

Abstract
The ventromedial hypothalamus (VMH) plays a complex role in glucose and energy homeostasis. The VMH is necessary for the counter-regulatory response to hypoglycaemia (CRR) that increases hepatic gluconeogenesis to restore euglycaemia. On the other hand, the VMH also restrains hepatic glucose production during euglycaemia and stimulates peripheral glucose uptake. The VMH is also important for the ability of oestrogen to increase energy expenditure. This latter function is mediated by VMH modulation of the lateral/perifornical hypothalamic area (lateral/perifornical hypothalamus) orexin neurones. Activation of VMH AMP-activated protein kinase (AMPK) is necessary for the CRR. By contrast, VMH AMPK inhibition favours decreased basal glucose levels and is required for oestrogen to increase energy expenditure. Specialised VMH glucose-sensing neurones confer the ability to sense and respond to changes in blood glucose levels. Glucose-excited (GE) neurones increase and glucose-inhibited (GI) neurones decrease their activity as glucose levels rise. VMH GI neurones, in particular, appear to be important in the CRR, although a role for GE neurones cannot be discounted. AMPK mediates glucose sensing in VMH GI neurones suggesting that, although activation of these neurones is important for the CRR, it is necessary to silence them to lower basal glucose levels and enable oestrogen to increase energy expenditure. In support of this, we found that oestrogen reduces activation of VMH GI neurones in low glucose by inhibiting AMPK. In this review, we present the evidence underlying the role of the VMH in glucose and energy homeostasis. We then discuss the role of VMH glucose-sensing neurones in mediating these effects, with a strong emphasis on oestrogenic regulation of glucose sensing and how this may affect glucose and energy homeostasis.
Keywords: AMP-activated protein kinase, estrogen, glucose-sensing neurones, hypoglycaemia, orexin
1 |. INTRODUCTION
The effects of oestrogen on glucose homeostasis may partly be mediated by direct actions on ventromedial hypothalamic (VMH) glucose-inhibited (GI) neurones. Glucose sensing and oestrogen regulation of VMH GI neurones are both dependent on the cellular fuel sensor, AMP-activated protein kinase (AMPK).1 Oestrogens also regulate energy homeostasis via AMPK-dependent VMH projections to the lateral/perifornical hypothalamus (LH/PFH) orexin neurones.2–5 In this review, we consider the mechanisms by which VMH oestrogen may regulate glucose and energy homeostasis. In so doing, we also discuss the putative role of VMH and LH/PFH glucose-sensing neurones with respect to the effects of oestrogen on metabolism.
2 |. VMH
The role of the VMH in glucose and energy homeostasis is complex. The VMH has been implicated in the control of glucose and energy homeostasis subsequent to the late 1800s.6 In the mid-1900s, the VMH was designated as the “satiety centre” based on lesion studies showing that VMH lesions led to hyperphagia, insulin resistance and obesity.7–9 However, later studies revealed that precise VMH lesions not damaging the surrounding circuitry produced insulin resistance and weight gain in the absence of hyperphagia.10 It was further suggested that the VMH contributes to neuronal circuits that raise circulating energy substrates (eg, glucose, free fatty acids), as well as to those that lower them. For example, anaesthetising the VMH with lidocaine raises baseline glucose levels at the same time as preventing glucose increases in response to metabolic needs such as hypoglycaemia and exercise.11–13 Thus, it is clear that the VMH cannot be relegated to a simplistic unidirectional role in metabolism. Instead, the VMH possesses opposing neurocircuits that together coordinate glucose and energy homeostasis in response to metabolic need.
2.1 |. The role of the VMH in glucose homeostasis
Glucose is the primary fuel for the brain. Thus, specialised brain circuitry has evolved to maintain sufficient glucose levels for neuronal function.14 The VMH plays an established role in at least two aspects of glucose homeostasis: the counter-regulatory response to hypoglycaemia (CRR) and hepatic glucose output.15 VMH AMP-activated protein kinase (AMPK) activity is a key factor in these two functions.16–18 More recent evidence suggests that the VMH is also involved in peripheral insulin sensitivity and glucose uptake into skeletal muscle and adipose.19 However, whether AMPK is involved in these latter functions remains unknown. The VMH possesses glucose-sensing neurones that likely confer the ability to sense and respond to changes in blood glucose levels.20,21
VMH glucose-sensing neurones change their action potential frequency in response to physiological changes in extracellular brain glucose concentration. Glucose-excited (GE) neurones increase, whereas glucose-inhibited (GI) neurones decrease, their action potential frequency as glucose levels increase.22 In addition to the VMH, GE and GI neurones are found throughout the hypothalamus, as well as in other brain regions (eg, amygdala).20,23–27 A direct causal role for VMH glucose-sensing neurones in the regulation of glucose homeostasis remains to be defined. However, significant circumstantial evidence strongly suggests that they play a key role in the CRR at the very least and are likely to contribute to other aspects of glucose homeostasis. This evidence is discussed below.
The mechanisms by which VMH GE and GI neurones sense glucose have been studied extensively, although questions still remain (Figure 1). GE neurones sense glucose similarly to pancreatic beta cells. That is, the pancreatic form of hexokinase IV, which catalyses the conversion of glucose to glucose 6-phosphate, is expressed in the hypothalamus and is implicated in both glucose sensing in vitro and glucose homeostasis in vivo.28–32 Moreover, individual GE (and GI) neurones express glucokinase and require this enzyme for glucose sensing.33,34 Glucokinase is not end-product limited, which enables the rate of glycolysis to increase as glucose levels rise. Increased glycolytic flux increases ATP synthesis. Increased ATP/ADP in turn closes the ATP sensitive potassium (KATP) channel leading to depolarisation and increased action potential frequency.35,36 Although this mechanism has been clearly demonstrated for VMH GE neurones, KATP channel independent glucose sensing has been described for GE neurones located elsewhere.24,25,37 Moreover, glucokinase inhibition only blocks glucose sensing in approximately 60% of VMH GE neurones.33,34,38 This suggests that, although it is critical for glucose sensing in the majority of VMH GE neurones, other mechanisms must also exist.
FIGURE 1.
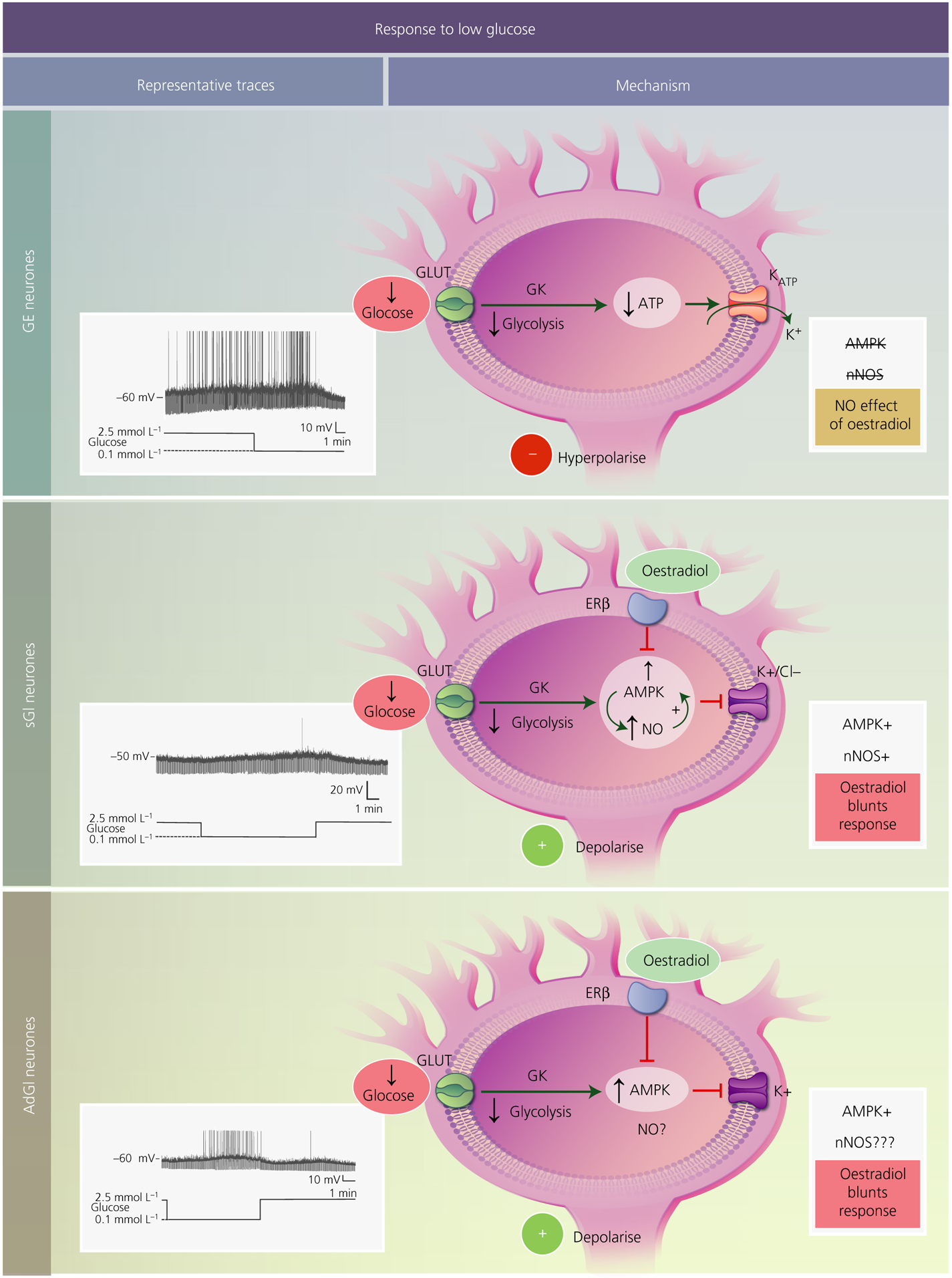
Mechanisms underlying the response of ventromedial hypothalamus (VMH) glucose-sensing neurones to low glucose. In all subtypes, glucose is transported into the cell most likely via a canonical glucose transporter (GLUT). Once inside the neurone, it is phosphorylated by the pancreatic form of glucokinase (GK) and proceeds through glycolysis. Decreased glucose leads to a reduction in ATP levels. For glucose-excited (GE) neurones (top), decreased ATP opens the ATP-sensitive potassium channel (KATP), leading to potassium efflux and hyperpolarisation. VMH GE neurones do not use AMP-activated protein kinase (AMPK) or neuronal nitric oxide synthase (nNOS) to sense glucose, nor are they regulated by oestradiol. Glucose-inhibited neurones are either constantly activated in low glucose (sustained or sGI neurones; middle) or show a transient activation that reverses when still in the presence of low glucose (adapting or AdGI neurones; lower). For sGI neurones, low glucose activates AMPK leading to phosphorylation of nNOS and increased NO production. NO, in turn, further activates AMPK, leading to closure of either potassium or chloride channels and cellular depolarisation. Oestradiol, through the oestrogen receptor (ER)β blunts activation in low glucose by inhibiting AMPK. Similar to sGI neurones, AdGI neurones require AMPK for potassium channel closure and activation in low glucose. Oestradiol similarly affects glucose sensitivity by blocking AMPK activation. It is not yet known whether nNOS is involved in glucose sensing by AdGI neurones. Representative current clamp recordings of each subtype of VMH glucose-sensing neurones in brain slices are shown to the left. The membrane potential in 2.5 mmol L−1 glucose is shown to the left of each trace. Upward deflections represent action potentials and downward deflections are the membrane voltage response to a constant hyperpolarising current pulse. Membrane resistance is directly proportional to these voltage responses. Thus, low glucose decreases membrane resistance in GE neurones, indicating ion channel opening (KATP). Conversely, low glucose increases resistance in sGI and AdGI neurones indicating closure of potassium or chloride channels. Interestingly, as demonstrated in the present study and in Santiago et al,1 AdGI neurones show a smaller transient activation when glucose levels return to 2.5 mmol L−1, suggesting that they are sensitive not only to glucose concentration per se, but also to the actual change in glucose level
