

Abstract
Cervical Cancer is one of the leading causes of cancer-associated mortality in women. The present study aimed to identify key genes and pathways involved in cervical cancer (CC) progression, via a comprehensive bioinformatics analysis. The GSE63514 dataset from the Gene Expression Omnibus database was analyzed for hub genes and cancer progression was divided into four phases (phases I–IV). Pathway enrichment, protein-protein interaction (PPI) and pathway crosstalk analyses were performed, to identify key genes and pathways using a criterion nodal degree ≥5. Gene pathway analysis was determined by mapping the key genes into the key pathways. Co-expression between key genes and their effect on overall survival (OS) time was assessed using The Cancer Genome Atlas database. A total of 3,446 differentially expressed genes with 107 hub genes were identified within the four phases. A total of 14 key genes with 11 key pathways were obtained, following extraction of ≥5 degree nodes from the PPI and pathway crosstalk networks. Gene pathway analysis revealed that CDK1 and CCNB1 regulated the cell cycle and were activated in phase I. Notably, the following terms, ‘pathways in cancer’, ‘focal adhesion’ and the ‘PI3K-Akt signaling pathway’ ranked the highest in phases II–IV. Furthermore, FN1, ITGB1 and MMP9 may be associated with metastasis of tumor cells. STAT1 was indicated to predominantly function at the phase IV via cancer-associated signaling pathways, including ‘pathways in cancer’ and ‘Toll-like receptor signaling pathway’. Survival analysis revealed that high ITGB1 and FN1 expression levels resulted in significantly worse OS. CDK1 and CCNB1 were revealed to regulate proliferation and differentiation through the cell cycle and viral tumorigenesis, while FN1 and ITGB1, which may be developed as novel prognostic factors, were co-expressed to induce metastasis via cancer-associated signaling pathways, including PI3K-Art signaling pathway, and focal adhesion in CC; however, the underlying molecular mechanisms require further research.
Keywords: cervical cancer, bioinformatics analysis, diagnosis, progression
Introduction
Cervical Cancer (CC) is a highly aggressive tumor and is one of the leading causes of cancer-associated mortality in women, with an estimated 570,000 new cases and 311,000 deaths in 2018 worldwide (1). Women with CC are considered to have a lower quality of life (2). The progression of CC, from normal cervical mucosal epithelium to cervical intraepithelial neoplasia (CIN) grade 1, 2, and 3, to CC (3) is associated with persistent high-risk human papillomavirus (HPV) infection (4). Furthermore, a number of risk factors, including early sexual activity (5), multiple sexual partners (6), long-term use of oral contraceptives (7), genetic factors [active oncogenes, including PIK3CA (8), ATAD2 (9) and CRNDE (10); tumor suppressor genes, including p53 (11), Ras association domain family 1 isoform A (12) and NOL7 (13)], tobacco use [current smoker, started smoking age ≤15 years, smoking duration ≥30 years, ≥20 cigarettes/day (14)] and other viral infections (such as HIV, herpes simplex virus (HSV) type II and bacterial infections caused by Chlamydia trachomatis) (15) have been associated with CC progression.
HPV infection plays a leading role in CC (16). The DNA of HPV integrates into the host cell genome [HPV16: q21-q31 of chromosome no. 13; HPV18: q24 of chromosome no. 8 (17)], disrupts the open reading frame and causes overexpression of E6 and E7 genes (18). It has been verified that E6 and E7 exert carcinogenic effects by binding to the cell cycle regulators, p53 and retinoblastoma (Rb) (19). While E6 and E7 proteins are upregulated, E6 can interact with its associated protein [E6-associated protein, E6AP (20)] to form a complex and bind to p53. This binding hydrolyzes p53 and results in the loss of p53-induced negative regulation of cell proliferation, thereby leading to unchecked cellular proliferation and malignant transformation (21). E7 has a high affinity for Rb, which controls the cell cycle. Binding of E7 to Rb can dissociate the Rb-E2F complex, thus releasing E2F to exert its role as a transcription factor, which leads to an uncontrolled cell cycle and cellular immortalization (22,23). Furthermore, centrosomes are central regulators of mitosis that are often increased in numbers in cancer cells (24). A previous study indicated that an abnormally increased number of centrosomes is associated with structural chromosomal abnormality in cervical lesions with high risk of HPV infection (25). Duensing and Münger (26) reported that abnormal number of centrosomes and associated spindle mitotic abnormality can be found in cells infected by the high-risk HPV16 E6 and E7 proteins, but not in cells infected by the low-risk HPV6. Although numerous experimental studies on genes [HPV16 L1 protein (27), sonic hedgehog (28) and FGFR4 (29)], and signaling pathways [Wnt/β-catenin signaling pathway (30), adenosinergic pathway (31) and ERK signal transduction (32)], as well as bioinformatics analyses have focused on microRNAs (33) and genes (34) associated with CC, and have provided an understanding of the pathophysiological mechanisms of the disease over the last decade, the underlying molecular mechanisms remain unclear.
The development of CC occurs over a number of years and its complexity presents clinical challenges in patients screening and treatment. Currently, The Bethesda System (35), which is a tool that is used to report Pap smear results for cervical cytologic diagnoses, provides useful data that allows research into the epidemiology, biology and pathology of cervical lesions; however, its diagnostic value remains poor (36). Instead, direct biopsy remains the gold standard for diagnosis. Nevertheless, invasive examinations may cause adverse psychological effects, including anxiety, depression or distress (37). Surgery, chemotherapy and radiotherapy (38) are the three major therapeutic strategies in the treatment of CC; however, their uses may be limited for various reasons. Surgery may be limited by the status and stage of patients, including late stage or tolerance to anesthesia (39), whereas chemotherapy is limited due to the lack of sensitivity and the development of drug resistance (40). In addition, radiotherapy can be limited by the maximum tolerated dose to adjacent normal tissues (41). Thus, it is essential to understand the underlying molecular mechanisms in the initiation and development of CC, in order to develop methods for its accurate diagnosis and effective treatment. A number of studies have reported that multiple genes [CXCL12 (42), FGFR4 (29) and SHH (43)], proteins [cyclin D1 (44), FOXO1 (45) and BASP1 (46)] and pathways [Toll-like signaling pathway (47), VEGF signaling pathway (48) and Wnt signaling pathway (30)] are involved in the natural progression of CC; however, few studies have investigated the fundamental pathological molecular mechanisms in the progression of CC (from normal, to CIN1, CIN2, CIN3, to cancer). Thus, the specific pathological processes remain unclear.
The present study provided a systematic investigation of the development of CC and further understanding of the associations between the four phases of CC progression, and thus revealed additional targets for the detection and treatment of CC. A flow diagram of the present study is presented in Fig. 1.
Figure 1.

Flow diagram of the present study. FC, fold change; DEGs, differentially expressed genes; STRING, Search Tool for the Retrieval of Interacting Genes/Proteins; GO, Gene Ontology; FDR, false discovery rate; KEGG, Kyto Encyclopedia of Genes and Genomes; PPI, protein-protein interaction.
Materials and methods
Identification of differentially expressed genes (DEGs)
The CC gene expression profile in the GSE63514 dataset, acquired using the GPL570 platform (Affymetrix Human Genome U133 Plus 2.0 Array) provided by den Boon in 2015 (49), was downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). The profile contained 128 cervical specimens, including: Normal (n=24), CIN1 (n=14), CIN2 (n=22), CIN3 (n=40) and cancer (n=28) samples. All samples were divided into four phases as follows: Phase I, normal to CIN1; phase II, CIN1 to CIN2; phase III, CIN2 to CIN3 and phase IV, CIN3 to cancer, and GEO2R tools (https://www.ncbi.nlm.nih.gov/geo/geo2r/) (50) within the limma package version 3.26.8 (51) were used to screen the DEGs at the four phases. The criteria fold change (FC) of expression >2 and P<0.05 were used to identify DEGs.
Identification of hub genes
The Search Tool for the Retrieval of Interacting Genes (STRING) database (52) and Cytoscape software (version 3.5.1) (53) were used to identify the hub genes in the four phases. The PPI network was constructed by searching for gene symbols and the minimum required interaction score was set at 0.7, to ensure high confidence in the results. The nodes that not connect to the major network were removed to decrease the error detection rate. CytoHubba (54), a plug-in for Cytoscape software, was used to investigate notable nodes in the interactome network using 12 topological algorithms, including Degree, Edge Percolated Component, Maximum Neighborhood Component, Density of Maximum Neighborhood Component and Maximal Clique Centrality, and centralities based on shortest paths, such as Bottleneck, EcCentricity, Closeness, Radiality, Betweenness, Clustering Coefficient and Stress. The genes that ranked in the top 10 for each topological algorithm were extracted and the duplication of each gene was calculated. Genes duplicated <2 times were excluded, in order to guarantee that the genes were associated with CC. The remaining genes were considered as hub genes in the four phases.
Functional enrichment analyses of GO and pathways
The functional features of the genes associated with the four phases were examined using WebGestalt (55) and ToppGene (56). In WebGestalt, over-representation analysis was selected as the enrichment method, Biological Process in GO as the functional database, gene symbol as the gene ID type and genome as the reference set for enrichment analysis. In ToppGene, two frequently used databases, Kyoto Encyclopedia of Genes and Genomes (KEGG; http://www.kegg.jp/) and BioCarta (https://www.biocarta.com/), were utilized to perform pathway enrichment analysis, to improve the reliability of the results. Pathways with a false discovery rate of P<0.05 were considered to indicate significantly enriched pathways.
Pathway crosstalk analysis
Pathway crosstalk analysis was performed (57), to investigate the interactions among the significantly enriched pathways. The pathways with either a false discovery rate of P>0.05 or <3 genes were removed as selection criteria. The number of shared genes between pairwise pathways was calculated and pairwise pathways with <2 overlapping genes were removed. The Jaccard Coefficient (JC) and the Overlap Coefficient (OC) parameters were calculated, to measure the overlap between the pathways. Specifically, JC=, while OC=, where A and B represent the gene numbers of the tested pathways. The interrelationships between pathways were visualized using Cytoscape software.
To determine the overall progression and further detect an association between two pathways, the KEGG and BioCarta databases were used to identify the upstream or downstream associations between pathways. Furthermore, the nodal degree was calculated using Centiscape (58) to identify key nodes. According to Han et al (59), key nodes are considered as those with a nodal degree ≥5.
Integration of the PPI network
The PPI network (60) was used to identify key proteins for the four phases of CC. As Protein Interaction Network Analysis (PINA) (https://omics.bjcancer.org/pina/) (61) is an integrated platform for protein interaction network construction, analysis and visualization, it can identify the associations between the queried genes based on integration of data from six public PPI databases: IntAct (62), MINT (63), BioGRID (64), DIP (65), HPRD (66) and MIPS MPact (67). Thus, the PINA4MS plug-in for Cytoscape software was used to construct the PPI network, to identify CC progression-associated genes. As PINA4MS requires UniProt accession numbers, the UniProt Retrieve/ID mapping tool (https://www.uniprot.org/uploadlists/) was used to input gene symbols. The key nodes for the PPI network were also extracted using a criterion of nodal degree ≥5.
