

Abstract
Herein we describe the development of a highly selective kinetic resolution of cyclobutanones via a Rh-catalyzed “cut-and-sew” reaction with selectivity factor up to 785. This reaction takes place at room temperature with excellent efficiency. Various trans-5,6-fused bicycles and C2-substituted cyclobutanones were obtained with excellent ee’s that can be further used as chiral building blocks. DFT calculations reveal the crucial roles of the DTBM-segphos ligand in stabilizing the rate- and enantioselectivity-determining C–C oxidative addition transition state via favorable ligand–substrate dispersion interactions.
While highly desirable, controlling both reactivity and stereochemistry constitutes a significant challenge during activation of inert chemical bonds. The recent advancement allows asymmetric C–H functionalization to emerge as a powerful tool for synthesis;1 by contrast, the corresponding asymmetric C–C cleavage/functionalization, though attractive for preparing chiral complex ring systems, has been much less developed.2 To date, the scope of transition metal-catalyzed asymmetric C–C activation3 has been primarily restricted to either the cleavage of an achiral C–C bond, e.g., an aryl–CN bond2b,c or the C1–C8 bond in benzocyclobutenones,2d,e or use of symmetrical substrates2f–n (Scheme 1a,b). Despite the fact that chiral unsymmetrical C– C bonds are common, the corresponding catalytic asymmetric transformation involving activation of these bonds remains elusive (Scheme 1c).
Scheme 1. Asymmetric C–C Activations.
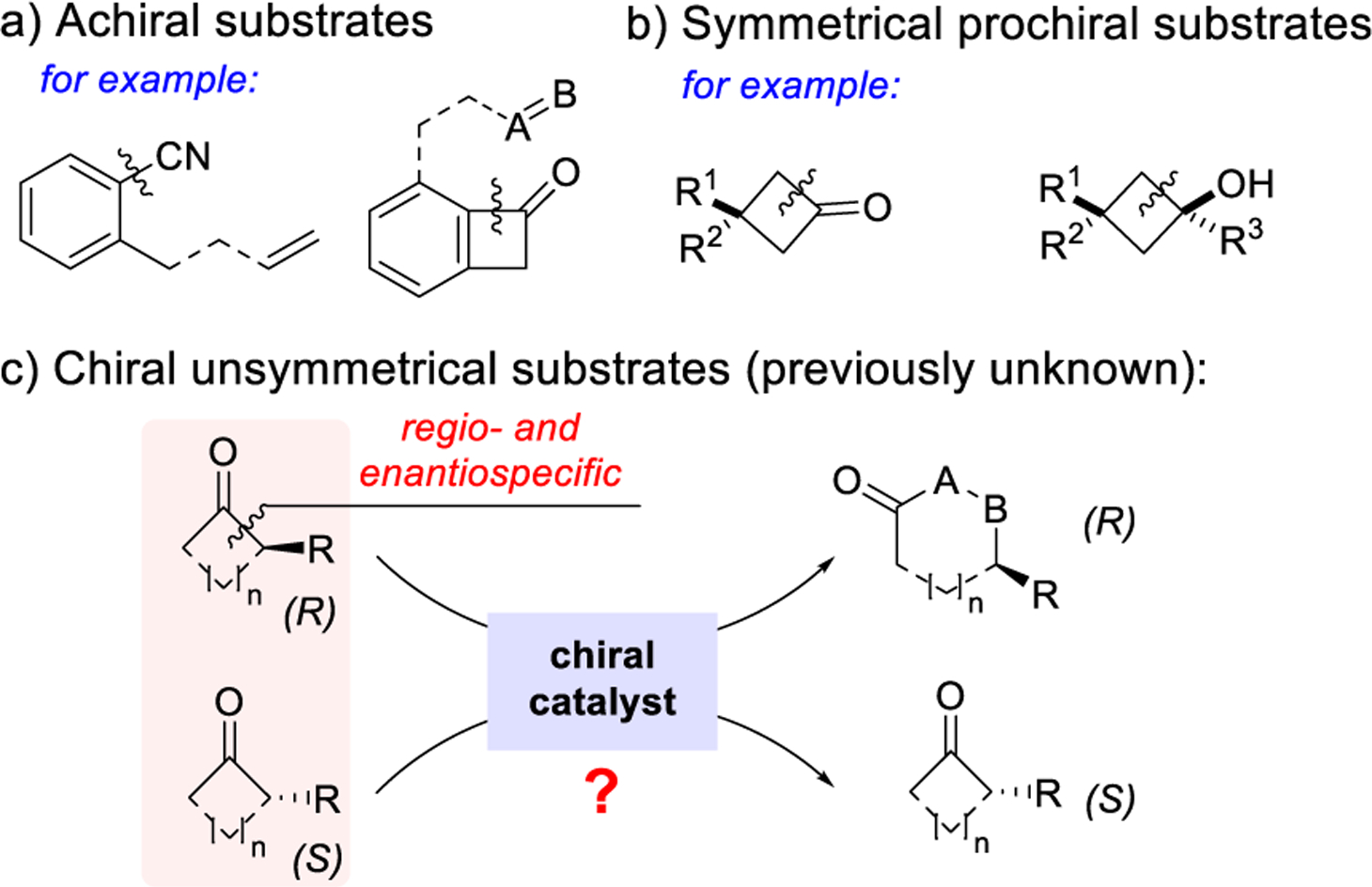
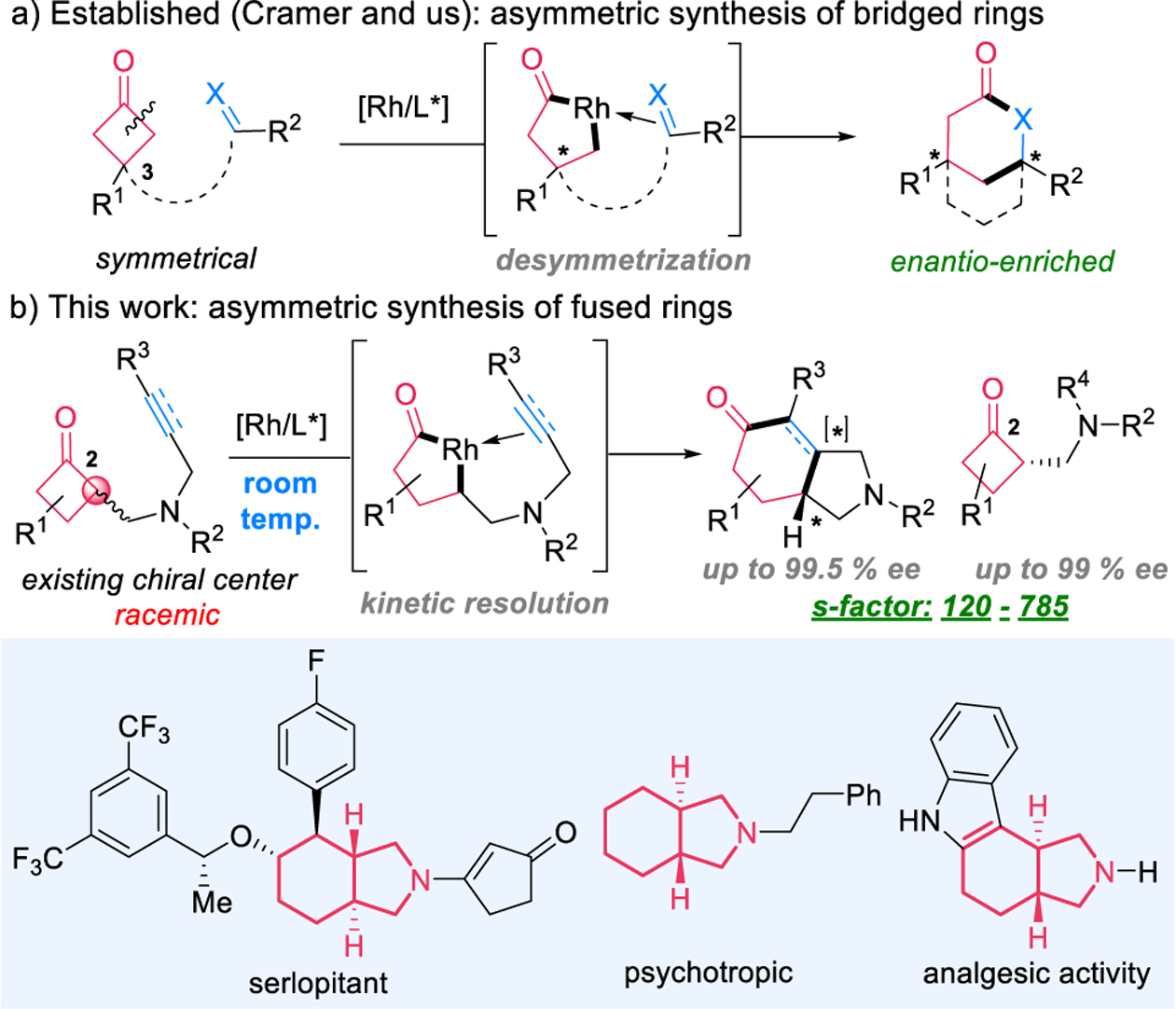
Our laboratory has an ongoing interest in developing a “cut-and-sew” approach for synthesis of various polycyclic structures widely found in bioactive compounds.3r This approach involves oxidative addition of a transition metal into the C–C bond of a cyclic ketone followed by migratory insertion of an unsaturated unit. In particular, the use of cyclobutanones as a common building block has been demonstrated in forming bridged and fused rings.4 The enantioselective synthesis of bridged-ring systems via desymmetrization of cyclobutanones has been reported by Cramer2f–h and us2i,j through intramolecular carboacylation of olefins, carbonyls, and allenes (Scheme 2a). However, enantioselective construction of fused rings, which would require substrates containing an existing C2-stereocenter, remained unknown and challenging. First, the catalyst needs to differentiate a pair of enantiomers (intermolecular recognition) instead of different π faces (e.g., olefins) or different sides of ketones (intramolecular recognition). Another issue is that cleavage of the less sterically hindered C–C bonds (the unproductive pathway) is generally more favorable.2j Hence, the desired reaction would have to address regio- and enantioselectivity problems simultaneously. Moreover, this type of reactions typically need high reaction temperatures (≥120 °C),5 which makes it difficult to control enantioselectivity. In this communication, we describe our preliminary results of a highly selective kinetic resolution of cyclobutanones via a Rh-catalyzed “cut-and-sew” reaction (Scheme 2b). Surprisingly, the reaction can operate at room temperature with the selectivity factor up to 785. This approach provides an asymmetric entry to the 5,6-fused bicycles6 often found in chiral bioactive compounds,7 as well as α-substituted cyclobutanones that are nontrivial to prepare enantioselectively without stoichiometric chiral auxiliaries.
