

Abstract
A distal-selective alkenyl C–H arylation method is reported through a directed palladium/norbornene (Pd/NBE) cooperative catalysis. The key is to use an appropriate combination of the directing group and the NBE cocatalyst. A range of acyclic and cyclic cis-olefins are suitable substrates, and the reaction is operated under air with excellent site-selectivity. Preliminary mechanistic studies are consistent with the proposed Pd/NBE-catalyzed C–H activation instead of the Heck pathway. Initial success on distal alkylation has also been achieved using MeI and methyl bromoacetate as electrophiles.
Trisubstituted olefins are prevalent structural motifs found in numerous natural products1 and other biologically active compounds;2 they often serve as versatile synthetic intermediates to access various other functional groups, such as epoxides, aziridines, and cyclopropanes as well as tertiary stereocenters.3 Typically, trisubstituted olefins are prepared through olefination of carbonyls, hydrofunctionalization of alkynes, cross couplings, olefin metathesis, etc. (Scheme 1a). Despite the efficacy of these established approaches, it is nontrivial to control regio- and stereoselectivity for synthesis of unsymmetrical trisubstituted olefins (compared to disubstituted olefins) in the absence of steric or electronic bias.4 Alternatively, direct functionalization of an unactivated alkenyl C–H bond in 1,2-disubstiuted olefins represents an attractive approach for preparing unsymmetrical trisubstituted alkenes. In particular, the use of directing groups (DGs) offers a convenient approach to enhance reactivity and site-selectivity for the alkenyl C–H activation due to proximity effect (Scheme 1b).5 To date, a number of transition metal complexes, e.g., Ru,6 Pd,7 Rh,8 Ir,9 Fe,10 and Co,11 have been reported to be efficient for functionalization of the proximal alkenyl C–H bonds. However, it would be attractive if a complementary approach could be developed to allow site-selective activation of the distal alkenyl C–H bonds, which would consequently enrich methods for regioselective synthesis of trisubstituted alkenes. Herein, we describe our initial development of a distal alkenyl C–H functionalization strategy via the palladium/norbornene (NBE) cooperative catalysis (Scheme 1c).
Scheme 1. Trisubstituted Olefins.

Recently, the Pd/NBE catalysis, originally discovered by Catellani, has emerged as a powerful tool for arene functionalization.12 In 2015, the Yu13 and our14 groups independently reported complementary methods for the Pd/NBE-catalyzed meta-selective C–H activation initiated by directed ortho-palladation. An intriguing question is whether such a distal C–H functionalization strategy could be extended beyond aromatic substrates, such as alkenes (Scheme 1c). Starting with a directed C–H palladation at the proximal alkene position, followed by NBE insertion and a second C–H palladation at the distal position, an alkenyl–norbornyl palladacycle II would be formed, which could react with electrophiles to install a functional group at the distal carbon. Subsequent NBE extrusion and reprotonation at the proximal position complete the catalytic cycle.
However, compared with arenes, several challenges could be anticipated for activating alkene C–H bonds with the proposed pathway. The primary concern comes from the more reactive olefin π bond. Due to the lack of stability by aromaticity, the C=C bond in alkenes could easily undergo various π-breaking transformations, such as reactions with oxidants or electrophiles, leading to undesired background reactions. Second, the π bond of alkenes is typically considered to be a better ligand for Pd than arenes; thus a number of Pd-mediated side reactions, including 2π-insertion and nucleophilic attack of coordinated olefins, could compete with the desired C–H palladation step. Third, after NBE insertion, the alkyl–Pd intermediate is known to form cyclopropanes easily with the alkene via migratory insertion, instead of palladation with the distal C–H bond.15 Hence, limited examples are known for the Catellani-type reaction of regular alkenyl substrates.16
To address these challenges, it would be beneficial to realize fast and reversible proximal C–H palladation with appropriate DGs. In addition, the structure of NBE could also be important to minimize undesired side reactions by serving as a reactive ligand. Moreover, the DG should be easily installable and removable. To explore the proposed distal alkene C–H functionalization, cis-alkene 1a with an “exo”-type DG17 based on oxime ethers18 was chosen as the initial substrate and arylation was studied as the model reaction (Table 1). After careful evaluation of various reaction parameters, ultimately, the desired C–H arylation product (3a) was obtained in 76% yield with Pd(OAc)2/N4 as the catalyst combination, 3-CF3-2-pyridone as the additive,19 and AgOAc as the halide scavenger in CHCl3 under air (see Supporting Information for detailed optimization). Other oxime ethers were also investigated as the DG (entry 2). While the aldehyde-derived ones (DG2–4) were much less reactive, the cyclohexanone-derived DG5 showed a comparable result. Control experiments indicate that both palladium and NBE are essential to this reaction, and diminished yield was observed in the absence of the pyridone additive (entries 3–5). Note that the normal Heck product (Z)-3a was formed dominantly in the absence of NBE (entry 5); in contrast, under the standard conditions the Heck reaction was significantly suppressed (vide infra, Scheme 2). In addition, simple NBE and other substituted NBEs also delivered the desired product (entry 6),20 although the imide-based N4 was most reactive.21 Moreover, replacement of AgOAc with the corresponding cesium salt was not effective under the standard conditions (entry 7).