Comprehensive gene-pathway analysis
To determine the molecular mechanisms and associations between the key genes and pathways, the gene-pathway network was constructed by examining the key pathways, in order to determine which pathway contained at least one of the key genes.
Co-expression and survival analysis for key genes
To identify the co-expression of key genes and their impact on OS time, the LinkedOmics database (68) was used, which was based on TCGA (69). The co-expression analysis was performed using Pearson correlation and OS analysis was assessed with Cox regression method. For survival analysis, samples were divided by the median value of the investigated gene. P<0.05 was considered to indicate a statistically significant difference for both the co-expression correlation and OS time.
Results
Identification of DEGs
Analysis of the GSE63514 dataset using GEO2R, with a criteria of ≥2 FC and P<0.05, identified a total of 3,446 DEGs for the four phases as follows: 446 DEGs in phase I, of which 76 were upregulated and 370 were downregulaged; 382 DEGs in phase II, of which 146 were upregulated and 236 were downregulated; 756 DEGs in phase III, of which 435 were upregulated and 321 were downregulated; 1,862 DEGs in phase IV, of which 816 were upregulated and 1046 were downregulated.
Identification of hub genes
Following removal of 2,256 irrelevant genes (Phase I, 265; Phase II, 197; Phase III, 603; Phase IV, 1191), 12 topological algorithms were used and the top 10 genes for each method were extracted. A total of 107 genes that appeared at least twice were conserved as hub genes, as presented in Table I. A total of 29 genes were identified in phase I, among which five genes were members of the kinesin family (KIF11, KIF15, KIF23, KIF4A and KIF14), and five genes were associated with meiosis and the maturation of oocytes [BUB1B (70), BUB1 (71), CCNA2 (72), CCNB1 (72) and CDK1 (73)], as well as other genes associated with inflammation and innate immune responses [STAT1 (74), GBP1 (75) and RHOA (76)]. A total of 25 hub genes were verified in phase II, among which the involvement of seven interferon-induced genes was identified (IFI44L, IFIT3, IFIF1, IFIF5, IFI44, IFIT2 and IFI6), and several pattern recognition receptor-associated genes [IRF7 (77), STAT1 (78) and CXCL10 (79)], as well as some genes involved in invasion and metastasis of cancer cells [HGF (80), IGF1 (81), KIT (82), FN1 (83) and CXCL12 (84)]. A number of common cancer-associated signaling pathway genes were identified in phase III [CCND1 (85), STAT1 (86) and VEGFA (87)]. A total of three C-X-C motif chemokine ligands (CXCL8, CXCL11 and CXCL4), two integrin subunits (ITGB1 and ITGA1), and one mitogen-activated protein (MAPK12) were identified in phase IV. Furthermore, PIK3CA (88) and FOS (89) participated in cancer-associated pathways in phase IV. The diversity of genes within the four phases demonstrated that CC progression is a complex process and its molecular mechanisms are not constant.
Table I.
Hub genes in phases I–IV.
| Phase | Gene | Regulation | Counts | LogFC | P-value |
|---|---|---|---|---|---|
| Phase I | CDK1 | + | 7 | 1.24 | <0.01 |
| KIF11 | + | 7 | 1.27 | 0.01 | |
| BUB1B | + | 6 | 1.13 | 0.02 | |
| BUB1 | + | 5 | 1.17 | 0.01 | |
| CCNA2 | + | 5 | 1.01 | <0.01 | |
| HLA-DPA1 | + | 5 | 1.49 | 0.03 | |
| CENPE | + | 5 | 1.36 | 0.02 | |
| RHOA | + | 4 | 1.47 | <0.01 | |
| KIF15 | + | 4 | 1.11 | <0.01 | |
| CCNB1 | + | 4 | 1.23 | 0.01 | |
| NDC80 | + | 4 | 1.47 | 0.01 | |
| TTK | + | 4 | 1.14 | <0.01 | |
| STAT1 | – | 4 | −1.47 | 0.03 | |
| CXCL10 | + | 4 | 2.47 | <0.01 | |
| KIF23 | + | 4 | 1.48 | 0.01 | |
| KIF4A | + | 3 | 1.00 | 0.05 | |
| PSMB9 | + | 3 | 1.12 | 0.03 | |
| GNG2 | + | 3 | 1.26 | 0.04 | |
| SPAG5 | + | 2 | 1.15 | 0.01 | |
| TRIP13 | + | 2 | 1.09 | 0.02 | |
| ANLN | + | 2 | 1.19 | 0.02 | |
| CDKN3 | + | 2 | 1.83 | <0.01 | |
| KIF14 | + | 2 | 1.15 | 0.01 | |
| MKI67 | + | 2 | 1.45 | 0.01 | |
| NUSAP1 | + | 2 | 1.22 | 0.02 | |
| NEK2 | + | 2 | 1.30 | 0.01 | |
| NCAPG | + | 2 | 1.18 | <0.01 | |
| DLGAP5 | + | 2 | 1.54 | <0.01 | |
| GBP1 | + | 2 | 1.45 | <0.01 | |
| Phase II | STAT1 | – | 9 | −1.03 | 0.02 |
| CXCL10 | – | 6 | −2.16 | 0.02 | |
| CXCL12 | – | 6 | −1.48 | 0.04 | |
| DCN | – | 6 | −1.37 | 0.01 | |
| CCL2 | – | 5 | −1.54 | 0.01 | |
| KIT | – | 5 | −1.26 | 0.05 | |
| IGF1 | – | 5 | −1.86 | <0.01 | |
| OAS2 | – | 4 | −1.37 | 0.01 | |
| IRF7 | – | 4 | −1.08 | 0.01 | |
| ISG15 | – | 4 | −1.83 | 0.02 | |
| FN1 | – | 4 | −1.34 | 0.04 | |
| HGF | – | 4 | −1.22 | 0.02 | |
| HERC6 | – | 3 | −1.71 | <0.01 | |
| MX2 | – | 3 | −1.86 | 0.01 | |
| IFIT3 | – | 3 | −1.79 | <0.01 | |
| IFIT1 | – | 3 | −2.96 | <0.01 | |
| GBP1 | – | 3 | −1.20 | 0.01 | |
| CDC6 | + | 3 | 1.40 | 0.01 | |
| IFIT5 | – | 2 | −1.11 | <0.01 | |
| IFI6 | – | 2 | −1.55 | 0.01 | |
| SP110 | – | 2 | −1.43 | <0.01 | |
| IFI44 | – | 2 | −1.87 | <0.01 | |
| DDX60 | – | 2 | −1.18 | <0.01 | |
| IFIT2 | – | 2 | −1.67 | <0.01 | |
| RSAD2 | – | 2 | −2.66 | <0.01 | |
| Phase III | BIRC5 | + | 9 | 1.13 | <0.01 |
| TOP2A | + | 8 | 1.36 | <0.01 | |
| KIF2C | + | 6 | 1.04 | <0.01 | |
| MCM10 | + | 6 | 1.16 | 0.01 | |
| VEGFA | + | 6 | 1.27 | <0.01 | |
| MAD2L1 | + | 5 | 1.03 | <0.01 | |
| KIF15 | + | 5 | 1.45 | <0.01 | |
| ASPM | + | 5 | 1.70 | <0.01 | |
| FOXM1 | + | 5 | 1.18 | 0.01 | |
| MX2 | + | 4 | 1.34 | 0.01 | |
| STAT1 | + | 4 | 1.20 | <0.01 | |
| PLXNA4 | – | 4 | −1.15 | 0.01 | |
| AR | – | 4 | −1.69 | <0.01 | |
| CCND1 | – | 3 | 1.20 | <0.01 | |
| OAS2 | + | 3 | 1.05 | <0.01 | |
| ACLY | + | 3 | 1.55 | 0.01 | |
| GNG2 | + | 3 | −1.21 | <0.01 | |
| RSAD2 | + | 2 | 1.85 | <0.01 | |
| ISG15 | + | 2 | 1.82 | <0.01 | |
| IFI35 | + | 2 | 1.35 | 0.01 | |
| IRF5 | + | 2 | 1.46 | <0.01 | |
| SAMHD1 | + | 2 | 1.05 | <0.01 | |
| MKI67 | + | 2 | 1.20 | <0.01 | |
| PLK4 | + | 2 | 1.03 | 0.01 | |
| AHCTF1 | + | 2 | 1.03 | <0.01 | |
| NUDC | + | 2 | 1.07 | <0.01 | |
| EXO1 | + | 2 | 1.29 | <0.01 | |
| PLXNA3 | + | 2 | 1.01 | 0.01 | |
| MMP9 | + | 2 | 2.55 | <0.01 | |
| PLAUR | + | 2 | 1.01 | <0.01 | |
| Phase IV | PIK3CA | + | 9 | 1.42 | <0.01 |
| CXCL8 | + | 8 | 1.28 | 0.05 | |
| ITGB1 | + | 8 | 2.20 | <0.01 | |
| PTK2 | + | 8 | 1.33 | <0.01 | |
| GNG2 | + | 6 | 1.18 | 0.05 | |
| ITGA1 | + | 6 | 1.33 | <0.01 | |
| GNG12 | – | 6 | −1.14 | <0.01 | |
| FOS | – | 5 | −1.13 | 0.05 | |
| EDN1 | + | 5 | 1.07 | <0.01 | |
| NMU | – | 4 | −2.23 | <0.01 | |
| LPAR5 | – | 4 | −1.36 | <0.01 | |
| STAT1 | + | 3 | 1.75 | <0.01 | |
| FN1 | + | 3 | 3.61 | <0.01 | |
| GSTM1 | – | 3 | −1.09 | 0.02 | |
| PLA2G4A | – | 3 | −1.41 | 0.02 | |
| CXCR4 | + | 2 | 1.58 | <0.01 | |
| HCAR3 | – | 2 | −1.60 | <0.01 | |
| S1PR5 | – | 2 | −1.56 | <0.01 | |
| CXCL5 | – | 2 | −2.10 | 0.01 | |
| NQO1 | – | 2 | −1.38 | 0.01 | |
| CXCL11 | + | 2 | 1.69 | 0.02 | |
| COMP | + | 2 | 1.62 | 0.01 | |
| MAPK12 | + | 2 | 1.38 | <0.01 |
+, upregulated; -, downregulated.
