

Abstract
The translation preinitiation complex (PIC) scans the mRNA for an AUG codon in a favorable context. Previous findings suggest that the factor eIF1 discriminates against non-AUG start codons by impeding full accommodation of Met-tRNAi in the P site of the 40S ribosomal subunit, necessitating eIF1 dissociation for start codon selection. Consistent with this, yeast eIF1 substitutions that weaken its binding to the PIC increase initiation at UUG codons on a mutant his4 mRNA and particular synthetic mRNA reporters; and also at the AUG start codon of the mRNA for eIF1 itself owing to its poor Kozak context. It was not known however whether such eIF1 mutants increase initiation at suboptimal start codons genome-wide. By ribosome profiling, we show that the eIF1-L96P variant confers increased translation of numerous upstream open reading frames (uORFs) initiating with either near-cognate codons (NCCs) or AUGs in poor context. The increased uORF translation is frequently associated with the reduced translation of the downstream main coding sequences (CDS). Initiation is also elevated at certain NCCs initiating amino-terminal extensions, including those that direct mitochondrial localization of the GRS1 and ALA1 products, and at a small set of main CDS AUG codons with especially poor context, including that of eIF1 itself. Thus, eIF1 acts throughout the yeast translatome to discriminate against NCC start codons and AUGs in poor context; and impairing this function enhances the repressive effects of uORFs on CDS translation and alters the ratios of protein isoforms translated from near-cognate versus AUG start codons.
Keywords: translation, initiation, accuracy, eIF1, yeast
INTRODUCTION
Accurate identification of the translation initiation codon in mRNA by ribosomes is crucial for the correct expression of cellular proteins, and it generally occurs in eukaryotic cells by a scanning mechanism. The 43S preinitiation complex (PIC), comprised of the small (40S) ribosomal subunit, methionyl initiator tRNA (Met-tRNAiMet) in a ternary complex (TC) with GTP-bound eIF2, and several other initiation factors, attaches to the 5′ end of the mRNA and scans the 5′UTR for an AUG start codon. In the scanning PIC, the TC is bound in a relatively unstable state, dubbed “POUT,” suitable for inspecting successive triplets in the P decoding site for complementarity with the anticodon of Met-tRNAi. Nucleotides surrounding the AUG, particularly at the −3 and +4 positions (relative to the AUG at +1 to +3)—the “Kozak context”—also influence start codon recognition. Hydrolysis of the GTP bound to eIF2 is stimulated by GTPase activating protein eIF5, but Pi release is blocked by eIF1, whose presence also prevents more stable binding of Met-tRNAiMet in the “PIN” state. Start-codon recognition triggers dissociation of eIF1 from the 40S subunit, which in concert with other events allows Pi release from eIF2-GDP·Pi and accommodation of Met-tRNAiMet in the PIN state of the 48S PIC (Supplemental Fig. S1). Subsequent dissociation of eIF2-GDP and other eIFs from the 48S PIC enables eIF5B-catalyzed subunit joining and formation of an 80S initiation complex with Met-tRNAiMet base-paired to AUG in the P site, ready for the elongation phase of protein synthesis (for reviews, see Hinnebusch 2014 and Hinnebusch 2017).
Although eIF1 enhances rapid TC loading in the POUT conformation, it physically impedes rearrangement to PIN during scanning at non-AUG codons by clashing with Met-tRNAi (Rabl et al. 2011; Lomakin and Steitz 2013; Hussain et al. 2014), thus requiring its displacement from the P site and subsequent dissociation from the 40S subunit to allow start codon selection (Supplemental Fig. S1; Maag et al. 2005; Cheung et al. 2007). Consequently, mutations that weaken eIF1 binding to the 40S subunit were found to increase initiation at the near-cognate UUG triplet present at the third codon of HIS4 mRNA and thereby suppress the reduced translation conferred by substitution of the AUG start codon by the his4-301 mutation, conferring the Sui− (suppressor of initiation codon) phenotype. They also elevate initiation at UUG codons in synthetic reporter mRNAs, presumably by increasing unscheduled dissociation of eIF1 from the PIC in the absence of a perfect codon:anticodon helix in the P site (Martin-Marcos et al. 2011, 2013). Moreover, decreasing wild-type (WT) eIF1 abundance increases initiation at near-cognates or AUGs in poor context, whereas overexpressing eIF1 suppresses these events (Valasek et al. 2004; Alone et al. 2008; Ivanov et al. 2010; Saini et al. 2010; Martin-Marcos et al. 2011). The mechanistic link between eIF1 abundance and initiation accuracy is exploited to negatively autoregulate eIF1 expression, as the AUG codon of the eIF1 mRNA itself (SUI1 in yeast) is in suboptimal context, and the frequency of its recognition is inversely related to eIF1 abundance (Ivanov et al. 2010; Martin-Marcos et al. 2011). The Sui− mutations that weaken 40S binding by eIF1 also relax discrimination against the poor context of the SUI1 AUG codon and elevate eIF1 expression, overcoming the autoregulation (Martin-Marcos et al. 2011). Studies using a reconstituted mammalian initiation system indicate that eIF1 also discriminates against mRNAs containing very short 5′UTRs (Pestova and Kolupaeva 2002), also known to disfavor initiation in yeast and mammalian cells (Hinnebusch 2011), possibly in response to a lack of stabilizing mRNA nucleotides occupying the mRNA exit channel of the 40S subunit.
The eIF1 mutation sui1-L96P was shown previously to suppress the reduced translation conferred by the AUG start codon mutation in his4-301 mRNA, conferring the Sui− phenotype, increasing by ∼18-fold the UUG:AUG ratio of expression from two matched HIS4-lacZ reporters differing only by an UUG or AUG start codon. In addition to relaxing discrimination against UUG start codons at HIS4, L96P conferred an ∼12-fold increase in expression of eIF1 itself, but a smaller, approximately fourfold increase in SUI1 mRNA, suggesting an approximately threefold increase in translational efficiency (TE). The latter was attributed primarily to relaxed discrimination against the suboptimal Kozak context of the SUI1 AUG codon because L96P had significantly less effect on eIF1 expression from a SUI1 mRNA altered to contain optimal Kozak context (Martin-Marcos et al. 2011). Thus, the eIF1-L96P substitution appears to diminish substantially eIF1 function in restricting initiation at both near-cognate (UUG) start codons and AUG codons in poor context. Biochemical analysis indicates that this defect results from impaired interactions of eIF1 with the scanning PIC owing to its weakened binding to the amino-terminal domain of eIF3c, the carboxy-terminal domain of eIF5, and the 40S subunit (Obayashi et al. 2017).
Although the role of yeast eIF1 in discriminating against near-cognate start codons or AUGs in poor context was well established for a small set of mRNAs, including HIS4, SUI1, and various synthetic reporter mRNAs, its global importance in restricting usage of near-cognate initiation codons and AUG codons in the poor context in living yeast cells was unknown. Accordingly, here we have used ribosome footprint profiling of the highly impaired Sui− variant eIF1-L96P to reveal the genome-wide function of eIF1 in promoting accurate start codon selection and proper expression of the yeast translatome.
RESULTS
eIF1-L96P decreases discrimination against near-cognate start codons genome-wide
To determine whether impairing eIF1 function reduces discrimination against suboptimal initiation sites throughout the yeast translatome, we conducted ribosomal footprint profiling of the strong Sui− variant sui1-L96P in parallel with an isogenic WT strain. In addition to decreasing the accuracy of start site selection, L96P reduces the rate of general translation initiation, decreasing the cell growth rate (Martin-Marcos et al. 2011) and reducing the abundance and average size of bulk polysomes, to a greater extent than observed for mutations substituting conserved Lys residues in direct contact with the 40S subunit, including sui1-K56E and sui1-K60E (Supplemental Fig. S2A–E; Martin-Marcos et al. 2013). The L96P mutation also confers an ≈3.5-fold larger increase in the HIS4-lacZ UUG:AUG initiation ratio relative to WT cells, versus compared to K56E and K60E (Martin-Marcos et al. 2011).
Ribosome profiling entails deep-sequencing of mRNA fragments protected from RNase digestion by translating 80S ribosomes that were arrested on mRNA by treating cell lysates with cycloheximide (Ribo-seq), conducted in parallel with total mRNA sequencing (RNA-seq) (Ingolia et al. 2012). The TE of each mRNA is calculated for each strain as the ratio of total sequencing reads for ribosome-protected fragments (RPFs) to total reads for mRNA fragments mapping to the coding sequences (CDS) of each gene; and is normalized to the average TE among all mRNAs (which is assigned a value of one). Thus, TE values are expressed relative to the average TE in that experiment. Changes in TE in the L96P mutant versus WT (ΔTEL96P) are expressed as the ratio TEL96P/TEWT, and significant changes were identified by DESeq analysis (Anders and Huber 2010) of data from biological replicates (see Materials and Methods for details). Both ribosome footprinting and RNA-seq results were highly reproducible between the two biological replicates analyzed for the L96P and WT strains (Pearson's R > 0.99) (Supplemental Fig. S3A–D).
In accordance with the increased steady-state level of eIF1 protein conferred by L96P (Martin-Marcos et al. 2011), RPF reads across the SUI1 CDS were increased by approximately sixfold in L96P versus WT cells (Fig. 1A, ΔRPF = 6.5). Consistent with previous results, L96P also increases the level of SUI1 mRNA (Fig. 1A, ΔmRNA = 5.5); although the nearly comparable increases in RPFs and mRNA reads for SUI1 do not indicate the approximately twofold increase in TE deduced previously from parallel measurements of steady-state eIF1 protein and SUI1 mRNA abundance (Martin-Marcos et al. 2011). Differences in normalization between the two approaches might account for this discrepancy. The increase in SUI1 mRNA levels in sui1-L96P cells is in line with previous observations indicating that translation initiation efficiency is an important determinant of mRNA stability in budding yeast (LaGrandeur and Parker 1999; Schwartz and Parker 1999; Chan et al. 2018).
FIGURE 1.
