

Abstract
Palladium/norbornene cooperative catalysis has emerged as a distinct approach to construct polyfunctionalized arenes from readily available starting materials. This Review provides a comprehensive overview of this field, including the early stoichiometric investigations, catalytic reaction developments, as well as the applications in the syntheses of bioactive compounds and polymers. The section of catalytic reactions is divided into two parts according to the reaction initiation mode: Pd(0)-initiated reactions and Pd(II)-initiated reactions.
Graphical Abstract

1. INTRODUCTION
Polysubstituted aromatics are ubiquitously found in pharmaceuticals and agrochemicals. In particular, benzene and pyridine represent the most frequently used ring systems from small molecule drugs.1 During the past decades, cross-coupling and nucleophilic aromatic substitutions (SNAr) clearly have become indispensable tools for preparing functionalized arenes from readily available aryl halides (haloarenes), and have been widely used in drug discovery and development.2,3 In a typical cross-coupling4 or SNAr reaction,5 a halogen substituent (X, or other leaving group) is replaced by a nucleophile at arene ipso position, which can be catalyzed by a transition metal (TM) such as palladium (Scheme 1A). While these reactions, particularly with the advancement of novel ligands and catalysts, can be highly efficient, the position of a newly introduced functional group (FG) is mainly dictated by the position of the halogen substituent. Hence, these arene-functionalization approaches largely rely on the availability of the corresponding aryl electrophiles (ArX).
Scheme 1.

Pd/NBE Catalysis: Merge of Ipso and Ortho Functionalizations
Besides using ArX as substrates, arene functionalization has also been frequently realized through substituting a less reactive C–H bond. Classical electrophilic aromatic substitution (EAS) is practical for electron-rich arenes; however, its site-selectivity is normally controlled by the inherent electronic bias of substrates.6 Recently, ortho metalation approaches, mediated by either stoichiometric organometallics (e.g., organolithium)7 or catalytic TMs,8,9 enabled broadly useful ortho C–H functionalization methods, which require assistance of a directing group (DG) (Scheme 1B). While examples of direct metalation at other positions of arenes with and without a DG have been reported,10–13 more general approaches remain to be uncovered.
Complementary to the aforementioned approaches, the palladium/norbornene (Pd/NBE) cooperative catalysis, also known as Catellani-type reactions, merge the merits of cross-coupling and ortho metalation, two powerful organic reactions, into one single transformation. The Pd/NBE chemistry, originally discovered by Catellani, allows simultaneous functionalization of both ortho and ipso positions of simple aryl halides (Scheme 1C). A nucleophile is coupled at the ipso position like cross-coupling reactions, while an electrophile is introduced at the ortho position, analogous to the ortho metalation approaches. Thus, the Pd/NBE catalysis holds the potential to introduce unusual strategies that can streamline synthesis of complex polysubstituted aromatic compounds. Beyond using aryl iodides as substrates, new advances on Pd(II)-initiated processes have emerged recently, which allow for site-selective functionalization of indoles, meta-C–H functionalization of arenes with ortho DGs and use of arylboron species as substrates.
This Review article was inspired by several excellent reviews and accounts in the field of Pd/NBE catalysis,14–22 and here will provide a comprehensive summary up to January 2019. It is structured in the following way: beginning with the section of early stoichiometric reactions, we will provide the historical background and describe the key studies of each step in the catalytic cycle. We will then discuss the development of catalytic reactions, including the classical Pd(0)-initiated reactions and the more recent Pd(II)-initiated reactions. Finally, the synthetic applications on preparing bioactive molecules and polymers will be summarized. This Review is focused on reactions using NBE as the cocatalyst, therefore catalytic reactions using NBE as stoichiometric reactants will not be included unless closely related.
2. STOICHIOMETRIC REACTIONS
2.1. Formation of Aryl-Norbornyl-Palladacycle (ANP) Intermediate
In a regular Mizoroki–Heck reaction, oxidative addition of an aryl halide to Pd(0) followed by olefin migratory insertion (e.g., ethylene) results in an alkyl-Pd(II) intermediate that would undergo fast β-hydrogen elimination to form styrene-type products.23,24 However, using a rigid olefin, such as NBE, instead could lead to a different scenario. For example, in 1974 Horino and co-workers isolated complex (1) from NBE migratory insertion into the Pd–Ph bond (Scheme 2).25 Through further reduction by LiAlD4, the deuterium-labeled product confirmed the exo cis migratory addition process, which was the reason for a difficult β-hydrogen elimination step.
Scheme 2.

Migratory Insertion of NBE into the Pd–Ph Bond
In 1982, Catellani and Chiusoli examined the reaction between bromobenzene and NBE using Pd(PPh3)4 as the catalyst (Scheme 3).26 In the presence of potassium acetate, a cyclic compound (2) containing two molecules of NBE was isolated, in which the ortho C–H bond of the arene was activated. Later, when potassium tert-butoxide was used instead, a different product 3 containing only one molecule of NBE, but two molecules of arenes, was formed.27 To explain the formation of these ortho functionalized arene products, an aryl-norbornyl-palladacycle (ANP) (4) was proposed to be the intermediate, which is formed via ortho palladation of Horino’s intermediate (1). Bases seemed to play an important role in the reactivity of ANP. In the presence of potassium phenoxide, however, reductive elimination from the same intermediate 4 to norbornyl benzocyclobutene 5 occurred, along with an elimination product (6).28 It is interesting to note that, when using the sodium salt of di-tert-butyl-p-cresol, 5 was selectively formed in 85% yield.
Scheme 3.

Pd-Catalyzed Interrupted Heck Reaction of Phenyl Bromide with NBE
To probe the presence of the ANP intermediate, Catellani and Chiusoli prepared complex 7 with 1 equiv of phenanthroline ligand (Scheme 4).29 The addition of potassium phenoxide led to complex 8, which was fully characterized by 1H NMR and confirmed by its reaction with NaBD4. In 1992, Catellani and Chiusoli further examined the arene substituent effect on the rate of the palladacycle formation step.30 They found that complex 10 was stable below −50 °C but would undergo cyclometalation to form 11 at −30 °C. Half conversion of 10 to 11 occurred in approximately 10, 100, and 240 min for R = OMe, H, NO2, respectively. Based on this trend, they proposed electrophilic aromatic substitution via a Wheland-type intermediate. However, more recent computational studies31–33 indicated that the palladation step during the ANP formation can also proceed through a concerted metalation–deprotonation (CMD) mechanism, particularly in catalytic reactions.
Scheme 4.

Stoichiometric Studies on the Cyclopalladation Step
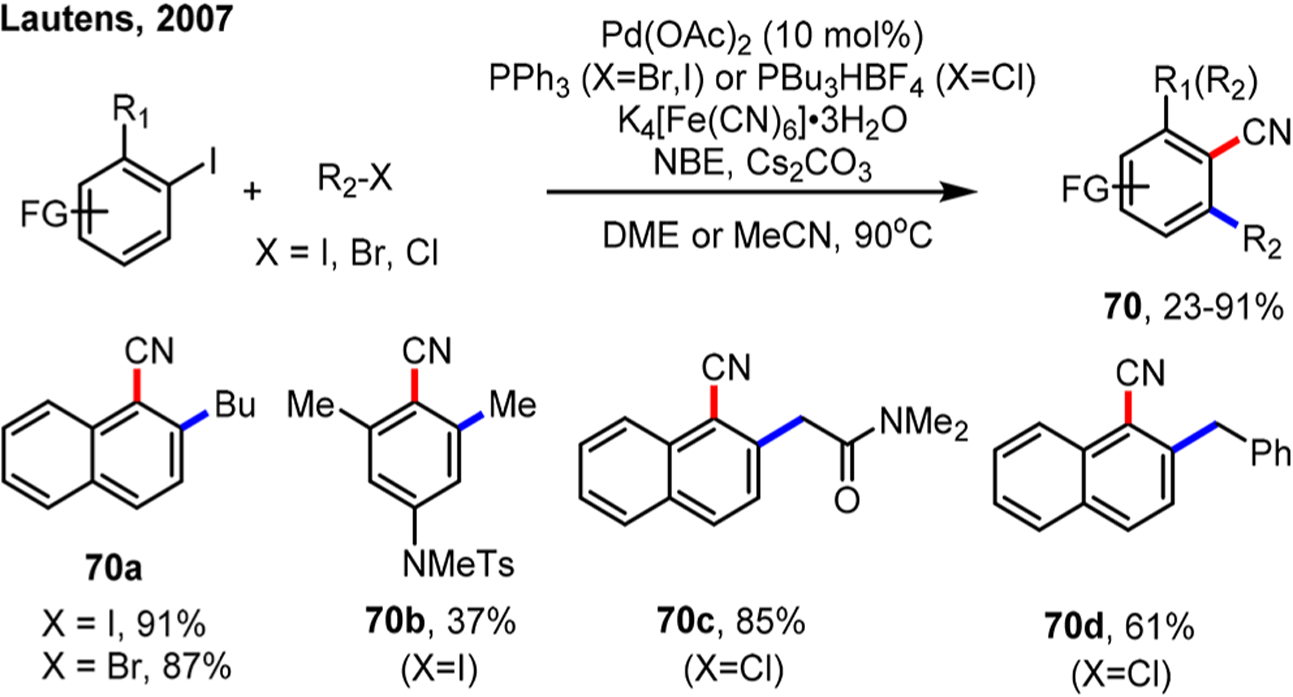
In 2011, Lautens and co-workers measured the kinetic isotopic effect (KIE) for the C–H metalation step (Scheme 5).34 The competition intermolecular KIE of 12 was determined to be 1.0, while the intramolecular KIE was found to be 4.2. The absence of a KIE in the intermolecular case suggested that ligand exchange with phenoxide could be rate-determining; the intramolecular KIE of 4.2 was consistent with a CMD mechanism.
Scheme 5.

Kinetic Isotopic Effect on the Cyclopalladation Step in Stoichiometric Studies
The structure of the migratory insertion product prior to palladacycle formation was also studied (Scheme 6). The Cheng group obtained a crystal structure of compound 14, prepared from the reaction between iodobenzene and norbornadiene.35 They found that the phenyl group was weakly bound to the palladium center in a η2 fashion, as the distance of Pd–Cipso and Pd–Cortho bonds were 2.43 and 2.59 Å, respectively. Interestingly, addition of excess ligands (e.g., PPh3, dppe, pyridine) did not replace the η2-coordinated arene. Presumably, the rigid skeleton of norbornadiene and the suitable cis arrangement of the phenyl ring and the palladium center contributed to the stabilization of the η2 interaction. The rotation of the aryl ring about the C–C bond was found to be rapid on the NMR time scale at 40 °C, but slow at the low temperature (−40 °C). Such a η2 interaction could play a role in the further C–H metalation step since it brings the palladium closer to the ortho C–H bond.
Scheme 6.

Formation of Intramolecular η2-Arene Palladium Species
Catellani later synthesized and characterized a series of dichloro-bridged arylbicycloheptylpalladium complexes with different substituents on the aryl ring.36 Among these complexes, the structure of ortho and para substituted complex 15 was characterized by X-ray crystallography. In addition, the rotation barrier for the ortho unsubstituted complex was determined by NMR to be ~17 kcal/mol. Interestingly, when an ortho methyl group was present in the complex, the palladium center favored a η1 interaction with the ipso carbon, according to both the NMR study and the X-ray structure. Further DFT calculation showed that the methyl substituent in complex 16 prefers anti relationship with respect to the C7 methylene due to steric repulsion, which would push the palladium to have a η1 interaction with the ipso carbon instead of a η2 interaction. Consequently, the η1 coordination mode likely puts the palladium in a suitable position for the β-carbon elimination reaction to extrude NBE.
2.2. Reaction with Electrophiles
The reactivity of the ANP intermediate with alkyl and aryl halides was then studied mainly by Catellani. As mentioned earlier, Catellani found that the reaction of bromobenzene and NBE gave product 3 in the presence of Pd(PPh3)4 and KOt Bu (vide supra, Scheme 3). It was anticipated that oxidative addition of bromobenzene to ANP was involved in the catalytic cycle. To further probe the intermediacy of a Pd(IV) species, Catellani found that complex 17 was formed upon addition of methyl iodide to the ANP complex 8 at −20 °C (Scheme 7).29 Complex 17 is stable at −20 °C, but would undergo reductive elimination to give 18 when the reaction was slowly warmed to room temperature. It is interesting to note that the methyl group exclusively migrated to the aromatic ring instead of the norbornyl group. Later, the Cheng group found that the analogous ortho methylation reaction using norbornadiene can also take place smoothly.37
Scheme 7.

Stoichiometric Reaction between ANP and Methyl Iodide
To elucidate the stereochemistry of the Pd(IV) complex formed, Catellani and Mann studied complex 19, prepared from the reaction with of ANP 8 with allyl bromide (Scheme 8).38 The nuclear Overhauser effects (NOE) were consistent with the stereochemistry drawn. Using p-nitrobenzyl bromide instead of allyl bromide, Catellani was able to isolate a similar Pd(IV) complex 20, which underwent further reductive elimination to give complex 21.39 It was proposed that, in such a model reaction, an initial rearrangement of ligands took place to put the aryl and benzyl groups at axial–equatorial, rather than equatorial cis positions (thus avoiding the need for an unlikely N–Pd–N widening), presumably by temporary dissociation of the bromide ligand. The relative stereochemistry of the oxidative addition step was later determined by the Lautens group in a catalytic reaction using an enantioenriched secondary alkyl halide (vide infra, Scheme 49).40 Since the product showed a net inversion of stereochemistry, the SN2 mechanism for the oxidative addition step was determined to be in accordance with the experiments.
Scheme 8.

Stoichiometric Reaction between ANP and Allyl/Benzyl Bromides
Scheme 49.

Synthesis of Bicyclic Structures by Type C Intermolecular Couplings
The reaction of ANP with aryl halides is more complicated based on studies of the catalytic reactions (Scheme 9). First, unlike the reaction with alkyl halides, the corresponding Pd(IV) complex from oxidative addition of ANP with aryl halides has not yet been observed. Second, the reaction outcomes depended on the substitution pattern in aryl halides: the reaction was unselective if an ortho-unsubstituted aryl halide was used. For example, when 1-bromo-4-fluorobenzene was allowed to react with NBE, two regioisomeric products were formed in a 1:3 ratio.27 The two products derived from aryl migration to either the norbornyl or the aryl site of ANP followed by ring closure. For example, compound 22a was formed through an initial C(sp2)–C(sp3) bond formation while 22b was derived from an initial C(sp2)–C(sp2) bond formation.
Scheme 9.

Unselective Reaction between Para-Substituted Aryl Bromides with NBE
Interestingly, the de Meijere group found that under Jeffery’s conditions, the reaction between iodobenzene and NBE gave a different product 23 (Scheme 10).41–43 Later, Catellani and Motti proposed that the reaction of iodobenzene with the initial ANP intermediate (4) took place via C(sp2)–C(sp3) bond formation to afford 24.44 Further migratory insertion of NBE and cyclopalladation resulted in new ANP 25. Interestingly, this time, the reaction of iodobenzene with ANP 25 occurred via C(sp2)–C(sp2) bond formation to afford 26. Further NBE extrusion and C–H annulation gave product 23. Nevertheless, it was interesting to note that the first and second ortho arylation proceeded through different selectivity.
Scheme 10.

Reaction between Iodobenzene with NBE under Jeffery’s Conditions
An important discovery made by Catellani was that, when aryl halides with ortho substituents (e.g., the n-butyl group in 27) were used as the substrates, arylation could occur selectively at the arene site (Scheme 11).44 Due to the presence of two ortho groups, the NBE moiety was then spontaneously extruded, and the final biphenyl product 28 was obtained by hydrogenolysis. In contrast, ortho unsubstituted substrates, such as meta or para substituted aryl halides, gave a mixture of products with arylation at aryl or norbornyl sites. Hence, Catellani attributed such a behavior to the steric effect caused by the presence of the ortho groups, which was later termed the “ortho effect”. Such an effect could also explain why intermediate 25 underwent the second arylation selectively through C(sp2)–C(sp2) bond formation in the aforementioned de Meijere’s case (Scheme 10).
Scheme 11.

Discovery of the “Ortho Effect”
2.3. NBE Extrusion
One of the most distinctive properties of NBE is its ability for reversible migratory insertion. Such a transformation was first discovered in a nickel system. In 1975, Porri and co-workers observed that NBE can reversibly insert into the Ni–allyl bond by switching the anionic ligands (Scheme 12).45 Acetate anions favored the NBE insertion product while halide anions favored the NBE extrusion product. One explanation is that the equilibrium exists in solution and different trans effect of the anionic ligands would shift the equilibrium.
Scheme 12.

Reversible Insertion of Nickel–Allyl Bond into NBE
In 1994, a seminal work by Catellani and Fagnola showed that complex 1 can react with p-fluorobenzylbromide under basic conditions to first give ortho alkylated complex 32, which can then undergo another ortho alkylation to afford complex 33 (Scheme 13).46 At this moment, NBE extrusion happened spontaneously to give an aryl-palladium complex (34). It was proposed that the NBE extrusion was driven by steric hindrance around the arene, and it could only happen after both ortho positions were substituted. Clearly, the NBE extrusion is not limited to ortho alkylation reactions (vide supra, Scheme 11). As discussed above, after selective aromatic C–H arylation of ortho-substituted complex 27, NBE extrusion occurred to afford product 28 after hydrogenolysis. It is interesting to note that a higher yield could be obtained if NBE was continuously removed by vacuum, suggesting an equilibrium of NBE insertion and extrusion. NBE extrusion could also take place after double ortho arylation (36), albeit in a lower yield due to the aforementioned “ortho effect”.
Scheme 13.

