

Abstract
Opioids target the μ-opioid receptor (MOR) to produce unrivaled pain management but their addictive properties can lead to severe abuse. We developed a whole animal behavioral platform for unbiased discovery of genes influencing opioid responsiveness. Using forward genetics in C. elegans, we identified a conserved orphan receptor, GPR139, with anti-opioid activity. GPR139 is coexpressed with MOR in opioid-sensitive brain circuits, binds to MOR and inhibits signaling to G proteins. Deletion of GPR139 in mice enhanced opioid-induced inhibition of neuronal firing to modulate morphine-induced analgesia, reward, and withdrawal. Thus, GPR139 could be a useful target for increasing opioid safety. These results also demonstrate the potential of C. elegans as a scalable platform for genetic discovery of GPCR signaling principles.
G protein coupled receptors (GPCRs) constitute the largest class of cell surface receptors and mediate sensory perception and cellular communication via hormones and neurotransmitters (1, 2). GPCRs function in various diseases and are prominent drug targets (3–5). There has been tremendous progress in understanding the molecular mechanisms of GPCR signaling stemming from identification of key components including G protein subunits, β-arrestins, downstream effectors, and regulatory proteins (6, 7). The majority of these components were discovered serendipitously leaving many critical questions about GPCR organization and function open. For one, many receptors are considered ‘orphan’ with poorly understood biology and unclear roles in cellular signaling (8, 9). Mechanisms that generate diverse physiological effects are not fully understood. Finally, how individual GPCRs adjust signaling in response to changes in the environment or circuit activity remains unclear.
Insufficient understanding of GPCR signaling hampers their targeting by drugs in a safe and effective manner. This is well illustrated by opioid analgesics that act on the μ-opioid receptor (MOR) and offer unsurpassed efficacy for pain management (10, 11). However, opioid drugs have substantial liabilities including dependence, tolerance and somatic side effects (12). Extensive investigation of MOR pharmacology led to the concept that activated MOR triggers distinct signaling events that differentially control various physiological reactions (13). As a result, identification of molecules that control MOR signaling in endogenous neural circuits remains critical, and could provide new pharmacological targets for increasing the efficacy and safety of opioid analgesics.
Opioid stimulation of MOR affects the nervous system and produces effects that are inherently behavioral in nature (14–16). Thus, screens for modulators of opioid signaling that use behavior as an ultimate readout could accelerate the relevance and translatability of discoveries. Fortunately, GPCR signaling is highly conserved and has been studied across mammalian and invertebrate model systems (17–19). Genetic studies in C. elegans have allowed discovery and evaluation of many conserved players in GPCR signaling, and elucidated their roles in neural circuits (19, 20). Furthermore, transgenic expression of mammalian GPCRs alters the behavior of C. elegans, and these heterologous GPCRs desensitize in response to ligands (21). C. elegans also has an opioid-like system that controls feeding behavior and responses to noxious stimuli (22, 23). These considerations prompted us to develop a transgenic C. elegans platform we used in an unbiased, forward genetic screen for regulators of MOR-controlled behavior.
Development of C. elegans platform for unbiased genetic discovery of opioid modulators
