

ABSTRACT
Phagocytosis is a dynamic process central to immunity and tissue homeostasis. Current methods for quantification of phagocytosis largely rely on indirect or static measurements, such as target clearance or dye uptake, and thus provide limited information about engulfment rates or target processing. Improved kinetic measurements of phagocytosis could provide useful, basic insights in many areas. We present a live-cell, time-lapse and high-content microscopy imaging method based on the detection and quantification of fluorescent dye ‘voids’ within phagocytes that result from target internalization to quantify phagocytic events with high temporal resolution. Using this method, we measure target cell densities and antibody concentrations needed for optimal antibody-dependent cellular phagocytosis. We compare void formation and dye uptake methods for phagocytosis detection, and examine the connection between target cell engulfment and phagolysosomal processing. We demonstrate how this approach can be used to measure distinct forms of phagocytosis, and changes in macrophage morphology during phagocytosis related to both engulfment and target degradation. Our results provide a high-resolution method for quantifying phagocytosis that provides opportunities to better understand the cellular and molecular regulation of this fundamental biological process.
KEY WORDS: Phagocytosis, Macrophage, High-content imaging, Phagolysosome, Time-lapse video, Quantification
Summary: Engulfment enumeration using high-content microscopy images of live cells phagocytosing in real time provides unprecedented high-resolution temporal data that allows new insights into this fundamental biological process.
INTRODUCTION
Phagocytosis is a fundamental cellular process whereby a cell engulfs large particles, such as microbes and other cells, which can serve many purposes, including nutrition, tissue remodeling and cellular defense (Davies et al., 2013; Flannagan et al., 2012; Sharma et al., 2018; Tauber, 2003). In large multicellular organisms, professional phagocytic cells, such as macrophages, provide innate immune protection against pathogens (Greenberg and Grinstein, 2002; Okabe and Medzhitov, 2016; Wynn et al., 2013). In more evolved animal species with adaptive immunity, phagocytic cells can engulf antibody-coated targets, including specifically targeted pathogens (Aderem and Underhill, 1999; Nimmerjahn and Ravetch, 2008; Zent and Elliott, 2017). In humans, monoclonal antibody (mAb) therapy takes advantage of this process of antibody-dependent cellular phagocytosis (ADCP) to treat various forms of cancer and autoimmune diseases, such as immune thrombocytopenia and hemolytic anemia (Imbach et al., 1981; Pottier et al., 1996; Semple and Freedman, 2005; Zent and Kay, 2010). Studies of mAb treatment in hematological malignancies, such as chronic lymphocytic leukemia (CLL), demonstrate an initial rapid loss of targeted cancer cells, which quickly (within hours) becomes resistant to further mAb therapy (Baig et al., 2014; Beurskens et al., 2012). Furthermore, more recent studies have demonstrated a pivotal role of macrophages in malignant cell clearance for the therapeutic efficacy of these treatments (Grandjean et al., 2016; Gul and van Egmond, 2015; Montalvao et al., 2013; Uchida et al., 2004; VanDerMeid et al., 2018; Weiskopf and Weissman, 2015).
However, phagocytosis is a highly dynamic process that requires rapid, large-scale alterations in phagocyte morphology to achieve target internalization and degradation. Thus, improvements in phagocytosis quantification methods are needed in order to achieve the requisite resolution for accurately assessing cellular engulfment kinetics. Historically, quantification of phagocytosis has been carried out either by fixing macrophages at specific time points after induction of phagocytosis for visualization by microscopy, measuring the amount of dye-labeled target material internalized by a phagocyte by imaging or flow cytometry (‘dye uptake’ assays), or by calculating the number of target cells remaining after a defined time of phagocytosis (‘cells remaining’ assays) (Fig. 1A) (Beletskii et al., 2005; Choy and Botelho, 2017; Kamen et al., 2019; Church et al., 2016; Gu et al., 2014; Lehmann et al., 2000; Sharma et al., 2014). As these conventional approaches are typically carried out on samples collected at relatively broad time intervals (e.g. 0.5–1.0 h intervals), they provide only coarse temporal resolution of phagocytic activity and fail to capture the minute-to-minute changes that occur during phagocytosis. Additionally, some of these strategies rely on indirect readouts to measure phagocytosis, such as dye uptake (derived from labeled target cells) or quantification of ‘free’ target cells remaining after co-incubation with phagocytes. Quantifying phagocytosis by target cell dye uptake, where phagocytosis is a function of the amount of target dye detected within isolated phagocytes, is indirect because the increase in target dye within the phagocyte could be due to passive dye transfer. Furthermore, target cell dye uptake cannot be used to precisely determine the number of phagocytic events due to variability in target labeling. There are more direct methods that utilize target fluorescence to measure phagocytosis, such as the use of fluorescent particles whose homogenous size, shape, and non-transferrable fixed fluorescent intensity allows one to more confidently calculate the number of phagocytic events. Fluorescent particles or beads, however, are inert non-degradable targets that are unsuitable for studying target processing and the process of target degradation following internalization. The advent of pH-sensitive dyes to label target cells (Beletskii et al., 2005; Kapellos et al., 2016; Kamen et al., 2019) allows high temporal resolution measurements of phagocytosis by reducing the background of ‘uneaten’ target cells, because dye intensity is only produced when the target cell reaches an acidic environment, such as found in the endolysosomal compartment. Thus, dye intensity can be measured repeatedly in a single sample without the need to isolate phagocytes. However, this method is an indirect measure of phagocytic engulfment events and will show an initial delay in phagocytosis kinetics due to the need for endolysosomal processing to produce a signal.
Fig. 1.

Establishment of live cell time-lapse high-content microscopy imaging method to measure phagocytic engulfment events. (A) Overview of phagocytosis measurement methods. (B) Imaging method workflow. (C) Representative images from an initial ADCP experiment with BMDMs co-cultured with CLL cells (10:1 ratio of target cells to BMDMs) and anti-CD52 (10 µg/ml) showing all color channels merged at an initial time point without target cells (left) and the 1-h time point (right) after target cells and mAb addition. Scale bar: 50 μm. The entire time-lapse is shown as Movie 1. (D) Macrophage binary mask application (green) illustrated for the same representative images shown in C. The entire time-lapse video with macrophage binary mask is shown as Movie 2. Yellow boxes (C,D) indicate the magnified region shown in Fig. 2. Macrophages were labeled with TAMRA (red) and target cells were labeled with CypHer5 (purple).