Scheme 2. Enantioselective “Cut-and-Sew” Reactions between Cyclobutanones and Unsaturated Bonds.
To explore the feasibility of the kinetic resolution approach, cyclobutanone 1a with a tethered olefin was chosen as the model substrate. After extensive studies, it was ultimately found that a combination of a cationic Rh complex and DTBM-segphos permitted a room-temperature carboacylation of 1a (Table 1). The kinetic resolution was highly selective: both product 2a and cyclobutanone 1a were obtained in nearly theoretical yields and excellent optical purity (98% ee) with selectivity factor8 of 458. The reaction was also diastereoselective: the trans-5,6-fused ring (2a) was observed as a single diastereomer. The absolute stereochemistry of product 2a was confirmed through X-ray crystallography, which shows that the S-enantiomer of the substrate reacted selectively. The cationic rhodium catalyst can be smoothly generated in situ from [Rh(C2H4)2Cl]2, (R)-DTBM-segphos, and AgSbF6 in 1,4-dioxane. It is noteworthy that fused ring formation via a cut-and-sew reaction between cyclobutanones and olefins has not been reported previously.6
Table 1.
Selected Conditions Optimizationa
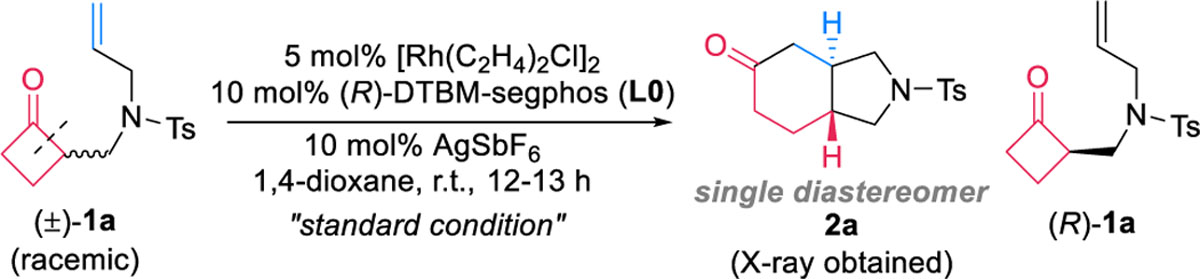 | |||||||
|---|---|---|---|---|---|---|---|
| entry | modification from “standard condition” | yield of 2a (ee)b + 1a (ee)b | s (conv.)c | ||||
| 1 | None | 46% | (98%) | 47% | (98%) | 458 | (50%) |
| 2 | w/o AgSbF6 | 10% | N/D | 89% | N/D | N/D | |
| 3 | w/o (R)-DTBM-segphos | 0% | N/A | 81% | N/A | N/A | |
| 4 | w/o [Rh(C2H4)2CI]2/AgSbF6 | 0% | N/A | 99% | N/A | N/A | |
| 5 | [Rh(coe)2CI]2 instead of [Rh(C2H4)2CI]2 | 44% | (99%) | 48% | (95%) | 747 | (49%) |
| 6 | [Rh(cod)CI]2 instead of [Rh(C2H4)2CI]2 | 0% | N/A | 99% | N/A | N/A | |
| 7 | [Rh(CO)2CI]2 instead of [Rh(C2H4)2CI]2 | 10% | N/D | 90% | N/D | N/D | |
| 8 | AgBF4 instead of AgSbF6 | 44% | (98%) | 45% | (96%) | 392 | (49%) |
| 9 | AgNTf2 instead of AgSbF6 | 45% | (98%) | 45% | (95%) | 371 | (49%) |
| 10 | L1 instead of L0 | 12% | N/D | 40% | N/D | N/D | |
| 11 | L2 instead of L0 | 10% | N/D | 67% | N/D | N/D | |
| 12 | L3 instead of L0 | 0% | N/A | 49% | N/A | N/A | |
| 13 | L4 instead of L0 | 0% | N/A | 95% | N/A | N/A | |
| 14 | L5 instead of L0 | 0% | N/A | 95% | N/A | N/A | |
| 15 | THF instead of 1,4-dioxane | 36% | (99%) | 52% | (94%) | 713 | (49%) |
| 16 | toluene instead of 1,4-dioxane | 0% | N/A | 90% | N/A | N/A | |
| 17 | 5 mol% [Rh] | 41% | (99%) | 50% | (86%) | 556 | (46%) |
 | |||||||
The reaction was run on a 0.1 mmol scale at room temperature for 12–13 h, and all the yields are isolated yields except for entries 10–14 (determined by 1H-NMR using 1,1,2,2-tetrachloroethane as internal standard).