NBE Extrusion after Double Ortho Functionalizations
3. CATALYTIC REACTIONS
3.1. General Considerations
3.1.1. General Catalytic Cycle.
Prior to the discussion of catalytic reactions, the general reaction mechanism is described first in this section. Based on the early studies of stoichiometric reactions (Section 2), the standard catalytic cycle typically contains four different stages (Scheme 14). The reaction is initiated by forming aryl-Pd(II) species 38 through either oxidative addition of ArX to Pd(0), C–H palladation, or transmetalation of an aryl nucleophile to Pd(II). Intermediate 38 then undergoes migratory insertion of NBE followed by C–H metalation to form the key ANP species 39. While ANP is a Pd(II) complex, it is quite electron-rich due to the two σ-donating carbon ligands. Thus, ANP 39 can further react with an external electrophile to introduce a functional group at the ortho position. In the last stage, the resulting intermediate 40 undergoes β-carbon elimination to extrude NBE, followed by termination to regenerate either the Pd(0) or Pd(II) catalyst. In this section, we classify the catalytic reactions according to the initial stage of the catalytic cycle (i.e., how intermediate 38 is formed). The first type is initiated by Pd(0) through oxidative addition into aryl halides or triflates while the second type is initiated by Pd(II) through N–H activation of indoles, C–H activation of arenes, or transmetalation with aryl nucleophiles. Recent variations on the classical Catellani reactions are closely related to these general processes, which include (1) enabling new pathways for generating aryl-Pd(II) species 38, (2) designing new electrophiles to selectively intercept ANP, and (3) designing new NBEs and ligands for selectivity/reactivity control.
Scheme 14.

General Catalytic Cycle for Pd/NBE Catalysis
Based on the general catalytic cycle in the Pd(0)-initiated reactions, one may assume that a plethora of transformations could be developed simply by switching different nucleophiles and electrophiles. However, the complexity of the Pd/NBE cooperative catalysis lies on the coexistence of various electrophiles and nucleophiles in a single reaction vessel. The success of this complex reaction depends on a number of selective and sequential reactions between different pairs of electrophiles and nucleophiles, which are modulated through different oxidation states of intermediates and subtle differences in electronic and steric effects among the different species. The early investigations of the Catellani reaction revealed that a variety of nucleophiles can be coupled with intermediate 41 in the termination step, which largely paralleled classical cross-coupling reactions.14–19 Generally, the reactivity of terminating nucleophiles needs to be carefully tuned, so that a premature termination with intermediate 38 would not happen. In many cases, the use of a masked nucleophile that can slowly generate the real nucleophile is preferable. On the other hand, the introduction of electrophiles at the ortho position seemed to be more challenging and quite limited. The key to develop new ortho functionalization is to enable selective reactions with the Pd(0) catalyst and the ANP intermediate 39: the electrophile employed should selectively oxidize the ANP intermediate instead of the Pd(0) catalyst, while the aryl halide substrate must selectively react with the Pd(0) instead of ANP.
Such selectivity could be made possible through differentiating the reactivity between ANP and the Pd(0) species (Scheme 15). We hypothesize that ANP is nucleophilic based on Catellani’s earlier works (vide supra, Schemes 7 and 8), but it is more sterically crowded and thus harder to undergo geometrical distortion. In contrast, Pd(0) is more flexible toward geometrical distortion. Therefore, aryl iodides containing a weak C–I bond should selectively react with Pd(0) under distortion control, and the electron-rich Pd(0) interacts with iodoarene’s π* orbital through back-donation.47 In contrast, a hard, more electrophilic external electrophile would tend to react with ANP. In addition, a coordinating moiety on the electrophile would help oxidative addition with ANP as the Pd(II) center is anticipated to be more Lewis acidic. For comparison, ANP would have a hard time to react with aryl iodides with ortho substituents due to its steric encumbrance. It should be noted that different electrophiles might interact with ANP in different ways and subtle changes on the additives might also affect reaction selectivity.
Scheme 15.

Selectivity Issue for Oxidative Addition of Pd(0) versus ANP
On the other hand, the Pd(II)-initiated reactions face a different type of challenge. The competition between aryl halide substrates and external electrophiles does not exist in this system, thus the scope of external electrophiles can be expected to be broader than the Pd(0)-initiated reactions. However, due to the use of aryl nucleophiles 37b as the substrates, their direct competing reactions with electrophiles become a notable issue, e.g., indole N-alkylation, directed ortho functionalization, and protodeboronation (vide infra). Other additional challenges will be discussed in the section 3.3.
Another interesting yet still challenging aspect is the control of enantioselectivity in the Pd/NBE catalysis. For asymmetric induction through ipso functionalization, the ligand effect in principle should parallel classical cross-coupling reactions. However, the difficulty could come from compatibility of chiral phosphine ligands in the Pd/NBE catalysis as many bidentate phosphines were found not suitable for these reactions. For asymmetric induction through ortho functionalization, the situation is more complicated. If a chiral phosphine ligand is used, at least two potential diastereomers of ANP could be formed considering the stereocenters introduced by NBE. Thus, a potential solution is to use chiral NBEs instead of chiral phosphine ligands (vide infra, Schemes 52 and 116). Since different oxidation states are involved in the catalytic cycle, it becomes hard to judge whether the phosphine ligand is bound to Pd all time, which could be another challenge for asymmetric induction.
Scheme 52.

Synthesis of 2,3-Dihydrobenzofurans by Ortho Alkylation of Aryl Iodides Using Epoxides
Scheme 116.
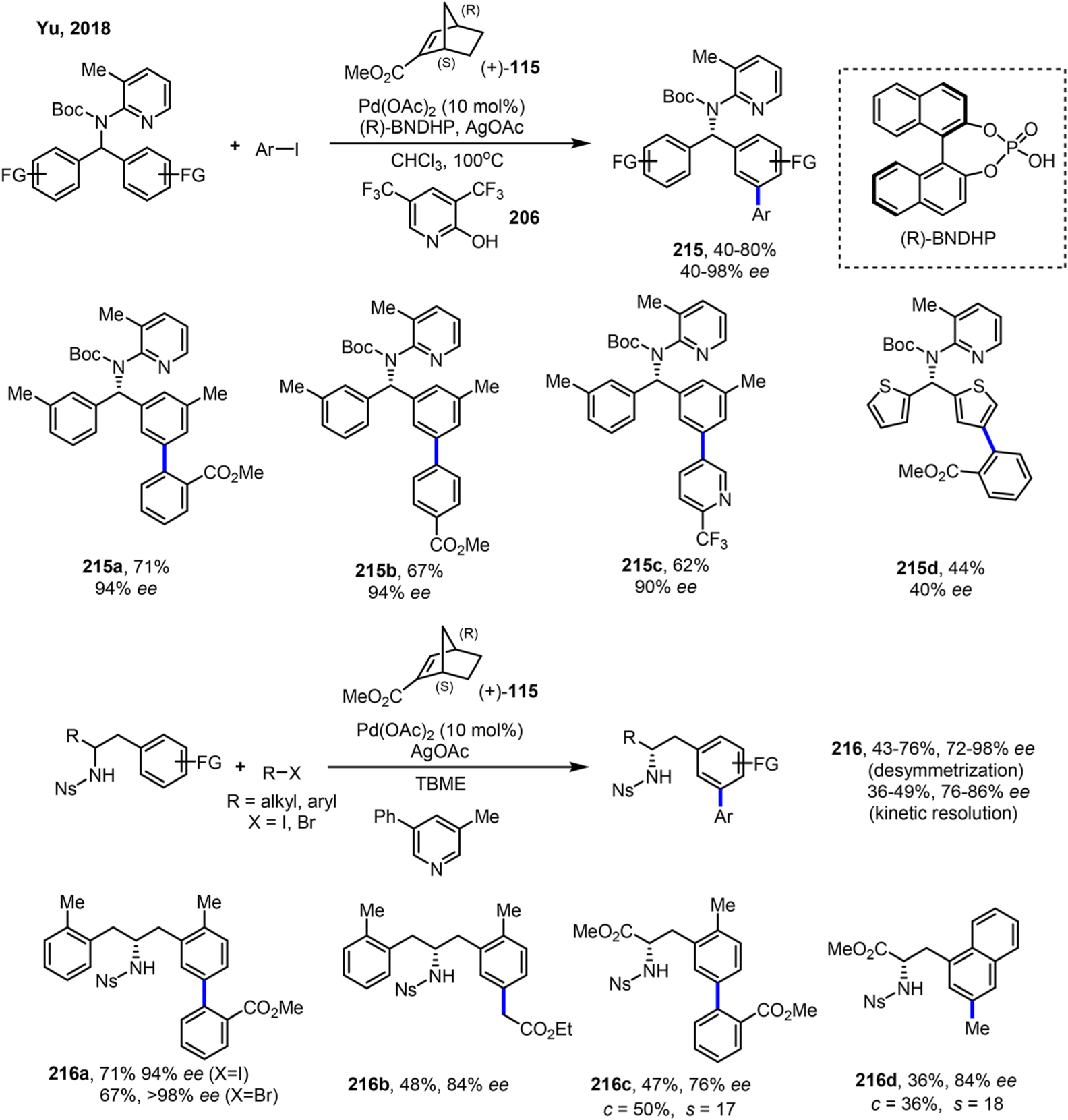
Enantioselective Remote Meta C–H Arylation and Alkylation of (Homo)Benzylamines with Pyridine-Type DGs
3.1.2. Why Is NBE Unique?
One intriguing aspect of the Pd/NBE catalysis is the reason that NBE is unique as a cocatalyst. Besides an olefin that is unable to undergo β-hydrogen elimination after X–Pd–Ar insertion, NBE has a strained, rigid, but not too sterically hindered [2.2.1] bicyclic scaffold, which allows for fast migratory insertion, convenient ortho C–H palladation, and reversible β-carbon elimination.
First, the migratory insertion rate of NBE is fast, thus suppressing the undesired direct ipso coupling of 37a with terminating nucleophiles in the Pd(0)-initiated reactions and undesired direct reaction of 37b (Scheme 14) with electrophiles in the Pd(II)-initiated reactions. Lautens measured the activation barrier for NBE migratory insertion rate using PPh3 as the ligand,34 which was found to be 17–18 kcal/mol for aryl palladium(II) species described in Scheme 16. The exceptionally high reactivity of NBE toward addition reactions can be explained from both thermodynamic and kinetic viewpoints. Thermodynamically, NBE has strain energy of 21.6 kcal/mol, while that of norbornane is 16.6 kcal/mol.48 The heat of hydrogenation was measured to be about 6 kcal/mol higher than that of cyclohexene,49 which is roughly the difference of ring strain between NBE and norbornane. Kinetically, the addition of NBE is accelerated because it is easier to distort this strained (predistorted) olefin to a pyramidalized transition state geometry (Figure 1). The alkene moiety in NBE is pyramidalized (instead of flat), resulting from mixing of the 2s orbital of the alkene carbon and the 2p orbital of the π bond.50,51 The out-of-plane bending angle (defined as the C—C=C—H dihedral angle) for NBE and norbornadiene is 7° and 2–4°, respectively.52
Scheme 16.

Measurement of the Migratory Insertion Barriers into NBE
Figure 1.

Out-of-Plane Bending Angles for NBE and norbornadiene.
Second, the rigid structure of NBE would reduce the distance between Pd and the arene ortho C–H bond after migratory insertion,36 thereby facilitating the C–H metalation step. The DFT calculation by Bi and co-workers showed that, when cesium carbonate was used as the base, the C–H metalation from such a NBE-directed species required an energy barrier of 21.1 kcal/mol, while the corresponding transition state derived from a flexible olefin (e.g., acrylate) exhibited a significantly higher barrier (Scheme 17).53
Scheme 17.
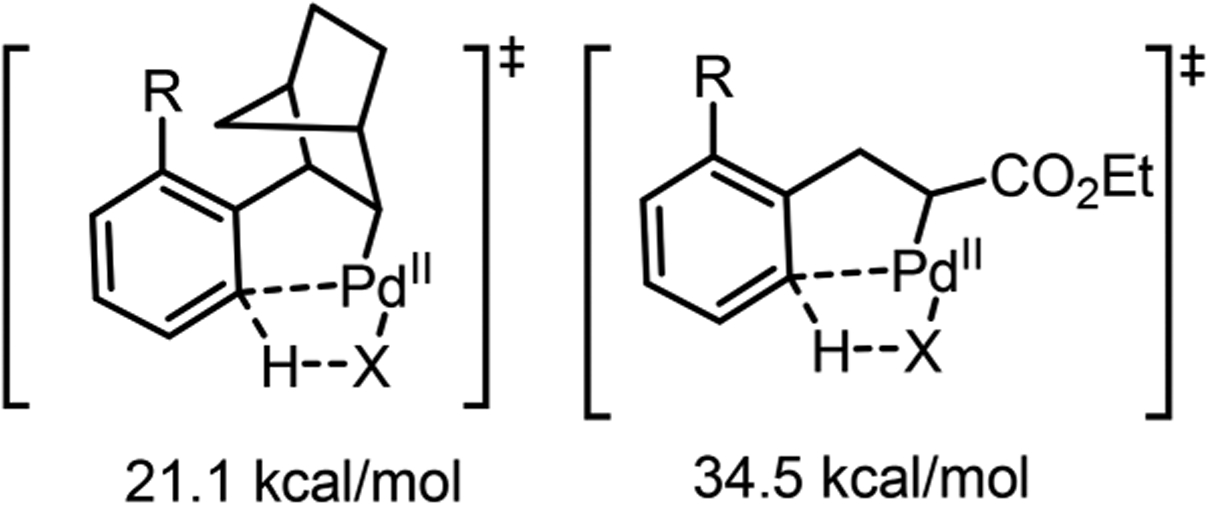
Activation Barrier of NBE or Acrylate Directed C–H Metalation
Finally, perhaps the most distinct feature for NBE is its facile β-carbon elimination reaction when both ortho positions are substituted. Presumably, the β-carbon elimination is kinetically facilitated by the η1 interaction between the palladium center and the ipso carbon, benefiting from the rigid structure of NBE. The NBE insertion and extrusion might be viewed as an equilibrium. While mono ortho substituted aryl palladium species favors NBE insertion, the two ortho substituents change the position of the equilibrium (Scheme 18). It should be pointed out that other reaction parameters, such as ligands and additives, could also have an influence on the equilibrium.
Scheme 18.

Equilibrium of NBE Insertion/Extrusion
It would be most informative to compare NBE with other potential “surrogates”. Norbornadiene seems to be a potential candidate, which has ring strain energy of 32.4 kcal/mol, making migratory insertion less reversible. In a stoichiometric study, Cheng employed complex 15 for C–H metalation and ortho methylation twice to give complex 42 with two ortho methyl groups (Scheme 19).37 However, norbornadiene extrusion did not take place spontaneously, which is in sharp contrast with the NBE case.46 Hence, in the catalytic reactions, use of norbornadiene generally gives lower yields compared to NBE if norbornadiene extrusion is required in the reaction.
Scheme 19.

Absence of Norbornadiene Extrusion in Stoichiometric Reactions
Acyclic olefins usually contain available β-hydrogens; however, palladacycle formation could still be favored over β-hydrogen elimination if a proper alkene is used (Scheme 20). For example, α,β-unsaturated sulfone 43 with a large isopropyl group can effectively slow down β-hydrogen elimination.54 The palladacycle formation and ortho arylation reaction took place selectively. However, after double ortho arylation, alkene extrusion did not occur; instead, another aromatic C–H activation followed by reductive elimination led to the final product. In addition, Lautens reported that, when a 1,1-disubstituted olefin was tethered to an aryl iodide (44), a similar ortho arylation took place to form a polycyclic ring.55
Scheme 20.

Acyclic Olefin Mediated Ortho Arylation
The 7-oxa or aza-[2.2.1]bicyclic alkenes could be possible options (Scheme 21). Migratory insertion, ANP formation, and oxidative addition are expected to have similar barriers. However, achieving β-carbon elimination and meanwhile suppressing β-oxygen/nitrogen elimination turned out to be challenging. However, if extrusion of 7-oxa/aza[2.2.1]bicyclic alkenes is not required in the catalytic cycle, successful examples have been reported where these bicyclic alkenes were incorporated in the final products.56 Yet, to the best of our knowledge, no example has been reported for using these alkenes as cocatalysts in the Catellani-type reactions, although an interesting Rh-catalyzed C–H amidation using 7-oxa[2.2.1]-bicyclic alkenes was reported.57
Scheme 21.