To study MOR signaling using a behavioral platform that can be scaled to cover an entire genome, we generated transgenic C. elegans expressing mammalian MOR throughout the nervous system (tgMOR, Fig. 1A, B). Because opioid agonists exert effects on motor activity in mammals, we assessed the effects of MOR activation on C. elegans locomotion. Exposure of tgMOR worms to fentanyl, a MOR agonist, reduced their movement (Fig. 1C; Suppl. Movies 1–10). Quantitation showed fentanyl inhibited thrashing of tgMOR animals over time (Fig. 1D). TgMOR animals rapidly recovered from paralysis in the presence of fentanyl indicating conservation of receptor desensitization mechanisms (Fig. 1D). Fentanyl did not affect non-transgenic wt animals, indicating that changes in motor activity result from activation of transgenic MOR (Fig. 1C and D; Suppl. Movies 1–10).
Fig. 1. Transgenic C. elegans platform for dissecting opioid signaling mechanisms.
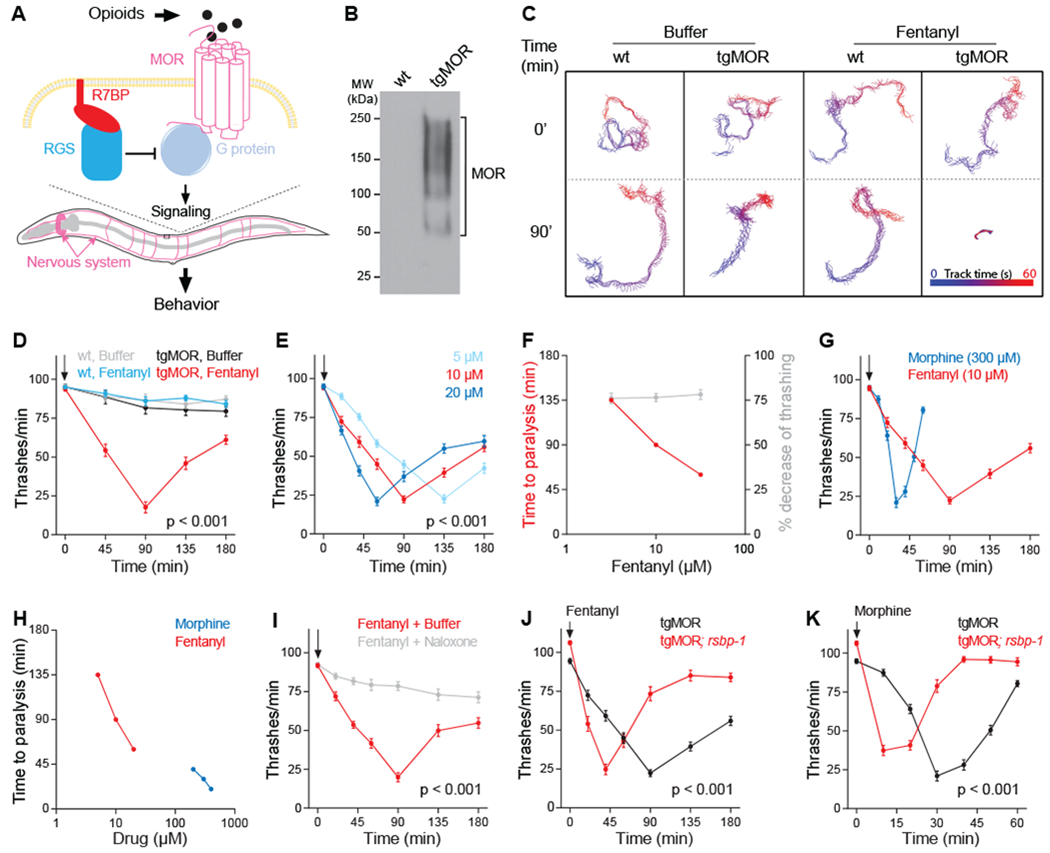
(A) Transgenic C. elegans model of MOR signaling (tgMOR). (B) Western blot showing expression of FLAG::MOR in the nervous system after immunoprecipitation. (C) Fentanyl inhibits thrashing of tgMOR. (D) Quantitation of fentanyl effects on tgMOR. (E) Time course of fentanyl doses on tgMOR. (F) Fentanyl dose response for tgMOR. (G) Time course for morphine and fentanyl on tgMOR. (H) Morphine and fentanyl dose response for tgMOR. (I) Naloxone blocks fentanyl effects on tgMOR. (J, K) Time courses showing tgMOR; rsbp-1 mutants are hypersensitive to (J) fentanyl and (K) morphine. Arrows denote drug application. If not indicated, opioids were used at the following concentrations: fentanyl (10μM), morphine (300μM) and naloxone (20μM). For all genotypes and drug conditions, means are shown from 30 or more animals obtained from three independent experiments. Error bars are S.E.M. Significance tested using two-way ANOVA. P values reported are for genotype/time interactions.
Higher concentrations of fentanyl accelerated response onset and subsequent recovery but the duration of paralysis was not affected (Fig. 1E and F; Fig. S1A and C). Similar behavior was also observed with morphine, a full MOR agonist with distinct pharmacological properties (Fig S1D and F) (24, 25). Consistent with morphine having lower potency on MOR (25), approximately 50-fold higher concentration of morphine was required for maximal effect compared to fentanyl (Fig. 1G and H). Although morphine produced a similar magnitude of effect as fentanyl, it had a distinct temporal profile with faster onset and more rapid recovery (Fig. 1G; Fig. S1D). Treatment of tgMOR animals with naloxone, a MOR antagonist, abolished the effect of fentanyl (Fig. 1I). Thus, our tgMOR platform can rapidly evaluate behavioral effects of opioids, and distinguish pharmacological effects and properties of different drugs.
To probe whether conserved molecular mechanisms control opioid signaling, we evaluated opioid-induced behavior in tgMOR animals lacking R7 Binding Protein 1 (RSBP-1). RSBP-1 is orthologous to mammalian Regulator of G protein signaling 7 Binding Protein (R7BP), a subunit of the complex that negatively regulates MOR signaling in mice (Fig. 1A) (26, 27). rsbp-1 loss-of-function mutants carrying tgMOR reached maximum paralysis and recovered more quickly than tgMOR animals treated with fentanyl (Fig. 1J; Fig. S1B) or morphine (Fig. 1K; Fig. S1E). Dose-response studies with tgMOR; rsbp-1 mutants showed a prominent left-ward shift in concentration dependence for both fentanyl and morphine (Fig. S1C and F). Thus, tgMOR; rsbp-1 mutants are hypersensitive to opioids, an outcome similar to R7BP deletion in mice (27). Taken together, these observations indicate that opioid signaling via MOR can be effectively modeled in C. elegans producing behavioral reactions mediated by conserved GPCR signaling machinery that functions independent of organism-specific neuronal circuitry.
Forward genetic screen identifies genes affecting behavioral sensitivity to opioids
The effects of opioids on tgMOR C. elegans and the molecular conservation of regulatory mechanisms prompted us to adopt this platform for an unbiased, forward genetic screen for regulators of opioid signaling (Fig 2A). We focused on identifying mutants with increased opioid sensitivity to uncover negative regulators of MOR signaling.
Fig. 2. Forward genetic screen with tgMOR platform identifies orphan receptor FRPR-13 as negative regulator of MOR signaling.