More than half of the VMH GI neurones also use glucokinase to sense glucose, although the downstream mechanisms are very different from those of GE neurones.38,39 There are at least three subtypes of VMH GI neurones. One subtype, located predominantly in the dorsomedial VMH, utilises a metabolically sensitive Cl− channel to respond to glucose. The other two subtypes, located in the ventrolateral VMH, use a metabolically sensitive K+ channel.1,22 The dorsomedial and one subtype of ventrolateral GI neurones show a sustained activation in low glucose (sustained or “sGI”). By contrast, activation in low glucose is transient with neuronal activity returning to the inhibited state despite continued exposure to low glucose in the other subtype of ventrolateral VMH GI neurones (adapting or AdGI).1
AMPK mediates glucose sensing in all three subtypes of VMH GI neurones. In this case, the increased ATP/ADP ratio inhibits AMPK. Decreased AMPK activity maintains Cl− or K+ channels in an open state, leading to neuronal inhibition. Closure of these channels activates VMH GI neurones in low glucose.1,39 The signalling pathway downstream from AMPK activation has been well established only for dorsomedial VMH sGI neurones, although it likely applies to ventrolateral sGI and AdGI neurones for the reasons discussed below. When glucose levels decline, AMPK is allosterically activated by AMP and phosphorylates neuronal nitric oxide synthase (nNOS) leading to increases in NO production. NO, through its receptor soluble guanylyl cyclase, increases the synthesis of cGMP. Increased cGMP leads to further activation of AMPK, possibly via activation of an upstream kinase that phosphorylates AMPK, rendering this enzyme more sensitive to allosteric activation by AMP. This increase in AMPK activity then leads to closure of Cl− channels and increased action potential frequency.39 As noted above, although AMPK is critical for glucose sensing in ventrolateral sGI and AdGI neurones, NO signalling has not been definitively demonstrated for these neurones using electrophysiology. However, we were unable to detect any subtype of VMH GI neurones in mice lacking nNOS using electrophysiological techniques.40 Moreover, nNOS inhibition blocked the detection of GI neurones using membrane potential dye in an isolated cell preparation taken from the entire VMH. Membrane potential sensitive dyes fluoresce when the membrane depolarises. However, fluorescence is significantly slower than the time required for an individual action potential. Thus, we defined a GI neurone as one that increased fluorescence when glucose was low and decreased fluorescence after glucose was raised to baseline. Therefore, it is not clear whether AdGI neurones are detectable using this technique.39 These observations at the very least are consistent with nNOS-mediated glucose sensing in dorsomedial and ventrolateral VMH sGI neurones and potentially in ventrolateral VMH AdGI neurones as well. The transmitter phenotype of nNOS expressing GI neurones is not clear. However, VMH nNOS neurones are largely glutamatergic and are known to co-express somatostatin.41,42 Below, we focus on the potential roles of VMH glucose-sensing neurones with respect to mediating VMH regulation of the different aspects of glucose homeostasis.
2.1.1 |. The role of the VMH and VMH glucose-sensing neurones in the CRR
The CRR is the sequential release of glucagon, epinephrine, corticosterone and growth hormone when glucose levels decline below 60 mg dL−1 to restore euglycaemia.43,44 Seminal observations at the end of the 20th century suggested a key role for the VMH in the CRR. VMH lesions abolished the hormonal response to hypoglycaemia.45 VMH glucoprivation rapidly increased blood glucose as a result of the release of CRR hormones.46 By contrast, VMH glucose infusion during peripheral hypoglycaemia blunted the CRR.47 A wide variety of studies now demonstrate a critical role for the VMH in the initiation of the CRR.16,40,48–58 The pathway acts via a polysynaptic connection from the VMH to the sympathetic splanchnic nerve that controls glucagon and epinephrine release from the pancreas and adrenal medulla, respectively.12,59,60 However, it is important to note that other brain areas, including the brainstem catecholamine neurones, as well as peripheral glucose sensors in the portal vein of the liver, are also critical for the CRR.61–69
Although a direct causal link between VMH glucose-sensing neurones and CRR initiation is lacking, the idea that such a link exists is strongly supported by substantial circumstantial evidence. On the one hand, treatments that alter the signalling pathways used by VMH glucose-sensing neurones similarly alter the CRR. For example, humans with diabetes resulting from glucokinase mutations (GCK-MODY) exhibit an increased glucose threshold for CRR initiation.70 Similarly, VMH glucokinase inhibition enhanced and activation reduced CRR hormone secretion in rats during insulin-induced hypoglycaemia.71 These data support a role for both VMH GE and GI neurones in the CRR in vivo. A role for GE neurones in particular is suggested by data showing that VMH KATP channel activation enhances the CRR.50 There is also ample evidence indicating that altering signalling pathways specific to VMH GI neurones also alters the CRR. For example, McCrimmon et al.16,72 found that down-regulating VMH AMPK suppresses CRR hormone release and VMH AMPK activation restores the defective CRR in rodents. We found that the CRR is blunted following VMH nNOS inhibition in vivo, as well as in mice lacking nNOS, whereas increasing VMH cGMP levels enhance the CRR.40 Finally, optogenetic activation of VMH nNOS neurones, which project to the bed nucleus of the stria terminalis, triggers the release of CRR hormones.73 Taken together, these data suggest that intact glucose-sensing signalling pathways in the VMH are required for hypoglycaemia detection and initiation of the full CRR.
On the other hand, the magnitude of the response of VMH glucose-sensing neurones to low glucose parallels the strength of the CRR. For example, repetitive episodes of insulin-induced hypoglycaemia lower the glucose threshold for initiation of the CRR in both rodents and humans. This serious condition, known as hypoglycaemia-associated autonomic failure (HAAF), is the major limiting factor in treating diabetes with intensive insulin therapy.43,44,74–76 We have found blunted inhibition of VMH GE neurones and activation of GI neurones by low glucose in rodents with HAAF.77,78 Moreover, restoration of glucose sensing by VMH GI neurones is associated with an improved CRR after HAAF. For example, treating rats by oral gavage in vivo or incubating VMH GI neurones in vitro with the antioxidant N-acetyl cysteine completely prevents the impaired CRR and reduces the activation of VMH GI neurones in low glucose after recurrent hypoglycaemia.57 Diabetes per se is associated with an impaired CRR and blunted activation of VMH GI neurones in low glucose. VMH overexpression of the antioxidant enzyme thioredoxin restores both the CRR and the detection of low glucose by VMH GI neurones in rats with type 1 diabetes mellitus.79 The above data are only examples of the parallels between glucose sensing by VMH GE and GI neurones and the CRR in vivo. Thus, although direct evidence indicating that VMH glucose-sensing neurones modulate the CRR is still lacking, the circumstantial evidence for this role cannot be discounted. It should also be noted that the above studies all focused on VMH sGI neurones. A putative role for VMH AdGI neurones is discussed below when oestrogenic regulation is discussed.
