

Abstract
Neutrophil elastase (NE) is a serine protease stored in the azurophilic granules of neutrophils and released into the extracellular milieu during inflammatory response or formation of neutrophil extracellular traps (NETs). Neutrophils release NETs to entrap pathogens by externalizing their cellular contents in a DNA framework decorated with anti-microbials and proteases, including NE. Importantly, excess NETs in tissues are implicated in numerous pathologies, including sepsis, rheumatoid arthritis, vasculitis, and cancer. However, it remains unknown how to effectively prevent NET formation. Here, we show that NE plays a major role during NET formation and that inhibition of NE is a promising approach for decreasing NET-mediated tissue injury. NE promoted NET formation by human neutrophils, and sivelestat, a small molecule inhibitor of NE, inhibited the formation of NETs in vitro. However, in a murine model of lipopolysaccharide-induced endotoxic shock, administration of free sivelestat did not exhibit any efficacy. To improve the efficacy of sivelestat in vivo, we have developed a nanoparticle system for delivering sivelestat. We demonstrate that nanoparticle-mediated delivery of sivelestat effectively inhibited NET formation, decreased the clinical signs of lung injury, reduced NE and other proinflammatory cytokines in serum, and rescued animals against endotoxic shock. Collectively, our data demonstrates that NE signaling can initiate NET formation and that nanoparticle-mediated inhibition of NE improves drug efficacy in prevention of NETs formation.
Keywords: neutrophil extracellular trap, nanoparticle, neutrophil elastase, sepsis
Sepsis is defined as a systemic inflammatory response due to infection, and its spectrum of diseases (severe sepsis and septic shock) affects around 1.5 million Americans every year [1]. In addition to mortality, the morbidity of sepsis is significant as many survivors of sepsis face severe limitations in performing their daily activities [2,3]. Moreover, sepsis places a considerable burden on the healthcare expenditure and is known as the most expensive condition treated in the U.S. hospitals costing $23 billion in 2013 [1,4]. Sepsis is driven by the propagation of hyper-inflammatory responses to infection [5–7], and currently, there is no specific treatment for sepsis. Clinical interventions include anti-inflammatory therapies, such as corticosteroids, administration of antibiotics, fluid resuscitation, and mechanical ventilation, and recent clinical trials for sepsis have all failed [8]. Anti-inflammatory therapies are utilized in sepsis because bacterial components are major drivers of the inflammatory response. For example, lipopolysaccharide (LPS), the endotoxin on the outer membrane of Gram-negative bacteria, is a potent activator of the acute inflammatory response [9,10]. Although LPS-induced endotoxic shock does not adequately mimic human sepsis [11], it is a very good model to study the pathophysiological features of the systemic inflammatory response that accompanies sepsis and could reveal novel therapeutic targets in this regard [12].
Neutrophils are the first responders to infection and play a critical role in host immune defense [13–15]. During bacterial infection, neutrophils can undergo programmed cell death, termed NETosis, by externalizing their cellular contents in a DNA framework decorated with antibacterial proteins and serine proteases [16]. These DNA-protein architectures extruded from neutrophils are called neutrophil extracellular traps (NETs), which mainly provide a physical fibrous network to entangle bacteria or other pathogens and enhance neutrophil antimicrobial activity [16–18]. However, NETs have been described as a double-edged sword [19] because excess NETs produced by overtly activated neutrophils have been implicated in promoting various human pathologies, including sepsis [20], rheumatoid arthritis [21], diabetes [22], vasculitis [23], and cancer [24]. Therefore, there have been recent efforts to prevent NET formation or eliminate excess NETs as potential therapeutic approaches [25–27].
Neutrophil elastase (NE) is a proteolytic enzyme stored in the azurophilic granules of neutrophils whose name is derived from its ability to degrade the extracellular matrix protein elastin. In addition to elastin, NE also degrades other cellular matrix proteins, including collagen and fibronectin [28,29]. The ability of elastase to destroy the extracellular matrix, particularly in the lungs, has been well known for over three decades, and deficiency of the endogenous NE inhibitor, α1-antitrypsin is associated with the development of lung emphysema [30]. NE is also known to induce cell proliferation and activate several cytokine and chemokine signaling pathways [31–34]. Endogenous proteinase inhibitors, such as α-1 antitrypsin, Elafin, and secretory leucocyte protease inhibitor (SLPI), are readily mobilized to counter NE activity. However, studies have shown that NE released from azurophil granules can bind tightly to the plasma membrane with its catalytic activity preserved, thereby shielding it from the activity of circulating endogenous inhibitors [35]. Additionally, it has been shown long before NETs were discovered that NE bound to DNA is insensitive to protease inhibitors [36,37]. Although the molecular mechanisms leading to NETs formation are not completely understood, recent studies have shown that reactive oxygen species (ROS) can initiate the translocation of NE from the granules to the nucleus where NE processes histones, leading to NET release into the extracellular space [38–40]. Hence, NE is pivotal in the process of NET formation, and inhibition of NE activity may provide a viable approach for preventing NETs formation. DNAse can degrade away the DNA backbone of NETs, but DNAse does not inhibit the activity of proteins decorated on NETs [41].
Several pharmaceutical companies have been investigating different NE inhibitors for use in the clinic [42]. However, there is currently no NE inhibitor approved by the FDA, indicating the challenge in demonstrating the clinical efficacy of NE inhibitors. For example, a highly selective and fully reversible oral inhibitor of NE produced by Astrazeneca (AZD9668) failed to show improvement in a recently conducted phase II clinical trial of patients with chronic obstructive pulmonary disease (COPD) – a disease in which NE has been implicated [43]. Sivelestat is a second-generation NE inhibitor discovered by ONO pharmaceuticals in Japan and is clinically available in Japan and South Korea for patients with acute lung injury (ALI) associated with systemic inflammatory response. A multi-national clinical trial of sivelestat on patients with ALI was deemed unsuccessful, and efforts to expand the use of sivelestat to other countries have failed [44]. Thus, there is a need for a new approach to improve the efficacy of sivelestat.
Nanoparticles have emerged as a veritable approach for the delivery of therapeutics to cells with the goal of achieving specificity and reducing systemic toxicity [45]. Since NE is stored in the granules of neutrophils, we hypothesized that nanoparticle-mediated delivery of NE inhibitor to neutrophils would improve its therapeutic efficacy. We previously described Interbilayer-Crosslinked Multilamellar Vesicles (ICMVs) as a new class of lipid-based nanoparticles with attractive features for targeted drug delivery [46]. Here, we show that ICMVs loaded with sivelestat (ICMV-Sive) are readily taken up by neutrophils and effectively inhibit NET formation in vitro. We also demonstrate that ICMV-Sive inhibits NET formation and extend animal survival in an in vivo model of endotoxic shock. Taken together, our data highlights the significance of NE in NET formation and suggests that nanoparticle-mediated delivery of sivelestat is a promising strategy for preventing NET formation in the context of endotoxic shock.
Materials and Methods
Animal experiments
The animal protocol was approved by the Institutional Animal Care and Use Committee at the University of Michigan, Ann Arbor. Four to six-week old female BALB/c mice were purchased from The Jackson Laboratory. Mice were maintained at the University of Michigan Animal Care facility under specific pathogen-free conditions. Mice were allowed to acclimatize for one week before experiments were started. LPS Escherichia coli 0111:B4 (Sigma Aldrich) was administered by intraperitoneal route (20 mg/Kg). Mice were injected with 50 mg/Kg sivelestat (Cayman Chemical) 1 hr after LPS challenge in the form of free drug or ICMV loaded drug. In some animals, blank ICMV were injected as a control. Animals challenged with LPS were monitored periodically for clinical signs and were assigned scores to indicate disease severity. Mice were monitored for movement, body condition, and alertness. Disease severity was scored in a semi-quantitative fashion as previously described [47] as follows: 0, = no abnormal clinical sign; 1, = ruffled fur but lively; 2, = ruffled fur, moving slowly, hunched, and sick; 3, = ruffled fur, squinted eye, hardly moving, down and very sick; 4, = moribund; and 5, = dead. A clinical score of 4 was used as the humane endpoint.