GO enrichment analysis of hub genes
To further identify the biological functions and locations of hub genes, GO enrichment analysis (90) was performed (Fig. 2). Hub genes were notably enriched in ‘biological regulation’, ‘metabolic process’ and ‘cellular component organization’ in phase I and II, while ‘responses to stimulus’ and ‘biological regulation’ were predominantly enriched at phases III–IV in biological process. For the cellular components, ‘nucleus’, ‘membrane-enclosed lumen’ and ‘macromolecular complex’ was enriched at phases I–III, while ‘chromosome’ and ‘membrane’ was identified in phases II and IV, respectively. ‘Protein binding’ was enriched at all four phases for Molecular Function. Furthermore, ‘nucleic acid binding’ and ‘hydrolase activity’ were enriched at phase II and III, while ‘ion binding’ was enriched at phase III and IV.
Figure 2.

Gene ontology enrichment analysis of hub genes for phase I, II, III and IV. The number in each phase represents the gene count.
Pathway enrichment analysis of hub genes
As presented in Table II, a total of 10 notably enriched pathways were identified at phase I, of which five pathways were associated with virus infections including, ‘influenza A’, ‘tuberculosis’, ‘herpes simplex infection’, ‘viral carcinogenesis’ and ‘Epstein-Barr virus infection’, and additional pathways involved in the ‘cell cycle’, ‘oocyte meiosis’ and ‘progesterone-mediated oocyte maturation’. Furthermore, the chemokine signaling pathway was also identified in phase I. The RIG-I-like receptor and Toll-like receptor signaling pathways were identified in phase II, and are associated with pattern-recognition receptors (91). In addition, several pathways, including ‘focal adhesion’, ‘Rap1 signaling pathway’, ‘Ras signaling pathway’, ‘PI3K-Akt signaling pathway’ and ‘Proteoglycans in cancer’ were associated with invasion and metastasis (92–96). The two common cancer-associated signaling pathways ‘Pathways in cancer’ and ‘Proteoglycans in cancer’, were enriched in phase III, while 70 pathways were significant enriched at phase IV (P<0.05). Apart from the common cancer-associated signaling pathways and virus infection pathways at phase IV, the ‘IL-17 signaling pathway’, ‘VEGF signaling pathway’ and ‘endocrine resistance’ also were also demonstrated to be associated with CC progression. Furthermore, the ‘AGE-RAGE signaling pathway in diabetic complications’ was also identified at phases II–IV.
Table II.
Pathway enrichment analysis for phases I–IV.
| Phase | Pathway | FDR | Involved genes |
|---|---|---|---|
| Phase I | Cell cycle | <0.001 | CDK1, TTK, CCNA2, BUB1, CCNB1, BUB1B |
| Progesterone-mediated oocyte maturation | 0.002 | CDK1, CCNA2, BUB1, CCNB1 | |
| Chemokine signaling pathway | 0.006 | RHOA, CXCL10, STAT1, GNG2 | |
| Oocyte meiosis | 0.018 | CDK1, BUB1, CCNB1 | |
| NOD-like receptor signaling pathway | 0.036 | RHOA, GBP1, STAT1 | |
| Influenza A | 0.036 | HLA-DPA1, CXCL10, STAT1 | |
| Tuberculosis | 0.037 | RHOA, HLA-DPA1, STAT1 | |
| Herpes simplex infection | 0.038 | CDK1, HLA-DPA1, STAT1 | |
| Viral carcinogenesis | 0.044 | RHOA, CDK1, CCNA2 | |
| Epstein-Barr virus infection | 0.044 | CDK1, HLA-DPA1, CCNA2 | |
| Phase II | Influenza A | <0.001 | CCL2, OAS2, IRF7, RSAD2, CXCL10, STAT1 |
| NOD-like receptor signaling pathway | <0.001 | GBP1, CCL2, OAS2, IRF7, STAT1 | |
| Herpes simplex infection | <0.001 | CCL2, OAS2, IRF7, IFIT1, STAT1 | |
| Pathways in cancer | 0.001 | HGF, IGF1, FN1, KIT, CXCL12, STAT1 | |
| Hepatitis C | 0.001 | OAS2, IRF7, IFIT1, STAT1 | |
| Cytokine-cytokine receptor interaction | 0.001 | HGF, CCL2, KIT, CXCL10, CXCL12 | |
| RIG-I-like receptor signaling pathway | 0.003 | IRF7, ISG15, CXCL10 | |
| Chemokine signaling pathway | 0.003 | CCL2, CXCL10, CXCL12, STAT1 | |
| Genes encoding secreted soluble factors | 0.003 | HGF, CCL2, IGF1, CXCL10, CXCL12 | |
| Proteoglycans in cancer | 0.004 | HGF, IGF1, FN1, DCN | |
| AGE-RAGE signaling pathway in diabetic complications | 0.005 | CCL2, FN1, STAT1 | |
| Toll-like receptor signaling pathway | 0.006 | IRF7, CXCL10, STAT1 | |
| Ensemble of genes encoding extracellular matrix and extracellular matrix-associated proteins | 0.008 | HGF, CCL2, IGF1, FN1, DCN, CXCL10, CXCL12 | |
| Measles | 0.009 | OAS2, IRF7, STAT1 | |
| PI3K-Akt signaling pathway | 0.015 | HGF, IGF1, FN1, KIT | |
| Focal adhesion | 0.024 | HGF, IGF1, FN1 | |
| Rap1 signaling pathway | 0.026 | HGF, IGF1, KIT | |
| Ras signaling pathway | 0.029 | HGF, IGF1, KIT | |
| Ensemble of genes encoding ECM-associated proteins including ECM-affilaited proteins, ECM regulators and secreted factors | 0.032 | HGF, CCL2, IGF1, CXCL10, CXCL12 | |
| Phase III | Pathways in cancer | 0.002 | BIRC5, CCND1, MMP9, AR, STAT1, GNG2, VEGFA |
| Bladder cancer | 0.006 | CCND1, MMP9, VEGFA | |
| Hepatitis B | 0.011 | BIRC5, CCND1, MMP9, STAT1 | |
| Pancreatic cancer | 0.012 | CCND1, STAT1, VEGFA | |
| Proteoglycans in cancer | 0.025 | PLAUR, CCND1, MMP9, VEGFA | |
| AGE-RAGE signaling pathway in diabetic complications | 0.027 | CCND1, STAT1, VEGFA | |
| Phase IV | Chemokine signaling pathway | <0.001 | GNG12, CXCL11, CXCL5, PIK3CA, CXCR4, PTK2, STAT1, CXCL8, GNG2 |
| Pathways in cancer | <0.001 | FN1, LPAR5, GNG12, ITGB1, PIK3CA, CXCR4, FOS, PTK2, STAT1, CXCL8, GNG2 | |
| Fluid shear stress and atherosclerosis | <0.001 | GSTM1, NQO1, MAPK12, PIK3CA, FOS, EDN1, PTK2 | |
| Signaling of Hepatocyte Growth Factor Receptor | <0.001 | ITGA1, ITGB1, PIK3CA, FOS, PTK2 | |
| PI3K-Akt signaling pathway | <0.001 | ITGA1, FN1, COMP, LPAR5, GNG12, ITGB1, PIK3CA, PTK2, GNG2 | |
| B Cell Survival Pathway | <0.001 | ITGA1, ITGB1, PIK3CA, FOS | |
| AGE-RAGE signaling pathway in diabetic complications | <0.001 | MAPK12, FN1, PIK3CA, EDN1, STAT1, CXCL8 | |
| Toll-like receptor signaling pathway | <0.001 | MAPK12, CXCL11, PIK3CA, FOS, STAT1, CXCL8 | |
| Aspirin Blocks Signaling Pathway Involved in Platelet Activation | <0.001 | PLA2G4A, ITGA1, ITGB1, PTK2 | |
| Erk and PI-3 Kinase Are Necessary for Collagen Binding in Corneal Epithelia | <0.001 | ITGA1, ITGB1, PIK3CA, PTK2 | |
| Pertussis | <0.001 | MAPK12, CXCL5, ITGB1, FOS, CXCL8 | |
| Focal adhesion | <0.001 | ITGA1, FN1, COMP, ITGB1, PIK3CA, PTK2 | |
| TNF signaling pathway | <0.001 | MAPK12, CXCL5, PIK3CA, FOS, EDN1 | |
| Leukocyte transendothelial migration | <0.001 | MAPK12, ITGB1, PIK3CA, CXCR4, PTK2 | |
| Regulation of actin cytoskeleton | <0.001 | ITGA1, FN1, GNG12, TGB1, PIK3CA, PTK2 | |
| VEGF signaling pathway | <0.001 | PLA2G4A, MAPK12, PIK3CA, PTK2 | |
| PTEN dependent cell cycle arrest and apoptosis | <0.001 | ITGB1, PIK3CA, PTK2 | |
| uCalpain and friends in Cell spread | <0.001 | ITGA1, ITGB1, PTK2 | |
| Trefoil Factors Initiate Mucosal Healing | <0.001 | ITGB1, PIK3CA, PTK2 | |
| Prolactin signaling pathway | <0.001 | MAPK12, PIK3CA, FOS, STAT1 | |
| Leishmaniasis | <0.001 | MAPK12, ITGB1, FOS, STAT1 | |
| Inhibition of Cellular Proliferation by Gleevec | <0.001 | PIK3CA, FOS, STAT1 | |
| CXCR4 Signaling Pathway | <0.001 | PIK3CA, CXCR4, PTK2 | |
| TPO Signaling Pathway | <0.001 | PIK3CA, FOS, STAT1 | |
| Bacterial invasion of epithelial cells | <0.001 | FN1, ITGB1, PIK3CA, PTK2 | |
| mCalpain and friends in Cell motility | <0.001 | ITGA1, ITGB1, PTK2 | |
| ECM-receptor interaction | <0.001 | ITGA1, FN1, COMP, ITGB1 | |
| Small cell lung cancer | <0.001 | FN1, ITGB1, PIK3CA, PTK2 | |
| EGF Signaling Pathway | <0.001 | PIK3CA, FOS, STAT1 | |
| IL-17 signaling pathway | <0.001 | MAPK12, CXCL5, FOS, CXCL8 | |
| PDGF Signaling Pathway | <0.001 | PIK3CA, FOS, STAT1 | |
| Amoebiasis | <0.001 | FN1, PIK3CA, PTK2, CXCL8 | |
| Endocrine resistance | <0.001 | MAPK12, PIK3CA, FOS, PTK2 | |
| Proteoglycans in cancer | <0.001 | MAPK12, FN1, ITGB1, PIK3CA, PTK2 | |
| Chagas disease (American trypanosomiasis) | <0.001 | MAPK12, PIK3CA, FOS, CXCL8 | |
| Agrin in Postsynaptic Differentiation | <0.001 | ITGA1, ITGB1, PTK2 | |
| Integrin Signaling Pathway | <0.001 | ITGA1, ITGB1, PTK2 | |
| Fc Epsilon Receptor I Signaling in Mast Cells | <0.001 | PLA2G4A, PIK3CA, FOS | |
| Cholinergic synapse | <0.001 | GNG12, PIK3CA, FOS, GNG2 | |
| Platelet activation | 0.001 | PLA2G4A, MAPK12, ITGB1, PIK3CA | |
| Osteoclast differentiation | 0.001 | MAPK12, PIK3CA, FOS, STAT1 | |
| Dopaminergic synapse | 0.001 | MAPK12, GNG12, FOS, GNG2 | |
| Hepatitis C | 0.001 | MAPK12, PIK3CA, STAT1, CXCL8 | |
| Cytokine-cytokine receptor interaction | 0.001 | CXCL11, CXCL5, CXCR4, ACKR3, CXCL8 | |
| Hepatitis B | 0.001 | PIK3CA, FOS, STAT1, CXCL8 | |
| Phospholipase D signaling pathway | 0.001 | PLA2G4A, LPAR5, PIK3CA, CXCL8 | |
| Shigellosis | 0.001 | MAPK12, ITGB1, CXCL8 | |
| Fc epsilon RI signaling pathway | 0.002 | PLA2G4A, MAPK12, PIK3CA | |
| Influenza A | 0.002 | MAPK12, PIK3CA, STAT1, CXCL8 | |
| Axon guidance | 0.002 | ITGB1, PIK3CA, CXCR4, PTK2 | |
| Integrin Signaling Pathway | 0.002 | ITGA1, PIK3CA, PTK2 | |
| Salmonella infection | 0.003 | MAPK12, FOS, CXCL8 | |
| MAPKinase Signaling Pathway | 0.003 | MAPK12, FOS, STAT1 | |
| Rap1 signaling pathway | 0.003 | MAPK12, LPAR5, ITGB1, PIK3CA | |
| Rheumatoid arthritis | 0.003 | CXCL5, FOS, CXCL8 | |
| Th1 and Th2 cell differentiation | 0.003 | MAPK12, FOS, STAT1 | |
| Circadian entrainment | 0.003 | GNG12, FOS, GNG2 | |
| Inflammatory mediator regulation of TRP channels | 0.003 | PLA2G4A, MAPK12, PIK3CA | |
| Ras signaling pathway | 0.003 | PLA2G4A, GNG12, PIK3CA, GNG2 | |
| Choline metabolism in cancer | 0.003 | PLA2G4A, PIK3CA, FOS | |
| Retrograde endocannabinoid signaling | 0.003 | MAPK12,GNG12,GNG2 | |
| T cell receptor signaling pathway | 0.004 | MAPK12, PIK3CA, FOS | |
| Th17 cell differentiation | 0.004 | MAPK12, FOS, STAT1 | |
| Serotonergic synapse | 0.004 | PLA2G4A, GNG12, GNG2 | |
| Toxoplasmosis | 0.004 | MAPK12, ITGB1, STAT1 | |
| Glutamatergic synapse | 0.004 | PLA2G4A, GNG12, GNG2 | |
| MAPK signaling pathway | 0.004 | PLA2G4A, MAPK12, GNG12, FOS | |
| Sphingolipid signaling pathway | 0.005 | S1PR5, MAPK12, PIK3CA | |
| NOD-like receptor signaling pathway | 0.012 | MAPK12, STAT1, CXCL8 | |
| cAMP signaling pathway | 0.018 | HCAR3, PIK3CA, FOS |
FDR, false discovery rate.