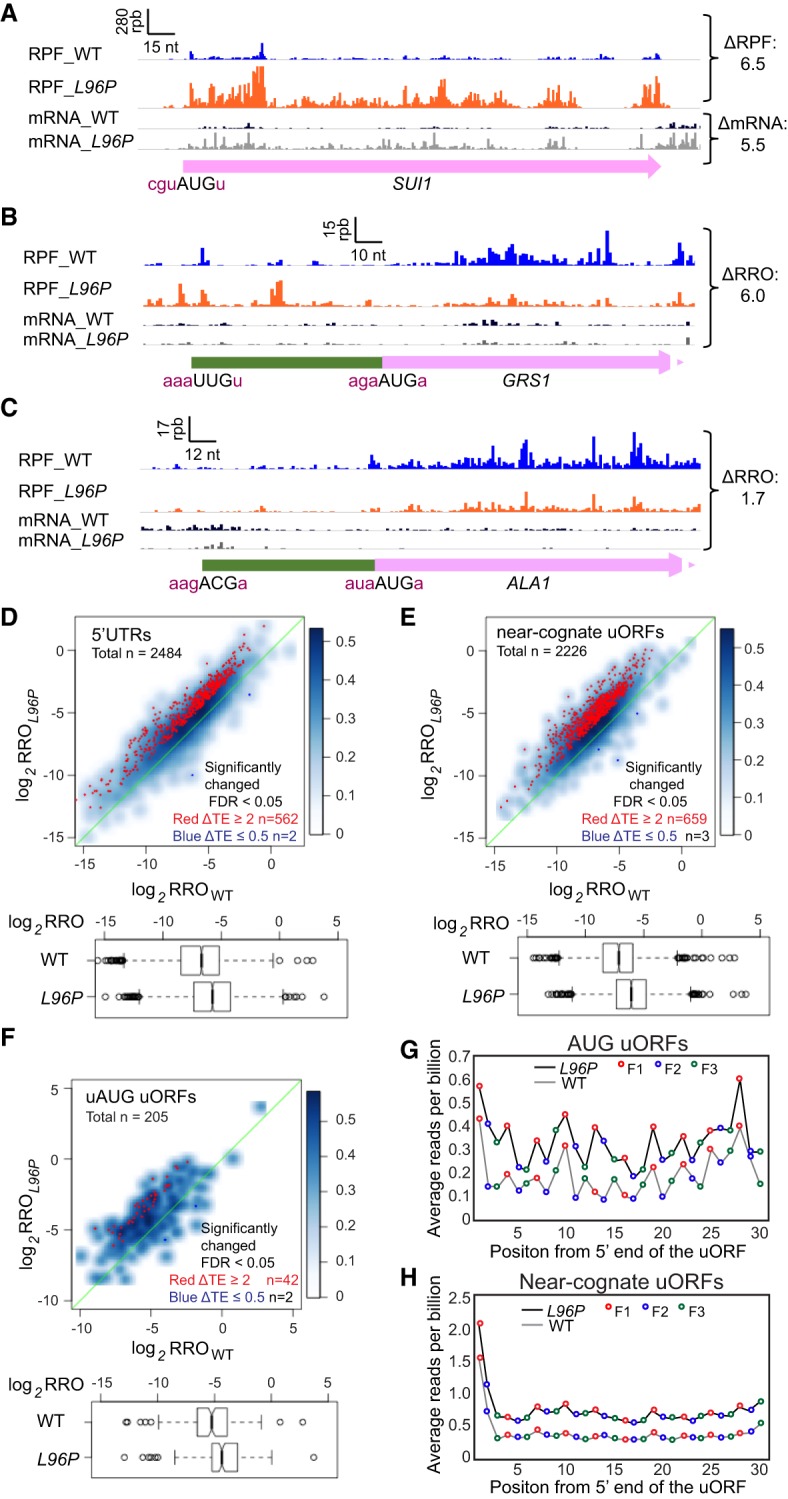
sui1-L96P increases the utilization of near-cognate and upstream AUG start codons. (A–C) Ribosome protected fragments (RPFs) and mRNA reads on the SUI1, GRS1, and ALA1 genes in WT and L96P cells in units of rpb (reads per billion mapped reads), showing schematically the position of the main CDS (pink), the CDS for amino-terminal extensions (green), and the −3 to −1 and +4 context nucleotides surrounding the AUG or near-cognate codon (NCC) (in brick red). ΔRPF and ΔmRNA give the ratios of RPFs and total mRNA fragments, respectively, in L96P versus WT cells. The Integrated Genomics Viewer (Broad Institute) was used to display ribosome/mRNA reads, with the rpb and nt scales indicated in each panel. For these and all other gene browser views, the reads per billion mapped reads (rpb) are plotted; the ribosome-protected-fragments are plotted to their predicted A-site positions, and the mRNA fragments are plotted to the 15th nt from their 5′ ends. (D–F) Scatter plots comparing log2 RRO (relative ribosome occupancy) values in L96P versus WT cells for 2484 5′UTRs (D), 2226 NCC-uORFs (E), and 205 uAUG uORFs (F) from genes with ≥32 RPF reads in the main CDS and ≥2 RPF reads in the 5′UTR or uORF in the averaged reads from four samples, two replicates of WT and two replicates of sui1-L96P. The sets of NCC_uORF and AUG_uORFs showing evidence of translation in current or previous ribosome profiling experiments were described previously (Martin-Marcos et al. 2017). 5′UTRs or uORFs showing significantly changed TE values in L96P versus WT cells of greater than or equal to twofold (at FDR < 0.05) are indicated in red or dark blue, and the numbers (n) of 5′UTRs or uORFs plotted are indicated. Notched box-plots below each scatterplot compare the distributions of log2 RRO values in WT versus L96P cells for the complete sets of 5′UTRs (D), NCC_uORFs (E), or AUG_uORFs (F). In these and other box-plots below, box limits indicate the 25th and 75th percentiles, whiskers extend 1.5 times the interquartile range from the 25th and 75th percentiles, outliers are represented by dots, and nonoverlapping notches indicate that two medians differ with ∼95% confidence. A fraction of outliers was omitted from the plots to expand the y-axis scale. (G–H) Three nucleotide periodicity of RPFs in subsets of the 205 translated AUG_uORFs (G, n = 123) and 2226 NCC_uORFs (H, n = 1394) described in panels F and E, respectively, in which the uORF length is >29 nt and the distance from the uORF start codon to the main CDS AUG is >29 nt. Black and gray lines show the average RPF reads per billion at each position relative to the 5′ end of these groups of uORFs. Reads at first, second, and third reading frames are labeled with red, blue, and green circles, respectively. (An AUG_uORF at YDR133C, YDR133C_52, was excluded because its high expression would dominate the plot.)
GRS1 and ALA1 are two well-studied genes whose main CDS begin with non-AUG start codons, which initiate amino-terminal extensions directing mitochondrial import of the encoded glycyl-tRNA synthetase and alanyl-tRNA synthetase, respectively; whereas the cytoplasmic isoforms initiate from downstream in-frame AUG codons (Chen et al. 2008; Chang et al. 2010). L96P dramatically increased the ratio of RPFs in the extension versus the main CDS (dubbed RRO for relative ribosome occupancy of the upstream CDS) by approximately sixfold (Fig. 1B, ΔRRO = 6.0); and it also conferred a modest increase in RRO of ∼1.7-fold at ALA1 (Fig. 1C, ΔRRO = 1.7). Based on other findings below, the different magnitudes of ΔRRO for the two genes might reflect the fact that the context of the GRS1 UUG matches closely the optimum Kozak consensus sequence in yeast in containing A's at −4 to −1, and U at +4, whereas the ALA1 ACG deviates from this consensus by lacking A's at −1 and −4 and containing A at +4 (Zur and Tuller 2013). Both genes also exhibit increased RPFs mapping upstream of the previously identified near-cognate (NCC) start codons (Chen et al. 2008; Chang et al. 2010) in L96P versus WT cells (Fig. 1B,C), which might represent elevated initiation from additional upstream NCC codons—shown below to be widespread in L96P cells.
To determine whether L96P confers a widespread relaxed discrimination against non-AUG start codons, we examined its effect on the translation of upstream open reading frames (uORFs) in 5′UTRs that initiate with any of the nine NCCs: GUG, UUG, CUG, ACG, AAG, AGG, AUC, AUU, AUA. Using bioinformatics, we previously identified 2430 such “NCC_uORFs” found in 1139 mRNAs that showed evidence of translation beginning at a discrete NCC start site in one or more ribosome profiling data sets from WT or various mutant strains obtained in our laboratory or published by others (Martin-Marcos et al. 2017) (see Materials and Methods), and which displayed sufficient ribosome occupancies in both the WT and L96P strains for quantitative analysis of changes in translation. We similarly identified a group of 290 translated AUG-initiated uORFs (AUG_uORFs) present in 262 mRNAs, of which 205 satisfied the minimal read thresholds described in Figure 1F; as well as the 5′UTR sequences of 2484 mRNAs showing evidence of translation at any location within their 5′UTRs in both the WT and L96P strains.
All three groups of upstream sequences, NCC_uORFs, AUG_uORFs, and 5′UTRs, show evidence of increased translation in the L96P mutant versus WT cells. Thus, plotting RROL96P versus RROWT values for each 5′UTR or uORF revealed that most of the data points fall above the diagonal, indicating generally elevated translation within 5′UTRs, and in the defined AUG or NCC uORFs described above, relative to the main CDSs in the L96P mutant (Fig. 1D–F, scatterplots in upper panels). Applying a false-discovery-rate (FDR) of <0.05, between ∼20%–30% of the NCC_uORFs (29%), AUG_uORFs (20%) and 5′UTRs (23%) displayed a significant increase in TE of greater than or equal to twofold in the L96P mutant (ΔTEL96P ≥ 2.0) (Fig. 1D–F, red dots in scatterplots); whereas only 2–3 members of each sequence group showed a significant reduction in TE in the mutant cells (ΔTEL96P ≤ 0.5) (Fig. 1D–F, blue dots in scatterplots). Consistent with this, box-plot analyses indicate significantly increased median RRO values in L96P versus WT cells for the groups of 5′UTRs, NCC_uORFs, and AUG_uORFs, with median RRO increases in the mutant (ΔRROL96P) of greater than or equal to twofold (Fig. 1D–F, box-plots in lower panels). (In these and other box-plots presented below, nonoverlapping notches indicate that the two medians differ with >95% confidence.) The increased ribosome occupancy observed across the group of AUG and NCC_uORFs in L96P versus WT cells is further illustrated by plotting the average occupancies at each nucleotide from the start codon of a subset of 123 of the 290 AUG_uORFs, and 1394 of the 2430 NCC_uORFs described above, with length >29 nt and distance from the uORF start codon to the main CDS AUG codon >29 nt, where the 3-nt periodicity of RPF occupancies is evident, at least at the higher read densities afforded by L96P (Fig. 1G,H).
The increased translation of both AUG and NCC uORFs in L96P cells was also observed by interrogating distinct sets of translated uORFs that were recently identified by application of a machine-learning algorithm to ribosome profiling data, which identified 438 AUG_uORFs and 544 NCC_uORFs that are conserved in sequence or position among different Saccharomyces yeast species (Spealman et al. 2018). Between 24%–27% of these uORFs belong to the groups of 290 AUG_uORFs and 2430 NCC_uORFs analyzed above (Supplemental Fig. S4A,B). Similar to our findings on the aforementioned uORF groups (Fig. 1D–F), the evolutionarily conserved uORFs exhibit approximately twofold increases in median RRO in L96P versus WT cells (Supplemental Fig. S4C,D, box-plots). Moreover, ∼20% of the NCC-uORFs exhibit a ΔTEL96P ≥ 2.0 at FDR < 0.05, whereas a relatively smaller proportion of conserved AUG_uORFs (∼11%) display a greater than or equal to twofold increase in TE in mutant versus WT cells (Supplemental Fig. S4C,D, red dots in scatterplots). Thus, we conclude that L96P evokes increased translation of a substantial fraction of both NCC_ and AUG_uORFs whose translation has been detected in distinct ribosome profiling data sets and by using different criteria to identify translated uORFs.
Among the 2226 NCC_uORFs and 205 AUG_uORFs that we identified bioinformatically and interrogated in Figure 1D–H, a small fraction are in the same reading frame as the main CDS and do not contain a stop codon prior to the CDS AUG; hence, they encode amino-terminal extensions (NTEs), as described above for GRS1 and ALA1 mRNAs (Fig. 1B,C). These NTEs include 188 of the 2226 NCC_uORFs and 6 of the 205 AUG_uORFs. A fraction of these 194 NTEs resemble GRS1 and ALA1 in showing elevated translation in L96P versus WT cells (Supplemental Fig. S5A–E upper); and similar to the complete sets of AUG_uORFs and NCC_uORFs, the group of 194 NTEs exhibits a significantly increased median RRO value in L96P versus WT cells (Supplemental Fig. S5E, lower). Interestingly, the NTEs encoded by YPK2 and TRZ1, which appear to exhibit increased translation in L96P cells (Supplemental Fig. S5C,D), were recently shown to contain a mitochondrial targeting sequence similar to the NTEs of GRS1 and ALA1 (Monteuuis et al. 2019).
eIF1-L96P selectively increases utilization of upstream AUG start codons with poor Kozak context
Considering that L96P increases the ratio of UUG:AUG initiation for a HIS4-lacZ reporter by more than an order of magnitude (Martin-Marcos et al. 2011), it might seem surprising that we observed marked increases in translation of both AUG_uORFs and NCC_ORFs in L96P cells (Fig. 1E,F; Supplemental Fig. S4C,D). However, we and others have noted previously that, as a group, the start codons of translated AUG_uORFs in yeast exhibit a poor match to the Kozak consensus context exhibited by the AUG codons of the most highly translated CDS in yeast (Arribere and Gilbert 2013; Martin-Marcos et al. 2017; Spealman et al. 2018). Given our previous demonstration that L96P reduces discrimination against the poor Kozak context at the eIF1 (SUI1) start codon (Fig. 1A; Martin-Marcos et al. 2011), we considered whether the increased translation of the group of AUG_uORFs conferred by L96P results primarily from overcoming poor context at these uORFs start codons.