Determined by chiral HPLC.
Calculated conversion (C) = eeSM/(eeSM + eePR); selectivity (s) = ln[(1 − C)(1 − eeSM)]/ln[(1 − C)(1 + eeSM)].
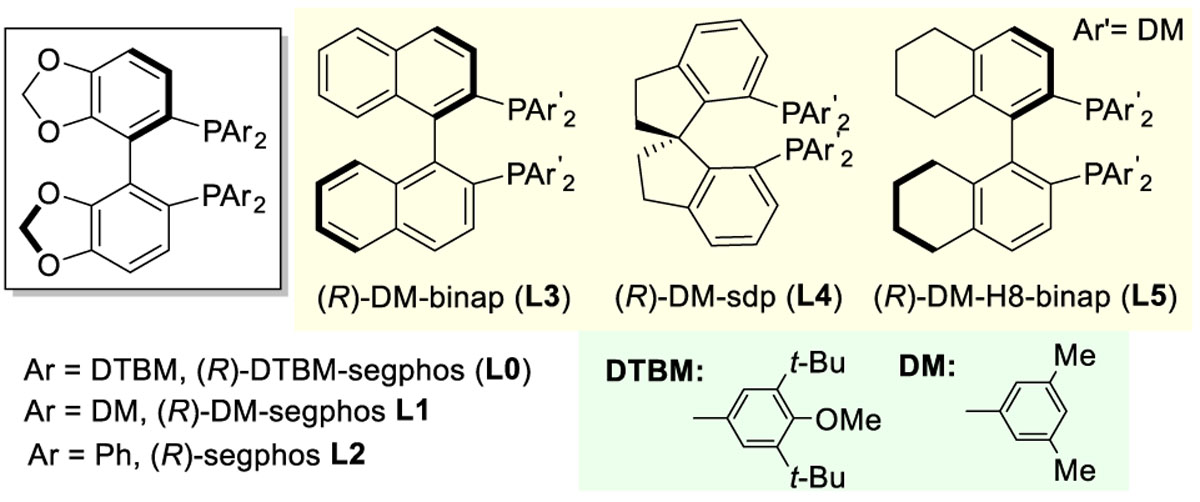
To gain more insights into this reaction, a series of control experiments were performed. Unsurprisingly, the rhodium, the silver salt, and the phosphine ligand were all essential for this transformation (Table 1, entries 2–4). The existing ligands on the rhodium precatalysts were found to be critical (entries 5–7). Ethylene and cyclooctene (COE) ligands, which undergo easier ligand substitution reactions, can both provide excellent yields and enantioselectivity; in contrast, stronger 1,5-cyclooctadiene (COD) and electron-deficient CO ligand led to much lower conversions. The counterions for the in situ-generated cationic rhodium catalysts can be extended to BF4 and NTf2, although the ee’s for the recycled cyclobutanone were slightly diminished with these catalysts (entries 8 and 9). DTBM-segphos was found to be crucial for this transformation. Based on the segphos backbone, switching the DTBM group to other aryl substituents significantly decreased the reactivity (entries 10 and 11). Chiral backbones other than segphos did not provide the desired product at room temperature (entries 12–14). Regarding the solvent used, 1,4-dioxane was superior, while THF gave some decomposition, and toluene afforded no desired product (entries 15 and 16). Lastly, comparably results could still be obtained with a halved catalyst loading (entry 17).
Next, we performed density functional theory (DFT) calculations to elucidate the origin of the unexpected high reactivity and enantioselectivity and the unique role of DTBM-segphos. The calculated energy profiles of the reaction with both enantiomers of 1a (Figure 1) indicated the rate- and enantioselectivity-determining step is the irreversible C–C oxidative addition because subsequent steps have lower energy barriers. Regarding the oxidative addition step, a relatively low activation barrier of 22.0 kcal/mol was observed for the more reactive enantiomer (S)-1a, which is consistent with the experimental reactivity at room temperature. The most favorable oxidative addition transition state (TS1) involves the cleavage of the more substituted C–C bond in (S)-1a and is stabilized by the weak coordination of the N-tosyl oxygen to the Rh center.6 The oxidative addition of the less substituted C–C bond (TS3) is kinetically disfavored due to the lack of such N-tosyl coordination. The C–C oxidative addition with the (R)-enantiomer of 1a (TS2) requires a much higher barrier, in agreement with the high selectivity factor observed in experiment. The origin of the enantioselectivity can be rationalized by the quadrant diagrams shown in Figure 2. The (S)-selective transition state (TS1) places the cyclobutanone moiety and the N-tosyl group on the substrate in quadrants I and III, which are less occupied by the C2-symmetric (R)-DTBM-segphos ligand. By contrast, in TS2, the cyclobutanone moiety is placed in the more occupied quadrant (II) and the N-Ts coordination cannot be achieved because the coordination site is blocked by the ligand in quadrant IV. The diastereoselectivity is determined in the subsequent alkene migratory insertion. This step strongly favors the formation of the trans-substituted pyrrolidine via TS4-trans. TS4-cis leading to the cis-diastereomer is 4.0 kcal/mol less stable because of the unfavorable steric repulsions about the forming C–C bond. The resulting seven-membered rhodacycle intermediate 8 undergoes facile C–C reductive elimination to yield product 2a and regenerate the Rh(I) catalyst.
Figure 1.