For biodistribution studies, groups of mice were injected with LPS (20 mg/Kg, i.p). After one hour, animals were injected with DiR-labelled ICMV-Sive intraperitoneally. Mice were sacrificed after 12 hrs, and major organs, including heart, liver, spleen, lungs and kidney, were harvested and imaged using the IVIS optical imaging system. For biochemical analysis of animal serum, LPS-challenged mice injected with free sivelestat or ICMV-Sive were sacrificed after 12 hrs. Blood was collected by cardiac puncture, and the levels of aspartate aminotransferase (AST) and creatinine in animal serum were measured.
Neutrophil isolation
Blood was collected from the peripheral vein of healthy volunteer donors into heparin tubes, and neutrophils were obtained by first spinning heparinized blood on Ficoll-Paque (Amersham Pharmacia Biotech, Piscataway, NJ) then subjecting the red blood cell (RBC) layer to 1.5% dextran sedimentation, followed by hypotonic lysis as previously described [48]. Isolated neutrophils were washed with PBS before use. Murine bone marrow-derived neutrophils were isolated, as previously described [49]. Briefly, femur and tibia were obtained, freed of muscle tissue and flushed with supplemented Hanks’ balanced saline solution (HBSS; 1X HBSS, 0.5% fetal bovine serum (FBS) and 20 mM HEPES) using 10 ml syringe and 30-gauge needle. Cells were pipetted up and down to obtain a single-cell suspension and centrifuged at 300 g for 5 mins. RBCs were lysed as above, and cells were then centrifuged on a discontinuous gradient of 52%, 69%, and 78% Percoll (GE Healthcare) diluted in HBSS (100% Percoll = 9 parts Percoll and 1 part 10X HBSS) and centrifuged (1500 g, 30 mins, without brake). Neutrophils from the 69%/78% interface were collected, washed in PBS, and resuspended in complete RPMI medium for use.
NET production, quantification, and microscopy
NETs were generated and quantified as previously described [16,50,51]. Briefly, to generate NETs, isolated neutrophils were resuspended in RPMI 1640 medium supplemented with 3% fetal bovine serum (NET medium) and 2 × 106 neutrophils per well were seeded on 6 well plates. The cells were activated for 4 hr with 100 nM phorbol 12-myristate 13-acetate (PMA; Sigma-Aldrich) at 37 °C. The supernatant was carefully removed, and the smear on the bottom of the wells was collected by vigorous agitation with fresh 2 ml of media. Samples were centrifuged at 100 g, and the NET supernatant was collected. The DNA content of the NET was quantified using a Take3 Trio micro-volume plate (Biotek Instruments) according to the manufacturer’s instructions. To quantify NETs, 0.1 × 106 neutrophils were plated in 96 well plates. The cells were incubated in NET medium in the presence or absence of free sivelestat or ICMV-Sive at the indicated concentration. NET formation was induced with 100 nM PMA or 5 μM recombinant human NE for 4 hours at 37 °C. NET was quantified by measuring the fluorescence intensity of extracellular DNA released in culture using 1 μM Sytox Green. The release of NE was also determined using 0.5 mM of the fluorogenic elastase substrate (Z-Ala-Ala-Ala-Ala)2Rhodamine110 (Cayman Chemical). Fluorescence intensity was measured using a fluorescent plate reader (Synergy Neo, BioTek Instruments). For fluorescence microscopy analysis, neutrophils were plated 20,000 cells/well and cultured as above to induce NETs. Cells were fixed with 4% paraformaldehyde (PFA), stained with 1 μM Sytox Green for 15 mins, and images were captured using a Nikon TiU microscope with attached CCD camera. In some experiments, images were analyzed using a size-restricted circle finding algorithm based on MATLAB script developed in-house. The area of each object in a given field of view was calculated. NET formation was also visualized using confocal microscopy. Neutrophils were seeded on 18 mm coverslips coated with 0.001% poly-L-lysine (Sigma-Aldrich). Neutrophils were then incubated for 4 hrs at 37 °C as described above in the presence/absence of sivelestat (20 μM) or DNAse (20 units/ml) with 100 nM PMA. Cells were fixed with 4% PFA and blocked with 10% FBS. DNA was stained with Sytox Green, and immunohistochemistry was performed with anti-NE IgG conjugated to Alexa Fluor 647 (Santa Cruz Biotechnology). The coverslips were mounted onto slides using Prolong Diamond Anti-fade media (Fisher Scientific), and images were acquired using a Nikon A1 confocal microscope.
Treatment of endothelial cell with NETs and flow cytometry
Human umbilical vein endothelial cells (HUVECs) were obtained from Angio-Proteomie. Cells were cultured in T75 flasks until confluent. Thereafter, 0.1 × 106 cells were seeded unto 12 well plates until 90% confluent. The cells were washed with PBS and cultured with NET supernatant containing 20 μg/ml DNA in the presence or absence of sivelestat (20 μM) for 12 hrs. The cells were trypsinized, washed twice with fluorescence-activated cell sorting (FACS) buffer containing 1% bovine serum albumin (BSA). The cells were incubated with anti-CD16/32 blocking antibodies and stained with CD54 (ICAM-1) antibody conjugated to allophycocyanin (APC) on ice for 20 mins. Cells were washed and resuspended in FACS buffer containing 1 μg/ml of 4’,6-diamidino-2-phenylindole (DAPI) and analyzed by flow cytometry. Flow cytometry data was analyzed using Flowjo software (Tree Star Inc, Ashland, OR).
Synthesis and characterization of ICMVs
ICMVs loaded with sivelestat were synthesized as previously reported with slight modifications [46,52]. Briefly, 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[4-(p-maleimidophenyl)butyramide] sodium salt (MPB) (Avanti Polar Lipids) were mixed in 1:1 molar ratios and dried under vacuum to produce thin films. In some experiments, 0.2 molar percent of the lipophilic fluorophores 1,1’-Dioctadecyl-3,3,3’,3’-Tetramethylindodicarbocyanine, 4-Chlorobenzenesulfonate Salt (DiD, Fisher Scientific) or 1,1’-dioctadecyl-3,3,3’,3’-tetramethylindotricarbocyanine iodide (DiR, Fisher Scientific) was added. The dried lipid films were hydrated in the presence of sivelestat to facilitate drug encapsulation. To induce vesicle fusion and crosslinking, CaCl2 (40 mM) and dithiothreitol (DTT; 15 mM) were added. The resulting ICMVs were centrifuged at 18000 X g for 5 min at 4 °C to pellet the particles, and the supernatant containing unloaded sivelestat was removed. Finally, the ICMV pellets were washed with DNA grade water (Fisher Scientific) and resuspended in PBS. To determine the amount of drug loaded in ICMVs, the particles were dissolved with methanol, and the drug concentration was determined by High Performance Liquid Chromatography (HPLC). Particle diameter and zeta potential were measured by dynamic light scattering (DLS) using a Malvern ZetaSizer Nano ZSP.
Assessment of neutrophil particle uptake
Isolated neutrophils were activated with 10 ng/ml TNF-α for 30 mins and incubated with ICMVs labeled with DID in 96 well plates or 18 mm slides coated with 0.001% poly-L-lysine for 1 hr. After incubation, cells were fixed with 4% PFA and washed twice to remove free particles. For flow cytometry analysis, cells were incubated with anti-CD16/32 blocking antibodies and stained with Ly6G antibody conjugated to Phycoerythrin (PE) on ice for 20 mins. For analysis by confocal microscopy, cells were incubated with Hoechst 33342 after fixing and observed under a confocal microscope.