Structures of Alternative Substituted NBEs
Due to these distinct features of NBE, the successful modification usually maintains its original skeleton. Currently, the effective variations are at the C1, C2, or C5 positions (Scheme 21). By and large, the reactivity of the C5-substituted NBEs is close to (yet sometimes slightly different from) simple unsubstituted NBE. The C1-substituted NBEs would inhibit C–H metalation, but promote β-carbon elimination, and thus they were developed for addressing the “ortho constraint” (vide infra, see Section 3.2.7). The C2-substituted NBEs are valuable in preventing side reactions, e.g., minimizing the undesired C–C reductive elimination from ANP to give norbornyl benzocyclobutene side-products. More detailed discussions will be provided in the following subsections.
3.2. Catalytic Reactions Initiated by Pd(0)
To date, various types of ortho/ipso difunctionalizations have been reported. The discovery time for each type of transformations is summarized in Table 1. The following subsections are arranged based on catalytic reactions involving different ortho functionalizations.
Table 1.
Summary of Discovery Time of Ortho/Ipso Difunctionalizations of Aryl Iodides
| ipso | orthoa | ||||
|---|---|---|---|---|---|
| alkylation | arylation | amination | acylation | alkoxycarbonylation | |
| Heck | 199758 | 2003138 | 2014180 | 2015191–193 | 2016204 |
| Suzuki | 200074 | 2003140 | 2014182 | 2015191,193 | |
| alkyne annulation | 2008109 | 2001137 | |||
| Sonogashira | 200476 | 201577,78 | 2018197 | ||
| C–N coupling | 2007107 | 2004161 | 2018189 | ||
| sp2 C–H activation | 200587 | 2009142 | 2018198 | ||
| hydrogenation | 200581 | 2005141 | 2013179 | 2015193 | 2016204 |
| cyanation | 200772 | 200772 | 2016184,185 | 2016196 | |
| enolate coupling | 201692 | 200990,91 | 2017186 | ||
| 1,2-addition | 200991 | ||||
| C–O coupling | 2018131 | 2012153 | |||
| carbene coupling | 201486 | 2014183 | |||
| borylation | 2015187 | 2019202 | |||
| thiolation | 2016200 | ||||
| sp3 C–H activation | 2018190 | ||||
The year in which the type of the ortho functionalization was first discovered in bold.
3.2.1. Ortho Alkylation of Aryl Iodides.
3.2.1.1. Intermolecular Couplings.
Based on their prior stoichiometric studies (vide supra, Scheme 13), Catellani and co-workers reported the first catalytic example of a Pd/NBE system in 1997, in which acrylates were added as the termination reagents to regenerate Pd(0) (Scheme 22).58 In this seminal work, alkyl iodides were used as the electrophile, which introduced alkyl groups at both of the ortho positions: iodobenzene and para-substituted aryl iodides were employed as the substrates, affording bis-ortho alkylated products. The optimized reaction conditions employed complex 1 as the catalyst, and the reaction proceeded at room temperature. While each step in the catalytic cycle was known conceptually at that time, it is remarkable that such a selective and catalytic process can be established. In particular, Pd(0) can selectively react with the aryl iodide instead of the alkyl iodide, while ANP can selectively react with the alkyl iodide instead of the aryl iodide.
Scheme 22.

First Catalytic Example in the Pd/NBE Catalysis
To obtain products with two different ortho alkyl groups, Catellani and Cugini then used ortho-substituted aryl iodides as the substrates (Scheme 23).59 In this case, Pd(OAc)2 was found to be a better precatalyst, and potassium acetate was added to facilitate NBE insertion60 and/or C–H palladation. The major side reaction was the direct reductive elimination from ANP to give the norbornyl benzocyclobutene derivative. Increasing the bulkiness of the ortho substituent led to lower conversion of substrates and formation of a greater amount of the norbornyl benzocyclobutene side product, which suggests that reductive elimination from ANP could be promoted by bulky ortho substituents. Low reactivity was observed (46c) when secondary alkyl halides were used, which indicated that the ANP intermediate could be sensitive to sterics of the electrophile. In addition, styrene was less reactive as a terminating olefin (46d).
Scheme 23.

Ortho Alkylation/Ipso Heck Reaction of Aryl Iodides
While these initial discoveries by Catellani are mechanistically interesting and synthetically appealing, it is noteworthy that an important change in conditions that have made the reaction practical and widely useful was reported by Lautens in 2000 (vide infra, Scheme 42).61 The introduction of phosphine ligands (e.g., PPh3 and P(2-furyl)3), as well as the use of Cs2CO3 as base and CH3CN as solvent, has greatly improved the generality of the reaction. Since then, this new set of conditions has been widely adopted in the Pd(0)-initiated catalytic reactions.
Scheme 42.

Ortho Alkylation/Ipso Intramolecular Heck Coupling Reaction of Aryl Iodides
Using such conditions, in 2006, Lautens expanded the substrate scope to hetereoaryl iodides (Scheme 24).62 This was the first time that 5-membered heterocycles were demonstrated to work in the Pd/NBE catalysis. Thiophene (47a), benzothiophene (47b), and N-protected indole (47c) were shown to be suitable heterocyclic cores in haloarene substrates, though 2-iodothiophene was not effective. The choice of the protecting group in the indole substrate (47c) was also important, as use of the methyl protecting group only gave the direct Heck product. Free primary alcohol63 and alkyl bromide64 were tolerated under modified reaction conditions.
Scheme 24.

Expansion of the Ortho Alkylation/Ipso Heck Reaction of Heteroaryl Iodides
Functionalized alkyl halides can also be successfully coupled. For example, Ferraccioli’s group showed that when alkyl halides containing a nitrogen nucleophile were used, further aza-Michael reactions could take place to give tetrahydroisoquinoline 48 (Scheme 25).65,66 When the reaction was stopped early, the major product was the uncyclized Heck product. Additional KOtBu was needed for forming the seven-membered ring. Later, the same group used 2-chloroamides to access isoquinolin-3-one 49 in a similar fashion.67
Scheme 25.

Ortho Alkylation/Ipso Heck/Aza-Michael Cascade of Aryl Iodides
In 2014, the Liu group reported the ortho trifluoroethylation via the Pd/NBE catalysis (Scheme 26).68 Electron-rich DavePhos was employed as the ligand to facilitate oxidative addition with CF3CH2I, which was assumed to be the rate-limiting step in this transformation. In a competition experiment, CF3CH2I was found much less reactive than isobutyl iodide but more reactive than neopentyl iodide. The reaction exhibited a broad substrate scope for both aryl iodides and olefins. Reactions of electron-neutral or -rich substrates proceeded smoothly, but strongly electron-withdrawing substituents, such as the trifluoromethyl group (52d), were detrimental to this reaction. In addition, 2-bromotoluene and simple iodobenzene (52e) were not suitable substrates.
Scheme 26.

Ortho Trifluoroethylation/Ipso Heck Reaction of Aryl Iodides
In 2015, conditions that can effectively couple sterically hindered secondary alkyl iodides intermolecularly were reported by Lautens, which were previously problematic electrophiles (Scheme 27).69 This reaction was most effective when tert-butyl acrylate was employed as the termination reagent, and both alkyl iodides and acrylates were used in excess. A variety of electron-rich and -deficient aryl iodides and acyclic and cyclic secondary alkyl iodides were suitable substrates. A chiral alkyl iodide (53c) was successfully coupled without significant loss of stereochemical information when MeCN was used as solvent.
Scheme 27.

Employing Secondary Alkyl Halides in the Pd/NBE catalysis
To incorporate a functionalized one-carbon alkyl unit into the ortho position, the Gu group used iodomethylsilane 54 as the electrophile (Scheme 28).70 The silyl moiety in the alkylated product 55 could be converted to other functional groups, e.g., an alcohol (56).
Scheme 28.

Ortho Silylmethylation/Ipso Heck Reaction of Aryl Iodides
The introduction of a methyl group could have a profound impact on the biological activity of a drug candidate.71 Besides Lauten’s first use of methyl iodide in the ipso cyanation reaction (vide infra, Scheme 37),72 another example was reported by Wilson on a pyridine substrate in 2016 (Scheme 29).73
Scheme 37.
Ortho Alkylation/Ipso Cyanation Reaction of Aryl Iodides
Scheme 29.
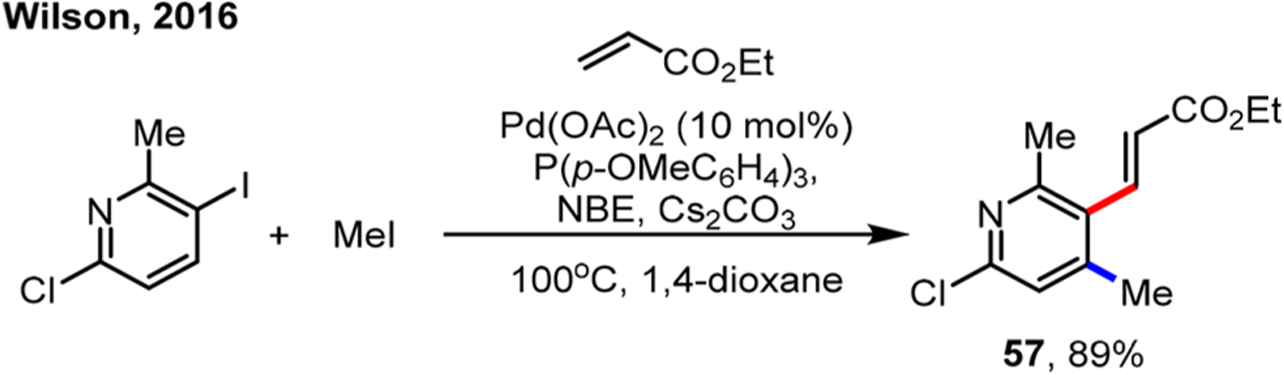
Ortho Methylation/Ipso Heck Reaction of Aryl Iodides
Besides the Heck quench at the ipso position, other types of cross-coupling reactions could also be employed as the terminating steps. In 2000, Catellani reported the first example of the ortho alkylation/ipso Suzuki–Miyaura reaction (Scheme 30).74 The reaction proceeded under mild reaction conditions using alkyl bromides as the alkylating reagent. When iodobenzene was used, the ortho dialkylation product 58a was observed. Note that isopropyl bromide (58d) was successfully coupled in this case, though an extended time (144 h) was required. Ortho-substituted arylboronic acids gave low reactivity under the optimized conditions.
Scheme 30.

Ortho Alkylation/Ipso Suzuki–Miyaura Reaction of Aryl Iodides
Recently, Gu and co-workers reported an enantioselective synthesis of biaryl atropisomers using the ortho alkylation/ipso Suzuki–Miyaura reaction (Scheme 31).75 The key was to use a specific P–N ligand containing both axial and center chirality. The ortho-substituted arylboronic acids were successfully coupled, and the aldehyde moiety could be further transformed to other functional groups.
Scheme 31.

Asymmetric Ortho Alkylation/Ipso Suzuki–Miyaura Reaction of Aryl Iodides
The Sonogashira coupling has also been employed as the termination step. In 2004, Catellani reported the first ortho alkylation-ipso-Sonogashira reaction (Scheme 32).76 However, high reactivity of terminal alkynes made it susceptible to generate multiple intermediates and consequently give various side products. For example, the direct Sonogashira coupling could compete with NBE insertion to give alkyne 61a; the alkyne could also intercept the aryl-norbornyl palladium intermediate after the NBE insertion step to give 61b as the side-product. Catellani found that the selectivity was improved when replacing K2CO3 with KOAc, probably owing to a more favorable NBE insertion or faster metalation to give the ANP intermediate. In addition, slow addition of both terminal alkynes and alkyl bromides suppressed side-reactions by maintaining a low concentration of reactive alkynes.
Scheme 32.

Ortho Alkylation/Ipso Sonogashira Reaction of Aryl Iodides
If a masked alkyne is used instead, the terminal alkyne would be generated slowly during the reaction, thereby maintaining a low concentration, which would avoid the slow addition operation. Based on their own77 and Chen’s78 work on the ortho amination/ipso Sonogashira reaction, the Gu group employed 1,1-dimethyl-2-alkynyol 62a as the masked alkyne reagent to slowly release terminal alkynes through the loss of acetone (Scheme 33).79 Interestingly, when aryl propiolic acid 62b was used, the esterification became the major side reaction. Both alkyl- and aryl-substituted alkynes were suitable coupling partners.
Scheme 33.
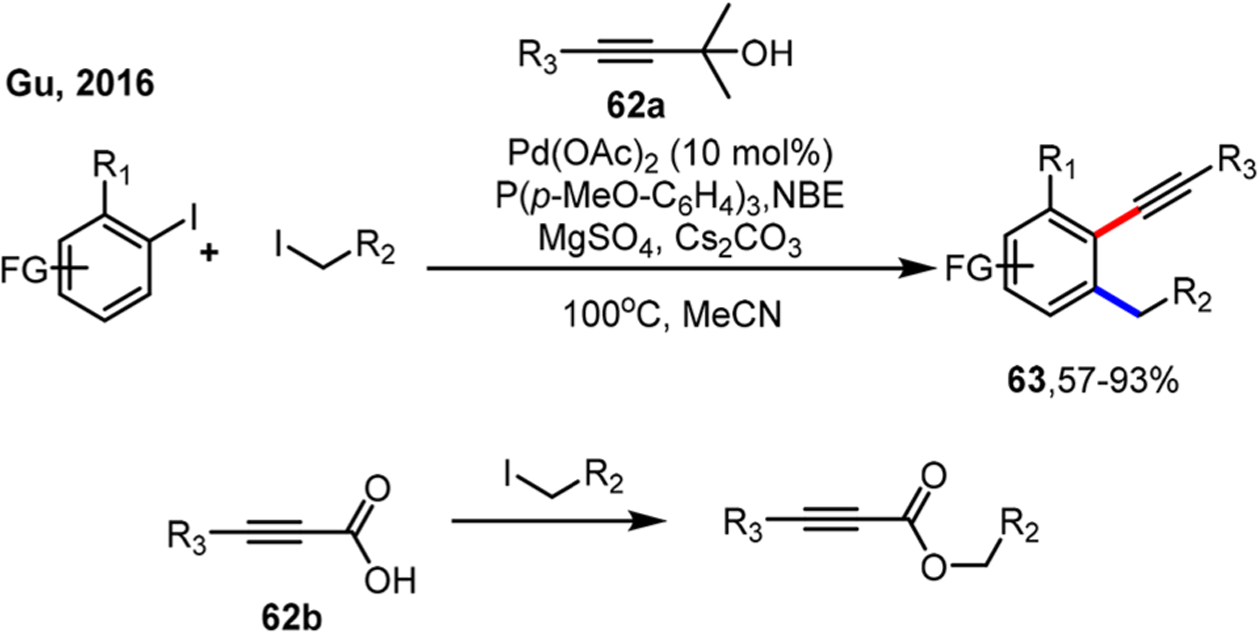
Ortho Alkylation/Ipso Sonogashira Reaction of Aryl Iodides Using Masked Alkynes
Although efficient, masked alkynes are usually synthesized from the corresponding terminal alkynes. Thus, it is still attractive to directly use terminal alkynes. Shortly after Gu’s work, Zhou found the acetylide anion could be maintained at a low concentration through carefully tuning the reaction conditions. As a consequence, good yields were obtained without slow addition of reagents (Scheme 34).80 Alkyl iodides, bromides, and chlorides were used in the reaction and a secondary alkyl iodide (64b) was successfully coupled. Other types of alkynes (64c and 64d) were also suitable under a modified condition.
Scheme 34.