2.1.2 |. The role of the VMH in hepatic glucose production during euglycaemia
In addition to triggering the release of gluconeogenic hormones during insulin-induced hypoglycaemia, the VMH also plays a role in hepatic glucose production during euglycaemia. As noted above, VMH inhibition raises baseline glucose levels at the same time as preventing increased glucose levels during exercise or hypoglycaemia, suggesting a bidirectional role for the VMH in glucose homeostasis.11–13 Moreover, VMH AMPK activation increases hepatic glucose production during euglycaemia, as well as in response to hypoglycaemia.18 On the other hand, glucose or insulin infusion, AMPK inhibition or KATP channel activation in the mediobasal hypothalamus, which contains the VMH and the adjacent arcuate nucleus, lower hepatic glucose production under euglycaemic conditions.18,80–82 Glucose and insulin suppression of hepatic glucose production is dependent on vagal afferents, whereas increased glucose production during the CRR is a result of the activation of sympathetic pathways (likely through the bed nucleus of the stria terminalis).80 Interestingly, to date, an anatomical pathway connecting the VMH and the brainstem parasympathetic centres has not been demonstrated. The simple explanation might be that activation of VMH GI neurones using AMPK is responsible for increased glucose production via bed nucleus of the stria terminalis (BNST) projections,73 whereas GE neurones regulate the hepatic vagus via projections to as yet undefined regions, lowering hepatic glucose production. An alternative explanation is that there is a balance between these TWO glucose-sensing subtypes involving mutual inhibition either directly or via downstream neurocircuits. For example, AMPK activation in the mediobasal hypothalamus blocks the glucose-induced inhibition of gluconeogenesis, suggesting that activating GE neurones is not sufficient to lower hepatic glucose output but that GI neurones must be simultaneously inhibited.18 Moreover, other questions exist. For example, as noted above, GE neurones respond to increased glucose by closing the KATP channel. Although GI neurones do not use the KATP channel to sense glucose, they do possess functional KATP channels. Indeed, lactate activates GI neurones by closing their KATP channels.83 Interestingly, the KATP channel activator, diazoxide, lowers hepatic glucose production when infused into the brain of non-diabetic rodents and humans. This effect in rodents is blocked by mediobasal hypothalamus administration of the KATP channel blocker glibenclamide.82 Given the dependence of VMH GE neurones on the KATP channel for glucose sensing, GE neurones might initially be considered to mediate these effects. However, the reality is clearly more complicated because these KATP channel modulators would also affect GI neurones. Finally, we found that VMH overexpression of the antioxidant enzyme thioredoxin restores the CRR and normal glucose sensing in GI neurones in diabetic animals at the same time as lowering their baseline blood glucose levels and reducing insulin requirements.79 Taken together, these data indicate a dual role for VMH GE and GI neurones in the regulation of hepatic glucose production during euglycaemia.
2.1.3 |. The role of the VMH in peripheral glucose uptake/insulin sensitivity
The VMH is important for increasing blood glucose during exercise. When lidocaine was injected into the VMH of exercising rats, hepatic glucose production and glycogenolysis was decreased compared to that in control rats.13 Conversely, injecting the VMH with alpha or beta adrenergic receptor antagonists during exercise increased blood glucose and free fatty acids.84 VMH stimulation elevates both circulating glucose levels and glucose uptake by skeletal muscle.85 Similarly, VMH microinjection of leptin and orexin increased glucose uptake in skeletal muscle.86–90 Activation of the VMH specific steroidogenic factor 1 (SF-1) neurones during a hyperinsulinaemic-euglycaemic clamp increased whole body glucose utilisation and increased glucose uptake in peripheral tissues such as red-type skeletal muscle and heart. This was accompanied by increased insulin-induced inhibition of genes associated with gluconeogenesis and the activity of glycogen phosphorylase a in the liver.19 Taken together, these data suggest an important role for the VMH in regulating peripheral insulin sensitivity and glucose utilisation. To date, a role for VMH AMPK in peripheral insulin sensitivity has not been demonstrated, nor have VMH glucose-sensing neurones been studied in the context of exercise. However, one might speculate that the SF-1 neurones for which activation leads to peripheral glucose uptake might comprise the GE neurones that are AMPK-independent and activated during euglycaemia and hyperglycaemia.
2.2 |. The role of the VMH in energy homeostasis
When VMH-lesioned rodents are pair-fed to sham-operated controls, they become obese, even in the absence of extra caloric input.10 Thus, VMH lesions cause “metabolic obesity” that is independent of hyperphagia. More recent studies have taken advantage of the fact that SF-1 expression in the adult brain is restricted to the VMH. SF-1 neurones project to autonomic centres including the parabrachial nucleus, locus ceruleus, rostral ventrolateral medulla and the nucleus tractus solitarious, as well as to other hypothalamic areas (eg, arcuate, paraventricular nucleus).60 Mice lacking SF-1 expression (SF-1 knockout mice) have altered VMH topology and die of adrenal insufficiency.91,92 However, when Majdic et al93 transplanted adrenal glands into SF-1 knockout mice to create a strain that lacked SF-1 expression only in the VMH, it was observed that these mice developed obesity. Postnatal SF-1 deletion in mice not only preserved VMH integrity, but also produced diet-induced obesity. This obesity was associated with leptin resistance and reduced energy expenditure as a result of decreased brown adipose tissue (BAT) thermogenesis.94 Optogenetic and chemogenetic activation of SF-1 neurones increase insulin sensitivity and whole-body glucose utilisation at the same time as suppressing food intake.19,95 These data provide strong support for a role of the VMH in both glucose and energy homeostasis and indicate a specific function in regulating whole body energy expenditure.
Increased thermogenesis in BAT and beige white adipose tissue (WAT) is one of the major factors mediating energy expenditure in the absence of physical exercise. The VMH has long been linked to regulation of BAT and, more recently, beige WAT thermogenesis. In the 1980s, it was found that VMH-lesions reduced BAT sympathetic nerve activity in response to acute cold.96 Conversely, VMH stimulation increases BAT thermogenesis.97 Recent evidence suggests that hormones such as oestrogen and 3,3′,5-triiodothyronine (T3) act in the VMH to stimulate the sympathetic activation of BAT thermogenesis.2,3,98,99 Interestingly, T3 action in the VMH is also required for the ability of this hormone to promote WAT browning and protect against obesity.98 A recent study by Wang et al100 showed that pharmacogenetic activation of VMH rat insulin promotor neurones in mice selectively increases sympathetic outflow to WAT and promoted browning. All of the above effects are dependent on VMH AMPK inhibition. Moreover, specific deletion of the AMPKα1 isoform in VMH SF-1 neurones prevents obesity on a high-fat diet by increasing BAT thermogenesis and WAT browning. SF-1 AMPKα1 deletion also improved glucose and lipid homeostasis.101,102 This VMH AMPK dependence suggests a potential role for VMH GI neurones in the regulation of energy, as well as glucose homeostasis. Interestingly, VMH endoplasmic reticulum stress causes obesity through sympathetic inhibition of BAT and alleviation of this endoplasmic reticulum stress by overexpressing the endoplasmic reticulum chaperone protein, GRP78, reverses ceramide-induced lipotoxicity in the VMH and consequent weight gain.103–105 These observations are consistent with our data showing that VMH overexpression of the antioxidant enzyme thioredoxin restored the CRR in diabetic rats at the same time as paradoxically lowering their baseline glucose levels and decreasing insulin requirements.79 Thus, regulation of VMH endoplasmic reticulum/oxidative stress is important for multiple aspects of glucose and energy homeostasis.
2.3 |. The effects of VMH oestrogen on energy homeostasis and regulation of GI neurones
The oestradiol receptor (ER)α is highly expressed in the ventrolateral subdivision of the VMH where it serves an important role in normal energy homeostasis in females. Silencing VMH ERα using RNA interference increased body weight, food intake and blood glucose levels at the same time as reducing energy expenditure.106 Furthermore, deletion of ERα in specific SF1 neurones leads to profound visceral obesity in female but not male mice fed normal or high-fat diets. This obesity was associated with glucose intolerance, decreased sympathetic activation of BAT and decreased BAT thermogenesis. However, these animals were not hyperphagic. This suggests that another subpopulation of VMH neurones might be responsible for the effects of oestrogen on food intake.107 These effects of VMH ERα recapitulate the effects of peripheral and central administration of 17β-oestradiol (E2). Moreover, central E2 decreases hypothalamic endoplasmic reticulum stress, suggesting a potential mechanism for the metabolic effects of this hormone.2
Interestingly, the effects of VMH bone morphogenic protein 8B (BMP8B) were shown to depend on oestrogen.3–5 It was found that VMH BMP8B increases BAT thermogenesis in females but not males. This effect is abolished in ovariectomised animals; however, when these animals are treated with E2, they respond normally to BMP8B. BMP8B injection into the VMH, but not the LH/PFH, decreased body weight and food intake in female rats and increased WAT browning, leading to increased energy expenditure. These effects of BMP8B are dependent on VMH AMPK inhibition. Such studies further demonstrated that VMH BMP8B increased orexin expression in the LH/PFH. Increased orexin expression was downstream of VMH AMPK inhibition and dependent on the LH/PFH glutamate vesicular transporter 2. It was concluded that the metabolic effects of BMP8B are mediated by VMH AMPK inhibition, which leads to activation of orexin neurones. It has also been demonstrated that orexin neurones project to the raphe pallidus to increase sympathetic activation of BAT.108,109 This orexinergic pathway may also be involved in browning of WAT because the raphe pallidus has been implicated in sympathetic innervation of WAT.100,110 These observations regarding the dependence of the effects of oestrogen on VMH AMPK inhibition, as well as its effects on endoplasmic reticulum stress, suggest that silencing of VMH GI neurones may be necessary for these hormonal effects.