Histology studies
For histopathological analysis, the right lung lobes were fixed in 4% PFA and embedded in paraffin. Five-micron sections were placed onto glass slides and stained with hematoxylin and eosin (H&E) for microscopy analysis. To identify NET formation in lungs in vivo, 5 μm sections of paraffin-embedded mouse lungs were prepared and mounted on glass slides. After dewaxing, samples were permeabilized with 0.1% Triton X-100 for 10 min and blocked with PBS containing 1% BSA and 0.1% Tween-20. The sections were incubated with primary antibodies – anti-citrullinated-histone H3 (1:100; Abcam) and anti-NE (1:50; Abcam), followed by detection with Alexa Fluor 488 goat anti-rat (1:500; Abcam) and Alexa Fluor 568 goat anti-rabbit (1:500; Abcam) secondary antibodies for 1 hr at room temperature. Samples were also stained with DAPI for DNA detection.
Statistical analysis
All data were plotted and analyzed using GraphPad Prism software version 5.0 (La Jolla, CA). A Kaplan Meier survival curve plot was used for the survival data, and the P values were determined using Mantel-Cox test. Differences between data sets were analyzed by performing one-way ANOVA followed by Tukey’s multiple-comparisons test. Differences were considered significant if P ≤ 0.05.
RESULTS
Sivelestat inhibits NET formation
As NETs are implicated in numerous human pathologies, there is significant interest to inhibit NET formation. We investigated the ability of sivelestat to inhibit NET formation. Activation of human neutrophils with phorbol 12-myristate 13-acetate (PMA) resulted in the formation of NETs as measured by extracellular DNA content with the cell impermeable dye, Sytox green (Fig. 1A). As expected, addition of DNAse to PMA-activated neutrophils resulted in a decreased Sytox green signal (Fig. 1A). Importantly, human neutrophils treated with both PMA and sivelestat exhibited a significantly decreased Sytox green signal, compared with the PMA control group (P < 0.001, Fig. 1A), suggesting strong inhibition of NET formation by sivelestat. We also quantified the release of NE during NET formation with a fluorogenic elastase substrate. NET formation by PMA-activated neutrophils resulted in a high elastase signal (Fig. 1B), but sivelestat significantly decreased the elastase signal from PMA-activated neutrophils (P < 0.001, Fig. 1B). To confirm these results, we visualized human neutrophils undergoing NET formation with confocal microscopy. As expected, human neutrophils activated with PMA released extracellular DNA, which appeared as fibrous strands (Fig. 1C). Sivelestat treatment effectively decreased these extracellular NET structures (Fig. 1C, Fig. S1). These results are in line with previous studies showing prevention of NET formation in NE knockout mice [38,41].
Figure 1. Inhibition of neutrophil elastase prevents NET formation.

Neutrophils were purified from human peripheral blood and activated with PMA in 96 well plates in the presence or absence of sivelestat (10 μM) or DNAse (20 units/ml) for 4 hrs. The release of extracellular DNA or neutrophil elastase was quantified by the addition of 1 μM Sytox Green and 0.5 mM of the elastase substrate (Z-Ala-Ala-Ala-Ala)2Rhodamine110, respectively, and analyzed using a fluorescent plate reader (A, B). Cells were seeded on microscopic slides, and immunofluorescence stain of NET formation was analyzed by confocal microscopy as the colocalization of extracellular DNA and neutrophil elastase (C). White arrows depict NETs. The data presented are representative of 3 independent experiments with similar results. ****, P < 0.0001 analyzed by one-way analysis of variance (ANOVA), followed by Tukey’s multiple-comparisons test. Scale bar = 50 <m.
Different signaling pathways have been shown to induce NETs, including PMA, LPS, IL-8, and microbes [53]. Given the broad-spectrum activities of NE and its ability to activate various cellular signaling pathways [31], we examined whether NE could induce NET formation. We cultured human neutrophils in the presence of recombinant human NE and quantified NET release by measuring the extracellular DNA content using the Sytox green assay. Indeed, we found that elastase treatment triggered robust NET formation (Fig. 2A). Importantly, addition of sivelestat during elastase treatment effectively reduced NET formation (P < 0.001, Fig. 2A,B). To further investigate the role of NE in NET formation, we isolated neutrophils from the bone marrow of wild type (WT) and NE knockout (KO) mice and activated them to form NETs. Consistent with previous reports [41], NE KO mice demonstrated impaired ability to form NETs, compared to WT mice (Fig. 2C, D). Overall, these results suggest that NE released during NET formation can induce de novo NETs, thereby constituting a feed-forward loop for the propagation of NET formation.
Figure 2. Neutrophil elastase induces NETs.

Human neutrophils were cultured in the presence of 5 μM recombinant human elastase for 4 hrs in the presence or absence of sivelestat. The release of extracellular DNA was quantified by the addition of 1 μM Sytox Green and analyzed using a fluorescence plate reader (A). For fluorescence microscopy, cells were fixed with 4% PFA before the addition of Sytox Green (B). Murine bone marrow-derived neutrophils from WT or NE KO mice were cultured with PMA to induce NETs. After 4 hrs, cells were fixed with 4% PFA and visualized under microscopy (C). Area of each object in microscopic field of view was calculated using an algorithm developed in-house (D). The data presented are representative of 3 independent experiments with similar results. **, P < 0.01; ****, P < 0.0001 analyzed by one-way analysis of variance (ANOVA), followed by Tukey’s multiple-comparisons test. Scale bar = 60 μm.
Sivelestat reduces NET-associated cytotoxicity and inflammatory responses
Cytotoxicity of NETs to various cells, including endothelial and epithelial cells, is a major factor in NET-associated pathologies [54,55], We investigated whether sivelestat could reverse NET-induced endothelial damage. NETs were harvested from activated human neutrophils and cultured with a monolayer of human umbilical vein endothelial cells (HUVECs) in the presence or absence of sivelestat. As expected, NETs induced endothelial cell death (Fig. 3A,B). Interestingly, NET-induced endothelial damage was reversed by sivelestat treatment (P < 0.0001, Fig. 3A,B). Additionally, NETs increased the expression of neutrophil adhesion molecule – Intercellular Adhesion Molecule - 1 (ICAM-1) on HUVECs, which was reversed by treatment with sivelestat although this was not statistically significant (Fig. 3C). The upregulation of ICAM-1 by NETs suggests that NETs can propagate the inflammatory response of endothelial cells. Therefore, we investigated the ability of sivelestat to inhibit inflammatory responses in neutrophils activated by another NET inducer – LPS. We isolated neutrophils from murine bone marrow and activated them with LPS in the presence or absence of sivelestat. LPS induced the production of NE and other pro-inflammatory cytokines and chemokines, including G-CFS, KC, TNF-α, and IL-6 (Fig. 3D–H). Importantly, sivelestat treatment significantly inhibited the production of NE, and inflammatory cytokines and chemokines from LPS-treated neutrophils (P < 0.0001, Fig. 3D–H). Taken together, our data show sivelestat prevents NET formation and NET-associated inflammatory responses.
Figure 3. Sivelestat inhibits NETs-induced endothelial damage and proinflammatory cytokine production.

Human umbilical vein endothelial cells (HUVECs) were cultured with NET supernatant derived from human neutrophils containing 20 μg/ml DNA in the presence or absence of sivelestat for 12 hrs. Cell viability (A, B) and the upregulation of endothelial adhesion molecule ICAM-1 (C) were analyzed by flow cytometry. Murine bone marrow-derived neutrophils were cultured with LPS 100 ng/ml in the presence or absence of sivelestat for 12 hrs. The levels of proinflammatory cytokines in the culture supernatant were analyzed by ELISA (D-H). The data presented are representative of 3 independent experiments with similar results. ****, p < 0.0001 analyzed by one-way analysis of variance (ANOVA), followed by Tukey’s multiple-comparisons test.