Ortho Alkylation/Ipso Sonogashira Reaction of Aryl Iodides Using Unprotected Alkynes
Perhaps the simplest ipso-functionalization is hydrogenation, a transformation that would lead to meta-substituted arenes. As early as in 1994, the stoichiometric ipso-hydrogenation was achieved by blowing hydrogen gas into the system.46 However, use of hydrogen gas is nonselective; therefore, a process that can slowly release hydride is preferable in the catalytic version. As an unexpected discovery, Wilhelm and Lautens found that ipso hydrogenation reaction could be achieved using alkyl boronic acids, e.g., iPrB(OH)2, as the terminating reagents (Scheme 35).81 It was proposed that, after transmetalation of iPrB(OH)2, β-hydrogen elimination took place instead of alkyl-aryl reductive elimination. Subsequent control experiments showed that the ipso hydrogenation products could be obtained without adding iPrB(OH)2 in some cases, and further deuterium study showed that alkyl halides could serve as an alternative hydride source (vide infra, Scheme 36). The substrate scope was later expanded to 3-iodothiophene by the same group.62
Scheme 35.
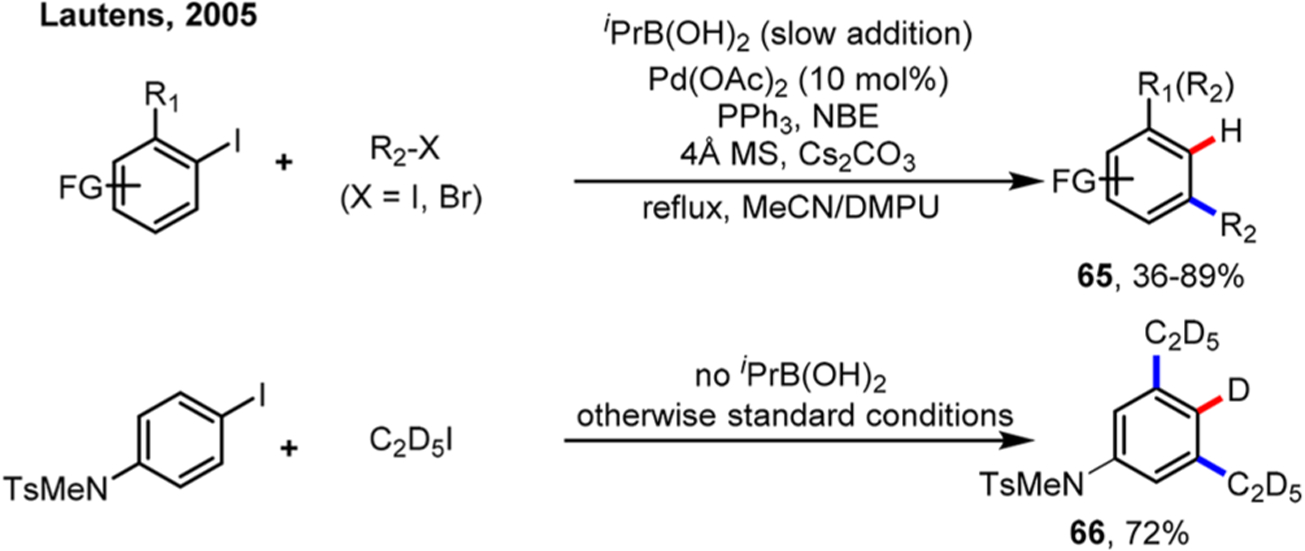
Ortho Alkylation/Ipso Hydrogenation Reaction of Aryl Iodides
Scheme 36.
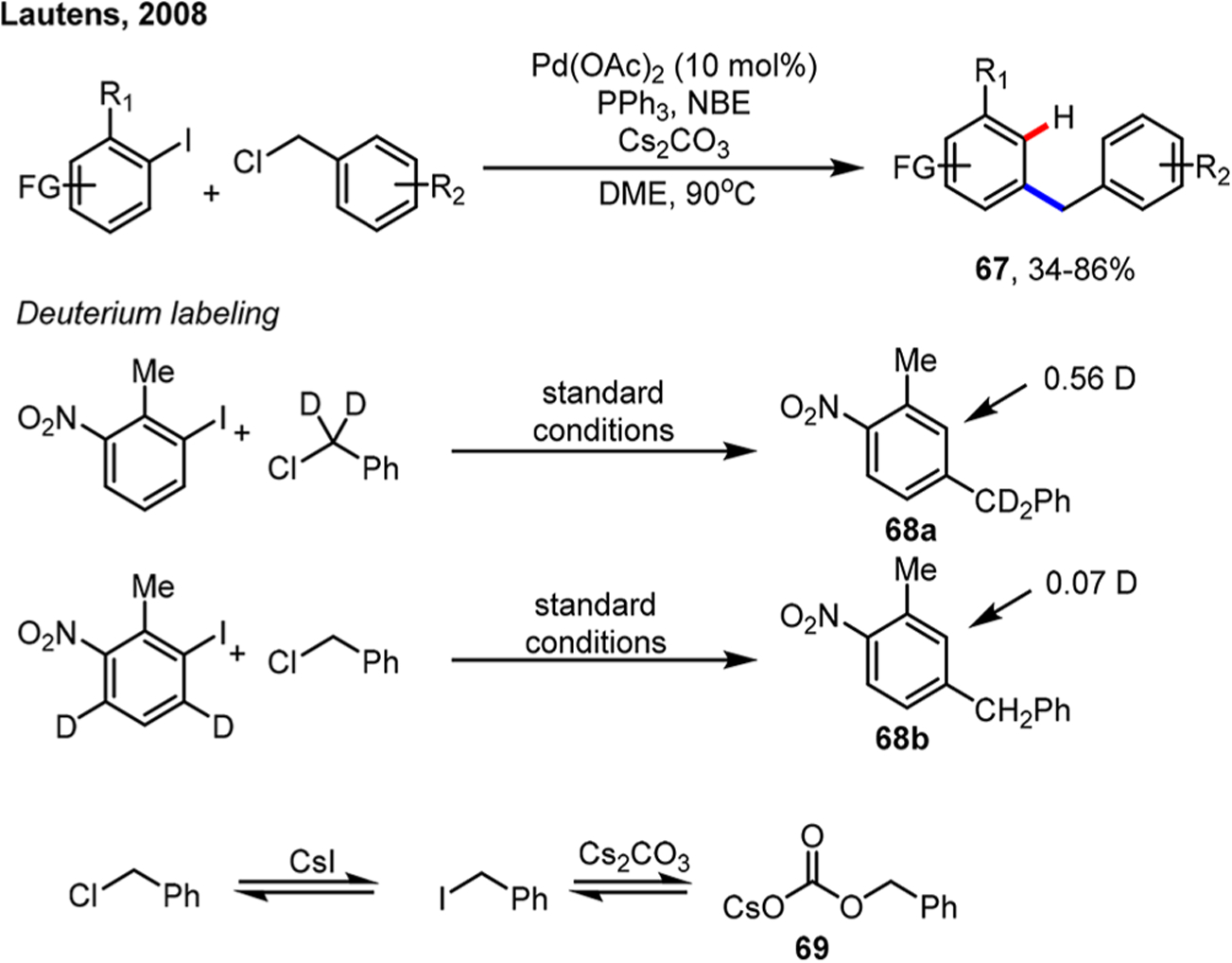
Ortho Benzylation/Ipso Hydrogenation Reaction of Aryl Iodides
When exploring the ortho benzylation reaction using benzyl chlorides, Martins and Lautens found that ipso hydrogenation products could be obtained without adding iPrB(OH)2 (Scheme 36).82 Deuterium labeling studies showed that multiple pathways could lead to the hydrogenation product: 56% of deuterium incorporation (68a) came from benzyl chloride. Interestingly, 7% of deuterium incorporation (68b) came from aryl iodide, presumably through protodepalladation of the final aryl-Pd(II) intermediate. Control experiments further showed the importance of iodide anion in the reduction process, and it was thus proposed that the benzyl carbonate 69, originated from in situ formed benzyl iodide, released benzyl alcohol through decarboxylation, which served as the real reductant.
Ipso cyanation has been achieved using metal cyanides as the nucleophile in the ortho alkylation reaction.83 The nature of metal cyanides was found to be critical:72 the less reactive potassium hexacyanoferrate was more effective than zinc cyanide by maintaining a low concentration of cyanide anion (Scheme 37). A variety of alkyl halides have been used, including methyl iodide (70b). Interestingly, alkyl chloride could also be coupled when more electron-rich PBu3 was used. Under the optimized conditions, α-chloroamides (70c), α-chloroesters, and benzyl chlorides (70d) all afforded the desired products.
Diazo compounds are known to serve as a surrogate for vinyl nucleophiles through carbene insertion followed by β-hydrogen elimination.84 They can be slowly generated from N-tosylhydrazones85 through the base-mediated Bamford–Stevens reaction, and are therefore suitable for the termination step in the Pd/NBE catalysis. In 2014, the Liang group reported the ipso vinylation/ortho alkylation using N-tosylhydrazones as the nucleophile (Scheme 38).86 The base employed was important, as it was needed for both the C–H metalation and diazo species generation steps. During Liang’s study, Cs2CO3 was found to be more effective than LiOtBu. Addition of water improved the yield, though the reason remains unknown. In addition, a one-pot procedure that directly used aromatic ketones without isolation of N-tosylhydrazones has also been developed.
Scheme 38.

Ortho Alkylation/Ipso Carbene Insertion Reaction of Aryl Iodides
Direct coupling of heteroarenes at the ipso position provides a straightforward way to form aryl–heteroaryl bonds. Although the intramolecular ipso-heteroarylation was developed by Lautens in 2005,87 its intermolecular counterpart was not developed until 2015. Zhou and co-workers found that, when using NaOH as the base, the relatively acidic protons in various five-membered heteroarenes could be reversibly removed, thereby slowly generating real nucleophiles (Scheme 39).88 As a result, different heteroarenes, such as benzoxazoles (72a), oxazoles (72b), and electron-deficient thiophenes (72c), could be coupled at the ipso position. Fast H/D exchange at the acidic C–H bonds in heteroarenes was observed in the presence of NaOH alone. Meta-substituted aryl iodides afforded a NBE-containing side-product (72d), probably due to a challenging β-carbon elimination step.
Scheme 39.

Ortho Alkylation/Ipso Direct Arylation Reaction of Aryl Iodides
Enolates have been established as viable cross-coupling partners to achieve the α-arylation of carbonyl compounds and nitriles.89 The Lautens and Catellani groups concurrently demonstrated the use of enolate coupling as the termination step in the ortho arylation reactions in 2009.90,91 In 2016, the Zhou group reported the first ortho alkylation/ipso enolate coupling reaction (Scheme 40).92 The key, again, was to use NaOH as the base. This suppressed unproductive side reactions such as ketone α-alkylation and self-aldol condensation. Secondary alkyl iodides (73b) could be coupled, albeit in a lower yield. In addition, tetraline product (73c) could be formed using 1-bromo-3-chloropropane through an intramolecular cyclization. Besides ketones, acetonitrile can also be coupled using LiOtBu as the base (73d), possibly due to a higher pKa of the α protons in nitriles compared to ketones.
Scheme 40.

Ortho Alkylation/Ipso Enolate Coupling Reaction of Aryl Iodides
3.2.1.2. Intramolecular Couplings.
As a three-component coupling reaction, the Pd/NBE catalysis can give cyclized products if at least two of the coupling partners are linked together (Scheme 41). Generally, there are four types of intramolecular couplings, depending on how these three components are tethered. Type A involves tethering the alkylation reagent with the termination nucleophile; Type B involves tethering aryl iodides with the termination nucleophile; Type C involves tethering aryl iodides with the alkylation reagent; Type D involves tethering aryl iodides with both the alkylation reagent and the termination nucleophile.
Scheme 41.

Four Types of Intramolecular Couplings
In the first type of intramolecular couplings (Type A), the ipso termination could be considered favored as the nucleophile would react intramolecularly after the ortho alkylation has occurred. Foreseeing the potential of constructing fused carbocycles, the Lautens group reported the first Pd/NBE-catalyzed annulation reaction using olefin-tethered alkyl halides in 2000 (Scheme 42).61,93 This was also the first time that a phosphine ligand was used in the Pd/NBE catalysis, which greatly improved the generality and reproducibility of the reaction. They also found that Cs2CO3 was superior to K2CO3. These changes in conditions have been widely adopted by the community in the development of other types of transformations. Regarding the substrate scope, both six- and seven-membered rings could be formed. Simple iodobenzene led to o,o′-dialkylated products (75c). Aryl iodides bearing ortho-chelating groups were generally poor substrates (75d), partially due to a competing self-dimerization reaction (vide infra, Scheme 57). Alkyl bromide 74 was more efficient than the corresponding iodide, and both Z and E enoates of 74a could afford the same product 75a. Unfortunately, aryl bromides and triflates were not suitable coupling partners in this reaction. Variations on the linkers and substitutions were later investigated by the same group.94,95
Scheme 57.

Homo Ortho Arylation/Ipso Heck reaction of Aryl Iodides
In 2018, Zhou and co-workers utilized the redox-relay Heck reaction as the termination step to access tetrahydronapthalene (76a–c) and indane (76d) scaffolds (Scheme 43).96,97 Preliminary studies of the asymmetric version was also described using a chiral phosphoramidite as the ligand, though the enantioselectivity and yield were moderate. The utility was demonstrated in a 4-step synthesis of (±)-eptazocine (see section 4.1).
Scheme 43.

Synthesis of Tetrahydronapthalenes or Indanes via Ortho Alkylation/Ipso Redox-Relay Heck Coupling Reaction of Aryl Iodides
Tethering alkyl halides with heteroarenes provides an opportunity construct fused heterocycles through ipso arylation (Scheme 44). Various hetereoarenes, including indole,87,98,99 pyrrole,100 pyrazole,100 imidazole,101 thiophene,102,103 furan,102 benzothiophene,102,103 azaindole,104 indazole,105 and triazole105 have been successfully coupled. Interestingly, when the C3-tethered unprotected indole was used, a spirocyclic indoline (77d) was formed via a dearomative cyclization reaction.106
Scheme 44.

Representative Examples of Ortho Alkylation/Ipso Intramolecular Direct Arylation Reaction of Aryl Iodides
The intramolecular Buchwald–Hartwig amination was also demonstrated as a suitable termination step by Lautens.107 Since bromoalkyl amines tend to self-cyclize under basic conditions, the protecting groups on the nitrogen were thus important. Although Boc, Bz, and Ts groups led to decomposition or aziridination, aryl (78a) and carbamates (78b) were suitable protecting groups (Scheme 45). Unexpectedly, 3-nitro-2-methyliodobenzene produced the corresponding indole as the product, possibly due to further dehydrogenation reaction. A secondary bromoalkylamine (78c) and bromopropylamine (78d) could also be coupled.
Scheme 45.

Ortho Alkylation/Ipso Intramolecular C–N Coupling Reaction of Aryl Iodides
Although enolate coupling was demonstrated to be an effective termination step,90–92 tertiary α-carbons of ketones cannot be coupled intermolecularly due to their low reactivity. In 2017, the Liang group showed that such ketones could be successfully coupled in an intramolecular fashion via Type A cyclization to construct spiral structures (Scheme 46).108
Scheme 46.

Ortho Alkylation/Ipso Intramolecular Enolate Coupling Reaction of Aryl Iodides
When alkyne-tethered alkyl halides were used, a complex catalytic cycle could take place, which may provide a variety of poly fused ring systems (Scheme 47). A common vinyl palladium intermediate 80 would be formed through carbopalladation of the alkyne. When R1 is a heteroaryl group (such as pyrrole109,110 or indole110) or R3 is an aryl/heteroaryl group,111 intermediate 80 could be terminated via intramolecular C–H annulation (81a–c). The Luan group recently reported that indole and arene dearomatization could also be used as the termination step (81d,e),112,113 whereas the intermolecular alkyne annulation could also be achieved. Otherwise (when neither R1 or R3 is an aryl group), further NBE insertion with 80 and annulation on the adjacent aromatic ring would occur (81f).114 Interestingly, chiral helical alkenes could be accessed when enantiopure bromoalkyl aryl alkynes were used (81g).115
Scheme 47.

Ortho Alkylation of Aryl Iodides Followed by Alkyne Insertion
To date, there are only two examples of Type B cyclization, which employed Heck reaction116 and Buchwald–Hartwig amination,117 respectively, as the termination steps to construct fused bicyclic structures (Scheme 48).
Scheme 48.

Synthesis of Bicyclic Structures by Type B Intermolecular Couplings
In Type C or Type D annulations, the alkylation reagent is tethered intramolecularly with aryl iodide substrates. The Lautens group reported the intramolecular ortho alkylation and intermolecular ipso Heck reaction (Scheme 49).118 The substrate scope was later extended to secondary alkyl halides, other types of linkers, and different ring sizes.40,119–122 Notably, the Lautens group studied the reaction with enantioenriched substrates and found that the annulation proceeded with an overall inversion of the stereocenter (84f) originating from the secondary alkyl halide.40 Considering that reductive elimination typically proceeds with retention of stereochemistry, this result suggests that oxidative addition of the secondary alkyl halide to ANP undergoes a SN2-like pathway. Interestingly, if the termination nucleophile is attached to the aryl halide, fused tricyclic structures could be formed (84b).123 Besides ipso Heck coupling, ipso cyanation83 and vinylation via carbene insertion86,124 have been established with aryl iodides and bromides under the intramolecular type-C reaction mode. Recently, the Liang group utilized a Pd/Cu cocatalyst system to synthesize polyfluoroarene-substituted benzofuran and benzopyran derivatives,125 where a polyflouoroaryl copper species was believed to be the key intermediate.126
In Type D intramolecular couplings, tricyclic products have been formed via either Heck or direct arylation as the termination step (Scheme 50).40,120
Scheme 50.