A well-studied AUG_uORF with an established regulatory function resides upstream of the main CDS of the CPA1 gene, where it inhibits translation of CPA1 mRNA in response to excess arginine (Werner et al. 1987). As shown for the corresponding uORF of the N. crassa arg-2 gene, the inhibition results from arginine-induced stalling during translation of the uORF peptide (Wei et al. 2012), which impedes the progression of scanning PICs to the downstream CDS (Gaba et al. 2001). At low arginine levels most of the scanning PICs bypass the CPA1 uORF, owing to its poor Kozak context, and translate the main CDS downstream instead (Werner et al. 1987). Importantly, we observed that the L96P mutation confers a marked increase in translation of the CPA1 uORF relative to the downstream CDS (Fig. 2A, ΔRRO = 4.5), consistent with relaxed discrimination against the poor context of an AUG_uORF in the eIF1 mutant.
FIGURE 2.

sui1-L96P broadly decreases discrimination against poor Kozak context of upstream AUG codons. (A) RPFs and mRNA reads on the CPA1 gene in WT and L96P cells are depicted as described in Figure 1A, with the uORF and main CDS shown in green and pink, respectively. (B) Notched box-plots showing the distributions and medians of ratios of log2 RRO values in L96P versus WT cells (ΔRROL96P) for the subset (n = 194) of 205 translated AUG_uORFs described in Figure 1F with at least 3 nt between the cap and the uORF (to allow calculation of context scores), and for the sets of 49 uORFs comprising the top (Better context) or bottom (Poor context) quartiles of these 194 AUG_uORFs ranked by AUG context score from highest to lowest. A fraction of outliers was omitted from the plots to expand the y-axis scale. (C) Notched box-plots showing the distributions and medians of AUG context scores for the same group of 194 uORFs described in B and the two sets of 49 uORFs comprising the top (Largest ΔRRO) or bottom (Smallest ΔRRO) quartiles of the 194 AUG_uORFs ranked by ΔRROL96P values high from largest to smallest. A fraction of outliers was omitted from the plots to expand the y-axis scale. (D) The sequence logo for positions −3 to −1 and +4 surrounding the AUG codons for the same groups of uORFs shown in C. The DNA rather than RNA sequence is depicted.
To examine whether L96P generally confers increased uORF translation of AUG_uORFs in poor context, we calculated the AUG context adaptation index AUGCAI (Miyasaka 1999) for 194 of the 205 AUG_uORFs described in Figure 1F that contain at least 3 nt between the cap and the uORF (needed to allow calculation of context scores). The AUGCAI quantifies the similarity between the context of a given AUG to that of the start codons of the 2% of all yeast genes with the highest ribosomal loads (Zur and Tuller 2013). AUGCAI scores among all yeast genes range from ∼0.16 (poorest) to ∼0.97 (best), with the most highly expressed genes exhibiting scores near the top of this range. After dividing a subset of 194 of the 205 AUG_uORFs defined in Figure 1F (for which AUGCAI scores could be calculated) into quartiles on the basis of descending AUGCAI scores, we determined that the median increase in uORF translation in L96P versus WT cells, measured as ΔRROL96P, is significantly greater for uORFs in the last quartile with lowest AUGCAI scores (Fig. 2B, col. 3, ΔRROL96P ≈ 3.0-fold) compared to the first quartile with highest AUGCAI scores (Fig. 2B, col. 2, ΔRROL96P ≈ 1.7-fold). Note however that the first quartile of uORFs with highest context scores has a median AUGCAI score of only 0.22, which is still at the low end of the corresponding range of AUGCAI scores for all main CDS, of between 0.09 and 1.0. Thus, the moderate increase in ΔRROL96P observed for this group of AUG_uORFs with the strongest contexts among AUG_uORFs (Fig. 2B, col. 2) is still consistent with our conclusion that L96P selectively increases translation of AUG_uORFs with poor-context AUG codons.
As an alternative approach to determining whether L96P confers a significantly larger increase in translation for the group of AUG_uORFs with the poorest Kozak context, we divided the uORFs into quartiles on the basis of descending ΔRROL96P values, and found that the first quartile with the largest ΔRROL96P values (indicating the greatest increases in uORF/CDS translation) have significantly lower median AUGCAI context scores for the uORF start codons compared to the last quartile (with the smallest ΔRROL96P values) (Fig. 2C). Consistent with the foregoing conclusions, a sequence logo for the initiation sites of a group of AUG_uORFs with largest ΔRROL96P values exhibits nonpreferred pyrimidines encoding the −3 position and little or no sequence preferences at the −2, −1, and +4 positions, which is also true for the entire group of AUG_uORFs; whereas the uORFs with the smallest ΔRROL96P values show essentially no sequence preferences at any of the 4 positions (Fig. 2D). The most highly translated genes in yeast show a strong preference for adenine at −3 to −1 and guanine or uridine at +4 (Zur and Tuller 2013). As thymidine and cytidine are the least prevalent nucleotides encoding the key −3 position of highly translated mRNAs, the elevated occurrence of these 2 nt at −3 for the AUG_uORFs with the largest increases in RROL96P supports the view that L96P preferentially relaxes discrimination against AUGs in poor Kozak context. As explained in Supplemental Figure S6B, this last conclusion also emerges from examining the AUG_uORFs found to be evolutionarily conserved in sequence or position (Spealman et al. 2018) that were analyzed in Supplemental Figure S4B.
Interestingly, L96P appears to have the opposite effect on the influence of Kozak context on the evolutionarily conserved NCC-initiated versus the AUG-initiated uORFs (described above in Supplemental Fig. S4A). Thus, the group of NCC_uORFs showing the largest increases in RROL96P values (Supplemental Fig. S6A, col. 2) has a significantly higher median context score compared to the NCC_uORFs with the smallest increases in RROL96P values (Supplemental Fig. S6A, col. 3) (P = 0.003 in Mann–Whitney U-test). It is noteworthy that the most highly up-regulated NCC_uORFs have a significantly higher median context score, of 0.34, compared to that of the conserved AUG_uORFs, of 0.23. Moreover, the sequence logo for these NCC_uORFs shows a small bias for the preferred A residues at the −3 and −1, positions (Supplemental Fig. S6C, row 1), not evident for the conserved AUG_ uORFs (Supplemental Fig. S6D, row 1). The tendency toward more favorable contexts for the up-regulated NCC_uORFs that is not observed for the translationally up-regulated AUG_uORFs is consistent with previous findings indicating that favorable context is more important for recognition of near-cognate versus AUG start codons (Chen et al. 2008; Chang et al. 2010). To explain why increased translation in L96P cells is relatively more dependent on good context for NCC versus AUG uORFs it could be proposed that the combination of a non-AUG start codon and extremely poor context is so detrimental for recognition by the scanning PIC that impairing the gatekeeper function of eIF1 by L96P has little impact on selection of such highly inefficient NCC start sites.
eIF1-L96P selectively increases utilization of main CDS AUG start codons with poor Kozak context
Having found that L96P generally relaxes discrimination against the AUG codons of uORFs, which tend to have relatively poor Kozak context, we next examined whether L96P reduces discrimination against the AUG codons of main CDSs in poor context throughout the translatome in the manner observed previously for translation of SUI1 mRNA encoding eIF1 itself (Fig. 1A; Martin-Marcos et al. 2011). The results in Figure 3B,C provide examples of two mRNAs, TEN1 and SWT1, that resemble SUI1 mRNA in possessing poor Kozak context and exhibiting increased TEs in L96P versus WT cells, whereas FIT3 mRNA contains optimum context and shows decreased TE in L96P cells (Fig. 3A). To determine the translatome-wide relationship between Kozak context and the impact of L96P on the translation of downstream CDSs, we first compared changes in TE for the main CDS in mutant versus WT cells (ΔTEL96P) for groups of genes with poor versus optimal contexts. Interestingly, L96P conferred a small, but significant increase in TE for the group of 99 among all mRNAs that contain nonpreferred bases at every position surrounding the AUG codon, ie. (C/U/G)−3(C/G/U)−2(G/C/U)−1(aug)(C/A)+4, (Shabalina et al. 2004; Zur and Tuller 2013) when compared to all 3650 mRNAs with adequate read-depth for analysis and containing annotated 5′UTR lengths of ≥5 nt (Fig. 3D, “Poor” vs. “All”). Consistent with this, a small increase in TE was also observed for a larger group of 598 mRNAs containing the least preferred bases C/U at the critical −3 position regardless of the sequence at the other context positions (Fig. 3D, “−3C/U” vs. “All”). In contrast, no significant change in TE was observed for 175 mRNAs with the preferred bases at all positions, that is, A−3A−2A−1(AUG)(G/U)+4, designated “Perfect” context, or for 3052 genes with A/G at −3 (the majority of mRNAs) (Fig. 3D, “Perfect” & “−3A/G” vs. “All”).
FIGURE 3.

sui1-L96P broadly decreases discrimination against poor Kozak context of main CDS AUG codons genome-wide. (A–C) RPFs and mRNA reads for the FIT3 (A), TEN1 (B) and SWT1 (C) genes in WT and L96P cells, as described in Figure 1A. (D) Notched box-plot of the distributions and medians of log2ΔTEL96P values for the CDSs of groups of genes with perfect or poor AUG context (as defined below the plot), preferred (A/G), or nonpreferred (C/U) bases at −3, and all 3650 genes with ≥10 RPF reads and ≥32 mRNA reads (average of four samples, two replicates of WT and two replicates of sui1-L96P) in the main CDS, and 5′UTR length >5 nt. (E) Logos of AUG context sequences for the 3650 genes in B, and the 100 genes showing the largest increase (TE_up) or decrease (TE_down) in CDS TE values in L96P versusWT. (F) Notched box-plot analysis of AUG context scores calculated for positions −6 to −1 and +4 to +6 for the “TE_up” and “TE_down” groups of genes defined in E. (G) Box-plot analysis of AUG context scores for the groups of genes exhibiting significantly increased (RPF_up) or decreased (RPF_down) RPFs in CDSs in L96P versus WT cells (FDR < 0.05), and for all 4035 genes. Before identifying the RPF_up and RPF_down groups, 258 mRNAs transcriptionally activated by Gcn4 were removed because sui1 alleles typically derepress GCN4 mRNA translation (Martin-Marcos et al. 2013), and we wished to exclude mRNAs with elevated RPFs owing to increased transcription. (H) Notched box-plot analysis of CDS ΔTEL96P values for the 3650 genes analyzed in D divided into deciles of equal size binned according to AUG context scores calculated as in F.
