

Abstract
Background
Does newborn screening for cystic fibrosis (CF) improve clinical outcomes, quality of life and survival?
Objectives
To examine whether newborn screening for CF prevents or reduces irreversible organ damage and improves clinical outcomes, quality of life and survival in people with CF without unacceptable adverse effects.
Search methods
We searched the Cochrane Cystic Fibrosis and Genetic Disorders Group Trials Register comprising references identified from electronic database searches, handsearches of relevant journals and abstract books of conference proceedings.
The Group's Trials Register last searched: June 2008.
Selection criteria
Randomised or quasi‐randomised controlled trials, published and unpublished, comparing screening to clinical diagnosis in people with CF.
Data collection and analysis
Two authors independently assessed trial eligibility and quality and independently extracted data. Allocation concealment was unclear in both studies and sequence generation adequate in one.
Main results
Searches identified six trials. Two trials involving 1,124,483 neonates (210 with CF) with a maximum follow up of 17 years were eligible for inclusion. Varying study designs, outcomes reported and summary measures precluded calculation of pooled estimates and only data from one study were analysed. Severe malnutrition was less common among screened participants. Compared with screened participants, the odds ratio of weight below the tenth percentile was 4.12 (95% CI 1.64 to 10.38) and for height was 4.62 (95% CI 1.69 to 12.61) in the control group.
At age seven, 88% of screened participants and 75% of controls had lung function parameters within normal limits of at least 89% predicted. At diagnosis chest radiograph scores were significantly better among screened participants; 33% of screened versus 50% of control participants had Wisconsin chest X‐ray (WCXR) scores over five (P = 0.097) and 24% of screened versus 45% of control participants had Brasfield chest X‐ray (BCXR) scores under 21 (P = 0.042)). Over time, chest radiograph scores were worse in the screened group (WCXR P = 0.017 and BCXR P = 0.041). Results were no longer significant after adjustment for genotype, pancreatic status, and Pseudomonas aeruginosa‐culture results. In screened participants colonisation with Pseudomonas aeruginosa occurred earlier. Estimates suggest diagnosis through screening is less expensive.
Authors' conclusions
Two randomised controlled trials assessing neonatal screening in CF were identified; data from one study were included. Nutritional benefits are apparent. Screening provides potential for better pulmonary outcomes, but confounding factors influenced long‐term pulmonary prognosis of people with CF. Screening seems less expensive than traditional diagnosis.
Plain language summary
Screening newborn babies for cystic fibrosis
In people with cystic fibrosis lung disease and malnutrition occur very early in life. These complications are suited to early treatment. Newborn screening may therefore improve outcomes for people with cystic fibrosis. We aimed to find out whether newborn screening prevents or reduces organ damage and improves clinical outcomes in people with CF without unacceptable adverse effects. This review includes two trials with 1,124,483 babies (210 with cystic fibrosis). The trials compared newborn screening to clinical diagnosis. We were only able to analyse data from one of the trials. This trial showed that severe malnutrition was less common among screened babies. Screened babies had better chest radiograph scores at diagnosis, but these scores became worse over time. The screened babies become colonised with Pseudomonas aeruginosa earlier. Costs for screening were less than costs for traditional diagnosis.
Background
Description of the condition
Cystic fibrosis (CF) is one of the most common life‐threatening autosomal recessive disorders among Caucasians, with an estimated incidence in Caucasians throughout the world of between 0.25 to 5 per 10,000 live births (Lewis 1995). CF is found in most other races but to a lesser frequency than in the Caucasian race. Pulmonary disease is the most important cause of severe disability and premature death in people with CF. Recent evidence suggests that pulmonary inflammation in CF occurs very early in life. Airway inflammation has been identified in affected infants by four weeks of age and it has been suggested that this can occur without prior infection (Balough 1995; Khan 1995). Bacterial lower respiratory tract infections, particularly those due to Staphylococcus aureus and Pseudomonas aeruginosa (P. aeruginosa), are common and associated with significant pulmonary deterioration. These are amenable to antibiotic therapy resulting in clearance of pathogens and reduction of inflammation (Armstrong 1997).
Description of the intervention
Neonatal screening for CF became feasible with the development in 1979 of a radioimmunoassay for immunoreactive trypsin (IRT) suitable for use on dried blood spots from newborns (Pollitt 1998). IRT levels are elevated in the first few weeks of life in babies with CF. However, in the first week of life, when most newborn blood samples are routinely taken; specificity of a single elevated IRT is low. Thus two or three stage protocols based on a second IRT test with or without subsequent deoxyribonucleic acid (DNA) analyses have been developed to reduce the false positive diagnoses associated with one stage IRT testing (Pollitt 1998).
How the intervention might work
Over the past 30 years there has been a marked increase in the life expectancy of people with CF such that in the United Kingdom predicted median life expectancy for the current cohort of CF infants is about 40 years (Elborn 1991). Most, but not all, of this increase has been attributed to improvements in therapy and it has been argued that therapeutic interventions administered before the onset of signs or symptoms may have the greatest long‐term benefit. This has led to the hypothesis that presymptomatic diagnosis, for example by newborn (neonatal) screening, and early treatment may prevent or reduce irreversible pulmonary damage and optimise early nutritional status and thereby improve outcome and quality of life in people with CF.
Why it is important to do this review
Despite acceptable test performance, the effectiveness of neonatal screening in reducing CF mortality and morbidity remains controversial. In 1983 the American Cystic Fibrosis Foundation identified a need for further information on the benefits and risks of early diagnosis and treatment before newborn screening could be recommended (Task Force 1983). A number of studies have been published from different countries. Dankert‐Roelse observed that those identified by neonatal screening had less pulmonary inflammation and less deterioration of lung function than those detected on clinical symptoms (Dankert‐Roelse 1995). Farrell found that neonatal screening provides the opportunity to prevent malnutrition in infants with CF (Farrell 1997). However, neonatal screening for CF may also be associated with adverse effects. False positive screening results may adversely affect the parents' relationship with their baby (Tluczek 1992; Baroni 1997). In addition, false reassurance among those with false‐negative screening tests may lead to delays in clinical diagnosis and loss of an opportunity to give genetic counselling at an appropriate time. There may be a risk of "labelling" those with mild disease who might not have presented clinically and exposing them to unnecessary treatments or interventions (Wilcken 1993).
Uncertainty regarding the effectiveness of newborn screening reflects the potential biases of non‐experimental comparisons of screened populations. Given the recognised cohort effect in CF mortality (and presumably morbidity), comparisons between earlier unscreened cohorts and later screened cohorts may incorrectly attribute the better outcome in screened cohorts to earlier diagnosis. Comparisons based on non‐experimental studies using concurrent controls may also be biased as non‐random assignment of centres or regions to screening may be confounded with social factors or access to medical care which may influence outcome. Thus careful assessment of the methodological quality of trials reporting evaluations of screening is crucial to their interpretation and entails modification of the criteria to those conventionally used to assess trials reporting interventions in clinically defined groups (Schulz 1995).
This review aims to establish whether there is evidence that early diagnosis of CF by means of neonatal screening, followed by current treatment, improves survival and long‐term morbidity, without unacceptable adverse effects.
Objectives
To analyse evidence for the effectiveness of neonatal screening from randomised controlled trials (RCTs) in CF.
To address whether neonatal screening for CF:
improves survival in CF;
reduces the number of respiratory complications and improves overall respiratory status in CF;
improves nutritional status in CF;
reduces long‐term complications of CF such as diabetes mellitus and liver cirrhosis;
is associated with significant adverse effects in the CF group diagnosed by screening (including delay in clinical diagnosis of 'missed' cases because of false‐negative tests and 'labelling' of those with mild disease);
is associated with significant adverse effects in the screened population (including psychological damage following false‐positive tests, interference with developing family relationships and misconceptions and miscommunication of results);
is a more economic way of achieving a diagnosis of CF than clinical methods.
Methods
Criteria for considering studies for this review
Types of studies
We aimed to identify all RCTs, published and unpublished. We included any trials where quasi‐randomisation methods were used, such as alternation.
Types of participants
Children screened for CF as well as parents and carers of children screened for CF.
Types of interventions
Any neonatal screening test, which enables infants with CF to be diagnosed before the age of three months and in most cases before the diagnosis becomes evident clinically, compared to clinical diagnosis and later treatment in people with CF.
Types of outcome measures
In the CF Group
Primary outcomes
Objective measures of lung function: forced vital capacity (FVC); forced expiratory volume at one second (FEV1); residual volume/total lung capacity (RV/TLC); forced expiratory flow 25‐75% (FEF25‐75%) (analysed as per cent predicted) in older children and adults
Chest radiograph scores
Nutritional status as noted by weight gain, body mass index, z score or other indices of nutritional state
Survival or age at death
Secondary outcomes
Number of respiratory exacerbations (defined using a validated criteria such as those stated by Fuchs (Fuchs 1994)) requiring treatment with systemic antibiotics
Occurrence of chronic pulmonary infection with any organism, defined as repeated positive cultures of the same micro‐organism of sputum or cough swabs
Number of hospital admissions, excluding those for assessment to confirm a presumptive positive screening result
Number of hospital days, excluding those for assessment to confirm a presumptive positive screening result
Number of respiratory complications such as cor pulmonale, haemoptysis and pneumothorax
Age at occurrence of diabetes mellitus requiring treatment with insulin
Age at occurrence of liver cirrhosis confirmed by ultrasound or histology
Cognitive function (post hoc change)
Quality of life (post hoc change)
Adverse effects in the CF group diagnosed by screening, including delay in clinical diagnosis of 'missed' cases because of false‐negative tests and 'labelling' of those with mild disease
We decided to examine cognitive function and quality of life as additional outcome measures. These were not initially included in the review, but trials have reported them in longer term analyses. We feel these outcomes reflect the changing aspirations of people with CF and their importance merits post hoc inclusion in the review.
In The Screened Group
Primary outcome
Adverse effects including psychological damage following false‐positive tests, interference with developing family relationships and misconceptions and miscommunication of results
Secondary outcome
Total medical costs of the screening process
We assessed whether diagnosing CF through neonatal screening was less expensive than when diagnosing clinically.
Search methods for identification of studies
Electronic searches
Relevant trials were identified from the Group's Cystic Fibrosis Trials Register using the terms: screening AND neonatal.
The Cystic Fibrosis Trials Register is compiled from electronic searches of the Cochrane Central Register of Controlled Trials (Clinical Trials) (updated each new issue of The Cochrane Library), quarterly searches of MEDLINE, a search of EMBASE to 1995 and the prospective handsearching of two journals, Pediatric Pulmonology and the Journal of Cystic Fibrosis. Unpublished work is identified by searching through the abstract books of three major cystic fibrosis conferences: the International Cystic Fibrosis Conference; the European Cystic Fibrosis Conference and the North American Cystic Fibrosis Conference. For full details of all searching activities for the register, please see the relevant sections of the Cystic Fibrosis and Genetic Disorders Group Module.
Date of the most recent search of the Group's Trials Register: June 2008.
Searching other resources
Additional RCTs were found from reference lists.
We also contacted the pharmaceutical companies which manufacture the screening tests for CF: Zeneca Diagnostics, AGEN Biomedical Limited, Dade Behring and EG&G Wallac.
