

Abstract
Traffic-related air pollution particulate matter (TRAP-PM) is associated with increased risk of Alzheimer Disease (AD). Rodent models respond to nano-sized TRAP-PM (nPM) with increased production of amyloid Aβ peptides, concurrently with oxidative damage. Because pro-Aβ processing of the amyloid precursor protein (APP) occurs on subcellular lipid rafts, we hypothesized that oxidative stress from nPM exposure would alter lipid rafts to favor Aβ production. This hypothesis was tested with J20 mice and N2a cells transgenic for hAPPswe (familial AD). Exposure of J20-APPswe mice to nPM for 150 hours caused increased lipid oxidation (4-HNE) and increased the pro-amyloidogenic processing of APP in lipid raft fractions in cerebral cortex; the absence of these changes in cerebellum parallels the AD brain region selectivity for Aβ deposits. In vitro, nPM induced similar oxidative responses in N2a-APPswe cells, with dose-dependent production of NO, oxidative damage (4-HNE, 3NT), and lipid raft alterations of APP with increased Aβ peptides. The antioxidant N-acetyl-cysteine (NAC) attenuated nPM-induced oxidative damage and lipid raft alterations of APP processing. These findings identify neuronal lipid rafts as novel targets of oxidative damage in the pro-amyloidogenic effects of air pollution.
Keywords: Aβ, lipid raft, oxidative stress, air pollutant, Alzheimer’s disease, N-acetyl-cysteine (NAC)
Graphical Abstract
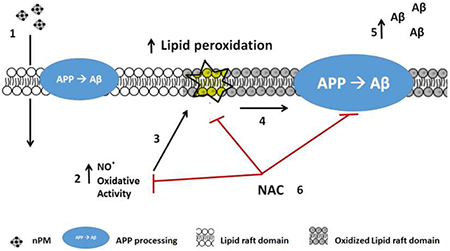
Traffic-related air pollutants (TRAP) of nano-particle size (nPM) increase the risk of Alzheimer Disease (AD). We propose a new molecular mechanism in the pro-amyloidogenic effect of TRAP. Exposure of mice or cells to nPM (1) increases the oxidative damage to cell membranes (2). In particular, the oxidation of lipid raft domains (3) alters APP processing (4) to increase production of Aβ fragments (5). Treatment with anti-oxidant N-acetyl-cysteine (NAC) attenuated nPM-induced oxidative damage to lipid rafts and pro-amyloidogenic alterations of APP processing. These findings identify lipid raft alterations of APP processing as a novel target of air pollution relevant to cognitive impairments.
Introduction
Alzheimer disease (AD) and accelerated cognitive aging are associated with traffic-related air pollution particulate matter (TRAP-PM) in several studies [1–5] and other reports critically reviewed in [6, 7]. However, the subcellular mechanisms of TRAP-PM induced neurodegeneration are obscure. We show lipid raft alterations that increase production of the neurotoxic amyloid peptide Aβ as a novel mechanism in air pollution toxicology.
Premature brain deposits of Aβ in children and young adults from a highly polluted Mexican city suggested increased production of brain Aβ as a possible mechanism in TRAP-PM associated AD risk [8]. Correspondingly, young wildtype rodents had increased levels of soluble Aβ peptides from exposure to various TRAP components: diesel exhaust [9], nickel nanoparticles [10], and TRAP-PM [11]. While these wildtype rodents do not acquire fibrillar deposits of the endogenous species-specific Aβ, mice carrying the APPswe mutation for familial AD together with human ApoE genes (EFAD genotype) responded to TRAP-nPM with increased Aβ oligomers and fibrillar Aβ plaque load [1]. In vitro, TRAP-nPM also increased Aβ production in neuronal N2a cells transgenic for APPswe [1]. TRAP-nPM is a subfraction of ultrafine PM, <0.2μ in size, collected from a local freeway, which is neurotoxic at low levels in vitro [12–14].
The present study analyzed subcellular mechanisms of nPM-induced Aβ increase with J20 mice and neuronal N2a cells transgenic for APPswe. Processing of the amyloid precursor protein (APP) occurs on lipid rafts, where neuronal APP is sequentially cleaved by the endoproteases α- or β-secretase, and by γ-secretase [15]. The resulting soluble APP fragments (sAPPα and sAPPβ) are then processed to Aβ38-43 peptides [16]. Pro-amyloidogenic processing is also characterized by an increased sAPPβ:sAPPα ratio [16]. Cerebral cortex was compared with the cerebellum, a brain region with minimal amyloid deposits and neurodegeneration in AD [8].
Because TRAP-nPM induces oxidative stress [12–14] and because the lipid oxidation product 4-HNE can increase neuronal Aβ levels [17], we considered oxidative mechanisms in altered APP processing. We also assayed nitric oxide (NO) which is rapidly induced by nPM in vitro together with 4-HNE [13, 14]. In vivo and in vitro models tested the hypothesis that nPM exposure would cause lipid raft oxidation and stimulate pro-amyloidogenic APP processing.
Materials and Methods
nPM collection and extraction.
Nano-sized particulate matter (nPM, <0.2 μm in diameter) was collected on Teflon filters by a High-Volume Ultrafine Particle (HVUP) Sampler [18] at 400 L/min flow, 150 meters downwind of the I-110 Freeway in central Los Angeles. These TRAP-PM0.2 particles are a mix of fresh ambient PM from vehicular traffic and industrial sources [19, 20]. Filter-trapped nPM were eluted by sonication into deionized water and stored at −20 °C. Their chemical composition matched prior studies, with enrichment of ions (NH4+, NO3−, SO42−), water-soluble organic compounds [12–14, 21] and elements [22, 23]. The nPM sample had 0.01 EU/μg PM endotoxins by Limulus assay. Controls for nPM extracts were sham-extracted sterile filters.
Animals.
In vivo experiments conformed to approved protocol (#20417) of the University of Southern California Institutional Animal Care and Use Committee (USC IACUC). Male J20-hAPPswe mice [24] were purchased from the Jackson Laboratory. Mice were ad libitum fed Purina Lab Chow (LabDiet, Hayward, CA) and sterile water. Under supervision of the USC Department of Animal Resources, mice were housed in groups of five at 22°C/30% humidity and light cycles of 0600–1800 hours with standard nesting material and allowed free movement. Just before nPM exposure, mice were transferred from home cages to exposure chambers that allowed free movement. Fighting in the novel environment of exposure chambers was minimized by maintaining the same home cage groups for exposure in the ‘home’ groups received from the Jackson Laboratory. These cage groups were randomly assigned to nPM or filtered air (control) exposure. Mice were exposed to 300 μg/m3 nPM, within mid-range used in other labs [25, 26]. Mice were exposed 5 h\ day, 3 days\week, for total consecutive 10 weeks (150 h), delivered to sealed exposure chambers [1, 12]. Experimental logistics limited the exposure study to 20 mice (10/exposure air). J20 mice had high baseline spontaneous mortality, as reported [27]: two mice died within 3 days of receipt; the remaining mice were equally distributed to experimental groups. General health was monitored daily and weight taken bi-weekly. There was no weight loss or evidence of respiratory distress in exposed or control mice. During the first three weeks of the study, two controls and one nPM exposed mice died unexpectedly, which decreased group size by the end of the 10-week exposure. No signs of illness were reported by the veterinarian staff; necropsy was not possible.
Mice were euthanized by isoflurane anesthesia (VETONE Fluriso, MWI, Boise, ID) following USC IACUC protocols, and perfused transcardially with phosphate-buffered saline (PBS). Hemisected brains were fixed in 4% paraformaldehyde (Sigma-Aldrich, Carlsbad, CA) and cryoprotected in 0.3% sodium azide (Sigma-Aldrich) for sagittal sectioning 0.5–2 mm from midline by vibratome, followed by immunohistochemistry Aβ, 4-HNE). The other hemisphere was dissected for brain regions of interest and frozen on dry ice. The complete cerebral cortex and cerebellum were pulverized on dry ice and homogenized for lipid raft isolation and Aβ ELISA (see below). Data analysis was blinded for exposure air type and analyzed collectively.
Immunohistochemistry.
