclib markedly decreased DNMT1 mRNA levels and consequently DNA methylation of ERVs in cancer cells.60
SV40 large T antigen protein and viral mimicry
The SV40 virus belongs to the polyomavirus family of small dsDNA viruses.61 SV40 virion encodes for early large T, middle and, small tumor antigens and the structural Vp capsid proteins. The SV40 large T antigen is a multifunctional protein that displays efficient ATPase, DNA, and RNA helicase activity. As a replicative helicase and initiator, large T protein assembles as a double-hexamer at the double-stranded DNA molecule to initiate genomic replication.61 Moreover, large T protein interacts with p53 and RB proteins.62,63 After binding to p53, T protein prevents it from activating p53-dependent genes and consequently to induce cell cycle arrest and apoptosis. After binding to RB1 and the retinoblastoma-related proteins p130 and p107, large T protein inhibits their interaction with E2F1, and thus its transcriptional activity.62,63 RB plays a role in heterochromatin formation and chromatin structure, in particular, by stabilizing histone methylation.62,63 A complex formed by RB and the EZH2 promotes histone methylation and DNA methylation at CpG islands.62,63 Thus, together they promote the silencing of microsatellites, LINEs, endogenous retroviruses, and transposons.62,63 Therefore, large T protein may circumvent these fundamental steps of genome regulation to promote cellular immortalization.
Infection by SV40 virus increases DNA repair and DNA-damage response (DDR) via activation of the ataxia-telangiectasia and Rad3-related (ATR) kinases.64–66 Cellular infections with the herpes simplex virus, human cytomegalovirus, and Epstein–Barr virus also cause DDR.67 However, in these cases, DNA repair response is mediated by the recombinase-activating enzymes RAG1 and RAG2.67 Ectopic expression of SV40 T gene alone in human fibroblasts triggers cytoplasmic and nuclear sensors that promote the activation of IRF1 and the production of IFNα/β.68–71 Both IFNs stimulate the expression of hundreds of ISGs, including IFN-stimulated gene 56 (isg56), 2’-5’ oligoadenylate synthetase-1 (oas1), radical S-adenosyl methionine domain-containing protein 2 ( rsad2), interferon alpha inducible protein 27 (Ifi27) interferon-induced GTP-binding protein (Mx1), and interferon-induced, double-stranded RNA-activated protein kinase or protein kinase R (EIF2AK2/PKR), as well as IRF7, IRF9, and RIG-1.68–71 Type II IFNs (IFNα and IFNβ) signal through the IFNα/β receptor (IFNAR) complex, which is a heterodimer of IFNAR1 and IFNAR2. Type III IFNγ signals through IFNγ receptor to activate the signal transducer and activator of transcription (STAT1/2) protein signaling pathways. These results suggest that DDMTi and SV40 large T protein by inducing the viral mimicry response reactive the innate response. In this context, their ability to induce the secretion of IFNs may be the most critical for their proinflammatory and antitumor effects. These events on epigenetic reprogrammability and reversion of phenotypic and functional features of CD8+ T-cell subsets are illustrated in Figure 2.
Figure 2.

Genetic and epigenetic regulators of CD8+ T-cell activation and differentiation into memory, effector, and exhausted T-cell populations. DNA demethylation at CpG sites of regulatory regions and locus and action of master transcriptional factors (TCF1, T-bet) result in the transcriptional repression or derepression of key genes associated with memory (sell, CCR7, IL7R), survival (BCL-2, CD127), proliferation (IL-2, IL-4), and effector functions (IFNs, GZMB, PRF1, KLRG1, GNLY). Effector T cells die upon antigen withdrawal or overactivation. DDMTi and SV40 large T antigen promote viral mimicry state and derepression of human endogenous retrovirus (HERVs) that activate the MDA5/RIG/MAVS innate immune response pathways. This leads to type I and III interferon-mediated synthesis of IGSs. A combination of viral mimicry agents and inhibitory antibodies to immune checkpoint molecules, such as CTLA-4, PD-1, PDL-1, and PDL-2, may reverse CD8+ T cell-exhaustion through epigenetic reprogramming.