Data collection and analysis
Selection of studies
The authors independently applied the inclusion criteria to all potential reports and selected trials for inclusion in the review. If there had been any disagreement, they would have resolved this by discussion.
Data extraction and management
The authors independently attempted to extract data from each RCT from text, tables and figures. They proposed to analyse the results of trials where quasi‐randomisation methods were used, such as alternation, separately.
In the CF Group, for the outcomes listed above, we proposed to group data into data measured annually but if reported at other time periods then consideration was given to examining these as well. If sufficient data were obtained we proposed to evaluate data for the group of participants with an early diagnosis due to the occurrence of meconium ileus, siblings of known participants and participants with a positive family history separately when possible. Similarly, we proposed to perform a separate analysis for newborns in whom the diagnosis of CF had been confirmed before and after four weeks of age.
Assessment of risk of bias in included studies
In order to establish a risk of bias for the included studies, the authors recorded the following details of methodological quality: concealment of allocation; generation of the randomisation sequence; whether the assessment of outcome was blinded; whether intention‐to‐screen analyses were possible from the available data; whether the method used to ascertain cases were similar in the screened and control groups; and whether the report documented the number of participants lost to follow up or subsequently excluded from the study.
Measures of treatment effect
The authors analysed outcomes both as continuous outcomes (e.g. FEV1, FVC, weight and body mass index) and as dichotomous or binary outcomes whenever possible. For continuous outcomes the authors recorded the mean change from baseline from each group and standard error and standard deviation. For dichotomous or binary outcomes, the authors calculated the odds ratio, risk ratio, differences in odds and risks and number needed to screen, as appropriate.
The authors also included longitudinal analysis of data in this review whenever possible. These included data from time of diagnosis until the end of the follow‐up. They reported methods of longitudinal analysis used in trials aimed to perform a meta‐analysis for longitudinal data.
Dealing with missing data
In order to allow an intention‐to‐screen analysis, the authors sought data on the number of participants with each outcome event, by allocated screened group, irrespective of identification of participants before the screening test results were known, e.g. through meconium ileus or family history, missed cases or subsequent exclusion from follow up.
Assessment of heterogeneity
If sufficient trials are included in future updates of the review, the authors will examine these for heterogeneity using the I2 statistic (Higgins 2003).
Data synthesis
As the authors have only been able to include data from a single trial in our analysis, they have used a fixed‐effect model. If, for future updates of this review, the authors are able to include more trials and establish a moderate to high degree of heterogeneity between these trials, they plan to analyse the data using a random‐effects model.
Subgroup analysis and investigation of heterogeneity
If, in the future, sufficient data are available and heterogeneity between trials is identified, the authors will investigate clinical heterogeneity by analysing the effects of screening separately for different age categories, occurrence of meconium ileus, genotype, pancreatic function, centre treatment and outcomes collected.
Sensitivity analysis
If sufficient trials are included in future updates of the review, the authors will investigate statistical heterogeneity, such as methodological issues e.g. trial design and control for bias.
Results
Description of studies
Results of the search
The searches identified six trials, of which two were eligible for inclusion within the review (UK Trial 1991; Wisconsin Trial 1998). A large number of reports based on these two trials were identified (UK Trial 1991; Wisconsin Trial 1998). Four trials identified by the searches were not eligible for inclusion (Carter 1987; Barlocco 1988; Dauphinais 1992; Lagoe 2005).
Included studies
Summary details are given in the 'Characteristics of included studies' section.
Over time more non‐randomised studies on the effect of newborn screening for CF in different countries all over the world have been published. In this review only RCTs were included.
The two included trials reported screening based on a population of 1,124,483 neonates; 552,354 (49.1%) of whom were allocated to the screened group and 572,129 (50.9%) to the control group (UK Trial 1991; Wisconsin Trial 1998). A total of 210 CF participants were included in the analysis. The participants ranged from 0 to 16 years of age. Length of follow up reported ranged from one year (Mischler 1998) to 16 years (Farrell 2003).
In the UK Trial, screening took place on alternate weeks from January 1985 to December 1989 in Wales and from January 1985 to October 1989 in the West Midlands (UK Trial 1991). Neonates born in the central Birmingham area were excluded from this trial, and all participants with meconium ileus were looked after in a central Birmingham hospital. We were concerned that it was not clear from the original paper whether these infants were included in the control arm of the study, which would lead to ascertainment bias. Having contacted the original investigators from the study, we received confirmation that any babies born in Central Birmingham were not screened because blood was collected with a capillary system, not on a 'card'. These babies were not included in the control population of the trial (Weller 2007).
As expected, the numbers of children diagnosed with CF in both arms of the UK Trial have increased with increasing follow up, hence numbers of affected children vary between reports of the same trial. Data from the most recent published report of this trial indicate that 230,076 neonates were randomised to be screened for CF and 234,510 to not receive a screening test (Doull 2001). After a repeated positive screening test, performed by measurement of immunoreactive trypsin, diagnosis of CF was confirmed by sweat testing. Control participants were diagnosed by clinical manifestations of the disease or family history and diagnosis was confirmed by sweat testing. Methods used to identify these cases were not stated.
At the most recent follow up, 86 children with CF were identified in the group randomised to screening, of whom 70 were detected by screening; an additional 16 children were screened but were not detected by screening (false negative diagnoses). Eight screened infants presented with meconium ileus while six had affected older siblings, two of whom were also among those with false negative screening results. Hence, the detection rate in this trial was 81% (58 out of 72), excluding those with a family history or meconium ileus. Median age at diagnosis in the screened group was eight weeks (range 3 to 22 weeks) reflecting variable diagnostic and management policies at the time of the trial (UK Trial 1991).
In the group randomised to 'no screening', 90 children were diagnosed as having CF of whom 19 had meconium ileus and 14 an affected older sibling (of whom one had both meconium ileus and an affected older sibling). Age of diagnosis in the unscreened group (excluding those with meconium ileus) ranged from 6 weeks to 22 months. In the original reports of clinical outcomes, nine children with a false‐negative screening result were included in the control group; hence the early reports of outcomes are not based on an intention‐to‐screen analysis. Comparing 58 screened infants with 44 (35 plus nine false negatives), the authors reported that mean (standard deviation (SD)) age at diagnosis in the screened participants was 9.1 (3.1) weeks and 50.7 (60.5) weeks in the control participants. Median ages were not reported. At the time of this published report 12 screened and seven control participants had reached four years of age (UK Trial 1991).
In the Wisconsin Trial, 650,341 neonates were screened for CF (Wisconsin Trial 1998). The screening test was performed on all infants but only half of the families were informed about the results in the neonatal period. Initially the trial used a two‐stage immune reactive trypsin protocol, but subsequently this was altered to immune reactive trypsin followed by DNA testing for the delta F508 mutation, reflecting genetic advances during the course of the trial. Data from the control participants were computer‐stored and the families and investigators were masked to the newborn screening results until the children reached four years of age, unless parents requested the information before then (Fost 1989). Confirmation of diagnosis by sweat testing took place after a positive screening result in the screened group during the neonatal period and in the control group after the child's fourth birthday when unmasking of the data revealed the children with a positive screening test, unless CF had been diagnosed earlier through other means. Thus, in comparison with the UK Trial, the total number of CF diagnoses in each arm was made at this point and did not increase with increasing follow up. However, due to differences in the beginning of the follow up between screened and control participants, data reported before the unblinding of the control data were subject to lead time bias. Participants with meconium ileus were excluded from analysis. Longitudinal data were analysed by a repeated‐measures analysis using generalised estimating equation (GEE) methods. Analyses were adjusted for different covariates as described under results.
In the screened arm there were 77 infants; 16 diagnosed through meconium ileus, two diagnosed through family history, 54 through screening test alone, and five with a false‐negative screening test (Wisconsin Trial 1998). The control group included 81 infants or children, 18 of whom had meconium ileus, 42 were diagnosed clinically before and 21 were diagnosed clinically after the unblinding of the control screening results at four years of age. Excluding participants with meconium ileus, mean (SD) age at diagnosis in the screened participants was reported as 13 (37) weeks and 107 (117) weeks in control participants. Median (range) age at diagnosis was seven (4 to 281) weeks in the screened and 28 (3 to 372) weeks in the control participants. At the time of the most recent published report for follow up, two screened and four control participants had reached 16 years. The following chest radiograph scores were used in this trial: Wisconsin chest X‐ray (WCXR) scores which range from 0 (normal) to 100 (most severe) and the Brasfield chest X‐ray (BCXR) scores which range from 25 (normal) to 3 (most severe).
Of the outcomes specified in the prior hypothesis of the review, nutritional status, chest radiograph scores, spirometry, number of hospital days and Shwachman‐Kulczycki scores were reported in both studies. However, the specific summary measures employed and radiograph methods varied between the two studies.
Conference abstracts reporting chest radiographic scores and spirometry have been published for both trials: radiographic methods varied. In the Wisconsin Trial nutritional status was summarised as z score for weight and height, proportion of participants with weight and height below the 10th percentile, weight percentile and proportion of participants with height below the fifth percentile, measured at age at diagnosis and during the follow up with a time interval of one year. The UK Trial reported SD scores for weight and height, measured from the age of one year at yearly intervals (UK Trial 1991). Number of hospital days in the Wisconsin Trial were reported as the mean (SD) number per person per year throughout the total follow‐up period and in UK Trial as the mean (SD) number at the ages of one and two years. Finally, Shwachman‐Kulczycki score was reported at age at diagnosis in the Wisconsin Trial but at the ages one and two years in the UK Trial. The number of respiratory exacerbations requiring treatment with systemic antibiotics, number of respiratory complications, the occurrence of diabetes mellitus and liver cirrhosis and adverse effects in the CF group diagnosed by screening were not reported in either included trial.
Excluded studies
Four trials identified by the searches were not eligible for inclusion (Barlocco 1988; Carter 1987; Dauphinais 1992; Lagoe 2005). There were five reports of one trial from one centre in Italy (Barlocco 1988); three reports from a trial in Connecticut, USA (Dauphinais 1992); and one report of a trial in the UK (Carter 1987) that were identified through searching abstract books and by contacting the authors. These failed to meet the inclusion criteria, as they were not RCTs. One trial looked at outcomes that were not part of our review (Lagoe 2005).
Risk of bias in included studies
In order to assess the risk of bias in the included studies, the methodological quality was examined using modified criteria suggested by Schulz (Schulz 1995). To clarify certain methodological terms used in this review, we have made the following glossary (Table 2).
1. Methodological terminology.
| Term | Definition |
| Ascertainment bias | A systematic error arising from the type of individuals or participants (mildly ill, moderately ill, severely ill) that the individual observer is seeing. Also, a systematic error arising from the diagnostic process. |
| Concealment of allocation | The process used to prevent foreknowledge of group assignment in a randomised controlled trial, which should be seen as distinct from blinding. The allocation process should be impervious to any influence by the individual making the allocation by having the randomisation process administered by someone who is not responsible for recruiting participants. |
| Intention‐to‐screen analysis | An intention‐to‐screen analysis is one in which all participants in a trial are analysed according to the intervention to which they were allocated, whether they received it or not. |
| Lead time bias | A systematic error arising when follow‐up of two groups does not begin at strictly comparable times. |
Allocation
Concealment of allocation and method used to generate the random sequence were assigned to one of three categories: adequate, unclear or inadequate. These corresponded to a low, unclear or high risk of bias.