Aβ amyloid deposits was immunostained with 4G8 antibody (residues 17–24 at N-terminal of APP (Covance, Princeton, NJ) followed by biotinylated secondary antibodies (1:250, Jackson Immunoresearch Laboratories, West Grove, PA). Lipid peroxidation was assayed with 4-HNE antibody (R&D Systems, Minneapolis, MN) followed by HRP-conjugated secondary antibody (dil. 1:250, Jackson Immunoresearch Laboratories). For Aβ IHC, sections were fixed in 70% formic acid/5 min. For both Aβ and 4-HNE IHC, endogenous peroxidases were blocked by 3% H2O2 and 10% methanol in TRIS-buffered saline (TBS), 30 min/22 °C. Sections were permeabilized in 0.1% Triton X-100 for 15 min, blocked by 30 min incubation in TBS with 2% BSA (RPI, Mt. Prospect, IL) and 0.1% Triton, and probed with primary antibodies. After rinsing with 0.1% Triton and TBS, sections were incubated with biotinylated secondary antibodies/1 h, followed by ABC peroxidase (Vector Laboratories, Inc., Burlingame, CA), and 3,3’-diaminobenzidine (DAB; Vector). Cortex sections were evaluated for % area covered Aβ load) by the ‘analyze particles’ function of NIH ImageJ software. Gray scale images were thresholded to discriminate immunopositive and negative Aβ zones. Total Aβ and 4-HNE loads represent the % of pixels with positive immunostaining. Neuron layers were defined by NeuN immunostaining (Millipore, Burlington, MA) and 4-HNE, and secondary fluorescent antibodies (AlexaFluor 488-donkey-anti-rabbit; 594-goat-anti-mouse, Invitrogen) (Supplementary Material Figure 1).
Cell culture.
Mouse neuroblastoma N2a cells carrying Swedish APP mutation APP (APPswe, K595N/M596L) [28, 29] were generously given by Dr. Huaxi Xu (Sanford/Burnham Institute, La Jolla, CA) and tested negative for mycoplasma contamination. Cells were grown in OptiMEM/DMEM medium (v:v) (Invitrogen, Foster City, CA) supplemented with 5% FBS (VWR) and 500 μg/ml G418 (RPI, selection agent), in a humidified incubator (37°C/5%CO2,). For cell treatment, TRAP-derived nPM was diluted (1,5,10 μg/ml) in Optimem/DMEM medium (v:v) with 5% FBS and 500 μg/ml G418, applied 24 h. N-acetylcysteine (NAC, Sigma-Aldrich) was diluted to 100 μM, and incubated 24 h. Data analysis was blinded for administered treatment. Experiments were repeated 3 times. For immuno/cytochemistry, two independent coverslips were analyzed in triplicate for each treatment and experiment.
ELISA.
Aβ peptides were assayed in brain lysate and cell culture media. Cerebral halfcortexes were homogenized in DEA buffer (0.2% diethylamine, 50 mM NaCl; 1 ml/200 mg tissue) with protease inhibitor cocktail (Sigma-Aldrich). After centrifugation (10,000g x 1h), supernatants were neutralized with Tris-HCl, pH 6.2 (‘Tris extract’). Pellets were resuspended in 1% SDS-PBS and centrifuged (10,000g x 1h) for supernatants (‘SDS extract’). Aβ40 and −42 were assayed by 4G8 Kit V-PLEX™ (Peptide Panel 1, Meso Scale Discovery, Rockville, MD).
MTT assay.
Mitochondrial reductase activity was assessed by oxidation of MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] (RPI). The MTT substrate was prepared at 5 mg/ml in PBS for 4 h in the dark. 0.1N acidic isopropanol was added to the cells solubilize the formazan reagent. Formazan was measured by absorbance at 570 nm, with 630 nm as background (Spectra Max M2 plate reader; Molecular Devices, Sunnyvale, CA). The upper nPM dose range of 10 μg/ml was identified as sublethal.
Viability Assay.
Cell viability/death rate was assessed using the LIVE/DEAD® Viability/Cytotoxicity Assay Kit (Thermo-Scientific). After 24h nPM treatment, cells were washed in PBS and incubated for 45 minutes at room temperature with 5μM EthD-1 (red, dead cells) and 5μM calcein AM (green, live cells). After washing in PBS and mounting in Fluoromount™ Aqueous Mounting Medium (Sigma), cells were inspected at 40X magnification on Nikon Eclipse TE300 microscope (Nikon Inc., Melville, NY). An average of 10 images/well/treatment was imaged. Percentage of dead cells (red) was calculated and averaged across experiments. An increased toxicity was observed at the highest concentration of 10 μg/ml nPM (Supplementary Material Figure 2).
Nitric oxide (NO).
Media nitrite was assayed by the Griess reagent [30] with NaNO2 as a standard, and untreated media as a control.
Immunocytochemistry
Cells were plated in 24-well-plate on coverslips (Electron Microscopy Science, Hatfield, PA) and exposed to nPM/24 h, fixed in 4% PFA/10 min, permeabilized with 1% NP-40/PBS, and blocked with 5% BSA. Primary antibodies were added at 4’C overnight (4-HNE, 1:500, R&D Systems; 3-NT, 1:100, rabbit; Millipore). Immunofluorescence was visualized using Alexa Fluor 488 antibodies (1:400, Invitrogen). Data analysis was blinded for treatment. Images were scored for intensity of expression: individual cells were identified, and intensity of signal normalized on cell size.
MitoSOX assay.
After nPM treatment for 24 h, cells were incubated with 5 μM MitoSOX (Invitrogen) at 37 °C/10 min, protected from light. Cells were washed and mounted with DAPI containing media (VECTASHIELD, Vector)[13].
Cholera toxin B cytochemistry.
After nPM treatment for 24 h, cells were fixed in 4% PFA/15 min, quenched in 50 mM NH4Cl/PBS/5 min, permeabilized in 0.1% Triton X-100/10 min and blocked in 1% BSA/10 min. Cells were incubated with fluorophore-conjugated cholera toxin B (CTx-B, 1:100, Invitrogen) at 37μC/30 min, washed in PBS, and mounted with DAPI (Vector) [31].
Lipid raft isolation.
For lipid raft isolation [32], cerebral cortex, cerebellum, N2a cells were homogenized in lysis buffer (25 mM Tris-HCl pH 8, 140 mM NaCl, 1% Triton X-100, 1 mM EDTA (EDS, Sigma-Aldrich), supplemented with 1 mM PMSF (Sigma-Aldrich), 1 mM Na3VO2, 10 mM NaF, and 1X Protease Inhibitor Cocktail. Lysates were homogenized through a 22G needle. Samples were adjusted to 40% Optiprep (Sigma-Aldrich) and loaded at the bottom of tubes (Quick-Seal® Polypropylene Tube, Beckman Coulter Inc., Brea, CA). A discontinuous gradient was layered on the top (30% and 5% Optiprep in lysis buffer). Samples were centrifuged at 400,000g/4h, 4μC, VTi 65.2 rotor. Fractions of 500 μl (8-9 fractions) were analyzed for lipid-raft microdomains by immunobloting for flotillin-1 (1:1000, Cell Signaling Technology, Danvers, MA) and caveolin-1 (1:1000, BD Bioscience, San Jose, CA) [33] (Supplementary Material Figure 3).
Western blots.
Samples of 20 μg protein were loaded on Novex NuPAGE 4-12% Bis-Tris protein gels (Thermo Scientific, Carlsbad, CA). Membranes were washed with PBS with 0.05% Tween-20 (PBST), then blocked with 5% milk/PBST or 5% BSA/PBST/1 h; incubated with primary antibodies\4°C: anti-amyloid precursor protein, APP (6E10, full length, APP, 1:1000, mouse; Covance); anti-BACE1 (β-secretase, 1:1000, rabbit; clone D10E5, Cell Signaling); anti-PS1 (presenilin 1, 1:1000, mouse, clone D39D1, Cell Signaling); anti-actin (1:10000, mouse, Sigma). Membranes were washed and incubated with HRP-conjugated secondary antibody (1:10000, Jackson ImmunoResearch). Chemiluminescence was detected by West Pico Chemiluminescent Substrate (Thermo Scientific).
Statistics.
Data for raw values or % control are shown as mean ± SEM. Data normality was assessed by D’Agostino-Pearson normality test. Statistical analysis used two-tailed t-test; one-way ANOVA with post-hoc analysis. p< 0.05 was accepted as statistically significant (GraphPad Software, San Diego, CA).
Results
Chronic nPM exposure of J20-hAPPswe mice increased Aβ, APP, oxidation in lipid rafts.
Cerebral cortex of male mice exposed to nPM for 150 total hours over 10 weeks had increased soluble Aβ and plaque Aβ load: total SDS-soluble Aβ40, +5-fold, p<0.05; Aβ42, +10-fold, p<0.05 (Fig. 1a); plaque Aβ, + 45%, p<0.05 (Fig. 1b). Lipid raft enriched subcellular fractions had increased APP (65%, p<0.05) (Fig. 1c, d). In contrast, no changes were detected in the endoproteases responsible for the pro-amyloidogenic processing of APP: BACE1 (β-secretase) or PS1 (catalytic subunit of γ-secretase) [34] (Fig. 1d). The cerebellum was analyzed because of its minimal amyloid deposits in EFAD mice [48] and humans [35, 36]. In contrast to cerebral cortex from the same mice, cerebellar lipid raft APP did not respond to nPM (Fig. 1e, f).