ICMVs delivering sivelestat inhibits NETs in vivo
Although Sivelestat is clinically available in Japan and Korea for the prevention of acute lung injury in patients, a recently concluded clinical trial aimed to expand its use in North America and Europe was declared unsuccessful [44]. Since sivelestat has poor pharmacokinetics and requires continuous infusion in human and animal models [56,57], we reasoned that targeted delivery of sivelestat to neutrophils would improve its efficacy in prevention of ALI and NET formation. We sought to use ICMVs for delivery of sivelestat to neutrophils. ICMVs encapsulating sivelestat were synthesized as previously described [46,52]. Briefly, dried lipid films containing DOPC, the anionic maleimide-headgroup lipid MPB and sivelestat dissolved in methanol were hydrated to make simple liposomes. After Ca2+-mediated vesicle fusion, dithiothreitol (DTT) was added to the vesicle suspension to crosslink maleimide headgroups of juxtaposed membranes and form ICMVs (Fig. 4A). Sivelestat was incorporated into ICMVs with an encapsulation efficiency of 60 ± 5% as determined by high-performance liquid chromatography (HPLC) (Fig. 4B). ICMVs loaded with sivelestat (ICMV-Sive) exhibited a homogenous hydrodynamic size of 266 ± 12 nm and a polydispersity (PDI) of 0.20 ± 0.04, as measured by dynamic light scattering (Fig. 4C) with a zeta potential of −41.8 ± 7.1 mV. ICMV-Sive incubated at 37 °C in 10% FBS released ~65% of sivelestat within 12 hrs (Fig. 4D).
Figure 4. Synthesis and characterization of sivelestat loaded ICMVs.

ICMVs were synthesized following the scheme shown in A. The size of sivelestat-loaded ICMVs was determined by dynamic light scattering analysis (B). Quantification of sivelestat encapsulated in ICMVs was analyzed by HPLC (C). Kinetics of sivelestat release from ICMVs was studied in media containing 10% FBS, and the drug release was quantified using LC-MS (D). The data presented are representative of 3 independent experiments with similar results.
To examine the ability of neutrophils to internalize ICMVs, murine neutrophils were incubated with fluorescently tagged ICMVs for 1 hr. Samples were then centrifuged and the supernatant discarded to remove free particles. Neutrophils readily phagocytosed ICMVs with > 65% of neutrophils associating with ICMVs within 1 hr of incubation (Fig. 5A). Confocal microscopy confirmed that ICMVs were internalized into neutrophils (Fig. 5B). Based on the efficient internalization of ICMVs by neutrophils, we hypothesized that ICMV-Sive would show an increased efficacy to inhibit NET formation, compared with free sivelestat. Previous studies have shown that activation of neutrophils increases their internalization ability [58]. To increase the uptake of the nanoparticles by neutrophils, cells were pretreated with TNF-α for 30 mins before incubation with ICMV-Sive or free drug for only 10 mins. Samples were then washed twice and the supernatant discarded to allow for the sole presence of internalized particles. NET formation was then induced with PMA for 4 hrs. Pre-treatment with ICMV-Sive significantly reduced the release of extracellular DNA (P < 0.05) and NE (P < 0.001) from PMA-treated neutrophils, compared to the free sivelestat treatment (Fig. 5C,D).
Figure 5. Inhibition of NET formation by ICMV-sivelestat.

Activated murine neutrophils were incubated for 1 hr with ICMVs labeled with DID. Cells were fixed with 4% PFA, washed twice and particle uptake was analyzed by flow cytometry (A, B) or confocal microscopy following staining with Hoechst 33342 (C). Activated neutrophils were cultured for 10 mins with blank ICMV, ICMVs loaded with sivelestat or free sivelestat. Cells were washed twice, followed by activation with 100 nM PMA for 4 hrs. The release of extracellular DNA or neutrophil elastase was quantified by the addition of 1 μM Sytox Green and 0.5 mM of the elastase substrate (Z-Ala-Ala-Ala-Ala)2Rhodamine110, respectively, and analyzed using a fluorescence plate reader (D, E). The data presented are representative of 3 independent experiments with similar results. *, P < 0.05; **, P < 0.01; ****, P < 0.0001 analyzed by one-way analysis of variance (ANOVA), followed by Tukey’s multiple-comparisons test. Scale bar = 50 μm.
To demonstrate the efficacy of ICMV-Sive in vivo, we used an LPS model of endotoxic shock [12]. LPS was injected into mice intraperitoneally, and after 1 hr of LPS injection, animals were administered i.p. with free sivelestat, blank ICMV, or ICMV-Sive. Animals were monitored for clinical signs and survival. At sacrifice, peritoneal lavage, blood, and lungs were collected for analysis. Strikingly, ICMV-Sive showed greater efficacy in the reduction of clinical signs (P < 0.001, Fig. 6A) and improvement in survival of mice (P < 0.05, Fig. 6B), compared to the free sivelestat control group. To investigate the ability of ICMV-Sive to inhibit NET formation in vivo, we performed immunofluorescence staining of paraffin-embedded mouse lung sections. NETs were identified by the co-localization of extracellular DNA, with NE and citrullinated-histone H3 (Cit-H3). ICMV-Sive showed greater reduction in NETs formation, compared to the free drug (Fig. 6C). Concomitantly, ICMV-Sive reduced lung injury as evidenced by reduced infiltration of inflammatory cells to the lungs, hemorrhage, and interstitial edema (Fig. 7A). This was accompanied by greater reduction of NE and other proinflammatory cytokines in the serum of the animals (Fig. 7B–D). To investigate the biodistribution and biosafety of ICMV-Sive, mice were challenged with LPS and 1 hr later, fluorescently labelled ICMV-Sive was administered. After 12 hrs, biodistribution of ICMV-Sive in major organs was examined using an in vivo imaging system (IVIS). ICMV-Sive mostly accumulated in the liver and spleen (Fig. 7E, F). Serum analysis indicated that injection of free sivelestat or ICMV-Sive did not elevate serum levels of AST and creatinine, compared with PBS-treated mice, indicating no major toxicity or abnormal liver and kidney functions in treated animals (Fig. 7G, H). Interestingly, ICMV-Sive also led to decreased neutrophil infiltration in the peritoneum (Fig. S2). Overall, our data indicated that nanoparticle-mediated delivery of sivelestat effectively decreased NET formation and clinical signs of lung injury in a murine model of LPS-induced endotoxic shock.
Figure 6. ICMV-sivelestat rescues mice from LPS-induced mortality.

Groups of mice (n = 10) were injected with LPS (20 mg/Kg) or PBS. After 1 hr, mice were administered i.p. with blank ICMV, ICMVs loaded with sivelestat or free sivelestat and monitored for clinical signs and survival (A, B). In a separate experiment, immunofluorescence staining was performed on lung sections from mice (n = 5) and analyzed by confocal microscopy. Staining depicts DAPI (blue), NE (green) and Cit-H3 (red). Colocalization of all three markers indicate NET formation. The data presented are representative of 3 independent experiments. (A) ***, P < 0.001 analyzed by one-way analysis of variance (ANOVA), followed by Tukey’s multiple-comparisons test. (B) *, P < 0.05, analyzed by log rank (Mantel-Cox) test. Scale bar = 50 μm.
Figure 7. ICMV-sivelestat prevents lung damage and proinflammatory cytokine production.