As an independent approach to examining whether L96P preferentially increases utilization of main CDS AUGs in poor context, we sorted genes on the magnitude of ΔTEL96P values and selected the subsets of mRNAs exhibiting either the largest increases or largest decreases in TE in mutant cells. Constructing sequence logos for positions −6 to −1 and +4 to +6 for these two groups revealed that the 100 mRNAs exhibiting the largest increases in TE in L96P versus WT cells (dubbed TE_up) do not exhibit the strong preference for A at −3 that characterizes all expressed mRNAs and that is also displayed by the 100 mRNAs showing the largest TE decreases (TE_down) (Fig. 3E, row 2 vs. rows 1 & 3). Consistent with this, calculating the AUGCAI context scores for these two sets of genes revealed that the TE_up mRNAs exhibit a median context score significantly below the median values of ∼0.47 for the TE_down mRNAs (P = 0.04 in Mann–Whitney U-test), which is close to the median score for all analyzed mRNAs (Fig. 3F). Finally, comparing ΔTEL96P values for 10 deciles of all 3650 mRNAs divided into bins of equal size according to AUG context scores revealed that only decile 1, with the lowest context scores, exhibits a significant increase in median TE in the mutant versus WT cells (Fig. 3H; first decile). Together, these results support the conclusion that L96P specifically increases the utilization of main CDSs AUGs with poor surrounding sequence contexts.
The inverse correlation between TE changes conferred by L96P and context scores just described was identified without taking into account whether the individual mRNAs under consideration exhibit statistically significant differences in TE between mutant and WT cells. Because too few such mRNAs were identified for rigorous analysis of their context scores, we examined two other groups of mRNAs exhibiting significant changes in ribosome occupancy across the CDS between mutant and WT cells (at FDR < 0.05). The 610 mRNAs showing significantly increased ribosome occupancy (RPF_up) exhibit significantly lower context scores compared to the group of 725 mRNAs with significantly decreased ribosome occupancies (RPF_down) in L96P versus WT cells (Fig. 3G, col. 2 vs. col. 3). These findings rigorously support our conclusion that L96P preferentially increases translation of main CDSs for those mRNAs containing lower than average context scores.
Together, the results in Figure 3 indicate that mRNAs whose main CDS AUG codons are in poor context tend to show increased TEs in the L96P mutant versus WT throughout the translatome. However, significant increases are limited largely to those mRNAs with the poorest matches to the optimum context in yeast (Fig. 3H). Because the majority of yeast mRNAs contain the preferred A at the −3 position relative to the AUG codon of the main CDS (Fig. 3E; Shabalina et al. 2004), the L96P mutation has relatively little effect on the TEs of most mRNAs. The decreased translation in L96P cells observed for the RPF_down subset of genes with highly favorable context (Fig. 3G, col. 3), might reflect increased competition for limiting initiation factors or 40S subunits owing to increased translation of mRNAs with less favorable contexts.
Increased 5′UTR translation conferred by L96P is associated with decreased CDS translation
We showed above that L96P increases the translation of a large number of both AUG_uORFs and NCC_uORFs (Fig. 1D–F). As discussed for CPA1 mRNA, elevated uORF translation appears to inhibit translation of the downstream main CDS (Fig. 2A), in agreement with the known translational repressive function of this uORF (Werner et al. 1987). Accordingly, we searched for other mRNAs in which altered uORF translation is associated with an inverse change in translation of the main CDS, and thus identified IDH2 (Fig. 4A), NOT3, and SEC1 (Supplemental Fig. S7A,B) as examples. Applying an FDR cut-off of <0.05, we identified 46 mRNAs exhibiting ΔTEL96P ≤ 0.5 for the main CDSs (Fig. 4B, all blue dots), of which two-thirds (30 mRNAs) also show evidence of elevated translation of the 5′UTR relative to the main CDS in L96P cells, as indicated by ΔRROL96P values of ≥2.0 (FDR < 0.05) (Fig. 4B, blue dots with orange circles). Thus, it is possible that the reductions in the main CDS TEs conferred by L96P for these 30 mRNAs are driven by increased 5′ UTR translation.
FIGURE 4.

Elevated upstream ribosome occupancies often associate with reduced translation in CDS. (A) RPFs and mRNA reads for IDH2 showing increased 5′UTR ribosome occupancy coupled with decreased CDS RPFs in L96P versus WT cells. (B) Scatter plot comparing log2TEL96P versus log2TEWT for CDS of 4242 genes with ≥10 RPF reads and ≥32 mRNA reads (average of four samples, two replicates of WT and two replicates of sui1-L96P). Genes with significant greater than or equal to twofold changes in CDS TE in L96P versus WT (FDR < 0.05) are highlighted by red and dark blue dots, respectively. Among them, genes with significantly elevated RRO ratios in the mutant vs WT (ΔRROL96P ≥ 2, FDR < 0.05) are denoted by orange circles. (C) Cumulative frequency distributions of CDS log2ΔTEL96P values for all 4242 genes (black line), or for genes with RRO values exceeding 1/32 in either WT (blue line) or L96P cells (red line). P-values are shown indicating the significance of differences between the distributions assigned using the Kolmogorov–Smirnov test. (D) Box-plot analysis of CDS ΔTEL96P values for the groups of mRNAs described in Figure 1D–F showing significantly increased TEs for 5′UTRs (5′UTR_up), NCC_uORFs (uNCC_up), or AUG_uORFs (uAUG_up). The numbers of mRNAs here are smaller than those in Figure 1E,F because of multiple uORFs residing in the same mRNAs. (E) Box-plot analysis of CDS ΔTEL96P values for all 4242 mRNAs analyzed in panels B–D, for 592 mRNAs lacking 5′UTR length annotations (NoA), and for eight bins of the 4242 mRNAs divided according to 5′UTR lengths, with the numbers of mRNAs in each bin indicated (n). (F,G) Same box-plot analysis as in E but excluding mRNAs with appreciable RRO values exceeding ∼0.03 in WT (E) or L96P cells (F).
We reasoned that if this last possibility was correct, then the inverse relationship between changes in 5′UTR versus CDS translation conferred by L96P should be the strongest for those mRNAs in which uORFs are translated at sufficiently high levels to impact the translation of the downstream CDS. To test this prediction, we identified mRNAs exhibiting comparable ribosome densities in 5′UTRs relative to main CDSs, with RRO values of >∼0.03 (one 5′UTR read per 32 CDS reads, a value similar to the mean ratio of uORF to CDS lengths of 0.037 for all translated uORFs), in either WT or L96P cells. Supporting our prediction, this group of mRNAs exhibits significantly greater CDS TE reductions conferred by L96P compared to all mRNAs (Fig. 4C, red & blue vs. black curves). Finally, we found that the groups of mRNAs highlighted with red dots in Figure 1D–F exhibiting significantly increased translation of either 5′UTRs (n = 562), NCC_uORFs (n = 659 in 311 mRNAs), or uAUG_uORFs (n = 42 in 36 mRNAs) all show significant CDS TE reductions in L96P versus WT cells, compared to all mRNAs (Fig. 4D), with the largest decreases in ΔTEL96P observed for the mRNAs with uAUG_uORFs (col. 4). Together, these analyses suggest that increased translation of uORFs that exhibit appreciable translation compared to the downstream CDS is frequently associated with reduced TEs for the CDSs in L96P cells.
Interestingly, the TE changes conferred by L96P appear to be associated with 5′UTR length, with greater reductions occurring with longer 5′UTR lengths (Fig. 4E), particularly for the group of ≈18% of all mRNAs containing 5′UTR lengths of >100 nt belonging to the last three bins of 5′UTR lengths analyzed in Figure 4E. Importantly, however, the greater TE reductions conferred by L96P for mRNAs with the longest 5′UTRs are largely eliminated if we exclude from the analysis all mRNAs with comparable ribosome densities in 5′UTRs versus CDS, that is, with RROs > ∼0.03 in either WT or L96P cells (Fig. 4F,G). We interpret these last findings to indicate that increasing 5′UTR length increases the probability of including upstream uORFs whose translation is elevated in L96P cells, which in turn decreases initiation at the downstream main CDS. This can explain why the relationship between ΔTEL96P and 5′UTR length is eliminated when considering only mRNAs lacking detectable 5′UTR translation. Thus, the tendency for mRNAs with long 5′UTRs to exhibit reduced relative TE in L96P versus WT cells provides additional evidence that TE reductions for main CDS conferred by L96P are driven largely by increased translation of inhibitory uORFs.
The increase in relative TE conferred by L96P for the two bins analyzed in Figure 4E encompassing the shortest 5′UTRs with lengths of <40 nt (cols. 3–4), remained in effect after excluding mRNAs with RROs > ∼0.03 (Fig. 4F,G, cols. 2–3), suggesting that it is unrelated to the absence of translated uORFs and might reflect shorter 5′UTRs per se. This possibility is consistent with observations in the reconstituted mammalian translation system indicating that eIF1 discriminates against AUGs located very close to the mRNA 5′ end (Pestova and Kolupaeva 2002), thus suggesting that disabling this function by L96P might confer a selective increase in TE for mRNAs with the shortest leaders.
In addition to the presence of uORFs, lengthy CDSs and the propensity for secondary structure in 5′UTRs are also associated with reduced initiation efficiency in WT cells (Kozak 1991; Kertesz et al. 2010; Hinnebusch 2011; Ding et al. 2012). However, we observed no relationship between ΔTEL96P and CDS length, except that the group of mRNAs with shortest CDS exhibit a slight tendency toward increased ΔTEL96P values (Supplemental Fig. S8A). But considering that mRNAs with shorter than average CDS also tend to have shorter 5′UTRs (Sen et al. 2016), the small increase in ΔTEL96P values for the first decile of CDS lengths analyzed in Supplemental Figure S8A might simply reflect the influence of short 5′UTR lengths already noted above (Fig. 4E). We also observed no relationship between ΔTEL96P values and 3′UTR lengths (Fig. 4; Supplemental Fig. S8B).
To examine the possible contribution of 5′UTR structure to the effects of L96P on CDS TEs, we interrogated a published database (Kertesz et al. 2010) wherein each transcribed nucleotide in 2679 different yeast transcripts was assigned a “parallel analysis of RNA structure” (PARS) score, based on its susceptibility in mRNA reannealed in vitro to digestion with nucleases specific for single-stranded or double-stranded RNA, with a higher PARS score denoting a higher probability of double-stranded conformation. In previous studies, we identified a significant tendency for mRNAs containing higher than average PARS scores in 5′UTR sequences to exhibit greater than average TE reductions in mutants defective for the RNA helicase Ded1 (Sen et al. 2016), which we could attribute by in vitro studies to unwinding of secondary structures in the 5′UTRs of such mRNAs by Ded1 (Gupta et al. 2018). In contrast, as described at length in Supplemental Figure S8C–I, we found no tendency for mRNAs with greater than average 5′UTR PARS scores to show larger than average TE reductions in response to the L96P substitution in eIF1. The fact that mRNAs with structured 5′UTRs display a heightened response to mutations in Ded1 but not to L96P is consistent with the large body of evidence indicating that eIF1 acts primarily in the assembly of 43S PICs (prior to mRNA recruitment) and in blocking recognition of poor initiation sites by the PIC (Hinnebusch 2011, 2017; Thakur and Hinnebusch 2018; Thakur et al. 2018), whereas Ded1 is crucial for overcoming the inhibitory effects of mRNA structures on PIC attachment to mRNA and subsequent scanning of 5′UTRs (Sen et al. 2015, 2019; Gao et al. 2016; Guenther et al. 2018; Gupta et al. 2018). In summary, our analyses suggest that very short 5′UTR lengths and the presence of well-translated uORFs are primary determinants of TE reductions for main CDSs conferred by the impairment of eIF1 by L96P.