Sequence generation was adequate in one trial and was so thought to have a low risk of bias (Wisconsin Trial 1998). In this trial blood samples were obtained from all newborn infants in Wisconsin and randomly divided into two equal groups depending on the last digit of the code number assigned to the specimen (Wisconsin Trial 1998). In the other trial, babies were allocated to either screening or non‐screening on alternate weeks, but the investigators do not state how the initial randomisation took place resulting in an unclear risk of bias (UK Trial 1991).
In the Wisconsin trial, the samples were sent to a centralised laboratory before being allocated to the screening or non‐screening group, thus preventing clinicians being able to foretell the allocation (Wisconsin Trial 1998). We therefore deemed there to be a low risk of bias from this trial. We considered allocation concealment to be inadequate in the trial using alternation and therefore deemed there to be a risk of bias (UK Trial 1991).
Blinding
Blinded assessment of outcome was recorded as either present or absent. RCTs were categorised according to whether double blinding had been reported or not. A higher number of people blinded in trials relates to a lower risk of bias.
Blinded assessment of outcome was not reported in either of the included studies, leading to an unclear risk of bias.
Incomplete outcome data
The reported 'Intention‐to‐screen' analysis was defined as complete, or analysed with less than 15% exclusions, or analysed with more than 15% exclusions. We regarded complete analysis or one with less than 15% exclusions to have a low risk of bias. We judged any analysis with more than 15% exclusions to be at risk of bias. If exclusions were not discussed, we judged there to be an unclear risk of bias.
Excluding participants with meconium ileus, performance of an intention‐to‐screen‐analysis was possible in one study (Wisconsin Trial 1998); and not possible in the other (UK Trial 1991).
Other potential sources of bias
CF is a highly variable disease clinically. Both experimental and non‐experimental comparisons may be subject to different types of bias. Firstly, there is ascertainment bias, this refers to whether the methods by which participants are entered into each group are comparable and can result in the screened group containing people with mild disease that would not have presented clinically. Similarly unscreened people with CF with severe disease may never be diagnosed because of dying at a young age. This is likely to result in biased comparisons of outcome in early life. This bias is likely to diminish but may not disappear with increasing duration of follow up. Ascertainment bias may be avoided by ensuring uniform probability of ascertainment in the screened and unscreened groups. The method to ascertain that cases in screened and control groups was categorised as similar yes or no, similarity means there is a low risk of bias.
The method to ascertain cases was similar in one study (Wisconsin Trial 1998) and not similar in the other (UK Trial 1991). Therefore, the Wisconsin Trial was thought to have a low risk of bias, whereas the the UK Trial was thought to have an increased risk of bias.
Secondly, lead time bias. This type of bias occurs especially when one group has been diagnosed earlier in the natural history of the disease than the other group. We considered both studies to have a potential risk from lead time bias (UK Trial 1991; Wisconsin Trial 1998).
Effects of interventions
Due to differences in study design (and the different summary measures of the outcomes employed), it was not possible to combine the data from both included trials.
Although the UK Trial met the inclusion criteria, we have not presented data from this trial in this review (UK Trial 1991). We decided to exclude these data from the table of comparisons for two reasons. Firstly, from the published data available at this time, performance of an intention‐to‐screen analysis was not possible in this trial and secondly the UK Trial was subject to ascertainment bias.
Therefore, we have only presented data from the Wisconsin Trial in this review (Wisconsin Trial 1998). Due to the lead time bias, only data after the unblinding of the control data at four years of age were analysed and reported here.
In the CF Group
Primary outcomes
1. Objective measures of lung function ‐ FVC, FEV1, RV/TLC, FEF25‐75% (analysed as per cent predicted)
Pulmonary function data from the Wisconsin Trial have recently been published (Farrell 2003). Differences in lung function between participant groups were reported only as P values, therefore at present the data cannot be entered in the data tables for this review. At seven years of age the % predicted FEV1 and FVC for the screened and control groups were not significantly different (P = 0.54). However, at this age 88% of the observations in the screened group and 75% of the observations in the control group were within the normal limits at 89% predicted or greater. Repeated measures analysis, adjusting for covariates CF center, sex, age, genotype, pancreatic status, and indicators of P. aeruginosa infection showed that over time until 16 years of age, there were no statistically significant differences between screened and non‐screened participants in % predicted FEV1 or FVC (P = 0.32) or in % predicted FEV1 (P = 0.18), FEF25‐75 (P = 0.37) and RV/TLC (P = 0.25).
2. Chest radiograph scores
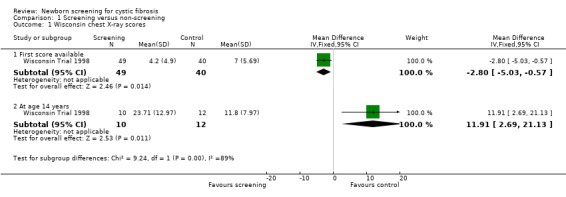
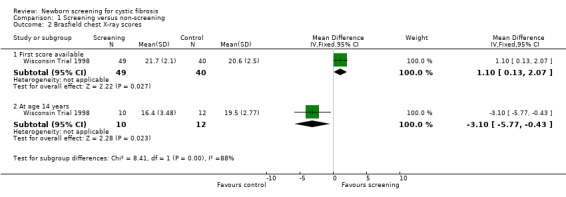
The first available WCXR and BCXR scores were significantly better among screened participants at an average age of 14.3 weeks in the screened group compared with 108 weeks for the control participants. Mean (SD) WCXR score in the screened group (49 participants) was 4.20 (4.90) and in the control group (40 participants) 7.00 (5.69), mean difference (MD) ‐2.80 (95% confidence interval (CI) ‐5.03 to ‐0.57). Mean (SD) BCXR score among screened participants was 21.70 (2.10) versus 20.60 (2.5) among control participants, MD 1.10 (95% CI 0.13 to 2.07). Before the unblinding of data, differences in first chest X‐ray scores were adjusted for differences in age at the time of diagnosis between groups, moreover differences were adjusted for genotype and pancreatic status. Compared with control participants, significantly fewer screened participants had discriminators of potentially irreversible lung disease; 33% of screened versus 50% of control participants had WCXR scores greater than five (P = 0.097) and 24% of screened versus 45% of control participants had BCXR scores less than 21 (P = 0.042). Measures of hyperinflation on the other hand were not significant between groups. Longitudinal assessment of chest radiograph abnormalities, analysed as described under lung function, using only the data recorded after five years of age revealed worse scores over time in the screened group (P = 0.017 for WCXR and P = 0.041 for BCXR). At 14 years of age 10 screened participants were compared with 12 control participants. Reasons for including only a limited amount of participants were not mentioned. Mean (SD) WCXR score was 23.71 (12.97) among screened participants and 11.80 (7.97) among control participants, MD ‐2.80 (95% CI ‐5.03 to ‐0.57) P = 0.015. Mean (SD) BCXR scores were 16.40 (3.48) in the screened group versus 19.50 (2.77) in the control group, MD ‐3.10 (95% CI ‐5.77 to ‐0.43) P = 0.025. Results, however, were no longer significant after adjustment for differences in genotype, pancreatic status, and P. aeruginosa‐culture results. Analysis of covariance revealed that P. aeruginosa infection explained most variability on chest radiograph scores.
3. Nutritional status
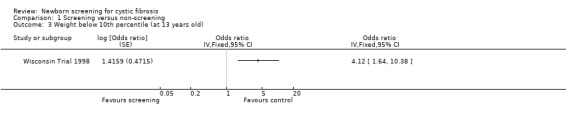
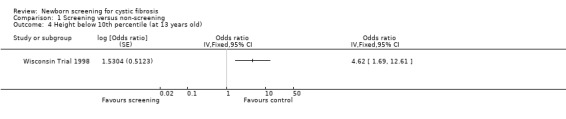
In the Wisconsin Trial, only mean values and standard deviations for nutritional data at the time of diagnosis were reported. At present therefore, data relating to outcomes at specified ages cannot be entered in the data tables for this review. However, in the most recently published study mean z scores for both weight and height over the first 13 years of life were reported using data available for 56 screened and 47 control participants who had not presented with meconium ileus (Farrell 2001). Repeated measures analysis, adjusted for the covariates age, sex, center, genotype, pancreatic status, and age at diagnosis, revealed a marginally significant difference in weight for age z scores while height for age z scores were significantly higher among screened compared with control participants up to the age of 13 years. Weight or height below the tenth percentile was used as an index of severe malnutrition. The odds ratio for the risk of a weight below the tenth percentile in the control group as compared with the screened group, was calculated in the meta‐analysis as being 4.12 (95% CI 1.64 to 10.38). Likewise the corresponding odds ratio for height was calculated in the meta‐analysis as being 4.62 (95% CI 1.69 to 12.61). These reported analyses were adjusted for age, sex, genotype and pancreatic status at diagnosis and the findings were similar when analyses were restricted to measurements taken after four years of age or included data obtained before the diagnosis for control participants as a result of the unblinding process.
In the Wisconsin Trial, vitamin E deficiency was assessed among 68 non‐meconium ileus participants; 37 screened and 31 control participants (Koscik 2003). Seventeen out of 37 participants of the screened participants, versus 17 out of 31 of the control participants had α‐Tocopherol levels less than 300 µg/dl, i.e. severe vitamin E deficiency, at the time of diagnosis. Those participants with severe vitamin E deficiency had worse cognitive development. However, these data were subject to lead time bias.
4. Survival or age at death
This was not reported in the Wisconsin Trial. In a recent publication from the UK Trial seven children died in their first five years of life, of whom three had presented with meconium ileus (UK Trial 1991). All four deaths in the 'low risk' children, that is, in those without either meconium ileus or an affected older sibling, were in children allocated to the unscreened arm of the trial. This was reported as significant (P < 0.05) using a Fisher's exact test. However, the authors suggest that newborn screening might have influenced only two of these four deaths since two of the children who died were in fact diagnosed clinically by seven weeks of age.
Secondary outcomes
1. Number of respiratory exacerbations requiring treatment with systemic antibiotics
Not reported in the Wisconsin Trial.
2. Occurrence of pulmonary colonisation with any organism
In the latest report of the Wisconsin Trial limited information on acquisition with P. aeruginosa was reported (Farrell 2003). Screened participants were culture positive for P. aeruginosa earlier than control participants (median time to culture positive status of 3.01 years, compared with 6.04 years; P = 0.007). More screened participants (19 out of 56) were treated in an old, small clinic that integrated young and old participants irrespective of their pulmonary status compared with control participants (7 out of 47). In the latest report of the Wisconsin Trial, 56 screened participants were evaluated for longitudinal development of P. aeruginosa infection (Li 2005). These individuals acquired nonmucoid P. aeruginosa at a median age of 1.0 years without a significant effect on chest X‐ray scores and lung function. At 16 years of age all participants were colonised with mucoid P. aeruginosa. Non‐screened participants were not included in this report.
3. Number of hospital admissions
Not reported in the Wisconsin Trial.
4.Number of hospital days
These data were reported in the Wisconsin Trial, but were subject to lead time bias and therefore not reported here.
5. Number of respiratory complications
Not reported in the Wisconsin Trial.