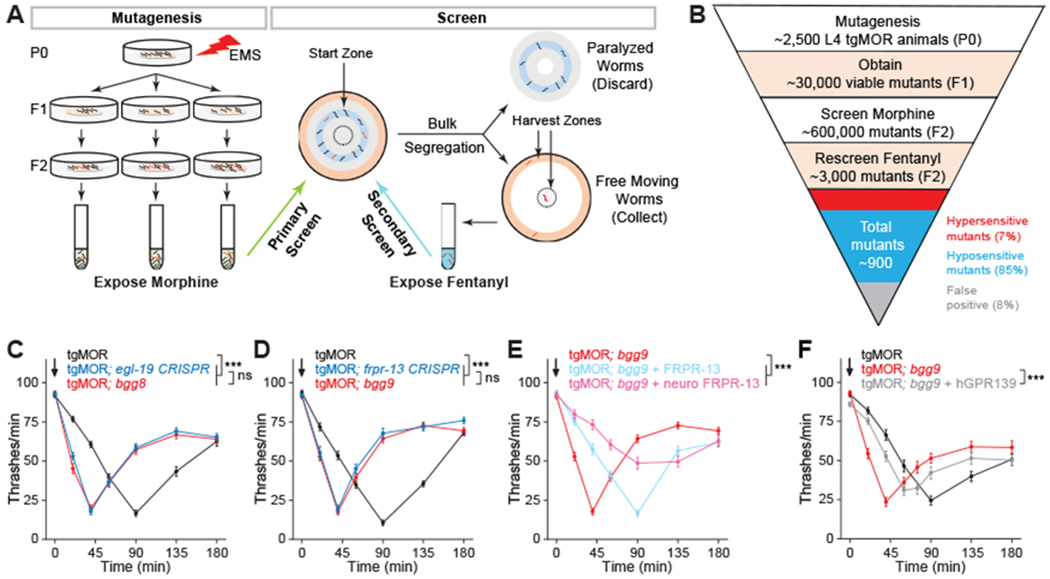
(A) Two-step genetic screen for tgMOR mutants with altered opioid sensitivity. (B) Outline of steps, generations, number of independent mutants isolated, and phenotypic categories observed for genetic screen with tgMOR. (C) tgMOR; bgg8 mutants are hypersensitive to fentanyl and CRISPR/Cas9 editing validates egl-19 as gene causing hypersensitivity. (D) tgMOR; bgg9 mutants are hypersensitive to fentanyl and CRISPR/Cas9 editing validates frpr-13 as gene causing hypersensitivity. (E) Transgenic expression of FRPR-13 using native or neuronal promoters reverses fentanyl hypersensitivity in tgMOR; bgg9 animals. (F) Transgenic expression of human GPR139 reverses fentanyl hypersensitivity in tgMOR; bgg9 animals. Arrows denote fentanyl (10μM) application. For all genotypes and drug conditions, means are shown from 30 or more animals obtained from three independent experiments. Error bars are S.E.M. Significance tested using two-way ANOVA. *** p<0.001 and ns = not significant
Key to the design of our screen was the observation that greater opioid response leads to faster paralysis and more rapid recovery. Thus, hypersensitive animals like tgMOR; rsbp-1 recover faster from the same drug dose than do tgMOR animals (Fig. 1J and K). As a result, bulk segregation on plates was used to isolate hypersensitive mutants based on their quicker recovery from opioid-induced paralysis and escape from the starting zone (Fig. 2A). Assay optimization with a mixture of tgMOR animals and hypersensitive tgMOR; rsbp-1 mutants showed that primary screening with morphine followed by secondary screening with fentanyl minimized false positive rates (Fig. 2A).
For the full-scale screen, we mutagenized ~2,500 tgMOR animals, evaluated ~600,000 progeny, and identified ~900 mutants with abnormal sensitivity to both morphine and fentanyl (Fig. 2B). Secondary evaluation in liquid thrashing assays with fentanyl eliminated false positives, identified mutants that lost opioid sensitivity, and confirmed a small number of hypersensitive mutants (Fig. 2B). We focused our efforts on comprehensive testing of opioid-induced behaviors for two mutants, tgMOR; bgg8 and tgMOR; bgg9. Both mutants had normal overall morphology and behavior in the opioid naïve state, but were paralyzed by fentanyl significantly faster than tgMOR worms (Fig. 2C and D). Additional dose-response studies showed a leftward shift in fentanyl-induced paralysis indicating that tgMOR; bgg8 and tgMOR; bgg9 mutants are hypersensitive to opioids (Fig. S2).
We mapped genetic lesions causing hypersensitivity by combining whole-genome sequencing with phenotypic selection (Fig. S3A). This process identified genomic regions of interest (3-5 Mb) that contained approximately 6 to 8 different lesions per mutant. To determine which lesion caused opioid hypersensitivity, we used CRISPR/Cas9 to edit single mutations into candidate genes of tgMOR animals (Fig. S3A).
For tgMOR; bgg8 animals, we identified a lesion in the calcium channel egl-19 that introduced a premature stop codon and likely resulted in loss of function (Fig. S3B). CRISPR/Cas9 editing the same egl-19 mutation into parental tgMOR animals confirmed egl-19 affects opioid sensitivity (Fig. 2C; Fig. S4A, B). Notably, egl-19 is homologous to L-type Ca2+ channels in mammals and extensive evidence indicates that L-type Ca2+ blockers potentiate the nociceptive properties of opioids in a clinical setting (28, 29). These observations demonstrate that our forward genetic screen identified conserved regulators of MOR signaling.
Another hypersensitive mutant, tgMOR; bgg9, contained a premature stop in frpr-13 which encodes an unstudied orphan GPCR (Fig. S3C). CRISPR/Cas9 editing of this lesion into tgMOR increased sensitivity to fentanyl, confirming that frpr-13 affects opioid sensitivity (Fig. 2D; Fig. S4C and D). Because the function of FRPR-13 is unknown, we further validated that it regulates opioid responses by transgenically expressing FRPR-13 in tgMOR; bgg9 mutants (Fig S3A). FRPR-13 expressed using the native frpr-13 promoter and Mos single copy insertion (MosSCI) reversed the hypersensitivity of frpr-13 (bgg8) mutants back to a normal response (Fig. 2E; Fig. S4E and F). Similarly, hypersensitivity of frpr-13 (bgg8) mutants was reversed when FRPR-13 was pan-neuronally expressed with MosSCI (Fig. 2E). Collectively, these results indicate that the FRPR-13 receptor alters sensitivity to opioids at a behavioral level.
FRPR-13/GPR139 negatively regulates MOR signaling.
Phylogenetic analysis revealed that FRPR-13 belongs to a large neuropeptide receptor group in C. elegans that is similar to two mammalian orphan GPCRs, GPR139 and GPR142 (Fig. S5). GPR139 and GPR142 are in a distinct subfamily of class A orphan receptors (30). Given that nothing is known about FRPR-13 and there is no prior connection between GPR139/142 and opioid signaling, we explored the functional conservation of these receptors. We focused on GPR139 because it is expressed in the central nervous system, whereas GPR142 is predominantly found in the periphery (31, 32). Transgenic expression of human GPR139 in tgMOR; bgg9 worms with disrupted FRPR-13 significantly reversed hypersensitivity to fentanyl (Fig. 2F). This indicates GPR139 is a functional ortholog of FRPR-13, and GPR139 can inhibit MOR signaling in vivo.