Recent advances in microscopy such as coverslip-displacement interferometry methods (e.g. ‘perfect focus’) to maintain focus over long time periods, LED light sources to precisely deliver and maintain desired light wavelengths, high-speed digital image acquisition with increased sensitivity, cellular dye-labeling reagents, imaging analysis software (such as improved algorithms to detect multiple specific objects in sequential images) and computational resources have allowed better visualization of cell processes such as phagocytosis (Montaño et al., 2018; Steinberg et al., 2007; Yeo et al., 2013). To improve upon current techniques and advance our understanding of phagocytosis kinetics, we combined some of these advances, including a stable-focusing method, programmable stages and filters, LEDs, imaging software, and pH-sensitive dyes to directly measure live-cell phagocytosis in real-time at a moderate to high-throughput with multiple samples and conditions. This resulting high-resolution measurement of discrete phagocytic events could be used to provide deeper insight into the general biological processes that coincide with cellular engulfment, as well as lead to the development of improved mAb therapies that rely on these processes as the principal mechanism of action.
RESULTS
Strategy for high-resolution measurement of discrete phagocytic events by live-cell time-lapse high-content microscopy imaging
We introduce here a new quantification strategy utilizing the dye exclusionary areas (‘voids’) that form when a dye-labeled phagocyte, in our example, a macrophage, internalizes target cells (Fig. 1A). This method complements previous ‘dye uptake’ and ‘cells remaining’ methods, and provides additional improved phagocytic measurements (Fig. S1). For our process (Fig. 1B), macrophages are seeded and allowed to adhere to the bottom of plastic tissue culture multiwell plates, and subsequently labeled with cell-permeable fluorescent dyes (e.g. Cell Tracker, eFluor670 or TAMRA-SE). Target cells, either unlabeled or labeled with pH-sensitive fluorescent dyes (e.g. CypHer5 or pHrodo), are then co-cultured with these macrophages. To initiate ADCP, mAb is added to these cultures. Live-cell imaging is performed using a Nikon Eclipse Ti Live-cell Imaging System with a motorized, environmentally controlled microscope stage programmed to image multiple fluorescence channels at multiple xy coordinates over specified time intervals using the ‘perfect focus’ feature of this microscope. The resulting image data is too vast to manually enumerate phagocytic events across hundreds of cells and time points easily. For example, a typical time-lapse video from a single xy field view contains ∼50–100 macrophages with as many as 20 internalization events per phagocyte. Furthermore, if we conduct a 4 h experiment with an image taken once every 2 min for 12 different xy positions (or wells), that would involve counting ∼1.4×106 individual engulfment events. Additionally, the use of automation reduces observer bias in the enumeration process. Therefore, we developed a user-guided, semi-automated process utilizing various functions within the Nikon NIS Elements imaging software to conduct robust enumeration of large numbers of phagocytic events in numerous macrophages across multiple samples for many hours. Finally, from these data, we developed and applied several index calculations to quantify phagocytosis.
Development of high-resolution temporal measurement of discrete phagocytic events reveals a delay in dye uptake
The difficulty in identifying and quantifying discrete phagocytic events in co-culture experiments by microscopy is illustrated in Fig. 1C and Movie 1. As is apparent in the merged image (phase and fluorescence overlaid) of the 60 min time point (Fig. 1C), non-phagocytosed target cells can cluster around individual macrophages, obscuring the macrophages and thus making identification and counting of discrete phagocytic events very difficult. Furthermore, one can appreciate, in this field of view, the laborious effort needed to enumerate phagocytic events for each individual macrophage across all frames over time. Our method overcomes these problems by first identifying and counting the macrophages in each image by creating a macrophage binary mask derived from the fluorescence signal from each macrophage in the field (Fig. 1D; Movie 2). The macrophage count allows us to normalize the measured engulfment events per field, and, therefore, compare levels of phagocytosis across a range of experimental conditions. Next, individual phagocytic events are counted as voids formed within dye-labeled macrophages, which is best illustrated by close-up images (Fig. 2; Movies 3–7). Even in close-up merged images (Fig. 2A, Movie 3), the difficulty in identifying internalized (versus bound) targets is apparent. To resolve internalized targets, we then separate the merged images into the component channels, as shown in Fig. 2A: differential phase contrast (Phase, Movie 4), macrophage dye label (TAMRA, Movie 5) and target cell dye label (CypHer5, Movie 6). In the Phase channel, non-phagocytosed cells obscure macrophages, making enumeration of phagocytic events extremely difficult. In the TAMRA channel, only the macrophages are prominently visible and the non-engulfed target cells no longer obstruct a clear view of the macrophage. In this situation, individual phagocytic events can be clearly visualized as dark voids within the labeled macrophages (Fig. 2A). As seen in Fig. 2A, CypHer5 signal from the target cells is often problematic for ascertaining individual phagocytic events due to diffuse signal within the macrophage, uneven labeling of target cells and signal from unengulfed target cells overlaying macrophages, as well as the delay in kinetics relative to actual phagocytic events due to the time required for pH compartment acidification (Kapellos et al., 2016; Neaga et al., 2013). Thus, void counts are a more sensitive and instantaneous measure of phagocytic uptake. Furthermore, imaging software can automatically count voids after generating a user-defined void binary mask (Fig. 2B; Movie 7), which provides high-resolution temporal data (Fig. 2C, left). The void count kinetics revealed a rapid initial rate of engulfment, a phenomenon suggested previously (Church et al., 2016), and then a subsequent decrease in voids, which implies an increased rate of target degradation that eventually outpaces additional target internalization. It is important to note that the high-throughput nature of our void counting method makes it impossible to individually assess some of the more subtle or transient phases of phagocytosis, such as phagosome sealing. While our method cannot specifically identify voids arising from incomplete phagosome sealing, the Nikon SpotDetection method we use to count the voids looks for circular events that are of a user-designated contrast threshold to the environment surrounding the void. This means the quantification will be biased towards closed voids nearer to the center of the macrophage cell body. Moreover, as discussed in detail below, we find a high degree of spatiotemporal correlation between voids and fluorescence from target cells labeled with the pH-sensitive dye pHrodo Red, indicating that the vast majority of voids detected arise from fully closed phagosomes.
Fig. 2.

Image close-ups from movies illustrate the void count of phagocytic engulfment events, and high-resolution measurements show a delay in dye uptake versus actual engulfment events. (A) Image close-ups of experiment shown in Fig. 1 at the initial time point (starting at the left) and subsequent 30-min intervals up to 90 min. Channels are shown together (Merged) as well as individually (Phase, TAMRA and CypHer5). Scale bar: 25 μm. (B) Voids binary mask (yellow) on the magnified TAMRA images shown in A as identified by the ‘Spot Detection’ function from the NIS-Elements image analysis software. (A,B) Videos of each magnified channel and merge, as well as voids binary mask application, are included in Movies 3–7. (C) Graphical representations of quantitative temporal data obtained from binary masks including void counts (left), dye uptake (center) and macrophage counts (right). Macrophages were labeled with TAMRA (red) and target cells were labeled with CypHer5 (purple).