Fig. 1.
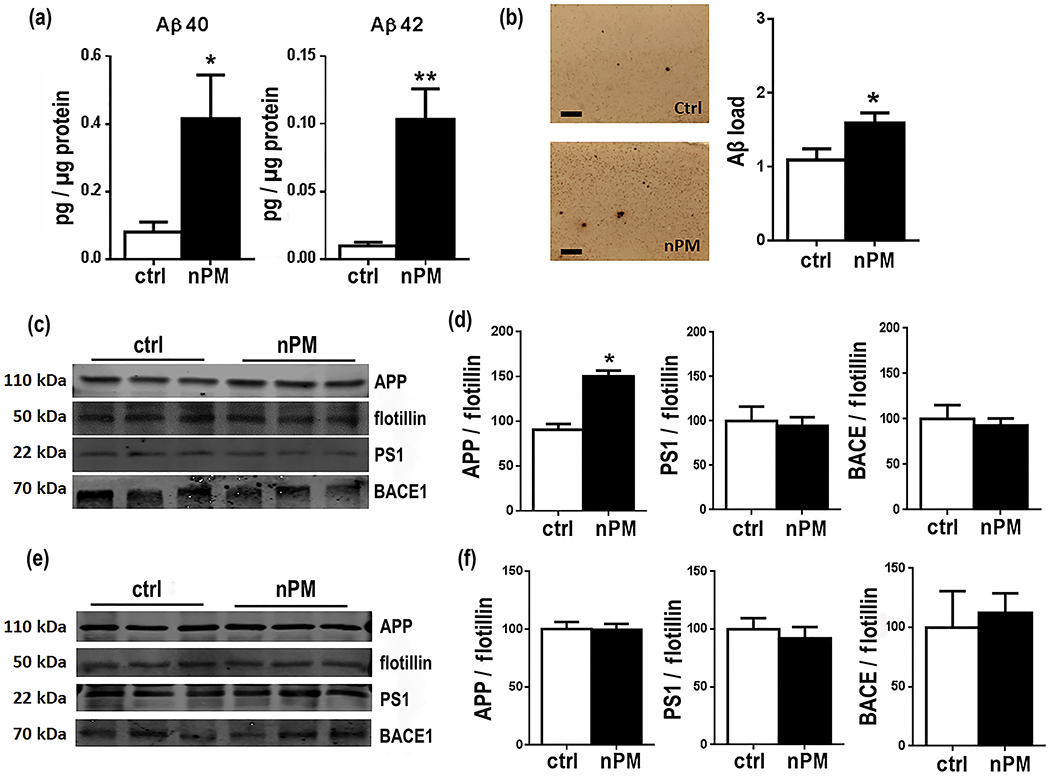
Chronic nPM exposure of J20 mice increased Aβ and APP in lipid raft fractions of cerebral cortex a cerebral cortex soluble Aβ peptides (0.1% SDS-PBS extract; ELISA): Aβ40, +5-fold, p<0.05; Aβ42, +10-fold, p<0.05. b Representative images of Aβ IHC and Aβ plaque load quantification in J20 cerebral cortex: +45%, p<0.05. c, d cerebral cortex lipid raft fractions for APP, PS1 and BACE1 (Western blots; flotillin loading control): nPM increased APP by +65%, p<0.05. e, f cerebellum lipid raft fractions: no response to nPM by APP, PS1, and BACE (Western blots). Scale bar: 100μm; mean ± SEM. * p<0.05, *** p<0.005, ****p<0.001. ctrl= 7, nPM= 8 mice.
The lipid oxidation marker 4-HNE was increased 2-fold (p<0.001) in cerebral cortex after chronic nPM exposure (Fig 2a, b). Neuronal cell bodies had 3-fold (p<0.005) more 4-HNE staining in cortical layers 2-4 (Fig. 2b). Neuronal localization of 4-HNE was shown by costaining with NeuN (Supplementary Material Figure 1). In contrast, cerebellar 4-HNE was not increased in neuropil or neuronal layers (Fig. 2c, d). Brain region specificity extended to lipid raft subcellular fractions, which had increased 4-HNE in cortex (20%, p<0.02), but not in cerebellum (Fig. 2e, f).
Fig. 2.
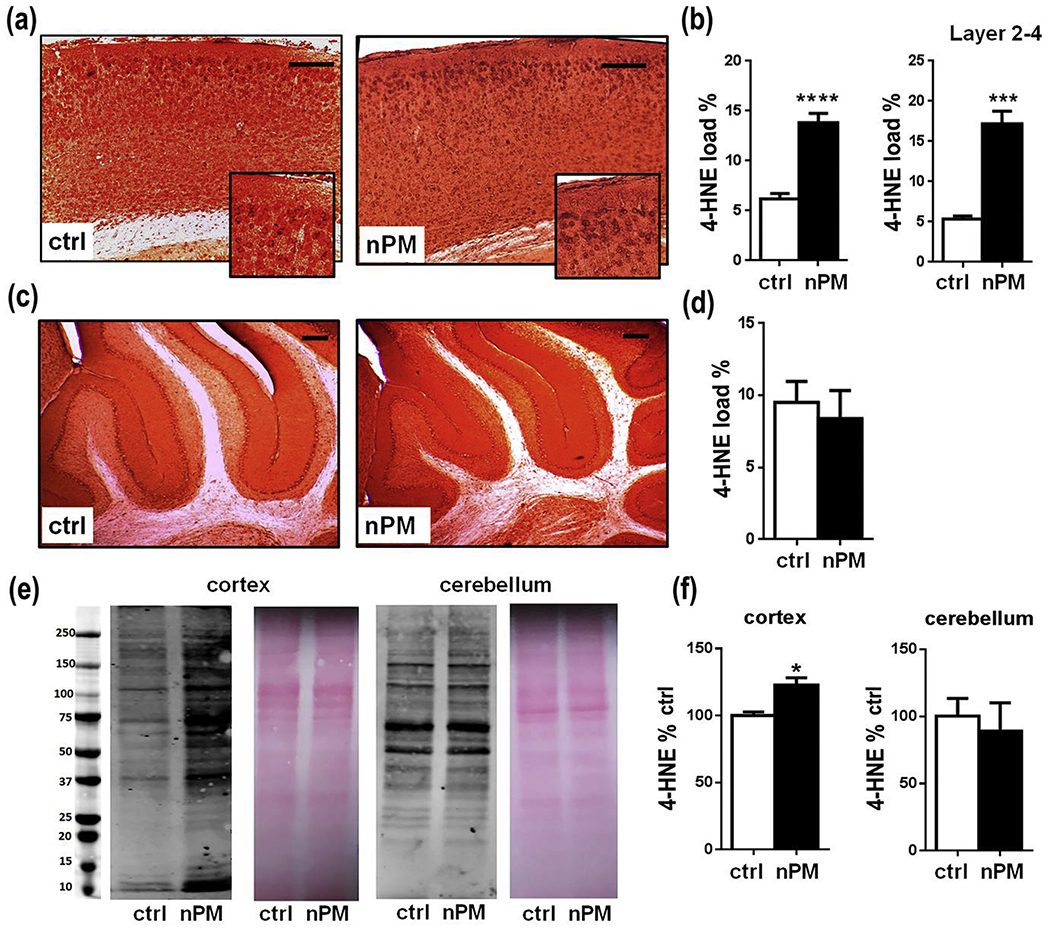
J20 mice exposed to chronic nPM had increased 4-HNE in cerebral cortex, but not cerebellum. a, c 4-HNE immunohistochemistry: a cerebral cortex. c cerebellum. b, d Quantification of 4-HNE immunostaining. b cerebral cortex: 2-fold increase of 4-HNE (p<0.001) from nPM exposure; b 4-HNE in cortical layers 2-4: d cerebellum 4-HNE, no change. e 4-HNE immunoblots of lipid raft enriched fractions, Ponceau red staining as a loading control for protein. f Quantified 4-HNE immunoblots of panel e: nPM exposure increased 4-HNE in lipid rafts from cortex (p < 0.05), but not cerebellum. ctrl= 7, nPM= 8 mice. Scale bar: 100 μm; mean ± SEM. * p<0.05, *** p<0.005, ****p<0.001.
In vitro nPM is pro-amyloidogenic
N2a neuroblastoma cells carrying the hAPPswe AD transgene were used to analyze pro-amyloidogenic responses to nPM. After 24 hours in vitro, cells responded to nPM with dose-dependent increases of Aβ levels in cell media (Fig. 3a) and cell lysates (Fig. 3b) at 5-10 μg/ml nPM, while 1 μg/ml nPM did not alter Aβ levels. Cell media had 2-3 fold more Aβ42 (p<0.05) and 60% more Aβ40 at 5-10 μg/ml nPM (p<0.01). Similarly, cellular Aβ40 (p<0.01) and Aβ42 (p<0.05) was increased 50% by 5 μg/ml. Total APP was decreased by 20% (p<0.05, Fig. 3c), while the APP fragment ratio of sAPPβ:sAPPα was increased by 25% (p<0.01) at higher nPM (Fig. 3d). Increased toxicity was observed at the highest concentration of 10 μg/ml nPM (Supplementary Material Figure 2).