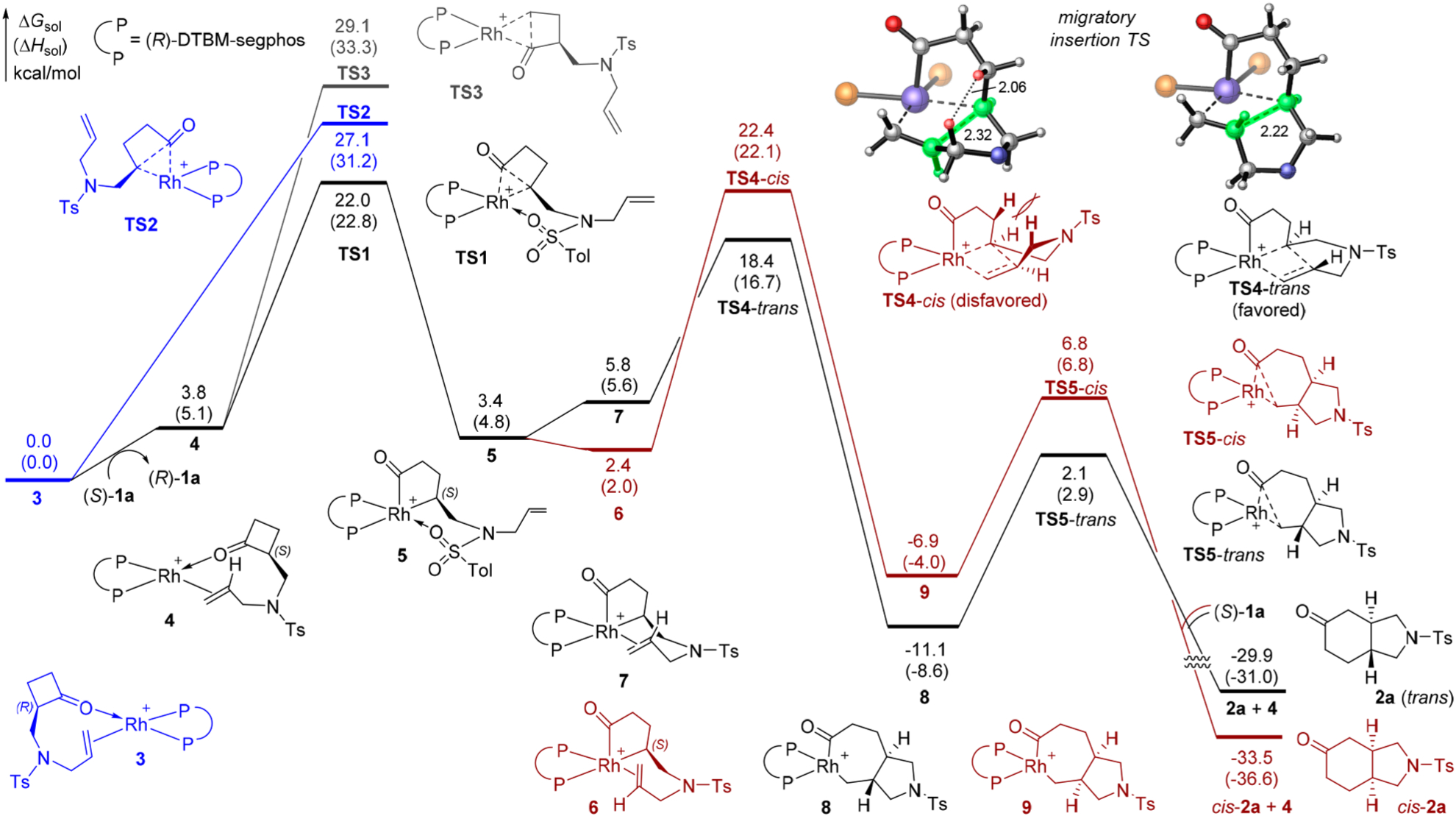
Computed energy profiles of the Rh-catalyzed C–C activation of (R)-1a (in blue) and (S)-1a (in black) at the M06/SDD-6–311+G(d,p), SMD(1,4-dioxane)//B3LYP/LANL2DZ-6–31G(d) level of theory. Distances are in angstroms.
Figure 2.
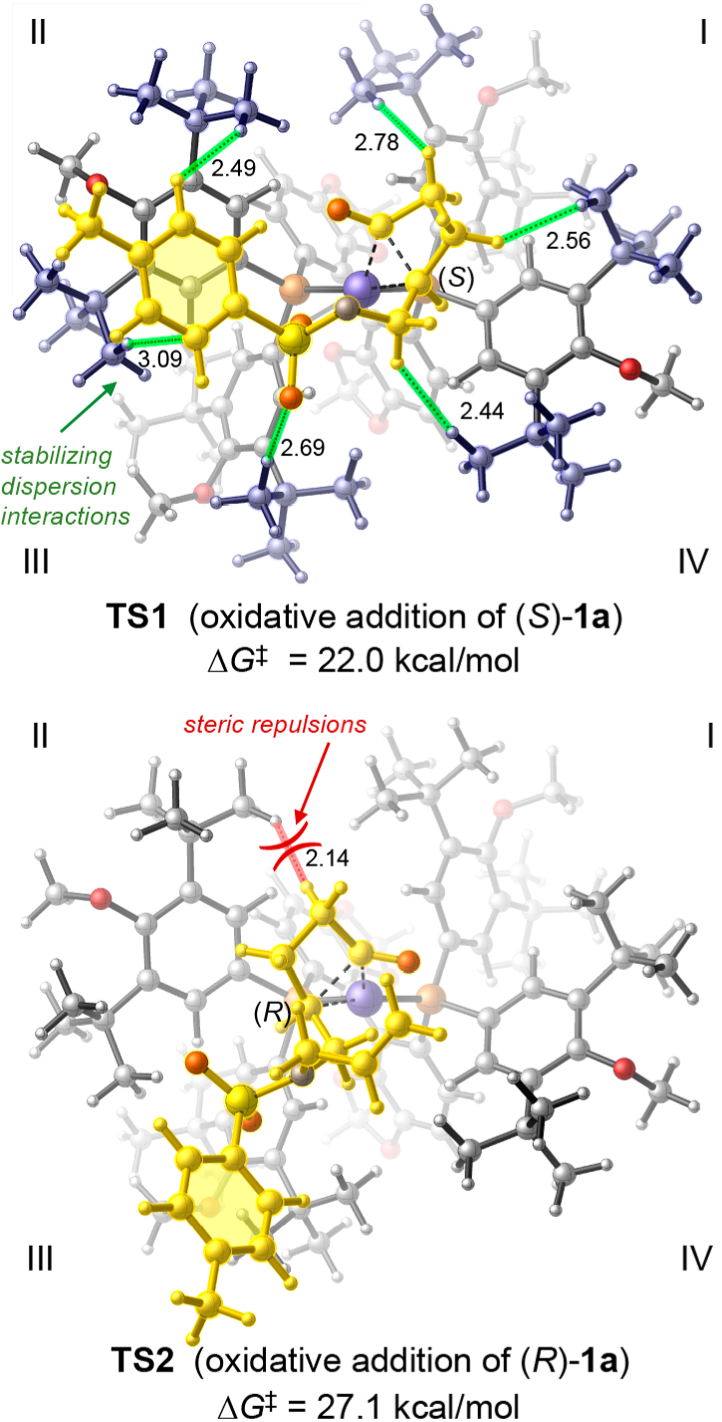
Oxidative addition transition states with the (R)-DTBM-segphos-supported Rh catalyst. The N-allyl group in TS1 is not shown for clarity.
In agreement with the low reactivity of the segphos (L2)-supported Rh catalyst, the rate-determining oxidative addition (TS6) requires a 3.3 kcal/mol higher barrier when the segphos ligand is employed in place of DTBM-segphos (Figure 3). To investigate the origin of the ligand effects on reactivity, we computed the dispersion interactions (ΔEdisp)9 between substrate (S)-1a and the bisphosphine ligand in the oxidative addition transition states (TS1 and TS6) and the catalyst resting states (3 and 10) using Grimme’s DFT-D3 method10 (see SI for details). Because TS1 is stabilized by various C–H/C–H and C–H/π interactions between the (R)-DTBM-segphos ligand and the substrate (Figure 2), the ΔEdisp of TS1 is 5.6 kcal/mol more favorable than that of the resting state 3, indicating a significant dispersion effect that promotes the oxidative addition.11 In the reaction with the smaller (R)-segphos ligand, the ΔEdisp of TS6 is only 3.6 kcal/mol more favorable than in the resting state 10. The weaker dispersion effects with (R)-segphos diminish the stabilization effects in the oxidative addition transition state, and thus lead to lower reactivity than in reactions with the (R)-DTBM-segphos ligand.
Figure 3.