Blimp-1, B-lymphocyte-induced maturation protein-1; DDMTi, DNA demethylating agents; GNLY, granulysin; GZMB, granzyme B; IFN, interferon; IGSs, interferon-stimulated genes; IL, interleukin; IL7R, interleukin 7 receptor; MAVS, mitochondrial antiviral-signaling protein; MDA5, melanoma differentiation-associated protein; PRF1, perforin; RIG I, retinoic acid-inducible gene I protein; RUNX, the Run-related domain for DNA binding and core-binding factor β complex; TCF1/Tcf-7, T-cell-specific high Mobility Group Box protein transcription factor 7; TOX, Thymocyte Selection Associated High Mobility Group Box protein; l to o = methylated (pin head closed in black), o = unmethylated (pin head opened in white).
Mouse model for immune cell immortalization and rejuvenation
Immortomouse is a transgenic BALB/c mouse strain containing genomic insertion of a transgene coding for one copy of SV40 T antigen, named the tsA58 gene.70 This gene encodes for a mutant temperature-sensitive nuclear phosphoprotein that is under the control of the class I antigen (H-2Kb) promoter of the mouse histocompatibility complex.72 The tsA58 protein expression is inducible by IFNγ under an in vitro permissive temperature of 33°C and it is unstable at 37°C. Thus, primary cell lines grow slowly or not at all at 37°C (nonpermissive temperature). Remarkably, a leakage in the expression of tsA58 protein is normally observed in the thymus, and the transgenic mice develop thymic hyperplasia instead of natural involution along their lifespan.72 Specifically bone marrow lymphoid cells, thymic fibroblasts, and epithelial cells contribute to overgrowth and enlargement of the gland. We have been investigating the significance of SV40 T expression in thymic and spleen lymphoid cell development in Balb/C immortomice.73 We confirmed that bone marrow naïve progenitor CD3+ T cells successfully develop into CD4+, CD8+ and Foxp3+ T-cell lineages despite the thymus hyperplasia.71 Next, we evaluated the innate and adaptive immune responses in Immortomice. To do this, we used a well-established protocol to measure in vivo cytotoxic activity of CD8+ T cells.74 First, Immortomice were immunized with adenovirus engineered to express beta-galactosidase protein. Next, splenic cells from naïve donor mice were pulsed with beta-galactosidase specific peptide TPHPARIGL, and subsequently these cells were tail injected in immunized SV40 T antigen Immortomice. Overall, the results confirmed that CD8+ T-cell population acquired memory and effector functions as estimated by lysing peptide-pulsed target cells at the cytotoxic rate similar to the control group. Consistent with our results, a recent report described the immunological features of DCs expressing a dexamathasone and doxycyclin inducible SV40 large T antigen.75 Isolated SV40 T-DCs were capable of developing an effective immune response in terms of cytokine production, antigen presentation, recruiting neutrophils, and T-cell polarization in in vitro and in vivo assays.75 Together, these data suggest that immortalized immune cells can normally develop innate and adaptive responses after infection or immunization. Finally, more research is needed to identify whether these immune cells under these theoretical considerations can resist the dysfunctional states in a different experimental setting. Previous safety evaluation in clinical studies have not warranted any potential risks related to SV40 T oncogenesis and tumorigenesis in accordance with the World Health Organization (WHO) regulations. Even though SV40 T mediates inhibition of p53 and Rb pathways, various parameters related to transforming properties such as cell growth and proliferation, apoptosis, and migration are not sufficient to induce oncogenic transformation in vivo.76,77 To achieve more tightly regulated control and safety of conditionally immortalized cells that could be reversed to normal cells, an ideal approach would be expression, via cell-specific gene promoter conditionally inducible, together with a suicide gene system for inactivation of immortalized dividing cells. For instance, the suicide gene herpes simplex virus thymidine kinase (HSV-TK) and the drug ganciclovir can provide effective and safe control to prevent adverse events from SV40 T transplanted cells in clinic medicine.78