Fig. 3.
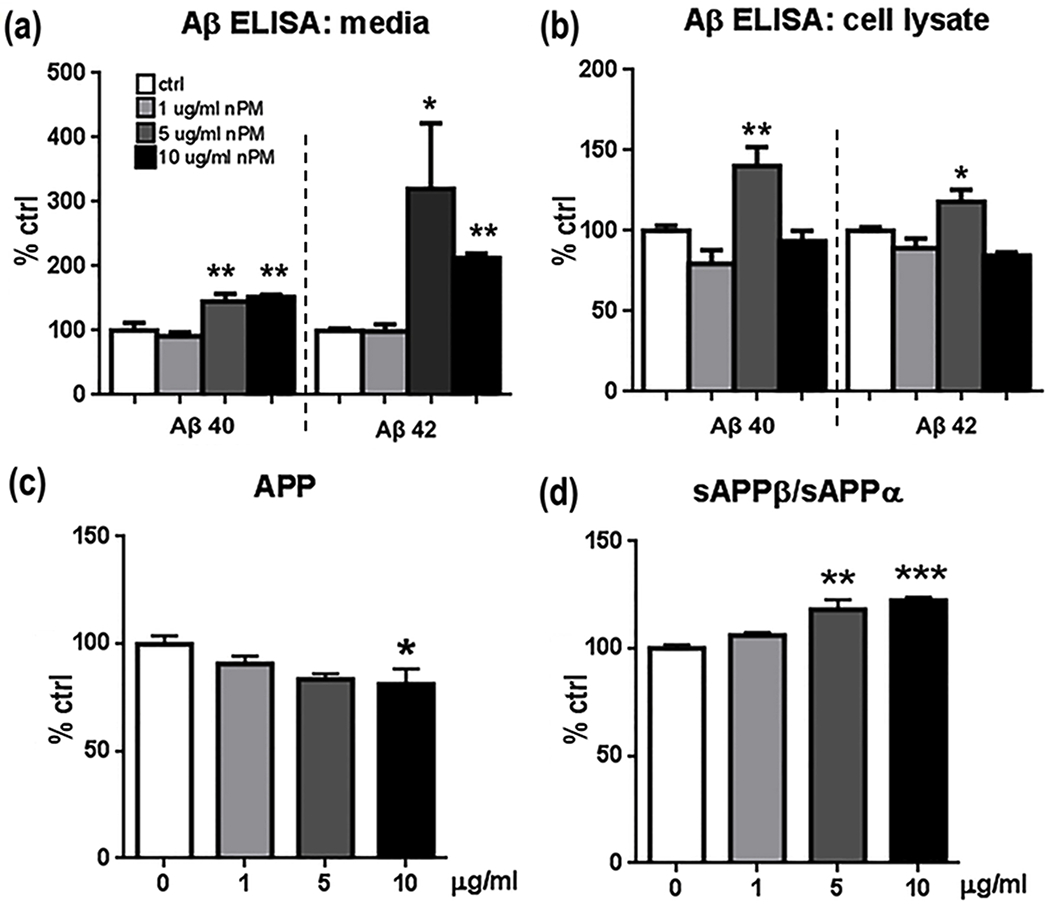
N2a-APP/swe cell responses of APP peptides to nPM at 1, 5 & 10 ug/ml for 24 h a Aβ 40, & −42 in conditioned media were increased by nPM at 5, 10 μg /ml, but not 1 μg /ml . (b). In cell lysates, 5 μg /ml nPM increased both Aβ 40 and Aβ 42 (* p<0,05, ** p<0.01, *** p<0.005, one-way ANOVA). c Western blots (6E10 antibody): nPM decreased total APP by 20% at 10 μg /ml nPM, * p<0.05). d Western blots (22C11 antibody): nPM (5-10 μg /ml) increased the ratio sAPPβ:sAPPα (** p<0.01, *** p<0.005). n=3 independent experiments, 3 samples each. Mean ± SEM. * p<0.05, ** p<0.01, ***p<0.005.
Lipid raft APP protein levels and lipid raft organization
Lipid raft subcellular fractions from N2a-APPswe cells responded to nPM with dose dependent decrease of full length APP (Fig.4a,b). The 50% decrease of APP in lipid rafts (5 μg/ml, p<0.05; 10 μg/ml, p<0.01) corresponded to increased Aβ40 and Aβ42 in the media (Fig. 3a, b), with inverse correlation of r2 −0.55 (p<0.005) (Supplementary Material Figure 4). Lipid raft fractions showed a trend for increased PS1: +50% at 1 μg/ml nPM to 2-fold at 10 μg/ml (p=0.06) (Fig.4c). However, nPM did not alter protein levels of BACE1 (Fig. 4d), flotillin-1(Fig. 4e), or APP mRNA (not shown).
Fig. 4.
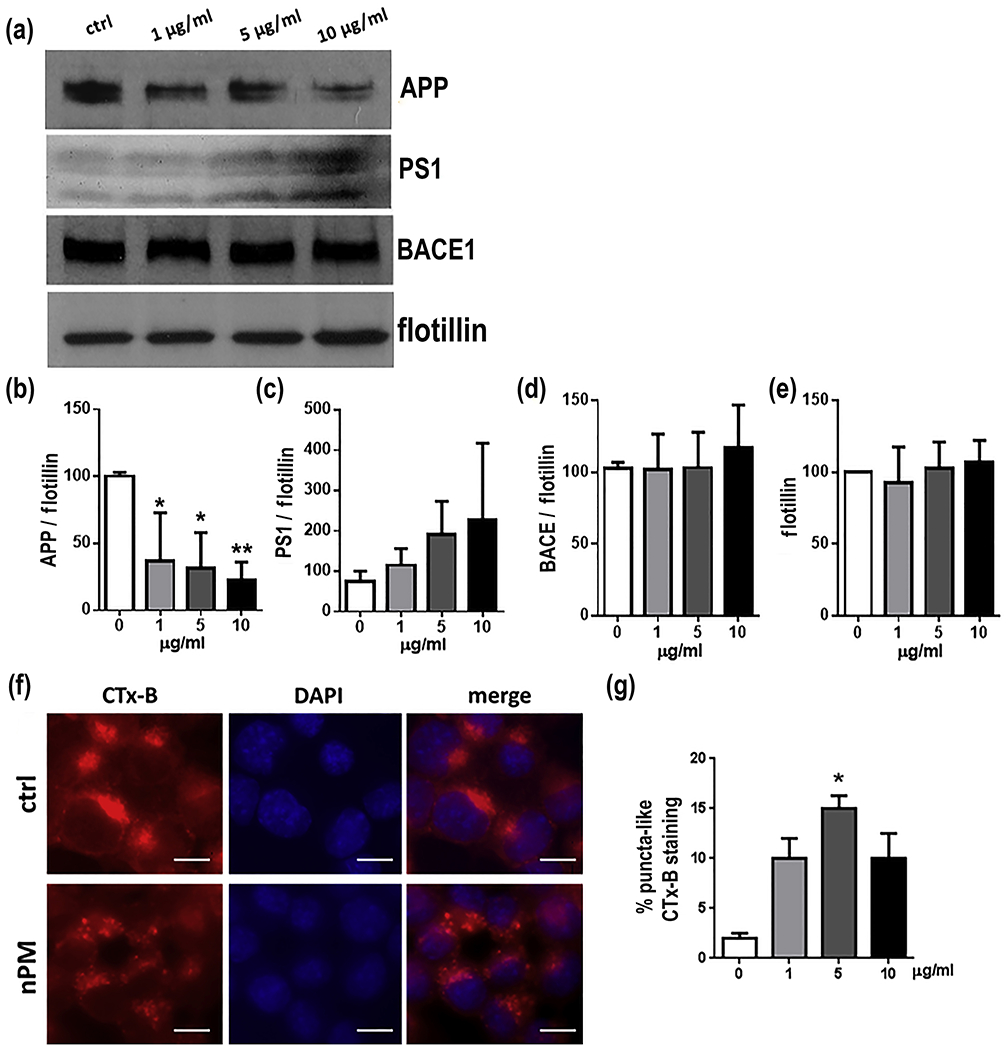
In vitro N2a-APP/swe cell lipid raft responses to nPM. a Western blots for APP-FL (full length), PS1, BACE1 and flotillin-1 in lipid raft b Total APP was decreased by nPM exposure with dose-dependence. c PS1 had a trend for increase at higher nPM (p=0.06). d BACE1 and e flotillin-1 did not respond to nPM. f Lipid raft fractions stained for cholera toxin B (Ctx-B), nuclear staining (DAPI, middle panels) and merged images; scale bar, 15 μm. Control (ctrl, globular, upper panels) and nPM exposed cells (puncta, lower panels). g nPM at all doses increased Ctx-B puncta-like staining of lipid rafts, with statistical significance at 5 μg/ml nPM. n=3 independent experiments, 3 samples each. Mean ± SEM * p<0.05, ** p<0.01, *** p<0.005.