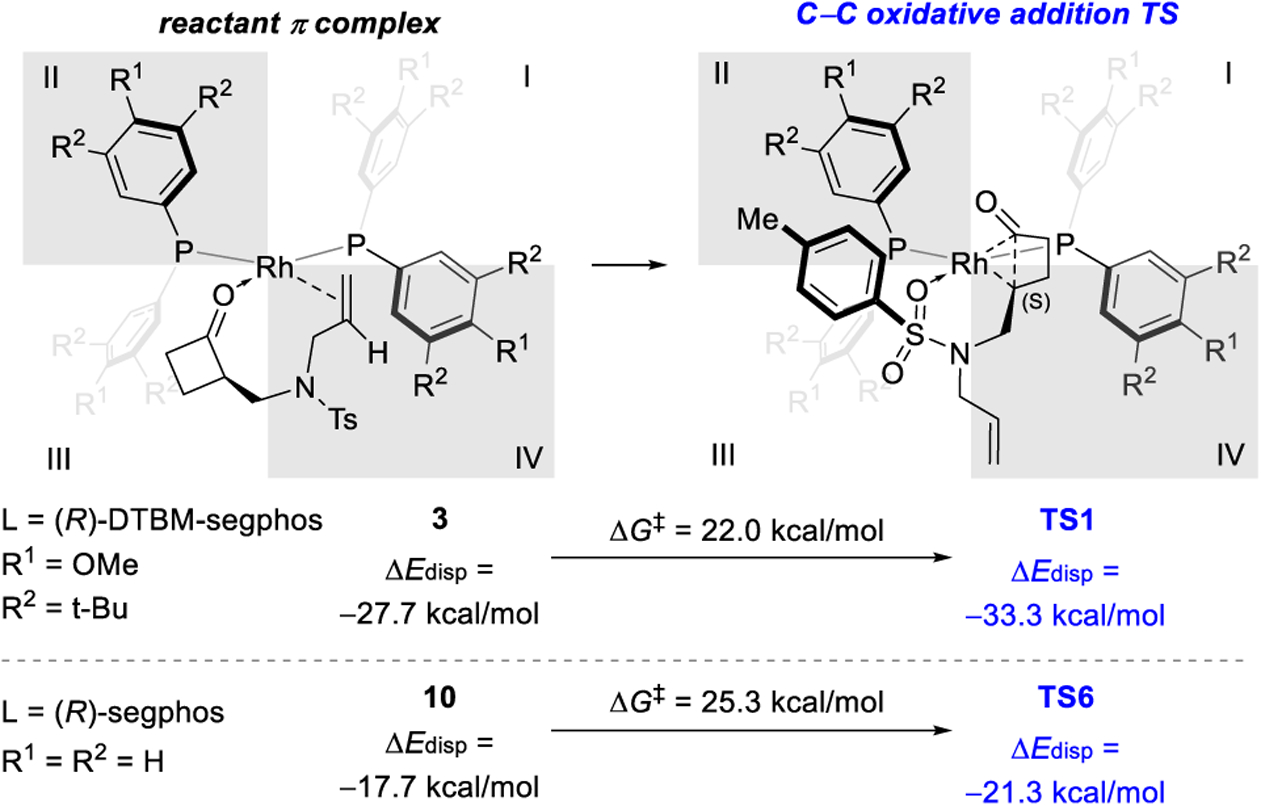
Ligand effects on reactivity. Dispersion interaction energies (ΔEdisp) between the bisphosphine ligand and the substrate were calculated with the DFT-D3 method.11
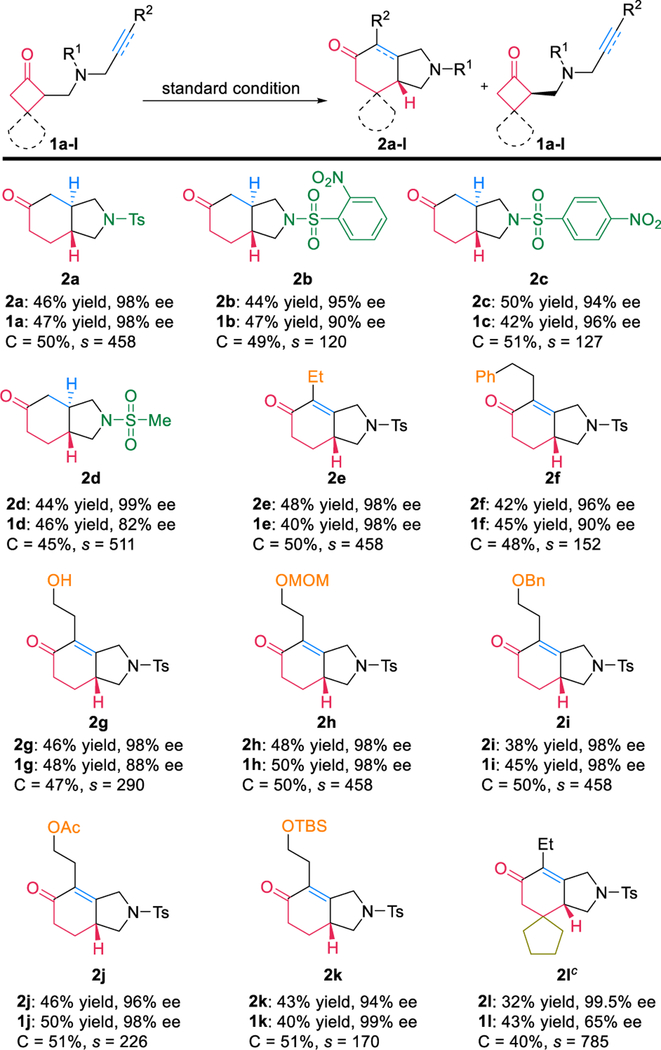
The substrate scope of the kinetic resolution was then investigated (Table 2). Given the importance of the sulfonyl oxygen coordination, the linkage can be changed to nosyl and methylsulfonyl groups with good yields and excellent enantioselectivity maintained (2b–2d).12 More substituted olefins were not reactive under the current conditions likely due to a slow migratory insertion step (the second highest energy barrier, Figure 1). Gratifyingly, alkyne-tethered cyclobutanones proved to be excellent substrates (2e–2l). Owing to the mild and redox-neutral reaction conditions, a number of labile groups were tolerated, including MOM- (2h), benzyl-(2i), acyl- (2j), and TBS-protected alcohols (2k), giving both good yields and excellent selectivity. Remarkably, free primary alcohol (2g) was well compatible. Substitution at the 3-position of cyclobutanone reduced the reactivity; however, at slightly elevated temperature (40 °C), the kinetic resolution still occurred with a good yield and excellent selectivity (2l).13
Table 2.
|
The reaction was run on a 0.1 mmol scale.
All yields are isolated yields; ee was determined by chiral HPLC; selectivity (s) = ln[(1 − C)(1 − eeSM)]/ln[(1 − C)(1 + eeSM)], calculated conversion (C) = eeSM/(eeSM + eePD).
Reaction was carried out with [Rh(CH2=CH2)2Cl]2 (10 mol%), (R)-DTBM-segphos (20 mol%), AgSbF6 (20 mol%), THF (1 mL) at 40 °C.