Future perspective
Current immunotherapies in humans rely on the isolation, engineering, or both, of dendritic cells and T-specific cell subtypes, which then require several rounds of in vitro expansion before being transferred into patients.1–4 Administration of exogenous cytokines IL-2 and IL-15 and the proinflammatory IL-12 have been used for expansion of CD4+, CD8+ T and NK cells for cell adoptive therapy.79,80 The efficacy of T cell immunotherapy based on CTLA-4 and PD-1/PD-L1 checkpoint blockade depends on a complex signaling mechanism that require inputs in receptor-mediated proliferation and differentiation stimulatory pathways and feedbacks of receptor-mediated inhibitory pathways.26,27,30 We know now that the cellular environment through secretion of IL-6, IL-10, IL-35, and TGF-β, and their separate pathways, as well as via direct inhibitory contact with other T cells, can tune down the TCR intracellular signaling pathways. Thus, many transduction and metabolic signaling pathways contribute to dysfunctional and exhausted CD8+ T cells that restrict the responsiveness to immunotherapies. The formation of memory T cells is a critical component of patient responses.36,49 The isolation of CD8+ T with stem-like unique signature among CD8+ T heterogeneous cell populations constitute a promising strategy combining immunotherapy and chemotherapies.2,20,36 Efficacy of epigenetic therapy with DNA demethylating agents depends on their incorporation into DNA of cycling cells during S phase.50 The passive demethylation of ERVs and CTAs activates the viral mimicry response in cancer cells and likely T-cell populations.12 DNMTi may also contribute to epigenetic reprogramming of genes for cytokines, cytotoxic factors, and cell cycle genes.35,36 We know that depending on schedules and doses DNA methyltransferase (DNMT) and HDAC inhibitors are highly cytotoxic and may deplete important T-cell lineages.12,50 Moreover, under continued stimulation the effector activities of CD8+ T cells are impaired by premature senescence caused by both shorting of telomeres, telomerase gene promoter inactivation, halt of telomerase reverse transcriptase activity, and, finally, activation-induced cell death.81 Targeted expression of SV40 T antigen may, in principle, overcome cellular senescence by hijacking pRB, p53, and DNA repair proteins, thus allowing continued DNA replication of exhausted CD8+ T cells. Therefore, ex vivo SV40 T antigen immortalization could assist to restore or preserve differentiation and cytotoxic features of CD8+ T cells. By answering these crucial questions, we could upgrade the efficacy and clinical outcomes of current immunotherapies in chronic infections and cancer.
Acknowledgments
We thank Gustavo Amarante-Mendes and Isabella Sampaio (University of Sao Paulo, Sao Paulo, Brazil), Daniel De Carvalho (Princess Margaret Cancer Center, Toronto, Canada), and Jorge Scutti (MD Anderson MD Anderson Cancer Center, Houston, TX, USA) for their collaboration and advice during this project.
Footnotes
Author contribution: JEB and MFDR were responsible for the study conception, selection and critical review of the papers, and manuscript preparation. All authors read and approved the final manuscript.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, proc 486048/2011 and 312206/2016-0), International Centre for Genetic Engineering and Biotechnology (CRP/BRA08-01), and Fundação de Amaparo a Pesquisa do Estado de São Paulo (FAPESP, proc 2018/08540-8).
Conflict of interest statement: The authors declare that there is no conflict of interest.
ORCID iD: José Belizário  https://orcid.org/0000-0002-3832-5279
https://orcid.org/0000-0002-3832-5279
Contributor Information
José Belizário, Department of Pharmacology, Institute Biomedical Sciences of the University of Sao Paulo, Avenida Lineu Prestes, 1524, São Paulo, CEP 05508-900, Brazil.
Maria Fernanda Destro Rodrigues, Postgraduate Program in Biophotonics Applied to Health Science, Nove de Julho University, São Paulo, SP, Brazil.