We used a panel of assays to evaluate how GPR139 influences MOR signaling in mammalian Human Embryonic Kidney (HEK) 293T cells. MOR activation drove rapid hyperpolarization of membrane potential upon reconstitution with the G protein-gated Inwardly Rectifying K+ (GIRK) channel (Fig. 3A and B). Introduction of GPR139 cDNA in equivalent concentrations to MOR inhibited morphine-induced hyperpolarization, whereas overexpression of GPR139 in high amounts nearly abolished GIRK activation (Fig. 3B and C). GPR139 co-immunoprecipitated with MOR indicating that these receptors can interact in a model cellular environment (Fig. 3D; Fig. S6A). The relevance of this interaction in an endogenous context remains to be established. We detected significant reduction of MOR at the cell surface when GPR139 was expressed at high levels suggesting that GPR139 can impede MOR trafficking (Fig. 3E and F). Yet, at stoichiometric levels, GPR139 had no effect on surface localization of MOR suggesting that GPR139 has other mechanisms to inhibit MOR (Fig. 3F). Indeed, at these lower stoichiometric levels GPR139 promoted association of the signaling inhibitor β-arrestin with MOR (Fig. 3G through I). This suggests that GPR139 has some constitutive activity that is sufficient to trigger β-arrestin recruitment. To further understand the implications of GPR139-MOR heteromerization and ensuing increased β-arrestin recruitment, we tested how GPR139 influenced MOR-mediated activation of G proteins with a Bioluminescence Resonance Energy Transfer (BRET) assay (Fig. 3J) (33). Morphine produced a rapid BRET response reflecting rearrangement in Gαo-Gβγ heterotrimers induced by MOR activation (Fig. 3K and L). Coexpression of GPR139 at low amounts inhibited MOR-induced G protein activation (Fig. 3K and L; Fig. S6B). This inhibitory effect was more pronounced if GPR139 was expressed at high amounts due to additional loss of MOR from the surface. Together, these results indicate that GPR139 can exert inhibitory effects on MOR in a cell-autonomous manner by affecting both receptor trafficking and signaling properties.
Fig. 3. GPR139 inhibits MOR signaling.
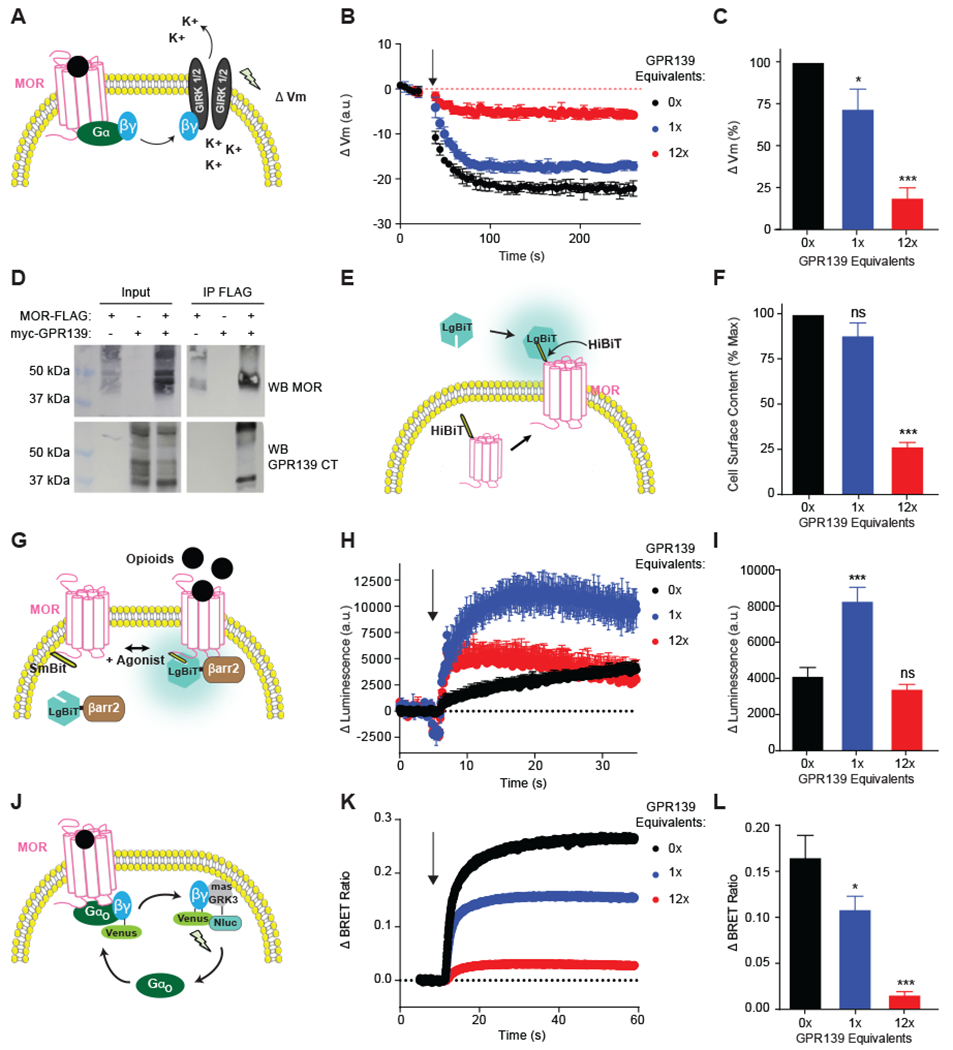
(A) Experimental design for evaluating MOR signaling via its effector GIRK. MOR activation leads to Gβγ subunit release, which opens GIRK channels to produce membrane hyperpolarization (Vm) that is measured with voltage sensitive dye. (B) Coexpression of GPR139 inhibits MOR-mediated kinetics of membrane potential change in response to morphine (0.1 μM). (C) Quantification shows GPR139 reduces morphine effects on Vm amplitude. (D) Co-immunoprecipitation of MOR-FLAG and myc-GPR139 following their coexpression. (E) Experimental design for evaluating cell surface abundance of MOR. HiBiT-tagged MOR complements the LargeBiT (LgBiT) nanoluciferase enzyme only at the plasma membrane. (F) Quantification of the maximal cell surface content of HiBiT-MOR indicates that GPR139 inhibits MOR surface localization only at high (12X) expression levels. (G) Experimental design for evaluating agonist-induced β-arrestin recruitment to MOR. Recruitment of β-arrestin2-LgBiT to SmBiT-MOR generates a functional nanoluciferase enzyme. (H) Effect of GPR139 coexpression on the kinetics of β-arrestin2-LgBiT recruitment induced by DAMGO (10 μM). (I) Quantification shows that low level GPR139 coexpression increases the extent of β-arrestin2 recruitment to MOR. (J) Experimental design for evaluating MOR signaling to G proteins by BRET assay that monitors MOR-mediated release of Gβγ subunits. (K) Effect of GPR139 coexpression on the kinetics of G protein activation by MOR in response to morphine (1 μM) application. (L) Quantification shows GPR139 coexpression reduces maximal BRET response of MOR-Gαo signaling. All experiments were performed in HEK293T cells. In all panels, means are shown from 3-5 independent experiments with 3-4 replicates each ± S.E.M. Significance tested by one-way ANOVA with Dunnett’s post-hoc test. *** p<0.001, ** p<0.01, * p<0.05. Arrows denote application of opioids.