Lipid rafts showed histochemical evidence of re-organization. Morphology of lipid raft was analyzed by localization of the cholera toxin subunit B (CTx-B), a high affinity marker for GM1 gangliosides [37]. Control cultures showed a globular homogeneous CTx-B staining, while treatment with nPM caused redistribution of CTx-B into multiple puncta (Fig. 4g, h), suggesting a re-organization of the lipid raft layer, as previously also reported for silica [31].
NO and markers of oxidative stress
Nitric oxide (NO) was induced within 15 minutes of nPM with dose-dependence (>4-fold, P<0.01 at 5 μg/ml) (Fig. 5a). Notably, NO was increased at 1 μg nPM/ml (+ 3-fold, p<0.05), which did not alter Aβ or MTT (not shown). Other biochemical markers of oxidative stress required higher nPM than for NO induction: MitoSOX (mitochondrial associated oxidants, 5 μg/ml, +3-fold, p<0.001; 10 μg/ml, + 2-fold, p<0.001), 3-nitrotyrosine (3NT; protein tyrosine nitration, +50%, p<0.05, at both 5 and 10μg/ml), and 4-HNE (lipid peroxidation; 5 μg/ml, +2-fold, p<0.005; 10 μg/ml, + 50%, p<0.01) (Fig. 5b–e). As observed for Aβ, 1 μg/ml nPM was below threshold.
Fig. 5.
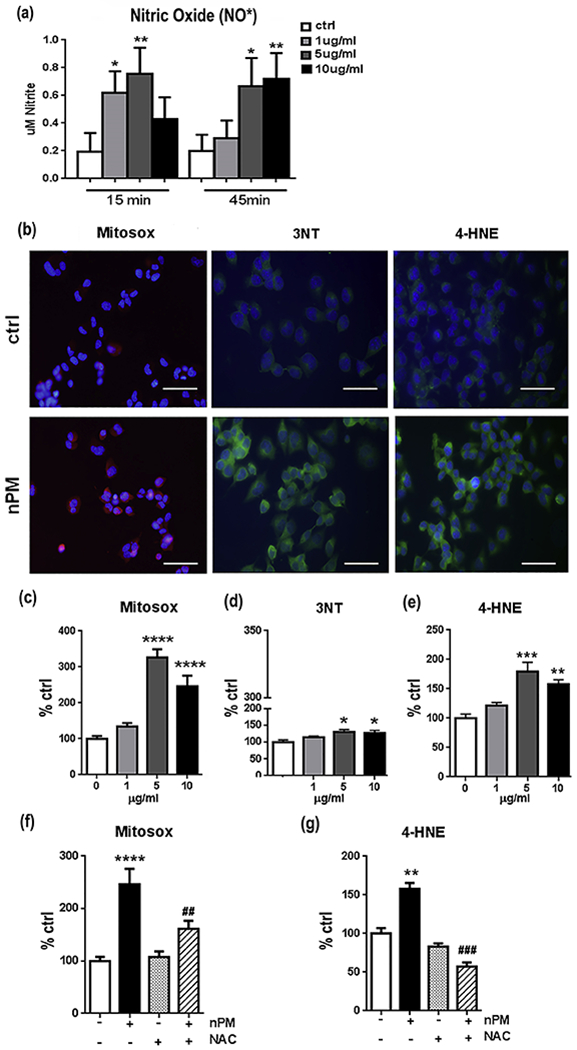
nPM induced NO and other markers of oxidative stress in N2a-APP/swe cells a NO• was assayed as nitrite (Griess reagent) in media. The NO increase was rapid by 15 min and was transient (not shown). b Histochemical images of oxidative stress markers after nPM exposure for 24 h c MitoSOX RED response to nPM, as immunofluorescent cell area. d 3NT. e 4-HNE. Responses were significant at 5-10 μg/ml, but not at 1 μg/ml. f, g NAC blocked response of MitoSOX and 4-HNE to 10 μg/ml nPM. n=3 independent experiments, 3 samples each. Mean ± SEM. * compared to ctrl, # compared to nPM alone * p<0.05, ** p<0.01, *** p<0.005, **** p<0.001, ## p<0.01, ### p<0.005. Scale bar= 50 μm
Inhibition of oxidative stress by NAC attenuated nPM-induced changes
The role of oxidative responses to nPM was analyzed with N-acetyl L-cysteine (NAC), a general antioxidant, which blocked pro-oxidative effects of nPM in brain slices [13] and of diesel PM0.2 in endothelia [38]. NAC treatment attenuated nPM induced oxidative responses as shown by inhibiting the increases in MitoSOX fluorescence (−50%, p<0.01) and 4-HNE (−2-fold, p<0.005) levels (Fig. 5f, g). For Aβ, 10 μM NAC attenuated nPM induced increases of Aβ40 and Aβ42 in culture media (Fig. 6a, hatched bars) and cell lysates (Fig. 6b, hatched bars). NAC alone did not alter Aβ in control cells not treated with nPM (open bars). For lipid raft APP, NAC treatment also attenuated nPM effects, maintaining APP at levels comparable to ctrl (Fig. 6c, d). Again, NAC alone did not alter APP levels. Lastly, NAC attenuated the nPM increase of lipid raft 4-HNE levels (−45%, p<0.05, Fig. 6e, f).
Fig. 6.
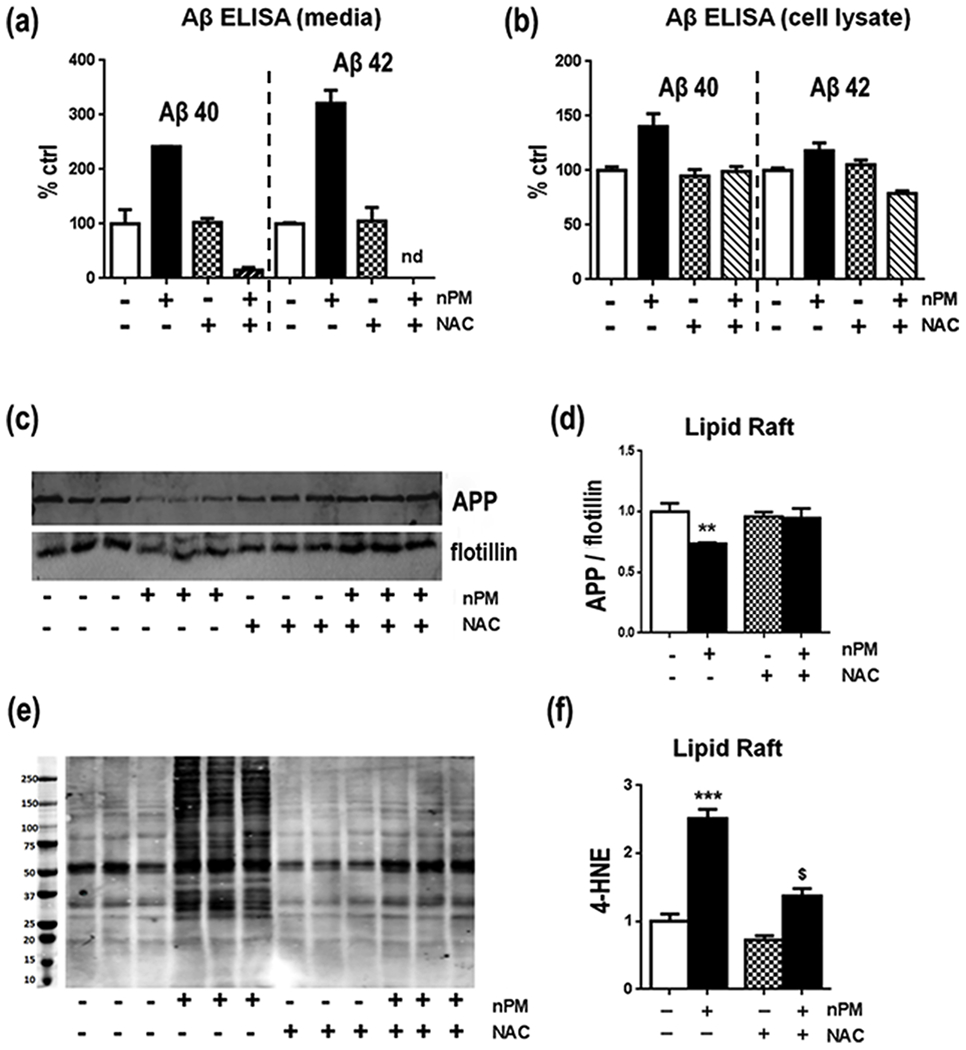
NAC attenuated nPM effects on N2A-APP/Swe cells for Aβ, APP and 4-HNE in lipid rafts a, b NAC at 10 μM attenuated the nPM induced Aβ in cell media and cell lysates. c Western blot of APP in lipid rafts. d APP/flotillin in lipid rafts: NAC blocked the nPM-induced APP decrease in lipid raft fractions vs controls. e Western blot of APP in lipid rafts. f NAC blocked the nPM-induced 4-HNE increase in lipid raft fractions vs controls. n=3 independent experiments, 3 samples each. Mean ± SEM. * p<0.05, ** p<0.01, *** p<0.005, # p<0.05, nd, not detected, * vs ctrl, # vs nPM.