References
- 1. Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov 2018; 8: 1–18. [DOI] [PubMed] [Google Scholar]
- 2. Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov 2015; 14: 561–584. [DOI] [PubMed] [Google Scholar]
- 3. Yang JC, Rosenberg SA. Adoptive T-cell therapy for cancer. Adv Immunol 2016; 130: 279–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Whiteside TL, Demaria S, Rodrigues-Ruiz ME, et al. Emerging opportunities and challenges in cancer immunotherapy. Clin Cancer Res 2016; 22: 1845–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Ann Rev Immunol 2009; 27: 591–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol 2013; 13: 227–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gray SM, Kaech SM, Staron MM. The interface between transcriptional and epigenetic control of effector and memory CD8+ T cell differentiation. Immunol Rev 2014; 261: 157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Henning AN, Roychoudhuri R, Restifo RP. Epigenetic control of CD8+ T cell differentiation. Nature Rev Immunol 2018; 18: 340–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xing Y, Hogquist KA. T-cell tolerance: central and peripheral. Cold Spring Harb Perspect Biol 2012; 4: a006957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schietinger A, Greenberg PD. Tolerance and exhaustion: defining mechanisms of T cell dysfunction. Trends Immunol 2014; 35: 51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dear AE. Epigenetic modulators and the new immunotherapies. N Engl J Med 2016; 374: 684–686. [DOI] [PubMed] [Google Scholar]
- 12. Jones PA, Ohtani H, Chakravarthy A, et al. Epigenetic therapy in immune-oncology. Nature Rev Cancer 2019; 19: 151–161. [DOI] [PubMed] [Google Scholar]
- 13. Guy CS, Vignali KM, Temirov J, et al. Distinct TCR signaling pathways drive proliferation and cytokine production in T cells. Nat Immunol 2013; 14: 262–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aliahmad P, Kadavallore A, de la Torre B, et al. TOX is required for development of the CD4 T cell lineage gene program. J Immunol 2011; 187: 5931–5940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. He X, Park K, Kappes DJ. The role of ThPOK in control of CD4/CD8 lineage commitment. Annu Rev Immunol 2010; 28: 295–320. [DOI] [PubMed] [Google Scholar]
- 16. Nikolich-Zugich J, Slifka MK, Messaoudi I. The many important facets of T-cell repertoire diversity. Nat Rev Immunol 2004; 4: 123–132. [DOI] [PubMed] [Google Scholar]
- 17. Rambold AS, Pearce EL. Mitochondrial dynamics at the interface of immune cell metabolism and function. Trends Immunol 2018; 39: 6–18. [DOI] [PubMed] [Google Scholar]
- 18. Mills EL, Kelly B, O’Neill LAJ. Mitochondria are the powerhouses of immunity. Nature Immunol 2017; 18: 488–498. [DOI] [PubMed] [Google Scholar]
- 19. Barber DL, Wherry EJ, Masopust D, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006; 439: 682–687. [DOI] [PubMed] [Google Scholar]
- 20. Sen DR, Kaminski J, Barnitz RA, et al. The epigenetic landscape of T cell exhaustion. Science 2016; 354: 1165–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 2015; 15: 486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bengsch B, Johnson AL, Kurachi M, et al. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8(+) T cell exhaustion. Immunity 2016; 45: 358–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Apetoh L, Smyth MJ, Drake CG, et al. Consensus nomenclature for CD8+ T cell phenotypes in cancer. OncoImmun 2015; 4: e998538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fuertes-Marraco SA, Neubert NJ, Verdeil G, et al. Inhibitory receptors beyond T cell exhaustion. Frontiers Immunol 2015; 6: 310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schietinger A, Philip M, Krisnawan VE, et al. Tumor-specific T cell dysfunction is a dynamic antigen-driven differentiation program initiated early during tumorigenesis. Immunity 2016; 45: 389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Philip M, Fairchild L, Sun L, et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 2017; 545: 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Buchbinder BI, Desai A. CTLA-4 and PD-1 pathways similarities, differences, and implications of their inhibition. Am J Clin Oncol 2016; 39: 98–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rowshanravan B, Halliday N, Sansom DM. CTLA-4: a moving target in immunotherapy. Blood 2018; 131: 58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tivol EA, Borriello F, Schweitzer AN, et al. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 1995; 3: 541–347. [DOI] [PubMed] [Google Scholar]
- 30. Sharpe AH and Pauken. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol 2018; 18: 153–167. [DOI] [PubMed] [Google Scholar]