Table 1.
Selected Optimization for the C–H Arylation

|
Unless otherwise noted, reactions were run on a 0.2 mmol scale. Yields were determined by 1H NMR analysis using CH2Br2 as the internal standard.
52% Heck product (Z)-3a observed.
Scheme 2. Mechanistic Studies.

With the optimized conditions in hand, the alkene scope was first investigated (Table 2). A range of styrenyl alkenes bearing both electron-deficient (3b) and -rich (3c) substituents afforded the desired products in good yields and excellent regio- and diastereoselectivity. Besides aliphatic alcohols, benzylic alcohol-derived substrates also reacted well. It is surprising that the phenyl-substituted substrate still gave the desired distal alkene-arylation product (3f) despite the potential side reaction on the arene, e.g., meta-arylation. It is likely that the chelation between the cis-alkene and the DG inhibited undesired ortho-palladation on the phenyl group. In addition, cis-1,2-dialkyl-substituted alkenes are competent substrates (3g–k). Apart from secondary alcohols, primary alcohol-derived substrates also reacted. Cyclic alkenes containing six- or eight-membered rings (3l, 3o) provided the desired products in moderate to good yields. Reactions with the more challenging linear alkenes derived from primary alcohols (3p, 3q) showed a slightly lower efficiency due to the flexibility of the substrates. Moreover, allylic and homoallylic protected amines (3r–t) worked well with a modified DG.
Table 2.
Alkene Scopea

|
Unless otherwise noted, reactions were run on a 0.2 mmol scale in a 4 mL vial under the standard conditions. Numbers are the isolated yields, and E/Z ratio was determined by 1H NMR analysis of the reaction crude.
36 h.
24 h.
DG6 = 3-methyl-2-pyridyl;
see Supporting Information for detailed conditions.
The scope of aryl iodides was then explored (Table 3). Aryl iodides with an ortho electron-withdrawing group proved to be most efficient, consistent with the typical Catellani arylation reaction.22 Interestingly, methyl 2,5-diiodobenzoate can be coupled to give the monoalkenylated product 4a in 63% yield, where the iodide ortho to the ester group reacted exclusively. Electron-deficient aryl iodides (4b–e, 4h) generally afforded the distal arylation products in good yield and excellent E/Z selectivity, while slightly electron-rich ones (4f and 4g) gave lower yields and moderate selectivity. Besides esters, other ortho groups, such as nitro (4d–f), ketone (4i), tertiary amide (4j), and Weinreb amide (4k), also delivered the desired products in good yields. Heteroarenes, such as thiophene (4g) and pyridine (4h), as well as iodide (4a), bromide (4e), and chloride (4c) substituents, were tolerated. Aryl iodides without an ortho electron-withdrawing group were much less reactive under the current conditions,23 likely due to the difficulty of the oxidative addition step between the aryl electrophile and the alkenyl–norbornyl palladacycle. Beyond arylation, under slightly modified conditions, the distal C–H alkylation with methyl iodide and methyl bromoacetate can be achieved in moderate yields (4l–n).