Pathway crosstalk analysis
The pathway crosstalk analysis results are presented in Fig. S1. A total of two major modules were identified in phase I, one of which was predominantly associated with inflammatory responses to viral infections, such as the NOD-like receptor signaling pathway and the chemokine signaling pathway, while the other module was associated with cycle regulation of oocytes, including cell cycle, progesterone-mediated oocyte maturation and oocyte meiosis (Fig. S1A). Similarly, the pathways were grouped into two modules in phase II. One module consisted of immune responses (RIG-I-like receptor and Toll-like receptor signaling pathways, and NOD-like receptor signaling pathway), which may trigger rapid activation of innate immunity by inducing the production of proinflammatory cytokines (97–99). The other module was predominantly involved in the regulation of cell proliferation and invasion (Ras signaling pathway, PI3K-Akt signaling pathway, Rap1 signaling pathway, focal adhesion and other cancer-associated signaling pathways) (Fig. S1B). Notably, the two modules in phases I and II were not independent as they were demonstrated to connect with each other via several signaling pathways. All pathways formed a cluster and common cancer-associated signaling pathways were indicated to play a critical role in phase III (Fig. S1C), while the connection between the pathways became highly complex in phase IV (Fig. S1D). The possible molecular mechanisms, such as inflammation caused by virus infections, pathways associated with cell invasion, IL-17 and VEGF signaling pathways, and endocrine resistance, are associated with one another. These results further verified the complexity of CC. A comprehensive combination of pathway crosstalk analysis containing 47 nodes and 105 edges among the four phases is presented in Fig. 3A. By analyzing the nodes with degrees ≥5, a subnetwork containing 11 key pathways was extracted (Fig. 3B). It indicates that the MAPK signaling pathway (degree=33), PI3K-Akt signaling pathway (degree=21) and focal adhesion (degree=15), which are ranked as the top three nodes and most interactive, may play critical roles in the progression of CC.
Figure 3.

Comprehensive pathways crosstalk analysis. (A) Combination of pathways crosstalk analysis for the four phases and the (B) subnetwork with nodal degree ≥5. Green, phase I; yellow, phase II; orange, phase III; red, phase IV. The arrow represents the up/downstream associations between the pathways. MAPK signaling pathway (degree=33), PI3K-Akt signaling pathway (degree=21) and Focal adhesion (degree=15) rank as the top three pathways, whereby the majority of other pathways transfer information with them.
PPI network analysis
A PPI network containing 51 nodes and 78 edges was constructed (Fig. 4A) by downloading the hub genes into the PINA database. Based on the description of a previous study (59), which defined the main nodes as nodes with degree >5, 14 key genes were identified from the PPI network (Fig. 4B). CDK1, FN1 and ITGB1 rank first, second and third, respectively, as the top three-degree levels (16, 12 and 8, respectively). STAT1 was the only gene demonstrated to be involved at all four phases. Furthermore, MMP9 presented self-regulating functions and was demonstrated to co-express with FN1 and ITGB1.
Figure 4.

Protein-protein interaction network analysis. (A) Protein-protein interaction network downloaded from The Protein Interaction Network Analysis platform and (B) subnetwork with nodal degree ≥5. Green, phase I; blue, phase II; yellow, phase II; red, phase IV. The nodal size represents the degree of each node. CDK1 (degree=16), FN1 (degree=12) and ITGB1 (degree=8) rank as the top three proteins. MMP9 was self-regulated and co-expressed with FN1 and ITGB1.
Comprehensive gene-pathway analysis
After mapping the key genes onto the key pathways using the KEGG and BioCarta databases, a potential gene-pathway flowchart, including eight key pathways and six key genes was constructed (Fig. 5). The results demonstrated the following: For phase I, CDK1 and CCNB1 participated in the regulation of the cell cycle, while CDK1 was also involved in viral carcinogenesis; for phases II–IV, ‘pathways in cancer,’ ‘focal adhesion’ and ‘PI3K-Akt signaling pathway’ were ranked the top three pathways according to the number of genes involved; FN1, ITGB1 and MMP9 may be associated with metastasis of tumor cells, and STAT1 participated in ‘pathways in cancer’ and ‘Toll-like receptor signaling pathway’, which functioned at a phase IV.
Figure 5.

Gene-pathway flowchart for the key genes and the pathways in phases I–IV. Circle, gene; rectangle, pathway. CDK1 and CCNB1 regulate the cell cycle and they are activated in phase I. For phases II–IV, ‘pathways in cancer’, ‘focal adhesion’ and ‘PI3K-Akt signaling pathway’ rank as the top three pathways according to the number of genes involved.
Co-expression network and survival analysis for key genes
By mining the data from LinkedOmics, the results of co-expression demonstrated that CDK1 had a significantly positive correlation with CCNB1 (P<0.0001), but negative correlation with FN1 (P=0.003) and MMP9 (P=0.001), respectively (Fig. 6). CCNB1 demonstrated a significantly negative correlation with ITGB1 (P=0.047); however, FN1, ITGB1 and MMP9 indicated a significantly positive correlation between each other (FN1 and ITGB1, P<0.001; FN1 and MMP9, P<0.0001; ITGB1 and MMP9, P=0.023). STAT1 was significantly positively correlated with MMP9 (P<0.0001).
Figure 6.
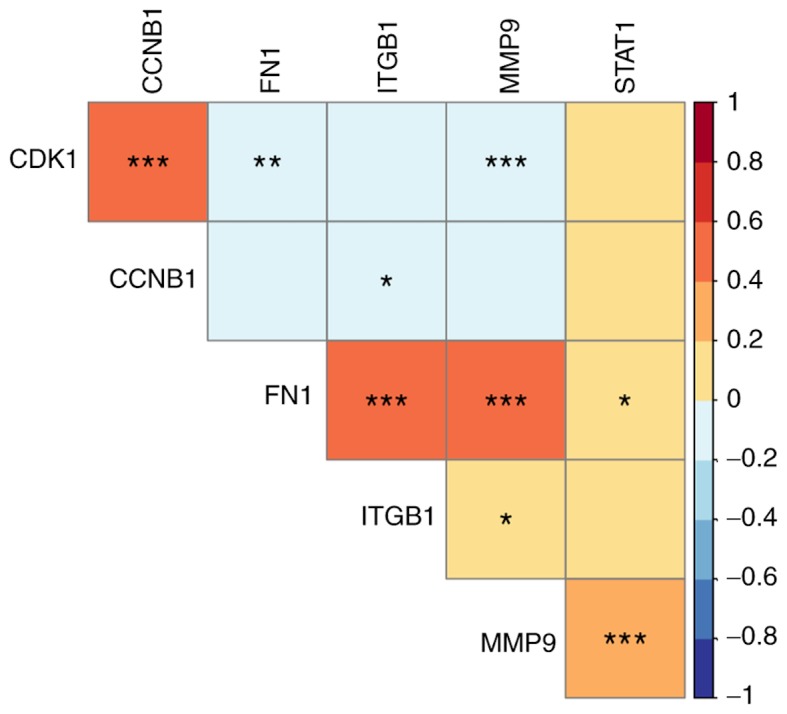
Co-expression analyses for the key genes. Positive correlation was detected between CDK1 and CCNB1; between FN1 and ITGB1, and MMP9, and between STAT1 and MMP9. Color in each grid represents the correlation coefficient between two genes. The values in the color legend represent the correlation coefficient. *P<0.05, **P<0.01, ***P<0.001.
Survival analysis indicated that patients with higher FN1 and ITGB1 expression levels had a significantly worse OS time (FN1, P=0.00080; ITGB1, P=0.00005; Fig. 7). However, CDK1, CCNB1, MMP9 and STAT1 were not demonstrated to have a significant effect on OS.