- 31. Lim EL, Cugliandolo FM, Rosner DR, et al. Phosphoinositide 3-kinase δ inhibition promotes antitumor responses but antagonizes checkpoint inhibitors. JCI Insight 2018; 3: pii: 120626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Doering TA, Crawford A, Angelosanto JM, et al. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity 2012; 37: 1130–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Scott AC, Dündar F, Zumbo P, et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 2019; 571: 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Seo H, Chen J, González-Avalos E, et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8+ T cell exhaustion. Proc Natl Acad Sci U S A 2019; 116: 12410–12415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kishton RJ, Sukumar M, Restifo NP. Metabolic regulation of T cell longevity and function in tumor immunotherapy. Cell Metab 2017; 26: 94–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Im SJ, Hashimoto M, Gerner MY, et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 2016; 1537: 417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Feselnfeld G. A brief history of epigenetics. Cold Spring Harb Perspect Biol 2014; 6: a018200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet 2012; 13: 343–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Scheer S, Ackloo S, Medina TS, et al. A chemical biology toolbox to study protein methyltransferases and epigenetic signaling. Nat Commun 2019; 10: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Buenrostro JD, Wu B, Chang HY, et al. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr Protoc Mol Biol 2015; 109: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Busslinger M, Tarakhovsky A. Epigenetic control of immunity. Cold Spring Harb Perspect Biol 2014; 6: a019307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Picascia A, Grimaldi V, Pignalosa O, et al. Epigenetic control of autoimmune diseases: from bench to bedside. Clin Immunol 2015; 157: 1–15. [DOI] [PubMed] [Google Scholar]
- 43. Youngblood B, Hale JS, Ahmed R. T-cell memory differentiation: insights from transcriptional signatures and epigenetics. Immunol 2013; 139: 277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Youngblood B, Hale JS, Kissick HT, et al. Effector CD8 T cells dedifferentiate into long-lived memory cells. Nature 2017; 552: 404–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Akondy RS, Fitch M, Edupuganti S, et al. Origin and differentiation of human memory CD8 T cells after vaccination. Nature 2017; 552: 362–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bengsch B, Ohtani T, Khan O, et al. Epigenomic-guided mass cytometry profiling reveals disease-specific features of exhausted CD8 T cells. Immunity 2018; 48: 1029–1045.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ahn E, Youngblood B, Lee J, et al. Demethylation of the PD-1 promoter is imprinted during the effector phase of CD8 T cell exhaustion. J Virol 2016; 90: 8934–8946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mognol GP, Spreafico R, Wong V, et al. Exhaustion-associated regulatory regions in CD8+ tumor-infiltrating T cells. Proc Natl Acad Sci U S A 2017; 114: E2776–E2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jadhav RR, Im SJ, Hu B, et al. Epigenetic signature of PD-1+ TCF1+ CD8 T cells that act as resource cells during chronic viral infection and respond to PD-1 blockade. Proc Natl Acad Sci U S A 2019; 116: 14113–14118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Agrawal K, Das V, Vyas P, et al. Nucleosidic DNA demethylating epigenetic drugs - A comprehensive review from discovery to clinic. Pharmacol Ther 2018; 188: 45–79. [DOI] [PubMed] [Google Scholar]
- 51. Roulois D, Yau HL, Singhania R, et al. DNA-demethylating agents target colorectal cancer cells by inducing viral Mimicry by endogenous transcripts. Cell 2015; 162: 961–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chiappinelli KB, Strissel PL, Desrichard A, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 2015; 162: 974–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Attermann AS, Bjerregaard AM, Saini SK, et al. Human endogenous retroviruses and their implication for immunotherapeutics of cancer. Ann Oncology 2018; 29: 2183–2191. [DOI] [PubMed] [Google Scholar]
- 54. Jacobs JL, Coyn CB. Mechanisms of MAVS regulation at the mitochondrial membrane. J Mol Biol 2013; 425: 5009–5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011; 34: 637–650. [DOI] [PubMed] [Google Scholar]
- 56. Yau HL, Ankur C, Almeida FA, et al. DNA demethylating agents enhance cytolytic activity of CD8+ T cells and anti-tumor immunity. bioRxiv 2017; 197236 DOI: 10.1101/197236. [DOI] [Google Scholar]
