

Abstract
Adenosine-to-inosine RNA editing is an essential post-transcriptional modification catalyzed by adenosine deaminase acting on RNA (ADAR)1 and ADAR2 in mammals. For numerous sites in coding sequences (CDS) and microRNAs, editing is highly conserved and has significant biological consequences, for example, by altering amino acid residues and target recognition. However, no comprehensive and quantitative studies have been undertaken to determine how specific ADARs contribute to conserved sites in vivo. Here, we amplified each RNA region with editing site(s) separately and combined these for deep sequencing. Then, we compared the editing ratios of all sites that were conserved in CDS and microRNAs in the cerebral cortex and spleen of wild-type mice, Adar1E861A/E861AIfih−/− mice expressing inactive ADAR1 (Adar1 KI) and Adar2−/−Gria2R/R (Adar2 KO) mice. We found that most of the sites showed a preference for one ADAR. In contrast, some sites, such as miR-3099-3p, showed no ADAR preference. In addition, we found that the editing ratio for several sites, such as DACT3 R/G, was up-regulated in either Adar mutant mouse strain, whereas a coordinated interplay between ADAR1 and ADAR2 was required for the efficient editing of specific sites, such as the 5-HT2CR B site. We further created double mutant Adar1 KI Adar2 KO mice and observed viable and fertile animals with the complete absence of editing, demonstrating that ADAR1 and ADAR2 are the sole enzymes responsible for all editing sites in vivo. Collectively, these findings indicate that editing is regulated in a site-specific manner by the different interplay between ADAR1 and ADAR2.
Keywords: ADAR1, ADAR2, coding sequence, microRNA, post-transcriptional modification
INTRODUCTION
RNAs are known to be subjected to multiple post-transcriptional modifications that affect the fate of each one. One such RNA modification is adenosine-to-inosine (A-to-I) RNA editing, which is widely conserved among many organisms, from nematodes to humans (Hundley and Bass 2010; Savva et al. 2012; Nishikura 2016). Although RNA editing sites were previously found by chance, the number of sites identified has dramatically increased after the introduction of deep sequencing technology (Li et al. 2009; Ramaswami et al. 2013; Sakurai et al. 2014), with an estimate of more than 100 million RNA editing sites present in human transcripts (Bazak et al. 2014; Picardi et al. 2015; Tan et al. 2017). Most of these sites are located in repetitive elements (REs) in noncoding regions, given that REs occasionally form double-stranded RNA (dsRNA) structures, which are required for adenosine-to-inosine RNA editing. As a consequence, RNA editing in REs alters dsRNA structure, which is indispensable in escaping recognition as non-self by the host immune system (Mannion et al. 2014; Liddicoat et al. 2015; Pestal et al. 2015). Although this role is conserved among mammals, RNA editing in REs occurs frequently in primates due to the abundance of REs, especially primate-specific Alu repeats (Neeman et al. 2005); it therefore follows that the total number of editing sites is much lower in rodents (Danecek et al. 2012; Li and Church 2013). In contrast, although their frequency is quite rare, RNA editing sites in protein-coding sequences (CDS) and microRNAs (miRNAs) are relatively conserved among mammals (Li et al. 2009; Li and Church 2013; Pinto et al. 2014; Nishikura 2016; Jinnah and Ulbricht 2019). Given that inosine is recognized as if it were guanosine by the translational machinery, RNA editing in CDS leads to recoding events that can potentially affect the functions of the corresponding proteins (Pullirsch and Jantsch 2010). Indeed, mutant mice expressing either an edited or unedited protein alone exhibit abnormal phenotypes, such as being more prone to seizures, hyperactivity, depression, and dysregulated vascular contractions (Higuchi et al. 2000; Kawahara et al. 2008a; Mombereau et al. 2010; Miyake et al. 2016; Jain et al. 2018). In addition, dysregulated RNA editing in CDS is linked to human diseases such as cancer and amyotrophic lateral sclerosis (Kawahara et al. 2004; Slotkin and Nishikura 2013; Peng et al. 2018). Furthermore, editing in miRNAs affects their expression and target recognition (Yang et al. 2005; Kawahara et al. 2007a,b, 2008b). Therefore, it is crucial to know the enzyme(s) responsible for each conserved site in order to understand the mechanisms underlying the tight regulation of RNA editing in vivo.
Adenosine deaminases acting on RNAs (ADARs) are the enzymes responsible for adenosine-to-inosine RNA editing, which requires a dsRNA structure for target recognition. In mammals, three ADARs have been identified (Hundley and Bass 2010; Nishikura 2016): while brain-specific ADAR3 appears enzymatically inactive, ADAR1 and ADAR2 are active enzymes that are ubiquitously expressed, although their expression level varies in a tissue-specific manner (George et al. 2005; Picardi et al. 2015; Huntley et al. 2016; Heraud-Farlow et al. 2017; Tan et al. 2017; Nakahama et al. 2018). For instance, ADAR2 is mainly localized in the nucleus, and highly expressed in the brain and aorta (Huntley et al. 2016; Tan et al. 2017; Jain et al. 2018; Nakahama et al. 2018). In contrast, ADAR1 has two isoforms: a short p110 isoform that is mainly localized in the nucleus and is highly expressed in the brain, and a long p150 isoform that is mainly localized in the cytoplasm and is highly expressed in the thymus and spleen, where ADAR2 is expressed at low level (Huntley et al. 2016; Nakahama et al. 2018). In addition to the different expression patterns of ADARs, it is known that ADAR1 and ADAR2 regulate RNA editing in a competitive manner in some cases, which has made it difficult to determine the contribution of each ADAR to the RNA editing of each conserved site in vivo (Kawahara et al. 2007b; Riedmann et al. 2008; Wahlstedt et al. 2009; Vesely et al. 2014; Picardi et al. 2015; Huntley et al. 2016; Tan et al. 2017). One simple solution to this problem may be by comparing the editing ratio of each site among wild-type (WT), Adar1-deficient (Adar1−/−) and Adar2-deficient (Adar2−/−) mice. However, Adar1−/− and Adar2−/− mice exhibit embryonic and early postnatal lethality, respectively (Higuchi et al. 2000; Hartner et al. 2004; Wang et al. 2004). Fortunately, the lethality of Adar2−/− mice can be rescued by the single substitution of adenine to guanine in an editing site in the coding region of Gria2 at the genomic DNA level. This leads to a change in one amino acid residue, from glutamine (Q) to arginine (R), which allows Adar2−/−Gria2R/R mice (which we termed Adar2 knockout [KO] mice) to survive until adulthood (Higuchi et al. 1993). In contrast, although the embryonic lethality of Adar1−/− mice can be rescued by concurrent knockout of the Ifih1 gene encoding MDA5, a cytoplasmic sensor for dsRNAs, Adar1−/− Ifih−/− mice still develop postnatal lethality for unknown reasons (Pestal et al. 2015). Therefore, several studies have tried to determine the ADAR responsible for each site by comparing primary neuronal cultures prepared from Adar1−/− mice and the brains of Adar2−/− mice (Riedmann et al. 2008), embryos collected from different Adar mutant mice (Vesely et al. 2012; Tan et al. 2017), or HeLa cells in which either ADAR1 or ADAR2 was knocked down (Nishimoto et al. 2008). However, in addition to the difficulties in comparing the different conditions, editing activity is relatively low in embryos and cultured cells. Of note, an editing-inactive E861A point mutation (Adar1E861A/E861A mice) in mutant mice is embryonically lethal, whereas it was recently reported that Adar1E861A/E861AIfih−/− mice (which we termed Adar1 KI mice) survive with a normal life-span (Liddicoat et al. 2015; Heraud-Farlow et al. 2017). Therefore, it is now possible to determine the contribution of each ADAR to the editing of conserved sites by comparing the editing ratios between wild-type (WT), Adar1 knock-in (KI), and Adar2 KO mice. However, although it is advantageous for the comprehensive identification of editing sites, total RNA-sequencing (RNA-seq) analysis is a poor method for accurate quantification of the editing ratio due to its limited sequencing depth (Tan et al. 2017). To compensate for this disadvantage, a microfluidic multiplex PCR and deep sequencing (mmPCR-seq) method was developed, in which PCR is performed with multiplex primers targeting each editing site followed by deep sequencing (Zhang et al. 2013). This method can quantify the editing ratio at many sites more accurately, although the amplification efficiency of each target is affected by the expression level of each RNA and primer design, which implies a lack of guarantee in obtaining the editing ratio of the sites targeted. In this regard, although the global dynamics of RNA editing (mostly in REs) were recently revealed by comparing editing ratios between WT, Adar1 KI, and Adar2 KO mice using the mmPCR-seq method for a limited number of samples (one WT mouse vs. two Adar1 KI mice), none of the sites in CDS were identified as an ADAR1 site in the brain for example (Tan et al. 2017). In addition, this analysis did not examine any editing sites in miRNAs. Therefore, a comprehensive and accurate quantification of the editing ratio at conserved sites is necessary to further understand the contribution of each ADAR to these sites in vivo.