Table 3.
Electrophile Scopea

|
Unless otherwise noted, reactions were run on a 0.2 mmol scale in a 4 mL vial under the standard conditions. Numbers are the isolated yields, and E/Z ratio was determined by 1H NMR analysis of the reaction crude.
110 °C.
24 h.
MeI was used.
Methyl bromoacetate was used.
A major competing reaction pathway is the directed Heck reaction (Scheme 2a). The aryl-Pd(II) species could undergo directed migratory insertion to the alkene followed by β-H elimination to give the Z-alkene product.24 It was interesting to observe that, by increasing the loading of the NBE cocatalyst, the undesired Heck pathway was significantly inhibited (Scheme 2b). To gain insights into the reaction mechanism, deuterium labeling studies were carried out. In the absence of the aryl iodide, upon addition of 3.0 equiv of CD3CO2D, 81% of the alkene substrate was recovered with 20% and 22% deuterium incorporation at the distal and proximal vinyl positions, respectively (Scheme 2c). This indicates reversible formation of C–H palladation intermediates I and II (vide supra, Scheme 1c). In addition, a four-membered-ring side product25 was obtained in 10% yield under these conditions, implying the formation of alkenyl–norbornyl palladacycle II. In the presence of the aryl iodide and CD3CO2D, deuterium incorporation was observed in the vinyl C–H bonds of both the arylation product and the recovered alkene substrate (Scheme 2c), which suggests protonation occurred at the proximal position. In addition, control experiments with the trans alkene substrate (E)-1n gave no desired arylation product, and Z/E isomerization was not observed under the standard reaction conditions (Scheme 2d), which rule out the pathways involving alkene isomerization-then-Heck or Heck-then-alkene isomerization.
From the view of practicality, the Pd loading can be reduced to 5 mol % on a larger scale (Scheme 3a). The arylation product can undergo various transformations (Scheme 3b). For example, the oxime-ether DG can be easily removed by treatment with Zn and acetic acid. The olefin moiety can be hydrogenated by Pd/C to deliver product 6. The Raney nickel-catalyzed condition can remove the DG and reduce the alkene simultaneously in a quantitative yield. Gratifyingly, the proximal position could undergo a further directed C–H/Heck coupling, which furnished an unsymmetrical tetrasubstituted alkene (7).26 The linear arylation products after saponification can be converted to the corresponding substituted phthalides27 in good yields (8–10, Scheme 3c). In addition, using methyl bromoacetate as the electrophile, a two-step 2-hydroxyethylation was realized in an efficient manner (Scheme 3d). Note that LiAlH4 chemoselectively reduced the ester moiety instead of the oxime DG. Finally, an initial silver-free condition was identified for the allyl amine substrate (Scheme 3e), which replaced AgOAc with CsOAc using methyl tert-butyl ether (MTBE) as solvent.
Scheme 3. Synthetic Utility.

In summary, a Pd/NBE-catalyzed strategy for distal C–H functionalization of alkenes is developed, which provides a convenient way to prepare trisubstituted olefins. The reaction is regio- and stereoselective and can operate in air. The use of easily removable exo-DGs in alkene C–H activation could have implications beyond this transformation. Further optimization of the distal alkylation reaction and expansion of the electrophile scope are the topics of the ongoing investigation.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the University of Chicago and NIGMS (1R01GM124414–01A1) for financial support. N.F. thanks the Metcalf fellowship for financial support. Mr. Renhe Li is acknowledged for checking the reproducibility of the experimental procedure.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.9b11479.