High-resolution temporal data for dye uptake can also be obtained from these images (Fig. 2C, center) by measuring the CypHer5 intensity within the macrophage binary mask. Compared to void count kinetics, there is a delay in reaching maximum dye intensity. This delay is likely due to the time required for phagolysosomal acidification to cause an increase in dye intensity. This difference between void count and dye uptake kinetics highlights the ability of the void method in measuring phagocytic engulfment events, without requiring phagolysosomal processing. Quantification of the macrophage binary mask revealed that the macrophage counts were consistent over time suggesting high accuracy and consistency between images (Fig. 2C, right; Fig. S2). To enable void count or dye uptake comparisons between samples, we normalized these measurements to the number of macrophages, to calculate a void or dye intensity index in subsequent experiments.
High-resolution measurements of phagocytic events provide detailed data to assess the effect of target cell numbers on ADCP
Using this high-resolution assay, we tested the effect of target cell numbers on ADCP by varying the target cell to effector macrophage ratio (T:E) from 2:1 to 80:1 (Fig. 3A,B). We found that regardless of T:E ratio, the overall shape of the void count traces and the time it took to reach the maximum void count were similar. The initial phagocytic rate, however, was much lower at a T:E ratio of 2:1 and peaked at a T:E of 10:1 (Fig. 3B). Quantification of total phagocytosis, using area under the curve (AUC) or void maximum, also showed that significantly less phagocytosis occurred at a 2:1 ratio and maximum phagocytosis peaked at 10:1 (Fig. 3B). Furthermore, as we approached a T:E ratio of 80:1, we saw a downward trend in overall phagocytosis, suggesting that the antibody may have become a limiting reagent due to the high number of target cells (Fig. S3, Movie 8). Our findings suggest, therefore, that an optimal T:E ratio for maximizing ADCP ranges somewhere between 5:1 and 40:1 and at ≤2:1 the lack of targets may limit the total number of engulfment events.
Fig. 3.

High-resolution measurements of phagocytic engulfment events used to assess effect of variations in target cell numbers or mAb concentrations. (A,B) Overall phagocytosis and its kinetics remain unchanged after sufficient target cell numbers are reached. Mouse BMDMs were co-cultured with varying target murine thymocytes to effector macrophage ratios (T:E ratios, ranging from 2:1 to 80:1) for 3 h in the presence of 10.0 µg/ml anti-CD90.2. (A) Normalized void counts (Void index) were calculated as described in text. The mean of two independent tests (T:E = 2:1) or mean+s.e.m. of three independent tests (all other T:E ratios) are shown. (B) These data were used to derive two kinetic measures: (1) the rate of void accumulation, or slope, of the initial 30 experimental minutes (top left) and (2) the time until the maximum void number was reached (top right), and two overall phagocytosis measures: (1) the AUC (bottom left), and (2) the maximum number of voids reached (bottom right) for each target to effector ratio tested. The mean of two independent tests (T:E = 2:1) or mean±s.e.m. of three independent tests (all other T:E ratios) are shown. (C,D) Decreasing mAb concentrations result in a lower overall phagocytosis and decreased kinetics. (C) Mouse BMDMs were co-cultured with murine thymocytes at a 10:1 T:E ratio, for 2 h in the presence of various anti-CD90.2 concentrations that started at 10.0 µg/ml and included three-fold titrations down to 0.12 µg/ml. The calculated void index is shown (mean+s.e.m. or three independent tests). (D) Kinetic and overall phagocytosis measures of these data (mean±s.e.m. of three independent tests) are shown as in B. *P≤0.05, **P≤0.01 (one-way ANOVA compared to 0.12 µg/ml using multiple comparisons).
Effect of mAb concentration on ADCP assessed by high-resolution measurements of phagocytic events
The optimal concentration for a given antibody to induce ADCP can vary greatly, and a great deal of research has been performed to increase antibody potency and efficacy by altering the antibody structure or glycosylation to enhance antigen or receptor affinities (Forero-Torres et al., 2012; Mössner et al., 2010; Tobinai et al., 2011). It is imperative, therefore, to have a sensitive method that can accurately assess the effects of antibody dose on phagocytosis. Using mouse thymocyte targets (10:1 T:E ratio), we tested the effect of various anti-CD90.2 mAb concentrations on phagocytosis of murine thymocytes by mouse bone-marrow-derived macrophages (BMDMs) (Fig. 3C,D). Interestingly, we observed slowed void accumulation and flattening of the void kinetic curves as antibody concentration decreased (Fig. 3C). We also found that all phagocytosis metrics improved with increasing antibody concentrations (Fig. 3D). Although we did not reach a saturating dose, the change in phagocytosis from 0 to 3.33 µg/ml was much greater than the change from 3.33 to 10 µg/ml, suggesting we were approaching an optimal concentration.
Phagolysosomal processing impacts dye uptake, but not void enumeration measurement
The SpotDetection method not only allows for the number of voids to be counted but can also be utilized to measure the relative levels of target fluorescence within the binary spot mask. We used the void binary mask to more accurately measure dye uptake instead of quantifying the target cell dye label in macrophage binary masks, which can be inaccurate due to non-engulfed target cells overlaying macrophages (Fig. S4A). Furthermore, we dilated the void binary masks to maximize the amount of target fluorescence captured within the binary mask and more reproducibly quantify target cell dye label from engulfed cells (Fig. S4A, data not shown). To test the role of phagolysosomal processing, we treated macrophages with or without bafilomycin A1 (Baf), a V-ATPase inhibitor that blocks phagolysosomal acidification (Fig. 4A–E) (Yoshimori et al., 1991). Target cells were labeled with a pH-sensitive dye, pHrodo Red, that increases in fluorescence intensity at low pH, such as found in the endolysosomal cellular compartments. As expected, the increase in target cell pHrodo Red intensity within the voids was nearly completely blocked with the addition of Baf (Fig. 4A,D,E, left; Movie 9). Void integrity was maintained for much longer after Baf treatment (Fig. 4B,E, right), confirming that the reduction in voids seen after peak void count is likely due to target degradation in the phagolysosomal compartment. Interestingly, Baf did not have a dramatic effect on initial rate, the maximum number of voids or time to void maximum (Fig. 4C, Fig. S4B), suggesting target processing does not play a large role in defining total cell engulfment.