In the ventrolateral VMH, we found that approximately 60% of the neuronal population were of the GI subtype. Of these, approximately half were K+ channel-dependent sGI and half were AdGI neurones. Moreover, 20% of ventrolateral VMH neurones were GE neurones, making 80% of the total neuronal population in this VMH subdivision glucose sensing.1 This overlap between ERα and glucose-sensing neurones in this location suggests that these neurones may play a role in the metabolic effects of oestrogen. In support of this, ventrolateral VMH sGI neurones are intrinsically sexually dimorphic. Ventrolateral VMH sGI neurones from pre-pubertal female mice showed a blunted activation in low glucose compared to males. Oestrogen blunted the activation in low glucose of both sGI and AdGI neurones from pre-pubertal males and females.1 As would be predicted from these observations, the response of sGI and AdGI neurones to low glucose in cycling females was greatest during dioestrus when oestrogen levels were lowest.111 The effects of oestrogen on the glucose sensitivity of ventrolateral VMH GI neurones were dependent on AMPK inhibition and were mediated by a membrane bound form of the oestrogen receptor. Interestingly, the effects of oestrogen on GI neurones were blocked by an Erβ antagonist and not an ERβ antagonist.1 The recovery of blood glucose levels in response to hypoglycaemia varied with oestrous cycle in a manner that paralleled the glucose sensitivity of ventrolateral VMH GI neurones.111 Accordingly, during dioestrous, when oestrogen levels are low, female mice increase blood glucose levels to euglycaemia after hypoglycaemia more quickly than in other phases of the oestrous cycle.111 By contrast, ventrolateral VMH GE neurones were neither sexually dimorphic, nor oestrogen sensitive. Moreover, of 12 ventrolateral VMH non-glucose-sensing neurones from males and females, oestrogen caused a slight depolarisation in one and was without effect in the other 11.1 The above data indicate that ventrolateral VMH sGI and AdGI neurones are a target of oestrogen and suggest a role in its metabolic effects, potentially via projections to the LH/PFH orexin neurones. These data also suggest that GE neurones are not involved in the effects of oestrogen on this metabolic pathway. However, it might be speculated that, if the balance between GE and GI neuronal activity is important to energy homeostasis, an oestrogen-induced increase in glucose inhibition of GI neurones would release any opposition to the influence of GE neurones in these processes.
3 |. LH/PFH OREXIN NEURONES
3.1 |. LH/PFH orexin neurones and glucose homeostasis
Within the brain, neurones that express orexin are only found in a small region extending from the lateral dorsomedial hypothalamus to the LH/PFH. Approximately half of the orexin neurones are sGI neurones.25,112,113 Interestingly, the mechanism by which they sense glucose is distinct from that of the VMH GI neurones. Although the metabolism of glucose leading to changes in AMPK activity is required for VMH GI neurones to sense glucose, the orexin neurones appear to sense the glucose molecule per se.112,114 Metabolism-independent glucose sensing was demonstrated using 2-deoxyglucose (2DG). Hexokinase converts 2DG to 2DG-6-phosphate, which then inhibits phosphoglucose isomerase, stopping glycolysis and depleting intracellular ATP.115 If glucose sensing is metabolism-dependent, 2DG should activate GI neurones. However, equimolar concentrations of 2DG inhibit orexin neurones to a similar degree as glucose.112,114 The ion channel that mediates the change in activity in response to glucose is an outwardly rectifying K+ channel; however, the signalling pathway linking glucose and this ion channel remains to be determined.112,116,117 It is possible that the “glucose receptor” is similar to the sweet taste receptor that has been described in the LH.118
The orexin neurones stimulate feeding during hypoglycaemia.119 They also contribute to the CRR via projections to the rostral ventrolateral medulla, leading to activation of the sympathetic splanchnic nerve and epinephrine release from the adrenal medulla.119–121 An elegant series of studies showed that orexin neurones also play a role in hypoglycaemia awareness.119,122 Hypoglycaemia awareness refers to the symptoms of hypoglycaemia (ie, sweating, anxiety) that alert an individual to falling glucose and prompt corrective behavioural responses. Similar to the CRR, recurrent hypoglycaemia also causes hypoglycaemia unawareness, which, in combination with HAAF, leads to dangerous decreases in blood glucose.123–126 Although the hormonal CRR and feeding responses to hypoglycaemia are easily measured in rodents, the lack of an animal model for hypoglycaemia awareness has significantly hampered the field. Otlivanchik et al122 utilised the standard conditioned place preference (CPP) test to train rats to prefer one side of a two-sided chamber which was associated with a food treat. They then injected sufficient insulin to induce hypoglycaemia when the rats were confined on the preferred side. The following day, they observed that the rats no longer preferred the side associated with the food treat after experiencing hypoglycaemia in that location. This indicated that the rats were “aware” of hypoglycaemia and found it aversive.122 This effect was blocked by orexin 1 receptor antagonism, indicating that it was mediated by orexin neurones. Interestingly, this effect was also blocked by recurrent hypoglycemic episodes in the home cage, indicating that this paradigm also models hypoglycaemia unawareness. The neurocircuitry that mediates hypoglycaemia awareness may overlap with the orexin projections to the tuberomammillary histamine neurones, which play a role in wakefulness and arousal.127–133
3.2 |. LH/PFH orexin neurones and energy homeostasis
There are two aspects of orexin physiology that are related to energy homeostasis. As discussed above, LH/PFH orexin neuronal activity is positively correlated with increased BAT thermogenesis and WAT browning.3–5 Orexin neurones, in the LH in particular, also play a role in the ingestion of palatable food.134–137 LH orexin neurones are activated by cues associated with reward and by the reward itself.134 The dopamine neurones within the ventral tegmental area (VTA) have been well studied with respect to different types of reward.138–147 LH orexin neurones project to the VTA dopamine neurones. Orexin application in the VTA increases the expression of NMDA and AMPA receptors on the plasma membrane. Moreover, orexin neurones co-release glutamate.139,148,149 This suggests that the net effect of orexin in the VTA is to enhance excitatory glutamate signalling. Recent data from our laboratory showed that the LH orexin-GI neurones are sensitive to circulating signals of energy balance and metabolic status. Ghrelin directly enhances, whereas leptin indirectly blunts, the activation of these neurones in low glucose. As would be predicted by these hormonal effects, fasting also enhances the activation of these neurones in low glucose. Increased activity of LH orexin neurones in low glucose activates VTA dopamine neurones.112 This indirect activation of VTA DA neurones is associated with increased NMDA and AMPA receptor current amplitude after 1 hour in low glucose, which persists for at least 1 hour after glucose is returned to baseline.150 The effect of low glucose on NMDA current amplitude is blocked by an orexin 1 receptor antagonist, suggesting that exposure to low glucose induces orexin-dependent glutamate plasticity. This is consistent with the fasting induced increase in the AMPA/NMDA receptor current ratio on VTA DA neurones, comprising an in vivo index of glutamate plasticity. These cellular effects translate into alterations in reward-based feeding behaviour. Accordingly, increasing LH glucose in rats that were weight restricted to 85% of their initial body weight decreased the desire to seek a palatable food reward in a standard CPP paradigm.150 These are the first data linking changes in brain glucose with feeding behaviour and suggesting a role for glucose-sensing neurones. The data also suggest a mechanism underlying the difficulty maintaining weight loss after dieting. That is, the hormonal changes associated with weight loss increase activation of orexin-GI neurones, especially after sleeping or between meals when glucose levels are lower. The increased orexinergic activity in turn causes a persistent excitation of the VTA dopamine neurones, which enhance the reward value of sugary fatty foods.
4 |. CONCLUSIONS
The VMH regulates multiple facets of physiological glucose and energy homeostasis in addition to correcting iatrogenic insulin-induced hypoglycaemia. VMH glucose-sensing neurones are likely candidates for enabling, in part, these actions (Figure 2). Specifically, AMPK-dependent VMH GI neurones are clearly linked to the CRR. However, they may also play a role in other AMPK-dependent VMH functions, including increasing basal hepatic glucose and inhibiting BAT thermogenesis and WAT browning. This latter function may result from inhibitory signalling to the LH/PFH orexin neurones. It is also possible that VMH GE neurones play an opposing role to the GI neurones. The effects of oestrogen on the glucose sensitivity of VMH GI neurones provide further support for a role of these neurones in physiological glucose and energy homeostasis. Finally, some of the roles of VMH GI neurones in energy homeostasis, especially with respect to oestrogen-mediated effects, may be mediated by the LH/PFH orexin neurones. LH orexin-GI neurones have a demonstrable link to reward-based feeding behaviour, in particular to drive the intake of rewarding fatty-sugary foods during glucose and energy deficit. Because orexin neurones also increase energy expenditure, it is counter-intuitive to assume that these latter neurones would be activated during low glucose. Thus, it is tempting to speculate that the orexin neurones stimulating BAT thermogenesis and WAT browning, which may be inhibited by VMH GI neurones, belong to the part of the orexin population that do not sense glucose. In conclusion, we have come a long way from describing the VMH and its glucose-sensing neurones as belonging to a “satiety centre”, although many unanswered questions still remain.
FIGURE 2.