In this study, we performed reverse-transcription (RT)-PCR for each RNA editing site followed by adjusting the amplicon length with a second round of PCR. After gel purification of each PCR product, similar amounts were combined for deep sequencing. Although this method is laborious, it yielded editing ratios with a high degree of accuracy for all sites examined. We applied this method to all RNA editing sites in CDS and miRNAs that are definitely or possibly conserved between humans and mice, and compared editing ratios in the cerebral cortex and spleen between WT, Adar1 KI, and Adar2 KO mice. Using this analysis, we found novel ADAR1 sites, such as UBE2O S/G and DACT3 R/G, as well as novel sites specific for ADAR2, including TMEM63B Q/R and SPEG S/G. ADAR2 edits DACT3 R/G and CDK13 Q/R sites only in the absence of ADAR1 activity in the cerebral cortex, whereas ADAR1 edits some sites, such as GABRA3 I/M, only in the absence of ADAR2 in the cerebral cortex. In addition, we found that the editing ratio for some sites was up-regulated in either Adar mutant mouse strain, that is, the DACT3 R/G site in the spleen of Adar2 KO mice and the +4 position of miR-376a-2-5p in the cerebral cortex of Adar1 KI mice, indicating that the presence or editing activity of one ADAR may negatively affect editing by the other ADAR in some cases. In contrast, we identified some sites, such as the serotonin (5-HT) 5-HT2CR receptor (5-HT2CR) B site, that required a coordinated interplay between ADAR1 and ADAR2 for efficient editing. Although most of the sites showed a preference for either ADAR1 or ADAR2, editing was preserved in both Adar KI and Adar2 KO mice at several other sites, such as in miR-3099-3p and miR-411-5p, which demonstrates a lack of preference for either ADAR. We also established Adar1 KI Adar2 KO mice for the first time and found that RNA editing was completely absent, which indicates that ADAR1 and ADAR2 are the sole editing enzymes in vivo. These findings indicate that RNA editing is regulated in a site-specific manner through the different interplay between ADAR1 and ADAR2. In addition, our comprehensive and quantitative data can act as a valuable resource for identifying the contribution of each ADAR to all conserved sites.
RESULTS AND DISCUSSION
Editing ratios for CDS and miRNA sites in the cerebral cortex are higher than for the spleen with a few exceptions
To comprehensively and accurately quantify editing ratios in conserved editing sites in humans and mice, we listed 69 sites in the CDS of 39 genes and 26 sites in 21 miRNAs, all of which are conserved or possibly conserved between humans and mice, in addition to some representative REs in introns and 3′ untranslated regions (3′UTR), and intronic self-editing sites in the Adar2 gene (Materials and Methods). Then, to obtain the editing ratios of as many sites as possible, we separately amplified each target site by RT-PCR (Fig. 1A). After adjusting the length of PCR products, and adding adaptor and barcode sequences with a second round of PCR, each PCR product was gel purified, and 30–200 PCR products were then combined and subjected to deep sequencing (Fig. 1A). We selected the cerebral cortex and spleen as representative tissues for this analysis because, as previously reported (Nakahama et al. 2018), ADAR1 p110 and ADAR2 are highly expressed in the cerebral cortex while cytoplasmic ADAR1 p150 is undetectable, and ADAR1 p150 is highly expressed in the spleen while ADAR2 is expressed at very low levels (Fig. 1B). PCR products containing the editing sites were amplified from these two tissues isolated from male WT, Adar1 KI, and Adar2 KO mice at 8 wk of age (n = 3 mice for each group). We found that the inactivation of ADAR1 or deletion of ADAR2 did not induce a compensatory up-regulation of the remaining ADAR (Fig. 1B). Compensatory up-regulation of Adar2 mRNA was also not reported in the brain and spleen of Adar1 KI mice (Heraud-Farlow et al. 2017). We successfully obtained editing ratios for all sites examined from the cerebral cortex (Supplemental Table S1). In contrast, we did not determine editing ratios at 19 sites in 11 genes in the spleen due to a lack of amplification of PCR products since most of these genes are expressed in a tissue-specific manner, such as the brain-specific Htr2c gene (Kawahara et al. 2007b, 2008a).
FIGURE 1.

Comparison of RNA editing ratios between the cerebral cortex and spleen of WT mice. (A) The protocol to create ion amplicon libraries for the evaluation of RNA editing ratios at multiple sites. After reverse-transcription using oligo(dT) primers or random hexamers, the first PCR was performed using cDNA (in green) that included an RNA editing site (shown as a red cross) and the first primers specific for each editing site (in yellow). Then, a second round of PCR was performed using an aliquot of the first PCR product as a template, with each second forward primer specific to the editing site and containing an A Adaptor (in light green), an Ion Xpress Barcode (in gray), editing site-specific sequences (in brown), and a reverse primer that contained a trP1 adaptor (in light blue); editing site-specific sequences (in brown) were also included. All second PCR products were designed to be 190 to 200 bp in length. After 50–300 PCR products were combined, the samples were sequenced using an Ion Torrent Personal Genome Machine (Ion PGM) system. (B) Immunoblot analysis of adenosine deaminase acting on RNA (ADAR)1 p110, ADAR1 p150, and ADAR2 expression in cerebral cortexes and spleens isolated from wild-type (WT), Adar1E861A/E861AIfih−/− mice (Adar1 KI) and Adar2−/− Gria2R/R (Adar2 KO) mice (n = 2 mice for each group). The expression of GAPDH is shown as a reference. (C) Validation of the methodology by referring to the editing ratios of known ADAR1 (AZIN1 serine/glycine [S/G]) and ADAR2 sites (Kv1.1 isoleucine/valine [I/V]). Editing ratios at each site in each indicated tissue isolated from WT, Adar1 knock-in (KI), and Adar2 knockout (KO) mice are displayed as the mean ± SEM (n = 3 mice for each group; Student's t-test, [*] P < 0.05, [**] P < 0.01, n.s., not significant). (D) Editing ratios of all sites examined were compared between cerebral cortexes and spleens isolated from WT mice. Values are displayed as the mean of values from three mice. The red squares, blue circles, green diamonds, and gray triangles represent editing sites in coding sequences (CDS), microRNAs (miRNAs), repetitive elements (REs), and introns, respectively. Editing ratios for sites that could only be amplified from the cerebral cortex are separately displayed in the “Cortex only” fraction. See Supplemental Charts to access an interactive version of this chart in which each editing site can be identified.
First, to validate the method, we focused on two representative editing sites, that is, AZIN1 S/G, a known ADAR1 site (Chen et al. 2013), and Kv1.1 I/V, a known ADAR2 site (Bhalla et al. 2004). We did not detect editing at the AZIN1 S/G site in the spleen of Adar1 KI mice, and no significant difference was observed between WT and Adar2 KO mice (Fig. 1C). In contrast, we found no significant difference in the editing ratio at the Kv1.1 I/V site in the cerebral cortex between WT and Adar1 KI mice, although the editing ratio was dramatically reduced to 1% in Adar2 KO mice (Fig. 1C). These results are clear evidence that ADAR1 and ADAR2 are the sole enzymes responsible for AZIN1 S/G and Kv1.1 I/V sites, respectively, and support our methodology. We further validated the methodology by randomly selecting 104 PCR products containing various editing sites from all mice examined and subjected these to resequencing. This analysis underscored the high reproducibility of technical replicates (Supplemental Table S2). In addition, we then again prepared PCR products for 40 randomly selected targets from the total RNA used in the first analysis, which were then subjected to sequencing. This analysis also revealed a high correlation between two independent analyses of the same RNAs (Supplemental Table S3), which further strengthened the validity of the methodology.
We then compared the editing ratios of all sites examined in the cerebral cortex and spleen in WT mice. We found that editing ratios in most CDS and miRNA sites in the cerebral cortex were higher than in the spleen (Fig. 1D; Supplemental Charts). This is in line with these sites being predominantly edited by ADAR2 and ADAR1 p110 in the nucleus. More specifically, editing in the CDS requires an editing (or exon) complementary sequence (ECS), which is usually located in an adjacent intron (Gerber and Keller 2001); therefore, these sites can be edited only in the nucleus. Notable exceptions were AZIN1 E/G and S/G sites, which are ADAR1 sites that were edited by 10% and 19%, respectively, in the spleen, whereas these sites were edited by <1% in the cerebral cortex of WT mice (Fig. 1C,D). These results indicate that the ECS of AZIN1 E/G and S/G sites may not require an intron and actually forms between exons. Indeed, although it was previously reported that the estimated dsRNA structure is formed with a portion of the downstream intron (Chen et al. 2013), the formation of a dsRNA structure only within the exon is also possible (Supplemental Fig. S1A). Furthermore, we compared the editing ratios of AZIN1 E/G and S/G sites between mature mRNA and precursor mRNA (pre-mRNA) in the spleen, which showed that only mature mRNA was edited at both sites (Supplemental Fig. S1B). Therefore, considering that the ECSs of the Kv1.1 I/V and the GABRA3 I/M sites are located within the same exon (Bhalla et al. 2004; Rula et al. 2008), AZIN1 E/G and S/G sites may be an additional case (and the first case as ADAR1 sites) in which a dsRNA structure forms within a single coding exon.