Fig. 4.

Differences between high-resolution phagocytic analysis of engulfment events and measurement of pH-sensitive dye uptake. (A–E) Inhibition of endolysosomal acidification blocks measurement of pH-sensitive dye uptake, but not phagocytic engulfment events. Mouse BMDMs were co-cultured with murine thymocytes for 4 h in the presence of 10.0 µg/ml anti-CD90.2 with or without Bafilomycin A1 (Baf). (A) Representative magnified images depicting pHrodo Red intensity of target thymocytes within Cell Tracker Deep Red (CTDR)-labeled macrophage voids, following phagocytosis in the presence or absence of Baf. Scale bars: 25 μm. (B) Plot of void index (mean+s.e.m. of three independent tests). (C) These data used to calculate the void rate in the first 30 min (left) and the void maximum (right). Data shown are mean±s.e.m. of three independent tests. (D) Dye uptake index based on pHrodo Red intensity (mean+s.e.m. of three independent tests). (E) Measurement of the AUC for dye uptake index (left) or void index (right) (mean±s.e.m. of three independent tests) illustrating the opposite effects of Baf on dye uptake versus phagocytic engulfment events. (F–H) ADCP leads to more phagocytic engulfment events than efferocytosis, but less dye uptake intensity. Mouse BMDMs were co-cultured for 1.5 h with either live thymocytes in the presence of 10.0 µg/ml anti-CD90.2 or dexamethasone-treated apoptotic thymocytes. (F) Representative magnified images depicting target cell pHrodo Red intensity within voids of CTDR-labeled macrophages following ADCP or efferocytosis of apoptotic cells. Scale bars: 25 μm. (G) ADCP void index (left y-axis) and dye intensity per void (right y-axis). Data shown are mean+s.e.m. of four and three independent tests, respectively. (H) Efferocytosis (Effero) void index (left y-axis) and dye intensity per void (right y-axis). Data shown are mean+s.e.m. of four and three independent tests, respectively. (I) Kinetic measurements of these data (mean±s.e.m. of four independent tests) as shown by void rate in the first experimental 30 min illustrating differences between ADCP and efferocytosis. *P≤0.05, **P≤0.01 (two-tailed Student's t-test).
We did observe the persistence of low intensity pHrodo signal in BafA1-treated macrophages (Fig. 4D), but do not believe this is due to BafA1-insensitive acidification events. Rather, we believe that the low level signal is due to a combination of additive background fluorescence in macrophages that have internalized multiple labeled targets and heterogeneity in target cell labeling where a small percentage of target cells display high baseline pHrodo intensity prior to engulfment, and therefore will provide substantial pHrodo signal upon engulfment even when target acidification is inhibited.
We also asked whether our methodology could capture, in real-time, the kinetics of void formation and phagolysosomal acidification for two distinct phagocytic pathways, ADCP and efferocytosis. We found that our method was able to assess the void and dye index kinetics simultaneously following ADCP and efferocytosis. Furthermore, by comparing the void index changes of both pathways, we discovered that efferocytosis occurs at a lower rate than ADCP (Fig. 4F–I; Fig. S4C). While we were not able to directly compare the rates of target degradation between ADCP and efferocytosis, due to these very different rates of phagocytosis, we did observe that each efferocytic phagolysosome appeared to show higher levels of phagosome acidification (i.e. higher pHrodo Red intensity per void) compared to ADCP (Fig. 4F–I; Movies 10,11). In efferocytosis, greater levels of acidification may be important for enabling the rapid degradation and turnover of potentially noxious dead cell materials that can spur immune activation and inflammation (Elliott and Ravichandran, 2010).
Macrophage mask parameter changes correlate with target cell engulfment and processing kinetics
Our high-content video imaging method allows more detailed analyses of phagocytosis. For example, we can utilize the areas measured from the macrophage binary mask to characterize gross changes in macrophage morphology during phagocytosis. While not a direct measure of macrophage ‘size’ or total volume, use of these ‘observed macrophage mask’ parameters enabled us to relate the changes in macrophage morphology with the void index during ADCP. We found that as void numbers increased, the observed macrophage mask parameters also increased, but as the internalized material was processed, the macrophage mask parameters began to decrease to baseline (Fig. 5A). If we overlaid the high-resolution temporal plots of change in macrophage mask parameters over the void index, the graphs were nearly indistinguishable (Fig. 5B). Kinetic measurements derived from these data, time to maximum and return to half maximum (for change in observed mask parameters or void index) are not significantly different (Fig. 5C). These similarities were lost if there was no antibody present (Fig. S5), which indicates that the relationship was dependent upon ADCP. In fact, there was a close linear correlation between changes in observed macrophage mask parameters and void index with an R2 value above 0.9 (Fig. 5D). These results reveal that, although phagocytosis causes the phagocyte cell body to spread as targets are processed, the macrophage can return to baseline, which suggests that factors governing cell morphology and spreading are flexible in regard to the amount a given cell engulfs.
Fig. 5.

High-resolution measurement of phagocytic engulfment events enables quantification of observed macrophage mask parameters. (A–D) Observed macrophage mask parameters increase and decrease proportionately to the number of phagocytic engulfment events. CTDR-labeled mouse BMDMs were co-cultured with murine thymocytes for 1.5 h in the presence of 10.0 µg/ml anti-CD90.2. (A) Representative magnified images depicting voids in macrophages over three different time-points (left column) and the corresponding macrophage mask parameter calculated using binary masking (right column). Scale bars: 25 µm. (B) Plots of change in observed macrophage mask parameters (ΔMask, left y-axis) and void index (right y-axis). Data shown are mean+s.e.m. of three independent tests. (C) Kinetic measurements of the time to reach maximum voids or observed macrophage mask parameter (left) and the time to return to the half-maximum voids or macrophage mask parameter (right) are comparable. Data shown are mean±s.e.m. of three independent tests. (D) Best fit line for linear regression analysis of mean void index (y-axis) versus mean change in macrophage mask parameter (ΔMask, x-axis) for each time point from B (R2=0.9213). *P≤0.05, **P≤0.01 (two-tailed Student's t-test).
DISCUSSION
Phagocytosis is an important functional readout for many phagocytic cell types that plays vital roles in a variety of contexts including basic cell functionality, polarization, infection and therapeutics (Davies et al., 2013; Flannagan et al., 2012; Sharma et al., 2018; Tauber, 2003). It is thus important to have a sensitive method that can reproducibly assess phagocytosis in real-time. Additionally, the ability of our imaging method to detect target processing could provide important new insights into connections between target engulfment and degradation, phagocyte heterogeneity, the processing of different cellular materials and the differential processing capacities of separate phagocytic pathways. As with any method there are, however, limitations. Our void counting method also still lacks the ability to discriminate between new voids and old voids and track individual void persistence. Real-time tracking of individual voids should be available in the near future using more sophisticated software programs.