Discussion
This is the first evidence that air pollutants alter lipid rafts in neuronal cells in vivo and in vitro, and the first evidence associating lipid raft oxidation from air pollutants with pro-amyloidogenic processes relevant to AD. The relation of pro-amyloidogenic processes to membrane oxidation to lipid rafts was shown in vivo and in vitro with J20 mice and N2a cells carrying the familial AD gene, APPswe, that increases Aβ42 production. The mouse and cell findings are summarized in Table 1. Traffic-related air pollution (TRAP) was modeled by exposure to nano-sized particulate material (nPM) collected from a local roadway site, which this lab has used for a decade [1, 12, 22].
Table 1:
Summary of nPM effects in vivo and in vitro
| In vitro : N2a-APP/swe | In vivo (hAPP/swe) | ||||
|---|---|---|---|---|---|
| 1 μg/ml -24h | 5 μg/ml -24h | 10 μg/ml -24h | nPM+NAC* | 150h | |
| Aβ40-42 Aβ plaque load |
nc | ↑ | ↑ | ↓ | ↑ |
| APP (lipid raft) | nc | ↓ | ↓ | ↑ | ↑ |
| NO | ↑ | ↑ | ↑ | ||
| MitoSOX | nc | ↑ | ↑ | ||
| 3NT | nc | ↑ | ↑ | ||
| 4-HNE | nc | ↑ | ↑ | ↓ | ↑ |
| CTx puncta | ↑ | ↑ | ↑ | ||
“↑” increase, “↓“ decrease, “nc” no change, “blank” data not available,
compared to nPM alone; NAC (N-acetyl L-cysteine). We propose a hierarchy of nPM effects, with lower dose inducing increases NO as first effect, but still below the threshold of oxidative stress required for changes in protein levels and increased production of Aβ peptides.
Exposure of J20-APPswe mice to TRAP-nPM for 10 weeks increased lipid oxidation (4-HNE) and increased APP and Aβ levels in cerebral cortex. A subpopulation of cortical neurons in layers 2-4 had 3-fold more 4-HNE staining. These changes in cerebral cortex, but not cerebellum, are the first indication of brain regional differences in pro-amyloidogenic effects of air pollutants, and correspond to the scarcity of amyloid plaques in the cerebellum of late stage AD [35].
Lipid rafts, the neuronal site of pro-amyloidogenic processing of APP by BACE1 and γ-secretase, also showed regional selectivity in subcellular fractions. Chronic nPM exposure increased APP and 4-HNE in lipid raft subcellular fractions of cerebral cortex, but not of cerebellum. The pro-amyloidogenic activity of TRAP-nPM can be generalized to both sexes, from the present data on male J20 mice and our prior studies of female EFAD [1].
Subcellular mechanisms were analyzed in vitro with N2A-APPswe. Acute nPM exposure caused lipid raft oxidation and increased ratio of the processed APP fragments sAPPβ:sAPPα, indicating increased pro-amyloidogenic APP processing. In lipid raft subcellular fractions, nPM increased Aβ peptides in cell lysates and media. Reciprocally, as nPM treatment decreased APP in lipid rafts, the levels of PS1 and Aβ were increased, consistent with increased pro-amyloidogenic APP processing. These responses to nPM were dose-dependent. Similarly, in primary cultures of cerebral cortex neurons, exposure to 10–40 μM H2O2 caused relocation of BACE1 with accumulation in APP positive-compartments, without altering levels of BACE1 protein [39]. We did not address colocalization of endogenous secretases and APP, because endogenous secretases were obscured by the strongly expressed APPswe transgene. The changes of lipid raft and APP, together with changes in APP and PS1 levels, show that nPM promotes pro-amyloidogenic APP processing in lipid rafts.
These data suggest a dose-dependent hierarchy of nPM effects in pro-amyloidogenic oxidative responses: the lowest dose of 1 μg/ml nPM that increased NO was below a threshold of oxidative stress needed to induce Aβ increase (Table 1). As observed in vitro, nPM effects on proteins arose at higher doses, identified as increased Aβ peptides released into the media at 5 μg/ml and by the lipid raft alterations. We note the opposite responses of APP levels on lipid rafts to nPM in vivo vs in vitro. While the APP:flotillin ratio was increased by chronic nPM exposure of J20-APPswe mice, in vitro the acute exposure of N2a-APPswe cells decreased the APP:flotillin ratio. The complex logistics of mouse exposure to nPM do not allow time course studies of lipid raft responses in vivo that might show multi-phasic changes. Despite in vivo and in vitro differences, there were similar responses to nPM for pro-amyloidogenic APP processing with increased oxidative stress.
Mechanisms in oxidative stress from TRAP-nPM include increased production of at least one species of free radical: nitric oxide (NO) was rapidly induced at the lowest nPM dose of 1 μg/ml, while higher doses increased protein tyrosine nitration (3NT), lipid oxidation (4-HNE), and mitochondrial oxidative stress (MitoSOX). A potential role of NO in human AD is indicated by 70% more tyrosine nitration of PS1 in AD brains and by an in vitro model of tyrosine nitration of PS1 in primary neurons exposed to NO [40, 41].
The antioxidant N-acetyl L-cysteine (NAC) showed the role of oxidative damage to nPM alterations of APP processing. In control N2a-APPswe cells not exposed to nPM, NAC alone did not alter APP. In cells exposed to nPM, NAC completely blocked increases of Aβ40 and Aβ42, as well as blocking the increase of 4-HNE and of mitochondrial oxidative responses (MitoSox). In lipid raft fractions, NAC prevented nPM-induced changes in APP levels and attenuated the 4-HNE response. A prior study treated SH-SY5Y cells with ethacrynoic acid (EA) to lower glutathione (GSH), causing >5-fold increases of HO-1 (oxidative stress marker) and of Aβ40-42 [42]. Paralleling our findings, the increase of PS1 fragments in lipid rafts was blocked by NAC. We acknowledge that in cell cultures levels of NAC are much higher than achievable in vivo. The antioxidant efficacy of NAC in cultured cells may be translatable to in vivo neuroprotection because NAC attenuated hyper-responsivity of human smokers to diesel exhaust [43]. NAC can potentially elevate cellular glutathione by providing cysteine, which is usually limiting. In cultured cells, moreover, NAC may act on external cell surfaces as a direct reductant, as well as by its conjugation with 4-HNE to attenuate reactions with membrane proteins.
We propose that air pollutants including TRAP, diesel exhaust, and nickel are pro-amyloidogenic through oxidative mechanisms that increase 4-HNE with cascading consequences. In SH-SY5Y neuronal cells, 4-HNE and other oxidation products of ω–3 polyunsaturated fatty acids (PUFAs) increased Aβ production, supporting our findings [44]. Moreover, AD brains had 2-fold more 4-HNE than age-matched controls [44], while 4-HNE forms adducts with Aβ that promote its aggregation into protofibrils [45]. Recall from above that PS1 is tyrosine nitration (3NT) in AD [40]. The neurodegenerative cascade from air pollution involves chronic increases of NO production and other oxidative stressors, leading to 4-HNE and other oxidation products in lipid raft that, in turn, increase Aβ production. Elevated 4-HNE may further compound neurodegenerative potential by forming adducts that enhance Aβ aggregation [45].