Most sites are preferentially edited by one ADAR
Next, we examined the editing retention rate in Adar1 KI and Adar2 KO mice, which was defined as the remaining editing levels in Adar mutant mice compared to WT mice. In most sites, the editing ratio was greatly reduced in either Adar1 KI or Adar2 KO mice, which shows that one Adar preferentially contributed to the editing of these sites (Fig. 2A–D; Supplemental Charts). In particular, while the majority of sites in representative REs were mainly edited by ADAR1, especially in the spleen, those in the CDS were preferentially edited by ADAR2, which included Kv1.1 I/V, FLNA Q/R, and CYFIP2 K/E, as well as many sites in glutamate receptor subunits (Fig. 1C; Supplemental Fig. S2A,B; Supplemental Table S1), as previously reported (Bhalla et al. 2004; Nishimoto et al. 2008; Riedmann et al. 2008; Stulić and Jantsch 2013). In addition, we showed for the first time that some sites, such as UNC80 S/G, mGluR4 Q/R, NOVA1 S/G, TMEM63B Q/R, and SPEG E/G, were ADAR2 sites (Supplemental Fig. S3A–D). ADAR2 also contributed predominantly to editing at the SPEG S/G site, especially in the spleen (Supplemental Fig. S3E). In contrast, a certain number of sites in the CDS were ADAR1-dependent, such as three sites in BLCAP and NEIL1 K/R, in addition to AZIN1 E/G and S/G sites (Figs. 1C, 3A,B), as previously reported (Nishimoto et al. 2008; Riedmann et al. 2008; Yeo et al. 2010; Chen et al. 2013). Furthermore, we identified that UBE2O S/G and DACT3 R/G were edited mainly by ADAR1 (Fig. 3C,D). The CDK13 Q/R site was reported to be edited by ADAR2 (Terajima et al. 2016), whereas the current analysis demonstrated that ADAR1 was the main contributor to the editing of this site (Fig. 3E). Interestingly, the editing ratios for BLCAP Y/C, DACT3 R/G, and CDK13 Q/R sites in the cerebral cortex were higher than those in the spleen where ADAR1 p150 was highly expressed (Fig. 3A,D,E). This demonstrates that nuclear ADAR1 p110 is the main contributor to this editing, given that their ECS are usually located in the adjacent intron (Levanon et al. 2005). Of note, although the deletion of ADAR2 did not affect editing of these sites in the cerebral cortex, we observed a significant level of RNA editing in Adar1 KI mice, demonstrating that ADAR2 edits these sites only in the absence of ADAR1 activity. In contrast, although GluA2 R/G, GluK2 I/V, and GluK2 Q/R sites are reported to be edited by both ADARs in vitro (Herb et al. 1996; Melcher et al. 1996; Nishikura 2016), we found that ADAR1 edits these sites only in the absence of ADAR2 (Supplemental Fig. S4A–C), indicating that ADAR2 is the main editor of these sites in vivo.
FIGURE 2.

Editing retention in Adar1 KI and Adar2 KO mice. (A–D) Values for the retention of editing in cerebral cortexes isolated from Adar1 knock-in (KI) (A) and Adar2 knockout (KO) (B) mice and those in spleens isolated from Adar1 KI (C) and Adar2 KO (D) mice are displayed. The mean editing ratios of wild-type (WT) mice are displayed on the vertical axis. The red squares, blue circles, green diamonds, and gray triangles represent editing sites in CDS, microRNAs (miRNAs), repetitive elements (REs), and introns, respectively. See Supplemental Charts to access an interactive version of these charts in which each editing site can be identified.
FIGURE 3.

Retention of RNA editing at known and novel ADAR1 sites in Adar1 KI and Adar2 KO mice. (A–E) Editing ratios for BLCAP tyrosine/cysteine (Y/C) (A), NEIL1 lysine/arginine (K/R) (B), UBE2O serine/glycine (S/G) (C), DACT3 arginine/glycine (R/G) (D), and CDK13 glutamine/arginine (Q/R) sites (E) in indicated tissues isolated from wild-type (WT), Adar1E861A/E861AIfih−/− mice (Adar1 knock-in [KI]), and Adar2−/−Gria2R/R (Adar2 knockout [KO]) mice are shown. Editing ratios are displayed as the mean ± SEM (n = 3 mice for each group; Student's t-test, [*] P < 0.05, [**] P < 0.01, [***] P < 0.001, n.s., not significant). The editing ratio of each mouse is also displayed as a circle on the right side of each column. Significant differences in editing ratios between the cerebral cortex and spleen in the same mutant mice are indicated by hashes (#) P < 0.05, (##) P < 0.01, (###) P < 0.001.
Regarding miRNA editing, we reconfirmed that sites in miR-423-5p, miR-376c-3p, and miR-151-3p were preferentially edited by ADAR1, whereas ADAR2 was the main enzyme responsible for sites in miR-27a-5p, miR-379-5p, and miR-99a-5p (Supplemental Fig. S5A–F; Supplemental Table S1; Kawahara et al. 2007a,b, 2008b; Vesely et al. 2014; Nishikura 2016). In addition, for the first time, this analysis identified ADAR2 as the enzyme responsible for the editing of miR-27b-5p (Supplemental Fig. S5G). It is noteworthy that most sites in the 3p-strand were edited by ADAR1, whereas ADAR2 was responsible for editing most of the sites in the 5p-strand, highlighting differences in accessibility between the two enzymes.
Several sites, especially in miRNAs, show no preference for a specific ADAR
To understand the degree of contribution of each ADAR on each editing site, we calculated the ADAR preference, in which a 0% preference indicated no contribution of either ADAR1 or ADAR2, while a 100% preference indicated only a single ADAR contribution to the editing of a certain site (Materials and Methods). When the ADAR preference was <50% for an ADAR, we termed it as a nonpreferred ADAR thereafter. This analysis demonstrated that most sites had a clear preference for either ADAR1 or ADAR2, although we found unique ADAR-preference patterns in some sites (Fig. 4A,B; Supplemental Charts). For instance, ADAR1 edited the GABRA3 I/M site only in the absence of ADAR2 activity in the cerebral cortex (Fig. 4A,C). However, both ADAR1 and ADAR2 contributed to this editing to a similar extent in the spleen (Fig. 4B,C). The GluA3 R/G site showed the opposite editing pattern, in which ADAR1 and ADAR2 similarly contributed to its editing in the cerebral cortex, whereas ADAR2 was the preferred enzyme in the spleen (Fig. 4A,B; Supplemental Fig. S4D). Such tissue-specific regulation may not only be due to the difference in dosage of ADARs and target mRNAs in each tissue, but also due to other regulatory factors, such as ADAR3 expression in the brain (Chen et al. 2000), post-transcriptional modification of ADARs (Desterro et al. 2005; Bavelloni et al. 2019), splicing efficiency (Licht et al. 2016) and protein–protein interactions (Rajendren et al. 2018).
FIGURE 4.

Contribution of each ADAR to each editing site. (A–B) ADAR preference in the cerebral cortex (A) and spleen (B). We compared values of the editing retention for each site between Adar1E861A/E861AIfih−/− mice (Adar1 knock-in [KI]) and Adar2−/−Gria2R/R (Adar2) knockout (KO) mice and defined the value of the editing retention in Adar1 KI as “A” and that in Adar2 KO as “B.” We calculated ADAR1 and ADAR2 preferences using the following formula: 100 × B/(A + B) and 100 × A/(A + B), respectively. Editing ratios in wild-type (WT) mice are shown on the vertical axes. In this figure, 100% of the ADAR preference indicates the sole contribution of a single ADAR to RNA editing at a certain site, and 50% indicates an equal contribution of both ADARs. The red squares, blue circles, green diamonds, and gray triangles represent editing sites in coding sequences (CDS), microRNAs (miRNAs), repetitive elements (REs), and introns, respectively. See Supplemental Charts to access an interactive version of these charts in which each editing site can be identified. (C,D) Editing ratios for the GABRA3 isoleucine/methionine (I/M) site (C), and the site in miR-3099-3p (D) in indicated tissues isolated from WT, Adar1 KI, and Adar2 KO mice are shown. Editing ratios are displayed as the mean ± SEM (n = 3 mice for each group; Student's t-test, [*] P < 0.05, [***] P < 0.001, n.s., not significant). The editing ratio of each mouse is also displayed as a circle on the right side of each column. Significant differences in editing ratios between the cerebral cortex and spleen in the same mutant mice are indicated by hashes (##) P < 0.01.
Next, we investigated the similarity in ADAR preference between the cerebral cortex and spleen. This analysis demonstrated a high correlation for most sites in CDS (Supplemental Fig. S6A,B; Supplemental Charts). For instance, we observed high ADAR1 and ADAR2 dependency for the three BLCAP sites, and the SPEG S/G site, respectively, in both the cerebral cortex and spleen. In contrast, although both ADAR1 and ADAR2 participated in the editing of representative REs in the cerebral cortex, ADAR1 was the preferred enzyme in the spleen (Fig. 4A,B; Supplemental Fig. S6A,B). As a result, we observed a relatively low similarity between the cerebral cortex and spleen with regard to the ADAR responsible for several RE sites examined, especially sites in the 3′UTR of Cds2 and Jmjd4 genes (Supplemental Fig. S5C; Supplemental Charts). Interestingly, the +7 position of miR-3099-3p was ∼90% edited in the cerebral cortex, with the editing ratio preserved in both Adar1 KI and Adar2 KO mice (Fig. 4D). Although this site was previously reported as a possible ADAR1 site (Vesely et al. 2014), this is the first case in which a highly edited site shows no preference for a specific ADAR (Fig. 4A). A similar phenomenon was also observed for miR-411-5p and miR-99b-3p-1 (Fig. 4A; Supplemental Fig. S7A,B). This may be the reason why the ADAR responsible for editing miR-411-5p was found not to be identical in past studies using Adar1−/− embryos in which the editing ratio is relatively low (Kawahara et al. 2008b; Vesely et al. 2014; Nishikura 2016). Taken together, although further studies are necessary to understand the context in which both ADARs can edit a substrate to the same extent, miR-3099-3p and miR-411-5p may be used as control substrates to analyze the difference in properties between ADAR1 and ADAR2 in vivo and in vitro.