6. The occurrence of diabetes mellitus
Not reported in the Wisconsin Trial.
7. The occurrence of liver cirrhosis
Not reported in the Wisconsin Trial.
8. Cognitive function (post hoc change)
Cognitive assessment data were obtained from 89 participants with CF from the Wisconsin Trial, aged 7 to 17 years (Koscik 2004). The Test of Cognitive Skills, Second Edition was administered to 42 screened and 47 control participants to generate the Cognitive Skills Index (CSI) and cognitive factor scores (verbal, non‐verbal and memory). Cognitive scores in the overall study population were similar to normative data: 102.5 (SD 16.6). The mean (SD) CSI scores were 104.4 (14.4) in the screened group and 99.8 (18.5) in the control group (P = 0.24). Mean (SD) scores for the 3 cognitive factors were: verbal: screened participants 104.6 (12.6) and control participants 101.8 (17.9) P = 0.45; non‐verbal: screened participants 106.4 (16.5) and control participants 100.7 (18.3) P = 0.17; and memory: screened participants 100.3 (15.1) and control participants 95.4 (19.5) P = 0.23. Low cognitive scores were found to be associated with low plasma α‐tocopherol levels. Subgroup analysis among participants with plasma α‐tocopherol levels less than 300 μg/dL reported worse scores among control participants: mean (SD) CSI scores for screened participants were 104.0 (16.2) versus 91.5 (15.1) for control patients, mean (SD). Verbal scores for screened participants were 104.8 (14.6) versus 93.0 (14.4) for control participants. Non‐verbal scores for screened participants were 103.4 (16.3) versus 94.7 (16.2); and finally memory scores for screened participants were 100.8 (18.0) and 89.1 (17.7) for control participants.
9. Quality of life (post hoc change)
In the Wisconsin Trial, quality of life was assessed using the Child Health Questionnaire (CHQ) among a subgroup of participants aged 10 to 15.5 years old (Koscik 2005). This questionnaire contains 87 items incorporated into 12 scales and has been validated for children 10 years and older. Seventy‐eight percent of all participants who were aged 10 years or older were included in this study. Reasons for exclusion were not mentioned. Koscik compared 15 screened participants to 21 control participants. Besides age at diagnosis, both groups differed significantly with respect to genotype (there were more participants with a ΔF508 mutation in the screened group) and lung function (average lung function measured as FEV1 and FEF25‐75 were worse among screened participants). Lung function did not differ anymore after controlling for P. aeruginosa status. CHQ scale scores were similar among screened and control participants except for the change in health scale, which showed better scores for the screened participants (median (range) scores 100 (50 to 100) for screened and 75 (25 to 100) for control participants (P = 0.02).
10. Adverse effects in the CF group diagnosed by screening
Not reported in the Wisconsin Trial.
In The Screened Group
Primary outcome
1. Adverse effects
The Wisconsin Trial examined adverse effects among parents in the screened population by comparing the 104 parents of infants with false‐positive diagnoses (who had the results communicated in the neonatal period) and parents in the control group of 18 children with false‐positive diagnoses who had the results communicated at the time of un‐masking of data aged four years. The exact timing of the administration of the questionnaire to each group was not clear. Misconception and miscommunication about screening results were reported as follows: lack of knowledge about neonatal screening ‐ 27% (screened) 67% (control); lack of knowledge about CF being among tests ‐ 75% (screened), 100% (control); incorrect interpretation of a positive IRT test ‐ 24% (screened), 89% control; and incorrect interpretation of a negative sweat test ‐ 12% (screened) infants, 6% (control).
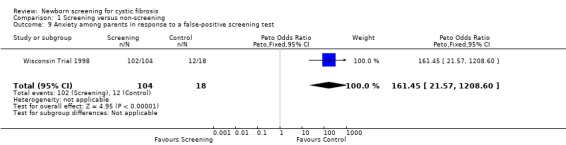
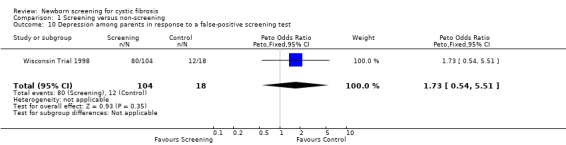
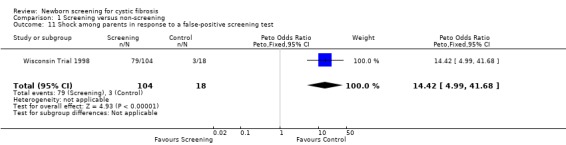
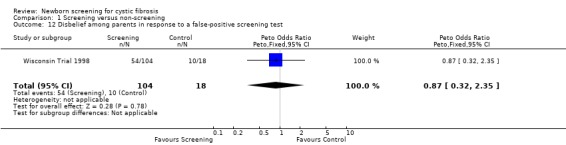
Negative emotional response of parents to a false‐positive screening test were reported as follows: anxiety ‐ 98% (screened), 67% control); depression ‐ 77% (screened), 67% (control); shock ‐ 76% (screened), 17% (control); disbelief ‐52% (screened), 56% (control); confusion ‐ 61% (screened), 72% (control); and anger ‐ 48% (screened), 67% (control).
Secondary outcome
1. The costs of the screening process
Analysis of screening costs from one year (1992) based on 70,000 births were reported as the cost per individual with CF diagnosed for two types of screening (Wisconsin Trial 1998):
for the IRT and sweat tests an estimated cost of US$7,613;
for the two tier screening IRT/DNA and sweat tests an estimated cost of US$7,403.
Costs for standard diagnostic methods, based on recognition through a positive family history or signs and symptoms and 1670 sweat tests performed, were reported as US$11,377 for each individual diagnosed with CF.
Another recent economic assessment of screening costs was reported for the year 2000 (Lee 2003). In that year 70,797 newborn IRT tests and 2926 DNA analyses (on blood of participants with the 4% highest daily IRT levels) were performed in Wisconsin (Wisconsin Trial 1998). The following costs were included in this economic assessment: the cost per test for the IRT/DNA screening and the cost per test for the sweat chloride testing, including technical fees and professional fees for interpretation of sweat test results. Expenses for counselling and costs for several 'sick visits' and a variety of diagnostic tests before being diagnosed with CF among non‐screened participants were not included in the analysis. The total costs of CF newborn screening (IRT and DNA) was estimated to be US$9025 per newly diagnosed individual with CF and US$2.66 per screened baby. These costs are comparable with costs for screening tests such as PKU. Assuming 20 newly diagnosed people with CF, the estimated annual cost per newly diagnosed CF child using the traditional method, based on costs of sweat testing, was US$16,846, or US$4.97 per newborn infant.
Additional estimated costs in screened children with a true negative screening test during infancy in whom sweat testing was performed later because of symptoms or signs suspect for CF were not reported. Because only mean values without standard deviations were reported, these data cannot be entered in the data tables for this review.
Discussion
The life expectancy of people with CF has improved enormously over the past 30 years. This systematic review aimed to establish whether there is evidence that early diagnosis of CF through neonatal screening, followed by current treatment, improves survival and long‐term morbidity, without unacceptable adverse effects. We have included two eligible trials, with limited evidence on the effects of neonatal screening. This reflects methodological issues in trial design as well as reporting of data.
Methodological issues include ascertainment bias, lead time bias and failure to analyse data on an intention‐to‐screen basis. Ascertainment bias was avoided in the Wisconsin Trial as all children were screened although results only made available to half in the neonatal period (unless requested by parents). This enabled uniform probability of ascertainment, and therefore comparable groups. The methodology used in the UK Trial did not address the issue of ascertainment bias, as a screening test was not performed on all individuals. Similarly, although control participants were identified through clinical manifestations of disease and sweat testing, the methods used to identify all clinically diagnosed cases were not stated. A further issue regarding case ascertainment was that children born in central Birmingham were excluded from the trial, and all people with CF in the region with meconium ileus are managed in central Birmingham.
Wald suggested that outcomes assessed before four years of age, when all screen‐detectable but as yet undiagnosed cases of CF in the control group in the Wisconsin Trial were revealed, should not be reported as they are subject to lead time bias (Wald 1998). In response to this, the Wisconsin trialists included analyses of the nutritional data restricted to data obtained after four years of age and demonstrated and reported persistence of the significant differences in height present in analyses including these data (Farrell 2001).
The validity of results of trials of screening interventions may also be compromised by failure to use an intention‐to‐screen analysis where all individuals are analysed in the group to which they were originally allocated. Excluding people with CF with meconium ileus, performance of an intention‐to‐screen‐analysis was possible in the Wisconsin Trial, but could not be made due to lack of data. An intention‐to‐screen analysis was not performed on data from the UK Trial because some of the false‐negative participants were analysed in the control group despite originally being allocated to the screened group. The issue associated with a lack of intention‐to‐screen analysis could be overcome if individual patient data were available; however these were not available for this version of the review.
Since screening only provides an opportunity for early treatment, the effect of screening is dependent on the available therapeutic options and care of people with CF at the time of study. Therefore studies of screening are not comparable with studies examining the effects of therapeutic interventions and are more difficult to interpret.
Regarding the pulmonary outcomes specified in this review, Chrispin‐Norman chest radiograph scores were reported in the UK Trial. In the Wisconsin Trial, screened participants at the time of diagnosis had better chest radiograph scores and more participants in the screened group had lung function tests within the normal limits at seven years of age, although differences in lung function parameters were not statistically significant between groups. Over time chest radiographs were worse in the screened groups and long‐term differences in lung function were statistically not significant between groups. However, despite adequate randomisation, the screened group contained more people with CF with pancreatic insufficiency, delta‐F508 genotypes and acquired P. aeruginosa at younger ages with more people with CF being treated at a small old clinic that integrated young and old people irrespective of their pulmonary status. Unfortunately, differences between screened and control participants regarding the several lung function parameters in the longitudinal analyses were not specified. Since only mean values for chest radiograph scores and pulmonary function were reported without standard deviations, these data cannot at present be entered in the data tables for this review.
Screening was associated with higher weight and height z scores in the Wisconsin Trial. After diagnosis all participants received a high energy intake (greater than 120% of daily requirement). Adequate weight gain was achieved at a significantly younger age among screened participants and screening was associated with a higher response rate within two years of diagnosis (Shoff 2006). In addition, fewer participants in the screened group had vitamin E deficiency at time of diagnosis. Vitamin E deficiency caused a worse cognitive function as studied among 89 participants aged 7 to 17 years (Koscik 2004). Overall, there was no statistically significant difference in cognitive function in children who were screened in the Wisconsin study compared to those diagnosed conventionally (Koscik 2004). Unfortunately, longitudinal nutritional data were represented only by mean values without standard deviations.
Overall, quality of life (QOL) did not differ significantly between screened and control participants from the Wisconsin Trial (Wisconsin Trial 1998). At the time this trial was designed no CF‐specific QOL measures validated for children and adolescents were available. Only a small proportion of participants were included in the QOL analysis and the reasons for exclusion of participants were not mentioned. A study using a CF questionnaire with a larger sample is underway (Koscik 2005).