Direct evidence that upstream AUGs or NCCs mediate changes in translation conferred by L96P
We sought next to demonstrate that changes in the translation of main CDSs deduced from ribosome profiling of the L96P mutant are conferred by increased recognition of NCC or poor-context AUG start codons by analyzing reporter mRNAs. Closer examination of the data in Figure 1E revealed that NCC_uORFs initiated by various near cognate triplets exhibit increased TEs in L96P cells (Fig. 5A), suggesting that L96P differs mechanistically from the eIF5 Sui− mutation SUI5, which is largely specific for UUG codons (Huang et al. 1997). The distribution of NCC start codons among the NCC_uORFs translationally up-regulated in L96P cells (Fig. 5A) is fairly similar to that of all translated NCC_uORFs (Fig. 5B), but shows a ≈1.3-fold enrichment for UUG and AAG codons, and 1.2- to 1.4-fold underrepresentation of AUU, AUA, AUG, and AGG codons. To determine whether the latter discrepancies might be dictated by the sequence contexts of the NCC start codons, we calculated the Kozak context scores for each of the nine NCCs for all 68,546 NCC uORFs encoded in the genome, for the 2199 translated NCC_uORFs (shown in Fig. 5B) and for the 649 NCC_uORFs whose translation was up-regulated in the L96P mutant (in Fig. 5A), for all such uORFs located ≥3 nt from the mRNA 5′ end, and compared the median context scores between the three groups of uORFs for each NCC. As shown in Supplemental Figure S9, the translated NCC uORFs have significantly higher context scores compared to all possible NCC uORFs in every case except AGG-initiated uORFs (cf. col. 2 vs. col. 1 for each NCC), indicating that, as expected, NCC uORF translation is stimulated by favorable context. However, the NCC uORFs whose translation is up-regulated by L96P do not have significantly different median context scores compared to the corresponding median scores for all translated uORFs (cf. col. 3 vs. col. 2 for each NCC). Thus, the enrichment or underrepresentation of particular NCC uORFs among the set of uORFs translationally up-regulated in L96P cells cannot be readily explained at the level of context scores.
FIGURE 5.
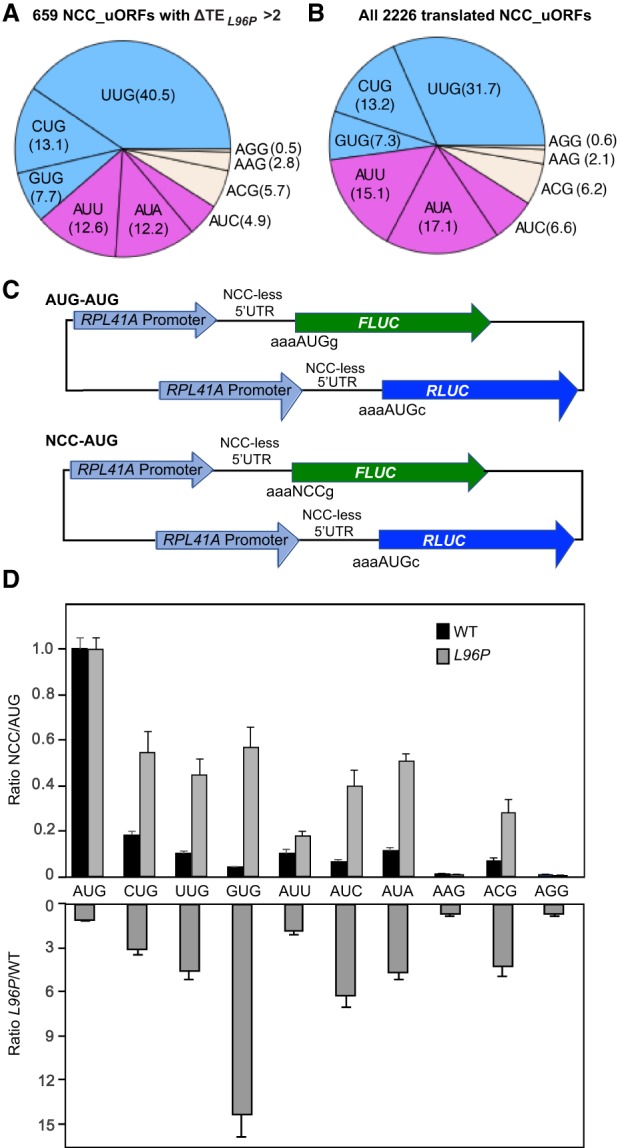
sui1-L96P increases the utilization of multiple near-cognate start codons. (A,B) Distributions of NCC start codons for the 659 NCC_uORFs exhibiting significantly increased uORF TEs in L96P vs. WT cells (A) and for all 2226 NCC_uORFs translated in our strains (B), described in Figure 1E. (C) Schematics of dual-luciferase reporter constructs with both FLUC and RLUC reporters containing the RPL41A promoter and RPL41A 5′UTR modified to lack all NCCs. The FLUC reporters contain either AUG (AUG–AUG constructs) or each of the nine different NCCs as start codons (NCC–AUG constructs). (D) Six transformants of WT or sui1-L96 strains harboring each of the 10 different NCC–AUG, or the control AUG–AUG, reporter constructs were cultured in SC-Ura at 30°C to A260 of ∼1.0, and firefly and renilla luciferase activities were assayed in the same WCEs. The mean ratio of FLUC/RLUC light units was calculated for each NCC–AUG construct and normalized by the mean FLUC/RLUC ratio produced by the control AUG–AUG construct in WT and L96P samples. The mean of normalized NCC/AUG reporter expression ratios (±SEM) are plotted in the upper panel, and the ratios of normalized NCC/AUG expression ratios between sui1-L96P and WT cells are plotted in the lower panel. The background signal in these experiments determined using cell extracts from a WT strain lacking a LUC reporter is ∼4.5 units; and expression of the AAG and AGG FLUC reporters in WT cells is much higher than this background level.
While it was shown previously that L96P increases initiation at a UUG start codon in HIS4 mRNA (Martin-Marcos et al. 2011), it was important to demonstrate that it would do so for other NCCs as well. To this end, we determined the effects of L96P on expression of a panel of firefly (FLUC) and renilla (RLUC) luciferase reporter genes, each driven by the same promoter (RPL41A) and containing a modified version of the RPL41A 5′UTR lacking all NCCs, but differing in the start codon (Fig. 5C). Expression of each FLUC reporter initiated by one of the nine different NCCs was normalized to that of the FLUC reporter with an AUG start codon to determine the NCC:AUG ratio, in both L96P and WT cells. As shown in Figure 5D, sui1-L96P increased the NCC:AUG initiation ratio for all of the NCCs except AAG and AGG, which are known to function poorly as start codons in WT cells (Kolitz et al. 2009) and are highly underrepresented among the NCC_uORFs translated in our strains (Fig. 5B). Apart from GUG and AUU, which displayed increases of NCC:AUG ratios of ∼15 and ∼1.5, respectively, the other five NCCs (CUG, UUG, AUC, AUA, and ACG) showed elevated NCC:AUG ratios of between approximately three- to approximately sixfold in L96P versus WT cells (Fig. 5D). (Note that the magnitude of the effects of L96P on reporter expression for the various NCCs (Fig. 5D) is not necessarily reflected in the proportions of NCC_uORFs showing elevated translation in L96P cells (Fig. 5A), as the latter depend on the frequencies of the different NCCs in all translated NCC_uORFs (Fig. 5B) and because any increase in NCC_uORF translation of twofold or more, regardless of the magnitude, was scored in constructing the chart in (Fig. 5A). Thus, our ribosome profiling and reporter data both indicate that sui1-L96P relaxes discrimination against a wide range of the NCCs, including single-base substitutions at all three nucleotide positions of the AUG start codon.
As shown in Figure 2A, increased translation of the uORF in CPA1 mRNA in L96P cells is associated with the decreased translation of the main CDS. To support this interpretation of the ribosome profiling data, we assayed matched CPA1-FLUC reporters with FLUC fused in-frame to the main CDS either containing or lacking the uORF ATG codon. Expression of the uORF-containing reporter is decreased in L96P mutant versus WT cells (Fig. 6A), in agreement with the ribosome profiling data in Figure 2A. Elimination of the uORF start codon leads to elevated expression in both strains, as expected from the removal of the inhibitory CPA1 uORF; moreover, the increase is relatively greater in the L96P strain with the result that expression of the mutated-uORF reporter is indistinguishable between the two strains (Fig. 6A). These findings support the interpretation that increased uORF translation in L96P cells resulting from relaxed discrimination against the poor-context uORF AUG codon decreases the fraction of scanning ribosomes able to access and translate the downstream CDS.
FIGURE 6.

Reporter gene analysis confirms that sui1-L96P decreases translation of the CPA1 and AHK1 main CDS by increasing uORF translation and increases utilization of NCC start codons for the GRS1 amino-terminal extension. (A) Plasmid-borne luciferase reporters with FLUC CDS fused to the 15th codon of the CPA1 CDS with either the WT uORF (top) or a mutated uORF with the AUG start codon altered to UAG (bottom) (depicted schematically on the left), were introduced into WT and sui1-L96P strains. Transformants were cultured in SC−Leu−Ura medium at 30°C, diluted to A600 ∼ 0.1, and cultured for ∼3 additional doublings. Firefly luciferase activities were assayed in WCEs and reported in relative light units (RLUs) per mg of total protein, as means (±SEM) determined from four independent transformants. (B) RPFs and mRNA reads on the AHK1 gene in WT and L96P cells, presented as in Figure 1A, showing the ΔRRO values for each of three nested uORFs all in the same reading frame (listed below the uORF schematics) and average ΔRRO for all 3 uORFs (shown on the right). (C,D) Plasmid-borne luciferase reporters with FLUC CDS fused to the 15th codon of the AHK1 CDS with either the WT 5′UTR (top) or mutated 5′UTR with an internal deletion of all three uORFs (bottom) (depicted on the left), were assayed for luciferase expression in WT and L96P transformants as in A. The AHK1-FLUC expression ratio for the WT versus truncated 5′UTR is plotted for WT and L96P cells. (E) GRS1-FLUC reporters with FLUC CDS (beginning at the second codon, i.e., lacking the AUG start codon) fused to the CDS for the amino-terminal extension (NTE) 6 nt downstream from the last in-frame NCC start codon (AUU), containing either the WT NTE CDS (first row) or mutated NTE CDSs (rows 2–4) with the indicated substitutions of the four in-frame NCC start codons (shown as gray vs. red ovals). Expression of firefly luciferase in WT or L96P transformants was determined as in A. Expression of the GRS1-FLUC_UUC(1–4m) construct, lacking all four NCC start codons, is approximately sixfold above the background signal of ∼4.5 units.
The AHK1 mRNA resembles CPA1 in showing elevated 5′ UTR translation associated with reduced translation of the main CDS in L96P cells (Fig. 6B). Three different NCC uORFs are present in the AHK1 5′UTR, all of which appear to exhibit elevated translation in L96P versus WT cells, associated with the decreased translation of the main CDS (Fig. 6B). Expression of a luciferase reporter with FLUC fused in-frame to the AHK1 main CDS shows only a small reduction in L96P versus WT cells (Fig. 6C, upper construct), ostensibly at odds with the approximately threefold reduction in the translation of the native AHK1 CDS indicated by ribosome profiling data (Fig. 6B). However, expression of the corresponding mutant reporter lacking all three uORFs shows a marked increase in L96P versus WT cells (Fig. 6C, lower construct), and correcting for this effect by normalizing expression of the WT reporter to the mutant-uORF reporter suggests an approximately threefold reduction in initiation at the AHK1 CDS AUG codon in L96P versus WT cells mediated by the uORFs (Fig. 6D). While this last result is consistent with the ribosome profiling data in Figure 6B for AHK1, it is unclear why the expression of the mutant-uORF is elevated by L96P and we cannot be certain that this effect would also apply to the WT reporter containing uORFs.