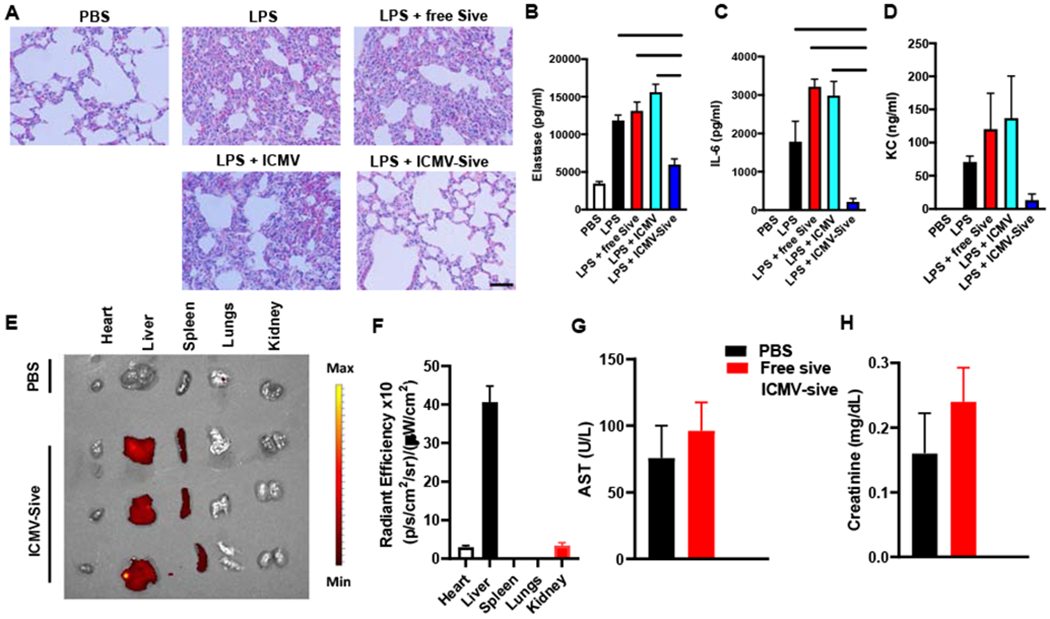
Groups of mice (n = 5) were injected with LPS (20 mg/Kg) or PBS. After 1 hr, mice were administered i.p. with blank ICMV, ICMV- sivelestat, or free sivelestat. Animals were sacrificed after 18 hr and H&E staining done on lung sections (A) (400x). The levels of NE, IL-6 and KC in animal serum were analyzed by ELISA (B-D). LPS-challenged mice were injected with ICMV-Sive labelled with DiR. Animals were sacrificed after 12 hr, and the level of fluorescence in the major organs, including heart, liver, spleen, lungs and kidney, were analyzed using IVIS imaging system (E, F). The levels of AST and creatinine in animal serum were also measured (G, H). The data presented are representative of 3 independent experiments. *, P <0.05; **, P < 0.01; ***, P < 0.001 analyzed by one-way analysis of variance (ANOVA), followed by Tukey’s multiple-comparisons test. Scale bar = 5000 μm.
Discussion
The formation of NETs by neutrophils was discovered as a novel mechanism of inhibiting microbial function [16]. However, excess NET formation has been implicated in the pathologies of several diseases from inflammation to cancer. Hence, there is significant interest to inhibit NET formation in order to limit bystander tissue injury. DNAse I was one of the first inhibitors of NETs formation that was described and is indeed clinically available for the treatment of cystic fibrosis, a disease in which lung damage is mediated by NET formation [59,60]. It is now known that DNAse does not inhibit the functions of NET-associated proteases and NETs treated with DNAse can still induce tissue injury [41,50]. Thus, it remains unknown how to effectively target and inhibit NETs.
Before NETs were discovered, Tkalcevic et al, reported that NE knockout mice are resistant to LPS-induced endotoxic shock [61]. Our findings are in line with the work of Tkalcevic et al and further highlights NETs formation as a major contributor to mortality in endotoxic shock. Consistent with the results of this study, Nakamura et al showed that mice deficient in SLP1, the endogenous inhibitor of neutrophil elastase are more susceptible to LPS-induced endotoxic shock [62]. Furthermore, Kolaczkowska et al, showed that NE knockout mice do not form NETs in a sepsis model [41]. Overall, the overwhelming evidence indicates that genetic and pharmacological inhibition of NE prevents NET formation.
NET formation has been shown to be triggered by different stimuli, including microbes, PMA, LPS, and cytokines. Indeed, the degree and kinetics of NET formation are dependent on the originating stimulus and the signaling pathway initiated [53,63]. Our data showed that NE induces NET formation in human neutrophils. Hence, NET formation induced by one stimulus can lead to the production of NE, which in turn promotes de novo NET formation and exacerbates tissue damage. This is in line with recent reports that NE signaling constitutes a feed-forward loop that drives NET formation [64]. It is speculated that NE contributes to the NET formation by activating the membrane pore-forming protein gasdermin D (GSDMD) [64]. Activated GSDMD perforates the granule membrane, increasing the release of NE and enabling the translocation of NE to the nucleus, where it processes histones and allows nuclear expansion [40,64].
Based on these findings, here we have sought an alternative strategy of inhibiting NET formation by targeting NE. We have shown that inhibition of NE signaling hinders the NET formation, reduces NET-mediated vascular damage, and alleviates the production of inflammatory cytokines. Since NE inhibition prevents the release of extracellular DNA and inhibits the function of NE, which is a major protease on NETs, we argue that NE inhibition is a promising approach for reducing NET-mediated tissue injury. Indeed, the role of NE in disease pathology has been well-documented [65,66]. For example, P aeruginosa, the most common pathogen in the lung of cystic fibrosis patients, has been shown to propagate tissue destruction by release of elastase [67,68]. In addition, sepsis is associated with higher levels of NE in serum [69,70]. However, despite concerted efforts for the development of NE inhibitors, none has been successful in clinical trials. Notable challenges in the clinical utility of NE inhibitors include the fact that NE bound to DNA in NETs is resistant to inhibitor activity [36]. Additionally, like sivelestat, most NE inhibitors function extracellularly and inhibit the action of NE released into the extracellular space.
To address the poor efficacy of NE inhibitors, we utilized ICMVs for neutrophil-targeted delivery of sivelestat, a potent NE inhibitor. We have previously shown that ICMVs have attractive features for drug delivery; compared to other lipid delivery systems such as liposomes, ICMVs exhibit greater encapsulation efficiency and greater retention of drug cargo in serum conditions [46]. In this study, we show that ICMV-mediated delivery of sivelestat promoted drug uptake by neutrophils and significantly improved the efficacy of sivelestat to inhibit NET formation, compared with free drug. In addition, we have shown that nanoparticle-mediated delivery of a NE inhibitor effectively rescued mice from LPS-induced endotoxic shock. Thus, our data suggest that nanoparticle-mediated delivery of NE is a viable strategy to inhibit NET formation and may significantly improve the efficacy of NE inhibitors.
In conclusion, we have demonstrated that inhibition of NE prevented NET formation and rescued animals from LPS-induced endotoxic shock. Further research is warranted to explore the therapeutic potential of NE inhibitors for not only sepsis but also other diseases, such as cancer, rheumatoid arthritis, and systemic lupus erythematosus, where NETs are known to play crucial pathogenic roles.
Supplementary Material
Acknowledgment
This work was supported in part by NIH (R01AI127070, R01EB022563, R01CA210273, R01CA223804, U01CA210152), MTRAC for Life Sciences Hub, UM Forbes Institute for Cancer Discovery Pilot Grant, and Emerald Foundation. E.B.O was supported by NSERC Postdoctoral Fellowship and CIHR Postdoctoral Fellowship. J.J.M. is a Young Investigator supported by the Melanoma Research Alliance (348774), DoD/CDMRP Peer Reviewed Cancer Research Program (W81XWH-16-1-0369), and NSF CAREER Award (1553831). Opinions interpretations, conclusions, and recommendations are those of the author and are not necessarily endorsed by the Department of Defense.