The Wisconsin Trial examined adverse effects among parents in the screened and control populations by comparing parents of infants with false‐positive diagnoses in each arm. It is difficult to assess how meaningful these results are as the timing of the administration of the questionnaire to each group was not clear. It seems there was a large time difference (four years) between the two groups of data. Also, there was no attempt to address the effects of screening on the false‐negative participants.
The direct medical costs of screening compared with clinical diagnosis were addressed in the Wisconsin study (Lee 2003). These suggest that in 2000 in the USA it cost almost US$8000 less to diagnose CF by screening rather than other methods. Costs have not been related to effect. Moreover, costs other than for sweat testing leading to the diagnosis of CF in non‐screened individuals were not included. Therefore, the difference in costs between an early diagnosis by screening and a traditional diagnosis might even be greater.
Although we were unable to perform a quantitative meta‐analysis due to the differences in study design and the reporting of outcomes in the two included trials, a future update of this review is planned based on individual patient data meta‐analyses. At present this review has identified existing randomised trials of screening and provides a summary of the data available.
Screening in these trials took place in the period between 1985 and 1994. With time treatment options for CF have improved and screening to establish an early diagnosis becomes more important, not only for prognosis of people with CF but also for research on early therapeutic interventions.
Authors' conclusions
Implications for practice.
Newborn screening for CF benefits growth and prevents malnutrition in people with CF. Malnutrition may adversely influence cognitive function. From the available RCTs at this time, pulmonary benefits from CF newborn screening are likely in early childhood. Results on long‐term pulmonary prognosis were biased by confounding factors such as infection and pancreatic status. The expense of CF newborn screening is similar to the costs for other screening tests such as phenylketonuria and is less expensive than when diagnosed clinically.
Implications for research.
Future trials should take account of methodological issues, but it may not be possible ethically to ensure uniform probability of ascertainment and therefore there will always be issues of ascertainment bias as well as lead time bias. The possibility of the use of randomised cluster trials in assessment of screening interventions has been suggested (Waters 1999) and may be useful.
What's new
| Date | Event | Description |
|---|---|---|
| 12 August 2009 | Amended | Contact details updated. |
History
Protocol first published: Issue 1, 1999 Review first published: Issue 3, 1999
| Date | Event | Description |
|---|---|---|
| 12 November 2008 | Amended | Converted to new review format. |
| 12 November 2008 | New search has been performed | A search of the Group's Cystic Fibrosis Trials Register did not identify any references for possible inclusion in the review. |
| 12 November 2008 | New citation required but conclusions have not changed | Marieke Merelle has stepped down as lead author on the review, but remains involved as a co‐author. Kevin Southern has taken on the role of lead author and guarantor for the review. |
| 20 February 2008 | New search has been performed | A search of the Cystic Fibrosis Trials Register identified four new references. Three of these were additional references to the already included Wisconsin Trial (Koscik 2006a; Koscik 2006b; Shoff 2006b). None of these references contained additional data which could be used in this review. The fourth reference was also excluded since the objective of this study was not part of our review (Lagoe 2005). |
| 21 February 2007 | Amended | Two authors have stepped down from an active role within the review (Carol Dezateux and Cath Lees), two new authors have joined the review team (Jeannette Dankert‐Roelse and Kevin Southern). Data from the original papers for weight and height below the 10th percentile have been analysed using the generic inverse variance method and are now presented graphically. |
| 21 February 2007 | New search has been performed | A search of the Cystic Fibrosis Trials Register identified five new references. All were additional references to the already included Wisconsin Trial (Braun 2006; Koscik 2004; Koscik 2005; Li 2005; Shoff 2006). |
| 25 February 2004 | New search has been performed | This update has identified three further publications from the UK trial (UK Trial 1991) and nine from the Wisconsin trial (Wisconsin Trial 1998). Doull presents revised total birth numbers in the screened and unscreened arms, updates figures for total CF diagnoses in the screened and unscreened arms, and reports mortality to five years in the UK trial (Doull 2001). The authors report seven deaths out of a total of 176 affected children. Three occurred in those with meconium ileus, while the remaining four all occurred in children in the unscreened arm who had no affected older sibling. Two of these children had been diagnosed clinically by seven weeks of age. Conference abstracts from both the UK and Wisconsin trials suggest that airway function as assessed by spirometry does not differ between screened and unscreened infants, but data have not been presented in a form that can be incorporated in this update. Data on pulmonary function, chest radiograph scores, screening costs and vitamin E deficiency were reported in the new reports of the Wisconsin trial. At seven years of age, differences in mean (standard deviation (SD)) % predicted FEV1/FVC (94 (1.8)% versus 95 (1.3)% screened versus control) were not statistically significant. At this age, 88% of screened participants and 75% of control participants had lung function parameters within the normal limits at 89% predicted or greater. Over time until 16 years of age, both groups showed mild pulmonary dysfunction. Differences in % predicted FEV1/FVC, % predicted FEV1 or FEF25‐75 and RV/TLC were not significant between screened and control participants. At time of diagnosis chest radiograph scores were significantly better among screened particiapnts. Longitudinal assessments between ages 5 and 16 years revealed worse scores over time in the screened group. Results, however, were no longer significant after adjustment for differences in genotype, pancreatic status, and P. aeruginosa‐culture results. Screened participants had a much longer duration of infection and were earlier colonised with P. aeruginosa than control participants. Although fewer screened patients had vitamin E deficiency, these data were subject to lead time bias. A more recent estimation of direct medical costs of screening suggested diagnosis through screening is less expensive than through traditional means. There is not substantive change following this update of the review. Seventeen references to the two included trials which were previously listed in 'Excluded Studies' (relating to outcomes not relevant to the review) have been moved to 'Included Studies'. |
| 10 April 2001 | New citation required and conclusions have changed | Substantive amendment |
Acknowledgements
We are grateful to Professor Ros Smyth, Jill Motley, Tracey Remmington, Olwen Beaven and the Cochrane Cystic Fibrosis and Genetic Disorders Editorial Group for their invaluable support; to Professor Carol Dezateux and Dr Cath Lees for their significant input into the earlier versions of this review; and to Drs G. Mastella, R.M. Dauphinais, G. Travert, P.M. Farrell, I. Doull and J.M Littlewood who kindly provided information regarding studies.
Data and analyses
Comparison 1. Screening versus non‐screening.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Wisconsin chest X‐ray scores | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.1 First score available | 1 | 89 | Mean Difference (IV, Fixed, 95% CI) | ‐2.80 [‐5.03, ‐0.57] |
| 1.2 At age 14 years | 1 | 22 | Mean Difference (IV, Fixed, 95% CI) | 11.91 [2.69, 21.13] |
| 2 Brasfield chest X‐ray scores | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.1 First score available | 1 | 89 | Mean Difference (IV, Fixed, 95% CI) | 1.10 [0.13, 2.07] |
| 2.2 At age 14 years | 1 | 22 | Mean Difference (IV, Fixed, 95% CI) | ‐3.10 [‐5.77, ‐0.43] |
| 3 Weight below 10th percentile (at 13 years old) | 1 | Odds ratio (Fixed, 95% CI) | Totals not selected | |
| 4 Height below 10th percentile (at 13 years old) | 1 | Odds ratio (Fixed, 95% CI) | Totals not selected | |
| 5 Lack of knowledge among parents about neonatal screening | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Totals not selected | |
| 6 Lack of knowledge among parents about CF being among tests | 1 | 122 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.23 [0.07, 0.77] |
| 7 Incorrect interpretation by parents of a positive IRT test | 1 | 122 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.06 [0.02, 0.16] |
| 8 Incorrect interpretation by parents of a negative sweat test | 1 | 122 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.87 [0.37, 9.38] |
| 9 Anxiety among parents in response to a false‐positive screening test | 1 | 122 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 161.45 [21.57, 1208.60] |
| 10 Depression among parents in response to a false‐positive screening test | 1 | 122 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.73 [0.54, 5.51] |
| 11 Shock among parents in response to a false‐positive screening test | 1 | 122 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 14.42 [4.99, 41.68] |
| 12 Disbelief among parents in response to a false‐positive screening test | 1 | 122 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.87 [0.32, 2.35] |
| 13 Confusion among parents in response to a false‐positive screening test | 1 | 122 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.61 [0.22, 1.71] |
| 14 Anger among parents in response to a false‐positive screening test | 1 | 122 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.48 [0.18, 1.30] |
1.1. Analysis.
Comparison 1 Screening versus non‐screening, Outcome 1 Wisconsin chest X‐ray scores.
1.2. Analysis.
Comparison 1 Screening versus non‐screening, Outcome 2 Brasfield chest X‐ray scores.
1.3. Analysis.
Comparison 1 Screening versus non‐screening, Outcome 3 Weight below 10th percentile (at 13 years old).
1.4. Analysis.
Comparison 1 Screening versus non‐screening, Outcome 4 Height below 10th percentile (at 13 years old).
1.5. Analysis.
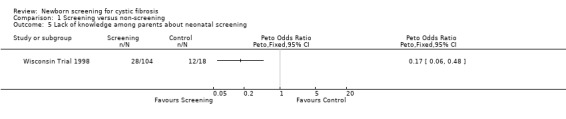
Comparison 1 Screening versus non‐screening, Outcome 5 Lack of knowledge among parents about neonatal screening.
1.6. Analysis.
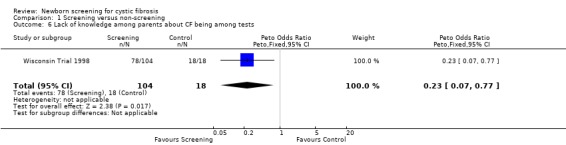
Comparison 1 Screening versus non‐screening, Outcome 6 Lack of knowledge among parents about CF being among tests.
1.7. Analysis.
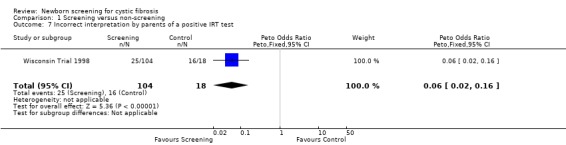
Comparison 1 Screening versus non‐screening, Outcome 7 Incorrect interpretation by parents of a positive IRT test.
1.8. Analysis.
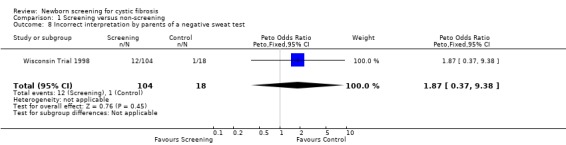
Comparison 1 Screening versus non‐screening, Outcome 8 Incorrect interpretation by parents of a negative sweat test.
1.9. Analysis.
Comparison 1 Screening versus non‐screening, Outcome 9 Anxiety among parents in response to a false‐positive screening test.
1.10. Analysis.
Comparison 1 Screening versus non‐screening, Outcome 10 Depression among parents in response to a false‐positive screening test.
1.11. Analysis.
Comparison 1 Screening versus non‐screening, Outcome 11 Shock among parents in response to a false‐positive screening test.
1.12. Analysis.
Comparison 1 Screening versus non‐screening, Outcome 12 Disbelief among parents in response to a false‐positive screening test.
1.13. Analysis.
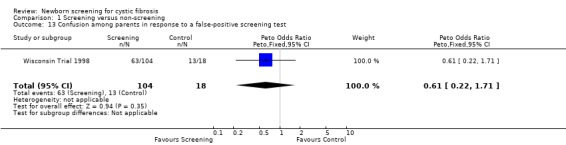
Comparison 1 Screening versus non‐screening, Outcome 13 Confusion among parents in response to a false‐positive screening test.