One could also envision the formation of voids due to cellular processes other than phagocytosis, such as macropinocytosis. We believe that our method is not appreciably affected by detection of macropinosomes for the following reasons. First, in the absence of target cells, few if any voids are observed suggesting very little void formation due to liquid uptake. Furthermore, our void detection method is set to detect dark spots roughly the diameter of our target cells (4–8 µm), which is a size largely inconsistent with micropinocytosis, which will give a range in sizes from ∼250 nm to 5 µm (Canton, 2018; Swanson and Watts, 1995). Finally, there is a tight spatial correlation between voids and target dye accumulation, again suggesting that void formation is the result of target cell internalization.
Another limitation of our void method is that it lacks the resolution to specifically identify material internalized via trogocytosis – the process of cellular ‘gnawing’ that is important to consider when assessing therapeutic mAb action. Trogocytosis typically results in the uptake of very small pieces of target membrane that can result in the loss of relevant target antigen without having a cytotoxic effect on the target (Joly and Hudrisier, 2003). Real-time methods to accurately assess trogocytosis are important to future mAb characterization and optimization efforts and, although our method currently lacks trogocytosis quantification capabilities, strategies like ours in combination with fluorescent antibodies or antigen to track target membrane uptake should prove useful in the future.
We believe our method greatly improves upon the standard strategies for measuring phagocytosis by quantifying individual engulfment events in real-time over a scale that previous techniques have not achieved. Furthermore, the high sensitivity and post-internalization assessments that our method allows ensure all phases of phagocytosis are captured, which could deepen our understanding of the entire phagocytic process. In conclusion, we propose that this new method could enhance our knowledge of the underlying mechanisms of numerous autoimmune diseases as well as improve current mAb therapeutics.
MATERIALS AND METHODS
Mice
Animal experiments were approved by the University of Rochester Animal Care and Use Committee and mice were housed under specific pathogen-free conditions. The C57BL/6J mouse strain was obtained from Jackson Labs (Bar Harbor, ME). Thymocytes and bone marrow were isolated from male and female mice of between 4 and 20 weeks of age. All animal procedures were reviewed and approved by the University of Rochester Committee on Animal Resources.
Tissue culture
Mouse bone marrow-derived macrophages (BMDMs) were prepared as previously described (Manzanero, 2012). Briefly, cells isolated from the femurs of both male and female C57BL6 mice between the ages of 5 and 15 weeks old were plated on untreated Petri dishes in RFHP10 [10% heat inactivated fetal bovine serum (Atlanta Biologicals), RPMI 1640, 10 mM HEPES, 1% penicillin-streptomycin-L-glutamine (Cellgro)] supplemented with 20 ng/ml macrophage colony-stimulating factor (M-CSF; BioLegend) and incubated for 5 days at 37°C and 5% CO2. On day 5, medium was exchanged for new M-CSF-supplemented RFHP10. The resulting BMDMs were harvested on days 6–9 by trypsinization with 0.25% trypsin and 0.1% EDTA (Corning), washed twice in RFHP10, re-plated at 5×104 cells/well in 24 well-tissue culture plates (Falcon) in 1 ml RFHP10, and incubated overnight at 37°C and 5% CO2 prior to phagocytosis assays.
Target cell preparation
Thymocytes were isolated from the thymus of C57BL6 mice by mashing the thymus in 10 ml RFHP10 through a 40 µM strainer, after using forceps and a Kimwipe to remove any residual blood from the organ. Following straining, the thymocytes were ready for use. Apoptotic thymocytes were prepared by 3–4 h treatment with 10 µM dexamethasone (Thermo Fisher Scientific) as previously described (Murphy et al., 2017). Human CLL cells were isolated from peripheral blood specimens of treatment naïve patients and purified to at least 90% CD5+CD19+ CLL cells and cultured as previously described (Baig et al., 2014; Church et al., 2016; VanDerMeid et al., 2018). All human specimen collection and usage was conducted with written informed consent after approval of the Institutional Research Subjects Review Board according to the ethical guidelines of the Declaration of Helsinki.
Dye labeling
Macrophages
Before dye labeling, BMDMs were washed twice in Hanks’ balanced salt solution (HBSS) without cations (HBSS−/−). The appropriate dye was then diluted in HBSS−/− and 500 µl were added to each well. In all of the experiments shown here, the macrophages were dye-labeled with either 2.5 µM 5-TAMRA, SE (TAMRA, Invitrogen) or 5 µM Cell Tracker Deep Red (CTDR, Thermo Fisher Scientific). After the addition of the dye BMDMs were incubated for ∼20 min at 37°C and 5% CO2 with tapping of the plate half-way through the incubation time. Following the 20 min incubation, the macrophages were washed with RFHP10 medium three times, then 500 µl was added to each well, and the plates were reincubated for ∼2 h at 37°C and 5% CO2 before use.
Target cells
Isolated target cells in RFHP10 were centrifuged at 450 g for 5 min and resuspended in ∼10 ml HBSS−/− followed by a second centrifugation. The resulting cell pellet was resuspended in ∼2 ml HBSS−/− and added to 2 ml 5 µM CypHer5E Mono NHS Ester (CypHer5, GE Life Sciences), or 10 µM pHrodo iFL Red STP ester (pHrodo Red, Thermo Fisher Scientific) diluted in HBSS−/− to reach a final dye concentration of 2.5 or 5 µM, respectively. After addition of dye, target cells were placed at 37°C for 20 min with inversion of the tube halfway through the incubation. RFHP10 was then added to the target cells to reach a final volume of 15 ml. Cells were recentrifuged and the cell pellet resuspended in 2–5 ml RFHP10.
Phagocytosis assays
Target cells were added at a 10:1 T:E ratio to dye-labeled macrophages unless otherwise indicated. For ADCP, mAb [anti-mouse CD90.2 (clone 30-H12, BioLegend) or (anti-human CD52, Alemtuzumab, Sanofi Genzyme) for murine thymocyte or human CLL cell targets, respectively] was added at 10 µg/ml final concentration unless otherwise specified. To inhibit phagolysosomal acidification in this assay, 10 µM Bafilomycin A1 (Millipore) was added to macrophages for 1 h prior to, and then continued after, adding target cells.