RNA editing is occasionally regulated by either competitive or coordinated interplay between ADAR1 and ADAR2
We observed that the editing ratio for some sites tended to be high in Adar mutant mice (Fig. 2A–D). This up-regulation was largely observed in sites that were edited by <40% in WT mice, regardless of the tissues examined, when the activity of the nonpreferred ADAR was lost. Of these sites, as previously reported (Kawahara et al. 2007b; Vesely et al. 2014), the editing ratios for the +4 position of miR-381-3p and the +6 position of miR-376b-3p, which were ADAR1-specific sites, were significantly up-regulated in the cerebral cortex of Adar2 KO mice (Fig. 5A,B). In addition, we found up-regulated editing for the DACT3 R/G site in the spleen and the +3 position of miR-99b-3p in the cerebral cortex of Adar2 KO mice for the first time (Figs. 3D, 5C). Interestingly, among multiple editing sites found in the mRNA of SON, only the S/S site (SON-2) was preferentially edited by ADAR1 and showed up-regulated editing in Adar2 KO mice, whereas no up-regulation was observed in other ADAR2 sites (Supplemental Fig. S8A–E). This antagonizing effect, which has been proposed for inactive ADAR3, may be attributed to an increased accessiblity of ADAR1 to the editing site upon loss of competition with nonpreferred ADAR2 (Chen et al. 2000; Kawahara et al. 2007b; Riedmann et al. 2008; Vesely et al. 2014; Tan et al. 2017). However, this is not the sole reason because we observed that editing at the +4 position of miR-376a-2-5p was significantly up-regulated in the cerebral cortex of Adar1 KI mice (Fig. 5D). In addition, this up-regulation was similarily observed in multiple tissues of both Adar1 KI and Adar2 KO mice (Tan et al. 2017). Furthermore, considering that the editing ratio for the +7 position of miR-3099-3p was not different among WT, Adar1 KI, and Adar2 KO mice (Fig. 4D), the presence of ADAR2 does not always inhibit access of ADAR1 to the editing site. These examples show that multiple factors, including the secondary structure surrounding the editing site, and the affinity and editing capacity of each ADAR, might affect negative regulation between ADAR1 and ADAR2, in addition to the relative dosage of each ADAR and the location of the editing site (miRNA and REs seem to be preferred targets). This requires further investigation.
FIGURE 5.

Sites with up-regulation of editing in either Adar1 KI or Adar2 KO mice. (A–D) Editing ratios for the +4 position of miR-381-3p (A), the +6 position of miR-376b-3p (B), the +3 position of miR-99b-3p (C), and the +4 position of miR-376a-2-5p (D) in indicated tissues isolated from wild-type (WT), Adar1E861A/E861AIfih−/− mice (Adar1 knock-in [KI]), and Adar2−/−Gria2R/R (Adar2 knockout [KO]) mice are shown. Editing ratios are displayed as the mean ± SEM (n = 3 mice for each group; Student's t-test, [*] P < 0.05, [**] P < 0.01, [***] P < 0.001). The editing ratio of each mouse is also displayed as a circle on the right side of each column.
In contrast to negative regulation, we found that some sites required coordinated interplay between ADAR1 and ADAR2 for efficient RNA editing. It has been reported that among the five editing sites of brain-specific 5-HT2CR encoded by the Htr2c gene, the A and B sites are mainly edited by ADAR1, while the E, C, and D sites are preferentially edited by ADAR2 (Burns et al. 1997; Liu et al. 1999; Nishikura 2016), as confirmed by this study (Fig. 6A). However, although editing at the B site almost disappeared in Adar1 KI mice, the deletion of ADAR2 also reduced the editing ratio significantly from 71% to 19% (Fig. 6A). This result demonstrates that coordinated interplay between ADAR1 and ADAR2, in which preceding editing by ADAR2 might alter the secondary structure, was required for efficient editing at this site. Indeed, the editing ratio of the B site in the rat brain was higher than that obtained by in vitro RNA editing assay with recombinant (r)ADAR1 (Liu et al. 1999). However, the B site editing was not increased by a simple combination of rADAR1 and rADAR2 (Chen et al. 2000). In addition, although subtle or nonsignificant, similar phenomena were also observed at the UBE2O S/G site in the cerebral cortex (ADAR1 site), the CACNA1D I/M site in the cerebral cortex, and the FLNB Q/R site in the spleen (ADAR2 sites), all of which do not have additional editing sites within the same dsRNA structure targeted by a different ADAR (Figs. 3C, 6B,C). Although the mechanism underlying coordinated interplay between ADAR1 and ADAR2 currently remains unknown, one possibility is that the nonpreferred ADAR has an additional editing site in the intronic ECS and that preceding editing of this site may alter the secondary structure, leading to efficient editing by the preferred ADAR. Intriguingly, preceding editing of the intronic F site of 5-HT2CR altered the editing pattern of some exonic sites (Flomen et al. 2004).
FIGURE 6.

Cases that require nonpreferred ADAR for efficient RNA editing. (A–C) Editing ratios of five sites in the serotonin 5-HT2CR (A), CACNA1D isoleucine/methionine (I/M) (B), and FLNB glutamine/arginine (Q/R) (C) sites in the indicated tissues isolated from wild-type (WT), Adar1E861A/E861AIfih−/− mice (Adar1 knock-in [KI]), and Adar2−/−Gria2R/R (Adar2 knockout [KO]) mice are shown. Editing ratios are displayed as the mean ± SEM (n = 3 mice for each group; Student's t-test, [*] P < 0.05, [**] P < 0.01, [***] P < 0.001, n.s., not significant). The editing ratio of each mouse is also displayed as a circle on the right side of each column.
We further analyzed the combination of editing within the same transcripts. As expected, the majority of Htr2c transcripts were edited only at the D site, with or without editing at the C site, in Adar1 KI mice (Fig. 7A). In contrast, editing at the A site, with or without editing at the B site, was detected in more than 60% of transcripts in Adar2 KO mice (Fig. 7A). Given that the two sites in CACNA1D are preferentially edited by ADAR2, no substantial alteration of the editing combination at the two sites was observed in Adar1 KI mice, whereas only unedited transcripts were observed in Adar2 KO mice (Fig. 7B). Conversely, no substantial alteration of the editing combination at the three sites in BLCAP was observed in the spleen of Adar2 KO mice, whereas more than 90% of transcripts were unedited in Adar1 KI mice (Fig. 7C). Intriguingly, ADAR2 edited either Y/C or Q/R sites, or both to some extent, but not the K/R site, which was the lowest edited site of BLCAP in the cerebral cortex of Adar1 KI mice, and therefore the combination pattern was different from that in the spleen. However, except for Htr2c transcripts, the multiple editing sites within the same dsRNA structure are usually targeted by one preferred ADAR. These results are in accordance with observations that the preferred dsRNA structure dictates substrate selectivity and that it differs between ADAR1 and ADAR2 (Lehmann and Bass 1999; Wang et al. 2018).
FIGURE 7.
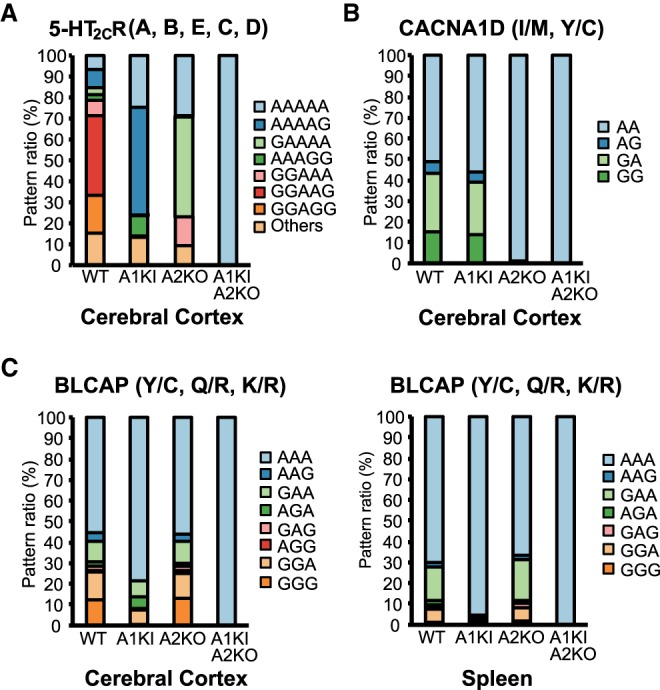
Comparison of the combined editing pattern in the same transcripts among Adar mutant mice. (A–C) The frequency of each combined editing pattern of the five sites (A, B, E, C, and D in order) in the serotonin 5-HT2CR (A), the two sites (isoleucine/methionine [I/M] and tyrosine/cysteine [Y/C] in order) in CACNA1D (B), and the three sites (Y/C, glutamine/arginine [Q/R] and lysine/arginine [K/R] in order) in BLCAP (C), in the indicated tissues isolated from wild-type (WT; n = 3 mice), Adar1E861A/E861AIfih−/− mice (Adar1 knock-in [KI]; n = 3 mice), Adar2−/−Gria2R/R (Adar2 knockout [KO]; n = 3 mice), and Adar1 KI Adar2 KO (n = 2 mice) mice is displayed as a percentage.
Adar1 KI Adar2 KO mice are viable with a complete absence of editing
Adar2 KO (Adar2−/−Gria2R/R) mice survive until adulthood (Higuchi et al. 1993). In addition, although Adar1E861A/E861A mice show embryonic lethality, Adar1 (Adar1E861A/E861A Ifih−/−) KI mice can survive with a normal life-span (Liddicoat et al. 2015; Heraud-Farlow et al. 2017). However, given that ADAR1-mediated RNA editing in CDS is independent of MDA5 activation and that the nonpreferred ADAR2 can edit many sites in CDS and miRNAs in the absence of preferred ADAR1 activity in compensation, we generated Adar1 KI Adar2 KO mice to examine whether RNA editing completely disappeared and to observe its phenotypic consequences. We found that Adar1 KI Adar2 KO mice were viable until adulthood and showed a significantly smaller body size compared to WT and Adar2 KO mice (Fig. 8A). However, this small body size was comparable to that of Adar1 KI mice, which is known to be small (Liddicoat et al. 2015; Heraud-Farlow et al. 2017). Mating of Adar1 KI Adar2 KO mice was difficult because of their small sizes; consequently, we successfully obtained pups from both male and female Adar1 KI Adar2 KO mice by in vitro fertilization, which indicated that Adar1 KI Adar2 KO mice were fertile. Therefore, although we could not exclude the possibility that additional abnormalities may have been detected by undertaking more detailed examinations, as reported in Adar2 KO mice that showed a myriad of subtle phenotypes (Horsch et al. 2011), a cumulative effect on critical phenotypes due to the inactivation of ADAR1 and the deletion of ADAR2 in mice was not apparent.