1.14. Analysis.
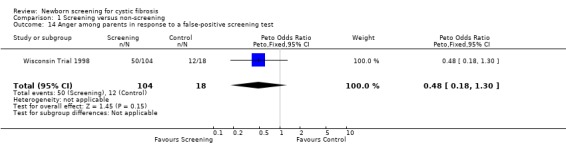
Comparison 1 Screening versus non‐screening, Outcome 14 Anger among parents in response to a false‐positive screening test.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
UK Trial 1991.
| Methods | Screening from January 1985 to December 1989 in Wales and from January 1985 to October 1989 in the West Midlands. Randomisation method: screening on alternate weeks. Control participants were identified by clinical manifestations of the disease. Validity: ascertainment bias and lead time bias. | |
| Participants | 230,076 screened and 234,510 not screened neonates (Doull 2001). 176 CF participants: 86 were in the screened group and 90 in the control group. Number of males and females was not reported. Infants born in the central Birmingham area, those with MI and those with an elder sibling with CF were excluded from analysis. In earlier reports 13 participants with a false‐negative screening result were incorrectly analysed as part of the control group. Age range 0 ‐ 4 years (data up to years are described in the study by Sebire (Sebire 1995)) and, in abstract only, to 10 years by Ryley (Ryley 2001). | |
| Interventions | Type of screening: IRT test followed by a second IRT test when the first IRT test was positive (IRT levels > 900 ng/ml). | |
| Outcomes | Outcomes included in this review: weight and height SD scores, carriage rates for P. aeruginosa and S. aureus, chest radiograph scores, lung function tests, number of hospital admissions, number of hospital days, survival and Shwachman score. | |
| Notes | Lung function tests are only reported in the study by Sebire (Sebire 1995) and, as an abstract only by Ryley (Ryley 2001). In both these reports there is only a description of the results given, no data are published. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Unclear risk | Randomisation method: screening on alternate weeks, but doesn't state how it was decided which group (screened or non‐screened) would be first. A change in attitude to the protocol in one district in the West Midlands led to the screening being stopped in October 1989, but it continued in Wales until December 1989. Babies born in the West Midlands between October and December 1989 are included in the unscreened group, but the investigators state that this 3‐month period was unlikely to have a significant effect to allocation over the whole 5‐year period. |
| Allocation concealment? | High risk | Alternation method ‐ screening on alternate weeks, investigators would be able to foresee which group babies would be allocated to. |
| Blinding? All outcomes | Unclear risk | Not reported in paper. |
| Incomplete outcome data addressed? All outcomes | High risk | ITT not possible. Infants born in the central Birmingham area, those with MI and those with an elder sibling with CF were excluded from analysis. |
| Free of selective reporting? | High risk | Lung function tests are only reported in the study by Sebire (Sebire 1995) and, as an abstract only by Ryley (Ryley 2001). In both these reports there is only a description of the results given, no data are published. |
| Free of other bias? | High risk | Ascertainment bias; lead time bias |
Wisconsin Trial 1998.
| Methods | Screening from 1985 until 1994. Randomisation method: terminal digit of blood spots. Two centres. Control participants were identified by presence of MI, family history of CF, development of symptoms or signs of CF, or positive result of screening test after unmasking of data at 4 years of age. Validity: lead time bias. | |
| Participants | 650,341 neonates entered the study; 325,171 were allocated to the screened group and 325,170 to the control group. 108 CF participants: 57 (63% male) were in the screened group and 51 (61% male) in the control group. Age range 0 ‐ 11 years. Participants with MI were excluded from analysis, except for the study by Farrell (Farrell 1995). | |
| Interventions | Type of screening: from April 15, 1985 to June 30, 1991: IRT testing on blood spots (levels > 180 ng/ml were considered positive) and from July 1, 1991 to June 30, 1994: IRT testing followed by DNA analysis for the ΔF508 mutant allele, when levels of IRT were > 110 ng/ml. | |
| Outcomes | Outcomes included in this review: lung function, chest radiograph scores, weight‐for‐age z score, height‐for‐age z score, weight percentile, height percentile (adjustments for these outcomes were made for difference in birth weight), Shwachman‐Kulczycki score, number of hospital days, occurrence of pulmonary colonisation with any organism, direct medical costs and adverse effects in the screened population. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Unclear risk | Randomisation method: terminal digit of code on blood spot specimen determined which group infant was assigned to, but no discussion of how it was decided which groups even numbers and odd numbers related to. |
| Allocation concealment? | Low risk | Specimens sent to a centralized laboratory to be randomised |
| Blinding? All outcomes | Unclear risk | Not reported in paper |
| Incomplete outcome data addressed? All outcomes | Unclear risk | Participants with MI were excluded from analysis, except for the study by Farrell (Farrell 1995). |
| Free of other bias? | High risk | Lead time bias |
CF: cystic fibrosis IRT: immunoreactive trypsin MI: meconium ileus P. aeruginosa: Pseudomonas aeruginosa S. aureus: Staphylococcus aureus SD: standard deviation
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Barlocco 1988 | Not a randomised controlled trial. |
| Carter 1987 | Not a randomised controlled trial. |
| Dauphinais 1992 | Not a randomised controlled trial. |
| Lagoe 2005 | The objective of this trial was not part of our review. |
Differences between protocol and review
Two secondary outcomes were later added to those for the CF Group, namely 'Cognitive function' and 'Quality of life'. These were not initially included in the review, but trials have reported them in longer term analyses. We feel these outcomes reflect the changing aspirations of people with CF and their importance merits post hoc inclusion in the review.
Contributions of authors
For the original review, Marieke Mérelle, Carol Dezateux, Cath Lees and Ad Nagelkerke independently assessed studies for inclusion in this review and assisted in writing of text.
Cath Lees independently extracted data from the included studies.
As from Issue 2, 2006 of The Cochrane Library, Carol Dezateux and Cath Lees have no longer actively participated in the review. Two new authors have joined the review team, Jeannette Dankert‐Roelse and Kevin Southern.
As from Issue 1, 2009 Kevin Southern acts as guarantor for the review.
Declarations of interest
None known.
Edited (no change to conclusions)
References
References to studies included in this review
UK Trial 1991 {published and unpublished data}
- Al‐Jader LN, Goodchild MC, Ryley HC, Harper PS. Attitudes of parents of cystic fibrosis children towards neonatal screening and antenatal diagnosis. Clinical Genetics 1990;38(6):460‐5. [DOI] [PubMed] [Google Scholar]
- Chatfield S, Owen G, Ryley H, Goodchild M, Weller P. Does early detection lead to an improved prognosis in cystic fibrosis neonates?. Acta Universitatis Carolinae Medica 1990;36(1‐4):96‐8. [PubMed] [Google Scholar]
- Chatfield S, Owen G, Ryley HC, Goodchild MC, Weller P. Evaluation of the usefulness of neonatal screening in cystic fibrosis [abstract]. Proceedings of the 14th Annual Meeting of the BAPEN & 27th Annual Meeting of the PRS; Oxford. 1989:A.
- Chatfield S, Owen G, Ryley HC, Williams J, Goodchild MC, Weller P. Neonatal screening for cystic fibrosis in Wales and the West Midlands: clinical assessment after five years of screening. Archives of Disease in Childhood 1991;66(1):29‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doull IJ, Ryley HC, Weller P, Goodchild MC. Cystic fibrosis‐related deaths in infancy and the effect of newborn screening. Pediatric Pulmonology 2001;31(5):363‐6. [DOI] [PubMed] [Google Scholar]
- Doull IJM, Ryley HC, Weller P, Goodchild MC. Death from cystic fibrosis in the first five years of life and the effect of newborn screening [abstract]. 13th International Cystic Fibrosis Congress; 2000 June 4‐8; Stockholm, Sweden. 2000; Vol. 112.
- Ryley HC, Deam SM, Williams J, Alfaham M, Weller PH, Goodchild MC, et al. Neonatal screening for cystic fibrosis in Wales and the West Midlands: I. Evaluation of immunoreactive trypsin test. Journal of Clinical Pathology 1988;41(7):726‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryley HC, Desai M, Weller P, Doull I. Clinical status of screened and unscreened CF children at age 10 years [abstract]. Abstracts of the 24th European Cystic Fibrosis Conference; 2001 June 6‐9; Vienna. 2001:P20.
- Sebire P, Desai M, Millar‐Jones L, Lacy D, Ryley HC, Weller PH, et al. Neonatal screening for cystic fibrosis: evidence in favour of screening from clinical scoring systems [abstract]. Proceedings of the 20th European Cystic Fibrosis Conference; 1995 June 18‐21; Brussels. 1995.
- Weller P. Personal communication 2007.
Wisconsin Trial 1998 {published and unpublished data}
- Braun AT, Farrell PM, Ferec C, Audrezet MP, Laxova A, Li Z, et al. Cystic fibrosis mutations and genotype‐pulmonary phenotype analysis. Journal of Cystic Fibrosis 2006;5(1):33‐41. [DOI] [PubMed] [Google Scholar]
- Douglas JA, Rock MJ, Lai HC, Splaingard ML, Becker MA, Zaremba KM, et al. Association of cognitive abilities and head circumference in patients with cystic fibrosis [abstract]. Pediatric Pulmonology 2000;Suppl 20:336. [Google Scholar]
- Farrell P, Kosorok M, Rock M, Splaingard M, Laxova A, Zeng L, et al. Pulmonary disease after delayed or with early diagnosis through neonatal screening [abstract]. Journal of Cystic Fibrosis 2002;1(Suppl 1):S16. [Google Scholar]
- Farrell PM. Improving the health of patients with cystic fibrosis through newborn screening Wisconsin Cystic Fibrosis Neonatal Screening Study Group. Advances in Pediatrics 2000;47:79‐115. [PubMed] [Google Scholar]
- Farrell PM, Koscik R, Laxova A, Mischler E, Splaingard M, Laessig R, et al. Neonatal screening for cystic fibrosis: comparison of single test for immunoreactive trypsinogen (IRT) vs IRT/DNA [abstract]. Pediatric Pulmonology 1994;Suppl 10:216‐7. [Google Scholar]
- Farrell PM, Koscik RE, Egmond A, Fosorok MR, Laxova A, Feenan L, et al. Early nutritional therapy in cystic fibrosis [abstract]. Pediatric Pulmonology 1995;Suppl 12:90‐1. [Google Scholar]
- Farrell PM, Kosorok MR, Laxova A, Shen G, Koscik RE, Bruns WT, et al. Nutritional benefits of neonatal screening for cystic fibrosis. New England Journal of Medicine 1997;337(14):963‐9. [DOI] [PubMed] [Google Scholar]
- Farrell PM, Kosorok MR, Rock MJ, Laxova A, Zeng L, Hoffman G, et al. Assessment of the benefits, risks and costs of cystic fibrosis screening in Wisconsin, USA [abstract]. Proceedings of the Fifth International Conference on Neonatal Screening for Cystic Fibrosis; 1998. 1998:239‐53.