Synthesis of Tricyclic Structures by Type D Intermolecular Coupling
3.2.1.3. Epoxides, Azirines, and Aziridines as Alkylation Reagents.
The previous subsections focus on ortho alkylation using alkyl halides. Alternative alkylation reagents that have been reported in the Pd/NBE catalysis include azirines, epoxides, or aziridines. These sterically less hindered and hard electrophiles are expected to selectively react with ANP rather than Pd(0) and can provide oxygen or nitrogen-containing heterocycles as products. Another advantage of using epoxides or aziridines for annulation reactions is that the termination nucleophiles are automatically “masked” by default, therefore minimizing direct ipso termination side reactions.
In 2010, the Lautens group reported the first annulation reaction using 2H-azirines for indole synthesis (Scheme 51).127 During their initial studies, they focused on the use of bifunctional α-haloimines for enabling the ortho alkylation/ipso N-arylation. Though effective, preparation of these α-haloimines was not trivial, leading them to explore 2H-azirines as an α-haloimine equivalent. Regarding the ortho alkylation step, it was proposed that the C–N oxidative addition to the ANP intermediate took place to give a Pd(IV) intermediate, followed by C–C reductive elimination.128 Due to the high reactivity of 2H-azirines (with a strain energy of 44–48 kcal/mol), some side reactions were also observed, such as dimerization of 2H-azirines and the further [3 + 2] reaction with the product to give polycyclic dihydroimidazoles.
Scheme 51.
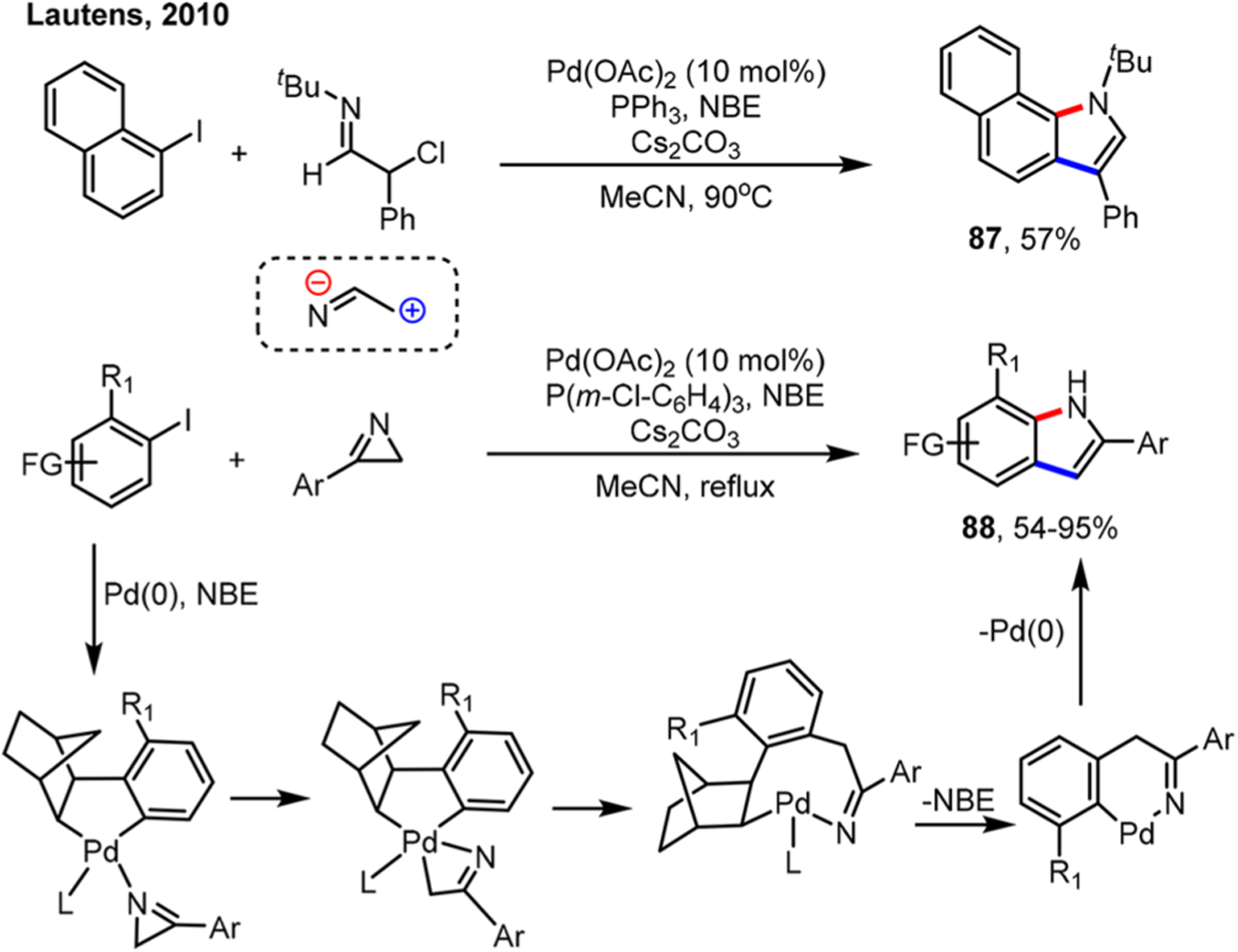
Synthesis of Indoles by Ortho Alkylation of Aryl Iodides Using 2H-Azirines
The use of abundant but less reactive epoxides129,130 in the Pd/NBE-catalyzed reactions was not reported until 2017. The Dong group developed an annulation between aryl iodides and epoxides to form 2,3-dihydrobenzofurans (Scheme 52).131 A polar aprotic solvent, DMF, was used, likely facilitating a SN2 type ring opening of epoxides. Sterically hindered Buchwald ligands were employed to circumvent undesired β-hydrogen elimination from intermediate 93 and facilitate the C–O bond formation step. Simple NBE provided the desired product but suffered from the multiple-NBE insertion pathway, the use of a bulky C2-substituted NBE 89 that is less reactive toward insertion suppressed such a side reaction. When an enantiopure epoxide was used, the enantiopure product was obtained with stereoretention (90c). Later, the catalytic asymmetric reaction via kinetic resolution of racemic epoxides with chiral NBE cocatalysts was explored by the same group.132 While the enantioselectivity was moderate (90d), it represents one of the first examples of chiral NBE-promoted asymmetric reactions in the Pd/NBE catalysis (for a chiral NBE-promoted asymmetric meta functionalization via a Pd(II)-initiated pathway, see Scheme 116). The synthetic utility of this method was demonstrated in the 4-step synthesis of insecticide fufenozide (see section 4.1).
In 2018, Zhou and co-workers independently reported the ortho alkylation/ipso Heck using epoxides as the electrophile (Scheme 53).133 Similarly, polar NMP as the solvent was essential for this reaction to produce the desired product. A novel C5-carboxylate-substituted NBE 94 (50 mol %) was used as both the cocatalyst and the base, which could also be conveniently removed after the reaction. In addition, through tethering the epoxide with an olefin, the intramolecular Heck termination led to a macrocycle (95b). Moreover, a one-pot oxa-Michael addition could be carried out under basic conditions to furnish the isochroman scaffold (96c). Such conditions were later applied to the synthesis of 2,3-dihydrobenzofurans.134
Scheme 53.
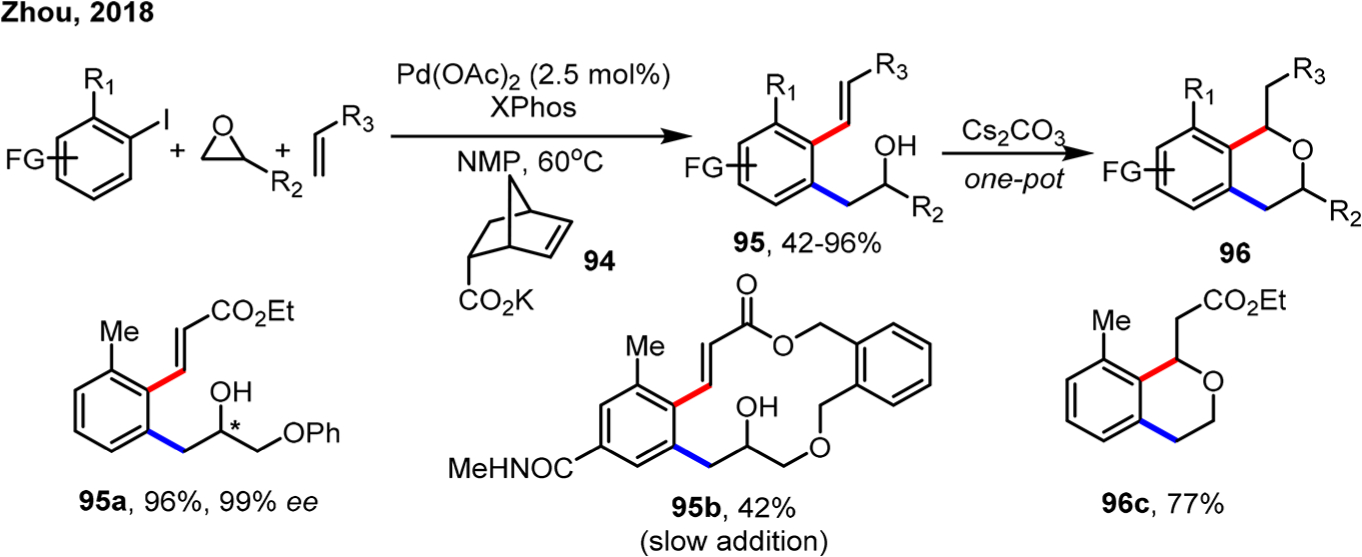
Synthesis of Isochromans by Ortho Alkylation of Aryl Iodides Using Epoxides
Analogous to epoxides, the Liang group developed an ortho alkylation method using aziridines to afford indolines in 2018 (Scheme 54).135 P(m-ClC6H4)3 was chosen as the optimal ligand and nonpolar toluene was used as the solvent. Interestingly, ortho-unsubstituted and electron-deficient aryl iodides could still give the desired products (97b), but simple iodobenzene was still not a suitable substrate. It is likely that the use of a bifunctional reagent might help the NBE extrusion step in the case of meta-substituted electron-deficient aryl iodides. For 1-alkyl aziridines, the C–N bond cleavage took place at the less sterically hindered site (97c); for 1-aryl aziridines, the C–N bond cleavage preferred to occur at the weaker benzylic position (97d). The difference of the regioselectivity could be explained by the relative easiness for the SN2 reactions. To further support the SN2 mechanism for ring opening, inversion of stereochemistry was observed at the benzylic position. In addition, their model stoichiometric reactions proved that NBE extrusion occurred from an 8-membered palladacycle to give a 6-membered palladacycle.
Scheme 54.
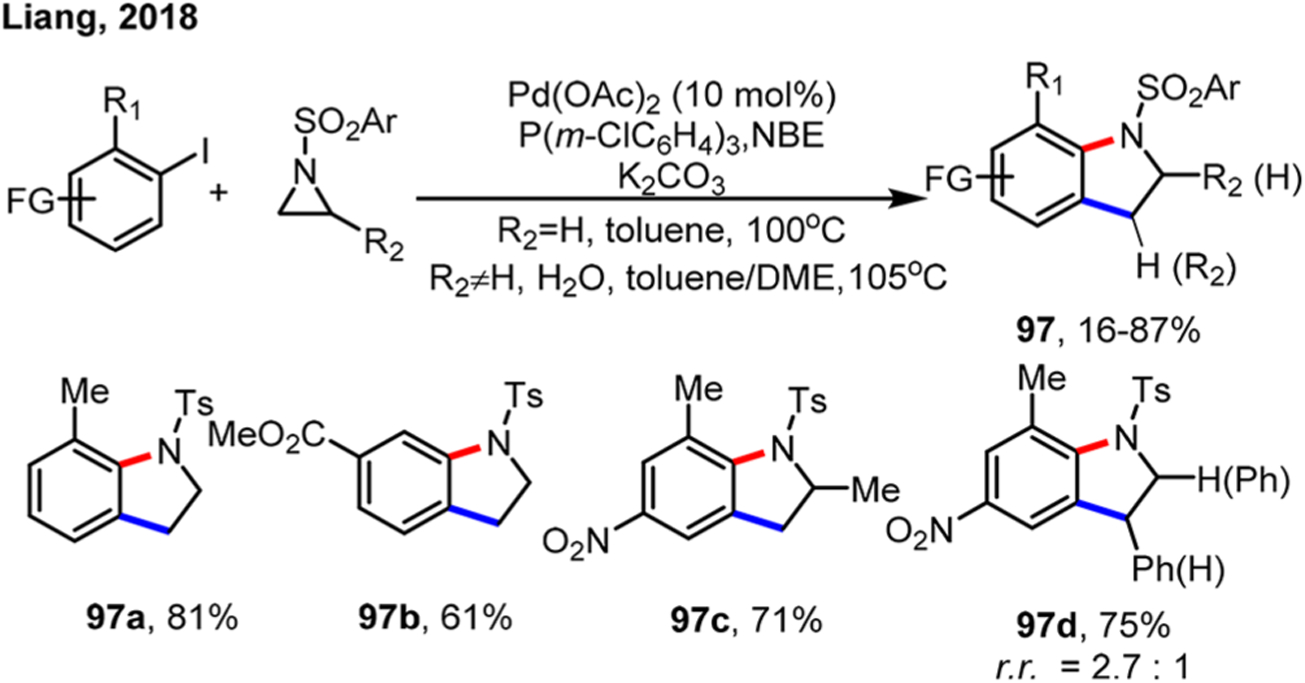
Synthesis of Indolines by Ortho Alkylation of Aryl Iodides Using Aziridines
Later, Zhou and co-workers achieved the ortho alkylation/ipso Heck reaction followed by aza-Michael addition using aziridines as the alkylation reagent to synthesize tetrahydroisoquinolines (Scheme 55).136 Excellent diastereoselectivity was observed during the 1,4-addition step (99b,c). Improved regioselectivity control was achieved with respect to 1-aryl aziridine substrates (99c).
Scheme 55.

Synthesis of Tetrahydroisoquinolines by Ortho Alkylation of Aryl Iodides Using Aziridines
3.2.2. Ortho Arylation of Aryl Iodides.
3.2.2.1. Intermolecular Couplings.
Besides alkyl-type electrophiles, the second class of electrophiles employed in the Pd/NBE catalysis are aryl halides. As discussed in the stoichiometric reactions section (vide supra, Scheme 9), the use of aryl halides as external electrophiles often led to more complicated outcomes, but the finding of the “ortho effect” was the key for selective aryl–aryl couplings. The Catellani group reported the first catalytic ortho arylation in 2001 using internal alkynes as the termination reagents (Scheme 56).137 A range of phenanthrene products (100) were obtained using diaryl alkynes. When the ortho substituent on aryl iodides was large, such as a tert-butyl group, no product was observed (100b). Alkylaryl alkynes gave a mixture of regioisomers (100c) together with some allene products; in contrast, dialkylacetylenes predominately gave the allene product (100d).
Scheme 56.

Synthesis of Phenanthrenes from Ortho-Substituted Aryl Iodides and Internal Alkynes
Analogous to the ortho alkylation reactions, olefins could be employed as the termination reagents (Scheme 57). In 2003, Catellani reported the first example of ortho arylation/ipso Heck using electron-poor, -rich, and -neutral olefins.138 Interestingly, ortho carbomethoxy-substituted aryl iodide gave the best yield (101b), likely due to its faster reaction with ANP. The reaction gave poor yields when a small ortho substituent was present (101c). Later, the same group demonstrated the first use of redox-relay Heck reaction as the termination step (102).139
Like the ortho alkylation cases, other types of cross-coupling reactions have been employed to functionalize the ipso position. For example, the Suzuki–Miyaura termination140 with arylboronic acids was achieved in the ortho arylation reaction (103a), affording o-terphenyl products (Scheme 58). While the ipso hydrogenation was achieved previously in the stoichiometric reactions,46 the use of dihydrogen gas led to a nonselective outcome in the catalytic reactions. Catellani therefore screened different hydrogen-transfer reagents and found that benzyl alcohol gave the best result (103b).141 In 2009, the same group reported one of the first examples of using enolate coupling as the termination step (103c),90 where the combination of 10% KOPh with K2CO3 was essential to maintain a suitable range of enolate concentration. They also reported using direct arylation as the termination step to afford teraryls (103d).142
Scheme 58.

Homo Ortho Arylation/Ipso Functionalizations of Aryl Iodides
The previous ortho arylation examples involved the homocoupling of aryl iodide substrates. It would be highly desirable if cross-couplings between two different aryl halides could be achieved; however, this would be more challenging because one aryl halide needs to selectively react with Pd(0) and the other has to selectively react with ANP. For example, the reaction between o-iodotoluene and o-iodoethylbenzene gave all four possible products with low selectivity.143 Catellani and co-workers found that, when replacing o-iodoethylbenzene with methyl o-iodobenzoate 104a, only three products were observed (Scheme 59).143 The predominance of the homocoupling product 105c suggested that methyl o-iodobenzoate is highly reactive to both Pd(0) and ANP. Interestingly, when the less reactive methyl o-bromobenzoate was used instead, the reaction selectively afforded 105b as the sole product.
Scheme 59.

Selectivity Problems in Cross Ortho Arylation
Although the exact reason remains to be uncovered, such selectivity could be tentatively explained as follows (Scheme 60). Compared to a C–I bond, a C–Br bond is shorter and stronger; thus, it generally requires more distortion in the transition state of oxidative addition. Thus, oxidative addition of aryl iodides with Pd(0) is favored by the low distortion energy, likely due to a more flexible Pd(0) complex and a weaker C–I bond. On the other hand, the Pd(II) center in ANP is considered to be more Lewis acidic and more rigid than the softer Pd(0) species; thus through chelation144 with the ester moiety, ANP would selectively react with aryl bromide 104b rather than the less coordinative 2-iodotoluene. It should be noted that these explanations still require further experimental and/or computational support, and caution must be taken when considering these simplified explanations since other factors like ligands and anions might also be crucial for achieving the desired selectivity.
Scheme 60.

Tentative Explanations on the Observed Selectivity
During their exploration of the reaction scope, Catellani and co-workers found that the “ortho effect” only required the aryl iodide to bear an ortho substituent, while an ortho group on the aryl bromide was not necessary (Scheme 61). Since electron-withdrawing or ortho chelating groups on haloarenes would increase their reactivity toward ANP, it is better to use electron-donating and nonchelating ortho substituents on the aryl iodide substrate to avoid their reactions with ANP. One exception is the ortho-CF3 substituted aryl iodide: the steric hindrance and noncoordinative nature of the CF3 moiety reduced its reactivity with ANP (106c, 106d). On the other hand, electron-withdrawing (106e) or ortho chelating groups (106f) on the aryl bromide part is preferable. Both electron-rich and -poor olefins could be coupled at the ipso position. Recently, gaseous ethylene was also successfully employed as the termination agent by Della Ca’ and Noël using the flow technique.145
Scheme 61.

Cross Ortho Arylation/Ipso Heck of Aryl Iodides
When an ortho heteroatom group is present in the aryl bromide part, it could act as not only a chelating group, but also a nucleophile to react with the enoate moiety formed at the ipso position (Scheme 62). An oxa-Michael reaction took place to give 6H-dibenzopyrans (107a) when o-bromophenol was used.146 The corresponding m-bromophenol or p-bromophenol did not react, showcasing the importance for ortho chelation when using electron-rich aryl bromides. The enantioselective version was later developed by Zhou and Catallani using a cinchona alkaloid as a cocatalyst.147 Similarly, o-bromoarene-sulfonylanilines afforded phenanthridine (107b) via a subsequent aza-Michael reaction.148 When trifluoroacetyl-protected 2-bromoanilines were used, hydrolytic cleavage of the trifluoroacetyl protecting group, followed by a retro-Mannich reaction, gave the phenanthridine product (107c).149 Methyl vinyl ketone was found to be an excellent termination reagent to promote the retro-Mannich reaction. In a similar manner, the reaction with bromobenzylamine provided dibenzo[c,e]azepines and their imine analogues (107d).150
Scheme 62.

Cross Ortho Arylation/Ipso Heck Followed by Michael Reaction
Other types of termination reactions have also been developed for cross-aryl couplings (Scheme 63). Cyanation with K4[Fe(CN)6]·3H2O was achieved by Lautens under microwave conditions to give aromatic nitrile products (108a).72 The same group later found that ipso hydrogenation (108b) could be achieved using 1,2-dimethoxyethane (DME) as the solvent and the reductant.151 Their deuterium labeling study unambiguously confirmed that the hydride source was the methylene hydrogen from DME. Catellani and co-workers reported the Suzuki–Miyaura quench with arylboronic acids (108c).152 Interestingly, the arylation selectivity could be improved when diethyl maleate was added as the ligand. If o-bromobenzyl alcohol was used, ipso hydrogenation occurred through transfer hydrogenation of the benzyl alcohol moiety,153 which gave o-biaryl carbaldeydes (108d) as the products.
Scheme 63.