Figure 7.

Survival analysis for the key genes. (A) FN1, (B) ITGB1, (C) CDK1 and (D) CCNB1, (E) MMP9 and (F) STAT1. ITGB1 and FN1 have significant effect on overall survival.
Discussion
To date, the occurrence and development of CC is hypothesized to be linked with persistent HPV infection (100); however, the specific molecular mechanisms require further investigation. In addition, although a number of studies are examining the molecular mechanisms of CC (101–104), the detailed pathological process remains unclear.
The results of the present study indicated that CDK1 had the highest degree, and participated in the cell cycle with CCNB1 and viral carcinogenesis in phase I. CDK1 is one of the major cell cycle regulatory proteins and operates at the center of the cell cycle regulatory network (105). It regulates the G-S phase transition and initiates DNA replication (106). Furthermore, as a core molecule at the M phase checkpoint, CDK1 plays a role in the regulation of G2 phase, at M phase in the cell cycle (107). The HPV infection pathway is regulated within the viral carcinogenesis pathway (19). E6 proteins inactivate p53 by binding (108), while p53 negatively regulates CDK1 transcription under normal physiological conditions (109). In addition, the activation of cyclin B1-CDK1 is the key event that initiates the start of mitosis (110). Centrosome separation can be regulated by CDK1 (111), and cyclin B1-CDK1 remains activated following centrosome separation (110). Hence, overexpression of CDK1 can cause dysfunction in cell cycle progression, failure of normal proliferation and differentiation, and thereby lead to malignant proliferation of cancer cells and the formation of CC. CCNB1 is a notable member of the cyclin family, a key initiator and a stringent quality control step of mitosis (112). It also plays a key role in the regulation of CDK1, and its phosphorylated substrates can promote the transition of the cell cycle from G2 to mitosis (113,114). Amplification of the HPV genome depends on prolongation of the G2 phase in the cell cycle (115). CCNB1 is a downstream target of STAT3, which is a key gene that regulates the proliferation and differentiation of CC cells (116). In cells with inactivated STAT3, CCNB1 expression is downregulated and amplification of the HPV genome is also decreased, resulting in decreased activity of CC cells (116). As the results of the present study demonstrated that CDK1 and CCNB1 occurred in phase I and functioned as regulators of proliferation and differentiation, they may be potential promoters of CIN and CC.
In the process of tumor invasion and metastasis, cancer cells can bind to ligands of the extracellular matrix (ECM) via integrins and degrade the basement membrane (BM) by secreting proteases via the pathways of focal adhesion and the PI3K-Akt signaling pathway (117). This degradation is also the prerequisite for stromal infiltration and cancer cell migration (118). ITGB1 belongs to the integrin family and FN1 is the ligand. The binding of ITGB1 and FN1 induces the phosphorylation of tyrosine and directly affects cytoskeleton reconstruction and signal transduction activities of the Ras-MAPK signaling pathway via the RAP1 signaling pathway, which initiates the expression of MMP genes (119). MMPs are a family of calcium and zinc-dependent proteases that degrade a variety of components of the ECM (120). Collagen type IV is the main scaffold in the BM of the ECM and also the main substrate of MMP9 (121). MMP9 can decompose the nestin in the BM to destroy the cells integrity and promote the invasion and metastasis of cancer cells (122). MMP9 expression in HPV-positive patients with CC is higher than in HPV-negative patients (123). Cardeal et al (124) reported that MMP9 is upregulated in human keratinocytes expressing the HPV16 E7 protein. This may be due to TIMP2, an inhibitor of MMP9, which could be downregulated by HPV16 E7. It was also demonstrated that HPV can directly regulate the activity of MMP9 in lung cancer cells (125). There may be an association between HPV infection and the MMP family, which may be beneficial in the diagnosis of cervical precancerous lesions and CC as MMP9 may be considered as a novel biomarker. However, the specific molecular mechanisms require further investigation. As FN1 and ITGB1 were targets of miR-9-3p (126) and FN1 promoted migration and invasion by upregulating MMP9 in cancer (127), it is not surprising that these three genes are co-expressed as a reaction triplet. Furthermore, since higher levels of FN1 and ITGB1 are significantly associated with lower OS rate, these two genes may be developed as novel prognostic factors for CC.
STAT1, the only gene that participates in all four phases, in the present study, is involved in the cancer pathway at phases II, III and IV. It has been reported that STAT1 is upregulated in both CIN1 and CC (128), and the results of the present study that STAT1 is upregulated in phase I, III and IV confirmed this finding. A previous study demonstrated that activated STAT1 plays a tumor suppressive role in breast cancer cells (129). Nevertheless, STAT1 also exerts tumor promoter effects under specific conditions (130). In some malignant diseases, including breast and lung cancers, STAT1 can act as an oncoprotein or a tumor suppressor of the same cell type based on the specific genetic background (130). In CC, STAT1 may have a protective effect in the early stages of HPV infection but may act as a proto-oncogene during the invasive phase of the disease (128). STAT1 can promote cancer cell death by activating p53 expression, and it plays a role in immunosurveillance, and the inhibition of angiogenesis and metastasis in cancer cells (130); however, STAT1 can also promote tumor invasion and metastasis in chronic inflammation (131). The effect of STAT1 in CC still remains unclear; therefore, further verification is required as it may be a key target for the treatment of CC.
The current study presented several limitations. First, as this was an in-silico study, the identification of DEGs may change with additional data, thus the results of subsequent analyses may change accordingly. Secondly, some genes were excluded to decrease the false-positive rate; however, these genes may also have a vital effect on CC. Thirdly, although several genes associated with HPV infection (TP53TG1, RAC1, PAK2 and LTBP2) were identified in DEGs, HPV infection was not observed in the pathway analyses. This may be due to the fact that an insufficient amount of hub genes were identified, or the genes had a low or moderate effect on the HPV infection pathway.
In conclusion, the present study revealed that CDK1, CCNB1, ITGB1, FN1, MMP9 and STAT1 played different roles in the progression of CC through different signaling pathways. CDK1 and CCNB1 served as regulators of proliferation and differentiation via regulation of the cell cycle and viral tumorigenesis, and initiated CIN and CC, whereas FN1, ITGB1 and MMP9 were co-expressed as a reaction triplet to trigger metastasis via cancer pathways, PI3K-Akt signaling pathway and focal adhesion. FN1 and ITGB1 may be novel prognostic factors for CC. STAT1 may have a protective effect in the early stage of HPV infection, but may also act as a proto-oncogene during the invasive stage; however, the specific molecular mechanisms require further investigation.
Supplementary Material
Acknowledgements
Not applicable.
Glossary
Abbreviations
- CC
cervical cancer
- CIN
cervical intraepithelial neoplasia
- GO
Gene Ontology
- PPI
protein-protein interaction
- TCGA
The Cancer Genome Atlas
- DEG
differentially expressed gene
- OS
overall survival
- STRING
Search Tool for the Retrieval of Interacting Genes
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- JC
jaccard coefficient
- OC
overlap coefficient
- PINA
protein interaction network analysis
Funding
No funding was received.
Availability of data and materials
All data generated and/or analyzed during this study are included in this published article. The datasets generated and/or analyzed during the current study are available in the GEO (https://www.ncbi.nlm.nih.gov/geo/) and LinkOmics (http://www.linkedomics.org/) repository.
Authors' contributions
YY, YF and WZ designed the present study and drafted the initial manuscript. KW and YL performed the literature review, acquired the data and performed the statistical analyses. All authors have read and approved the manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Muliira RS, Salas AS, O'Brien B. Quality of life among female cancer survivors in Africa: An integrative literature review. Asia Pac J Oncol Nurs. 2017;4:6–17. doi: 10.4103/2347-5625.199078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gravitt PE, Winer RL. Natural history of HPV infection across the lifespan: Role of viral latency. Viruses. 2017;9:E267. doi: 10.3390/v9100267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, Snijders PJ, Peto J, Meijer CJ, Muñoz N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol. 1999;189:12–19. doi: 10.1002/(SICI)1096-9896(199909)189:1<12::AID-PATH431>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 5.Louie KS, de Sanjose S, Diaz M, Castellsagué X, Herrero R, Meijer CJ, Shah K, Franceschi S, Muñoz N, Bosch FX, International Agency for Research on Cancer Multicenter Cervical Cancer Study Group Early age at first sexual intercourse and early pregnancy are risk factors for cervical cancer in developing countries. Br J Cancer. 2009;100:1191–1197. doi: 10.1038/sj.bjc.6604974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu ZC, Liu WD, Liu YH, Ye XH, Chen SD. Multiple sexual partners as a potential independent risk factor for cervical cancer: A meta-analysis of epidemiological studies. Asian Pac J Cancer Prev. 2015;16:3893–3900. doi: 10.7314/APJCP.2015.16.9.3893. [DOI] [PubMed] [Google Scholar]
- 7.Smith JS, Green J, Berrington de Gonzalez A, Appleby P, Peto J, Plummer M, Franceschi S, Beral V. Cervical cancer and use of hormonal contraceptives: A systematic review. Lancet. 2003;361:1159–1167. doi: 10.1016/S0140-6736(03)12949-2. [DOI] [PubMed] [Google Scholar]