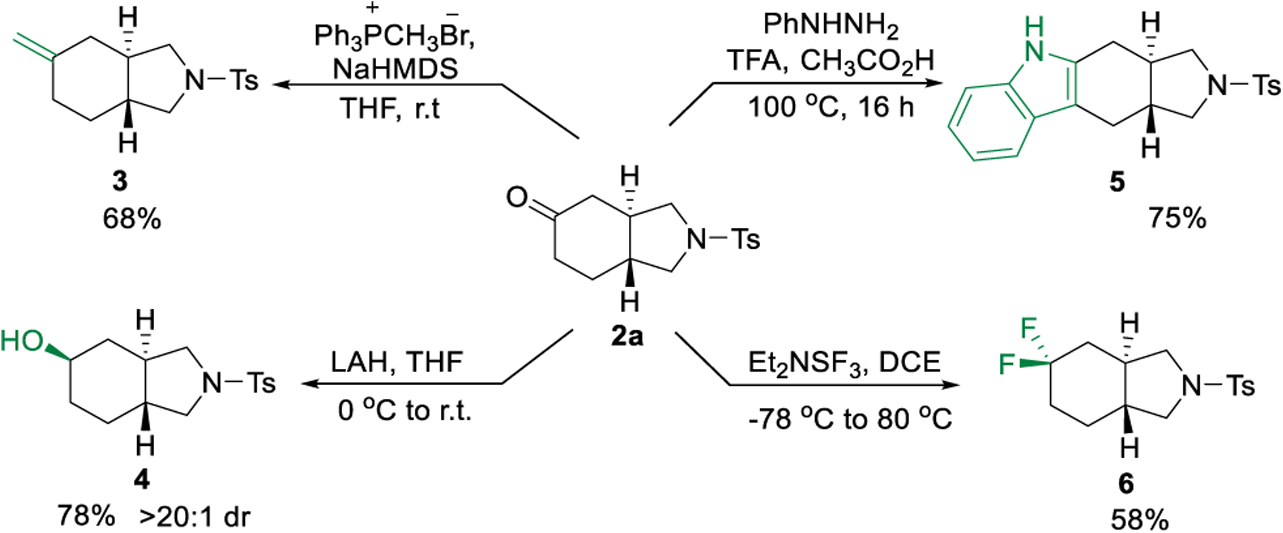
The enantio-enriched trans-5,6-fused bicyclic product (2a) can be conveniently transformed to a variety of other structural motifs (Scheme 3).14 For example, the ketone moiety underwent smooth olefination to give alkene 3 in 68% yield. LAH reduction afforded the secondary alcohol (4) with excellent diastereoselectivity. In addition, Fischer indole synthesis could be employed to efficiently generate tetracycle 5 in a good yield. Moreover, the gem-difluoro compound (6) was obtained upon treatment of 2a with (diethylamino)sulfur trifluoride (DAST).
Scheme 3. Transformations of Bicyclic Product 2a.
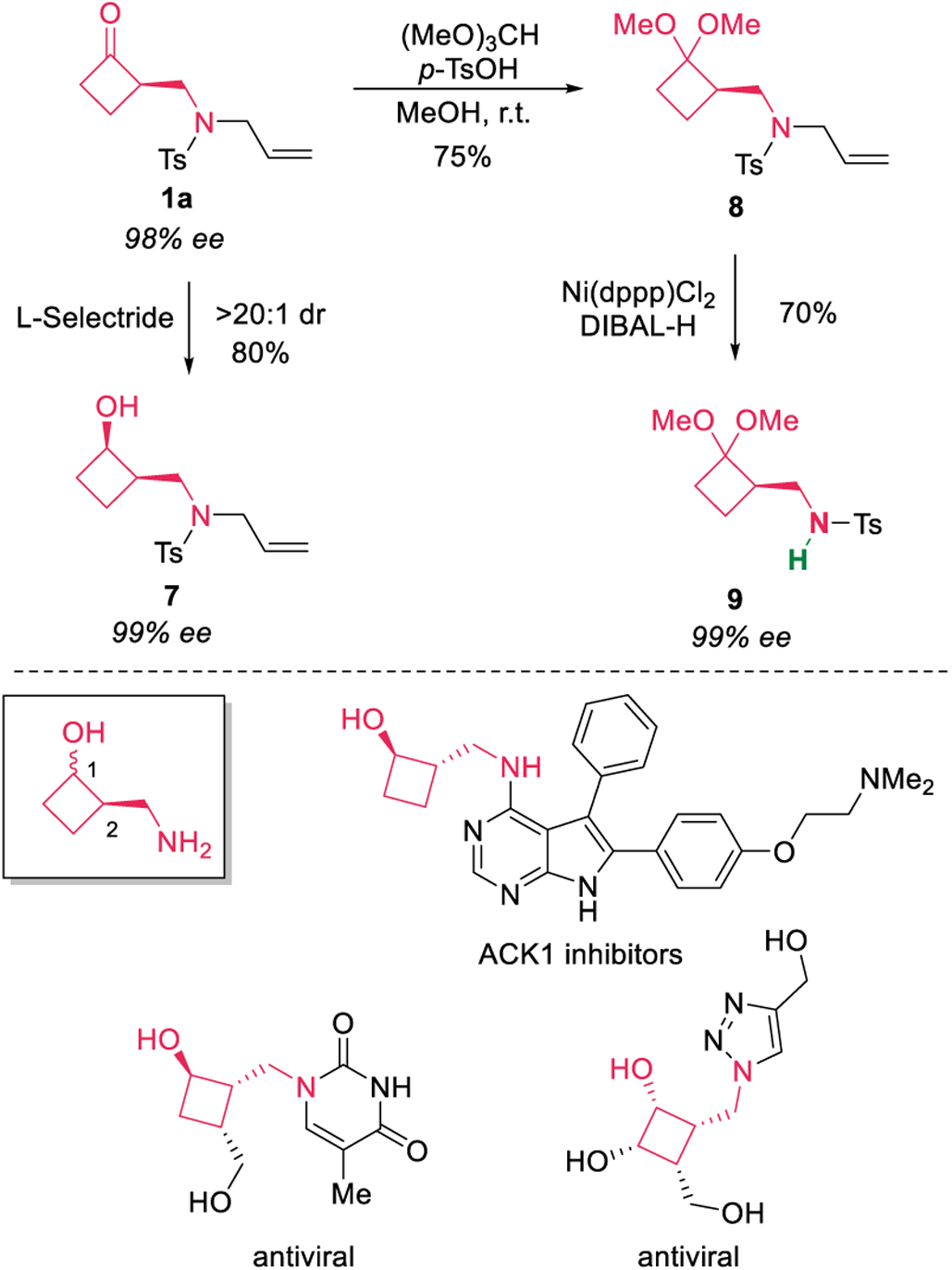
On the other hand, the obtained enantio-enriched cyclobutanone (1a) could serve as a versatile precursor to access other chiral four-membered ring scaffolds (Scheme 4). For example, the ketone moiety could be reduced to an alcohol (7) diastereoselectively. In addition, after the ketal protection, the allyl moiety can be removed via a Ni-catalyzed isomerization. Notably, no loss of enantio-purity was observed in these reactions. The 2-aminomethyl-1-cyclobutanol moiety has been found in a number of pharmaceutical agents.15
Scheme 4. Transformations of Enantio-enriched Cyclobutanone 1a.
In summary, we describe the discovery of kinetic resolutions/asymmetric transformations of cyclobutanones with existing stereocenters via a Rh-catalyzed “cut-and-sew” reaction. The reaction takes place at room temperature and tolerates many functional groups. High efficiency and excellent enantio- and diastereoselectivity have been obtained. The enantio-enriched trans-5,6-fused ring products and C2-substituted cyclobutanones could serve as useful building blocks for the asymmetric synthesis of bioactive compounds. As elucidated by the DFT study, the reaction mechanism and the origin of the ligand effect for enhanced reactivity and enantioselectivity could have implications for other C–C activation reactions. The development of related stereo-convergent reactions is ongoing in our laboratory.16
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge NIGMS (2R01GM109054, G.D.) and NSF (CHE-1654122, P.L.) for funding. We thank Dr. Alexander Filatov for X-ray structures. Chiral Technologies is thanked for their generous donation of chiral HPLC columns. DFT calculations were performed at the Center for Research Computing at the University of Pittsburgh and the Extreme Science and Engineering Discovery Environment (XSEDE) supported by NSF.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b09344.
Experimental procedures, spectral data, computational details, additional computational results, and Cartesian coordinates (PDF)
Crystallographic data for 2a (CIF)
The authors declare no competing financial interest.