- 57. Mullins CS, Linnebacher M. Endogenous retrovirus sequences as a novel class of tumor-specific antigens: an example of HERV-H env encoding strong CTL epitopes. Cancer Immunol Immunother 2012; 61: 1093–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sheng W, LaFleur MW, Nguyen TH, et al. LSD1 ablation stimulates anti-tumor immunity and enables checkpoint blockade. Cell 2018; 174: 549–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Liddicoat BJ, Piskol R, Chalk AM, et al. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 2015; 349: 1115–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Goel S, DeCristo MJ, Watt AC, et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017; 548: 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. An P, Sáenz Robles MT, Pipas JM. Large T antigens of polyomaviruses: amazing molecular machines. Ann Rev Microbial 2012; 66: 213–236. [DOI] [PubMed] [Google Scholar]
- 62. Sadasivam S, DeCaprio JA. The DREAM complex: master coordinator of cell cycle-dependent gene expression. Nat Rev Cancer 2013; 13: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ishak CA, Marshall AE, Passos DT, et al. An RB-EZH2 complex mediates silencing of repetitive DNA Sequences. Mol Cell 2016; 64: 1074–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Boichuk S, Hu L, Hein J, et al. Multiple DNA damage signaling and repair pathways deregulated by simian virus 40 large T antigen. J Virol 2010; 84: 8007–8020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hauster S, Ulrich T, Wuster S, et al. Loss of LIN9, a member of DREAM complex, cooperates with SV40 large T antigen to induce genomic instability an anchorage-independent growth. Oncogene 2012; 31: 1859–1868. [DOI] [PubMed] [Google Scholar]
- 66. Banerjee P, DeJesus R, Gjoerup O, et al. Viral interference with DNA repair by targeting of the single-stranded DNA binding protein RPA. PLoS Pathog 2013; 9: e1003725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hollingworth R, Grand RJ. Modulation of DNA damage and repair pathways by human tumour viruses. Viruses 2015; 7: 2542–2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Cantalupo PG, Sáenz-Robles MT, Baldwin A, et al. Cell-type specific regulation of gene expression by simian virus 40 T antigens. Virology 2009; 386: 183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Giacobbi NS, Gupta T, Coxon A, et al. Polyomavirus T antigens activate an antiviral state. Virology 2015; 476: 377–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Rathi AV, Cantalupo PG, Sarkar SN, et al. Induction of interferon-stimulated genes by Simian virus 40 T antigens. Virology 2010; 406: 202–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Forero A, Giacobbi NS, McCormick KD, et al. Simian virus 40 large T antigen induces IFN-stimulated genes through ATR kinase. J Immunol 2014; 192: 5933–5942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jat PS, Noble MD, Ataliotis P, et al. Direct derivation of conditionally immortal cell lines from an H-2Kb-tsA58 transgenic mouse. Proc Natl Acad Sci U S A 1991; 88: 5096–5100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Belizário JE, Brandão W, Rossato C, et al. Thymic and post-thymic regulation of naïve CD4+ T cell lineage fates in humans and mice models. Mediat Inflamm 2016; 2016: 9523628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Clemente T, Dominguez MR, Vieira NJ, et al. In vivo assessment of specific cytotoxic T lymphocytes killing. Methods 2013; 61: 105–109. [DOI] [PubMed] [Google Scholar]
- 75. Richter C, Thieme S, Bandola J, et al. Generation of inducible immortalized dendritic cells with proper immune function in vitro and in vivo. PLoS One 2013; 8: e62621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sachsenmeier KF, Pipas JM. Inhibition of Rb and p53 is insufficient for SV40 T-antigen transformation. Virology 2001; 283: 40–48. [DOI] [PubMed] [Google Scholar]
- 77. Hein J, Boichuk S, Wu J, et al. Simian virus 40 large T antigen disrupts genome integrity and activates a DNA damage response via Bub1 binding. J Virol 2009; 83: 117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Greco R, Oliveira G, Stanghellini MT, et al. Improving the safety of cell therapy with the TK-suicide gene. Front Pharmacol 2015; 6: 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kared H, Camous X, Larbi A. T cells and their cytokines in persistent stimulation of the immune system. Curr Opin Immunol 2014; 28: 79–85. [DOI] [PubMed] [Google Scholar]
- 80. Doedens AL, Rubinstein MP, Gross ET, et al. Molecular programming of tumor-infiltrating CD8+ T cells and IL15 resistance. Cancer Immunol Res 2016; 4: 799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Bellon M, Nicot C. Telomere dynamics in immune senescence and exhaustion triggered by chronic viral infection. Viruses 2017; 9: pii: E289. [DOI] [PMC free article] [PubMed] [Google Scholar]