Cross Ortho Arylation/Ipso Functionalizations of Aryl Iodides
When the carboxyl group was used as the chelating group, a further decarboxylation reaction could occur to give intermediate 109, which serves as the common intermediate for further functionalization (Scheme 64). In 2017, Kwong and Lin reported a net π-extension reaction of aryl halides using 2-halobenzoic acids and norbornadiene to afford product 110.154 The products were formed through reductive elimination from a 109-like intermediate, followed by a retro-Diels–Alder reaction. In 2018, Kwong and Fu group extended this reaction by adding alkynes to achieve a regioselective aromatic π-extension reaction (111).155 Instead of reductive elimination, in this case, 109 underwent NBE extrusion and alkyne insertion/annulation. In the same year, Yang and Liang reported that 109 could undergo another arylation with 2-bromobenzoic acid, followed by norbornene extrusion, another decarboxylation, and reductive elimination to give triphenylenes 112 as products.156
Scheme 64.

π-Extension Reaction Using 2-Halobenzoic Acids
3.2.2.2. The “Ortho Effect” in Ortho Arylation Reactions.
As discussed in section 2.2, the outcome of the reaction between ANP and the haloarene electrophile depends on the substituent pattern of the ANP aryl ring. If the ANP intermediate is derived from an aryl iodide with an ortho substituent, selective aryl–aryl bond formation (rather than aryl–norbornyl bond formation) would occur.44 Otherwise, the arylation would be unselective. Such a phenomenon was termed by Catellani as the “ortho effect”. While the “ortho effect” successfully helped the development of the ortho arylation reactions, its origin remained unknown for a long time.
To date, two possible pathways have been proposed for the reaction of ANP with the haloarene electrophile. The first one involves a Pd(IV) pathway, where ANP directly undergoes oxidative addition with haloarene. The intermediacy of Pd(IV) has been directly observed with alkyl electrophiles,38 however not in with aryl electrophiles. In a related case, Vicente reported isolation of a Pd(IV) complex by oxidative addition of 2-iodobenzoic acid to a Pd(II) complex,144 proving the feasibility of such a process. The alternative pathway involves a transmetalation process, where oxidative addition of the haloarene electrophile with a separate Pd(0) species forms an aryl-Pd(II) species, followed by dinuclear transmetalation between the aryl-Pd(II) species and ANP. Such a pathway has been supported by DFT calculation on a simplified system by Cardens and Echavarren.157
In 2011, Catellani, Deret, and Malacria performed the DFT calculation using M06 hybrid functional to study the origin of the “ortho effect” (Scheme 65).158 First, they found that both pathways could dominate depending on the substituent pattern on the aryl moiety in ANP. Without the ortho substituent, the transmetalation pathway is favored over the Pd(IV) pathway by 8–10 kcal/mol for different substrates, in agreement with the Echavarren’s finding. In contrast, the Pd(IV) pathway becomes the preferred pathway when an ortho substituent is present in the aryl moiety of ANP by 1–7 kcal/mol. The steric clash between the ortho substituent and NBE (114a and 114b) greatly increases the barrier for the transmetalation pathway, while the Pd(IV) pathway remains with a similar barrier. Second, they found that, for the ortho-unsubstituted ANP, the energy difference between the two coupling modes, i.e., C(sp2)–C(sp2) or C(sp2)–C(sp3), in the transmetalation pathway is small (1.5–2.0 kcal/mol), rendering an unselective arylation. In contrast, the reductive elimination step from the Pd(IV) intermediate 113 is highly selective, favoring the C(sp2)–C(sp2) coupling by 4–12 kcal/mol over the C(sp2)–C(sp3) coupling.
Scheme 65.

DFT Calculations on the Origin of the “Ortho Effect”
Although understanding the ortho effect allows choosing appropriate substrates for successful reactions, it is still highly desirable to use common ortho-unsubstituted aryl iodides as substrates for selective cross couplings. In 2015, the Yu group found that 2-carbomethoxy-substituted NBE 115 could afford a “normal” bis ortho arylation product of iodobenzene rather than the NBE-containing product (Scheme 66).159 It is likely that the undesired C(sp2)–C(sp3) coupling was inhibited due to the steric repulsion of the carbomethoxy substituent on NBE 115. In this case, both ortho positions were arylated to form a self-trimer structure. Very recently, the Dong group employed bridgehead-substituted NBE 117 to achieve mono-ortho arylation of ortho-unsubstituted aryl iodides via cross couplings.160 Presumably, the presence of the bridgehead substituent on NBE not only inhibited the undesired arylation on the NBE part but also promoted the NBE extrusion (for a detailed discussion, see section 3.2.7).
Scheme 66.

Overcoming Ortho Effect with Substituted NBEs
3.2.2.3. Intramolecular Couplings.
Since the cross ortho arylation between an aryl iodide and an aryl bromide requires an ortho chelating group on the aryl bromide, the chelating moiety could be conveniently used as the termination agent. Such intramolecular couplings could be viewed as a Type A annulation (vide supra, Scheme 41). However, caution should be taken in these types of transformations, since an alternative pathway involving ipso coupling, followed by oxidative addition into ArBr and C–H annulation could potentially give the same product in the absence of NBE.
The first example of such a transformation was reported by Catellani, in which the ortho amide group served both as a chelating group and a nucleophile for the ipso termination (Scheme 67).161 A range of 6-phenanthridinones (119a) was afforded in moderate to excellent yields. Notably, this was also the first time when the Buchwald–Hartwig amination was employed as the termination step. Surprisingly, in the absence of NBE, self-dimerization of o-bromoarylcarboxamide took place to give condensed pyridones.162 Similarly, o-bromo-N-tosylani-line afforded carbazole products (119b). Interestingly, when acetamide was used instead of sulfonamide, deprotected carbazole products were observed, and carbazomycin A was synthesized using this method (see section 4.1).163
Scheme 67.

Synthesis of Fused Rings via Ortho-Arylation Ipso-C–N or C–O Coupling
Lautens employed o-chloro-N-silylimines to achieve ipso C–N coupling and provide phenanthridine products (119c).164 N-Silylimines are more stable than the corresponding unprotected imines and the silyl group was cleaved under the reaction conditions.165 Interestingly, the reaction scope was broad, as N-silylketimine, N-silylamidines, and even N-unsubstituted ketimines were all suitable substrates. To avoid using unstable imines, Malacria synthesized phenanthridines (119d) from benzyamines and aryl iodides.166 Subsequent oxidation using dioxygen provided the aromatic products. Control experiments showed no product formation when NBE was omitted, ruling out the alternative pathway involving amination of the aryl iodide followed by intramolecular ring closure. Assoanine and pratosine were later synthesized using this method (see section 4.1).167 Besides ipso C–N coupling, ipso C–O coupling has also been developed. In 2012, Catellani reported the first example of such transformations in the synthesis of dibenzopyrans (119e) using o-bromobenzyl alcohols as the coupling partner.153 In this case, no additional phosphine ligand was added, but a tertiary alcohol had to be used to avoid β-hydrogen elimination. Similarly, the use of o-bromophenol provided dibenzofurans (119f).168
Due to the difficulty of the C–O bond reductive elimination using o-bromophenols, the intermediate after NBE extrusion could be trapped for further functionalization. In 2017, the Luan group successfully intercepted such an intermediate through alkyne insertion and dearomatization to achieve a [2+2+1] spiroannulation using 1-bromo-2-naphthol and an internal alkyne (Scheme 68).169
Scheme 68.

Dearomatizing [2+2+1] Annulation of Bromonaphthols with Aryl Iodides and Alkynes
In 2018, Yamamoto reported an annulation with 4-iodo-2-quinolones via ortho arylation/ipso C–O coupling (Scheme 69).170 The presence of NBE was found to be essential for this reaction. DFT calculations showed the importance of the amide group serving as a directing moiety in the CMD step.
Scheme 69.

Ortho Arylation/Ipso C–O Coupling of 4-Iodo-2-Quinolones
It is important to note that, although the chelating group in the aryl bromide part could lower the barrier for the oxidative addition with ANP, it might increase the barrier and change the regioselectivity in the subsequent reductive elimination step.171 Discovered by Malacria and Lacôte, the reaction of 2-iodotoluene and 2-bromophenylacetamide gave a NBE-containing dihydrophenanthrene product (122a) (Scheme 70).172 This product came from the arylation at the norbornyl site, which was abnormal when considering the ortho effect. Interestingly, the addition of excess water switched the selectivity back to the normal aryl–aryl coupling product 122b, albeit followed by a dearomatization step. According to their DFT studies, the Pd(IV) pathway was still favored over the transmetalation pathway, in accordance with the ortho effect. However, the presence of the chelating group forced a distorted octahedral geometry in the Pd(IV) intermediate 123, which subtly changed the reductive elimination selectivity. With the addition of water, the chelation could be partially released, restoring the normal selectivity. The nature of the primary amide chelating group also plays an important role, as other chelating groups did not show such an effect.
Scheme 70.

Switching Regioselectivity for Reductive Elimination by Chelation
Another unusual example is the use of 2-bromoaniline (Scheme 71). The unexpected C(sp2)–C(sp3) coupling followed by ortho C–N coupling gave NBE-containing products 124, even though ortho substituted aryl iodides were used.173 Such a transformation became synthetically useful when norbornadiene was used instead of NBE, so that the product (124) could be further transformed into dibenzoazepines (125) via a retro-Diels–Alder reaction. Ortho-unsubstituted aryl iodides reacted similarly. These results are in sharp contrast with the case of aforementioned N-protected 2-bromoanilines (119b). The DFT calculation showed that the electron-rich aniline ring is nucleophilic and nicely matched the LUMO around the norbornyl C(sp3) position; in contrast, the protected aniline is less electron-rich; thus it provided the normal aryl–aryl coupling product.
Scheme 71.

Formation of Dibenzoazepine Derivatives by Ortho Arylation of Aryl Iodides with o-Bromoanilines
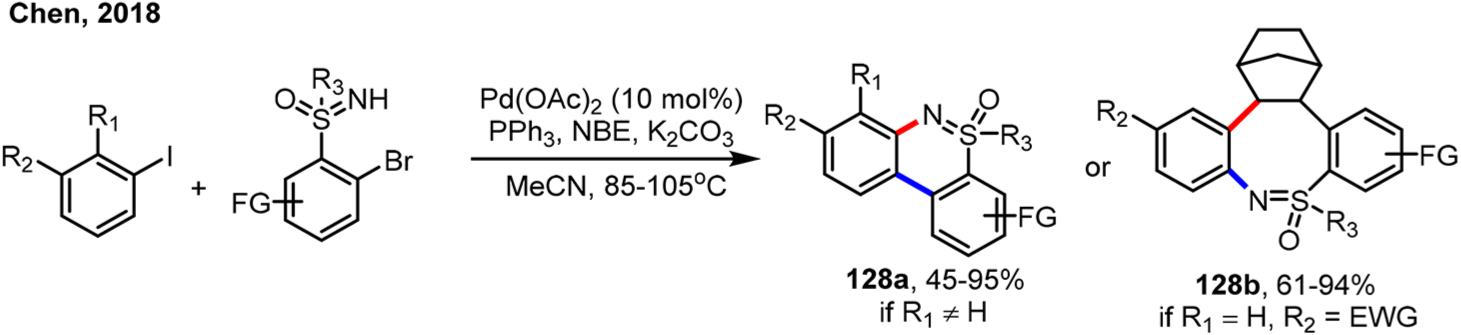
Very recently, the Chen group reported the ortho arylation of iodoarenes using 2-bromo-NH-sulfoximines as the arylation reagent (Scheme 72).174 When ortho-substituted aryl iodides were used, the expected ortho arylation, followed by ipso C–N coupling, afforded cyclic sulfoximines (128a). In contrast, when meta-substituted aryl iodides were used, NBE-containing products (128b) were obtained instead, resulting from the C(sp2)–C(sp3) coupling. These results could be nicely explained by the ortho effect, where the transmetalation pathway took place when ortho unsubstituted aryl iodides were used.
Scheme 72.
Divergent Preparation of Heterocyclic Sulfoximines
Ketone, ester, and aldehyde-derived aryl bromides can also serve as bifunctional reagents (Scheme 73). The aryl-Pd(II) intermediates are usually considered to be electrophilic especially in the C–C cross-coupling reactions; however, sometimes they could also show nucleophilic characteristics when interacting with carbon–heteroatom multiple bonds.175 Lautens first showed such dual reactivity in the Pd/NBE catalysis using 2′-chloroaceteophenone as a reagent.91 Using DME as the solvent with excess water, ipso 1,2-addition occurred to give 9H-fluoren-9-ol 129a. It is likely that water acted as a proton source and DME behaved as a reductant. In contrast, in anhydrous acetonitrile, phenanthren-9-ol 129b was obtained as a result of the ipso enolate coupling. Besides ketones, 1,2-addition to esters, aldehydes, and N-sulfinyl imines as the termination step has also been demonstrated (129c).
Scheme 73.