To probe the physiological relevance of inhibitory influences of GPR139 on opioid signaling in the mammalian nervous system, we used mouse models. GPR139 was expressed in brain regions implicated in opioid actions on reward, analgesia and withdrawal (Fig. 4A and E; Fig. S7) (31, 32). GPR139 was extensively coexpressed with MOR in a number of neuronal populations in these areas, most prominently in medial habenula (MHb) and locus coeruleus (LC) (Fig. 4A and E; Fig. S7; Table S1). To test the role of GPR139 on opioid modulation we obtained Gpr139 knockout mice (Gpr139−/−, Fig. S8). We performed patch clamp recordings of MHb neurons in brain slices with drugs that block synaptic communication and circuit activity. In slices from Gpr139+/+ animals, MOR activation resulted in dose-dependent inhibition of spontaneous firing (Fig. 4B and C). Firing of MHb neurons from Gpr139−/− mice was significantly reduced by low-level MOR activation that did not cause an effect in Gpr139+/+ neurons (Fig. 4B and C). Furthermore, Gpr139−/− neurons showed more pronounced net inhibition by DAMGO, a synthetic enkephalin-mimetic peptide (Fig. 4D; Fig. S9A and B). Recovery upon drug washout was delayed in Gpr139−/− neurons which indicates greater susceptibility to opioid inhibition (Fig. S9C). Hypersensitivity to morphine-induced inhibition of firing in LC neurons also occurred in Gpr139−/− mice (Fig. 4F and G). GPR139 ablation resulted in increased basal firing rates selectively in LC but not MHb neurons (Fig. S9D). Taken as a whole, these findings indicate that GPR139 counteracts MOR cell-autonomously in endogenous physiologically relevant neuronal settings as well as reconstituted systems.
Fig. 4. GPR139 inhibits opioid modulation of neuronal firing.
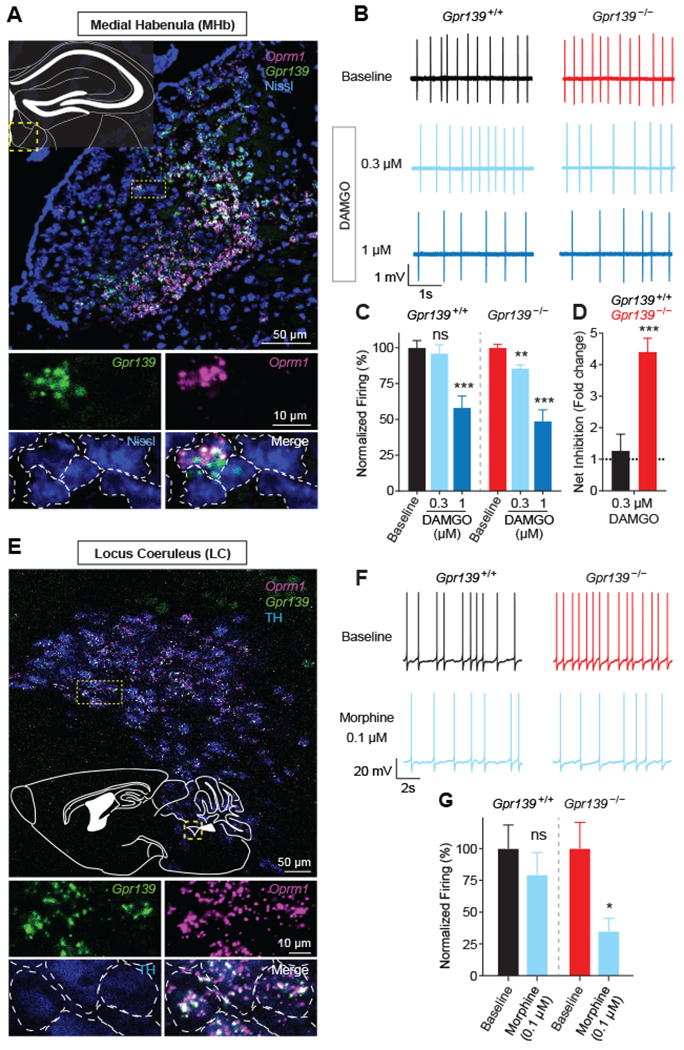
(A) In situ hybridization showing extensive coexpression of MOR mRNA (Oprm1) and Gpr139 in medial habenula (MHb) neurons. (B) Representative traces showing changes in MHb neuron firing in response to different doses of DAMGO in Gpr139+/+ and Gpr139−/− mice. (C) Quantification of normalized firing frequency in MHb neurons shows responsiveness to low DAMGO concentration (0.3μM) in Gpr139−/− but not Gpr139+/+ (n = 11 cells from 6 mice per genotype). (D) Quantification shows MHb neurons from Gpr139−/− animals have increased net inhibition of neuronal firing following DAMGO treatment. (E) In situ hybridization showing Oprm1 and Gpr139 coexpression in locus coeruleus (LC) neurons. (F) Representative traces showing changes in LC neuron firing in response to morphine in Gpr139+/+ and Gpr139−/− mice. (G) Quantification indicates morphine inhibits firing of LC neurons from Gpr139−/− mice but not Gpr139+/+ animals (n = 7-9 cells from 4-6 mice per genotype). All results were reported as mean ± SEM. Significance tested using unpaired Students’ t test. *p < 0.05; **p < 0.01; ***p < 0.001, ns = not significant
GPR139 modulates behavioral responses to opioids