Finally, we sought to confirm that the elevated translation of the in-frame amino-terminal extension of the GRS1 CDS in L96P cells indicated by the ribosome profiling data (Fig. 1B) can be attributed to relaxed discrimination against the near-cognate UUG start codon. As shown in Figure 6E (construct 1), a GRS1-FLUC reporter in which FLUC is fused in-frame to the amino-terminal extension upstream of the main CDS AUG codon, exhibits a marked increase in expression in L96P versus WT cells, in agreement with the ribosome profiling data in Figure 1B. Eliminating the UUG start codon for the amino-terminal extension (Chang and Wang 2004) has the expected effect of reducing GRS1-FLUC expression in WT cells; however, the mutant reporter expression is still increased in sui1-L96P versus WT cells (Fig. 6E, construct 2). Noting the presence of three additional in-frame NCCs downstream from the UUG start codon, we generated additional mutant constructs lacking these NCCs in the presence or absence of the UUG codon. Expression of the construct lacking all four NCCs was extremely low in both strains (Fig. 6E, construct 4), indicating that the residual expression of the single UUG mutant (construct 2) was conferred by initiation at one or more of the three downstream NCCs, and that these NCC-initiation events are elevated in the L96P strain. Removal of only the three downstream NCCs conferred only a slight reduction in expression compared to the WT construct, and expression remained strongly responsive to L96P (Fig. 6E, cf. constructs 3 and 1), consistent with the conclusion that the UUG is the major initiation site for the amino-terminal extension. These results support the interpretation of the ribosome profiling data that the increased translation of the GRS1 amino-terminal extension in L96P cells results primarily from relaxed discrimination against the near-cognate UUG codon. The profiling data in Figure 1B also suggested increased translation from other NCCs present upstream of the UUG start codon in L96P cells. Having found however that eliminating the four NCCs discussed above essentially abolished GRS1-FLUC expression (Fig. 6E, construct 4), we presume that the additional upstream NCCs are not in the same reading frame as GRS1.
DISCUSSION
Previous studies have shown that eIF1 mutations that reduce its binding to the 40S subunit or other components of the scanning PIC, including the sui1-L96P mutation studied here, increase the frequency of aberrant initiation at a UUG start codon relative to initiation at an AUG codon on HIS4 or synthetic reporter mRNAs—the Sui− phenotype—and also increase initiation at the SUI1 AUG codon in its native, poor Kozak context but not when surrounded by the optimal context defined for yeast mRNAs (Cheung et al. 2007; Martin-Marcos et al. 2011, 2013). This relaxed discrimination against near-cognate or suboptimal AUG codons has been attributed to inappropriate dissociation of eIF1 from the scanning PIC when a perfect match between the start codon and the anticodon of Met-tRNAi is lacking, or in the absence of surrounding context nucleotides favorable for start codon recognition. This explanation is supported by biochemical analysis of Sui− eIF1 variants showing that they weaken eIF1 binding to the 40S subunit, and accelerate eIF1 dissociation from PICs reconstituted with mRNA containing a UUG start codon, compared to WT eIF1 (Cheung et al. 2007; Martin-Marcos et al. 2013). It was unclear however whether a Sui− variant of eIF1 would have a global effect on the accuracy of start codon recognition in vivo.
Here we showed that eIF1-L96P, one of the strongest known Sui− eIF1 mutants (Martin-Marcos et al. 2011), shown to be impaired for eIF1 interactions with eIF3c, eIF5, and the 40S (Obayashi et al. 2017), increases utilization of the near-cognate start codons found at GRS1 and ALA1 that initiate translation of the amino-terminal extensions of these proteins, relative to the sequences downstream encoding the shorter protein isoforms initiated from internal AUG codons (Fig. 1B,C). Reporter assays demonstrated that the increased translation of the amino-terminal CDS of GRS1 in L96P cells is largely dependent on its 5′-proximal UUG start codon (Fig. 6E). The L96P mutation also increased the translation of hundreds of NCC_uORFs with start codons representing all nine near-cognate triplets (Figs. 1E, 5A). Reporter assays provided direct evidence that L96P increases initiation at all near-cognate start codons except for the most poorly recognized AAG and AGG triplets (Fig. 5D; Kolitz et al. 2009).
Interestingly, the L96P mutation also increased the translation of many AUG-initiated uORFs, which we attributed to the fact that the majority of these uORFs exhibit poor Kozak context for their AUG codons. Supporting this interpretation, the increase in translation conferred by L96P tends to be greater for the AUG_uORFs with the poorest contexts compared to those with better (albeit still relatively poor) contexts. We also observed that L96P increased translation of the main CDSs of a small fraction of mRNAs with poor-context AUG start codons, including TEN1 and SWT1 mRNAs (Fig. 3B,C), which thus resemble the SUI1 mRNA encoding eIF1 (Fig. 1A). Unlike the AUG_uORFs, the AUG start codons of the majority of main CDSs in yeast mRNAs are in good context, containing adenine at the critical −3 residue; consequently, there are far fewer main CDS whose translation is increased by L96P compared to the more widespread increase in translation observed for AUG_uORFs.
There were considerably more instances of mRNAs showing reduced versus elevated translation of their main CDSs in response to L96P (Fig. 4A,B), and we provided strong bioinformatic evidence that this decrease is associated with elevated uORF translation in these mRNAs, including both AUG_uORFs and NCC_uORFs (Fig. 4C,D), which is in keeping with the known inhibitory function of uORFs (Hinnebusch et al. 2016). Direct evidence for this mechanism was provided for CPA1 mRNA, and possibly also AHK1 mRNA, both containing AUG_uORFs with poor-context start codons whose translation is elevated in L96P cells (Figs. 2A, 6B), by showing that decreased translation of the corresponding reporter mRNAs in L96P cells was eliminated by removing the uORFs (Fig. 6A,C). Thus, we conclude that reducing the stringency of start codon recognition by an eIF1 mutation can engender an indirect reduction in the translation of the main CDS of an mRNA by increasing the translation of an uORF initiating with an NCC or poor-context AUG start codon. A similar mechanism underlies the negative autoregulatory loop controlling eIF5 expression in mammalian cells, as the mRNA encoding eIF5 contains a uORF with a poor-context AUG codon whose translation is elevated in response to increased eIF5 abundance, a condition known to reduce initiation accuracy in cells, leading in turn to reduced translation of the downstream eIF5 CDS (Loughran et al. 2012).
While this work was in progress, a study on the effects of depleting or eliminating WT eIF1 in cultured mammalian cells was reported (Fijałkowska et al. 2017), which described certain results similar to ours, but other findings that appear to differ from our observations on the eIF1-L96P mutant. In cells depleted or devoid of mammalian eIF1, numerous mRNAs with NCC-uORFs showed increased ratios of RPFs in uORFs versus main CDS (the parameter RRO defined here), in agreement with our findings on NCC-uORFs in the L96P mutant (Fig. 1E). Moreover, it was found that increased translation of the group of NCC_uORFs on depletion of mammalian eIF1 was associated with a modest reduction in average TE for the main CDSs, consistent with the notion that eliminating eIF1 favors initiation at NCC_uORFs with commensurate reduced initiation at downstream CDSs. We reached the same conclusion here, as the L96P mutation tends to reduce the CDS TEs of mRNAs harboring NCC-uORFs whose translation is elevated in the mutant (Fig. 4D). However, ostensibly at odds with our findings, it was observed that mRNAs showing decreased main CDS TEs on mammalian eIF1 depletion tend to have poor AUG contexts compared to those showing elevated CDS TEs. In contrast, we observed increased TEs in L96P cells for the subset of main CDS with the poorest AUG context scores (Fig. 3D–H). Furthermore, unlike our finding that increased initiation at AUG_uORFs is associated with decreased main CDS translation in L96P cells (Fig. 4D), depletion of mammalian eIF1 generally reduced, rather than increased, the uORF:CDS translation ratios (RRO values) for mRNAs harboring AUG_uORFs, having this effect only for NCC_uORFs. Thus, in contrast to our findings for L96P, depletion of eIF1 from mammalian cells appeared to preferentially increase utilization of NCC start codons at uORFs while having little effect on initiation at AUG codons at either uORFs or main CDS. It seems unlikely that the relatively larger response of AUG_uORFs in yeast L96P cells results from overall poorer contexts for AUG_uORF start codons in yeast, as both AUG_uORFs and NCC-uORFs have equally poor contexts in mammals, well below that found for main CDS AUG codons (Fijałkowska et al. 2017). Although increased utilization of poor-context AUG codons at uORFs or main CDS was not broadly observed on mammalian eIF1 depletion, it did confer the increased expression of eIF1B (an apparent eIF1 paralog) and decreased expression of eIF5 (Fijałkowska et al. 2017) expected from relaxed discrimination against the poor-context AUG start codons of the eIF1 main CDS and the eIF5 inhibitory uORF, respectively (Loughran et al. 2012).
One possibility to explain the discrepancy noted above might be that the L96P mutation produces a greater reduction in yeast eIF1 function compared to the ≈50% depletion of WT eIF1 protein achieved in the mammalian cells (Fijałkowska et al. 2017), making it relatively harder to detect a widespread reduced discrimination against poor-context AUGs in the eIF1-depleted mammalian cells. Consistent with this possibility, the eIF1-depleted mammalian cells displayed no growth defect, whereas the yeast L96P mutant grows slowly (Martin-Marcos et al. 2011) and shows a marked reduction in bulk translation (Supplemental Fig. S2D). Another factor could be that inactivation of yeast eIF1 by L96P appears to confer a quantitatively larger increase in the utilization of NCCs versus poor-context AUGs. Whereas L96P increased initiation between fivefold and 20-fold for different UUG-initiated reporter mRNAs (Fig. 5D; Martin-Marcos et al. 2011) and for the GRS1 UUG start codon (Fig. 1B), it rarely increased utilization of poor-context AUG codons by twofold and many such mRNAs show reduced not elevated TEs in L96P cells (Fig. 3D, col. 3), thus suggesting that another feature(s) of these mRNAs besides context contributes substantially to their TEs. If comparable differential effects on NCC versus poor-context AUG utilization occurred in the eIF1-depleted mammalian cells, it would help explain why they did not exhibit a detectable widespread increase in initiation at poor-context AUGs.
In summary, our findings indicate that eIF1 functions throughout the yeast translatome to suppress initiation at both near-cognate start codons and at AUG start codons with poor Kozak context, both at main CDS and uORFs. This activity of eIF1 prevents abnormally high expression of the amino-terminally extended isoforms encoded by GRS1 and ALA1 initiated by NCCs, but has limited effects on the translation of the vast majority of main CDS because they contain AUG start codons in a good context. In contrast, the ability of eIF1 to discriminate against poor start codons serves to prevent excessive initiation at uORFs initiated by either NCC or AUG codons—as the latter tend to contain poor Kozak context—minimizing the inhibitory effects of the uORFs and thereby promoting the translation of the downstream main CDS.
MATERIALS AND METHODS
Yeast strains and plasmids
Isogenic yeast strains containing WT or mutant alleles of SUI1 were constructed previously (Martin-Marcos et al. 2011) and are described in Table 1. Plasmids used in this work are listed in Table 2 and were generated as follows.
Table 1.
Yeast strains used in this study

Table 2.