- Farrell PM, Kosorok MR, Rock MJ, Laxova A, Zeng L, Lai HC, et al. Early diagnosis of cystic fibrosis through neonatal screening prevents severe malnutrition and improves long‐term growth. Pediatrics 2001;107(1):1‐13. [DOI] [PubMed] [Google Scholar]
- Farrell PM, Kosorok MR, Rock MJ, Wisconsin Cystic Fibrosis Neonatal Screening Study Group. Assessments of the benefits, risks, and costs of cystic fibrosis newborn screening in Wisconsin [abstract]. Morbidity and Mortality Weekly Report 1997;46(No. RR‐16):8‐9. [Google Scholar]
- Farrell PM, Kosorok MR, Zeng L, Rock MJ, Splaingard M, Laxova A, et al. Assessing pulmonary outcomes associated with early diagnosis through neonatal screening [abstract]. Abstracts of the 24th European Cystic Fibrosis Conference; 2001 June 6‐9; Vienna. 2001:P36.
- Farrell PM, Lai H, Li Z, Kosorok MR, Laxova A, Green CG, et al. Evidence on improved outcomes with early diagnosis of cystic fibrosis through neonatal screening: enough is enough!. Journal of Pediatrics 2005;147(3 Suppl):S30‐6. [DOI] [PubMed] [Google Scholar]
- Farrell PM, Laxova A, Splaingard M, Rock MJ, Zeng L, Kosorok MR, et al. Lung disease severity after diagnosis of cystic fibrosis through neonatal screening [abstract]. Pediatric Pulmonology 2000;Suppl 20:308. [Google Scholar]
- Farrell PM, Li Z, Kosorok M, Laxova A, Green CG, Collins J, et al. Bronchopulmonary disease in children with cystic fibrosis after early or delayed diagnosis. American Journal of Respiratory and Critical Care Medicine 2003;168(9):1100‐8. [DOI] [PubMed] [Google Scholar]
- Farrell PM, Li Z, Kosorok MR, Laxova A, Collins J. Mucoid pseudomonas aeruginosa infection, antibiotic resistance, and lung disease progression in children with cystic fibrosis [abstract]. Journal of Cystic Fibrosis 2005;4(Suppl):S56. [DOI] [PubMed] [Google Scholar]
- Farrell PM, Li Z, Kosorok MR, Laxova A, Green CG, Collins J, Lai HC, et al. Longitudinal evaluation of bronchopulmonary disease in children with cystic fibrosis. Pediatric Pulmonology 2003;36(3):230‐40. [DOI] [PubMed] [Google Scholar]
- Farrell PM, Mischler EH. Newborn screening for cystic fibrosis. Advances in Pediatrics 1992;39:35‐70. [PubMed] [Google Scholar]
- Farrell PM, Shen G, Splaingard M, Colby CE, Laxova A, Kosorok MR, et al. Acquisition of Pseudomonas aeruginosa in children with cystic fibrosis [abstract]. Pediatrics 1997;100(5):886. [DOI] [PubMed] [Google Scholar]
- Farrell PM, Wisconsin Cystic Fibrosis Neonatal Screening Study Group. Assessment of the benefits and costs of CF neonatal screening in Wisconsin [abstract]. Pediatric Pulmonology 1997;Suppl 14:192‐3. [Google Scholar]
- Farrell PM, Wisconsin Cystic Fibrosis Neonatal Screening Study Group. Comparison of newborn screening methods and use of the sweat test for diagnosis of cystic fibrosis [abstract]. Morbidity and Mortality Weekly Report 1997;46(No. RR‐16):11‐2. [Google Scholar]
- Fost N, Farrell PM. A prospective randomized trial of early diagnosis and treatment of cystic fibrosis: a unique ethical dilemma. Clinical Research 1989;37(3):495‐500. [PubMed] [Google Scholar]
- Gregg RG, Simantel A, Farrell PM, Koscik R, Kosorok MR, Laxova A, et al. Newborn screening for cystic fibrosis in Wisconsin: comparison of biochemical and molecular methods. Pediatrics 1997;99(6):819‐24. [DOI] [PubMed] [Google Scholar]
- Hassemer DJ, Laessig RH, Hoffman GL, Farrell PM. Laboratory quality control issues related to screening newborns for cystic fibrosis using immunoreactive trypsin. Pediatric Pulmonology 1991;Suppl 7:76‐83. [DOI] [PubMed] [Google Scholar]
- Koscik R, Douglas J, Zaremba K, Lai H, Rock M, et al. Quality of life in children with cystic fibrosis [abstract]. Pediatric Pulmonology 2003;Suppl 25:358‐9. [Google Scholar]
- Koscik R, Kosorok MR, Zaremba KM, Laxova A, Lai H, Douglas JA, et al. Early vitamin E deficiency and subsequent cognitive function: A potential benefit of neonatal screening for cystic fibrosis [abstract].. Pediatric Pulmonology 2003;Suppl 25:359. [Google Scholar]
- Koscik RL, Douglas JA, Zaremba K, Rock MJ, Splaingard ML, Laxova A, et al. Quality of life of children with cystic fibrosis. Journal of Pediatrics 2005;147(3 Suppl 1):S64‐8. [DOI] [PubMed] [Google Scholar]
- Koscik RL, Farrell PM, Kosorok MR, Zaremba KM, Laxova A, Lai HC, et al. Cognitive function of children with cystic fibrosis: deleterious effect of early malnutrition. Pediatrics 2004;113(6):1549‐58. [DOI] [PubMed] [Google Scholar]
- Koscik RL, Shoff S, Lai H, Zaremba K, Laxova A, Rock M, et al. Is recovery of birth weight Z‐score within 2 years of diagnosis related to quality of life in later childhood [abstract]. Pediatric Pulmonology 2006;41(Suppl 29):400. [Google Scholar]
- Koscik RL, Zaremba K, Laxova A, Gershan W, Rock M, Farrell P. Quality of life in childhood and adolescence: do patients with CF and their parents agree? [abstract]. Pediatric Pulmonology 2006;41(Suppl 29):399. [Google Scholar]
- Kosorok MR, Farrell PM, Wisconsin Cystic Fibrosis Neonatal Screening Study Group. Design and execution of the Wisconsin Cystic Fibrosis Newborn Screening trial [abstract]. Morbidity and Mortality Weekly Report 1997;46(No. RR‐16):8. [Google Scholar]
- Kosorok MR, Jalaluddin M, Farrell PM, Shen G, Colby CE, Laxova A, et al. Comprehensive analysis of risk factors for acquisition of Pseudomonas aeruginosa in young children with cystic fibrosis. Pediatric Pulmonology 1998;26(2):81‐8. [DOI] [PubMed] [Google Scholar]
- Lai HC, Chen ST, Koscik RE, Farrell PM, Kosorok MR. Occurrence of poor growth at diagnosis of Cystic Fibrosis [abstract]. Pediatric Pulmonology 1995;Suppl 12:311. [Google Scholar]
- Lai HC, Kosorok MR, Laxova A, Davis LA, FitzSimmon SC, Farrell PM. Nutritional status of patients with cystic fibrosis with meconium ileus: a comparison with patients without meconium ileus and diagnosed early through neonatal screening.. Pediatrics 2000;105(1 Pt 1):53‐61. [DOI] [PubMed] [Google Scholar]
- Lai HC, Kosorok MR, Laxova A, Farrell PM. Nutritional status of CF patients with meconium ileus (MI): a comparison with non‐MI patients diagnosed early through neonatal screening [abstract]. Pediatric Pulmonology 1998;Suppl 17:356. [DOI] [PubMed] [Google Scholar]
- Lee DS, Rosenberg MA, Peterson A, Makholm L, Hoffman G, Laessig RH, et al. Analysis of costs of diagnosing cystic fibrosis with a newborn screening program. Journal of Paediatrics 2003;142(6):617‐23. [DOI] [PubMed] [Google Scholar]
- Li Z, Kosorok MR, Farrell PM, Laxova A, West SHE, Green CG, et al. Longitudinal development of mucoid Pseudomonas aeruginosa infection and lung disease progression in children with cystic fibrosis. JAMA 2005;293(5):581‐8. [DOI] [PubMed] [Google Scholar]
- Li Z, Lai HJ, Kosorok MR, Laxova A, Rock MJ, Splaingard ML, Farrell PM. Longitudinal pulmonary status of cystic fibrosis children with meconium ileus. Pediatric Pulmonology 2004;38(4):277‐84. [DOI] [PubMed] [Google Scholar]
- Marcus MS, Sondel SA, Farrell PM, Laxova A, Carey PM, Langhough R, et al. Nutritional status of infants with cystic fibrosis associated with early diagnosis and intervention. American Journal of Clinical Nutrition 1991;54(3):578‐85. [DOI] [PubMed] [Google Scholar]
- Mischler E, Farrell P, Bruns T, Rock M, Tluczek A, Colby H, et al. Wisconsin experience with newborn screening for cystic fibrosis: no conclusions yet! [abstract]. Pediatric Pulmonology 1988;Suppl 2:43‐4. [Google Scholar]
- Mischler E, Farrell PM, Splaingard M, Laxova A, Koscik R. Pulmonary epidemiology over 10 years of cystic fibrosis in a screened population [abstract]. Pediatric Pulmonology 1995;Suppl 12:285. [Google Scholar]
- Mischler E, Rock M, Farrell P, Bruns T, Palta M, Fost N, et al. Wisconsin experience with screening and clinical psychosocial description of patients with false positive screen [abstract]. Pediatric Pulmonology 1987;Suppl:81‐3. [Google Scholar]
- Mischler EH, Marcus MS, MS, Sondel SA, Laxova A, Carey P, Langhough R, et al. Nutritional assessment of infants with cystic fibrosis diagnosed through screening. Pediatric Pulmonology 1991;Suppl 7:56‐63. [DOI] [PubMed] [Google Scholar]
- Mischler EH, Wilfond BS, Fost N, Laxova A, Reiser C, Sauer CM, et al. Cystic fibrosis newborn screening: impact on reproductive behavior and implications for genetic counselling. Pediatrics 1998;102(1 Pt 1):44‐52. [DOI] [PubMed] [Google Scholar]
- Rock MJ, Davis L, Marcus M, Lai HC, Douglas J, Kosorok M, et al. Early nutritional intervention in infants with CF [abstract]. Pediatric Pulmonology 1999;Suppl 19:102‐3. [Google Scholar]
- Rock MJ, Mischler EH, Farrell PM, Wei LJ, Bruns WT, Hassemer DJ, et al. Newborn screening for cystic fibrosis is complicated by age‐related decline in immunoreactive trypsinogen levels. Pediatrics 1990;85(6):1001‐7. [PubMed] [Google Scholar]
- Shoff S, Lai H. Recovery of birth weight Z‐score within 2 years of diagnosis is positively associated with pulmonary status at age 6 years [abstract]. Pediatric Pulmonology 2006;41(Suppl 29):388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoff SM, Ahn HY, Davis L, Lai HC, the Wisconsin CF Neonatal Screening Group. Temperal associations among energy intake, plasma linoleic acid, and growth improvement in response to treatment initiation after diagnosis of cystic fibrosis. Pediatrics 2006;117(2):391‐400. [DOI] [PubMed] [Google Scholar]
- Tluczek A, Mischler EH, Bowers B, Peterson NM, Morris ME, Farrell PM, et al. Psychological impact of false‐positive results when screening for cystic fibrosis. Pediatric Pulmonology 1991;Suppl 7:29‐37. [DOI] [PubMed] [Google Scholar]
- Tluczek A, Mischler EH, Farrell PM, Fost N, Peterson NM, Carey P, et al. Parents' knowledge of neonatal screening and response to false positive cystic fibrosis testing. Journal of Developmental and Behavioral Pediatrics 1992;13(3):181‐6. [PubMed] [Google Scholar]
- Egmond AW, Kosorok MR, Koscik R, Laxova A, Farrell PM. Effect of linoleic acid intake on growth of infants with cystic fibrosis. American Journal of Clinical Nutrition 1996;63(5):746‐52. [DOI] [PubMed] [Google Scholar]
- Wilfond BS, Farrell PM, Laxova A, Mischler E. Severe hemolytic anemia associated with vitamin E deficiency in infants with cystic fibrosis. Clinical Pediatrics 1994;33(1):2‐7. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Barlocco 1988 {published and unpublished data}
- Barlocco EG, Benetazzo D, Borgo G, Braggion C, Canciani M, Caprini D, et al. Historical course of neonatal screening for cystic fibrosis (CF) in Veneto and surrounding areas. Evaluation over 12.8 years [abstract]. Proceedings of the Second International Conference on Neonatal Screening for Cystic Fibrosis; 1987. 1987:5‐12.