FIGURE 8.

Complete absence of RNA editing in Adar1 KI Adar2 KO mice. (A) The body weights of wild-type (WT; n = 4 mice), Adar1E861A/E861A Ifih−/− mice (Adar1 knock-in [KI]; n = 6 mice), Adar2−/−Gria2R/R (Adar2 knockout [KO]; n = 9 mice), and Adar1 KI Adar2 KO (n = 11 mice) mice at 10 d of age are displayed as the mean ± SEM (Student's t-test, [**] P < 0.01, n.s., not significant). (B) Editing ratios of all sites examined in the cerebral cortex were compared between WT and Adar1 KI Adar2 KO mice. Values are displayed as the mean of the values of three WT and two Adar1 KI Adar2 KO mice. The red squares, blue circles, green diamonds, and gray triangles represent editing sites in coding sequences (CDS), microRNAs (miRNAs), repetitive elements (REs), and introns, respectively.
Finally, using Adar1 KI Adar2 KO mice, we analyzed the editing ratios of all sites examined in Adar1 KI and Adar2 KO mice. This analysis demonstrated a complete loss of RNA editing in both the cerebral cortex and spleen of Adar1 KI Adar2 KO mice (Fig. 8B). Using total RNA-sequencing (RNA-seq) analysis, we further examined whether editing at certain sites was maintained in these mutant mice. However, although more than 3000 known editing sites were detected in WT mice, editing sites were not detected in Adar1 KI Adar2 KO mice except for the GluA2 Q/R site, which was knocked-in at the genomic DNA level (Supplemental Table S4). These results highlight that ADAR1 and ADAR2 are the enzymes responsible for all editing sites in vivo and that critical editing sites differ between ADAR1 and ADAR2.
In summary, we have provided comprehensive and quantitative data for RNA editing in all sites that are known to be conserved or may be conserved in the CDS and miRNAs of WT, Adar1 KI, Adar2 KO, and Adar1 KI Adar2 KO mice. This can act as an invaluable resource for furthering our understanding of how Adar preferentially contributes to the editing of each site. In addition, total RNA-seq data obtained from ADAR1 KI Adar2 KO mice can contribute to excluding false-positive sites. We have demonstrated that editing is regulated by a site-specific mechanism related to the interplay between ADAR1 and ADAR2, which sometimes cooperatively edited certain sites; however, in other sites the nonpreferred ADAR had an inhibitory role on editing by the preferred ADAR. These findings were obtained by comparing the editing ratios of each specific site between WT, Adar1 KI, and Adar2 KO mice. We sometimes found that the ADAR responsible for editing a specific site, such as the CDK13 Q/R site, was different to the one previously reported. This observation may be attributable to differences in materials (cultured cells, embryos, and tissues at different ages), in which substrates and ADARs have a unique expression signature, and/or the methodology (knockdown, knockout, and knock-in) used. In this regard, although it may be difficult with regard to ADAR1, which is believed to have critical functions other than RNA editing (Liddicoat et al. 2015; Pestal et al. 2015), comparing editing ratios between Adar2 KO and Adar2 KI mice will provide valuable information on how the absence or presence of one inactive ADAR differentially affects the editing of each site catalyzed by the other ADAR. Finally, given that RNA editing at each site is affected by many factors, including secondary structure, the presence of neighboring and opposing nucleotides, the tissue-specific relative dosage of each ADAR and regulators such as ADAR3 and aminoacyl tRNA synthetase complex-interacting multifunctional protein 2 (AIMP2) (Riedmann et al. 2008; Kuttan and Bass 2012; Tan et al. 2017; Wang et al. 2018), further studies are required to elucidate which factors determine the interplay between ADAR1 and ADAR2.
MATERIALS AND METHODS
Mouse administration
Mice were maintained on a 12-h/12-h light–dark cycle at a temperature of 23 ± 1.5°C with a humidity of 45 ± 15% as previously described (Nakahama et al. 2018). All experimental procedures that involved mice were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of Osaka University.
Mutant mice
Cryopreserved spermatozoa of Adar2 KO mice were obtained from the Mutant Mouse Resource and Research Center (RRID: MMRRC_034679-UNC) and in vitro fertilization was performed at the Institute of Experimental Animal Sciences Faculty of Medicine, Osaka University. Adar1E861A/+ mice that harbor a heterozygous editing-inactive E861A point mutation were generated by genome editing using the CRISPR/Cas9 system at The Genome Editing Research and Development Center, Graduate School of Medicine, Osaka University. Briefly, pronuclear-stage mouse embryos (CLEA Japan Inc.) were electroporated to introduce Cas9 mRNA, single guide RNA (sgRNA: TCCGGGAGATGATTTCGGCA) and single-stranded donor oligonucleotides (ssODN: ATGGCTGTGCCCATCTTGCTTACCTGATGAAGCCCCTCCGGGAGATGATTGCGGCATGGCAGTCATTGACCGTCTCTCCCTTCAGGCTGAGAGAGTCCCCTTT); these introduced a point mutation at the corresponding codon (underlined G in the target nucleotide). Mouse embryos that developed to the two-cell stage were transferred into the oviducts of female surrogates. Adar1E861A/E861A mice were then established by crossing with Adar1E861A/+ mice. We did not find a difference in phenotypes between the obtained Adar1E861A/E861A mice and Adar1E861A/E861A mice that were previously established by a conventional method (Liddicoat et al. 2015; Nakahama et al. 2018). Furthermore, Adar1 KI mice were obtained by crossing Adar1E861A/+ mice with Ifih−/− mice as reported previously (Nakahama et al. 2018). To establish Adar1 KI Adar2 KO mice, we generated Adar1E861A/+Ifih−/−Adar2−/−Gria2R/R mice. During this procedure, repeated backcrossing was required to induce homologous recombination, given that the Adar1 and Gria2 genes localize to the same chromosome. Finally, we performed in vitro fertilization using sperm and ova collected from Adar1E861A/+ Ifih−/−Adar2−/−Gria2R/R mice. Genotyping of the Adar1 gene was performed by direct Sanger sequencing of the PCR products amplified from the region, including the point mutation. All mice used in experiments were in a C57BL/6J background.
Western blot analysis
Tissue lysates from mouse cerebral cortex and spleen were prepared and stored at −80°C until use as described previously (Miyake et al. 2016). Lysates were then separated using sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE), transferred to a polyvinylidene difluoride (PVDF) membrane (Bio-Rad, Hercules) and immunoblotted with primary antibodies using a SNAP i.d 2.0 Protein Detection System (Merck Millipore) as previously described (Nakahama et al. 2018). The primary antibodies used were as follows: mouse monoclonal anti-ADAR1 antibody (15.8.6; Santa Cruz Biotechnology), mouse monoclonal anti-ADAR2 antibody (1.3.1; Santa Cruz Biotechnology), and mouse monoclonal anti-GAPDH (M171-3; MBL).
Total RNA preparation
Total RNA was extracted from the cerebral cortices and spleens collected from 8-wk-old male mice using TRIzol reagent (Thermo Fisher Scientific) in accordance with the manufacturer's protocol. The RNA concentration was measured using a NanoDrop One (Thermo Fisher Scientific) and stored at −80°C until use after adjustment to 1 µg/µL.
Selection of conserved RNA editing sites
To list the conserved editing sites in CDS and miRNAs, we initially selected A-to-I RNA editing sites that were conserved in humans and mice (Kawahara et al. 2008b; Chiang et al. 2010; Maas et al. 2011; Alon et al. 2012; Danecek et al. 2012; Ekdahl et al. 2012; Gu et al. 2012; Daniel et al. 2014; Pinto et al. 2014; Ramaswami and Li 2014; Vesely et al. 2014; Nishikura 2016; Terajima et al. 2016). Next, by Sanger sequencing, we examined RNA editing, in a preliminary manner, in the cerebral cortex and spleen for sites reported to be possibly edited in mice and that may be conserved in humans (HIST2H2AB L/L, HIST2H2AC N/S, ZNF397 N/D, miR-542-3p, miR-574-5p, and miR-708-3p) (Cattenoz et al. 2013; Vesely et al. 2014; Hosaka et al. 2019). We observed possible RNA editing only at HIST2H2AB L/L and HIST2H2AC N/S sites, which were therefore included in the list. We further examined RNA editing, in a preliminary manner, for sites reported to be present only in humans but that are possibly conserved in mice (AR T/A, RHOQ N/S, NCSTN S/G, TNRC18 E/G, XKR6 R/G, BEST1 I/V, GIPC1 T/A, and GIPC1 P/P sites, and miR-200b and miR-455) (Martinez et al. 2008; Han et al. 2014; Sakurai et al. 2014; Nishikura 2016; Wang et al. 2017). We subsequently detected possible RNA editing in only miR-200b and miR-455, which were included in the list. Finally, the NEIL1 K/K site was reported to be present in only human cancer cells (Anadón et al. 2015). However, this site is adjacent to a conserved K/R site, and therefore was included as a possible conserved site in the list. Consequently, we listed 69 sites in the CDS of 39 genes and 26 sites in 21 miRNAs as sites that were definitely or possibly conserved, and these were used for subsequent analysis (Supplemental Table S1). In addition, several representative REs in the intron and 3′UTR (Nakahama et al. 2018), and in intronic self-editing sites in the Adar2 gene (Rueter et al. 1999), were included as references.