To understand how GPR139 influences opioid actions in vivo, we evaluated mouse behavior. Deletion of GPR139 had no overt effects on animal health and body composition (Fig. S10A–C). Gpr139−/− mice also had normal baseline learning (Fig. 5A), nociception (Fig. 5B), locomotor activity (Fig. S10D), habituation to a novel environment (Fig. S10E) and motor coordination (Fig S10F). However, responses of Gpr139−/− mice to morphine were increased. When tested in a conditioned place preference (CPP) paradigm, Gpr139−/− mice showed augmented responses to the rewarding effects of morphine (Fig. 5A) in agreement with increased opioid sensitivity of Gpr139−/− MHb neurons (Fig. 4B through D), a region involved in drug reward (34). Similarly, Gpr139−/− mice exhibited significantly increased morphine analgesia in thermal (Fig. 5B and C; Fig. S11A) and mechanical (Fig. S11B) pain paradigms. This augmentation was evident from increases in both maximal response and effect duration across multiple morphine doses (Fig. 5B and C; S11A and B). Thus, deletion of GPR139 broadly increases sensitivity to the acute effects of morphine. Termination of chronic morphine administration caused lower somatic withdrawal in mice lacking GPR139 across a spectrum of measures (Fig. 5D; Fig. S12). The diminished withdrawal observed in Gpr139−/− mice may be related to observed changes in baseline firing rate seen in Gpr139−/− LC neurons (Fig. 4F; Fig. S9D), a neuronal population involved in opioid withdrawal (35).
Fig. 5. GPR139 controls behavioral sensitivity of mice to opioid administration.
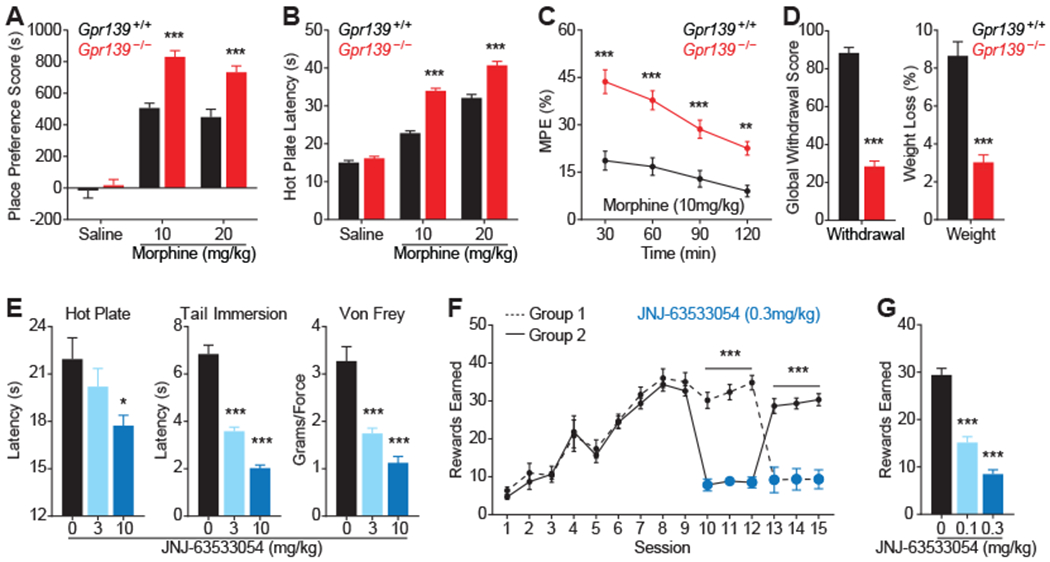
(A) Conditioned place preference paradigm showing increased reward in Gpr139−/− mice. (B) Hot plate assay showing increased dose-dependent, anti-nociceptive effects of morphine in Gpr139−/− mice. (C) Gpr139−/− animals have increased duration of morphine analgesia in hot plate assay. (D) Gpr139−/− mice have decreased behavioral responses and weight loss to naloxone-precipitated somatic withdrawal following chronic morphine exposure. Global score reflects aggregate measure of several withdrawal signs (diarrhea, jumps, dog shakes, paw tremor, back walking, tremor and ptosis). (E) Augmentation of GPR139 function by JNJ63533054 decreases analgesia induced by morphine (10 mg/kg) across pain models. (F) Activation of GPR139 by JNJ63533054 inhibits morphine intake (0.3 mg/kg/infusion) in self-administration task. (G) Quantification of JNJ63533054 effects on morphine self-administration. Significance tested using two-way ANOVA or Student’s t-test. Animal numbers for each test provided in Methods. *** p<0.001, * p<0.05
To test the translational relevance of our findings, we examined the effects of JNJ-63533054, a surrogate ligand that facilitates GPR139 actions (32). Administration of JNJ-63533054 dose-dependently diminished morphine analgesia in both thermal and mechanical pain paradigms (Fig 5E). These effects were not observed in Gpr139−/− mice, indicating specificity of JNJ-63533054 actions (Fig. S13). To determine if activating GPR139 affects reward, we examined the effects of JNJ-63533054 in a morphine self-administration paradigm. Following escalation of morphine intake, wild-type mice were divided into 2 groups with alternating exposure to JNJ-63533054. Administration of drug suppressed morphine intake (Fig. 5F; Fig. S14). The effect of JNJ-63533054 was dose-dependent and completely reversible upon cessation of exposure (Fig. 5F; Fig. S14). Overall, these in vivo results indicate that GPR139 negatively regulates a number of responses to acute opioid exposure, and potentiates withdrawal from chronic opioid administration.