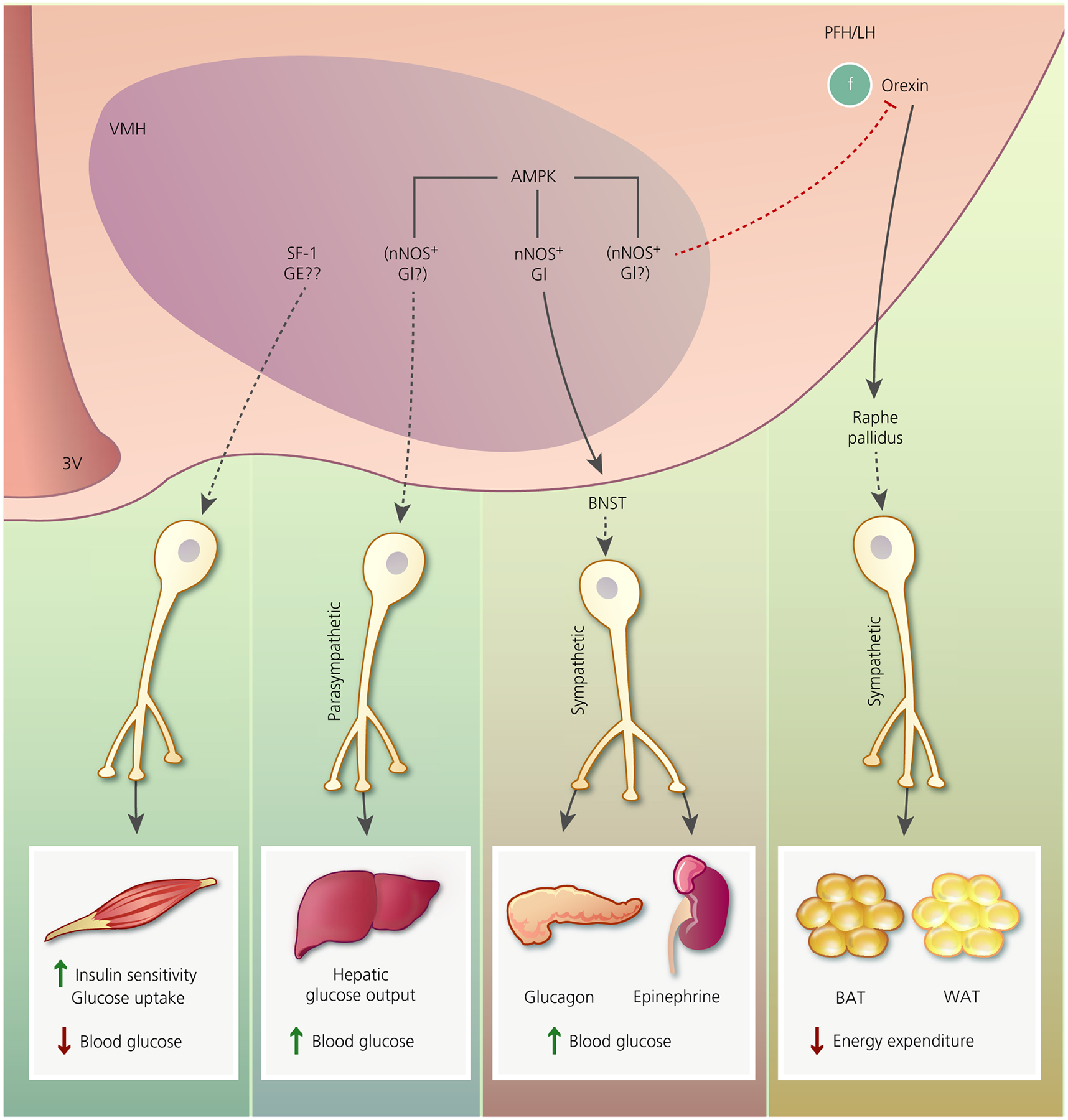
Putative pathways by which ventromedial hypothalamus (VMH) glucose-sensing neurones may regulate glucose and energy homeostasis. Activation of steroidogenic factor 1 (SF-1) neurones increases skeletal muscle insulin sensitivity and glucose uptake, leading to decreased blood glucose. We speculate that this might involve glucose-excited (GE) neurones. VMH AMP-activated protein kinase (AMPK) activation increases hepatic glucose output via a parasympathetically mediated mechanism, leading to increased blood glucose. During hypoglycaemia, VMH AMPK activation restores euglycaemia by increasing glucagon and epinephrine secretion. This effect is mediated by activation of the sympathetic nervous systems via likely projections to the bed nucleus of the stria terminalis (BNST). By contrast, VMH AMPK activation is associated with blunted brown fat (BAT) thermogenesis and white fat (WAT) browning. This effect is dependent on projections from orexin neurones in the perifornical and lateral hypothalamus (PFH/LH) to the raphe pallidus. Oestrogen activation of thermogenesis via this pathway requires VMH AMPK inhibition. VMH nitric oxide synthase (nNOS)-glucose-inhibited (GI) neurones have a demonstrated role in glucagon and epinephrine secretion during hypoglycaemia. Based on the effects of VMH AMPK described above, we speculate that VMH nNOS-GI neurones may play a role in hepatic glucose output and BAT/WAT thermogenesis. We further speculate that, because oestrogen blunts activation of VMH GI neurones by inhibiting AMPK, sGI and/or AdGI neurones may mediate some of the effects of oestrogen. 3V, third ventricle
Funding information
National Institutes of Health, Grant/Award Number: R01DK10367; Juvenile Diabetes Research Foundation, Grant/Award Number: 3-SRA-2017-488-S-B
REFERENCES
- 1.Santiago AM, Clegg DJ, Routh VH. Estrogens modulate ventrolateral ventromedial hypothalamic glucose-inhibited neurons. Mol Metab. 2016;5:823–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.González-García I, Contreras C, Estévez-Salguero Á, et al. Estradiol regulates energy balance by ameliorating hypothalamic ceramide-induced ER stress. Cell Rep. 2018;25:413–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martínez de Morentin PB, González-García I, Martins L, et al. Estradiol regulates brown adipose tissue thermogenesis via hypothalamic AMPK. Cell Metab. 2014;20:41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martins L, Seoane-Collazo P, Contreras C, et al. A functional link between AMPK and orexin mediates the effect of BMP8B on energy balance. Cell Rep. 2016;16:2231–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Whittle AJ, Carobbio S, Martins L, et al. BMP8B increases brown adipose tissue thermogenesis through both central and peripheral actions. Cell. 2012;149:871–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mayer J, Thomas DW. Regulation of food intake and obesity. Science. 1967;156:328–337. [DOI] [PubMed] [Google Scholar]
- 7.Hetherington AW, Ranson SW. Hypothalamic lesions and adiposity in the rat. Anat Rec. 1940;78:149–172. [Google Scholar]
- 8.Hetherington AW, Ranson SW. The spontaneous activity and food intake of rats with hypothalamic lesions. Am J Physiol. 1942;136:609–617. [Google Scholar]
- 9.Hetherington AW, Ranson SW. The relation of various hypothalamic lesions to adiposity in the rat. J Comp Neurol. 1942;76: 475–499. [Google Scholar]
- 10.Cox JE, Powley TL. Intragastric pair feeding fails to prevent VMH obesity or hyperinsulinemia. Am J Physiol. 1981;240:E566–E572. [DOI] [PubMed] [Google Scholar]
- 11.Steffens A, Mogenson G, Stevenson J. Blood glucose, insulin, and free fatty acids after stimulation and lesions of the hypothalamus. Am J Physiol. 1972;222:1446–1452. [DOI] [PubMed] [Google Scholar]
- 12.Steffens AB, Scheurink AJ, Luiten PG, Bohus B. Hypothalamic food intake regulating areas are involved in the homeostasis of blood glucose and plasma FFA levels. Physiol Behav. 1988;44:581–589. [DOI] [PubMed] [Google Scholar]
- 13.Vissing J, Wallace JL, Scheurink AJ, Galbo H, Steffens AB. Ventromedial hypothalamic regulation of hormonal and metabolic responses to exercise. Am J Physiol. 1989;256:R1019–R1026. [DOI] [PubMed] [Google Scholar]
- 14.Cryer PE. Glucose counterregulation in man. Diabetes. 1981;30:261–264. [DOI] [PubMed] [Google Scholar]
- 15.Shimazu T, Minokoshi Y. Systemic glucoregulation by glucose-sensing neurons in the ventromedial hypothalamic nucleus (VMH). J Endo Soc. 2017;1:449–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McCrimmon RJ, Shaw M, Fan X, et al. Key role for AMP-activated protein kinase in the ventromedial hypothalamus in regulating counterregulatory hormone responses to acute hypoglycemia. Diabetes. 2008;57:444–450. [DOI] [PubMed] [Google Scholar]
- 17.McCrimmon RJ, Shaw M, Fan X, et al. AMP-activated protein kinase (AMPK): a key mediator of hypoglycemia-sensing in the ventromedial hypothalamus (VMH). Am Diabet Assoc Scientif Sess. 2006;62-OR. [Google Scholar]
- 18.Yang CS, Lam CKL, Chari M, et al. Hypothalamic AMP-activated protein kinase regulates glucose production. Diabetes. 2010;59:2435–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coutinho EA, Okamoto S, Ishikawa AW, et al. Activation of SF1 neurons in the ventromedial hypothalamus by dreadd technology increases insulin sensitivity in peripheral tissues. Diabetes. 2017;66:2372–2386. [DOI] [PubMed] [Google Scholar]
- 20.Oomura Y, Kimura K, Ooyama H, Maeo T, Iki M, Kuniyoshi N. Reciprocal activities of the ventromedial and lateral hypothalamic area of cats. Science. 1964;143:484–485. [DOI] [PubMed] [Google Scholar]
- 21.Oomura Y, Ono H, Ooyama H, Wayner MJ. Glucose and osmosensitive neurons of the rat hypothalamus. Nature. 1969;222:282–284. [DOI] [PubMed] [Google Scholar]
- 22.Song Z, Levin BE, McArdle JJ, Bakhos N, Routh VH. Convergence of pre- and postsynaptic influences on glucosensing neurons in the ventromedial hypothalamic nucleus. Diabetes. 2001;50:2673–2681. [DOI] [PubMed] [Google Scholar]
- 23.Beall C, Hamilton D, Gallagher J, et al. Mouse hypothalamic GT1–7 cells demonstrate AMPK-dependent intrinsic glucose-sensing behaviour. Diabetologia. 2012;55:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Melnick IV, Price CJ, Colmers WF. Glucosensing in parvocellular neurons of the rat hypothalamic paraventricular nucleus. Eur J Neurosci. 2011;34:272–282. [DOI] [PubMed] [Google Scholar]
- 25.Burdakov D, Gerasimenko O, Verkhratsky A. Physiological changes in glucose differentially modulate the excitability of hypothalamic melanin-concentrating hormone and orexin neurons in situ. J Neurosc. 2005;25:2429–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fioramonti X, Contie S, Song Z, Routh VH, Lorsignol A, Penicaud L. Characterization of glucosensing neuron subpopulations in the arcuate nucleus: integration in NPY and POMC networks? Diabetes. 2007;56:1219–1227. [DOI] [PubMed] [Google Scholar]
- 27.Zhou L, Podolsky N, Sang Z, et al. The medial amygdalar nucleus: a novel glucose-sensing region that modulates the counterregulatory response to hypoglycemia. Diabetes. 2010;59:2646–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jetton TL, Liang Y, Pettepher CC, et al. Analysis of upstream glucokinase promoter activity in transgenic mice and identification of glucokinase in rare neuroendocrine cells in the brain and gut. J Biol Chem. 1994;269:3641–3654. [PubMed] [Google Scholar]
- 29.Mobbs CV, Kow LM, Yan XJ. Brain glucose-sensing mechanisms: ubiquitous silencing by aglycemia vs. hypothalamic neuroendocrine responses. Am J Physiol. 2001;281:E649–E654. [DOI] [PubMed] [Google Scholar]
- 30.Yang XJ, Kow LM, Funabashi T, Mobbs CV. Hypothalamic glucose sensor: similarities to and differences from pancreatic beta-cell mechanisms. Diabetes. 1999;48:1763–1772. [DOI] [PubMed] [Google Scholar]
- 31.Yang XJ, Kow LM, Pfaff DW, Mobbs CV. Metabolic pathways that mediate inhibition of hypothalamic neurons by glucose. Diabetes. 2004;53:67–73. [DOI] [PubMed] [Google Scholar]
- 32.Yang X-j, Mastaitis J, Mizuno T, Mobbs CV. Glucokinase regulates reproductive function, glucocorticoid secretion, food intake, and hypothalamic gene expression. Endocrinology. 2007;148:1928–1932. [DOI] [PubMed] [Google Scholar]
- 33.Dunn-Meynell AA, Routh VH, Kang L, Gaspers L, Levin BE. Glucokinase is the likely mediator of glucosensing in both glucose-excited and glucose-inhibited central neurons. Diabetes. 2002;51:2056–2065. [DOI] [PubMed] [Google Scholar]
- 34.Kang L, Dunn-Meynell AA, Routh VH, et al. Glucokinase is a critical regulator of ventromedial hypothalamic neuronal glucosensing. Diabetes. 2006;55:412–420. [DOI] [PubMed] [Google Scholar]