- 8.Ma YY, Wei SJ, Lin YC, Lung JC, Chang TC, Whang-Peng J, Liu JM, Yang DM, Yang WK, Shen CY. PIK3CA as an oncogene in cervical cancer. Oncogene. 2000;19:2739–2744. doi: 10.1038/sj.onc.1203597. [DOI] [PubMed] [Google Scholar]
- 9.Zheng L, Li T, Zhang Y, Guo Y, Yao J, Dou L, Guo K. Oncogene ATAD2 promotes cell proliferation, invasion and migration in cervical cancer. Oncol Rep. 2015;33:2337–2344. doi: 10.3892/or.2015.3867. [DOI] [PubMed] [Google Scholar]
- 10.Bai X, Wang W, Zhao P, Wen J, Guo X, Shen T, Shen J, Yang X. LncRNA CRNDE acts as an oncogene in cervical cancer through sponging miR-183 to regulate CCNB1 expression. Carcinogenesis. 2019 Oct 12; doi: 10.1093/carcin/bgz166. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 11.Bremer GL, Tieboschb AT, van der Putten HW, de Haan J, Arends JW. p53 tumor suppressor gene protein expression in cervical cancer: Relationship to prognosis. Eur J Obstet Gynecol Reprod Biol. 1995;63:55–59. doi: 10.1016/0301-2115(95)02225-V. [DOI] [PubMed] [Google Scholar]
- 12.Cohen Y, Singer G, Lavie O, Dong SM, Beller U, Sidransky D. The RASSF1A tumor suppressor gene is commonly inactivated in adenocarcinoma of the uterine cervix. Clin Cancer Res. 2003;9:2981–2984. [PubMed] [Google Scholar]
- 13.Hasina R, Pontier AL, Fekete MJ, Martin LE, Qi XM, Brigaudeau C, Pramanik R, Cline EI, Coignet LJ, Lingen MW. NOL7 is a nucleolar candidate tumor suppressor gene in cervical cancer that modulates the angiogenic phenotype. Oncogene. 2006;25:588–598. doi: 10.1038/sj.onc.1209070. [DOI] [PubMed] [Google Scholar]
- 14.Roura E, Castellsagué X, Pawlita M, Travier N, Waterboer T, Margall N, Bosch FX, de Sanjosé S, Dillner J, Gram IT, et al. Smoking as a major risk factor for cervical cancer and pre-cancer: Results from the EPIC cohort. Int J cancer. 2014;135:453–466. doi: 10.1002/ijc.28666. [DOI] [PubMed] [Google Scholar]
- 15.Husain RS, Ramakrishnan V. Global variation of human papillomavirus genotypes and selected genes involved in cervical malignancies. Ann Glob Health. 2015;81:675–683. doi: 10.1016/j.aogh.2015.08.026. [DOI] [PubMed] [Google Scholar]
- 16.Soto D, Song C, McLaughlin-Drubin ME. Epigenetic alterations in human papillomavirus-associated cancers. Viruses. 2017;9:E248. doi: 10.3390/v9090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mincheva A, Gissmann L, zur Hausen H. Chromosomal integration sites of human papillomavirus DNA in three cervical cancer cell lines mapped by in situ hybridization. Med Microbiol Immunol. 1987;176:245–256. doi: 10.1007/BF00190531. [DOI] [PubMed] [Google Scholar]
- 18.Senapati R, Senapati NN, Dwibedi B. Molecular mechanisms of HPV mediated neoplastic progression. Infect Agent Cancer. 2016;11:59. doi: 10.1186/s13027-016-0107-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Münger K, Baldwin A, Edwards KM, Hayakawa H, Nguyen CL, Owens M, Grace M, Huh K. Mechanisms of human papillomavirus-induced oncogenesis. J Virol. 2004;78:11451–11460. doi: 10.1128/JVI.78.21.11451-11460.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martinez-Zapien D, Ruiz FX, Poirson J, Mitschler A, Ramirez J, Forster A, Cousido-Siah A, Masson M, Vande Pol S, Podjarny A, et al. Structure of the E6/E6AP/p53 complex required for HPV-mediated degradation of p53. Nature. 2016;529:541–545. doi: 10.1038/nature16481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell. 1993;75:495–505. doi: 10.1016/0092-8674(93)90384-3. [DOI] [PubMed] [Google Scholar]
- 22.Sherr CJ. The Pezcoller lecture: Cancer cell cycles revisited. Cancer Res. 2000;60:3689–3695. [PubMed] [Google Scholar]
- 23.Martin LG, Demers GW, Galloway DA. Disruption of the G1/S transition in human papillomavirus type 16 E7-expressing human cells is associated with altered regulation of cyclin E. J Virol. 1998;72:975–985. doi: 10.1128/JVI.72.2.975-985.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Löffler H, Fechter A, Matuszewska M, Saffrich R, Mistrik M, Marhold J, Hornung C, Westermann F, Bartek J, Krämer A. Cep63 recruits Cdk1 to the centrosome: Implications for regulation of mitotic entry, centrosome amplification, and genome maintenance. Cancer Res. 2011;71:2129–2139. doi: 10.1158/0008-5472.CAN-10-2684. [DOI] [PubMed] [Google Scholar]
- 25.Skyldberg B, Fujioka K, Hellström AC, Sylvén L, Moberger B, Auer G. Human papillomavirus infection, centrosome aberration, and genetic stability in cervical lesions. Mod Pathol. 2001;14:279–284. doi: 10.1038/modpathol.3880303. [DOI] [PubMed] [Google Scholar]
- 26.Duensing S, Münger K. The human papillomavirus type 16 E6 and E7 oncoproteins independently induce numerical and structural chromosome instability. Cancer Res. 2002;62:7075–7082. [PubMed] [Google Scholar]
- 27.Chaiwongkot A, Niruthisard S, Kitkumthorn N, Bhattarakosol P. Quantitative methylation analysis of human papillomavirus 16 L1 gene reveals potential biomarker for cervical cancer progression. Diagn Microbiol Infect Dis. 2017;89:265–270. doi: 10.1016/j.diagmicrobio.2017.08.010. [DOI] [PubMed] [Google Scholar]
- 28.Sharma A, De R, Javed S, Srinivasan R, Pal A, Bhattacharyya S. Sonic hedgehog pathway activation regulates cervical cancer stem cell characteristics during epithelial to mesenchymal transition. J Cell Physiol. 2019 Feb 4; doi: 10.1002/jcp.28231. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 29.Li YP, Zhang L, Zou YL, Yu Y. Association between FGFR4 gene polymorphism and high-risk HPV infection cervical cancer. Asian Pac J Trop Med. 2017;10:680–684. doi: 10.1016/j.apjtm.2017.07.008. [DOI] [PubMed] [Google Scholar]
- 30.Zhang J, Gao Y. CCAT-1 promotes proliferation and inhibits apoptosis of cervical cancer cells via the Wnt signaling pathway. Oncotarget. 2017;8:68059–68070. doi: 10.18632/oncotarget.19155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mora-García ML, Ávila-Ibarra LR, García-Rocha R, Weiss-Steider B, Hernández-Montes J, Don-López CA, Gutiérrez-Serrano V, Titla-Vilchis IJ, Fuentes-Castañeda MC, Monroy-Mora A, et al. Cervical cancer cells suppress effector functions of cytotoxic T cells through the adenosinergic pathway. Cell Immunol. 2017;320:46–55. doi: 10.1016/j.cellimm.2017.09.002. [DOI] [PubMed] [Google Scholar]
- 32.Song ZC, Ding L, Ren ZY, Sun XS, Yang Q, Wang L, Feng MJ, Liu CL, Wang JT. Effects of Src on cervical cancer cells proliferation and apoptosis through ERK signal transduction pathway. Zhonghua Liu Xing Bing Xue Za Zhi. 2017;38:1246–1251. doi: 10.3760/cma.j.issn.0254-6450.2017.09.021. (In Chinese; Abstract available in Chinese from the publisher) [DOI] [PubMed] [Google Scholar]
- 33.Xu Z, Zhou Y, Shi F, Cao Y, Dinh TLA, Wan J, Zhao M. Investigation of differentially-expressed microRNAs and genes in cervical cancer using an integrated bioinformatics analysis. Oncol Lett. 2017;13:2784–2790. doi: 10.3892/ol.2017.5766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen SZ, Ma WL, Zheng WL. Screening for cervical cancer-related genes and their bioinformatics analysis. Nan Fang Yi Ke Da Xue Xue Bao. 2008;28:585–588. (In Chinese) [PubMed] [Google Scholar]
- 35.Nayar R, Wilbur DC. The bethesda system for reporting cervical cytology: A historical perspective. Acta Cytol. 2017;61:359–372. doi: 10.1159/000477556. [DOI] [PubMed] [Google Scholar]
- 36.Shoji T, Takatori E, Takeuchi S, Yoshizaki A, Uesugi N, Sugai T, Sugiyama T. Clinical significance of atypical glandular cells in the bethesda system 2001: A comparison with the histopathological diagnosis of surgically resected specimens. Cancer Invest. 2014;32:105–109. doi: 10.3109/07357907.2014.880453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O'Connor M, Gallagher P, Waller J, Martin CM, O'Leary JJ, Sharp L, Irish Cervical Screening Research Consortium (CERVIVA) Adverse psychological outcomes following colposcopy and related procedures: A systematic review. BJOG. 2016;123:24–38. doi: 10.1111/1471-0528.13462. [DOI] [PubMed] [Google Scholar]
- 38.Haque N, Uddin AFMK, Dey BR, Islam F, Goodman A. Challenges to cervical cancer treatment in Bangladesh: The development of a women's cancer ward at Dhaka Medical College Hospital. Gynecol Oncol Rep. 2017;21:67–72. doi: 10.1016/j.gore.2017.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roque DR, Wysham WZ, Soper JT. The surgical management of cervical cancer: An overview and literature review. Obstet Gynecol Surv. 2014;69:426–441. doi: 10.1097/OGX.0000000000000089. [DOI] [PubMed] [Google Scholar]
- 40.Marin JJ, Romero MR, Blazquez AG, Herraez E, Keck E, Briz O. Importance and limitations of chemotherapy among the available treatments for gastrointestinal tumours. Anticancer Agents Med Chem. 2009;9:162–184. doi: 10.2174/187152009787313828. [DOI] [PubMed] [Google Scholar]
- 41.Chen HHW, Kuo MT. Improving radiotherapy in cancer treatment: Promises and challenges. Oncotarget. 2017;8:62742–62758. doi: 10.18632/oncotarget.18409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lecavalier-Barsoum M, Chaudary N, Han K, Pintilie M, Hill RP, Milosevic M. Targeting CXCL12/CXCR4 and myeloid cells to improve the therapeutic ratio in patient-derived cervical cancer models treated with radio-chemotherapy. Br J Cancer. 2019;121:249–256. doi: 10.1038/s41416-019-0545-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang F, Ren CC, Liu L, Chen YN, Yang L, Zhang XA, Wang XM, Yu FJ. SHH gene silencing suppresses epithelial-mesenchymal transition, proliferation, invasion, and migration of cervical cancer cells by repressing the hedgehog signaling pathway. J Cell Biochem. 2018;119:3829–3842. doi: 10.1002/jcb.26414. [DOI] [PubMed] [Google Scholar]