Plasmids used in this study

To construct the AUG–AUG and NCC–AUG dual-luciferase reporters depicted in Figure 5C, the FLUC and RLUC CDSs were each placed 3′ of the promoter and NCC-depleted 5′UTR of RPL41A and inserted separately into plasmid YCplac33 (Gietz and Sugino 1988) in opposite orientations. For the AUG–AUG construct, 386 bp upstream of the RPL41A CDS containing the promoter, 5′ UTR, and ATG codon, were amplified by PCR from yeast genomic DNA using primers FJO201 and FJO202 (sequences of these and other primers listed in Table 3), thereby replacing its three near-cognate start codons at positions −3 (ATC), −9 (ATT), and −12 (ATC) with noncognate triplets AAC, AAA, and AAC, respectively; and the FLUC CDS beginning at the second codon followed by the HIS3 transcription terminator were amplified from plasmid pRaugFFuug (Kolitz et al. 2009) using primers FJO203 and FJO204. Equimolar amounts of the two PCR fragments were mixed and reamplified using primers FJO201 and FJO204, and the resulting fusion PCR product was digested with SacI and BamHI and cloned between the same sites in YCplac33. A second reporter gene containing the RPL41A promoter, NCC-depleted 5′UTR, RLUC CDS, and CYC1 terminator was generated in a similar manner using primers FJO205 to FJO208 and inserted in the opposite orientation into the HindIII and BamHI sites of the intermediate plasmid just described, producing the AUG–AUG plasmid pFJZ1052. To generate the various NCC–AUG reporters, the RPL41A promoter and NCC-depleted 5′UTR in FJZ1052 was PCR-amplified using FJO201 as the forward primer and the appropriate member of the set FJO209 to FJO217 as a reverse primer to replace the ATG start codon with the desired NCC codon. The resulting PCR products were digested with SacI and XmaI and cloned between the same sites in pFJZ1052 to generate plasmids pFJZ1053 to pFJZ1061, respectively.
Table 3.
Oligonucleotide primers used in this study

The WT CPA1-FLUC construct depicted in Figure 6A was constructed by PCR-amplifying 786 bp of native CPA1 upstream sequences and the first 45 bp of CDS sequences from WT yeast genomic DNA using primers FJO218–9, digesting the product with SacI and XmaI, and inserting it between the same sites in pFJZ1052, to generate pFJZ1064. The ATG start codon in the CPA1 uORF of pFJZ1064 was changed to TAG by a fusion PCR strategy using primers FJO218/FJO220 to produce the first PCR fragment, primers FJO221/FJO219 for the second fragment, and primers FJO218/FJO219 for the fusion of the two fragments. The final product was digested with SacI and XamI and inserted between the same sites in pFJZ1064 to generate pFJZ1065.
Luciferase reporters AHK1-FLUC and AHK1-FLUC_5′UTR_ truncated depicted in Figure 6C were generated in a similar manner to that just described. Thus, 697 bp of native AHK1 upstream sequences and the first 45 bp of CDS sequences were amplified from WT yeast DNA using primers FJO222 and FJO223, and inserted between the SacI and XamI sites in pFJZ1052 to generate pFJZ1066. The AHK1 native sequence in pFJZ1066 was PCR-amplified using primers FJO222 and FJO224 to remove 168 nt from the AHK1 5′UTR, position −5 to −172 from the main ATG, and the resulting product was inserted between the SacI and XamI sites in pFJZ1066 to generate pFJZ1067.
To construct the GRS1-FLUC reporter of Figure 6E, 728 bp of native GRS1 upstream sequence was amplified from yeast genomic DNA using primers FJO225 and FJO226 and inserted between the SacI and XamI sites in pFJZ1052 to generate pFJZ1068. The GRS1 sequence in pFJZ1068 was subsequently amplified using forward primer FJO225 and reverse primers FJO227, FJO228, or FJO229 to generate the three NCC-mutated versions of the NTE indicated in Figure 6E. The resulting PCR products were inserted between the SacI and XamI sites in pFJZ1068 to generate plasmids pFJZ1069, pFJZ1070, and pFJZ1071.
Biochemical assays using yeast cell extracts
Luminescence expressed from dual-luciferase reporters was measured as described previously (Kolitz et al. 2009). Analysis of polysome profiles was conducted as previously described (Walker et al. 2013).
Ribosome footprint profiling and RNA-seq
Ribosome profiling was conducted essentially as described previously (Martin-Marcos et al. 2017) on yeast strains PMY30 (WT SUI1) and PMY33 (sui1-L96P), examining two biological replicates of each strain, cultured in SC-Leu at 30°C. As before, cells were not treated with cycloheximide before harvesting, and cycloheximide was added only to the lysis buffer at 5× the standard concentration. Moreover, RNase-trimmed ribosomes were purified by velocity sedimentation through sucrose gradients prior to extraction of mRNA; and Illumina Ribo-Zero Gold rRNA Removal Kit (Yeast) was used on linker-ligated mRNA in lieu of poly(A) selection. Generation, processing, and analysis of sequence libraries of RPFs or total mRNA fragments, including Wiggle track normalization for viewing RPF or RNA reads in the IGV browser, were conducted exactly as described previously (Martin-Marcos et al. 2017).
DESeq (Anders and Huber 2010) was used to identify significant changes in mRNA, RPFs, or TEs between the mutant and WT strains, a statistical package well-suited to identifying changes in mRNA expression or translation with a very low incidence of false positives using results from only two biological replicates for each of the strains being compared, provided the replicates are highly correlated (as they were in our case [Supplemental Fig. S3; Zhang et al. 2014; Lamarre et al. 2018]). Briefly, DESeq operates by pooling information about variances of read counts across the thousands of genes analyzed to model count variances for genes of similar expression levels. The modeled variances are used in the framework of a generalized linear model (GLM) to identify expression changes and place confidence intervals on the magnitude of changes, and also to exclude genes showing aberrantly high variability. Transcriptional and translational changes are analyzed together in a GLM by including library type (mRNA-seq or Ribo-seq) as one of the variables, in addition to genotype, in a multifactor design. The TE emerges as the effect of the Ribo-seq library type against the mRNA-seq baseline, and significant interactions of TE with the genotype indicate translational control by the gene product under consideration (Ingolia 2016). Genes with less than 128 total mRNA reads, or less than 40 total RPF reads, in the four samples combined (two replicates of both WT and mutant strains) were excluded from the calculation of TE values.
As described previously (Martin-Marcos et al. 2017), uORFs with evidence of translation in WT and eIF1-L96P cells were identified by first using the yassour-uorf program of Brar et al. (2012) to identify all potential uORFs within annotated 5′UTRs initiating with either AUG or a near-cognate codon, and then quantifying the RPFs mapping to the +1 and −1 codons of all putative uORFs. A uORF was judged to be translated if the +1 to −1 RPF ratio exceeded four and the total RPF counts at +1 and −1 exceeded 15, and also if the reads in the zero frame are at least 50% of the reads in all three frames. As described previously (Martin-Marcos et al. 2017), this analysis was conducted on multiple ribosome profiling data sets to obtain a compendium of uORFs showing evidence of translation in one or more profiling studies. After excluding uORFs shorter than three codons, we identified 564 AUG-initiated uORFs and 5497 near-cognate uORFs. In the second step, we validated ∼51% and ∼44% of the AUG uORFs and near-cognate uORFs, respectively, by using a distinct uORF identification tool, RibORF (Ji et al. 2015), based on the criteria of 3-nt periodicity and uniformity of read distribution across uORF codons. Applying a moderately stringent probability of prediction of >0.5, RibORF confirmed that 290 AUG uORFs and 2430 near-cognate uORFs show evidence of translation in the data sets from which they were first identified by the yassour-uorf program. A bed file was generated containing the sequence coordinates of every uORF and combined with a bed file containing the coordinates of the 5′UTR, main CDS, and 3′UTR of each gene, and used to obtain RPF counts for 5′UTRs, uORFs, and main CDS in each strain examined, excluding the first and last nucleotide triplets of 5′UTRs, the first and last codons of uORFs, and the first 20 codons of main CDS. mRNA read counts were determined for all codons of the main CDS. DESeq (Anders and Huber 2010) was again used for differential expression analysis of changes in TE, RPFs, or RRO values, to impose cutoffs for minimum read numbers and to remove outliers for uORFs.
For all notched box-plots, constructed using a web-based tool at http://shiny.chemgrid.org/boxplotr/, the upper and lower boxes contain the second and third quartiles and the band gives the median. If the notches in two plots do not overlap, there is roughly 95% confidence that their medians are different.
The AUG context adaptation index (context score) (Miyasaka 1999) was calculated as AUGCAI = (w−6 × w−5 × w−4 × w−3 × w−2 × w−1 × w+1 × w+2 × w+3)1/9, where wi is the fractional occurrence of that particular base, normalized to the most prevalent base, present in the ith position of the context among the ∼270 most highly expressed yeast genes, taken from the matrix of frequencies and relative adaptiveness (w) of the nucleotide in the AUG context of this group of ∼270 reference genes (Zur and Tuller 2013). The context scores range from ∼0.16 (poorest) to ∼0.97 (best) among all yeast genes.
DATA DEPOSITION
Sequencing data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSM2895472–GSM2895475 in the GSE108334 records for ribosome profiling, and GSE138599 for mRNA sequencing data.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Tingfen Yan for help with bioinformatics, Nicholas Ingolia, Thomas Dever, and Nicholas Guydosh for many helpful discussions about data analysis and interpretation of results, and Ivaylo Ivanov for critical reading of the manuscript. This work was supported in part by the Intramural Research Program of the National Institutes of Health.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.073536.119.