- 35.Ashford MLJ, Boden PR, Treherne JM. Tolbutamide excites rat glucoreceptive ventromedial hypothalamic neurones by indirect inhibition of ATP-sensitive K+ channels. Br J Pharmacol. 1990;101:531–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ashford MLJ, Boden PR, Treherne JM. Glucose-induced excitation of hypothalamic neurones is mediated by ATP-sensitive K+ channels. Pfugers Arch. 1990;415:479–483. [DOI] [PubMed] [Google Scholar]
- 37.Roland AV, Moenter SM. Glucosensing by GnRH neurons: inhibition by androgens and involvement of AMP-activated protein kinase. Mol Endocrinol. 2011;25:847–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kang L, Routh VH, Kuzhikandathil EV, Gaspers L, Levin BE. Physiological and molecular properties of rat hypothalamic ventromedial nucleus glucosensing neurons. Diabetes. 2004;53:559. [DOI] [PubMed] [Google Scholar]
- 39.Murphy BA, Fakira KA, Song Z, Beuve A, Routh VH. AMP-activated protein kinase (AMPK) and nitric oxide (NO) regulate the glucose sensitivity of ventromedial hypothalamic (VMH) glucose-inhibited (GI) neurons. Am J Physiol. 2009;297:C750–C758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fioramonti X, Marsollier N, Song Z, et al. Ventromedial hypothalamic nitric oxide production is necessary for hypoglycemia detection and counterregulation. Diabetes. 2010;59:519–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chachlaki K, Malone SA, Qualls-Creekmore E, et al. Phenotyping of nNOS neurons in the postnatal and adult female mouse hypothalamus. J Comp Neurol. 2017;525:3177–3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yamada K, Emson PC, Hokfelt T. Immunohistochemical mapping of nitric oxide synthase in the rat hypothalamus and colocalization with neuropeptides. J Chem Neuroanat. 1996;10:295–316. [DOI] [PubMed] [Google Scholar]
- 43.Amiel SA, Sherwin RS, Simonson DC, Tamborlane WV. Effect of intensive insulin therapy on glycemic thresholds for counterregulatory hormone release. Diabetes. 1988;37:901–907. [DOI] [PubMed] [Google Scholar]
- 44.Amiel SA, Tamborlane WV, Simonson DC, Sherwin RS. Defective glucose counterregulation after strict glycemic control of insulin-dependent diabetes mellitus. N Engl J Med. 1987;316:1376–1383. [DOI] [PubMed] [Google Scholar]
- 45.Borg WP, During MJ, Sherwin RS, Borg MA, Brines ML, Shulman GI. Ventromedial hypothalamic lesions in rats suppress counterregulatory responses to hypoglycemia. J Clin Invest. 1994;93:1677–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Borg WP, Sherwin RS, During MJ, Borg MA, Shulman GI. Local ventromedial hypothalamus glucopenia triggers counterregulatory hormone release. Diabetes. 1995;44:180–184. [DOI] [PubMed] [Google Scholar]
- 47.Borg MA, Sherwin RS, Borg WP, Tamborlane WV, Shulman GI. Local ventromedial hypothalamus glucose perfusion blocks counterregulation during systemic hypoglycemia in awake rats. J Clin Invest. 1997;99:361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chan O, Cheng H, Herzog R, et al. Increased GABAergic tone in the ventromedial hypothalamus contributes to suppression of counterregulatory reponses after antecedent hypoglycemia. Diabetes. 2008;57:1363–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fan X, Chan O, Ding Y, Zhu W, Mastaitis J, Sherwin R. Reduction in SGLT1 mRNA expression in the ventromedial hypothalamus improves the counterregulatory responses to hypoglycemia in recurrently hypoglycemic and diabetic rats. Diabetes. 2015;64:3564–3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McCrimmon RJ, Evans ML, Fan X, et al. Activation of ATP-sensitive K+ channels in the ventromedial hypothalamus amplifies counterregulatory hormone responses to hypoglycemia in normal and recurrently hypoglycemic rats. Diabetes. 2005;54:3169–3174. [DOI] [PubMed] [Google Scholar]
- 51.McCrimmon RJ, Song Z, Cheng H, et al. Corticotrophin-releasing factor receptors within the ventromedial hypothalamus regulate hypoglycemia- induced hormonal counterregulation. J Clin Invest. 2006;116:1723–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Szepietowska B, Horvath TL, Sherwin RS. Role of synaptic plasticity and EphA5-EphrinA5 interaction within the ventromedial hypothalamus in response to recurrent hypoglycemia. Diabetes. 2014;63:1140–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Szepietowska B, Zhu W, Chan O, Horblitt A, Dziura J, Sherwin RS. Modulation of β-Adrenergic receptors in the ventromedial hypothalamus influences counterregulatory responses to hypoglycemia. Diabetes. 2011;60:3154–3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tong Q, Ye C, McCrimmon RJ, et al. Synaptic glutamate release by ventromedial hypothalamic neurons is part of the neurocircuitry that prevents hypoglycemia. Cell Metab. 2007;5:383–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhu W, Czyzyk D, Paranjape SA, et al. Glucose prevents the fall in ventromedial hypothalamic GABA that is required for full activation of glucose counterregulatory responses during hypoglycemia. Am J Physiol. 2010;298:E971–E977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Diggs-Andrews KA, Zhang X, Song Z, Daphna-Iken D, Routh VH, Fisher SJ. Brain insulin action regulates hypothalamic glucose sensing and the counterregulatory response to hypoglycemia. Diabetes. 2010;59:2271–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fioramonti X, Deak A, Deshpande S, et al. Hypothalamic S-nitrosylation contributes to the counter-regulatory response impairment following recurrent hypoglycemia. Plos One. 2013;8:e68709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reno CM, Puente EC, Sheng Z, et al. Brain GLUT4 knockout mice have impaired glucose tolerance, decreased insulin sensitivity, and impaired hypoglycemic counterregulation. Diabetes. 2017;66:587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Canteras NS, Simerly RB, Swanson LW. Organization of projections from the ventromedial nucleus of the hypothalamus: a phaseolus vulgaris-leucoagglutinin study in the rat. J Comp Neurol. 1994;348:41–79. [DOI] [PubMed] [Google Scholar]
- 60.Lindberg D, Chen P, Li C. Conditional viral tracing reveals that steroidogenic factor 1-positive neurons of the dorsomedial subdivision of the ventromedial hypothalamus project to autonomic centers of the hypothalamus and hindbrain. J Comp Neurol. 2013;521:3167–3190. [DOI] [PubMed] [Google Scholar]
- 61.Routh VH, Donovan CM, Ritter S. 2. Hypoglycemia detection. Transl Endocrinol Metab. 2012;3:47–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bohland M, Matveyenko AV, Saberi M, Khan AM, Watts AG, Donovan CM. Activation of hindbrain neurons is mediated by portal-mesenteric vein glucosensors during slow-onset hypoglycemia. Diabetes. 2014;63:2866–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Donovan CM, Hamilton-Wessler M, Halter JB, Bergman RN. Primacy of liver glucosensors in the sympathetic response to progressive hypoglycemia. Proc Natl Acad Sci USA. 1994;91:2863–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Donovan CM, Watts AG. Peripheral and central glucose sensing in hypoglycemic detection. Physiology. 2014;29:314–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jokiaho AJ, Donovan CM, Watts AG. The rate of fall of blood glucose determines the necessity of forebrain-projecting catecholaminergic neurons for male rat sympathoadrenal responses. Diabetes. 2014;63:2854–2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li A-J, Wang Q, Elsarelli MM, Brown RL, Ritter S. Hindbrain catecholamine neurons activate orexin neurons during systemic glucoprivation in male rats. Endocrinology. 2015;156:2807–2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li A-J, Wang Q, Ritter S. Selective pharmacogenetic activation of catecholamine subgroups in the ventrolateral medulla elicits key glucoregulatory responses. Endocrinology. 2018;159:341–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ritter S, Li A-J, Wang Q, Dinh TT. Minireview: the value of looking backward: the essential role of the hindbrain in counterregulatory responses to glucose deficit. Endocrinology. 2011;152:4019–4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhou C, Teegala SB, Khan BA, Gonzalez C, Routh VH. Hypoglycemia: role of hypothalamic glucose-inhibited (GI) neurons in detection and correction. Front Physiol. 2018;9:192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chakera AJ, Hurst PS, Spyer G, et al. Molecular reductions in glucokinase activity increase counter-regulatory responses to hypoglycemia in mice and humans with diabetes. Mol Metab. 2018;17:17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Levin BE, Becker TC, Eiki J-i, Zhang BB, Dunn-Meynell AA. Ventromedial hypothalamic glucokinase is an important mediator of the counterregulatory response to insulin-induced hypoglycemia. Diabetes. 2008;57:1371–1379. [DOI] [PubMed] [Google Scholar]