- 44.Gu J, Zhang X, Yang Z, Wang N. Expression of cyclin D1 protein isoforms and its prognostic significance in cervical cancer. Cancer Manag Res. 2019;11:9073–9083. doi: 10.2147/CMAR.S224026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chay DB, Han GH, Nam S, Cho H, Chung JY, Hewitt SM. Forkhead box protein O1 (FOXO1) and paired box gene 3 (PAX3) overexpression is associated with poor prognosis in patients with cervical cancer. Int J Clin Oncol. 2019;24:1429–1439. doi: 10.1007/s10147-019-01507-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang H, Wang Y, Zhang B, Xiong S, Liu L, Chen W, Tan G, Li H. High brain acid soluble protein 1(BASP1) is a poor prognostic factor for cervical cancer and promotes tumor growth. Cancer Cell Int. 2017;17:97. doi: 10.1186/s12935-017-0452-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang X, Cheng Y, Li C. The role of TLRs in cervical cancer with HPV infection: A review. Signal Transduct Target Ther. 2017;2:17055. doi: 10.1038/sigtrans.2017.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frumovitz M, Sood AK. Vascular endothelial growth factor (VEGF) pathway as a therapeutic target in gynecologic malignancies. Gynecol Oncol. 2007;104:768–778. doi: 10.1016/j.ygyno.2006.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.den Boon JA, Pyeon D, Wang SS, Horswill M, Schiffman M, Sherman M, Zuna RE, Wang Z, Hewitt SM, Pearson R, et al. Molecular transitions from papillomavirus infection to cervical precancer and cancer: Role of stromal estrogen receptor signaling. Proc Natl Acad Sci USA. 2015;112:E3255–E3264. doi: 10.1073/pnas.1509322112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, et al. NCBI GEO: Archive for functional genomics data sets-update. Nucleic Acids Res. 2013;41:D991–D995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–D452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, Lin CY. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;8(Suppl 4):S11. doi: 10.1186/1752-0509-8-S4-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang J, Vasaikar S, Shi Z, Greer M, Zhang B. WebGestalt 2017: A more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 2017;45:W130–W137. doi: 10.1093/nar/gkx356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009;37:W305–W311. doi: 10.1093/nar/gkp427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hu Y, Pan Z, Hu Y, Zhang L, Wang J. Network and pathway-based analyses of genes associated with Parkinson's disease. Mol Neurobiol. 2017;54:4452–4465. doi: 10.1007/s12035-016-9998-8. [DOI] [PubMed] [Google Scholar]
- 58.Scardoni G, Petterlini M, Laudanna C. Analyzing biological network parameters with CentiScaPe. Bioinformatics. 2009;25:2857–2859. doi: 10.1093/bioinformatics/btp517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Han JD, Berlin N, Hao T, Goldberg DS, Berriz GF, Zhang LV, Dupuy D, Walhout AJ, Cusick ME, Roth FP, Vidal M. Evidence for dynamically organized modularity in the yeast protein-protein interaction network. Nature. 2004;430:88–93. doi: 10.1038/nature02555. [DOI] [PubMed] [Google Scholar]
- 60.Kanwal A, Fazal S. Construction and analysis of protein-protein interaction network correlated with ankylosing spondylitis. Gene. 2018;638:41–51. doi: 10.1016/j.gene.2017.09.049. [DOI] [PubMed] [Google Scholar]
- 61.Wu J, Vallenius T, Ovaska K, Westermarck J, Mäkelä TP, Hautaniemi S. Integrated network analysis platform for protein-protein interactions. Nat Methods. 2009;6:75–77. doi: 10.1038/nmeth.1282. [DOI] [PubMed] [Google Scholar]
- 62.Kerrien S, Alam-Faruque Y, Aranda B, Bancarz I, Bridge A, Derow C, Dimmer E, Feuermann M, Friedrichsen A, Huntley R, et al. IntAct-source resource for molecular interaction data. Nucleic Acids Res. 2007;35:D561–D565. doi: 10.1093/nar/gkl958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chatr-aryamontri A, Ceol A, Palazzi LM, Nardelli G, Schneider MV, Castagnoli L, Cesareni G. MINT: The Molecular INTeraction database. Nucleic Acids Res. 2007;35:D572–D574. doi: 10.1093/nar/gkl950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Breitkreutz BJ, Stark C, Reguly T, Boucher L, Breitkreutz A, Livstone M, Oughtred R, Lackner DH, Bähler J, Wood V, et al. The BioGRID interaction database: 2008 update. Nucleic Acids Res. 2008;36:D637–D640. doi: 10.1093/nar/gkm1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Salwinski L, Miller CS, Smith AJ, Pettit FK, Bowie JU, Eisenberg D. The database of interacting proteins: 2004 update. Nucleic Acids Res. 2004;32:D449–D451. doi: 10.1093/nar/gkh086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Peri S, Navarro JD, Amanchy R, Kristiansen TZ, Jonnalagadda CK, Surendranath V, Niranjan V, Muthusamy B, Gandhi TK, Gronborg M, et al. Development of human protein reference database as an initial platform for approaching systems biology in humans. Genome Res. 2003;13:2363–2371. doi: 10.1101/gr.1680803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Güldener U, Münsterkötter M, Oesterheld M, Pagel P, Ruepp A, Mewes HW, Stümpflen V. MPact: The MIPS protein interaction resource on yeast. Nucleic Acids Res. 2006;34:D436–D441. doi: 10.1093/nar/gkj003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vasaikar SV, Straub P, Wang J, Zhang B. LinkedOmics: Analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 2018;46:D956–D963. doi: 10.1093/nar/gkx1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cancer Genome Atlas Research Network. Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM. The cancer genome atlas pan-cancer analysis project. Nat Genet. 2013;45:1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gasca S, Pellestor F, Assou S, Loup V, Anahory T, Dechaud H, De Vos J, Hamamah S. Identifying new human oocyte marker genes: A microarray approach. Reprod Biomed Online. 2007;14:175–183. doi: 10.1016/S1472-6483(10)60785-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schwab MS, Roberts BT, Gross SD, Tunquist BJ, Taieb FE, Lewellyn AL, Maller JL. Bub1 is activated by the protein kinase p90(Rsk) during Xenopus oocyte maturation. Curr Biol. 2001;11:141–150. doi: 10.1016/S0960-9822(01)00045-8. [DOI] [PubMed] [Google Scholar]
- 72.Ouandaogo ZG, Frydman N, Hesters L, Assou S, Haouzi D, Dechaud H, Frydman R, Hamamah S. Differences in transcriptomic profiles of human cumulus cells isolated from oocytes at GV, MI and MII stages after in vivo and in vitro oocyte maturation. Hum Reprod. 2012;27:2438–2447. doi: 10.1093/humrep/des172. [DOI] [PubMed] [Google Scholar]
- 73.Li J, Qian WP, Sun QY. Cyclins regulating oocyte meiotic cell cycle progression†. Biol Reprod. 2019;101:878–881. doi: 10.1093/biolre/ioz143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kaplan MH. STAT signaling in inflammation. JAKSTAT. 2013;2:e24198. doi: 10.4161/jkst.24198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Qiu X, Guo H, Yang J, Ji Y, Wu CS, Chen X. Down-regulation of guanylate binding protein 1 causes mitochondrial dysfunction and cellular senescence in macrophages. Sci Rep. 2018;8:1679. doi: 10.1038/s41598-018-19828-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bros M, Haas K, Moll L, Grabbe S. RhoA as a key regulator of innate and adaptive immunity. Cells. 2019;8:E733. doi: 10.3390/cells8070733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Erdely A, Antonini JM, Salmen-Muniz R, Liston A, Hulderman T, Simeonova PP, Kashon ML, Li S, Gu JK, Stone S, et al. Type I interferon and pattern recognition receptor signaling following particulate matter inhalation. Part Fibre Toxicol. 2012;9:25. doi: 10.1186/1743-8977-9-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Luu K, Greenhill CJ, Majoros A, Decker T, Jenkins BJ, Mansell A. STAT1 plays a role in TLR signal transduction and inflammatory responses. Immunol Cell Biol. 2014;92:761–769. doi: 10.1038/icb.2014.51. [DOI] [PubMed] [Google Scholar]
- 79.Smolen KK, Ruck CE, Fortuno ES, III, Ho K, Dimitriu P, Mohn WW, Speert DP, Cooper PJ, Esser M, Goetghebuer T, et al. Pattern recognition receptor-mediated cytokine response in infants across 4 continents. J Allergy Clin Immunol. 2014;133:818–826.e4. doi: 10.1016/j.jaci.2013.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Xiang C, Chen J, Fu P. HGF/Met signaling in cancer invasion: The impact on cytoskeleton remodeling. Cancers (Basel) 2017;9:E44. doi: 10.3390/cancers9050044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lei T, Ling X. IGF-1 promotes the growth and metastasis of hepatocellular carcinoma via the inhibition of proteasome-mediated cathepsin B degradation. World J Gastroenterol. 2015;21:10137–10149. doi: 10.3748/wjg.v21.i35.10137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cho S, Kitadai Y, Yoshida S, Tanaka S, Yoshihara M, Yoshida K, Chayama K. Deletion of the KIT gene is associated with liver metastasis and poor prognosis in patients with gastrointestinal stromal tumor in the stomach. Int J Oncol. 2006;28:1361–1367. [PubMed] [Google Scholar]
- 83.Li B, Shen W, Peng H, Li Y, Chen F, Zheng L, Xu J, Jia L. Fibronectin 1 promotes melanoma proliferation and metastasis by inhibiting apoptosis and regulating EMT. Onco Targets Ther. 2019;12:3207–3221. doi: 10.2147/OTT.S195703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liu Y, Ren CC, Yang L, Xu YM, Chen YN. Role of CXCL12-CXCR4 axis in ovarian cancer metastasis and CXCL12-CXCR4 blockade with AMD3100 suppresses tumor cell migration and invasion in vitro. J Cell Physiol. 2019;234:3897–3909. doi: 10.1002/jcp.27163. [DOI] [PubMed] [Google Scholar]
- 85.Chen Y, Jiang J, Zhao M, Luo X, Liang Z, Zhen Y, Fu Q, Deng X, Lin X, Li L, et al. microRNA-374a suppresses colon cancer progression by directly reducing CCND1 to inactivate the PI3K/AKT pathway. Oncotarget. 2016;7:41306–41319. doi: 10.18632/oncotarget.9320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Thomas SJ, Snowden JA, Zeidler MP, Danson SJ. The role of JAK/STAT signalling in the pathogenesis, prognosis and treatment of solid tumours. Br J Cancer. 2015;113:365–371. doi: 10.1038/bjc.2015.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stacker SA, Achen MG. The VEGF signaling pathway in cancer: The road ahead. Chin J Cancer. 2013;32:297–302. doi: 10.5732/cjc.012.10319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shi X, Wang J, Lei Y, Cong C, Tan D, Zhou X. Research progress on the PI3K/AKT signaling pathway in gynecological cancer (Review) Mol Med Rep. 2019;19:4529–4535. doi: 10.3892/mmr.2019.10121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li X, Jiang S, Tapping RI. Toll-like receptor signaling in cell proliferation and survival. Cytokine. 2010;49:1–9. doi: 10.1016/j.cyto.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gene Ontology Consortium. Gene ontology consortium: Going forward. Nucleic Acids Res. 2015;43:D1049–D1056. doi: 10.1093/nar/gku1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kawai T, Akira S. Toll-like receptor and RIG-1-like receptor signaling. Ann N Y Acad Sci. 2008;1143:1–20. doi: 10.1196/annals.1443.020. [DOI] [PubMed] [Google Scholar]