Discussion
We developed a C. elegans behavioral platform for the unbiased genetic discovery of GPCR signaling modulators. Transgenic GPCR expression endows animals with the ability to respond to a foreign chemical modality, akin to chemogenetic approaches used to interrogate mammalian circuitry and behavior (36). The platform displays cardinal features of behavioral responses to receptor activation, allowing phenotypic interrogation of signaling pathways using intact neuronal circuitry in vivo. Use of behavior as an ultimate readout provides a high degree of relevance and potential translational validity. Characterization of tgMOR C. elegans revealed cross-species conservation of critical GPCR signaling elements. This transgenic platform also demonstrated utility in uncovering biology for an opioid receptor signaling network. The scalable nature of our screens may permit further exploration of signaling mechanisms for GPCRs of interest. Additionally, this approach could be adapted for different neuronal circuitry, behavioral readouts, and other GPCRs thereby expanding potential opportunities for discovery.
Using forward genetic screening we identified an evolutionarily conserved orphan receptor system with anti-opioid activity: FRPR-13 in C. elegans and its mammalian ortholog GPR139. Although the full spectrum of GPR139 effects on cellular physiology and mechanisms of suppressing MOR action remain to be elucidated, our examination indicates that some of these actions involve direct inhibitory influences of GPR139 on MOR signaling. Opposing crosstalk between GPCRs is an intriguing concept (37), and our study now adds the poorly understood GPR139 orphan receptor to a growing realm of molecules that oppose MOR (38–40). Notably, α-Melanocyte Stimulating Hormone (a peptide derived from the same precursor as the MOR ligand, β-endorphin) was reported as one endogenous ligand for GPR139 (41). This further argues for the physiological significance of the GPR139-MOR connection and indicates that GPR139 might affect homeostatic control of the endogenous opioid signaling system. Whether GPR139 modulates endogenous opioid function remains to be determined.
Our results suggest that GPR139 could potentially be exploited pharmacologically for increasing safety and efficacy of opioid pharmacotherapy. While our study focused on the anti-opioid effects of GPR139, its widespread expression in the nervous system may indicate that this orphan receptor has additional roles in shaping neuronal physiology independent of MOR.
Materials and Methods
For detailed description of all procedures and methods refer to Supplementary Materials.
Supplementary Material
Acknowledgments
We wish to thank Ms. Natalia Martemyanova for producing and maintaining mice examined in this study and members of the Grill and Martemyanov labs for valuable input. We thank Drs. Thomas Bannister and Xiaohong Pan for providing JNJ-63533054, Dr. Brian Ackley for helpful discussions and Max Planck Florida Institutes’ microscopy core facility. Knockout mice were made by the KOMP Repository (www.komp.org) at the University of California Davis (U42RR024244).
Funding: This work was supported by an NIH Cutting Edge Basic Research Award (R21DA040406) to Drs. Grill and Martemyanov, DA036596 to Dr. Martemyanov, and an NIH Center of Biomedical Research Excellence Grant (P20GM103638) to University of Kansas Genome Sequencing Core.
Footnotes
Competing Interests: B.G. and K.A.M have filed a patent on the utility of GPR139 as a drug target.
Data and Material availability: All data are available in the manuscript or the supplementary material. Reagents developed in this study are freely available upon request.
References and Notes
- 1.Lefkowitz RJ, Seven transmembrane receptors: something old, something new. Acta physiologica (Oxford, England) 190, 9–19 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Offermanns S, G-proteins as transducers in transmembrane signalling. Prog Biophys Mol Biol 83, 101–130 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Hauser AS et al. , Pharmacogenomics of GPCR Drug Targets. Cell 172, 41–54 e19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sriram K, Insel PA, G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol Pharmacol 93, 251–258 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Santos R et al. , A comprehensive map of molecular drug targets. Nat Rev Drug Discov 16, 19–34 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gainetdinov RR, Premont RT, Bohn LM, Lefkowitz RJ, Caron MG, Desensitization of G protein-coupled receptors and neuronal functions. Annu Rev Neurosci 27, 107–144 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Neer EJ, G proteins: critical control points for transmembrane signals. Protein Sci 3, 3–14 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Civelli O et al. , G protein-coupled receptor deorphanizations. Annual review of pharmacology and toxicology 53, 127–146 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Insel PA et al. , G Protein-Coupled Receptor (GPCR) Expression in Native Cells: “Novel” endoGPCRs as Physiologic Regulators and Therapeutic Targets. Mol Pharmacol 88, 181–187 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Todd KH, A Review of Current and Emerging Approaches to Pain Management in the Emergency Department. Pain and therapy 6, 193–202 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Volkow ND, McLellan AT, Opioid Abuse in Chronic Pain--Misconceptions and Mitigation Strategies. The New England journal of medicine 374, 1253–1263 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Kreek MJ, Zhou Y, Butelman ER, Levran O, Opiate and cocaine addiction: from bench to clinic and back to the bench. Curr Opin Pharmacol 9, 74–80 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Violin JD, Crombie AL, Soergel DG, Lark MW, Biased ligands at G-protein-coupled receptors: promise and progress. Trends Pharmacol Sci 35, 308–316 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Koob GF, Volkow ND, Neurobiology of addiction: a neurocircuitry analysis. The lancet. Psychiatry 3, 760–773 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Charbogne P, Kieffer BL, Befort K, 15 years of genetic approaches in vivo for addiction research: Opioid receptor and peptide gene knockout in mouse models of drug abuse. Neuropharmacology 76 Pt B, 204–217 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mazei-Robison MS, Nestler