Preparation of ion amplicon libraries for RNA editing sites
After 1 µg of total RNA from each tissue was treated with DNase I (Thermo Fisher Scientific) at 37°C for 20 min, cDNA was synthesized by reverse-transcription (RT) using a SuperScript III First-Strand Synthesis System (Thermo Fisher Scientific) according to the manufacturer's instructions (Fig. 1A). Random hexamers were used as RT primers for editing sites in introns and miRNAs, while oligo(dT) primers were used for sites in mRNAs to avoid possible contamination of pre-mRNA fragments. A first round of PCR using 30 cycles was performed using cDNA, Phusion Hot Start High-Fidelity DNA Polymerase (Thermo Fisher Scientific) and first primers that were editing-site specific (Fig. 1A; Supplemental Table S5). A second round of PCR using 20 to 25 cycles was then performed using an aliquot of the first PCR product as a template and second primers that were editing-site specific; an A adaptor (5′-CCATCTCATCCCTGCGTGTCTCCGACTCAG-3′), an Ion Xpress Barcode and a trP1 adaptor (5′-CCTCTCTATGGGCAGTCGGTGAT-3′) were in forward and reverse primers, respectively (Fig. 1A; Supplemental Table S5). All second PCR products were designed to be 190 to 200 bp in length. After gel purification, the concentration of each PCR product was measured using a NanoDrop One and then equal amounts of 50–300 PCR products were combined. After a quality check using a 2100 Bioanalyzer (Agilent Technologies) with a High Sensitivity DNA kit, the resultant amplicon library samples were subjected to deep sequencing using an Ion Torrent Personal Genome Machine (Ion PGM) system (Thermo Fisher Scientific) at the CoMIT Omics Center, Graduate School of Medicine, Osaka University.
Quantification of the RNA editing ratio with ion amplicon sequencing reads
An RNA editing ratio for each site was calculated with its read data generated by an Ion PGM. For each amplicon sequence, the prefix of length k, which we termed the k-prefix, was taken and compared with the k-prefix of a read generated by the sequencer. Note that the k-prefix of an amplicon sequence was derived from a specific primer sequence within the amplicon, meaning that the k-prefixes derived from all amplicon sequences were unique. Also, k was set to six in this study. If the k-prefix of the read was identical to that of the amplicon, it was investigated to determine whether a known editing site in the amplicon was edited. This was repeated until all sequence reads were scanned. Using this simple process, implemented with an in-house script, we calculated the editing ratio by dividing the number of edited reads by that of the total reads for each site. We set the minimum threshold for the number of total reads to 1000. Then, the mean editing ratio at each site was calculated using the editing ratios obtained from three WT, three Adar1 KI, three Adar2 KO, and two Adar1 KI Adar2 KO mice.
Calculation of editing retention
To determine how much editing was retained in tissues from Adar1 KI and Adar2 KO mice, the mean editing ratio of each mutant mouse was divided by that of WT mice to calculate the value for the retention of editing at each site. We only considered sites with a more than 5% editing ratio in WT mice for this analysis.
Calculation of ADAR preference
To quantify to what extent each ADAR is responsible for the editing of each site, we compared the value of the editing retention for each site in Adar1 KI and Adar2 KO mice. We defined the value of the editing retention in Adar1 KI as “A” and that in Adar2 KO as “B”. We then calculated ADAR1 and ADAR2 preferences using the following formulas: 100 × B/(A + B) and 100 × A/(A + B), respectively. In this calculation, 50% indicates an equal contribution by both ADARs. We only considered sites with more than a 5% editing ratio in WT mice for this analysis.
Similarity of ADAR preference between tissues
To express a similarity or difference in the ADAR preference between the cerebral cortex and spleen, we compared the value of the ADAR preference between these two tissues, and defined the small value as “C” and the large one as “D”. We calculated the similarity of the ADAR preference using the following formula: 100 − (D − C). In this calculation, 100% indicates the contribution of the same ADAR between the cerebral cortex and spleen to a certain editing site, while 0% indicates the contribution of different ADARs between the cerebral cortex and spleen. We only considered sites with more than a 5% editing ratio in WT mice for this analysis.
Quantification of RNA editing ratio by Sanger sequencing
The editing ratio was analyzed by Sanger sequencing as described previously with minor modifications (Miyake et al. 2016). In brief, 100 ng of each total RNA was incubated with 0.1 U/µL DNase I (Thermo Fisher Scientific) at 37°C for 20 min and then the denatured RNAs were reverse transcribed into cDNAs using a SuperScript III First-Strand Synthesis System (Thermo Fisher Scientific) with random hexamers. PCR was performed with Phusion Hot Start High-Fidelity DNA Polymerase (Thermo Fisher Scientific) and the following primers: Azin1-Fw1 (5′-GATGAGCCAGCCTTCGTGT-3′) and Azin1-Rv1 (5′-TGGTTCGTGGAAAGAATCTGC-3′) for mature mRNA, and Azin1-Fw2 (5′-TGAGACTTATGCCTGATCGTTG-3′) and Azin1-Rv2 (5′-CCAGCAAATCTAAACTGTCACTCA-3′) for pre-mRNA. After gel purification, each RT-PCR product was directly sequenced using the following primer: 5′-CAAGGAAGATGAGCCTCTGTTT-3′. The editing ratio was determined as the % ratio of the “G” peak over the sum of the “G” and “A” peaks of the sequencing chromatogram.
Total RNA-sequencing analysis
After ribosomal RNAs were removed from total RNA using a Ribo-Zero rRNA Removal Kit (Illumina, San Diego), a strand-specific RNA library was prepared using SureSelect Strand Specific RNA (Agilent) in accordance with the manufacturer's instructions as previously described (Nakahama et al. 2018). The library samples were then subjected to deep sequencing using an Illumina NovaSeq 6000 with 100-bp paired-end reads at Macrogen.
Genome-wide identification of editing sites
We adopted a genome-wide approach to identify editing sites with total RNA-seq reads as previously described (Nakahama et al. 2018) but with modifications. In brief, sequence reads were mapped onto a reference mouse genome (NCBIM37/mm9) with a spliced aligner HISAT2 (Kim et al. 2015). The mapped reads were then processed by adding read groups, and sorting and marking duplicates with the tools AddOrReplaceReadGroups and MarkDuplicates compiled in GATK4 (McKenna et al. 2010). GATK SplitNCigarReads, BaseRecalibrator, and ApplyBQSR were used to split “n” trim and reassign mapping qualities, which output analysis-ready reads for the subsequent variant calling. The GATK HaplotypeCaller was run for variant detection, in which the stand-call-conf option was set to 20.0 and the dont-use-soft-clipped-bases option was used. The results of variant calling were further filtered by GATK VariantFiltration using Fisher strand values (FS) > 30.0 and quality by depth values (QD) < 2.0 as recommended by the GATK developer for RNA-seq analysis. The remaining variants that were expected to be of high quality were annotated with ANNOVAR software (Wang et al. 2010). Among these variants, we picked up known editing sites registered in the rigorously annotated database of A-to-I RNA editing (RADAR) (Ramaswami and Li 2014). Finally, A-to-I editing ratios in each sample were calculated by dividing the allelic depth by the read depth for the editing sites shown in the annotated results.
Analysis of dsRNA structure
Potential secondary dsRNA structure was calculated using Mfold (Zuker 2003).
Statistical analyses
A two-tailed Student's t-test was used as indicated in each figure legend. All values are displayed as the mean ± standard error of the mean (SEM). Nonsignificance is displayed as n.s., while statistical significance is displayed as P < 0.05 (* or #), P < 0.01 (** or ##) or P < 0.001 (*** or ###).
DATA DEPOSITION
The RNA-seq data used in this study are available through the DNA Data Bank of Japan (DDBJ) under accession number DRA007927. The mfold web server is open source software for the prediction of nucleic acid folding and hybridization (http://unafold.rna.albany.edu/?q=mfold/RNA-Folding-Form).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank all the staff in the Genome Editing Research and Development Center, the Center for Medical Research and Education and the CoMIT Omics Center, Graduate School of Medicine, Osaka University, for technical support. Computations were partially performed on the National Institute of Genetics (NIG) supercomputer at the Research Organization of Information and Systems (ROIS) National Institute of Genetics, Japan. This work was supported by Grants-in-Aid KAKENHI (17K19352 and 19K22580 to Y. Kawahara; 18K15186 and 15K19126 to T.N.; 18K11526 and 15K00401 to Y. Kato) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan and by grants from SENSHIN Medical Research Foundation, The Mochida Memorial Foundation for Medical and Pharmaceutical Research (to Y. Kawahara), Nagao Memorial Fund (to T.N.), and the Takeda Science Foundation (to Y. Kawahara and T.N.). P.H.C.C. was supported by a MEXT scholarship.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.072728.119.