Ortho Arylation/Intramolecular Ipso 1,2-Addition of Aryl Iodides
Although most intermolecular couplings involving ortho arylation belong to Type A, a few examples of Type B annulation have also been reported (Scheme 74). The first example was described by Lautens in 2010, where phenoxazines and dihydrodibenzoxazepines were obtained through the ortho arylation/ipso C–N coupling (130).117 In 2013, the Gu group applied the ortho arylation/ipso intramolecular Heck reaction with iodopyrroles in the total synthesis of rhazinal (see section 4.1).176 In 2017, the Luan group used alkyne-tethered aryl iodides as substrates, which enabled an intramolecular alkyne insertion and dearomatization (131) for constructing the cores of polyketide natural products, dalesconols A and B.169
Scheme 74.
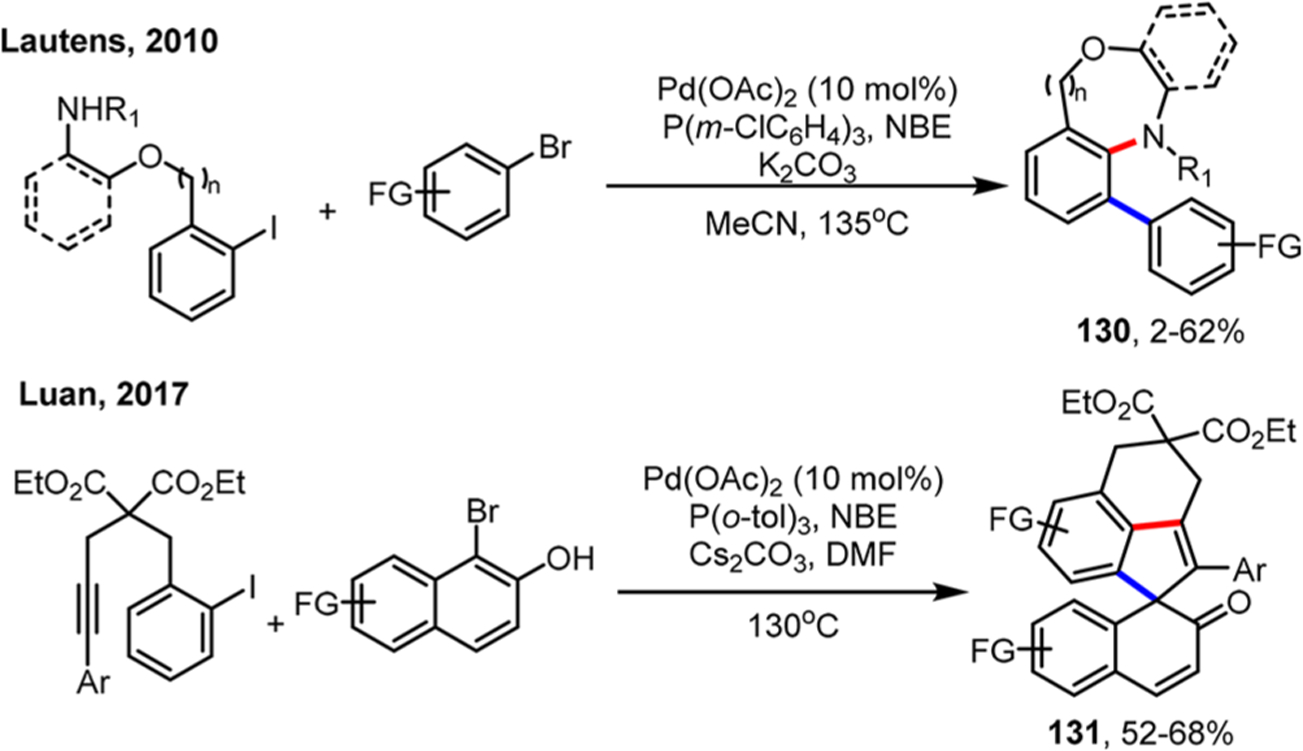
Type B Intramolecular Coupling in Ortho Arylation of Aryl Iodides
3.2.3. Ortho Amination of Aryl Iodides.
Despite the early discovery of ortho alkylation (1997) and arylation (2001), the introduction of heteroatoms at the arene ortho position in the Pd/NBE catalysis was not reported until 2013. The Dong group found that O-benzoylhydroxylamines177,178 can serve as an excellent external electrophile to trap ANP, which led to the development of ortho amination reactions (Scheme 75).179 The electrophilic nitrogen moiety in O-benzoylhydroxylamines is expected to have a strong interaction with the nucleophilic ANP, and the benzoate part might act as both a leaving group and a chelating moiety. For comparison, N-chloroamines only gave a complex mixture. Isopropanol was employed as the reductant to deliver hydrogen at the ipso positions, and the deuterium labeling study confirmed the hydride transfer mechanism. The three reactants were used in near equimolar ratio with only 25 mol % of NBE. The scope of aryl iodides is also quite broad: some traditionally challenging substrates could be successfully coupled. For example, 1-fluoro-2-iodobenzene (132b) that was known to have difficulties in the β-carbon elimination step143 due to the small size of fluorine can work in the ortho amination reaction. Methyl 2-iodobenzoate usually afforded self-arylation dimer in other cases,143 and its success (132c) shows that O-benzoylhydroxylamines are more reactive and/or selective toward ANP. Acyclic secondary O-benzoylhydroxylamines could be coupled (132d) but primary O-benzoylhydroxylamines were not suitable. When aryl iodides with para substituents or small meta substituents were used, ortho/ortho′ bisamination took place, similar to the ortho alkylation chemistry.
Scheme 75.
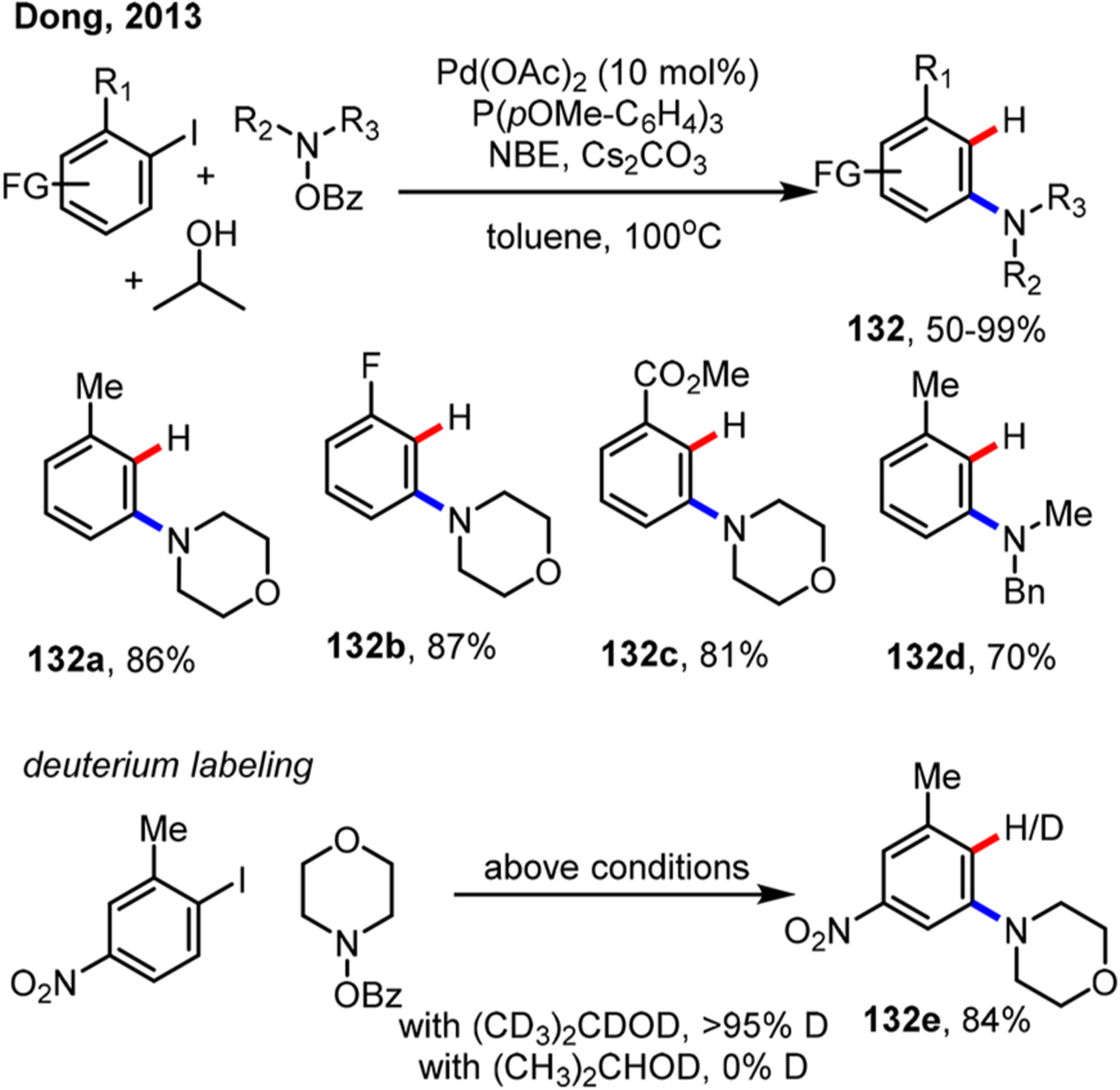
Ortho Amination/Ipso Hydrogenation of Aryl Iodides
Subsequently (2014–2018), other types of ortho amination reactions with different ipso terminations have been developed (Scheme 76). For example, Heck coupling with electron-deficient180 or -rich181 olefins (133a, 133f), Suzuki–Miyaura coupling with arylboronic esters (133b)182 or methyl boronic acids (133i),73 carbene coupling (133c),183 Sonogashira coupling (133d, 133e),77,78 cyanation (133g, 133h),184,185 and enolate coupling (133j)186 have been successfully demonstrated at the ipso position using the similar strategies developed in the aforementioned ortho alkylation/arylation reactions.
Scheme 76.

Ortho Amination/Ipso Functionalizations of Aryl Iodides
Interestingly, the Ritter group reported the first example of the ipso borylation reaction using bis(pinacolato)diboron as the termination reagent (Scheme 77).187 The merit of such a transformation was demonstrated in the diverse and convenient derivatizations of the formed B(pin) moiety. For example, the C–B bond can be converted into C–N (134b), C–Cl, C–Br (134c), and C–I and C–OH (134d) bonds, which are hard to access via direct terminations. The major side reaction was the ipso hydrogenation, and the reductant was the HBpin generated in the reaction. The synthetic utility was demonstrated in the modular syntheses of Abilify and Flunixin (see section 4.1).
Scheme 77.

Ortho Amination/Ipso Borylation and Derivatization of Aryl Iodides
Besides intermolecular reactions, Type B intramolecular couplings have also been reported based on ortho amination (Scheme 78). Using phenol-tethered aryl iodides, the Luan group developed an intercepted ortho amination terminated by phenol dearomatization,188 in which norbornadiene was incorporated into the product (135). In 2018, Lautens reported the first example of the Pd/NBE-catalyzed intermolecular ipso amidation reaction (136).189 The key was the use of Pd(PPh3)4 as the catalyst, preventing intermolecular ipso termination; in contrast, the use of RuPhos gave the direct ipso termination side products (no ortho amination). Very recently, the Liang group developed the first ipso termination using intramolecular sp3 C–H activation (137).190 The use of catalytic pivalic acid was important to lower the activation barrier for palladation of unactivated aliphatic C–H bonds, as shown by their DFT calculations.
Scheme 78.

Type B Intramolecular Couplings in Ortho Amination
3.2.4. Ortho Acylation of Aryl Iodides.
Aromatic ketones are widely found in pharmaceuticals, agrochemicals, organic electrons, and polymers; thus, it is attractive to develop efficient and site-selective methods for arene acylation. In 2015, the Liang,191 Gu,192 and Dong193 groups concurrently reported the Pd/NBE-catalyzed ortho acylation reaction of aryl iodides (Scheme 79). The work by Liang and Gu focused on using the Heck reaction to functionalize the ipso position. Symmetrical acyl anhydrides were used by Liang, and in situ generated acyl anhydrides from acyl chlorides were used by Gu. Good functional group tolerance was observed for both cases. In addition, both aromatic and aliphatic acyl electrophiles were compatible, although α-nonbranched aliphatic anhydrides usually gave reduced yields (139c).
Scheme 79.

Initial Reports of Ortho Acylation Reaction
The Dong group developed an ortho acylation/ipso hydrogenation method, in which a bifunctional mixed anhydride was designed to provide the acyl electrophile and slowly release isopropanol reductant. The acyl–O bond was selectively cleaved instead of the carboxyl–O bond, probably owing to the higher electrophilicity of the former. An amide-substituted NBE (141) was found to be more effective than simple NBE. Both electron-rich and deficient (hetero)aryl iodides were suitable substrates. Para-substituted aryl iodides gave bis-ortho acylation along with a tandem cyclization product. Aryl (142a), heteroaryl, and alkyl (142b) carboxylic anhydrides were suitable substrates. While the scope was focused on ipso hydrogenation, ipso Heck (142c) and Suzuki–Miyaura (142d) couplings were also demonstrated as single examples. The synthetic utility of the ortho acylation/ipso hydrogenation method was demonstrated in the synthesis of ketoprofen (see section 4.1).
Similar to the N-benzoyloxyamines, the electrophilic acyl moiety should also have strong interactions with ANP; in addition, the carboxylate leaving group could further decrease the oxidative addition barrier through chelating effects. DFT calculations have been performed to understand the reaction between benzoic anhydride and ANP,53 which was found to be the rate-determining step (Scheme 80). The benzoate leaving group acts as a chelating group and the C–O bond is cleaved via a five-membered transition state (143a). Interestingly, the iodide ligated transition state was found to be more reactive than the P(2-furyl)3-ligated (143b) one. In addition, oxidative addition of the P(2-furyl)3-ligated Pd(0) with aryl iodides was more favorable than with anhydrides by 10 kcal/mol.
Scheme 80.

DFT Calculations on the Reaction of ANP with Anhydrides
Very recently, the Gu group utilized the in situ generated mixed-anhydrides to successfully expand the scope of the acyl moieties that can be installed (Scheme 81).194 The Yamaguchi reagent (2,4,6-trichlorobenzoyl chloride) was added to convert carboxylic acids to mixed anhydrides during the reaction. Notably, α-nonbranched aliphatic anhydrides can be smoothly coupled, possibly due to an increased electrophilicity. Pivalic acid cannot be coupled using this protocol, likely owing to the bulkiness of the anhydride reagent.
Scheme 81.

Expanding Scope of Ortho Acylation of Aryl Iodides Using Mixed Anhydride
Besides Heck,191,192 Suzuki–Miyaura,191,193,195 and hydrogenation193 quenches in the ortho acylation, other termination reactions have also been developed for ipso functionalization (Scheme 82). For example, ipso cyanation196 of ortho-substituted aryl iodides was achieved by Chen using CuCN as the cyanide source (145a). Other transformations like Sonogashira coupling (145b)197 and direct heteroarylation (145c)198 have also been realized with the ortho acylation chemistry.
Scheme 82.

Different ipso Functionalizations in ortho Acylation Reaction
In addition to the use of anhydrides as the acylating reagent, thioesters were also found to be suitable electrophiles. Based on studies of the Liebeskind–Srogl reaction,199 the Gu group first demonstrated the use of thioesters in the Pd/NBE-catalyzed ortho acylation/ipso thiolation reaction in 2016 (Scheme 83).200 The addition of CuI dramatically improved the yield from 10% to 85% with 1-iodonaphthalene. The Cu(I) salt was proposed to serve as a C(O)–S bond activator, and its exact role remains to be disclosed. Thioesters derived from aromatic, heteroaromatic, and aliphatic acids can all be coupled. A reaction using two different thioesters in one pot did not give cross-products (147). In 2018, the same group reported the related ortho acylation/ipso selenation reaction (148) using acyl selenides.201 In this case, CuI was found to be detrimental to the reaction.
Scheme 83.

Ortho Acylation/Ipso Thiolation and Selenation Reaction of Aryl Iodides
Besides intermolecular reactions, Type A intramolecular couplings have also been reported (Scheme 84). In 2015, the Dong group found the acrylic acid-derived anhydride provided indenone products (149) likely through an ortho acylation/ipso intermolecular Heck annulation.193 The scope of this transformation was later expanded to various other α,β-unsaturated acid anhydrides,202 and the reaction was applied in the syntheses of pauciflorol F and acredinone A (see section 4.1). Employing the in situ generated mixed anhydride strategy, the Gu group also reported the ipso intermolecular Heck using carboxylic acidtethered olefins,194 forming five- and six-membered rings (150). Highly electrophilic fluorinated imidoyl chlorides were used by Chen and Zhu as a new type of electrophiles, and subsequent intermolecular ipso C(sp2)–H activation afforded the phenanthridine products (151).203
Scheme 84.

Ipso Intermolecular Couplings in Ortho Acylation Reaction
3.2.5. Ortho Alkoxycarbonylation and Aminocarbonylation of Aryl Iodides.
Aryl carboxylic acid derivatives are not only commonly found in biologically important molecules, but also serve as a versatile C1 unit to access other types of functional groups, e.g., CHO, CH2OH, CN, etc. In 2016, the first example of an ortho alkoxycarbonylation reaction was realized using a modified mixed anhydride by the Dong group (Scheme 85).204 By taking advantage of the fact that ANP is sensitive to the sterics of the electrophile, selective cleavage of the less bulky C(carboxyl)–O bonds was achieved with 2,6-dimethylbenzoate-derived anhydrides (153). A Heck reaction was used as the ipso termination. Preliminary results on ortho alkoxycarbonylation/ipso hydrogenation (152b) and ortho aminocarbonylation/ipso Heck reactions (152c) were also achieved by similar strategies. When tethering the anhydride with an olefin, macrolactone products were formed in moderate to good yields, which represents a Type A annulation.
Scheme 85.

Ortho Alkoxylcarbonylation Reaction of Aryl Iodides and Its Extension
In 2016, Jiao and co-workers reported the ortho aminocarbonylation/ipso C(sp2)−H activation reaction using aryl carbamic chlorides as the electrophiles (Scheme 86).33 Phenanthridinones were formed as the major products. When one substituent on the nitrogen is an allyl group, an ipso Heck reaction was favored to afford lactams 158. Subsequent KIE, Hammett plot, and computational studies suggested that the rate-limiting step was the oxidative addition of ANP with aryl carbamic chlorides.
Scheme 86.
Ortho Aminocarbonylation/Ipso Intramolecular Coupling Reactions
3.2.6. Ortho Functionalizations of Aryl Bromides and Triflates.
From the viewpoint of practicality, aryl bromides are usually cheaper and more accessible than the corresponding aryl iodides. However, in contrast to the wide use of aryl bromides in cross-coupling reactions, aryl iodides are dominatingly used in the Pd/NBE chemistry. Such a constraint could be attributed to the slower oxidative addition of Pd(0) with aryl bromides or triflates, which makes external electrophiles more competitive for oxidation with Pd(0).
In the case of ortho alkylation reactions, two Type C intramolecular couplings (161 and 163) have been reported using aryl bromides as substrates (Scheme 87).83,124 The efficiencies were generally lower than the corresponding aryl iodides. Another special example was reported in the case of the ortho amination reaction (162), where the use of Ag2CO3 as the additive and dppe as the ligand were important for the success of the reaction.179
Scheme 87.

Ortho Alkylation/Type C Intramolecular Couplings of Aryl Bromides
Using aryl bromides or triflates in ortho arylation appears to be harder due to the selectivity issue in the initial oxidative addition of Pd(0). In 2011, Lautens and co-workers demonstrated the first example of using aryl triflates in the ortho arylation/ipso C–N coupling (Scheme 88) and applied this reaction in the synthesis nitidine and NK109 (see section 4.1).205 Remarkable selectivity was observed in this reaction, though the exact reason is unclear. In a similar endeavor, Maestri and Malacria reported their synthesis of this class of products through coupling of aryl triflates with bromobenzylamines.206
Scheme 88.
Ortho Arylation of Aryl Triflates
In 2018, the Liu and Dong groups systematically studied the use of aryl bromides in the Pd/NBE catalysis, particularly in the intermolecular settings (Scheme 89).207 Bidentate phosphines with a flexible backbone (e.g., dCypb and DPEPhos) were found to generally give higher yields. The DFT study suggested that the use of flexible bidentate ligands not only accelerated the oxidative addition into aryl bromides, but also facilitated the NBE extrusion step. Different ipso termination reactions, including hydrogenation (165a), borylation (165e), Heck (165b), Suzuki–Miyaura (165c), and Sonogashira (165d) couplings, have been demonstrated to be effective with various aryl and heteroaryl bromides. The availability of this method allowed for two consecutive Pd/NBE-catalyzed difunctionalizations to construct penta-substituted aromatic compounds (166). Other ortho functionalizations, such as acylation (167a) and alkylation (167b), have also been achieved using aryl bromides as substrates (Scheme 90).
Scheme 89.

Ortho Amination of Aryl Bromides
Scheme 90.

Other Ortho Functionalizations of Aryl Bromides
Beside the difficulty in achieving selective oxidative addition of Pd(0) with aryl bromides, the steps after ortho functionalizations could also be affected due to the presence of different halide anions. It was found that the ratio between products 168a and 168b depended on the halogen substitution of the substrate (Scheme 91).207 The aryl bromide gave more premature termination product (168b) than the aryl iodide. Interestingly, the addition of 20 mol % CsI improved the selectivity for the desired product (168a), which indicated that the halide anion might influence the steps after the ortho amination with ANP. Later, the Della Ca’ group also observed that the presence of potassium iodide was crucial for improving the yield with aryl bromides.208
Scheme 91.

Halide Effect in the Pd/NBE Catalysis
Very recently, Cushman disclosed the synthesis of benzo-[1,6]naphthyridinones from 4-bromoquinolines (Scheme 92).209 Less than 2% of the product was obtained in the absence of NBE, suggesting that this reaction went through an ortho arylation pathway. Interestingly, although two aryl bromides were used, the self-dimerization of o-bromobenzamides did not take place.
Scheme 92.