EJ, Opiate-induced molecular and cellular plasticity of ventral tegmental area and locus coeruleus catecholamine neurons. Cold Spring Harb Perspect Med 2, a012070 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hao N, Behar M, Elston TC, Dohlman HG, Systems biology analysis of G protein and MAP kinase signaling in yeast. Oncogene 26, 3254–3266 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Frooninckx L et al. , Neuropeptide GPCRs in C. elegans. Front Endocrinol (Lausanne) 3, 167 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koelle MR, Neurotransmitter signaling through heterotrimeric G proteins: insights from studies in C. elegans. WormBook : the online review of C. elegans biology, 1–78 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bargmann CI, Marder E, From the connectome to brain function. Nat Methods 10, 483–490 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Salom D et al. , Heterologous expression of functional G-protein-coupled receptors in Caenorhabditis elegans. FASEB J 26, 492–502 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheong MC, Artyukhin AB, You YJ, Avery L, An opioid-like system regulating feeding behavior in C. elegans. eLife 4, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mills H et al. , Opiates Modulate Noxious Chemical Nociception through a Complex Monoaminergic/Peptidergic Cascade. J Neurosci 36, 5498–5508 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Virk MS, Williams JT, Agonist-specific regulation of mu-opioid receptor desensitization and recovery from desensitization. Molecular pharmacology 73, 1301–1308 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Williams JT et al. , Regulation of mu-opioid receptors: desensitization, phosphorylation, internalization, and tolerance. Pharmacological reviews 65, 223–254 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anderson GR et al. , R7BP complexes with RGS9-2 and RGS7 in the striatum differentially control motor learning and locomotor responses to cocaine. Neuropsychopharmacology 35, 1040–1050 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Terzi D, Cao Y, Agrimaki I, Martemyanov KA, Zachariou V, R7BP modulates opiate analgesia and tolerance but not withdrawal. Neuropsychopharmacology 37, 1005–1012 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vaupel DB, Lange WR, London ED, Effects of verapamil on morphine-induced euphoria, analgesia and respiratory depression in humans. J Pharmacol Exp Ther 267, 1386–1394 (1993). [PubMed] [Google Scholar]
- 29.Dogrul A, Yesilyurt O, Isimer A, Guzeldemir ME, L-type and T-type calcium channel blockade potentiate the analgesic effects of morphine and selective mu opioid agonist, but not to selective delta and kappa agonist at the level of the spinal cord in mice. Pain 93, 61–68 (2001). [DOI] [PubMed] [Google Scholar]
- 30.Kakarala KK, Jamil K, Sequence-structure based phylogeny of GPCR Class A Rhodopsin receptors. Molecular phylogenetics and evolution 74, 66–96 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Matsuo A et al. , Molecular cloning and characterization of a novel Gq-coupled orphan receptor GPRg1 exclusively expressed in the central nervous system. Biochem Biophys Res Commun 331, 363–369 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Liu C et al. , GPR139, an Orphan Receptor Highly Enriched in the Habenula and Septum, Is Activated by the Essential Amino Acids L-Tryptophan and L-Phenylalanine. Mol Pharmacol 88, 911–925 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Masuho I et al. , Distinct profiles of functional discrimination among G proteins determine the actions of G protein-coupled receptors. Sci Signal 8, ra123 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mathis V, Kenny PJ, From controlled to compulsive drug-taking: The role of the habenula in addiction. Neurosci Biobehav Rev, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rasmussen K, Beitner-Johnson DB, Krystal JH, Aghajanian GK, Nestler EJ, Opiate withdrawal and the rat locus coeruleus: behavioral, electrophysiological, and biochemical correlates. J Neurosci 10, 2308–2317 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Urban DJ, Roth BL, DREADDs (designer receptors exclusively activated by designer drugs): chemogenetic tools with therapeutic utility. Annual review of pharmacology and toxicology 55, 399–417 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Selbie LA, Hill SJ, G protein-coupled-receptor cross-talk: the fine-tuning of multiple receptor-signalling pathways. Trends Pharmacol Sci 19, 87–93 (1998). [DOI] [PubMed] [Google Scholar]
- 38.Roszer T, Banfalvi G, FMRFamide-related peptides: anti-opiate transmitters acting in apoptosis. Peptides 34, 177–185 (2012). [DOI] [PubMed] [Google Scholar]
- 39.Toll L, Bruchas MR, Calo G, Cox BM, Zaveri NT, Nociceptin/Orphanin FQ Receptor Structure, Signaling, Ligands, Functions, and Interactions with Opioid Systems. Pharmacol Rev 68, 419–457 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hebb AL, Poulin JF, Roach SP, Zacharko RM, Drolet G, Cholecystokinin and endogenous opioid peptides: interactive influence on pain, cognition, and emotion. Prog Neuropsychopharmacol Biol Psychiatry 29, 1225–1238 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Nohr AC et al. , The orphan G protein-coupled receptor GPR139 is activated by the peptides: Adrenocorticotropic hormone (ACTH), alpha-, and beta-melanocyte stimulating hormone (alpha-MSH, and beta-MSH), and the conserved core motif HFRW. Neurochemistry international 102, 105–113 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Minevich G, Park DS, Blankenberg D, Poole RJ, Hobert O, CloudMap: a cloud-based pipeline for analysis of mutant genome sequences. Genetics 192, 1249–1269 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paix A, Folkmann A, Rasoloson D, Seydoux G, High Efficiency, Homology-Directed Genome Editing in Caenorhabditis elegans Using CRISPR-Cas9 Ribonucleoprotein Complexes. Genetics 201, 47–54 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Masuho I et al. , Distinct profiles of functional discrimination among G proteins determine the actions of G protein-coupled receptors. Science signaling 8, ra123 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Levitt ES, Williams JT, Morphine desensitization and cellular tolerance are distinguished in rat locus ceruleus neurons. Mol Pharmacol 82, 983–992 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.