REFERENCES
- Alon S, Mor E, Vigneault F, Church GM, Locatelli F, Galeano F, Gallo A, Shomron N, Eisenberg E. 2012. Systematic identification of edited microRNAs in the human brain. Genome Res 22: 1533–1540. 10.1101/gr.131573.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anadón C, Guil S, Simó-Riudalbas L, Moutinho C, Setien F, Martínez-Cardús A, Moran S, Villanueva A, Calaf M, Vidal A, et al. 2015. Gene amplification-associated overexpression of the RNA editing enzyme ADAR1 enhances human lung tumorigenesis. Oncogene 35: 4407 10.1038/onc.2015.469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavelloni A, Focaccia E, Piazzi M, Raffini M, Cesarini V, Tomaselli S, Orsini A, Ratti S, Faenza I, Cocco L, et al. 2019. AKT-dependent phosphorylation of the adenosine deaminases ADAR-1 and -2 inhibits deaminase activity. FASEB J 33: 9044–9061. 10.1096/fj.201800490RR [DOI] [PubMed] [Google Scholar]
- Bazak L, Haviv A, Barak M, Jacob-Hirsch J, Deng P, Zhang R, Isaacs FJ, Rechavi G, Li JB, Eisenberg E, et al. 2014. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res 24: 365–376. 10.1101/gr.164749.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla T, Rosenthal JJC, Holmgren M, Reenan R. 2004. Control of human potassium channel inactivation by editing of a small mRNA hairpin. Nat Struct Mol Biol 11: 950–956. 10.1038/nsmb825 [DOI] [PubMed] [Google Scholar]
- Burns CM, Chu H, Rueter SM, Hutchinson LK, Canton H, Sanders-Bush E, Emeson RB. 1997. Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature 387: 303–308. 10.1038/387303a0 [DOI] [PubMed] [Google Scholar]
- Cattenoz PB, Taft RJ, Westhof E, Mattick JS. 2013. Transcriptome-wide identification of A>I RNA editing sites by inosine specific cleavage. RNA 19: 257–270. 10.1261/rna.036202.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CX, Cho DS, Wang Q, Lai F, Carter KC, Nishikura K. 2000. A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. RNA 6: 755–767. 10.1017/S1355838200000170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Li Y, Lin CH, Chan THM, Chow RKK, Song Y, Liu M, Yuan Y, Fu L, Kong KL, et al. 2013. Recoding RNA editing of AZIN1 predisposes to hepatocellular carcinoma. Nat Med 19: 209–216. 10.1038/nm.3043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang HR, Schoenfeld LW, Ruby JG, Auyeung VC, Spies N, Baek D, Johnston WK, Russ C, Luo S, Babiarz JE, et al. 2010. Mammalian microRNAs: experimental evaluation of novel and previously annotated genes. Genes Dev 24: 992–1009. 10.1101/gad.1884710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Nellåker C, McIntyre RE, Buendia-Buendia JE, Bumpstead S, Ponting CP, Flint J, Durbin R, Keane TM, Adams DJ. 2012. High levels of RNA-editing site conservation amongst 15 laboratory mouse strains. Genome Biol 13: r26 10.1186/gb-2012-13-4-r26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel C, Silberberg G, Behm M, Öhman M. 2014. Alu elements shape the primate transcriptome by cis-regulation of RNA editing. Genome Biol 15: R28 10.1186/gb-2014-15-2-r28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desterro JMP, Keegan LP, Jaffray E, Hay RT, O'Connell MA, Carmo-Fonseca M. 2005. SUMO-1 modification alters ADAR1 editing activity. Mol Biol Cell 16: 5115–5126. 10.1091/mbc.e05-06-0536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekdahl Y, Farahani HS, Behm M, Lagergren J, Öhman M. 2012. A-to-I editing of microRNAs in the mammalian brain increases during development. Genome Res 22: 1477–1487. 10.1101/gr.131912.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flomen R, Knight J, Sham P, Kerwin R, Makoff A. 2004. Evidence that RNA editing modulates splice site selection in the 5-HT2C receptor gene. Nucleic Acids Res 32: 2113–2122. 10.1093/nar/gkh536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George CX, Wagner MV, Samuel CE. 2005. Expression of interferon-inducible RNA adenosine deaminase ADAR1 during pathogen infection and mouse embryo development involves tissue-selective promoter utilization and alternative splicing. J Biol Chem 280: 15020–15028. 10.1074/jbc.M500476200 [DOI] [PubMed] [Google Scholar]
- Gerber AP, Keller W. 2001. RNA editing by base deamination: more enzymes, more targets, new mysteries. Trends Biochem Sci 26: 376–384. 10.1016/S0968-0004(01)01827-8 [DOI] [PubMed] [Google Scholar]
- Gu T, Buaas FW, Simons AK, Ackert-Bicknell CL, Braun RE, Hibbs MA. 2012. Canonical A-to-I and C-to-U RNA editing is enriched at 3′UTRs and microRNA target sites in multiple mouse tissues. PLoS One 7: e33720 10.1371/journal.pone.0033720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Kim H, Shin J, Jeong E, Lee W, Kim KY, Park SY, Lee D, Won J, Jeong S, et al. 2014. RNA editing in RHOQ promotes invasion potential in colorectal cancer. J Exp Med 211: 613–621. 10.1084/jem.20132209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartner JC, Schmittwolf C, Kispert A, Müller AM, Higuchi M, Seeburg PH. 2004. Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J Biol Chem 279: 4894–4902. 10.1074/jbc.M311347200 [DOI] [PubMed] [Google Scholar]
- Heraud-Farlow JE, Chalk AM, Linder SE, Li Q, Taylor S, White JM, Pang L, Liddicoat BJ, Gupte A, Li JB, et al. 2017. Protein recoding by ADAR1-mediated RNA editing is not essential for normal development and homeostasis. Genome Biol 18: 166 10.1186/s13059-017-1301-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herb A, Higuchi M, Sprengel R, Seeburg PH. 1996. Q/R site editing in kainate receptor GluR5 and GluR6 pre-mRNAs requires distant intronic sequences. Proc Natl Acad Sci 93: 1875 10.1073/pnas.93.5.1875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi M, Single FN, Köhler M, Sommer B, Sprengel R, Seeburg PH. 1993. RNA editing of AMPA receptor subunit GluR-B: a base-paired intron-exon structure determines position and efficiency. Cell 75: 1361–1370. 10.1016/0092-8674(93)90622-W [DOI] [PubMed] [Google Scholar]
- Higuchi M, Maas S, Single FN, Hartner J, Rozov A, Burnashev N, Feldmeyer D, Sprengel R, Seeburg PH. 2000. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 406: 78–81. 10.1038/35017558 [DOI] [PubMed] [Google Scholar]
- Horsch M, Seeburg PH, Adler T, Aguilar-Pimentel JA, Becker L, Calzada-Wack J, Garrett L, Götz A, Hans W, Higuchi M, et al. 2011. Requirement of the RNA-editing enzyme ADAR2 for normal physiology in mice. J Biol Chem 286: 18614–18622. 10.1074/jbc.M110.200881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosaka T, Yamashita T, Teramoto S, Hirose N, Tamaoka A, Kwak S. 2019. ADAR2-dependent A-to-I RNA editing in the extracellular linear and circular RNAs. Neurosci Res 147: 48–57. 10.1016/j.neures.2018.11.005 [DOI] [PubMed] [Google Scholar]
- Hundley HA, Bass BL. 2010. ADAR editing in double-stranded UTRs and other noncoding RNA sequences. Trends Biochem Sci 35: 377–383. 10.1016/j.tibs.2010.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntley MA, Lou M, Goldstein LD, Lawrence M, Dijkgraaf GJ, Kaminker JS, Gentleman R. 2016. Complex regulation of ADAR-mediated RNA-editing across tissues. BMC Genomics 17: 61 10.1186/s12864-015-2291-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M, Mann TD, Stulić M, Rao SP, Kirsch A, Pullirsch D, Strobl X, Rath C, Reissig L, Moreth K, et al. 2018. RNA editing of Filamin A pre-mRNA regulates vascular contraction and diastolic blood pressure. EMBO J 37: e94813 10.15252/embj.201694813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinnah H, Ulbricht RJ. 2019. Using mouse models to unlock the secrets of non-synonymous RNA editing. Methods 156: 40–45. 10.1016/j.ymeth.2018.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, Ito K, Sun H, Aizawa H, Kanazawa I, Kwak S. 2004. RNA editing and death of motor neurons. Nature 427: 801–801. 10.1038/427801a [DOI] [PubMed] [Google Scholar]
- Kawahara Y, Zinshteyn B, Chendrimada TP, Shiekhattar R, Nishikura K. 2007a. RNA editing of the microRNA-151 precursor blocks cleavage by the Dicer–TRBP complex. EMBO Rep 8: 763–769. 10.1038/sj.embor.7401011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, Zinshteyn B, Sethupathy P, Iizasa H, Hatzigeorgiou AG, Nishikura K. 2007b. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science 315: 1137–1140. 10.1126/science.1138050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, Grimberg A, Teegarden S, Mombereau C, Liu S, Bale TL, Blendy JA, Nishikura K. 2008a. Dysregulated editing of serotonin 2C receptor mRNAs results in energy dissipation and loss of fat mass. J Neurosci 28: 12834–12844. 10.1523/JNEUROSCI.3896-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, Megraw M, Kreider E, Iizasa H, Valente L, Hatzigeorgiou AG, Nishikura K. 2008b. Frequency and fate of microRNA editing in human brain. Nucleic Acids Res 36: 5270–5280. 10.1093/nar/gkn479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL. 2015. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12: 357–360. 10.1038/nmeth.3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuttan A, Bass BL. 2012. Mechanistic insights into editing-site specificity of ADARs. Proc Natl Acad Sci 109: E3295–E3304. 10.1073/pnas.1212548109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann KA, Bass BL. 1999. The importance of internal loops within RNA substrates of ADAR1. J Mol Biol 291: 1–13. 10.1006/jmbi.1999.2914 [DOI] [PubMed] [Google Scholar]
- Levanon EY, Hallegger M, Kinar Y, Shemesh R, Djinovic-Carugo K, Rechavi G, Jantsch MF, Eisenberg E. 2005. Evolutionarily conserved human targets of adenosine to inosine RNA editing. Nucleic Acids Res 33: 1162–1168. 10.1093/nar/gki239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JB, Church GM. 2013. Deciphering the functions and regulation of brain-enriched A-to-I RNA editing. Nat Neurosci 16: 1518–1522. 10.1038/nn.3539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JB, Levanon EY, Yoon J, Aach J, Xie B, LeProust E, Zhang K, Gao Y, Church GM. 2009. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science 324: 1210–1213. 10.1126/science.1170995 [DOI] [PubMed] [Google Scholar]