REFERENCES
- Alone PV, Cao C, Dever TE. 2008. Translation initiation factor 2γ mutant alters start codon selection independent of Met-tRNA binding. Mol Cell Biol 28: 6877–6888. 10.1128/MCB.01147-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol 11: R106 10.1186/gb-2010-11-10-r106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arribere JA, Gilbert WV. 2013. Roles for transcript leaders in translation and mRNA decay revealed by transcript leader sequencing. Genome Res 23: 977–987. 10.1101/gr.150342.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brar GA, Yassour M, Friedman N, Regev A, Ingolia NT, Weissman JS. 2012. High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science 335: 552–557. 10.1126/science.1215110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan LY, Mugler CF, Heinrich S, Vallotton P, Weis K. 2018. Non-invasive measurement of mRNA decay reveals translation initiation as the major determinant of mRNA stability. Elife 7: e32536 10.7554/eLife.32536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KJ, Wang CC. 2004. Translation initiation from a naturally occurring non-AUG codon in Saccharomyces cerevisiae. J Biol Chem 279: 13778–13785. 10.1074/jbc.M311269200 [DOI] [PubMed] [Google Scholar]
- Chang CP, Chen SJ, Lin CH, Wang TL, Wang CC. 2010. A single sequence context cannot satisfy all non-AUG initiator codons in yeast. BMC Microbiol 10: 188 10.1186/1471-2180-10-188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SJ, Lin G, Chang KJ, Yeh LS, Wang CC. 2008. Translational efficiency of a non-AUG initiation codon is significantly affected by its sequence context in yeast. J Biol Chem 283: 3173–3180. 10.1074/jbc.M706968200 [DOI] [PubMed] [Google Scholar]
- Cheung YN, Maag D, Mitchell SF, Fekete CA, Algire MA, Takacs JE, Shirokikh N, Pestova T, Lorsch JR, Hinnebusch AG. 2007. Dissociation of eIF1 from the 40S ribosomal subunit is a key step in start codon selection in vivo. Genes Dev 21: 1217–1230. 10.1101/gad.1528307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Shah P, Plotkin JB. 2012. Weak 5′-mRNA secondary structures in short eukaryotic genes. Genome Biol Evol 4: 1046–1053. 10.1093/gbe/evs082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fijałkowska D, Verbruggen S, Ndah E, Jonckheere V, Menschaert G, Van Damme P. 2017. eIF1 modulates the recognition of suboptimal translation initiation sites and steers gene expression via uORFs. Nucleic Acids Res 45: 7997–8013. 10.1093/nar/gkx469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaba A, Wang Z, Krishnamoorthy T, Hinnebusch AG, Sachs MS. 2001. Physical evidence for distinct mechanisms of translational control by upstream open reading frames. EMBO J 20: 6453–6463. 10.1093/emboj/20.22.6453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Putnam AA, Bowers HA, Guenther UP, Ye X, Kindsfather A, Hilliker AK, Jankowsky E. 2016. Coupling between the DEAD-box RNA helicases Ded1p and eIF4A. Elife 5: e16408 10.7554/eLife.16408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz RD, Sugino A. 1988. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74: 527–534. 10.1016/0378-1119(88)90185-0 [DOI] [PubMed] [Google Scholar]
- Guenther UP, Weinberg DE, Zubradt MM, Tedeschi FA, Stawicki BN, Zagore LL, Brar GA, Licatalosi DD, Bartel DP, Weissman JS, et al. 2018. The helicase Ded1p controls use of near-cognate translation initiation codons in 5′ UTRs. Nature 559: 130–134. 10.1038/s41586-018-0258-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta N, Lorsch JR, Hinnebusch AG. 2018. Yeast Ded1 promotes 48S translation pre-initiation complex assembly in an mRNA-specific and eIF4F-dependent manner. Elife 7: e38892 10.7554/eLife.38892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch AG. 2011. Molecular mechanism of scanning and start codon selection in eukaryotes. Microbiol Mol Biol Rev 75: 434–467. 10.1128/MMBR.00008-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch AG. 2014. The scanning mechanism of eukaryotic translation initiation. Annu Rev Biochem 83: 779–812. 10.1146/annurev-biochem-060713-035802 [DOI] [PubMed] [Google Scholar]
- Hinnebusch AG. 2017. Structural insights into the mechanism of scanning and start codon recognition in eukaryotic translation initiation. Trends Biochem Sci 42: 589–611. 10.1016/j.tibs.2017.03.004 [DOI] [PubMed] [Google Scholar]
- Hinnebusch AG, Ivanov IP, Sonenberg N. 2016. Translational control by 5′-untranslated regions of eukaryotic mRNAs. Science 352: 1413–1416. 10.1126/science.aad9868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Yoon H, Hannig EM, Donahue TF. 1997. GTP hydrolysis controls stringent selection of the AUG start codon during translation initiation in Saccharomyces cerevisiae. Genes Dev 11: 2396–2413. 10.1101/gad.11.18.2396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain T, Llácer JL, Fernández IS, Munoz A, Martin-Marcos P, Savva CG, Lorsch JR, Hinnebusch AG, Ramakrishnan V. 2014. Structural changes enable start codon recognition by the eukaryotic translation initiation complex. Cell 159: 597–607. 10.1016/j.cell.2014.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT. 2016. Ribosome footprint profiling of translation throughout the genome. Cell 165: 22–33. 10.1016/j.cell.2016.02.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS. 2012. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat Protoc 7: 1534–1550. 10.1038/nprot.2012.086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov IP, Loughran G, Sachs MS, Atkins JF. 2010. Initiation context modulates autoregulation of eukaryotic translation initiation factor 1 (eIF1). Proc Natl Acad Sci 107: 18056–18060. 10.1073/pnas.1009269107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Z, Song R, Regev A, Struhl K. 2015. Many lncRNAs, 5′UTRs, and pseudogenes are translated and some are likely to express functional proteins. Elife 4: e08890 10.7554/eLife.08890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kertesz M, Wan Y, Mazor E, Rinn JL, Nutter RC, Chang HY, Segal E. 2010. Genome-wide measurement of RNA secondary structure in yeast. Nature 467: 103–107. 10.1038/nature09322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolitz SE, Takacs JE, Lorsch JR. 2009. Kinetic and thermodynamic analysis of the role of start codon/anticodon base pairing during eukaryotic translation initiation. RNA 15: 138–152. 10.1261/rna.1318509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. 1991. A short leader sequence impairs the fidelity of initiation by eukaryotic ribosomes. Gene Expr 1: 111–115. [PMC free article] [PubMed] [Google Scholar]
- LaGrandeur T, Parker R. 1999. The cis acting sequences responsible for the differential decay of the unstable MFA2 and stable PGK1 transcripts in yeast include the context of the translational start codon. RNA 5: 420–433. 10.1017/S1355838299981748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamarre S, Frasse P, Zouine M, Labourdette D, Sainderichin E, Hu G, Le Berre-Anton V, Bouzayen M, Maza E. 2018. Optimization of an RNA-seq differential gene expression analysis depending on biological replicate number and library size. Front Plant Sci 9: 108 10.3389/fpls.2018.00108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomakin IB, Steitz TA. 2013. The initiation of mammalian protein synthesis and mRNA scanning mechanism. Nature 500: 307–311. 10.1038/nature12355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loughran G, Sachs MS, Atkins JF, Ivanov IP. 2012. Stringency of start codon selection modulates autoregulation of translation initiation factor eIF5. Nucleic Acids Res 40: 2898–2906. 10.1093/nar/gkr1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maag D, Fekete CA, Gryczynski Z, Lorsch JR. 2005. A conformational change in the eukaryotic translation preinitiation complex and release of eIF1 signal recognition of the start codon. Mol Cell 17: 265–275. 10.1016/j.molcel.2004.11.051 [DOI] [PubMed] [Google Scholar]
- Martin-Marcos P, Cheung YN, Hinnebusch AG. 2011. Functional elements in initiation factors 1, 1A, and 2β discriminate against poor AUG context and non-AUG start codons. Mol Cell Biol 31: 4814–4831. 10.1128/MCB.05819-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Marcos P, Nanda J, Luna RE, Wagner G, Lorsch JR, Hinnebusch AG. 2013. β-hairpin loop of eIF1 mediates 40S ribosome binding to regulate initiator tRNAMet recruitment and accuracy of AUG selection in vivo. J Biol Chem 288: 27546–27562. 10.1074/jbc.M113.498642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Marcos P, Zhou F, Karunasiri C, Zhang F, Dong J, Nanda J, Kulkarni SD, Sen ND, Tamame M, Zeschnigk M, et al. 2017. eIF1A residues implicated in cancer stabilize translation preinitiation complexes and favor suboptimal initiation sites in yeast. Elife 6: e31250 10.7554/eLife.31250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyasaka H. 1999. The positive relationship between codon usage bias and translation initiation AUG context in Saccharomyces cerevisiae. Yeast 15: 633–637. [DOI] [PubMed] [Google Scholar]
- Monteuuis G, Miścicka A, Świrski M, Zenad L, Niemitalo O, Wrobel L, Alam J, Chacinska A, Kastaniotis AJ, Kufel J. 2019. Non-canonical translation initiation in yeast generates a cryptic pool of mitochondrial proteins. Nucleic Acids Res 47: 5777–5791. 10.1093/nar/gkz301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obayashi E, Luna RE, Nagata T, Martin-Marcos P, Hiraishi H, Singh CR, Erzberger JP, Zhang F, Arthanari H, Morris J, et al. 2017. Molecular landscape of the ribosome pre-initiation complex during mRNA scanning: structural role for eIF3c and its control by eIF5. Cell Rep 18: 2651–2663. 10.1016/j.celrep.2017.02.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestova TV, Kolupaeva VG. 2002. The roles of individual eukaryotic translation initiation factors in ribosomal scanning and initiation codon selection. Genes Dev 16: 2906–2922. 10.1101/gad.1020902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabl J, Leibundgut M, Ataide SF, Haag A, Ban N. 2011. Crystal structure of the eukaryotic 40S ribosomal subunit in complex with initiation factor 1. Science 331: 730–736. 10.1126/science.1198308 [DOI] [PubMed] [Google Scholar]
- Saini AK, Nanda JS, Lorsch JR, Hinnebusch AG. 2010. Regulatory elements in eIF1A control the fidelity of start codon selection by modulating tRNAiMet binding to the ribosome. Genes Dev 24: 97–110. 10.1101/gad.1871910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz DC, Parker R. 1999. Mutations in translation initiation factors lead to increased rates of deadenylation and decapping of mRNAs in Saccharomyces cerevisiae. Mol Cell Biol 19: 5247–5256. 10.1128/MCB.19.8.5247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen ND, Zhou F, Ingolia NT, Hinnebusch AG. 2015. Genome-wide analysis of translational efficiency reveals distinct but overlapping functions of yeast DEAD-box RNA helicases Ded1 and eIF4A. Genome Res 25: 1196–1205. 10.1101/gr.191601.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen ND, Zhou F, Harris MS, Ingolia NT, Hinnebusch AG. 2016. eIF4B stimulates translation of long mRNAs with structured 5′ UTRs and low closed-loop potential but weak dependence on eIF4G. Proc Natl Acad Sci 113: 10464–10472. 10.1073/pnas.1612398113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen ND, Gupta N SKA, Preiss T, Lorsch JR, Hinnebusch AG. 2019. Functional interplay between DEAD-box RNA helicases Ded1 and Dbp1 in preinitiation complex attachment and scanning on structured mRNAs in vivo. Nucleic Acids Res 47: 8785–8806. 10.1093/nar/gkz595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabalina SA, Ogurtsov AY, Rogozin IB, Koonin EV, Lipman DJ. 2004. Comparative analysis of orthologous eukaryotic mRNAs: potential hidden functional signals. Nucleic Acids Res 32: 1774–1782. 10.1093/nar/gkh313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spealman P, Naik AW, May GE, Kuersten S, Freeberg L, Murphy RF, McManus J. 2018. Conserved non-AUG uORFs revealed by a novel regression analysis of ribosome profiling data. Genome Res 28: 214–222. 10.1101/gr.221507.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur A, Hinnebusch AG. 2018. eIF1 loop 2 interactions with Met-tRNAi control the accuracy of start codon selection by the scanning preinitiation complex. Proc Natl Acad Sci 115: E4159–E4168. 10.1073/pnas.1800938115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur A, Marler L, Hinnebusch AG. 2018. A network of eIF2β interactions with eIF1 and Met-tRNAi promotes accurate start codon selection by the translation preinitiation complex. Nucleic Acids Res 47: 2574–2593. 10.1093/nar/gky1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valasek L, Nielsen KH, Zhang F, Fekete CA, Hinnebusch AG. 2004. Interactions of eukaryotic translation initiation factor 3 (eIF3) subunit NIP1/c with eIF1 and eIF5 promote preinitiation complex assembly and regulate start codon selection. Mol Cell Biol 24: 9437–9455. 10.1128/MCB.24.21.9437-9455.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker SE, Zhou F, Mitchell SF, Larson VS, Valasek L, Hinnebusch AG, Lorsch JR. 2013. Yeast eIF4B binds to the head of the 40S ribosomal subunit and promotes mRNA recruitment through its N-terminal and internal repeat domains. RNA 19: 191–207. 10.1261/rna.035881.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Wu C, Sachs MS. 2012. The arginine attenuator peptide interferes with the ribosome peptidyl transferase center. Mol Cell Biol 32: 2396–2406. 10.1128/MCB.00136-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner M, Feller A, Messenguy F, Piérard A. 1987. The leader peptide of yeast gene CPA1 is essential for the translational repression of its expression. Cell 49: 805–813. 10.1016/0092-8674(87)90618-0 [DOI] [PubMed] [Google Scholar]
- Zhang ZH, Jhaveri DJ, Marshall VM, Bauer DC, Edson J, Narayanan RK, Robinson GJ, Lundberg AE, Bartlett PF, Wray NR, et al. 2014. A comparative study of techniques for differential expression analysis on RNA-seq data. PLoS One 9: e103207 10.1371/journal.pone.0103207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zur H, Tuller T. 2013. New universal rules of eukaryotic translation initiation fidelity. PLoS Comput Biol 9: e1003136 10.1371/journal.pcbi.1003136 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.