Experimental procedures; spectral data (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).For selected syntheses of natural products that contain trisubstituted olefins, see:; (a) Wipf P; Kim S Total Synthesis of the Enantiomer of the Antiviral Marine Natural Product Hennoxazole A. J. Am. Chem. Soc 1995, 117, 558–559. [Google Scholar]; (b) White JD; Carter RG; Sundermann KF; Wartmann M Total Synthesis of Epothilone B, Epothilone D, and cis- and trans-9,10-Dehydroepothilone D. J. Am. Chem. Soc 2001, 123, 5407–5413. [DOI] [PubMed] [Google Scholar]; (c) Smith AB III; Qiu Y; Jones DR; Kobayshi K Total Synthesis of (−)-Discodermolide. J. Am. Chem. Soc 1995, 117, 12011–12012. [Google Scholar]
- (2).For selected syntheses of drugs that contain trisubstituted olefins, see:; (a) Ishikawa H; Suzuki T; Hayashi Y High-Yielding Synthesis of the Anti-Influenza Neuramidase Inhibitor (−)-Oseltamivir by Three “One-Pot” Operations. Angew. Chem., Int. Ed 2009, 48, 1304–1307. [DOI] [PubMed] [Google Scholar]; (b) Wang H; Zheng X; Cia Z; Yu O; Zheng S; Zhu T Synthesis and Evaluation of an Injectable Everolimus Prodrug. Bioorg. Med. Chem. Lett 2017, 27, 1175–1178. [DOI] [PubMed] [Google Scholar]; (c) Nemoto H; Shiraki M; Nagamochi M; Fukumoto K A Concise Enantiocontrolled Total Synthesis of (−)-α-Bisabolol and (+)-4-epi-α-Bisabolol. Tetrahedron Lett. 1993, 34, 4939–4942. [Google Scholar]
- (3).(a) Verendel JJ; Pamies O; Dieguez M; Andersson PG Asymmetric Hydrogenation of Olefins Using Chiral Crabtree-type Catalysts: Scope and Limitations. Chem. Rev 2014, 114, 2130–2169. [DOI] [PubMed] [Google Scholar]; (b) Etayo P; Vidal-Ferran A Rhodium-Catalysed Asymmetric Hydrogenation as a Valuable Synthetic Tool for the Preparation of Chiral Drugs. Chem. Soc. Rev 2013, 42, 728–754. [DOI] [PubMed] [Google Scholar]
- (4).Nguyen TT; Koh MJ; Mann TJ; Schrock RR; Hoveyda AH Synthesis of E- and Z-Trisubstituted Alkenes by Catalytic Cross-Metathesis. Nature 2017, 552, 347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).For alkenyl C–H activation reviews, see:; (a) Shang X; Liu Z Transition Metal-Catalyzed Cvinyl–Cvinyl Bond Formation via Double Cvinyl–H Bond Activation. Chem. Soc. Rev 2013, 42, 3253–3260. [DOI] [PubMed] [Google Scholar]; (b) Wang K; Hu F; Zhao Y; Wang J Directing Group-Assisted Transition-Metal-Catalyzed Vinylic C-H bond Functionalization. Sci. China: Chem 2015, 58, 1252–1265. [Google Scholar]; (c) Maraswami M; Loh T-P Transition-Metal-Catalyzed Alkenyl sp2 C–H Activation: A Short Account. Synthesis 2019, 51, 1049–1062. [Google Scholar]
- (6).For selected examples, see:; (a) Trost BM; Imi K; Davies IW Elaboration of Conjugated Alkenes Initiated by Insertion into a Vinylic C-H Bond. J. Am. Chem. Soc 1995, 117, 5371–5372. [Google Scholar]; (b) Kakiuchi F; Tanaka Y; Sato T; Chatani N; Murai S Catalytic Addition of Olefinic C–H Bonds to Olefins. Chem. Lett 1995, 24, 679–680. [Google Scholar]; (c) Zhang J; Loh T-P Ruthenium- and Rhodium-Catalyzed Cross-Coupling Reaction of Acrylamides with Alkenes: Efficient Access to (Z, E)-Dienamides. Chem. Commun 2012, 48, 11232–11234. [DOI] [PubMed] [Google Scholar]; (d) Hu XH; Zhang J; Yang XF; Xu YH; Loh T-P Stereo- and Chemoselective Cross-Coupling between Two Electron-Deficient Acrylates: An Efficient Route to (Z, E)-Muconate Derivatives. J. Am. Chem. Soc 2015, 137, 3169–3172. [DOI] [PubMed] [Google Scholar]