- 92.Gkretsi V, Stylianopoulos T. Cell adhesion and matrix stiffness: Coordinating cancer cell invasion and metastasis. Front Oncol. 2018;8:145. doi: 10.3389/fonc.2018.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang YL, Wang RC, Cheng K, Ring BZ, Su L. Roles of Rap1 signaling in tumor cell migration and invasion. Cancer Biol Med. 2017;14:90–99. doi: 10.20892/j.issn.2095-3941.2016.0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Campbell PM, Der CJ. Oncogenic Ras and its role in tumor cell invasion and metastasis. Semin Cancer Biol. 2004;14:105–114. doi: 10.1016/j.semcancer.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 95.Sun F, Wang J, Sun Q, Li F, Gao H, Xu L, Zhang J, Sun X, Tian Y, Zhao Q, et al. Interleukin-8 promotes integrin β3 upregulation and cell invasion through PI3K/Akt pathway in hepatocellular carcinoma. J Exp Clin Cancer Res. 2019;38:449. doi: 10.1186/s13046-019-1455-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sanderson RD. Heparan sulfate proteoglycans in invasion and metastasis. Semin Cell Dev Biol. 2001;12:89–98. doi: 10.1006/scdb.2000.0241. [DOI] [PubMed] [Google Scholar]
- 97.Loo YM, Gale M., Jr Immune signaling by RIG-I-like receptors. Immunity. 2011;34:680–692. doi: 10.1016/j.immuni.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nyati KK, Prasad KN. Role of cytokines and Toll-like receptors in the immunopathogenesis of Guillain-Barré syndrome. Mediators Inflamm. 2014;2014:758639. doi: 10.1155/2014/758639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Saxena M, Yeretssian G. NOD-Like receptors: Master regulators of inflammation and cancer. Front Immunol. 2014;5:327. doi: 10.3389/fimmu.2014.00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Crosbie EJ, Einstein MH, Franceschi S, Kitchener HC. Human papillomavirus and cervical cancer. Lancet. 2013;382:889–899. doi: 10.1016/S0140-6736(13)60022-7. [DOI] [PubMed] [Google Scholar]
- 101.Balasubramaniam SD, Balakrishnan V, Oon CE, Kaur G. Key molecular events in cervical cancer development. Medicina (Kaunas) 2019;55:E384. doi: 10.3390/medicina55070384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lin M, Ye M, Zhou J, Wang ZP, Zhu X. Recent advances on the molecular mechanism of cervical carcinogenesis based on systems biology technologies. Comput Struct Biotechnol J. 2019;17:241–250. doi: 10.1016/j.csbj.2019.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ye J, Yin L, Xie P, Wu J, Huang J, Zhou G, Xu H, Lu E, He X. Antiproliferative effects and molecular mechanisms of troglitazone in human cervical cancer in vitro. Onco Targets Ther. 2015;8:1211–1218. doi: 10.2147/OTT.S79899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yugawa T, Kiyono T. Molecular mechanisms of cervical carcinogenesis by high-risk human papillomaviruses: Novel functions of E6 and E7 oncoproteins. Rev Med Virol. 2009;19:97–113. doi: 10.1002/rmv.605. [DOI] [PubMed] [Google Scholar]
- 105.Jones MC, Askari JA, Humphries JD, Humphries MJ. Cell adhesion is regulated by CDK1 during the cell cycle. J Cell Biol. 2018;217:3203–3218. doi: 10.1083/jcb.201802088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hochegger H, Dejsuphong D, Sonoda E, Saberi A, Rajendra E, Kirk J, Hunt T, Takeda S. An essential role for Cdk1 in S phase control is revealed via chemical genetics in vertebrate cells. J Cell Biol. 2007;178:257–268. doi: 10.1083/jcb.200702034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Vassilev LT. Cell cycle synchronization at the G2/M phase border by reversible inhibition of CDK1. Cell Cycle. 2006;5:2555–2556. doi: 10.4161/cc.5.22.3463. [DOI] [PubMed] [Google Scholar]
- 108.Shaikh F, Sanehi P, Rawal R. Molecular screening of compounds to the predicted protein-protein interaction site of Rb1-E7 with p53-E6 in HPV. Bioinformation. 2012;8:607–612. doi: 10.6026/97320630008607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yun J, Chae HD, Choy HE, Chung J, Yoo HS, Han MH, Shin DY. p53 negatively regulates cdc2 transcription via the CCAAT-binding NF-Y transcription factor. J Biol Chem. 1999;274:29677–29682. doi: 10.1074/jbc.274.42.29677. [DOI] [PubMed] [Google Scholar]
- 110.Lindqvist A, van Zon W, Karlsson Rosenthal C, Wolthuis RM. Cyclin B1-Cdk1 activation continues after centrosome separation to control mitotic progression. PLoS Biol. 2007;5:e123. doi: 10.1371/journal.pbio.0050123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Crasta K, Huang P, Morgan G, Winey M, Surana U. Cdk1 regulates centrosome separation by restraining proteolysis of microtubule-associated proteins. EMBO J. 2006;25:2551–2563. doi: 10.1038/sj.emboj.7601136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Fang Y, Yu H, Liang X, Xu J, Cai X. Chk1-induced CCNB1 overexpression promotes cell proliferation and tumor growth in human colorectal cancer. Cancer Biol Ther. 2014;15:1268–1279. doi: 10.4161/cbt.29691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Krek W, Nigg EA. Differential phosphorylation of vertebrate p34cdc2 kinase at the G1/S and G2/M transitions of the cell cycle: Identification of major phosphorylation sites. EMBO J. 1991;10:305–316. doi: 10.1002/j.1460-2075.1991.tb07951.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Morgan DO. Principles of CDK regulation. Nature. 1995;374:131–134. doi: 10.1038/374131a0. [DOI] [PubMed] [Google Scholar]
- 115.Banerjee NS, Wang HK, Broker TR, Chow LT. Human papillomavirus (HPV) E7 induces prolonged G2 following S phase reentry in differentiated human keratinocytes. J Biol Chem. 2011;286:15473–15482. doi: 10.1074/jbc.M110.197574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Morgan EL, Wasson CW, Hanson L, Kealy D, Pentland I, McGuire V, Scarpini C, Coleman N, Arthur JSC, Parish JL, et al. STAT3 activation by E6 is essential for the differentiation-dependent HPV18 life cycle. PLoS Pathog. 2018;14:e1006975. doi: 10.1371/journal.ppat.1006975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Stewart DA, Cooper CR, Sikes RA. Changes in extracellular matrix (ECM) and ECM-associated proteins in the metastatic progression of prostate cancer. Reprod Biol Endocrinol. 2004;2:2. doi: 10.1186/1477-7827-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chiu CF, Ho MY, Peng JM, Hung SW, Lee WH, Liang CM, Liang SM. Raf activation by Ras and promotion of cellular metastasis require phosphorylation of prohibitin in the raft domain of the plasma membrane. Oncogene. 2013;32:777–787. doi: 10.1038/onc.2012.86. [DOI] [PubMed] [Google Scholar]
- 119.Matter ML, Ruoslahti E. A signaling pathway from the alpha5beta1 and alpha(v)beta3 integrins that elevates bcl-2 transcription. J Biol Chem. 2001;276:27757–27763. doi: 10.1074/jbc.M102014200. [DOI] [PubMed] [Google Scholar]
- 120.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 121.Sand JM, Larsen L, Hogaboam C, Martinez F, Han M, Røssel Larsen M, Nawrocki A, Zheng Q, Karsdal MA, Leeming DJ. MMP mediated degradation of type IV collagen alpha 1 and alpha 3 chains reflects basement membrane remodeling in experimental and clinical fibrosis-validation of two novel biomarker assays. PLoS One. 2013;8:e84934. doi: 10.1371/journal.pone.0084934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bokhari AA, Baker TM, Dorjbal B, Waheed S, Zahn CM, Hamilton CA, Maxwell GL, Syed V. Nestin suppression attenuates invasive potential of endometrial cancer cells by downregulating TGF-β signaling pathway. Oncotarget. 2016;7:69733–69748. doi: 10.18632/oncotarget.11947. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 123.da Silva Cardeal LB, Brohem CA, Corrêa TC, Winnischofer SM, Nakano F, Boccardo E, Villa LL, Sogayar MC, Maria-Engler SS. Higher expression and activity of metalloproteinases in human cervical carcinoma cell lines is associated with HPV presence. Biochem Cell Biol. 2006;84:713–719. doi: 10.1139/o06-084. [DOI] [PubMed] [Google Scholar]
- 124.Cardeal LB, Boccardo E, Termini L, Rabachini T, Andreoli MA, di Loreto C, Longatto Filho A, Villa LL, Maria-Engler SS. HPV16 oncoproteins induce MMPs/RECK-TIMP-2 imbalance in primary keratinocytes: Possible implications in cervical carcinogenesis. PLoS One. 2012;7:e33585. doi: 10.1371/journal.pone.0033585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Shiau MY, Fan LC, Yang SC, Tsao CH, Lee H, Cheng YW, Lai LC, Chang YH. Human papillomavirus up-regulates MMP-2 and MMP-9 expression and activity by inducing interleukin-8 in lung adenocarcinomas. PLoS One. 2013;8:e54423. doi: 10.1371/annotation/a50fa759-93fb-444d-9523-9d4a805ef898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ding Y, Pan Y, Liu S, Jiang F, Jiao J. Elevation of MiR-9-3p suppresses the epithelial-mesenchymal transition of nasopharyngeal carcinoma cells via down-regulating FN1, ITGB1 and ITGAV. Cancer Biol Ther. 2017;18:414–424. doi: 10.1080/15384047.2017.1323585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wang J, Deng L, Huang J, Cai R, Zhu X, Liu F, Wang Q, Zhang J, Zheng Y. High expression of Fibronectin 1 suppresses apoptosis through the NF-κB pathway and is associated with migration in nasopharyngeal carcinoma. Am J Transl Res. 2017;9:4502–4511. [PMC free article] [PubMed] [Google Scholar]
- 128.Rajkumar T, Sabitha K, Vijayalakshmi N, Shirley S, Bose MV, Gopal G, Selvaluxmy G. Identification and validation of genes involved in cervical tumourigenesis. BMC Cancer. 2011;11:80. doi: 10.1186/1471-2407-11-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Koromilas AE, Sexl V. The tumor suppressor function of STAT1 in breast cancer. JAKSTAT. 2013;2:e23353. doi: 10.4161/jkst.23353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhang Y, Liu Z. STAT1 in cancer: Friend or foe? Discov Med. 2017;24:19–29. [PubMed] [Google Scholar]
- 131.Akram M, Kim KA, Kim ES, Shin YJ, Noh D, Kim E, Kim JH, Majid A, Chang SY, Kim JK, Bae ON. Selective inhibition of JAK2/STAT1 signaling and iNOS expression mediates the anti-inflammatory effects of coniferyl aldehyde. Chem Biol Interact. 2016;256:102–110. doi: 10.1016/j.cbi.2016.06.029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated and/or analyzed during this study are included in this published article. The datasets generated and/or analyzed during the current study are available in the GEO (https://www.ncbi.nlm.nih.gov/geo/) and LinkOmics (http://www.linkedomics.org/) repository.