Live-cell imaging acquisition
Live-cell imaging was performed using a stage-top environmental chamber that maintained the cells at 37°C in 5% CO2 mounted onto a Nikon Ti-Eclipse inverted microscope. Image capture was conducted using the NIS Elements imaging software. Within each experiment a single image xy coordinate was selected for each well for up to 12 wells on a 24-well tissue culture plate. The imaging field chosen was that nearest to the middle of each well (for optimal focus) but where BMDM cell density still allowed for sufficient space between cells. An image loop, where images were captured for each coordinate entered, was taken every 1.5 to 2 min for 2–12 h depending on the experiment. For each coordinate, a single image was taken for phase contrast and each of the appropriate fluorescent filters [APC (CypHer5 or CTDR), and TRITC (TAMRA or pHrodo Red)]. For every experiment, at least three images were taken before antibody addition (i.e. prior to ADCP) or before apoptotic target cell addition (i.e. prior to efferocytosis) to measure background for each condition. There is a short time delay after background acquisition to account for subsequent additions.
Macrophage masking
The number of macrophages was calculated for each image using a binary mask layer that was user-defined in NIS-Elements software by thresholding based on dye intensity color of the macrophage dye label and area of the individual macrophage mask. Values for the macrophage binary layer were adjusted for each experiment based on the cell staining intensity. Cells with diameters ≤10 µm were excluded to remove dead cells and cell debris. Once these values were determined for an individual experiment, the macrophage binary mask layer was automatically calculated for each frame using a NIS-Elements custom macro (available upon request). Besides enumeration, the binary masks provide additional data, including mean dye intensities and area.
Void masking
Voids were quantified and counted within the NIS-Elements imaging software using the Spot Detection function with user-selected parameters: round-shaped and clustered dark spots. For each experiment, user-adjusted values varied for diameter (∼4 µm) and spot contrast value, which was set to maximize detection of ‘true voids’ and minimize detection of ‘false voids’ caused by issues such as cell architecture and cell spacing. Because the macrophage dye label intensity varied between experiments, the experimental spot contrast values varied between 15 and 100. Again, once these parameters and values were determined for Spot Detection, the void binary mask layer could be calculated automatically for each frame using a custom NIS-Elements macro.
Phagocytosis quantification
Void index
To allow comparisons within and across experiments, we normalized phagocytic void enumeration by using a void index calculation:
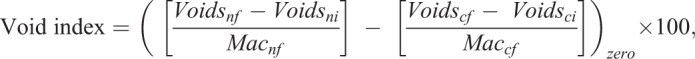 |
where: Voidsnf=Void count of test sample (n) in frame number f; Voidsni=Average void count of test sample (n) at initial frame(s); Macnf=Mac number of test sample (n) in frame number f; Voidscf=Void count of control (c) in frame number f; Voidsci=Void count of control (c) in frame number f; Maccf=Mac number of control (c) in frame number f; and zero=> negative values converted into zero.
The void index provides a value that removes background and accounts for macrophage number in each frame. The first bracketed term provides the initial normalization by subtracting the average number of voids from the initial frames of the experiment (Voidsni), when no phagocytosis occurs, from the raw void count quantified for each image frame taken thereafter (Voidsnf) and then dividing by the number of macrophages in that frame (Macnf). This value is then subtracted from a control sample in the second bracketed term, which is calculated in the same way as the sample in the first bracketed term, but whose values are derived from a control experiment run at the same time (for ADCP, the control sample has no added antibody; for efferocytosis, the control sample has no added apoptotic cells). Negative background subtracted values were set to zero, and then each frame was multiplied by 100. Thus, the void index corresponds to the normalized void count per 100 macrophages.
Dye intensity index
The dye intensity index was calculated as described for the void index but utilized the mean intensity for the target cell dye label channel (e.g. TRITC for pHrodo Red) contained within the dilated void mask.
Macrophage observed mask parameters
Macrophage observed mask parameters were calculated through the utilization of the macrophage binary mask layer, which also collects the total thresholded area that approximately corresponds to the total macrophage area, within each frame. The change in total macrophages observed mask parameters (ΔMask) is calculated by subtracting the data for each frame from the average of the initial background frames.
Other metrics
Void rate is the slope of the void index over the first experimental 30 min. Time to void maximum was the time, in minutes, to reach the maximum void index value for each condition. The AUC was calculated over the entire experimental time using Prism8 (GraphPad). Void maximum was the highest normalized void index value reached for a given experimental condition.
Statistical analyses
We found that all our data was normally distributed using the Shapiro–Wilk test. Therefore, parametric statistical testing was applied in all experiments. Statistical significance of differences in mean values±s.e.m. was calculated using either an unpaired, two-tailed Student's t-test or one-way ANOVA where indicated. P-values less than 0.05 were considered statistically significant. Outlier samples were excluded where indicated using Grubb's testing with an α value=0.05.
Supplementary Material
Acknowledgements
We would like to thank members of the Elliott laboratory and Center for Vaccine Biology and Immunology and Joshua M. Zent (Department of Biomedical Engineering, Medical Scientist Training Program, The Ohio State University) for critical discussions, Derick R. Peterson, Ph.D. (Director, Biostatistics Shared Resource, Wilmot Cancer Institute, University of Rochester Medical Center) for statistical consultations, CLL patients at the Wilmot Cancer Institute for their participation in our research efforts and donation of specimens, Dr. Paul M. Barr, Sharon Lewinski RN and Tania Orzol NP for assistance with specimen collection, and Chris Langsdorf (Thermo Fisher Scientific) for samples and advice for dye-labeling cells.
Footnotes
Competing interests
The authors declare no conflict of interest. While we thank our funding sources for their support, none of these organizations, including Acerta Pharma LLC, contributed to the concept or design of this project, nor did they contribute to the data acquisition or data analysis presented here.
Author contributions
Conceptualization: C.C.C., J.J.P., M.R.E., C.S.Z.; Methodology: C.C.C., J.J.P., M.R.E.; Software: C.C.C., J.J.P.; Validation: C.C.C.; J.J.P. Formal analysis: C.C.C., J.J.P.; Investigation: C.C.C., J.J.P.; Resources: C.C.C., J.J.P., H.E.W., F.R.-E., K.R.V., C.S.Z., M.R.E.; Data curation: C.C.C., J.J.P.; Writing - original draft: C.C.C., J.J.P.; Writing - review & editing: C.C.C., J.J.P., C.S.Z., M.R.E.; Visualization: C.C.C., J.J.P.; Supervision: C.S.Z., M.R.E.; Project administration: C.S.Z., M.R.E.; Funding acquisition: M.R.E., C.S.Z.