- 72.McCrimmon RJ, Fan X, Cheng H, et al. Activation of AMP-activated protein kinase within the ventromedial hypothalamus amplifies counterregulatory hormone responses in rats with defective counterregulation. Diabetes. 2006;55:1755–1760. [DOI] [PubMed] [Google Scholar]
- 73.Faber CL, Matsen ME, Velasco KR, et al. Distinct neuronal projections from the hypothalamic ventromedial nucleus mediate glycemic and behavioral effects. Diabetes. 2018;67:2518–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cryer PE. Hypoglycemia-associated autonomic failure in diabetes. Am J Physiol. 2001;281:E1115–E1121. [DOI] [PubMed] [Google Scholar]
- 75.Cryer PE. Mechanisms of hypoglycemia-associated autonomic failure and its component syndromes in diabetes. Diabetes. 2005;54:3592–3601. [DOI] [PubMed] [Google Scholar]
- 76.Cryer PE. The barrier of hypoglycemia in diabetes. Diabetes. 2008;57:3169–3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fioramonti X, Song Z, Vazirani RP, Beuve A, Routh VH. Hypothalamic NO in hypoglycemia detection and counter-regulation: a two edged sword. Antioxid Redox Signal. 2010;14: 505–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Song Z, Routh VH. Recurrent hypoglycemia reduces the glucose sensitivity of glucose-inhibited neurons in the ventromedial hypothalamus nucleus (VMN). Am J Physiol. 2006;291:R1283–R1287. [DOI] [PubMed] [Google Scholar]
- 79.Zhou C, Routh VH. Thioredoxin-1 overexpression in the ventromedial nucleus of the hypothalamus (VMH) preserves the counterregulatory response to hypoglycemia during type 1 diabetes mellitus in male rats. Diabetes. 2018;67:120–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pocai A, Lam TKT, Gutierrez-Juarez R, et al. Hypothalamic KATP channels control hepatic glucose production. Nature. 2005;434:1026–1031. [DOI] [PubMed] [Google Scholar]
- 81.Pocai A, Obici S, Schwartz GJ, Rossetti L. A brain-liver circuit regulates glucose homeostasis. Cell Metab. 2005;1:53–61. [DOI] [PubMed] [Google Scholar]
- 82.Kishore P, Boucai L, Zhang K, et al. Activation of K(ATP) channels suppresses glucose production in humans. J Clin Invest. 2011;121:4916–4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Song Z, Routh VH. Differential effects of glucose and lactate on glucosensing neurons in the ventromedial hypothalamic nucleus. Diabetes. 2005;54:15–22. [DOI] [PubMed] [Google Scholar]
- 84.Scheurink AJ, Steffens AB, Benthem L. Central and peripheral adrenoceptors affect glucose, free fatty acids, and insulin in exercising rats. Am J Physiol. 1988;255:R547–R556. [DOI] [PubMed] [Google Scholar]
- 85.Fujikawa T, Castorena CM, Lee S, Elmquist JK. The hypothalamic regulation of metabolic adaptations to exercise. J Neuroendocrinol. 2017;29:e12533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Haque MS, Minokoshi Y, Hamai M, Iwai M, Horiuchi M, Shimazu T. Role of the sympathetic nervous system and insulin in enhancing glucose uptake in peripheral tissues after intrahypothalamic injection of leptin in rats. Diabetes. 1999;48:1706–1712. [DOI] [PubMed] [Google Scholar]
- 87.Minokoshi Y, Haque MS, Shimazu T. Microinjection of leptin into the ventromedial hypothalamus increases glucose uptake in peripheral tissues in rats. Diabetes. 1999;48:287–291. [DOI] [PubMed] [Google Scholar]
- 88.Shiuchi T, Haque MS, Okamoto S, et al. Hypothalamic orexin stimulates feeding-associated glucose utilization in skeletal muscle via sympathetic nervous system. Cell Metab. 2009;10:466–480. [DOI] [PubMed] [Google Scholar]
- 89.Shiuchi T, Toda C, Okamoto S, et al. Induction of glucose uptake in skeletal muscle by central leptin is mediated by muscle β2-adrenergic receptor but not by AMPK. Sci Rep. 2017;7:15141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Toda C, Shiuchi T, Kageyama H, et al. Extracellular signal-regulated kinase in the ventromedial hypothalamus mediates leptin-induced glucose uptake in red-type skeletal muscle. Diabetes. 2013;62:2295–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Davis AM, Seney ML, Stallings NR, Zhao L, Parker KL, Tobet SA. Loss of steroidogenic factor 1 alters cellular topography in the mouse ventromedial nucleus of the hypothalamus. J Neurobiol. 2004;60:424–436. [DOI] [PubMed] [Google Scholar]
- 92.Schlosser DA, Parker KL, Luo X, Ikeda Y. Steroidogenic factor 1 is the essential transcript of the mouse Ftz-F1 gene. Mol Endocrinol. 1995;9:1233–1239. [DOI] [PubMed] [Google Scholar]
- 93.Majdic G, Young M, Gomez-Sanchez E, et al. Knockout mice lacking steroidogenic factor 1 are a novel genetic model of hypothalamic obesity. Endocrinology. 2002;143:607–614. [DOI] [PubMed] [Google Scholar]
- 94.Kim KW, Zhao L, Donato J, et al. Steroidogenic factor 1 directs programs regulating diet-induced thermogenesis and leptin action in the ventral medial hypothalamic nucleus. Proc Natl Acad Sci USA. 2011;108:10673–10678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Viskaitis P, Irvine EE, Smith MA, et al. Modulation of SF1 neuron activity coordinately regulates both feeding behavior and associated emotional states. Cell Rep. 2017;21:3559–3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Niijima A, Rohner-Jeanrenaud F, Jeanrenaud B. Role of ventromedial hypothalamus on sympathetic efferents of brown adipose tissue. Am J Physiol. 1984;247:R650–R654. [DOI] [PubMed] [Google Scholar]
- 97.Perkins MNRN, Stock MJ, Stone TW. Activation of brown adipose tissue thermogenesis by the ventromedial hypothalamus. Nature. 1981;289:401–402. [DOI] [PubMed] [Google Scholar]
- 98.Martínez-Sánchez N, Moreno-Navarrete JM, Contreras C, et al. Thyroid hormones induce browning of white fat. J Endocrinol. 2016;232:351–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Martínez-Sánchez N, Seoane-Collazo P, Contreras C, et al. Hypothalamic AMPK-ER stress-JNK1 axis mediates the central actions of thyroid hormones on energy balance. Cell Metab. 2017;26:212–229.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang B, Li A, Li X, et al. Activation of hypothalamic RIP-Cre neurons promotes beiging of WAT via sympathetic nervous system. EMBO Rep. 2018;19:e44977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Seoane-Collazø P, Fernø J, Gonzalez F, et al. Hypothalamic-autonomic control of energy homeostasis. Endocrine. 2015;50:276–291. [DOI] [PubMed] [Google Scholar]
- 102.Seoane-Collazo P, Roa J, Rial-Pensado E, et al. SF1-specific AMPKα1 deletion protects against diet-induced obesity. Diabetes. 2018;67:2213–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Contreras C, González-García I, Martínez-Sánchez N, et al. Central ceramide-induced hypothalamic lipotoxicity and ER stress regulate energy balance. Cell Rep. 2014;9:366–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Contreras C, González-García I, Seoane-Collazo P, et al. Reduction of hypothalamic endoplasmic reticulum stress activates browning of white fat and ameliorates obesity. Diabetes. 2017;66:87–99. [DOI] [PubMed] [Google Scholar]
- 105.Liñares-Pose L, Rial-Pensado E, Estévez-Salguero Á, et al. Genetic targeting of GRP78 in the VMH improves obesity independently of food intake. Genes. 2018;9:357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Musatov S, Chen W, Pfaff DW, et al. Silencing of estrogen receptor α in the ventromedial nucleus of hypothalamus leads to metabolic syndrome. Proc Natl Acad Sci USA. 2007;104:2501–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Xu Y, Nedungadi Thekkethil P, Zhu L, et al. Distinct hypothalamic neurons mediate estrogenic effects on energy homeostasis and reproduction. Cell Metab. 2011;14:453–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cerri M, Morrison SF. Activation of lateral hypothalamic neurons stimulates brown adipose tissue thermogenesis. Neuroscience. 2005;135:627–638. [DOI] [PubMed] [Google Scholar]
- 109.Tupone D, Madden CJ, Cano G, Morrison SF. An orexinergic projection from perifornical hypothalamus to raphe pallidus increases rat brown adipose tissue thermogenesis. J Neurosci. 2011;31:15944–15955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wiedmann NM, Stefanidis A, Oldfield BJ. Characterization of the central neural projections to brown, white, and beige adipose tissue. FASEB J. 2017;31:4879–4890. [DOI] [PubMed] [Google Scholar]