REFERENCES
- (1).For selected reviews on stereoselective C–H functionalizations, see:; (a) Giri R; Shi B-F; Engle KM; Maugel N; Yu J-Q Transition Metal-Catalyzed C–H Activation Reactions: Diastereoselectivity and Enantioselectivity. Chem. Soc. Rev 2009, 38, 3242–3272. [DOI] [PubMed] [Google Scholar]; (b) Doyle MP; Duffy R; Ratnikov M; Zhou L Catalytic Carbene Insertion into C–H Bonds. Chem. Rev 2010, 110, 704–724. [DOI] [PubMed] [Google Scholar]; (c) Newton CG; Wang S-G; Oliveira CC; Cramer N Catalytic Enantioselective Transformations Involving C–H Bond Cleavage by Transition-Metal Complexes. Chem. Rev 2017, 117, 8908–8976. [DOI] [PubMed] [Google Scholar]; (d) Woźniak Ł; Cramer N Enantioselective C–H Bond Functionalizations by 3d Transition-Metal Catalysts. Trends in Chemistry 2019, 1, 471–484. [Google Scholar]
- (2).For a recent review, see:; (a) Souillart L; Parker E; Cramer N Asymmetric Transformations via C–C Bond Cleavage In C-C Bond Activation; Dong G, Ed.; Springer: Berlin, Heidelberg, 2014; pp 163–193. For representative examples through oxidative addition into C–C bonds, see: [DOI] [PubMed] [Google Scholar]; (b) Watson MP; Jacobsen EN Asymmetric Intramolecular Arylcyanation of Unactivated Olefins via C–CN Bond Activation. J. Am. Chem. Soc 2008, 130, 12594–12595. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Nakao Y; Ebata S; Yada A; Hiyama T; Ikawa M; Ogoshi S Intramolecular Arylcyanation of Alkenes Catalyzed by Nickel/AlMe2Cl. J. Am. Chem. Soc 2008, 130, 12874–12875. [DOI] [PubMed] [Google Scholar]; (d) Xu T; Ko HM; Savage NA; Dong G Highly Enantioselective Rh-Catalyzed Carboacylation of Olefins: Efficient Syntheses of Chiral Poly-Fused Rings. J. Am. Chem. Soc 2012, 134, 20005–20008. [DOI] [PubMed] [Google Scholar]; (e) Deng L; Xu T; Li H; Dong G Enantioselective Rh-Catalyzed Carboacylation of C=N Bonds via C–C Activation of Benzocyclobutenones. J. Am. Chem. Soc 2016, 138, 369–374. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Parker E; Cramer N Asymmetric Rhodium(I)-Catalyzed C–C Activations with Zwitterionic Bis-phospholane Ligands. Organometallics 2014, 33, 780–787. [Google Scholar]; (g) Souillart L; Parker E; Cramer N Highly Enantioselective Rhodium(I)-Catalyzed Activation of Enantiotopic Cyclobutanone C–C Bonds. Angew. Chem., Int. Ed 2014, 53, 3001–3005. [DOI] [PubMed] [Google Scholar]; (h) Souillart L; Cramer N Highly Enantioselective Rhodium(I)-Catalyzed Carbonyl Carboacylations Initiated by C–C Bond Activation. Angew. Chem., Int. Ed 2014, 53, 9640–9644. [DOI] [PubMed] [Google Scholar]; (i) Ko HM; Dong G Cooperative Activation of Cyclobutanones and Olefins Leads to Bridged Ring Systems by a Catalytic [4 + 2] Coupling. Nat. Chem 2014, 6, 739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Zhou X; Dong G 4 + 1) vs (4 + 2): Catalytic Intramolecular Coupling between Cyclobutanones and Trisubstituted Allenes via C–C Activation. J. Am. Chem. Soc 2015, 137, 13715–13721. [DOI] [PMC free article] [PubMed] [Google Scholar]; For representative examples through β-carbon elimination, see:; (k) Matsuda T; Shigeno M; Makino M; Murakami M Enantioselective C–C Bond Cleavage Creating Chiral Quaternary Carbon Centers. Org. Lett 2006, 8, 3379–3381. [DOI] [PubMed] [Google Scholar]; (l) Matsuda T; Shigeno M; Murakami M Asymmetric Synthesis of 3,4-Dihydrocoumarins by Rhodium-Catalyzed Reaction of 3-(2-Hydroxyphenyl)-cyclobutanones. J. Am. Chem. Soc 2007, 129, 12086–12087. [DOI] [PubMed] [Google Scholar]; (m) Liu L; Ishida N; Murakami M Atom- and Step-Economical Pathway to Chiral Benzobicyclo[2.2.2]octenones through Carbon-Carbon Bond Cleavage. Angew. Chem., Int. Ed 2012, 51, 2485–2488. [DOI] [PubMed] [Google Scholar]; (n) Matsumura S; Maeda Y; Nishimura T; Uemura S Palladium-Catalyzed Asymmetric Arylation, Vinylation, and Allenylation of tert-Cyclobutanols via Enantioselective C–C Bond Cleavage. J. Am. Chem. Soc 2003, 125, 8862–8869. [DOI] [PubMed] [Google Scholar]; (o) Seiser T; Cramer N Enantioselective C–C Bond Activation of Allenyl Cyclobutanes: Access to Cyclohexenones with Quaternary Stereogenic Centers. Angew. Chem., Int. Ed 2008, 47, 9294–9297. [DOI] [PubMed] [Google Scholar]
- (3).For selected reviews on C–C bond activation, see:; (a) Crabtree RH The Organometallic Chemistry of Alkanes. Chem. Rev 1985, 85, 245–269. [Google Scholar]; (b) Jones WD The Fall of the C-C Bond. Nature 1993, 364, 676–677. [Google Scholar]; (c) Murakami M; Ito Y Cleavage of Carbon–Carbon Single Bonds by Transition Metals. Top. Organomet. Chem 1999, 3, 97. [Google Scholar]; (d) Rybtchinski B; Milstein D Metal Insertion into C–C Bonds in Solution. Angew. Chem., Int. Ed 1999, 38, 870–883. [DOI] [PubMed] [Google Scholar]; (e) Jun C-H Transition Metal-Catalyzed Carbon-Carbon Bond Activation. Chem. Soc. Rev 2004, 33, 610–618. [DOI] [PubMed] [Google Scholar]; (f) Necas D; Kotora M Rhodium-Catalyzed C-C Bond Cleavage Reactions. Curr. Org. Chem 2007, 11, 1566–1591. [Google Scholar]; (g) Park YJ; Park J-W; Jun C-H Metal–Organic Cooperative Catalysis in C–H and C–C Bond Activation and Its Concurrent Recovery. Acc. Chem. Res 2008, 41, 222–234. [DOI] [PubMed] [Google Scholar]; (h) Murakami M; Matsuda T Metal-Catalysed Cleavage of Carbon-Carbon Bonds. Chem. Commun 2011, 47, 1100–1105. [DOI] [PubMed] [Google Scholar]; (i) Seiser T; Saget T; Tran DN; Cramer N Cyclobutanes in Catalysis. Angew. Chem., Int. Ed 2011, 50, 7740–7752. [DOI] [PubMed] [Google Scholar]; (j) Korotvicka A; Necas D; Kotora M Rhodium-catalyzed C–C Bond Cleavage Reactions - An Update. Curr. Org. Chem 2012, 16, 1170–1214. [Google Scholar]; (k) Chen F; Wang T; Jiao N Recent Advances in Transition-Metal-Catalyzed Functionalization of Unstrained Carbon–Carbon Bonds. Chem. Rev 2014, 114, 8613–8661. [DOI] [PubMed] [Google Scholar]; (l) Dermenci A; Coe JW; Dong G Direct Activation of Relatively Unstrained Carbon-Carbon Bonds in Homogeneous Systems. Org. Chem. Front 2014, 1, 567–581. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Dong G C–C bond activation; Springer-Verlag: Berlin, 2014; Vol. 346. [Google Scholar]; (n) Murakami M; Ishida N, Fundamental Reactions to Cleave Carbon–Carbon σ-Bonds with Transition Metal Complexes In Cleavage of Carbon-Carbon Single Bonds by Transition Metals; Wiley-VCH Verlag GmbH & Co. KGaA: 2015; p 1. [Google Scholar]; (o) Souillart L; Cramer N Catalytic C–C Bond Activations via Oxidative Addition to Transition Metals. Chem. Rev 2015, 115, 9410–9464. [DOI] [PubMed] [Google Scholar]; (p) Fumagalli G; Stanton S; Bower JF Recent Methodologies That Exploit C–C Single-Bond Cleavage of Strained Ring Systems by Transition Metal Complexes. Chem. Rev 2017, 117, 9404–9432. [DOI] [PubMed] [Google Scholar]; (q) Kim D-S; Park W-J; Jun C-H Metal–Organic Cooperative Catalysis in C–H and C–C Bond Activation. Chem. Rev 2017, 117, 8977–9015. [DOI] [PubMed] [Google Scholar]; (r) Chen P.-h.; Billett BA; Tsukamoto T; Dong G “Cut and Sew” Transformations via Transition-Metal-Catalyzed Carbon–Carbon Bond Activation. ACS Catal. 2017, 7, 1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).For seminal examples, see:; (a) Murakami M; Itahashi T; Ito Y Catalyzed Intramolecular Olefin Insertion into a Carbon–Carbon Single Bond. J. Am. Chem. Soc 2002, 124, 13976–13977. [DOI] [PubMed] [Google Scholar]; (b) Zhou X; Zafar I; Dong G Catalytic Intramolecular Decarbonylative Coupling of 3-Aminocyclobutenones and Alkenes: a Unique Approach to [3.1.0] Bicycles. Tetrahedron 2015, 71, 4478–4483. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Deng L; Jin L; Dong G Fused-Ring Formation by an Intramolecular “Cut-and-Sew” Reaction between Cyclobutanones and Alkynes. Angew. Chem., Int. Ed 2018, 57, 2702–2706. See also refs 2f–j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).For C–C activation via a β-carbon elimination pathway, the reaction temperatures are typically lower. For examples, see refs 2l–o.
- (6).The same type of products could also be obtained via a (3+1+2) cycloaddition with aminomethylcyclopropanes by Bower, though the asymmetric version has yet been reported:; Wang G-W; McCreanor NG; Shaw MH; Whittingham WG; Bower JF New Initiation Modes for Directed Carbonylative C–C Bond Activation: Rhodium-Catalyzed (3 + 1 + 2) Cycloadditions of Aminomethylcyclopropanes. J. Am. Chem. Soc 2016, 138, 13501–13504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) Speck K; Magauer T The Chemistry of Isoindole Natural Products. Beilstein J. Org. Chem 2013, 9, 2048–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rateb ME; Houssen WE; Schumacher M; Harrison WTA; Diederich M; Ebel R; Jaspars M Bioactive Diterpene Derivatives from the Marine Sponge Spongionella sp. J. Nat. Prod 2009, 72, 1471–1476. [DOI] [PubMed] [Google Scholar]; (c) Abe S; Tanaka N; Kobayashi J Prenylated Acylphloroglucinols, Chipericumins A–D, from Hypericum chinense. J. Nat. Prod 2012, 75, 484–488. [DOI] [PubMed] [Google Scholar]; (d) Ständer S; Kwon P; Hirman J; Perlman AJ; Weisshaar E; Metz M; Luger TA Serlopitant Reduced Pruritus in Patients with Prurigo Nodularis in a Phase 2, Randomized, Placebo-Controlled Trial. J. Am. Acad. Dermatol 2019, 80, 1395–1402. [DOI] [PubMed] [Google Scholar]
- (8).Kagan HB; Fiaud JC In Topics in Stereochemistry; Eliel EL; Wilen SH; Allinger NL, Eds.; Interscience: New York, 1988; Vol. 18, pp 249–330. [Google Scholar]
- (9).(a) Lu G; Liu RY; Yang Y; Fang C; Lambrecht DS; Buchwald SL; Liu P Ligand–Substrate Dispersion Facilitates the Copper-Catalyzed Hydroamination of Unactivated Olefins. J. Am. Chem. Soc 2017, 139, 16548–16555. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Thomas AA; Speck K; Kevlishvili I; Lu Z; Liu P; Buchwald SL Mechanistically Guided Design of Ligands That Significantly Improve the Efficiency of CuH-Catalyzed Hydroamination Reactions. J. Am. Chem. Soc 2018, 140, 13976–13984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Grimme S; Antony J; Ehrlich S; Krieg H A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys 2010, 132, 154104. [DOI] [PubMed] [Google Scholar]
- (11).For examples of dispersion-effects-promoted reactivity, see:; (a) Ref 9. [Google Scholar]; (b) Neel AJ; Hilton MJ; Sigman MS; Toste FD Exploiting non-covalent π interactions for catalyst design. Nature 2017, 543, 637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lu Q; Neese F; Bistoni G London dispersion effects in the coordination and activation of alkanes in σ-complexes: a local energy decomposition study. Phys. Chem. Chem. Phys 2019, 21, 11569–11577. [DOI] [PubMed] [Google Scholar]
- (12).Carbon, oxygen, and N -acyl linkers were unsuccessful with this catalytic system.
-
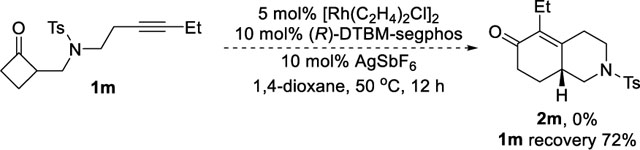
(13).The substrate with a homopropargyl group on the NTs linker failed to deliver the 6,6-fused product even at elevated temperature:
- (14).Bower also showed several derivatizations in the Supporting Information of ref 6, including Wolff–Kishner reduction, Grignard addition, etc.
- (15).(a) Jiao X; Kopecky DJ; Liu J; Liu J; Jaen JC; Cardozo MG; Sharma R; Walker N; Wesche H; Li S; Farrelly E; Xiao S-H; Wang Z; Kayser F Synthesis and optimization of substituted furo[2,3-d]-pyrimidin-4-amines and 7H-pyrrolo[2,3-d]pyrimidin-4-amines as ACK1 inhibitors. Bioorg. Med. Chem. Lett 2012, 22, 6212–6217. [DOI] [PubMed] [Google Scholar]; (b) Marsac Y; Nourry A; Legoupy S; Pipelier M; Dubreuil D; Aubertin A-M; Bourgougnon N; Benhida R; Huet F Synthesis of mono- and polyhydroxylated cyclobutane nucleoside analogs. Tetrahedron 2005, 61, 7607–7612. [Google Scholar]; (c) Tran TTT; Ngo NT; Dinh TH; Vo-Thanh G; Legoupy S Synthesis of Novel Triazolo Cyclobutane Nucleoside Analogs. Bull. Korean Chem. Soc 2015, 36, 1390–1395. [Google Scholar]
- (16).Attempts to realize the dynamic kinetic asymmetric transformation through adding a base or acid to epimerize the C2 stereocenter were not fruitful under the current conditions.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.