Data availability
The data supporting the findings of this study are available within the article and its Supplementary Information files. All relevant data can be provided by the authors upon reasonable request.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare no competing financial interest.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Hajj J, Blaine N, Salavaci J, Jacoby D, The "Centrality of Sepsis": A Review on Incidence, Mortality, and Cost of Care., Healthc. (Basel, Switzerland). 6 (2018). doi: 10.3390/healthcare6030090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Pandharipande PP, Girard TD, Jackson JC, Morandi A, Thompson JL, Pun BT, Brummel NE, Hughes CG, Vasilevskis EE, Shintani AK, Moons KG, Geevarghese SK, Canonico A, Hopkins RO, Bernard GR, Dittus RS, Ely EW, BRAIN-ICU Study Investigators, Long-term cognitive impairment after critical illness., N. Engl. J. Med 369 (2013) 1306–16. doi: 10.1056/NEJMoa1301372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Iwashyna TJ, Ely EW, Smith DM, Langa KM, Long-term cognitive impairment and functional disability among survivors of severe sepsis., JAMA. 304 (2010) 1787–94. doi: 10.1001/jama.2010.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Arefian H, Heublein S, Scherag A, Brunkhorst FM, Younis MZ, Moerer O, Fischer D, Hartmann M, Hospital-related cost of sepsis: A systematic review., J. Infect 74 (2017) 107–117. doi: 10.1016/j.jinf.2016.11.006. [DOI] [PubMed] [Google Scholar]
- [5].Okeke EB, Uzonna JE, In Search of a Cure for Sepsis: Taming the Monster in Critical Care Medicine., J. Innate Immun 8 (2016) 156–70. doi: 10.1159/000442469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Faix JD, Biomarkers of sepsis., Crit. Rev. Clin. Lab. Sci 50 (2013) 23–36. doi: 10.3109/10408363.2013.764490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gibot S, Béné MC, Noel R, Massin F, Guy J, Cravoisy A, Barraud D, De Carvalho Bittencourt M, Quenot J-P, Bollaert P-E, Faure G, Charles P-E, Combination Biomarkers to Diagnose Sepsis in the Critically Ill Patient, Am. J. Respir. Crit. Care Med 186 (2012) 65–71. doi : 10.1164/rccm.201201-0037OC. [DOI] [PubMed] [Google Scholar]
- [8].Marshall JC, Why have clinical trials in sepsis failed?, Trends Mol. Med 20 (2014) 195–203. doi: 10.1016/j.molmed.2014.01.007. [DOI] [PubMed] [Google Scholar]
- [9].Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F, Inflammatory caspases are innate immune receptors for intracellular LPS, Nature 514 (2014) 187–192. doi: 10.1038/nature13683. [DOI] [PubMed] [Google Scholar]
- [10].Freudenberg MA, Tchaptchet S, Keck S, Fejer G, Huber M, Schütze N, Beutler B, Galanos C, Lipopolysaccharide sensing an important factor in the innate immune response to Gram-negative bacterial infections: Benefits and hazards of LPS hypersensitivity, Immunobiology 213 (2008) 193–203. doi: 10.1016/J.IMBIO.2007.11.008. [DOI] [PubMed] [Google Scholar]
- [11].Guillon A, Preau S, Aboab J, Azabou E, Jung B, Silva S, Textoris J, Uhel F, Vodovar D, Zafrani L, de Prost N, Radermacher P, T.R.C. of the F.I.C.S. (Société de R. de L. Translational Research Committee of the French Intensive Care Society (Société de Réanimation de Langue Française), Preclinical septic shock research: why we need an animal ICU., Ann. Intensive Care. 9 (2019) 66. doi: 10.1186/s13613-019-0543-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Doi K, Leelahavanichkul A, Yuen PST, Star RA, Animal models of sepsis and sepsis-induced kidney injury., J. Clin. Invest 119 (2009) 2868–78. doi: 10.1172/JCI39421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bardoel BW, Kenny EF, Sollberger G, Zychlinsky A, The Balancing Act of Neutrophils, Cell Host Microbe 15 (2014) 526–536. doi: 10.1016/j.chom.2014.04.011. [DOI] [PubMed] [Google Scholar]
- [14].Nathan C, Neutrophils and immunity: challenges and opportunities, Nat Rev Immunol 6 (2006) 173–182. doi:nri1785[pii]10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- [15].Ley K, Hoffman HM, Kubes P, Cassatella MA, Zychlinsky A, Hedrick CC, Catz SD, Neutrophils: New insights and open questions, Sci. Immunol 3 (2018) eaat4579. doi: 10.1126/SCIIMMUNOL.AAT4579. [DOI] [PubMed] [Google Scholar]
- [16].Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A, Neutrophil extracellular traps kill bacteria., Science. 303 (2004) 1532–5. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- [17].Sollberger G, Tilley DO, Zychlinsky A, Neutrophil Extracellular Traps: The Biology of Chromatin Externalization, Dev. Cell 44 (2018) 542–553. doi: 10.1016/j.devcel.2018.01.019. [DOI] [PubMed] [Google Scholar]
- [18].Castanheira FVS, Kubes P, Neutrophils and NETs in modulating acute and chronic inflammation., Blood. 133 (2019) 2178–2185. doi: 10.1182/blood-2018-11-844530. [DOI] [PubMed] [Google Scholar]
- [19].Kaplan MJ, Radic M, Neutrophil extracellular traps: double-edged swords of innate immunity., J. Immunol 189 (2012) 2689–95. doi: 10.4049/jimmunol.1201719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Czaikoski PG, Mota JMSC, Nascimento DC, Sônego F, e S FV. Castanheira PH Melo GT Scortegagna RL Silva R Barroso-Sousa FO Souto A Pazin-Filho F Figueiredo JC Alves-Filho FQ Cunha, Neutrophil Extracellular Traps Induce Organ Damage during Experimental and Clinical Sepsis, PLoS One. 11 (2016) e0148142. doi: 10.1371/journal.pone.0148142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sur Chowdhury C, Giaglis S, Walker UA, Buser A, Hahn S, Hasler P, Enhanced neutrophil extracellular trap generation in rheumatoid arthritis: analysis of underlying signal transduction pathways and potential diagnostic utility, Arthritis Res. Ther 16 (2014) R122. doi: 10.1186/ar4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, Kahn CR, Wagner DD, Diabetes primes neutrophils to undergo NETosis, which impairs wound healing, Nat. Med 21 (2015) 815–819. doi: 10.1038/nm.3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kambas K, Chrysanthopoulou A, Vassilopoulos D, Apostolidou E, Skendros P, Girod A, Arelaki S, Froudarakis M, Nakopoulou L, Giatromanolaki A, Sidiropoulos P, Koffa M, Boumpas DT, Ritis K, Mitroulis I, Tissue factor expression in neutrophil extracellular traps and neutrophil derived microparticles in antineutrophil cytoplasmic antibody associated vasculitis may promote thromboinflammation and the thrombophilic state associated with the disease., Ann. Rheum. Dis 73 (2014) 1854–63. doi: 10.1136/annrheumdis-2013-203430. [DOI] [PubMed] [Google Scholar]