While the fossil fuel sources of air pollutants receive great attention, TRAP also contains airborne PM from the earth’s crust that may contribute to neurotoxicity. For example, silica particles (SiO2) induced Aβ and oxidative stress in N2a cells [46]. Macrophages also responded to silica PM with increased production of superoxide and cytokines (IL-1B, TNFα) and lipid raft re-organization of CTx-B by co-author Forman [31] that resembled these nPM CTx-B induced changes. Moreover, SiO2 increased oxidative stress, increased intracellular Aβ42 and phosphorylation of tau in both human SK-N-SH and N2a cells [46]. Because TRAP-PM2.5 and cigarette smoke show super-additive synergies for several age-related pathologies [47], we anticipate synergies of airborne PM from silica and TRAP on lipid raft APP processing that may model the combination of desert dust and TRAP in some Asian cities.
We recognize caveats of in vivo and in vitro models for air pollution neurotoxicology. Our studies exposed mice to 300 μg/m3 re-aerosolized suspensions of TRAP-nPM concentrated from ambient sources at one urban site [12], which is within the range of the 200-300 μg/m3 used by other labs for chronic rodent exposure [25, 26]. While some Asian cities experience surges of PM2.5 exceeding 300 μg/m3, most US urban sites are below 30 μg/m3. Future studies will address different doses and exposure duration. Another issue is how much of inhaled air pollution particulates reach the brain [48]. Some nasally inhaled nano-scale PM does reach the forebrain, presumably through olfactory neurons [14, 47, 49]. In fact, we do not know how much of the neurodegenerative impact of air pollution is direct “nose-to-brain” vs indirect from lungs which receive most inhalants. Besides the portion of lung-entrapped PM that may enter the circulation, there is evidence for systemic inflammatory responses to air pollutants [50]. Lastly, in vitro studies add nPM suspensions to cultured cells at higher levels than possible for in vivo lab exposure models of re-aerosolized suspensions. Thus, the lowest levels of nPM that induced NO in vitro (1 μg/ml) must far exceed ambient brain exposure. Nonetheless, the present models showed good agreement for nPM induced 4-HNE and Aβ production in vivo and in vitro.
These data show that air pollution particulate matter can increase Aβ production by inducing oxidative changes in lipid rafts where APP is processed, as observed in vivo and in vitro. These data identify lipid raft alterations of APP processing as a novel target of air pollution relevant to cognitive impairments. The efficacy of N-acetylcysteine in blocking these responses suggests a pharmacological approach to mechanisms in air pollution.
Supplementary Material
Highlights:
Exposure to traffic-related air pollutants (TRAP) of nano-particle size (nPM) increases pro-amyloidogenic processing of APP, in vivo and in vitro.
Exposure of mice or cells to TRAP increases the oxidative damage to cell membranes, as shown by the increased levels of 4-HNE on lipid rafts, both in vivo and in vitro.
In vitro treatment with antioxidant N-acetyl-cysteine (NAC) attenuated nPM-induced oxidative damage to lipid rafts and pro-amyloidogenic alterations of APP processing.
Lipid raft alterations of APP processing represents a novel target of air pollution.
Acknowledgments:
We thank Prof. Myron Goodman (USC) for use of the ultracentrifuge. This study was supported by Cure Alzheimer’s Fund and National Institute on Aging R21AG050201 and R01AG051521and P50AG05142-31 to CEF, and National Institute of Environmental Health Sciences (NIEHS) ES023864 to HJF
Funding: This study was supported by Cure Alzheimer’s Fund and National Institute on Aging R21AG050201 and R01AG051521 and P50AG05142-31 to CEF, and National Institute of Environmental Health Sciences (NIEHS) ES023864 to HJF.
Abbreviation:
- APP
Amyloid precursor protein
- Aβ
amyloid Beta
- AD
Alzheimer Disease
- NAC
N-acetyl-cysteine
- hAPPswe
human Swedish APP mutation
- BACE1
β-secretase
- CTx-B
cholera toxin subunit B
- N2a
neuroblastoma cells
- PS1
presenilin 1, gamma secretase sub-unit
- NO
nitric oxide
- nPM
nano particulate matter
- sAPP
soluble APP
- TRAP-PM
Traffic-related air pollution particulate matter
- 3NT
3-nitro-tyrosine
- 4-HNE
4-Hydroxy-2-nonenal, lipid peroxidation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cacciottolo M, et al. , Particulate air pollutants, APOE alleles and their contributions to cognitive impairment in older women and to amyloidogenesis in experimental models. Transl Psychiatry, 2017. 7(1): p. e1022 DOI: 10.1038/tp.2016.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen H, et al. , Exposure to ambient air pollution and the incidence of dementia: A population-based cohort study. Environ Int, 2017. 108: p. 271–277 DOI: 10.1016/j.envint.2017.08.020. [DOI] [PubMed] [Google Scholar]
- 3.Zhang X, Chen X, and Zhang X, The impact of exposure to air pollution on cognitive performance. Proc Natl Acad Sci U S A, 2018. 115(37): p. 9193–9197 DOI: 10.1073/pnas.1809474115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calderon-Garciduenas L, et al. , Hallmarks of Alzheimer disease are evolving relentlessly in Metropolitan Mexico City infants, children and young adults. APOE4 carriers have higher suicide risk and higher odds of reaching NFT stage V at </=40 years of age. Environ Res, 2018. 164: p. 475–487 DOI: 10.1016/j.envres.2018.03.023. [DOI] [PubMed] [Google Scholar]
- 5.Calderon-Garciduenas L, Reynoso-Robles R, and Gonzalez-Maciel A, Combustion and friction-derived nanoparticles and industrial-sourced nanoparticles: The culprit of Alzheimer and Parkinson’s diseases. Environ Res, 2019. 176: p. 108574 DOI: 10.1016/j.envres.2019.108574. [DOI] [PubMed] [Google Scholar]
- 6.Paul KC, et al. , Ambient Air Pollution, Noise, and Late-Life Cognitive Decline and Dementia Risk. Annu Rev Public Health, 2019. 40: p. 203–220 DOI: 10.1146/annurev-publhealth-040218-044058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peters R, et al. , Air Pollution and Dementia: A Systematic Review. J Alzheimers Dis, 2019. 70(s1): p. S145–S163 DOI: 10.3233/JAD-180631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Calderon-Garciduenas L, et al. , Long-term air pollution exposure is associated with neuroinflammation, an altered innate immune response, disruption of the blood-brain barrier, ultrafine particulate deposition, and accumulation of amyloid beta-42 and alpha-synuclein in children and young adults. Toxicol Pathol, 2008. 36(2): p. 289–310 DOI: 10.1177/0192623307313011. [DOI] [PubMed] [Google Scholar]
- 9.Levesque S, et al. , Air pollution & the brain: Subchronic diesel exhaust exposure causes neuroinflammation and elevates early markers of neurodegenerative disease. J Neuroinflammation, 2011. 8: p. 105 DOI: 10.1186/1742-2094-8-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim SH, et al. , Rapid doubling of Alzheimer’s amyloid-beta40 and 42 levels in brains of mice exposed to a nickel nanoparticle model of air pollution. F1000Res, 2012. 1: p. 70 DOI: 10.12688/f1000research.1-70.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhatt DP, et al. , A pilot study to assess effects of long-term inhalation of airborne particulate matter on early Alzheimer-like changes in the mouse brain. PLoS One, 2015. 10(5): p. e0127102 DOI: 10.1371/journal.pone.0127102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morgan TE, et al. , Glutamatergic neurons in rodent models respond to nanoscale particulate urban air pollutants in vivo and in vitro. Environ Health Perspect, 2011. 119(7): p. 1003–9 DOI: 10.1289/ehp.1002973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davis DA, et al. , Urban air pollutants reduce synaptic function of CA1 neurons via an NMDA/NOpathway in vitro. J Neurochem, 2013. 127(4): p. 509–19 DOI: 10.1111/jnc.12395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng H, et al. , Nanoscale Particulate Matter from Urban Traffic Rapidly Induces Oxidative Stress and Inflammation in Olfactory Epithelium with Concomitant Effects on Brain. Environ Health Perspect, 2016. 124(10): p. 1537–1546 DOI: 10.1289/EHP134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ehehalt R, et al. , Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J Cell Biol, 2003. 160(1): p. 113–23 DOI: 10.1083/jcb.200207113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chow VW, et al. , An overview of APP processing enzymes and products. Neuromolecular Med, 2010. 12(1): p. 1–12 DOI: 10.1007/s12017-009-8104-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paola D, et al. , Oxidative stress induces increase in intracellular amyloid beta-protein production and selective activation of betaI and betaII PKCs in NT2 cells. Biochem Biophys Res Commun, 2000. 268(2): p. 642–6 DOI: 10.1006/bbrc.2000.2164. [DOI] [PubMed] [Google Scholar]