Funding
This work is supported by grants to M.R.E. (National Institutes of Health, AI114554); M.R.E./C.C.C. (Acerta Pharma LLC, 005656), and M.R.E./C.S.Z. (University of Rochester Research Award), and by generous gifts from Elizabeth Aaron (C.S.Z.). J.J.P. was supported in part by a University of Rochester Immunology Training T32 Grant from NIH (AI007285). Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.237883.supplemental
References
- Aderem A. and Underhill D. M. (1999). Mechanisms of phagocytosis in macrophages. Annu. Rev. Immunol. 17, 593-623. 10.1146/annurev.immunol.17.1.593 [DOI] [PubMed] [Google Scholar]
- Baig N. A., Taylor R. P., Lindorfer M. A., Church A. K., LaPlant B. R., Pettinger A. M., Shanafelt T. D., Nowakowski G. S. and Zent C. S. (2014). Induced resistance to ofatumumab-mediated cell clearance mechanisms, including complement-dependent cytotoxicity, in chronic lymphocytic leukemia. J. Immunol. 192, 1620-1629. 10.4049/jimmunol.1302954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beletskii A., Cooper M., Sriraman P., Chiriac C., Zhao L., Abbot S. and Yu L. (2005). High-throughput phagocytosis assay utilizing a pH-sensitive fluorescent dye. Biotechniques 39, 894-897. 10.2144/000112001 [DOI] [PubMed] [Google Scholar]
- Beurskens F. J., Lindorfer M. A., Farooqui M., Beum P. V., Engelberts P., Mackus W. J. M., Parren P. W. H. I., Wiestner A. and Taylor R. P. (2012). Exhaustion of cytotoxic effector systems may limit monoclonal antibody-based immunotherapy in cancer patients. J. Immunol. 188, 3532-3541. 10.4049/jimmunol.1103693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canton J. (2018). Macropinocytosis: new insights into its underappreciated role in innate immune cell surveillance. Front. Immunol. 9, 2286 10.3389/fimmu.2018.02286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy C. H. and Botelho R. J. (2017). Quantifying phagocytosis by immunofluorescence and microscopy. Methods Mol. Biol. 1519, 43-53. 10.1007/978-1-4939-6581-6_4 [DOI] [PubMed] [Google Scholar]
- Church A. K., VanDerMeid K. R., Baig N. A., Baran A. M., Witzig T. E., Nowakowski G. S. and Zent C. S. (2016). Anti-CD20 monoclonal antibody-dependent phagocytosis of chronic lymphocytic leukaemia cells by autologous macrophages. Clin. Exp. Immunol. 183, 90-101. 10.1111/cei.12697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies L. C., Jenkins S. J., Allen J. E. and Taylor P. R. (2013). Tissue-resident macrophages. Nat. Immunol. 14, 986-995. 10.1038/ni.2705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott M. R. and Ravichandran K. S. (2010). Clearance of apoptotic cells: implications in health and disease. J. Cell Biol. 189, 1059-1070. 10.1083/jcb.201004096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannagan R. S., Jaumouillé V. and Grinstein S. (2012). The cell biology of phagocytosis. Annu. Rev. Pathol. 7, 61-98. 10.1146/annurev-pathol-011811-132445 [DOI] [PubMed] [Google Scholar]
- Forero-Torres A., de Vos S., Pohlman B. L., Pashkevich M., Cronier D. M., Dang N. H., Carpenter S. P., Allan B. W., Nelson J. G., Slapak C. A. et al. (2012). Results of a phase 1 study of AME-133v (LY2469298), an Fc-engineered humanized monoclonal anti-CD20 antibody, in FcgammaRIIIa-genotyped patients with previously treated follicular lymphoma. Clin. Cancer Res. 18, 1395-1403. 10.1158/1078-0432.CCR-11-0850 [DOI] [PubMed] [Google Scholar]
- Grandjean C. L., Montalvao F., Celli S., Michonneau D., Breart B., Garcia Z., Perro M., Freytag O., Gerdes C. A. and Bousso P. (2016). Intravital imaging reveals improved Kupffer cell-mediated phagocytosis as a mode of action of glycoengineered anti-CD20 antibodies. Sci. Rep. 6, 34382 10.1038/srep34382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg S. and Grinstein S. (2002). Phagocytosis and innate immunity. Curr. Opin. Immunol. 14, 136-145. 10.1016/S0952-7915(01)00309-0 [DOI] [PubMed] [Google Scholar]
- Gu B. J., Sun C., Fuller S., Skarratt K. K., Petrou S. and Wiley J. S. (2014). A quantitative method for measuring innate phagocytosis by human monocytes using real-time flow cytometry. Cytometry A 85, 313-321. 10.1002/cyto.a.22400 [DOI] [PubMed] [Google Scholar]
- Gul N. and van Egmond M. (2015). Antibody-dependent phagocytosis of tumor cells by macrophages: a potent effector mechanism of monoclonal antibody therapy of cancer. Cancer Res. 75, 5008-5013. 10.1158/0008-5472.CAN-15-1330 [DOI] [PubMed] [Google Scholar]
- Imbach P., Barandun S., d'Apuzzo V., Baumgartner C., Hirt A., Morell A., Rossi E., Schöni M., Vest M. and Wagner H. P. (1981). High-dose intravenous gammaglobulin for idiopathic thrombocytopenic purpura in childhood. Lancet 1, 1228-1231. 10.1016/S0140-6736(81)92400-4 [DOI] [PubMed] [Google Scholar]
- Joly E. and Hudrisier D. (2003). What is trogocytosis and what is its purpose? Nat. Immunol. 4, 815 10.1038/ni0903-815 [DOI] [PubMed] [Google Scholar]
- Kamen L., Myneni S., Langsdorf C., Kho E., Ordonia B., Thakurta T., Zheng K., Song A. and Chung S. (2019). A novel method for determining antibody-dependent cellular phagocytosis. J. Immunol. Methods 468, 55-60. 10.1016/j.jim.2019.03.001 [DOI] [PubMed] [Google Scholar]
- Kapellos T. S., Taylor L., Lee H., Cowley S. A., James W. S., Iqbal A. J. and Greaves D. R. (2016). A novel real time imaging platform to quantify macrophage phagocytosis. Biochem. Pharmacol. 116, 107-119. 10.1016/j.bcp.2016.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann A. K., Sørnes S. and Halstensen A. (2000). Phagocytosis: measurement by flow cytometry. J. Immunol. Methods 243, 229-242. 10.1016/S0022-1759(00)00237-4 [DOI] [PubMed] [Google Scholar]