- (7).For leading references, see:; (a) Zhou H; Xu Y; Chung W; Loh T-P Palladium-Catalyzed Direct Arylation of Cyclic Enamides with Aryl Silanes by sp2 C-H Activation. Angew. Chem., Int. Ed 2009, 48, 5355–5357. [DOI] [PubMed] [Google Scholar]; (b) Pankajakshan S; Xu Y; Cheng JK; Low MT; Loh T-P Palladium-Catalyzed Direct C-H Arylation of Enamides with Simple Arenes. Angew. Chem., Int. Ed 2012, 51, 5701–5705. [DOI] [PubMed] [Google Scholar]; (c) Liang Q; Yang C; Meng F-F; Jiang B; Xu Y-H; Loh T-P Chelation versus Non-Chelation Control in the Stereoselective Alkenyl sp2 C-H Bond Functionalization Reaction. Angew. Chem., Int. Ed 2017, 56, 5091–5095. [DOI] [PubMed] [Google Scholar]; (d) Liu M; Yang P; Karunananda MK; Wang Y; Liu P; Engle KMC (alkenyl)–H Activation via Six-Membered Palladacycles: Catalytic 1,3-Diene Synthesis. J. Am. Chem. Soc 2018, 140, 5805–5813. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Luo Y-C; Yang C; Qiu S-Q; Liang Q-J; Xu Y-H; Loh T-P Palladium(II)-Catalyzed Stereo-specific Alkenyl C–H Bond Alkylation of Allylamines with Alkyl Iodides. ACS Catal. 2019, 9, 4271–4276. [Google Scholar]
- (8).For selected examples, see:; (a) Besset T; Kuhl N; Patureau FW; Glorius F RhIII-Catalyzed Oxidative Olefination of Vinylic C-H Bonds: Efficient and Selective Access to Di-unsaturated α-Amino Acid Derivatives and Other Linear 1,3-Butadienes. Chem. - Eur. J 2011, 17, 7167–7171. [DOI] [PubMed] [Google Scholar]; (b) Wang H; Beiring B; Yu D; Collins KD; Glorius F [3]Dendralene Synthesis: Rhodium(III)-Catalyzed Alkenyl C-H Activation and Coupling Reaction with Allenyl Carbinol Carbonate. Angew. Chem., Int. Ed 2013, 52, 12430–12434. [DOI] [PubMed] [Google Scholar]; (c) Boultadakis-Arapinis M; Hopkinson MN; Glorius F Using Rh(III)-Catalyzed C–H Activation as a Tool for the Selective Functionalization of Ketone-Containing Molecules. Org. Lett 2014, 16, 1630–1633. [DOI] [PubMed] [Google Scholar]; (d) Lei Z; Ye J; Sun J; Shi Z Direct Alkenyl C–H Functionalization of Cyclic Enamines with Carboxylic Acids via Rh Catalysis Assisted by Hydrogen Bonding. Org. Chem. Front 2014, 1, 634–638. [Google Scholar]
- (9).For selected examples, see:; (a) Zhang YJ; Skucas E; Krische MJ Direct Prenylation of Aromatic and α,β-Unsaturated Carboxamides via Iridium-Catalyzed C-H Oxidative Addition-Allene Insertion. Org. Lett 2009, 11, 4248–4250. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ye K; He H; Liu W-B; Dai L; Helmchen G; You S-L Iridium-Catalyzed Allylic Vinylation and Asymmetric Allylic Amination Reactions with o-Aminostyrenes. J. Am. Chem. Soc 2011, 133, 19006–19014. [DOI] [PubMed] [Google Scholar]