- 18.Misra C, et al. , A high flow rate, very low pressure drop impactor for inertial separation of ultrafine from accumulation mode particles. Journal of Aerosol Science, 2002. 33(5): p. 735–752 10.1016/S0021-8502(01)00210-5. [DOI] [Google Scholar]
- 19.Hasheminassab S, et al. , Source apportionment and organic compound characterization of ambient ultrafine particulate matter (PM) in the Los Angeles Basin. Atmospheric Environment, 2013. 79: p. 529–539 DOI: 10.1016/j.atmosenv.2013.07.040. [DOI] [Google Scholar]
- 20.Saffari A, et al. , Seasonal and spatial variation of trace elements and metals in quasi-ultrafine (PM(0).(2)(5)) particles in the Los Angeles metropolitan area and characterization of their sources. Environ Pollut, 2013. 181: p. 14–23 DOI: 10.1016/j.envpol.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 21.Cheng H, et al. , Urban traffic-derived nanoparticulate matter reduces neurite outgrowth via TNFalpha in vitro. J Neuroinflammation, 2016. 13: p. 19 DOI: 10.1186/s12974-016-0480-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Woodward NC, et al. , Traffic-related air pollution impact on mouse brain accelerates myelin and neuritic aging changes with specificity for CA1 neurons. Neurobiol Aging, 2017. 53: p. 48–58 DOI: 10.1016/j.neurobiolaging.2017.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Babadjouni R, et al. , Nanoparticulate matter exposure results in neuroinflammatory changes in the corpus callosum. PLoS One, 2018. 13(11): p. e0206934 DOI: 10.1371/journal.pone.0206934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mullan M, et al. , A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of beta-amyloid. Nat Genet, 1992. 1(5): p. 345–7 DOI: 10.1038/ng0892-345. [DOI] [PubMed] [Google Scholar]
- 25.Allen JL, et al. , Early postnatal exposure to ultrafine particulate matter air pollution: persistent ventriculomegaly, neurochemical disruption, and glial activation preferentially in male mice. Environ Health Perspect, 2014. 122(9): p. 939–45 DOI: 10.1289/ehp.1307984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coburn JL, et al. , Acute exposure to diesel exhaust impairs adult neurogenesis in mice: prominence in males and protective effect ofpioglitazone. Arch Toxicol, 2018. 92(5): p. 1815–1829 DOI: 10.1007/s00204-018-2180-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dubal DB, et al. , Life extension factor klotho prevents mortality and enhances cognition in hAPP transgenic mice. J Neurosci , 2015. 35(6): p. 2358–71 DOI: 10.1523/JNEUROSCI.5791-12.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cai D, et al. , Presenilin-1 regulates intracellular trafficking and cell surface delivery of beta-amyloid precursor protein. J Biol Chem, 2003. 278(5): p. 3446–54 DOI: 10.1074/jbc.M209065200. [DOI] [PubMed] [Google Scholar]
- 29.Thinakaran G, et al. , Metabolism of the “Swedish” amyloid precursor protein variant in neuro2a (N2a) cells. Evidence that cleavage at the “beta-secretase” site occurs in the golgi apparatus. J Biol Chem, 1996. 271(16): p. 9390–7. [DOI] [PubMed] [Google Scholar]
- 30.Ignarro LJ, et al. , Oxidation of nitric oxide in aqueous solution to nitrite but not nitrate: comparison with enzymatically formed nitric oxide from L-arginine. Proc Natl Acad Sci U S A, 1993. 90(17): p. 8103–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Premasekharan G, et al. , Iron-mediated lipid peroxidation and lipid raft disruption in low-dose silica-induced macrophage cytokine production. Free Radic Biol Med, 2011. 51(6): p. 1184–94 DOI: 10.1016/j.freeradbiomed.2011.06.018. [DOI] [PubMed] [Google Scholar]
- 32.Macdonald JL and Pike LJ, A simplified methodfor the preparation of detergent-free lipid rafts. J Lipid Res, 2005. 46(5): p. 1061–7 DOI: 10.1194/jlr.D400041-JLR200. [DOI] [PubMed] [Google Scholar]
- 33.Otto GP and Nichols BJ, The roles of flotillin microdomains--endocytosis and beyond. J Cell Sci, 2011. 124(Pt 23): p. 3933–40 DOI: 10.1242/jcs.092015. [DOI] [PubMed] [Google Scholar]
- 34.Laudon H, Winblad B, and Naslund J, The Alzheimer’s disease-associated gamma-secretase complex: functional domains in the presenilin 1 protein. Physiol Behav, 2007. 92(1–2): p. 115–20 DOI: 10.1016/j.physbeh.2007.05.037. [DOI] [PubMed] [Google Scholar]
- 35.Catafau AM, et al. , Cerebellar Amyloid-beta Plaques: How Frequent Are They, and Do They Influence 18F-Florbetaben SUV Ratios? J Nucl Med, 2016. 57(11): p. 1740–1745 DOI: 10.2967/jnumed.115.171652. [DOI] [PubMed] [Google Scholar]
- 36.Youmans KL, et al. , APOE4-specific changes in Abeta accumulation in a new transgenic mouse model of Alzheimer disease. J Biol Chem, 2012. 287(50): p. 41774–86 DOI: 10.1074/jbc.M112.407957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chinnapen DJ, et al. , Rafting with cholera toxin: endocytosis and trafficking from plasma membrane to ER. FEMS Microbiol Lett, 2007. 266(2): p. 129–37 DOI: 10.1111/j.1574-6968.2006.00545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Appleyard WJ, National Health Service White Paper. Will it work for patients? Arch Dis Child, 1989. 64(4): p. 643–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tan JL, et al. , Mild oxidative stress induces redistribution of BACE1 in non-apoptotic conditions and promotes the amyloidogenic processing of Alzheimer’s disease amyloid precursor protein. PLoS One, 2013. 8(4): p. e61246 DOI: 10.1371/journal.pone.0061246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guix FX, et al. , Modification of gamma-secretase by nitrosative stress links neuronal ageing to sporadic Alzheimer’s disease. EMBO Mol Med, 2012. 4(7): p. 660–73 DOI: 10.1002/emmm.201200243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Picon-Pages P, Garcia-Buendia J, and Munoz FJ, Functions and dysfunctions of nitric oxide in brain. Biochim Biophys Acta Mol Basis Dis, 2019. 1865(8): p. 1949–1967 DOI: 10.1016/j.bbadis.2018.11.007. [DOI] [PubMed] [Google Scholar]
- 42.Oda A, Tamaoka A, and Araki W, Oxidative stress up-regulates presenilin 1 in lipid rafts in neuronal cells. J Neurosci Res, 2010. 88(5): p. 1137–45 DOI: 10.1002/jnr.22271. [DOI] [PubMed] [Google Scholar]
- 43.Carlsten C, et al. , Antioxidant N-acetylcysteine diminishes diesel exhaust-induced increased airway responsiveness in person with airway hyper-reactivity. Toxicol Sci, 2014. 139(2): p. 479–87 DOI: 10.1093/toxsci/kfu040. [DOI] [PubMed] [Google Scholar]
- 44.Grimm MO, et al. , Oxidized Docosahexaenoic Acid Species and Lipid Peroxidation Products Increase Amyloidogenic Amyloid Precursor Protein Processing. Neurodegener Dis, 2016. 16(1-2): p. 44–54 DOI: 10.1159/000440839. [DOI] [PubMed] [Google Scholar]
- 45.Siegel SJ, et al. , The oxidative stress metabolite 4-hydroxynonenal promotes Alzheimer protofibril formation. Biochemistry, 2007. 46(6): p. 1503–10 DOI: 10.1021/bi061853s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang X, et al. , Uptake of silica nanoparticles: neurotoxicity and Alzheimer-like pathology in human SK-N-SH and mouse neuro2a neuroblastoma cells. Toxicol Lett, 2014. 229(1): p. 240–9 DOI: 10.1016/j.toxlet.2014.05.009. [DOI] [PubMed] [Google Scholar]
- 47.Forman HJ and Finch CE, A critical review of assays for hazardous components of air pollution. Free Radic Biol Med, 2018. 117: p. 202–217 DOI: 10.1016/j.freeradbiomed.2018.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gonzalez-Maciel A, et al. , Combustion-Derived Nanoparticles in Key Brain Target Cells and Organelles in Young Urbanites: Culprit Hidden in Plain Sight in Alzheimer’s Disease Development. J Alzheimers Dis, 2017. 59(1): p. 189–208 DOI: 10.3233/JAD-170012. [DOI] [PubMed] [Google Scholar]
- 49.Oberdorster G, et al. , Translocation of inhaled ultrafine particles to the brain. Inhal Toxicol, 2004. 16(6-7): p. 437–45 DOI: 10.1080/08958370490439597. [DOI] [PubMed] [Google Scholar]
- 50.Mumaw CL, et al. , Microglial priming through the lung-brain axis: the role of air pollution-induced circulating factors. FASEB J, 2016. 30(5): p. 1880–91 DOI: 10.1096/fj.201500047. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.