- Manzanero S. (2012). Generation of mouse bone marrow-derived macrophages. Methods Mol. Biol. 844, 177-181. 10.1007/978-1-61779-527-5_12 [DOI] [PubMed] [Google Scholar]
- Montalvao F., Garcia Z., Celli S., Breart B., Deguine J., Van Rooijen N. and Bousso P. (2013). The mechanism of anti-CD20-mediated B cell depletion revealed by intravital imaging. J. Clin. Invest. 123, 5098-5103. 10.1172/JCI70972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montaño F., Grinstein S. and Levin R. (2018). Quantitative phagocytosis assays in primary and cultured macrophages. Methods Mol. Biol. 1784, 151-163. 10.1007/978-1-4939-7837-3_15 [DOI] [PubMed] [Google Scholar]
- Mössner E., Brünker P., Moser S., Püntener U., Schmidt C., Herter S., Grau R., Gerdes C., Nopora A., van Puijenbroek E. et al. (2010). Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood 115, 4393-4402. 10.1182/blood-2009-06-225979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy P. S., Wang J., Bhagwat S. P., Munger J. C., Janssen W. J., Wright T. W. and Elliott M. R. (2017). CD73 regulates anti-inflammatory signaling between apoptotic cells and endotoxin-conditioned tissue macrophages. Cell Death Differ. 24, 559-570. 10.1038/cdd.2016.159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neaga A., Lefor J., Lich K. E., Liparoto S. F. and Xiao Y. Q. (2013). Development and validation of a flow cytometric method to evaluate phagocytosis of pHrodo™ bioparticles® by granulocytes in multiple species. J. Immunol. Methods 390, 9-17. 10.1016/j.jim.2011.06.027 [DOI] [PubMed] [Google Scholar]
- Nimmerjahn F. and Ravetch J. V. (2008). Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 8, 34-47. 10.1038/nri2206 [DOI] [PubMed] [Google Scholar]
- Okabe Y. and Medzhitov R. (2016). Tissue biology perspective on macrophages. Nat. Immunol. 17, 9-17. 10.1038/ni.3320 [DOI] [PubMed] [Google Scholar]
- Pottier Y., Pierard I., Barclay A., Masson P. L. and Coutelier J.-P. (1996). The mode of action of treatment by IgG of haemolytic anaemia induced by an anti-erythrocyte monoclonal antibody. Clin. Exp. Immunol. 106, 103-107. 10.1046/j.1365-2249.1996.d01-818.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple J. W. and Freedman J. (2005). Autoimmune pathogenesis and autoimmune hemolytic anemia. Semin. Hematol. 42, 122-130. 10.1053/j.seminhematol.2005.04.002 [DOI] [PubMed] [Google Scholar]
- Sharma L., Wu W., Dholakiya S. L., Gorasiya S., Wu J., Sitapara R., Patel V., Wang M., Zur M., Reddy S. et al. (2014). Assessment of phagocytic activity of cultured macrophages using fluorescence microscopy and flow cytometry. Methods Mol. Biol. 1172, 137-145. 10.1007/978-1-4939-0928-5_12 [DOI] [PubMed] [Google Scholar]
- Sharma N., Akkoyunlu M. and Rabin R. L. (2018). Macrophages—common culprit in obesity and asthma. Allergy 73, 1196-1205. 10.1111/all.13369 [DOI] [PubMed] [Google Scholar]
- Steinberg B. E., Scott C. C. and Grinstein S. (2007). High-throughput assays of phagocytosis, phagosome maturation, and bacterial invasion. Am. J. Physiol. Cell Physiol. 292, C945-C952. 10.1152/ajpcell.00358.2006 [DOI] [PubMed] [Google Scholar]
- Swanson J. A. and Watts C. (1995). Macropinocytosis. Trends Cell Biol. 5, 424-428. 10.1016/S0962-8924(00)89101-1 [DOI] [PubMed] [Google Scholar]
- Tauber A. I. (2003). Metchnikoff and the phagocytosis theory. Nat. Rev. Mol. Cell Biol. 4, 897-901. 10.1038/nrm1244 [DOI] [PubMed] [Google Scholar]
- Tobinai K., Ogura M., Kobayashi Y., Uchida T., Watanabe T., Oyama T., Maruyama D., Suzuki T., Mori M., Kasai M. et al. (2011). Phase I study of LY2469298, an Fc-engineered humanized anti-CD20 antibody, in patients with relapsed or refractory follicular lymphoma. Cancer Sci. 102, 432-438. 10.1111/j.1349-7006.2010.01809.x [DOI] [PubMed] [Google Scholar]
- Uchida J., Hamaguchi Y., Oliver J. A., Ravetch J. V., Poe J. C., Haas K. M. and Tedder T. F. (2004). The innate mononuclear phagocyte network depletes B lymphocytes through Fc receptor-dependent mechanisms during anti-CD20 antibody immunotherapy. J. Exp. Med. 199, 1659-1669. 10.1084/jem.20040119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanDerMeid K. R., Elliott M. R., Baran A. M., Barr P. M., Chu C. C. and Zent C. S. (2018). Cellular cytotoxicity of next-generation CD20 monoclonal antibodies. Cancer Immunol. Res. 6, 1150-1160. 10.1158/2326-6066.CIR-18-0319 [DOI] [PubMed] [Google Scholar]
- Weiskopf K. and Weissman I. L. (2015). Macrophages are critical effectors of antibody therapies for cancer. mAbs 7, 303-310. 10.1080/19420862.2015.1011450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn T. A., Chawla A. and Pollard J. W. (2013). Macrophage biology in development, homeostasis and disease. Nature 496, 445-455. 10.1038/nature12034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo J. C., Wall A. A., Stow J. L. and Hamilton N. A. (2013). High-throughput quantification of early stages of phagocytosis. Biotechniques 55, 115-124. 10.2144/000114075 [DOI] [PubMed] [Google Scholar]
- Yoshimori T., Yamamoto A., Moriyama Y., Futai M. and Tashiro Y. (1991). Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J. Biol. Chem. 266, 17707-17712. [PubMed] [Google Scholar]
- Zent C. S. and Elliott M. R. (2017). Maxed out macs: physiologic cell clearance as a function of macrophage phagocytic capacity. FEBS J. 284, 1021-1039. 10.1111/febs.13961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zent C. S. and Kay N. E. (2010). Autoimmune complications in chronic lymphocytic leukaemia (CLL). Best Pract. Res. Clin. Haematol. 23, 47-59. 10.1016/j.beha.2010.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.