- 111.Santiago AM, Clegg DJ, Routh VH. Ventromedial hypothalamic glucose sensing and glucose homeostasis vary throughout the estrous cycle. Physiol Behav. 2016;167:248–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sheng Z, Santiago AM, Thomas MP, Routh VH. Metabolic regulation of lateral hypothalamic glucose-inhibited orexin neurons may influence midbrain reward neurocircuitry. Mol Cell Neurosci. 2014;62:30–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yamanaka A, Beuckmann CT, Willie JT, et al. Hypothalamic orexin neurons regulate arousal according to energy balance in mice. Neuron. 2003;38:701–713. [DOI] [PubMed] [Google Scholar]
- 114.Gonzalez JA, Jensen LT, Fugger L, Burdakov D. Metabolism-independent sugar sensing in central orexin neurons. Diabetes. 2008;57:2569–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.McComb RB, Yushok WD. Metabolism of ascites tumor cells. IV enzymatic reactions involved in adenosinetriphosphate degradation induced by 2-deoxyglucose. Cancer Res. 1964;24:198–205. [PubMed] [Google Scholar]
- 116.Burdakov D, Jensen LT, Alexopoulos H, et al. Tandem-pore K+ channels mediate inhibition of orexin neurons by glucose. Neuron. 2006;50:711–722. [DOI] [PubMed] [Google Scholar]
- 117.González JA, Jensen LT, Doyle SE, et al. Deletion of TASK1 and TASK3 channels disrupts intrinsic excitability but does not abolish glucose or pH responses of orexin/hypocretin neurons. Eur J Neurosci. 2009;30:57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ren X, Zhou L, Terwilliger R, Newton S, De Araujo IE. Sweet taste signaling functions as a hypothalamic glucose sensor. Front Integr Neurosci. 2009;3:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Otlivanchik O, Le Foll C, Levin BE. Perifornical hypothalamic orexin and serotonin modulate the counterregulatory response to hypoglycemic and glucoprivic stimuli. Diabetes. 2015;64:226–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Korim WS, Bou Farah L, McMullan S, Verberne AJM. Orexinergic activation of medullary premotor neurons modulates the adrenal sympathoexcitation to hypothalamic glucoprivation. Diabetes. 2014;63:1895–1906. [DOI] [PubMed] [Google Scholar]
- 121.Korim WS, Llewellyn-Smith IJ, Verberne AJM. Activation of medulla-projecting perifornical neurons modulates the adrenal sympathetic response to hypoglycemia: involvement of orexin type 2 (OX2-R) receptors. Endocrinology. 2016;157:810–819. [DOI] [PubMed] [Google Scholar]
- 122.Otlivanchik O, Sanders NM, Dunn-Meynell A, Levin BE. Orexin signaling is necessary for hypoglycemia-induced prevention of conditioned place preference. Am J Physiol. 2016;310:R66–R73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dagogo-Jack S, Rattarasarn C, Cryer PE. Reversal of hypoglycemia unawareness, but not defective glucose counterregulation, in IDDM. Diabetes. 1994;43:1426–1434. [DOI] [PubMed] [Google Scholar]
- 124.Fanelli CG, Epifano L, Rambotti AM, et al. Meticulous prevention of hypoglycemia normalizes the glycemic thresholds and magnitude of most of neuroendocrine responses to, symptoms of, and cognitive function during hypoglycemia in intensively treated patients with short-term IDDM. Diabetes. 1993;42:1683–1689. [DOI] [PubMed] [Google Scholar]
- 125.Gulanski BI, Feyter HMD, Page KA, et al. Increased brain transport and metabolism of acetate in hypoglycemia unawareness. J Clin Endocrinol Metab. 2013;98:3811–3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Martín-Timón I, del Cañizo-Gómez FJ. Mechanisms of hypoglycemia unawareness and implications in diabetic patients. World J Diabet. 2015;6:912–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Eriksson KS, Sergeeva O, Brown RE, Haas HL. Orexin/hypocretin excites the histaminergic neurons of the tuberomammillary nucleus. J Neurosci. 2001;21:9273–9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Eriksson KS, Sergeeva OA, Selbach O, Haas HL. Orexin (hypocretin)/dynorphin neurons control GABAergic inputs to tuberomammillary neurons. Eur J Neurosci. 2004;19:1278–1284. [DOI] [PubMed] [Google Scholar]
- 129.Miklós IH, Kovács KJ. Functional heterogeneity of the responses of histaminergic neuron subpopulations to various stress challenges. Eur J Neurosci. 2003;18:3069–3079. [DOI] [PubMed] [Google Scholar]
- 130.Mochizuki T, Arrigoni E, Marcus JN, et al. Orexin receptor 2 expression in the posterior hypothalamus rescues sleepiness in narcoleptic mice. Proc Natl Acad Sci USA. 2011;108:4471–4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Torrealba F, Yanagisawa M, Saper CB. Colocalization of orexin a and glutamate immunoreactivity in axon terminals in the tuberomammillary nucleus in rats. Neuroscience. 2003;119:1033–1044. [DOI] [PubMed] [Google Scholar]
- 132.Wang Y-Q, Li R, Wu X, et al. Fasting activated histaminergic neurons and enhanced arousal effect of caffeine in mice. Pharmacol Biochem Behav. 2015;133:164–173. [DOI] [PubMed] [Google Scholar]
- 133.Williams RH, Chee MJS, Kroeger D, et al. Optogenetic-mediated release of histamine reveals distal and autoregulatory mechanisms for controlling arousal. J Neurosci. 2014;34:6023–6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Aston-Jones G, Smith RJ, Sartor GC, et al. Lateral hypothalamic orexin/hypocretin neurons: a role in reward-seeking and addiction. Brain Res. 2010;1314:74–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Cason A, Aston-Jones G. Attenuation of saccharin-seeking in rats by orexin/hypocretin receptor 1 antagonist. Psychopharmacology. 2013;228:499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cason A, Aston-Jones G. Role of orexin/hypocretin in conditioned sucrose-seeking in rats. Psychopharmacology. 2013;226:155–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Cason AM, Smith RJ, Tahsili-Fahadan P, Moorman DE, Sartor GC, Aston-Jones G. Role of orexin/hypocretin in reward-seeking and addiction: implications for obesity. Physiol Behav. 2010;100:419–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Baimel C, Borgland SL. Orexin signaling in the VTA gates morphine-induced synaptic plasticity. J Neurosci. 2015;35: 7295–7303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Borgland SL, Taha SA, Sarti F, Fields HL, Bonci A. Orexin A in the VTA is critical for the induction of synaptic plasticity and behavioral sensitization to cocaine. Neuron. 2006;49:589–601. [DOI] [PubMed] [Google Scholar]
- 140.Cao J-L, Covington HE III, Friedman AK, et al. Mesolimbic dopamine neurons in the brain reward circuit mediate susceptibility to social defeat and antidepressant action. J Neurosci. 2010;30:16453–16458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Dela Cruz JAD, Coke T, Bodnar RJ. Simultaneous detection of c-fos activation from mesolimbic and mesocortical dopamine reward sites following naive sugar and fat ingestion in rats. J Vis Exp. 2016;114:53897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Fields HL, Hjelmstad GO, Margolis EB, Nicola SM. Ventral tegmental area neurons in learned appetitive behavior and positive reinforcement. Annu Rev Neurosci. 2007;30:289–316. [DOI] [PubMed] [Google Scholar]
- 143.Liu S, Globa AK, Mills F, et al. Consumption of palatable food primes food approach behavior by rapidly increasing synaptic density in the VTA. Proc Natl Acad Sci USA. 2016;113:2520–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mahler SVVE, Beckley JT, Keistler CR, et al. Designer receptors show role for ventral pallidum input to ventral tegmental area in cocaine seeking. Nat Neurosci. 2014;4:577–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mansvelder HD, McGehee DS. Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron. 2000;27:349–357. [DOI] [PubMed] [Google Scholar]
- 146.Skibicka KP, Hansson C, Alvarez-Crespo M, Friberg PA, Dickson SL. Ghrelin directly targets the ventral tegmental area to increase food motivation. Neuroscience. 2011;180:129–137. [DOI] [PubMed] [Google Scholar]
- 147.Tsai H-C, Zhang F, Adamantidis A, et al. Phasic firing in dopaminergic neurons is sufficient for behavioral conditioning. Science. 2009;324:1080–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Balcita-Pedicino JJ, Sesack SR. Orexin axons in the rat ventral tegmental area synapse infrequently onto dopamine and γ-aminobutyric acid neurons. J Comp Neurol. 2007;503:668–684. [DOI] [PubMed] [Google Scholar]
- 149.Bonci A, Borgland S. Role of orexin/hypocretin and CRF in the formation of drug-dependent synaptic plasticity in the mesolimbic system. Neuropharmacology. 2009;56(Suppl 1):107–111. [DOI] [PubMed] [Google Scholar]
- 150.Teegala SB, Sheng Z, Dalal MS, Hirschberg PR, Beck KD, Routh VH. Lateral hypothalamic orexin glucose-inhibited neurons may regulate reward-based feeding by modulating glutamate transmission in the ventral tegmental area. Brain Res. 2018. May 19 pii: S0006–8993(18)30282–8. 10.1016/j.brainres.2018.05.025. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]