- [24].Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, Upadhyay P, Uyeminami DL, Pommier A, Küttner V, Bružas E, Maiorino L, Bautista C, Carmona EM, Gimotty PA, Fearon DT, Chang K, Lyons SK, Pinkerton KE, Trotman LC, Goldberg MS, Yeh JT-H, Egeblad M, Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice, Science (80-. ). 361 (2018) eaao4227. doi: 10.1126/science.aao4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Knight JS, Subramanian V, O’Dell AA, Yalavarthi S, Zhao W, Smith CK, Hodgin JB, Thompson PR, Kaplan MJ, Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice., Ann. Rheum. Dis 74 (2015) 2199–206. doi: 10.1136/annrheumdis-2014-205365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kusunoki Y, Nakazawa D, Shida H, Hattanda F, Miyoshi A, Masuda S, Nishio S, Tomaru U, Atsumi T, Ishizu A, Peptidylarginine Deiminase Inhibitor Suppresses Neutrophil Extracellular Trap Formation and MPO-ANCA Production, Front. Immunol 7 (2016) 227. doi: 10.3389/fimmu.2016.00227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Louttit C, Park KS, Moon JJ, Bioinspired nucleic acid structures for immune modulation, Biomaterials. 217 (2019) 119287. doi: 10.1016/j.biomaterials.2019.119287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Korkmaz B, Moreau T, Gauthier F, Neutrophil elastase, proteinase 3 and cathepsin G: Physicochemical properties, activity and physiopathological functions, Biochimie. 90 (2008) 227–242. doi: 10.1016/j.biochi.2007.10.009. [DOI] [PubMed] [Google Scholar]
- [29].Pipoly DJ, Crouch EC, Degradation of native type IV procollagen by human neutrophil elastase. Implications for leukocyte-mediated degradation of basement membranes., Biochemistry. 26 (1987) 5748–54. doi: 10.1021/bi00392a025. [DOI] [PubMed] [Google Scholar]
- [30].Carp H, Miller F, Hoidal JR, Janoff A, Potential mechanism of emphysema: alpha 1- proteinase inhibitor recovered from lungs of cigarette smokers contains oxidized methionine and has decreased elastase inhibitory capacity., Proc. Natl. Acad. Sci. U. S. A 79 (1982) 2041–5. doi: 10.1073/pnas.79.6.2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Pham CTN, Neutrophil serine proteases: specific regulators of inflammation, Nat. Rev. Immunol 6 (2006) 541–550. doi: 10.1038/nri1841. [DOI] [PubMed] [Google Scholar]
- [32].Meyer-Hoffert U, Wingertszahn J, Wiedow O, Human Leukocyte Elastase Induces Keratinocyte Proliferation by Epidermal Growth Factor Receptor Activation, J. Invest. Dermatol 123 (2004) 338–345. doi: 10.1111/j.0022-202X.2004.23202.x. [DOI] [PubMed] [Google Scholar]
- [33].Devaney JM, Greene CM, Taggart CC, Carroll TP, O’Neill SJ, McElvaney NG, Neutrophil elastase up-regulates interleukin-8 via toll-like receptor 4, FEBS Lett 544 (2003) 129–132. doi: 10.1016/S0014-5793(03)00482-4. [DOI] [PubMed] [Google Scholar]
- [34].Zhao P, Lieu T, Barlow N, Sostegni S, Haerteis S, Korbmacher C, Liedtke W, Jimenez-Vargas NN, Vanner SJ, Bunnett NW, Neutrophil Elastase Activates Protease-activated Receptor-2 (PAR2) and Transient Receptor Potential Vanilloid 4 (TRPV4) to Cause Inflammation and Pain., J. Biol. Chem 290 (2015) 13875–87. doi: 10.1074/jbc.M115.642736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Owen CA, Campbell MA, Sannes PL, Boukedes SS, Campbell EJ, Cell surface-bound elastase and cathepsin G on human neutrophils: a novel, non-oxidative mechanism by which neutrophils focus and preserve catalytic activity of serine proteinases., J. Cell Biol 131 (1995) 775–89. doi: 10.1083/jcb.131.3.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Belorgey D, Bieth JG, DNA binds neutrophil elastase and mucus proteinase inhibitor and impairs their functional activity., FEBS Lett 361 (1995) 265–8. [DOI] [PubMed] [Google Scholar]
- [37].Belorgey D, Bieth JG, Effect of polynucleotides on the inhibition of neutrophil elastase by mucus proteinase inhibitor and alpha 1-proteinase inhibitor., Biochemistry. 37 (1998) 16416–22. doi: 10.1021/bi981536o. [DOI] [PubMed] [Google Scholar]
- [38].Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A, Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps., J. Cell Biol 191 (2010) 677–91. doi: 10.1083/jcb.201006052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hakkim A, Fuchs TA, Martinez NE, Hess S, Prinz H, Zychlinsky A, Waldmann H, Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation., Nat. Chem. Biol 7 (2011) 75–7. doi: 10.1038/nchembio.496. [DOI] [PubMed] [Google Scholar]
- [40].Metzler KD, Goosmann C, Lubojemska A, Zychlinsky A, Papayannopoulos V, A Myeloperoxidase-Containing Complex Regulates Neutrophil Elastase Release and Actin Dynamics during NETosis, Cell Rep 8 (2014) 883–896. doi: 10.1016/j.celrep.2014.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kolaczkowska E, Jenne CN, Surewaard BGJ, Thanabalasuriar A, Lee W-Y, Sanz M-J, Mowen K, Opdenakker G, Kubes P, Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature., Nat. Commun 6 (2015) 6673. doi: 10.1038/ncomms7673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].von Nussbaum F, Li VM-J, Neutrophil elastase inhibitors for the treatment of (cardio)pulmonary diseases: Into clinical testing with pre-adaptive pharmacophores, Bioorg. Med. Chem. Lett 25 (2015) 4370–4381. doi: 10.1016/j.bmcl.2015.08.049. [DOI] [PubMed] [Google Scholar]
- [43].Vogelmeier C, Aquino TO, O’Brien CD, Perrett J, Gunawardena KA, A randomised, placebo-controlled, dose-finding study of AZD9668, an oral inhibitor of neutrophil elastase, in patients with chronic obstructive pulmonary disease treated with tiotropium., COPD. 9 (2012) 111–20. doi: 10.3109/15412555.2011.641803. [DOI] [PubMed] [Google Scholar]
- [44].Zeiher BG, Artigas A, Vincent J-L, Dmitrienko A, Jackson K, Thompson BT, Bernard G, STRIVE Study Group, Neutrophil elastase inhibition in acute lung injury: results of the STRIVE study., Crit. Care Med 32 (2004) 1695–702. [DOI] [PubMed] [Google Scholar]
- [45].Bao G, Mitragotri S, Tong S, Multifunctional Nanoparticles for Drug Delivery and Molecular Imaging, Annu. Rev. Biomed. Eng 15 (2013) 253–282. doi: 10.1146/annurev-bioeng-071812-152409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Moon JJ, Suh H, Bershteyn A, Stephan MT, Liu H, Huang B, Sohail M, Luo S, Um SH, Khant H, Goodwin JT, Ramos J, Chiu W, Irvine DJ, Interbilayer-crosslinked multilamellar vesicles as synthetic vaccines for potent humoral and cellular immune responses., Nat. Mater 10 (2011) 243–51. doi: 10.1038/nmat2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Okeke EB, Okwor I, Mou Z, Jia P, Uzonna JE, CD4+CD25+ regulatory T cells attenuate lipopolysaccharide-induced systemic inflammatory responses and promotes survival in murine Escherichia coli infection, Shock. 40 (2013) 65–73. doi: 10.1097/SHK.0b013e318296e65b. [DOI] [PubMed] [Google Scholar]