- (10).For selected examples, see:; (a) Ilies L; Matsubara T; Ichikawa S; Asako S; Nakamura E Iron-Catalyzed Directed Alkylation of Aromatic and Olefinic Carboxamides with Primary and Secondary Alkyl Tosylates, Mesylates, and Halides. J. Am. Chem. Soc 2014, 136, 13126–13129. [DOI] [PubMed] [Google Scholar]; (b) Monks BM; Fruchey ER; Cook SP Iron-Catalyzed C(sp2)-H Alkylation of Carboxamides with Primary Electrophiles. Angew. Chem., Int. Ed 2014, 53, 11065– 11069. [DOI] [PubMed] [Google Scholar]; (c) Ilies L; Ichikawa S; Matsubara T; Nakamura E Iron-Catalyzed Directed Alkylation of Alkenes and Arenes with Alkylzinc Halides. Adv. Synth. Catal 2015, 357, 2175–2179. [Google Scholar]; (d) Cera G; Haven T; Ackermann L Expedient Iron-Catalyzed C-H Allylation/Alkylation by Triazole Assistance with Ample Scope. Angew. Chem., Int. Ed 2016, 55, 1484–1488. [DOI] [PubMed] [Google Scholar]
- (11).For selected examples, see:; (a) Gensch T; Vasquez-Cespedes S; Yu D-G; Glorius F Org. Lett 2015, 17, 3714–3717. [DOI] [PubMed] [Google Scholar]; (b) Wang H; Zhang S; Wang Z; He M; Xu K Cobalt-Catalyzed MonoselectiveOrtho-C–H Functionalization of Carboxamides with Organoaluminum Reagent. Org. Lett 2016, 18, 5628–5631. [DOI] [PubMed] [Google Scholar]; (c) Li T; Shen C; Sun Y; Zhang J; Xiang P; Lu X; Zhong G Cobalt-Catalyzed Olefinic C–H Alkenylation/Alkylation Switched by Carbonyl Groups. Org. Lett 2019, 19, 7772–7777. [DOI] [PubMed] [Google Scholar]
- (12).For selected reviews, see:; (a) Ye J; Lautens M Palladium-Catalysed Norbornene-Mediated C–H Functionalization of Arenes. Nat. Chem 2015, 7, 863–870. [DOI] [PubMed] [Google Scholar]; (b) Della Ca’ N; Fontana M; Motti E; Catellani M. Pd/Norbornene: A Winning Combination for Selective Aromatic Functionalization via C–H Bond Activation. Acc. Chem. Res 2016, 49, 1389–1400. [DOI] [PubMed] [Google Scholar]; (c) Wang J; Dong G Palladium/Norbornene Cooperative Catalysis. Chem. Rev 2019, 119, 7478– 7528. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Cheng H-G; Chen S; Chen R; Zhou Q Palladium (II)-Initiated Catellani-Type Reactions. Angew. Chem., Int. Ed 2019, 58, 5832–5844. [DOI] [PubMed] [Google Scholar]
- (13).Wang X-C; Gong W; Fang L-Z; Zhu R-Y; Li S; Engle KM; Yu J-Q Ligand-Enabled meta-C–H Activation Using a Transient Mediator. Nature 2015, 519, 334–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Dong Z; Wang J; Dong G Simple Amine-Directed Meta-Selective C–H Arylation via Pd/Norbornene Catalysis. J. Am. Chem. Soc 2015, 137, 5887–5890. [DOI] [PubMed] [Google Scholar]
- (15).(a) Catellani M; Chiusoli GP Competitive Processes in Palladium-Catalyzed C-C Bond Formation. J. Organomet. Chem 1982, 233, C21–C24. [Google Scholar]; (b) Khanna A; Premachandra IDUA; Sung PD; Van Vranken DL Palladium-Catalyzed Catellani Amino-cyclopropanation Reactions with Vinyl Halides. Org. Lett 2013, 15, 3158–3161. [DOI] [PubMed] [Google Scholar]
- (16).For alkenyl Catellani-type reactions:; (a) Wang J; Dong Z; Yang C; Dong G Modular and Regioselective Synthesis of All-Carbon Tetrasubstituted Olefins Enabled by an Alkenyl Catellani Reaction. Nat. Chem 2019, 11, 1106–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yamamoto Y; Murayama T; Jiang J; Yasui T; Shibuya M The Vinylogous Catellani Reaction: a Combined Computational and Experimental Study. Chem. Sci 2018, 9, 1191–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Xu Y; Dong G sp3 C–H Activation viaexo-type Directing Groups. Chem. Sci 2018, 9, 1424–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).For earlier works, see:; (a) Desai LV; Stowers KJ; Sanford MS Insights into Directing Group Ability in Palladium-Catalyzed CH Bond Functionalization. J. Am. Chem. Soc 2008, 130, 13285– 13293. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ren Z; Mo F; Dong G Catalytic Functionalization of Unactivated sp3 C–H Bonds via exo-Directing Groups: Synthesis of Chemically Differentiated 1,2-Diols. J. Am. Chem. Soc 2012, 134, 16991–16994. [DOI] [PubMed] [Google Scholar]
- (19).For the first use of substituted 2-pyridone as the concerted metalation deprotonation promotor, see:; Wang P; Farmer ME; Huo X; Jain P; Shen P-X; Ishoey M; Bradner JE; Wisniewski SR; Eastgate MD; Yu J-Q. Ligand-Promoted Meta-C–H Arylation of Anilines, Phenols, and Heterocycles. J. Am. Chem. Soc 2016, 138, 9269–9276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).For the first use of a substituted NBE in the Pd/NBE catalysis, see:; (a) Dong Z; Wang J; Ren Z; Dong G Ortho C-H Acylation of Aryl Iodides by Palladium/Norbornene Catalysis. Angew. Chem., Int. Ed 2015, 54, 12664–12668.For the first use of a substituted NBE in the C–H activation-initiated Pd/NBE catalysis, see: [DOI] [PubMed] [Google Scholar]; (b) Shen P-X; Wang X-C; Wang P; Zhu R-Y; Yu J-Q Ligand-Enabled Meta-C–H Alkylation and Arylation Using a Modified Norbornene. J. Am. Chem. Soc 2015, 137, 11574–11577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Liu Z-S; Qian G; Gao Q; Wang P; Cheng H-G; Wei Q; Liu Q; Zhou Q Palladium/Norbornene Cooperative Catalysis to Access Tetrahydronaphthalenes and Indanes with a Quaternary Center. ACS Catal. 2018, 8, 4783–4788. [Google Scholar]
- (22).(a) Motti E; Della Ca’ N; Deledda S; Fava E; Panciroli F; Catellani M. Palladium-Catalyzed Unsymmetrical Aryl Couplings in Sequence Leading to O-Teraryls: Dramatic Olefin Effect on Selectivity. Chem. Commun 2010, 46, 4291–4293. [DOI] [PubMed] [Google Scholar]; (b) Martins A; Candito DA; Lautens M Palladium-Catalyzed Reductive ortho-Arylation: Evidence for the Decomposition of 1,2-Dimethoxyethane and Subsequent Arylpalladium (II) Reduction. Org. Lett 2010, 12, 5186–5188. [DOI] [PubMed] [Google Scholar]
-
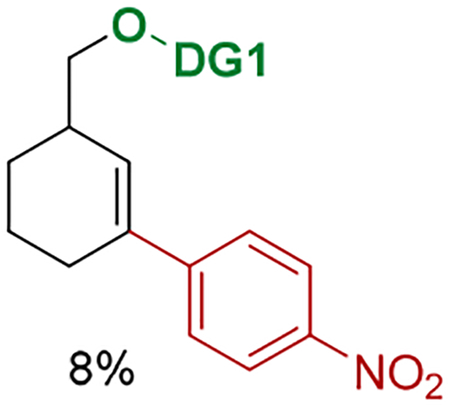
(23).For example, p-nitrophenyl iodide gave the desired product albeit in 8% yield.
- (24).Romine AM; Yang KS; Karunannanda MK; Chen JS; Engle KM Synthetic and Mechanistic Studies of a Versatile Heteroaryl Thioether Directing Group for Pd(II) Catalysis. ACS Catal. 2019, 9, 7626–7640. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
(25).A typical side product was observed in this reaction, which supports a Catellani pathway.

- (26).Attempts to realize a direct distal/proximal difunctionalization of the alkene substrat
- (27).León A; Del-Ángel M; Ávila JL; Delgado G Phthalides: Distribution in Nature, Chemical Reactivity, Synthesis, and Biological Activity In Progress in the Chemistry of Organic Natural Products; Kinghorn AD, Falk H, Gibbons S, Kobayashi J, Eds. Springer: Cham, 2017; Vol. 104, pp 127–246. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.