- Licht K, Kapoor U, Mayrhofer E, Jantsch MF. 2016. Adenosine to inosine editing frequency controlled by splicing efficiency. Nucleic Acids Res 44: 6398–6408. 10.1093/nar/gkw325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, Walkley CR. 2015. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349: 1115–1120. 10.1126/science.aac7049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Emeson RB, Samuel CE. 1999. Serotonin-2C receptor pre-mRNA editing in rat brain and in vitro by splice site variants of the interferon-inducible double-stranded RNA-specific adenosine deaminase ADAR1. J Biol Chem 274: 18351–18358. 10.1074/jbc.274.26.18351 [DOI] [PubMed] [Google Scholar]
- Maas S, Godfried Sie CP, Stoev I, Dupuis DE, Latona J, Porman AM, Evans B, Rekawek P, Kluempers V, Mutter M, et al. 2011. Genome-wide evaluation and discovery of vertebrate A-to-I RNA editing sites. Biochem Biophys Res Commun 412: 407–412. 10.1016/j.bbrc.2011.07.075 [DOI] [PubMed] [Google Scholar]
- Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, Nellåker C, Vesely C, Ponting CP, McLaughlin PJ, et al. 2014. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep 9: 1482–1494. 10.1016/j.celrep.2014.10.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez HD, Jasavala RJ, Hinkson I, Fitzgerald LD, Trimmer JS, Kung H, Wright ME. 2008. RNA editing of androgen receptor gene transcripts in prostate cancer cells. J Biol Chem 283: 29938–29949. 10.1074/jbc.M800534200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melcher T, Maas S, Herb A, Sprengel R, Seeburg PH, Higuchi M. 1996. A mammalian RNA editing enzyme. Nature 379: 460–464. 10.1038/379460a0 [DOI] [PubMed] [Google Scholar]
- Miyake K, Ohta T, Nakayama H, Doe N, Terao Y, Oiki E, Nagatomo I, Yamashita Y, Abe T, Nishikura K, et al. 2016. CAPS1 RNA editing promotes dense core vesicle exocytosis. Cell Rep 17: 2004–2014. 10.1016/j.celrep.2016.10.073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mombereau C, Kawahara Y, Gundersen BB, Nishikura K, Blendy JA. 2010. Functional relevance of serotonin 2C receptor mRNA editing in antidepressant- and anxiety-like behaviors. Neuropharmacology 59: 468–473. 10.1016/j.neuropharm.2010.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahama T, Kato Y, Kim JI, Vongpipatana T, Suzuki Y, Walkley CR, Kawahara Y. 2018. ADAR1-mediated RNA editing is required for thymic self-tolerance and inhibition of autoimmunity. EMBO Rep 19: e46303 10.15252/embr.201846303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neeman Y, Dahary D, Levanon EY, Sorek R, Eisenberg E. 2005. Is there any sense in antisense editing? Trend Genet 21: 544–547. 10.1016/j.tig.2005.08.005 [DOI] [PubMed] [Google Scholar]
- Nishikura K. 2016. A-to-I editing of coding and non-coding RNAs by ADARs. Nat Rev Mol Cell Biol 17: 83–96. 10.1038/nrm.2015.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimoto Y, Yamashita T, Hideyama T, Tsuji S, Suzuki N, Kwak S. 2008. Determination of editors at the novel A-to-I editing positions. Neurosci Res 61: 201–206. 10.1016/j.neures.2008.02.009 [DOI] [PubMed] [Google Scholar]
- Peng X, Xu X, Wang Y, Hawke DH, Yu S, Han L, Zhou Z, Mojumdar K, Jeong KJ, Labrie M, et al. 2018. A-to-I RNA editing contributes to proteomic diversity in cancer. Cancer Cell 33: 817–828.e817. 10.1016/j.ccell.2018.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, Stetson DB. 2015. Isoforms of RNA-editing enzyme ADAR1 independently control nucleic acid sensor MDA5-driven autoimmunity and multi-organ development. Immunity 43: 933–944. 10.1016/j.immuni.2015.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picardi E, Manzari C, Mastropasqua F, Aiello I, D'Erchia AM, Pesole G. 2015. Profiling RNA editing in human tissues: towards the inosinome Atlas. Sci Rep 5: 14941 10.1038/srep14941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto Y, Cohen HY, Levanon EY. 2014. Mammalian conserved ADAR targets comprise only a small fragment of the human editosome. Genome Biol 15: R5 10.1186/gb-2014-15-1-r5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullirsch D, Jantsch MF. 2010. Proteome diversification by adenosine to inosine RNA editing. RNA Biol 7: 205–212. 10.4161/rna.7.2.11286 [DOI] [PubMed] [Google Scholar]
- Rajendren S, Manning AC, Al-Awadi H, Yamada K, Takagi Y, Hundley HA. 2018. A protein–protein interaction underlies the molecular basis for substrate recognition by an adenosine-to-inosine RNA-editing enzyme. Nucleic Acids Res 46: 9647–9659. 10.1093/nar/gky800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami G, Li JB. 2014. RADAR: a rigorously annotated database of A-to-I RNA editing. Nucleic Acids Res 42: D109–D113. 10.1093/nar/gkt996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami G, Zhang R, Piskol R, Keegan LP, Deng P, O'Connell MA, Li JB. 2013. Identifying RNA editing sites using RNA sequencing data alone. Nat Methods 10: 128–132. 10.1038/nmeth.2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedmann EM, Schopoff S, Hartner JC, Jantsch MF. 2008. Specificity of ADAR-mediated RNA editing in newly identified targets. RNA 14: 1110–1118. 10.1261/rna.923308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueter SM, Dawson TR, Emeson RB. 1999. Regulation of alternative splicing by RNA editing. Nature 399: 75–80. 10.1038/19992 [DOI] [PubMed] [Google Scholar]
- Rula EY, Lagrange AH, Jacobs MM, Hu N, Macdonald RL, Emeson RB. 2008. Developmental modulation of GABAA receptor function by RNA editing. J Neurosci 28: 6196–6201. 10.1523/JNEUROSCI.0443-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai M, Ueda H, Yano T, Okada S, Terajima H, Mitsuyama T, Toyoda A, Fujiyama A, Kawabata H, Suzuki T. 2014. A biochemical landscape of A-to-I RNA editing in the human brain transcriptome. Genome Res 24: 522–534. 10.1101/gr.162537.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savva YA, Rieder LE, Reenan RA. 2012. The ADAR protein family. Genome Biol 13: 252 10.1186/gb-2012-13-12-252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin W, Nishikura K. 2013. Adenosine-to-inosine RNA editing and human disease. Genome Med 5: 105 10.1186/gm508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stulić M, Jantsch MF. 2013. Spatio-temporal profiling of Filamin A RNA-editing reveals ADAR preferences and high editing levels outside neuronal tissues. RNA Biol 10: 1611–1617. 10.4161/rna.26216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan MH, Li Q, Shanmugam R, Piskol R, Kohler J, Young AN, Liu KI, Zhang R, Ramaswami G, Ariyoshi K, et al. 2017. Dynamic landscape and regulation of RNA editing in mammals. Nature 550: 249–254. 10.1038/nature24041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terajima H, Yoshitane H, Ozaki H, Suzuki Y, Shimba S, Kuroda S, Iwasaki W, Fukada Y. 2016. ADARB1 catalyzes circadian A-to-I editing and regulates RNA rhythm. Nat Genet 49: 146–151. 10.1038/ng.3731 [DOI] [PubMed] [Google Scholar]
- Vesely C, Tauber S, Sedlazeck FJ, von Haeseler A, Jantsch MF. 2012. Adenosine deaminases that act on RNA induce reproducible changes in abundance and sequence of embryonic miRNAs. Genome Res 22: 1468–1476. 10.1101/gr.133025.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vesely C, Tauber S, Sedlazeck FJ, Tajaddod M, Av H, Jantsch MF. 2014. ADAR2 induces reproducible changes in sequence and abundance of mature microRNAs in the mouse brain. Nucleic Acids Res 42: 12155–12168. 10.1093/nar/gku844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlstedt H, Daniel C, Ensterö M, Öhman M. 2009. Large-scale mRNA sequencing determines global regulation of RNA editing during brain development. Genome Res 19: 978–986. 10.1101/gr.089409.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Miyakoda M, Yang W, Khillan J, Stachura DL, Weiss MJ, Nishikura K. 2004. Stress-induced apoptosis associated with null mutation of ADAR1 RNA editing deaminase gene. J Biol Chem 279: 4952–4961. 10.1074/jbc.M310162200 [DOI] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. 2010. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38: e164 10.1093/nar/gkq603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Xu X, Yu S, Jeong KJ, Zhou Z, Han L, Tsang YH, Li J, Chen H, Mangala LS, et al. 2017. Systematic characterization of A-to-I RNA editing hotspots in microRNAs across human cancers. Genome Res 27: 1112–1125. 10.1101/gr.219741.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Park S, Beal PA. 2018. Selective recognition of RNA substrates by ADAR deaminase domains. Biochemistry 57: 1640–1651. 10.1021/acs.biochem.7b01100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Chendrimada TP, Wang Q, Higuchi M, Seeburg PH, Shiekhattar R, Nishikura K. 2005. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat Struct Mol Biol 13: 13 10.1038/nsmb1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo J, Goodman RA, Schirle NT, David SS, Beal PA. 2010. RNA editing changes the lesion specificity for the DNA repair enzyme NEIL1. Proc Natl Acad Sci 107: 20715–20719. 10.1073/pnas.1009231107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Li X, Ramaswami G, Smith KS, Turecki G, Montgomery SB, Li JB. 2013. Quantifying RNA allelic ratios by microfluidic multiplex PCR and sequencing. Nat Methods 11: 51–54. 10.1038/nmeth.2736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31: 3406–3415. 10.1093/nar/gkg595 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.