- [48].Okeke EB, Mou Z, Onyilagha N, Jia P, Gounni AS, Uzonna JE, Deficiency of Phosphatidylinositol 3-Kinase δ Signaling Leads to Diminished Numbers of Regulatory T Cells and Increased Neutrophil Activity Resulting in Mortality Due to Endotoxic Shock., J. Immunol 199 (2017) 1086–1095. doi: 10.4049/jimmunol.1600954. [DOI] [PubMed] [Google Scholar]
- [49].Boxio R, Bossenmeyer-Pourié C, Steinckwich N, Dournon C, Nüsse O, Mouse bone marrow contains large numbers of functionally competent neutrophils., J. Leukoc. Biol 75 (2004) 604–11. doi: 10.1189/jlb.0703340. [DOI] [PubMed] [Google Scholar]
- [50].Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, Lohmeyer J, Preissner KT, Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones., PLoS One 7 (2012) e32366. doi: 10.1371/journal.pone.0032366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU, Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps, Cell Death Differ 16 (2009) 1438–1444. doi: 10.1038/cdd.2009.96. [DOI] [PubMed] [Google Scholar]
- [52].Li AV, Moon JJ, Abraham W, Suh H, Elkhader J, Seidman MA, Yen M, Im E-J, Foley MH, Barouch DH, Irvine DJ, Generation of Effector Memory T Cell-Based Mucosal and Systemic Immunity with Pulmonary Nanoparticle Vaccination, Sci. Transl. Med 5 (2013) 204ra130–204ra130. doi: 10.1126/scitranslmed.3006516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Kenny EF, Herzig A, Krüger R, Muth A, Mondal S, Thompson PR, Brinkmann V, von Bernuth H, Zychlinsky A, Diverse stimuli engage different neutrophil extracellular trap pathways., Elife 6 (2017). doi: 10.7554/eLife.24437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Schreiber A, Rousselle A, Becker JU, von Mässenhausen A, Linkermann A, Kettritz R, Necroptosis controls NET generation and mediates complement activation, endothelial damage, and autoimmune vasculitis., Proc. Natl. Acad. Sci. U. S. A 114 (2017) E9618–E9625. doi: 10.1073/pnas.1708247114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Carmona-Rivera C, Zhao W, Yalavarthi S, Kaplan MJ, Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2., Ann. Rheum. Dis 74 (2015) 1417–24. doi: 10.1136/annrheumdis-2013-204837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Aikawa N, Ishizaka A, Hirasawa H, Shimazaki S, Yamamoto Y, Sugimoto H, Shinozaki M, Taenaka N, Endo S, Ikeda T, Kawasaki Y, Reevaluation of the efficacy and safety of the neutrophil elastase inhibitor, Sivelestat, for the treatment of acute lung injury associated with systemic inflammatory response syndrome; a phase IV study., Pulm. Pharmacol. Ther 24 (2011) 549–54. doi: 10.1016/j.pupt.2011.03.001. [DOI] [PubMed] [Google Scholar]
- [57].Miyazaki Y, Inoue T, Kyi M, Sawada M, Miyake S, Yoshizawa Y, Effects of a Neutrophil Elastase Inhibitor (ONO-5046) on Acute Pulmonary Injury Induced by Tumor Necrosis Factor Alpha (TNF α ) and Activated Neutrophils in Isolated Perfused Rabbit Lungs, Am. J. Respir. Crit. Care Med 157 (1998) 89–94. doi: 10.1164/ajrccm.157.1.9612021. [DOI] [PubMed] [Google Scholar]
- [58].Klebanoff SJ, Vadas MA, Harlan JM, Sparks LH, Gamble JR, Agosti JM, Waltersdorph AM, Stimulation of neutrophils by tumor necrosis factor., J. Immunol 136 (1986) 4220–5. [PubMed] [Google Scholar]
- [59].Fuchs HJ, Borowitz DS, Christiansen DH, Morris EM, Nash ML, Ramsey BW, Rosenstein BJ, Smith AL, Wohl ME, Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group., N. Engl. J. Med 331 (1994) 637–42. doi: 10.1056/NEJM199409083311003. [DOI] [PubMed] [Google Scholar]
- [60].Papayannopoulos V, Staab D, Zychlinsky A, Neutrophil elastase enhances sputum solubilization in cystic fibrosis patients receiving DNase therapy., PLoS One. 6 (2011) e28526. doi: 10.1371/journal.pone.0028526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Tkalcevic J, Novelli M, Phylactides M, Iredale JP, Segal AW, Roes J, Impaired immunity and enhanced resistance to endotoxin in the absence of neutrophil elastase and cathepsin G, Immunity. 12 (2000) 201–210. doi: 10.1016/S1074-7613(00)80173-9. [DOI] [PubMed] [Google Scholar]
- [62].Nakamura A, Mori Y, Hagiwara K, Suzuki T, Sakakibara T, Kikuchi T, Igarashi T, Ebina M, Abe T, Miyazaki J, Takai T, Nukiwa T, Increased susceptibility to LPS-induced endotoxin shock in secretory leukoprotease inhibitor (SLPI)-deficient mice., J. Exp. Med 197 (2003) 669–74. doi: 10.1084/jem.20021824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Chapman EA, Lyon M, Simpson D, Mason D, Beynon RJ, Moots RJ, Wright HL, Caught in a Trap? Proteomic Analysis of Neutrophil Extracellular Traps in Rheumatoid Arthritis and Systemic Lupus Erythematosus., Front. Immunol 10 (2019) 423. doi: 10.3389/fimmu.2019.00423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, Menninger S, Eickhoff J, Nussbaumer P, Klebl B, Krüger R, Herzig A, Zychlinsky A, Gasdermin D plays a vital role in the generation of neutrophil extracellular traps., Sci. Immunol 3 (2018) eaar6689. doi: 10.1126/sciimmunol.aar6689. [DOI] [PubMed] [Google Scholar]
- [65].Chalmers JD, Moffitt KL, Suarez-Cuartin G, Sibila O, Finch S, Furrie E, Dicker A, Wrobel K, Elborn JS, Walker B, Martin SL, Marshall SE, Huang JT-J, Fardon TC, Neutrophil Elastase Activity Is Associated with Exacerbations and Lung Function Decline in Bronchiectasis, Am. J. Respir. Crit. Care Med 195 (2017) 1384–1393. doi: 10.1164/rccm.201605-1027OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Döring G, The Role of Neutrophil Elastase in Chronic Inflammation, Am. J. Respir. Crit. Care Med 150 (1994) S114–S117. doi: 10.1164/ajrccm/150.6_Pt_2.S114. [DOI] [PubMed] [Google Scholar]
- [67].Suter S, The role of bacterial proteases in the pathogenesis of cystic fibrosis., Am. J. Respir. Crit. Care Med 150 (1994) S118–22. doi: 10.1164/ajrccm/150.6_Pt_2.S118. [DOI] [PubMed] [Google Scholar]
- [68].Amitani R, Wilson R, Rutman A, Read R, Ward C, Burnett D, Stockley RA, Cole PJ, Effects of human neutrophil elastase and Pseudomonas aeruginosa proteinases on human respiratory epithelium., Am. J. Respir. Cell Mol. Biol 4 (1991) 26–32. doi: 10.1165/ajrcmb/4.1.26. [DOI] [PubMed] [Google Scholar]
- [69].Wang T, Zhu Z, Liu Z, Yi L, Yang Z, Bian W, Chen W, Wang S, Li G, Li A, Martin GS, Zhu X, Plasma Neutrophil Elastase and Elafin as Prognostic Biomarker for Acute Respiratory Distress Syndrome, SHOCK. 48 (2017) 168–174. doi: 10.1097/SHK.0000000000000845. [DOI] [PubMed] [Google Scholar]
- [70].Mahmoud El-Lahouny DM Khalifa KA Hosny MR, Serum polymorphonuclear leukocyte elastase enzyme level in neonatal sepsis, Menoufia Med. J 31 (2018) 970. doi: 10.4103/MMJ.MMJ_31_17. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.