- Barlocco EG, Benetazzo D, Borgo G, Braggion C, Canciani M, Caprini D, et al. The study of factors affecting the causes of cystic fibrosis [abstract]. Proceedings of the Second International Conference on Neonatal Screening for Cystic Fibrosis; 1987. 1987:13‐8.
- Barlocco EG, Mastella G, Borgo G, Braggion C, Cazzola G, Conforti M. Medical care and genetic factors in the outcome of the CF patients diagnosed by neonatal screening [abstract]. Proceedings of the Third International Conference on Neonatal Screening for Cystic Fibrosis; 1988. 1988:153‐65.
- Mastella G, Barlocco EG, Antonacci B, Borgo G, Braggion C, Cazolla G, et al. Is neonatal screening advantageous? The answer of a wide 15 years follow‐up study [abstract]. Proceedings of the Third International Conference on Neonatal Screening for Cystic Fibrosis; 1988. 1988:127‐42.
- Mastella G, Castellani C, Faraguna D, Miano A, Zanolla L. Long‐term outcome in patients with cystic fibrosis identified through newborn screening [abstract]. Pediatric Pulmonology 1995;Suppl 12:88‐9. [Google Scholar]
Carter 1987 {unpublished data only}
- Carter E, Johnson S. A comparison of progress of children with cystic fibrosis diagnosed by immunoreactive trypsin screening or by clinical means [abstract]. Proceedings of the Second International Conference on Neonatal Screening for Cystic Fibrosis; 1987. 1987:65‐6.
Dauphinais 1992 {published data only}
- Dauphinais RM. A cost analysis of blood‐spot screening newborns for cystic fibrosis. Journal of Clinical Immunoassay 1992;15(2):121‐5. [Google Scholar]
- Dauphinais RM. Comparative costs in diagnosing cystic fibrosis by blood‐spot screening vs. non‐screening [abstract]. Proceedings of the Fourth International Conference of Neonatal Screening for Cystic Fibrosis; 1990. 1990:12.
- Dauphinais RM, Jezyk PD. IRT screening of newborns for cystic fibrosis: Connecticut experience [abstract]. Proceedings of the Third International Conference on Neonatal Screening for Cystic Fibrosis; 1988; Caen, France. 1988:230.
Lagoe 2005 {published data only}
- Lagoe E, Labella S, Arnold G, Rowley PT. Cystic fibrosis newborn screening: a pilot study to maximize carrier screening. Genetic Testing 2005;9(3):255‐60. [DOI] [PubMed] [Google Scholar]
Additional references
Armstrong 1997
- Armstrong DS, Grimwood K, Carlin JB, Carzino R, Gutierrez JP, Hull J, et al. Lower airway inflammation in infants and young children with cystic fibrosis. American Journal of Respiratory and Critical Care Medicine 1997;156(4 Pt 1):1197‐204. [DOI] [PubMed] [Google Scholar]
Balough 1995
- Balough K, McCubbin M, Weinberger M, Smits W, Ahrens R, Fick R. The relationship between infection and inflammation in the early stages of lung disease from cystic fibrosis. Pediatric Pulmonology 1995;20(2):63‐70. [DOI] [PubMed] [Google Scholar]
Baroni 1997
- Baroni MA, Anderson YE, Mischler E. Cystic fibrosis newborn screening: Impact of early screening results on parenting stress. Pediatric Nursing 1997;23(2):143‐51. [PubMed] [Google Scholar]
Dankert‐Roelse 1995
- Dankert‐Roelse JE, Meerman GJ. Long term prognosis of patients with cystic fibrosis in relation to early detection by neonatal screening and treatment in a cystic fibrosis centre. Thorax 1995;50(7):712‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Doull 2001
- Doull IJ, Ryley HC, Weller P, Goodchild MC. Cystic fibrosis‐related deaths in infancy and the effect of newborn screening. Pediatric Pulmonology 2001;31(5):363‐6. [DOI] [PubMed] [Google Scholar]
Elborn 1991
- Elborn JS, Shale DJ, Britton JR. Cystic fibrosis: Current survival and population estimates to the year 2000. Thorax 1991;46(12):881‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Farrell 1997
- Farrell PM, Kosorok MR, Laxova A, Shen G, Koscik RE, Bruns T, et al. Nutritional benefits of neonatal screening for cystic fibrosis. New England Journal of Medicine 1997;337(14):963‐9. [DOI] [PubMed] [Google Scholar]
Farrell 2001
- Farrell PM, Kosorok MR, Rock MJ, Laxova A, Zeng L, Lai HC, et al. Early diagnosis of cystic fibrosis through neonatal screening prevents severe malnutrition and improves long‐term growth. Pediatrics 2001;107(1):1‐13. [DOI] [PubMed] [Google Scholar]
Farrell 2003
- Farrell PM, Li Z, Kosorok M, Laxova A, Green CG, Collins J, et al. Bronchopulmonary disease in children with cystic fibrosis after early or delayed diagnosis. American Journal of Respiratory and Critical Care Medicine 2003;168(9):1100‐8. [DOI] [PubMed] [Google Scholar]
Fost 1989
- Fost N, Farrell PM. A prospective randomized trial of early diagnosis and treatment of cystic fibrosis: a unique ethical dilemma. Clinical Research 1989;37(3):495‐500. [PubMed] [Google Scholar]
Fuchs 1994
- Fuchs HJ, Borowitz DS, Christiansen DH, Morris EM, Nash ML, Ramsey BW, et al. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. New England Journal of Medicine 1994;331:637‐42. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Khan 1995
- KhanTZ, Wagener JS, Bost T, Martinez J, Accurso FJ, Riches DW. Early pulmonary inflammation in infants with cystic fibrosis. American Journal of Respiratory and Critical Care Medicine 1995;151(4):1075‐82. [DOI] [PubMed] [Google Scholar]
Koscik 2003
- Koscik R, Kosorok MR, Zaremba KM, Laxova A, Lai H, Douglas JA, et al. Early vitamin E deficiency and subsequent cognitive function: A potential benefit of neonatal screening for cystic fibrosis [abstract]. Pediatric Pulmonology 2003;Suppl 25:359. [Google Scholar]
Koscik 2004
- Koscik RL, Farrell PM, Kosorok MR, Zaremba KM, Laxova A, Lai HC, et al. Cognitive function of children with cystic fibrosis: deleterious effect of early malnutrition. Pediatrics 2004;113(6):1549‐58. [DOI] [PubMed] [Google Scholar]
Koscik 2005
- Koscik RL, Douglas JA, Zaremba K, Rock MJ, Splaingard ML, Laxova A, et al. Quality of life of children with cystic fibrosis. Journal of Pediatrics 2005;147(3 Suppl 1):S64‐8. [DOI] [PubMed] [Google Scholar]
Lee 2003
- Lee DS, Rosenberg MA, Peterson A, Makholm L, Hoffman G, Laessig RH, et al. Analysis of costs of diagnosing cystic fibrosis with a newborn screening program. Journal of Pediatrics 2003;142(6):617‐23. [DOI] [PubMed] [Google Scholar]
Lewis 1995
- Lewis PA. The epidemiology of cystic fibrosis. In: Hodson ME, Geddes DM editor(s). Cystic Fibrosis. London: Chapman & Hall Medical, 1995:1‐5. [Google Scholar]
Li 2005
- Li Z, Kosorok MR, Farrell PM, Laxova A, West SHE, Green CG, et al. Longitudinal development of mucoid Pseudomonas aeruginosa infection and lung disease progression in children with cystic fibrosis. JAMA 2005;293(5):581‐8. [DOI] [PubMed] [Google Scholar]
Mischler 1998
- Mischler EH, Wilfond BS, Fost N, Laxova A, Reiser C, Sauer CM, et al. Cystic fibrosis newborn screening: impact on reproductive behavior and implications for genetic counselling. Pediatrics 1998;102(1 Pt 1):44‐52. [DOI] [PubMed] [Google Scholar]
Pollitt 1998
- Pollitt RG. Screening for cystic fibrosis. Seminars in Neonatology 1998;3:9‐15. [Google Scholar]
Schulz 1995
- Schulz K, Chalmers I, Hayes RJ, Altman DG. Empirical evidence of bias: Dimensions of methodological quality associated with estimates of treatment effects in controlled trials. Journal of the American Medical Association 1995;273(5):408‐12. [DOI] [PubMed] [Google Scholar]
Shoff 2006
- Shoff SM, Ahn HY, Davis L, Lai HC, the Wisconsin CF Neonatal Screening Group. Temperal associations among energy intake, plasma linoleic acid, and growth improvement in response to treatment initiation after diagnosis of cystic fibrosis. Pediatrics 2006;117(2):391‐400. [DOI] [PubMed] [Google Scholar]
Task Force 1983
- Ad hoc task force on neonatal screening, Cystic Fbrosis Foundation. Neonatal screening for cystic fibrosis [position paper]. Pediatrics 1983;72(5):741‐5. [PubMed] [Google Scholar]
Tluczek 1992
- Tluczek A, Mischler EH, Farrell PM, Fost N, Peterson NM, Carey P, et al. Parents' knowledge of neonatal screening and response to false positive cystic fibrosis testing. Journal of Developmental and Behavioral Pediatrics 1992;13(3):181‐6. [PubMed] [Google Scholar]
Wald 1998
- Wald NJ, Morris JK. Neonatal screening for cystic fibrosis. No evidence yet of any benefit. BMJ 1998;316(7129):404‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Waters 1999
- Waters DL, Wilcken B, Irwig L, Esperen P, Mellis C, Simpson JM, et al. Clinical outcomes of newborn screening for cystic fibrosis. Archives of Disease in Childhood 1999;80(1):F1‐F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Weller 2007
- Weller P. Personal communication 2007.
Wilcken 1993
- Wilcken B. Newborn screening for cystic fibrosis: Its evolution and a review of the current situation. Screening 1993;2:43‐62. [Google Scholar]