Synthesis of Benzo[1,6]naphthyridinones from 4-Bromoquinolines
3.2.7. The “Ortho Constraint” and Development of Bridgehead-Substituted NBEs.
One long-standing limitation in the Pd/NBE catalysis is the requirement of an ortho substituent in aryl halide substrates in order to achieve mono ortho functionalization. Generally, if ortho unsubstituted aryl iodides are employed, mono ortho functionalized products cannot be obtained, but instead, either NBE-containing side-products or bis-functionalized products would be formed (Scheme 93). This phenomenon is termed as the “ortho constraint”.160 To be more specific, if para-substituted aryl iodides were used, bis-ortho-functionalized products were usually the major products for the ortho alkylation,58 amination,179 acylation,193 and alkoxycarbonylation reactions.204 As a special case, the ortho arylation reactions would be more complicated due to the aforementioned selectivity problem on Ar–Ar vs Ar–norbornyl couplings, also known as the “ortho effect” (vide supra, section 3.2.2.2). As for the meta substituted aryl iodides, if the meta substituent is small (e.g., a methoxy group), bis-ortho-functionalized products could sometimes be observed.81 In other cases, NBE-containing side-product(s) caused by unsuccessful β-carbon elimination were usually formed.88,160 The origin of such an “ortho constraint” is closely related to the aforementioned early finding by Catellani that the NBE extrusion only occurred easily when both ortho substituents were present.46
Scheme 93.

“Ortho Constraint” in the Pd/NBE Catalysis
Yet, there were still some special cases when such an “ortho constraint” was not obeyed (Scheme 94). The first example was reported in 2009,120 where Lautens successfully employed ortho-unsubstituted aryl iodides in a Type D intramolecular reaction (170). This experiment showed that, under certain reaction conditions, NBE extrusion could occur with only one ortho substituent present. NBE extrusion could be described as a thermodynamically unfavorable but kinetically feasible process; thus, the equilibrium could be shifted if a facile intramolecular termination step is present. For the same reason, the use of bifunctional reagents also led to the success in two other special cases (vide supra, 76b and 97b in Schemes 43 and 54, respectively).96,135 However, the success of this approach is largely relied on the structure of bifunctional reagents, and in many other cases, bifunctional reagents could not afford the desired products for ortho-unsubstituted aryl iodides. For intermolecular ipso couplings, Ranu and co-workers reported a single example of using meta-CF3-substituted iodobenzene in a successful ortho amination reaction (171).185 In contrast, other meta substituents gave a mixture of products. It is noteworthy that such meta-CF3-substituted aryl halides were not always successful in other Pd/NBE-catalyzed transformations.204,207
Scheme 94.

Violation of the “Ortho Constraint” in Special Cases
The cause for the “ortho constraint” is attributed to a facile second C–H metalation instead of β-carbon elimination. Aiming to develop a general catalyst-controlled approach to address the “ortho constraint”, the Dong group in 2018 designed and synthesized a class of bridgehead-substituted NBEs (Scheme 95).160 The hypothesis was that, by installing proper group(s) at C1 bridgehead position(s), the steric repulsion between R1 and E and/or between R2 and L would destabilize the transition state for the second C–H metalation and meanwhile the β-carbon elimination can be promoted through controlling the orientation of the aryl group to form the η1 complex.
Scheme 95.

Controlling Relative Rates for C–H Metalation versus β-Carbon Elimination Using Modified NBEs
Alkyl C1-substituted NBE 117 was found to be most selective and reactive. A range of ortho-unsubstituted aryl iodides, previously problematic substrates, can now be employed to provide mono ortho-functionalized products effectively. Such bridgehead-substituted NBEs proved to be general in various ortho/ipso bis-functionalizations (Scheme 96). Different ipso terminations, like Heck coupling (172a), hydrogenation (172b), Sonogashira coupling (172c), and Suzuki–Miyaura coupling (172d), can be employed. Besides ortho amination, ortho acylation (172e) and arylation (172f) can also be achieved. The same group also used such C1-substituted NBE 117 for the annulation reaction to synthesize indenone (172g), where the simple NBE failed to deliver the desired product.202
Scheme 96.

Mono Ortho Functionalization of Aryl Iodides without Ortho Substituents
Para-substituted aryl iodides and simple iodobenzene are more challenging substrates to give mono ortho functionalization due to a fast second C–H metalation step. In this case, the double bridgehead-substituted NBE afforded the desired products in high mono/di-selectivity (Scheme 97). The DFT calculation supports the effect of the presence of bridgehead substituents in NBE in retarding the second C–H metalation step while promoting the β-carbon elimination step. Mono ortho functionalization of para-substituted aryl iodides with electron-donating or withdrawing groups (EDGs or EWGs) has also been demonstrated.
Scheme 97.

Mono Ortho Functionalization of Iodobenzene
The synthetic utility of this method was illustrated in the arene functionalization at the site complementary to the EAS reactions. A sequence of electrophilic iodination followed by ortho functionalization allowed for installing FGs at positions para to EWGs or meta to EDGs. For example, site-selective amination of strychnine at the C5 position was achieved in two steps (175), while the conventional approach required a seven-step sequence (Scheme 98).210
Scheme 98.

Complementary Functionalization of Strychnine
3.2.8. Other Types of Ortho Functionalizations.
Sometimes even though the external electrophile could selectively react with the ANP intermediate, NBE might still get stuck into the product when the side-reaction pathways are faster than the desired β-carbon elimination reaction. In these cases, NBE functions as a reactant instead of a catalyst. In addition, since NBE is not extruded during the reaction, the ortho substituent of aryl halides is generally not required. Though strictly speaking these reactions do not fall into the category of the Pd/NBE cooperative catalysis, they are still discussed here due to the relevance of the reaction pathways.
In 2014, Shi and co-workers reported an ortho amination reaction using diaziridinones as the external electrophile (Scheme 99).211 In their proposed mechanism, the ANP intermediate undergoes oxidative addition with diaziridinones, which subsequently releases tert-butyl isocyanate (tBuNCO) to form the indoline structure (176). If norbornadiene was used instead, the products could be further transformed into indoles through a retro-Diels–Alder reaction. Later, it was shown that the use of anilines or ureas could lead to similar products.212 Recently, disilanes was also found to be able to intercept the ANP intermediate by the Zhang,213 Cheng,56,214 and Liang215 groups. Due to the facile C–Si reductive elimination, NBE-containing bis-silylated products were obtained. Interestingly, if 2,3-dicarbomethoxy-7-oxanorbornadiene was used instead of NBE, retro-Diels–Alder reaction occurred to give 177 as the final products. When vinyl bromides were utilized as the electrophiles, an intramolecular Heck reaction instead of NBE extrusion led to products 178, after the ortho alkenylation step.216
Scheme 99.

Other Types of Ortho Functionalizations of Aryl Iodides
3.3. Catalytic Reactions Initiated by Pd(II)
3.3.1. N–H Bond Activation-Initiated 2-Functionalization of Indoles.
In 2011, Jiao and Bach reported a Pd/NBE-catalyzed C2-alkylation of unprotected indoles with alkyl bromides, which represents the first Pd(II)-initiated Pd/NBE-catalyzed reaction (Scheme 100).217 The reaction is highly regioselective and unprotected indoles were used directly as substrates. Addition of water greatly enhanced the reaction rate. The major side reactions were base-mediated formation of N- or 3-alkylindoles; thus, a weaker base, e.g., K2CO3, was found to be more effective than Cs2CO3. KHCO3 could also be used when more acidic indoles were employed as substrates (179b). The reaction was sensitive to steric hindrance of the alkyl bromides, and secondary alkyl bromides did not react. Aryl bromides and aryl iodides (179b) were tolerated because Pd(0) was in principle not involved. The N-methylindole failed to give any product (179c), while 3-methylindole exhibited moderate reactivity (179d).218 This method has been successfully applied in various natural product syntheses (see section 4.1).
Scheme 100.
2-Alkylation of Indoles via Pd/NBE Catalysis
Regarding the proposed catalytic cycle, the reaction is initiated by Pd(II)-mediated N–H activation of indole, followed by NBE insertion and C2-palladation to give the key pallacycle intermediate (182) (Scheme 101).218 Oxidative addition of 182 with alkyl bromide, followed by reductive elimination and NBE extrusion, would afford intermediate 184, which then undergoes protodepalladation to afford the 2-alkylindole product and regenerate the Pd(II) catalyst. The structure of such a five-membered palladacycle was unambiguously prepared and characterized by single crystal X-ray crystallography (Scheme 102). In addition, such a structure could be trapped using β-iodostryene as the electrophile (186) (vide supra, Scheme 99).
Scheme 101.

Catalytic Cycle for 2-Alkylation of Indoles
Scheme 102.

Evidence for the Formation of the N-NBE Type Palladacycle
In 2013, Bach developed the 2-alkylation of electron-deficient pyrroles (Scheme 103).219 Given that pyrroles are generally more electron-rich and less acidic than indoles, electron-withdrawing substituents were necessary for this reaction. In this case, addition of water did not significantly affect the yield. They further achieved C2 alkylation of Boc-protected tryptophan (189).220 The presence of air seemed to be advantageous in this case as it prevented reduction of Pd(II) to Pd(0).
Scheme 103.
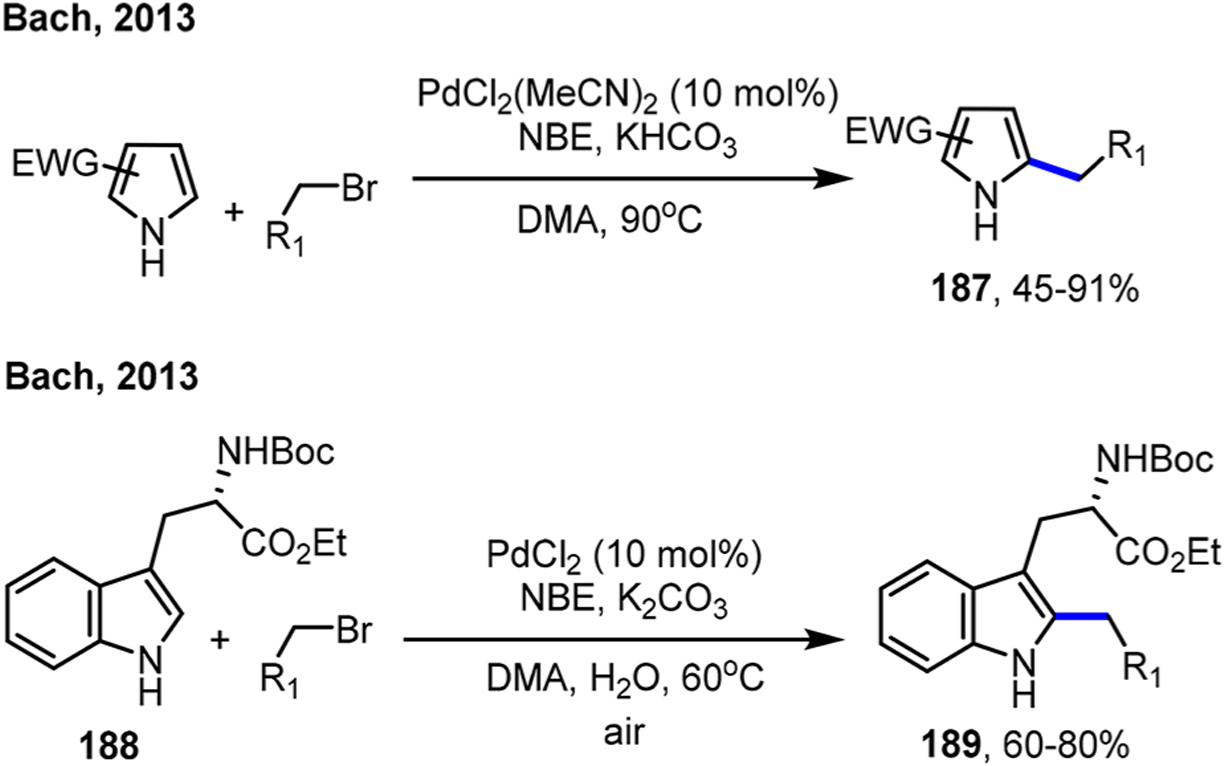
Reaction Extension to 2-Alkylation of Pyrroles and Indoles
Besides alkylation, one example using iodobenzene as the external electrophile was reported by Bach for C2-arylation of indoles (Scheme 104).217 In 2017, Jiang and Xue further extended the substrate scope for this reaction.221 Iodobenzene gave a higher yield than bromobenzene, and chlorobenzene was unreactive. Unlike the Pd(0)-initiated cross arylation reactions, electron-deficient aryl electrophiles were not required, although they would generally give higher yields than the more electron-rich ones.
Scheme 104.

2-Arylation of Indoles via the Pd/NBE Catalysis
In 2018, the Liu group reported a related C2 trifluoroethylation of indoles owing to the interest of 2-trifluoroethylindole moieties in medicinal chemistry (Scheme 105).222 Dibenzoyl-methane (dbm) was found to be the most effective ligand. The reaction tolerated different substitutions in the indole ring (191a,b), but having a substitution at the C7 position (191c) or an EWG at the C3 position significantly reduced the reactivity. The utility of this transformation was demonstrated in the late-stage trifluoroethylation of some bioactive molecules.
Scheme 105.

2-Trifluoroethylation of Indoles via Pd/NBE Catalysis
3.3.2. C–H Bond Activation-Initiated Meta Functionalization of Arenes Using a Directing Group.
3.3.2.1. Meta-Arylation and Alkylation of Arenes.
As described in the general catalytic cycle (Scheme 14), the aryl-Pd(II) intermediate (38) can not only be generated from oxidative addition with ArX but also come from a directed ortho palladation process, which is present in numerous Pd-catalyzed ortho-functionalizations of arenes. In 2015, the Yu223 and Dong224 groups independently reported the Pd/NBE-catalyzed meta-selective arene C–H activation reactions using ortho-DGs (Scheme 106). In this transformation, NBE relays the palladium from the initial ortho position to the meta position; upon meta functionalization with an electrophile and NBE extrusion via β-carbon elimination, the resulting aryl-Pd(II) species undergoes protodepalladation (the reverse process of ortho palladation) to generate the Pd(II) catalyst. Based on such a catalytic cycle, the nature of the ortho DG is expected to play a critical role because it needs to be strong enough to direct ortho palladation but not too strong to inhibit NBE insertion and protodepalladation.
Scheme 106.

General Reaction Pathway for the Meta-Functionalization of Arenes Using Ortho-DGs
In 2015, Yu and co-workers reported the meta-selective C–H alkylation and arylation using an amide DG (Scheme 107).223 The pyridine-type ligand 196 was found most efficient. Different alkyl halides without β-hydrogens could be smoothly coupled, such as methyl iodide (195a), ethyl iodoacetate and benzyl bromides. Ethyl iodide gave a low yield (195b). Meta-methylation of dihydrobenzofurans was also effective (195c). A meta arylation method was also successfully developed using electron-deficient aryl iodides with an ortho chelating group (195d) or multiple electron-withdrawing groups.
Scheme 107.

Meta-Alkylation/Arylation of Phenylacetic-Acid-Derived Amides
Concurrently, the Dong group developed the meta arylation of N,N-dialkyl benzyl amines (Scheme 108).224 Under mildly acidic conditions, the dimethylamine moiety can direct reversible ortho metalation; such a DG could be easily installed and transformed into other FGs, such as benzyl chloride or aldehyde. AsPh3 was used as the ligand along with a “cocktail” of acetate salts. Heteroarene substrates, such as pyrrole 197b and pyridine 197c, were also amenable to this transformation. Ortho chelating groups were needed in the aryl iodide part to facilitate the oxidative addition with ANP.
Scheme 108.
Meta-Arylation of Simple Tertiary Benzyl Amines
Alkyl halides with β-hydrogens and aryl iodides without ortho coordinating groups were problematic electrophiles because reductive elimination from the ANP intermediate to form the norbornyl benzocyclobutene would become the major competitive pathway. To address the scope limitation, the Yu group found 2-carbomethoxy-substituted NBE 115 could effectively suppress formation of the benzocyclobutene side-product (Scheme 109).159 Together with an elaborated quinoline ligand 199, alkyl halides with β-hydrogens (198a and 198b) and regular aryl iodides without ortho coordinating groups (198c and 198d) were successfully coupled. Yet, the extension to heterocycles was unsuccessful under these conditions. When the reaction was performed in the presence of 10 equiv of deuterated acetic acids, the observed 70% D-incorporation at the ortho position is consistent with the protodepalladation pathway. Interestingly, both ortho and meta positions were deuterated in the recovered starting material, indicating the reversibility of the meta-C–H activation step.
Scheme 109.

Expanding Scope of Meta-Alkylation/Arylation with a Modified NBE
In 2016, Zhao and Shi reported an interesting meta arylation of oxalyl amide-protected β-arylethylamines (Scheme 110).225 It was demonstrated that aryl iodides without ortho chelating groups could be coupled with regular NBE. Remarkably, electron-rich aryl iodides could also be coupled (202b), albeit in a moderate yield. Meta-arylation of thiophenes (202c) was also successful. Although the exact reason is unclear, the unique DG employed here and/or the absence of ancillary ligands might play a key role in promoting the desired arylation instead of forming the benzocyclobutene side-product.
Scheme 110.
Meta-Arylation of Oxalyl Amide-Protected β-Arylethylamines
To expand the types of DGs that can be employed, the Yu group developed a versatile 3-acetylamino-2-hydroxypyridine class of ligands (204) to achieve meta arylation of a wide range of substrates, including aniline, phenol, and 2-benzyl heterocycle derivatives (Scheme 111).226 These DGs allowed for forming 6- or 7-membered palladacycles. The utility was demonstrated in the meta arylation of a lenalidomide-derived substrate. In addition, silver-free conditions were also established using CsOAc in place of AgOAc in t-Amyl–OH.
Scheme 111.

Meta-Arylation of Anilines, Phenols, and Heterocycles with Pyridine-Type DGs
The Yu group further found that electron-deficient 2-pyridone 206 can effectively promote meta C–H arylation with pyridine-based DGs (Scheme 112),227,228 which likely served as an efficient X-ligand to promote the CMD process.229 The ring size of the initially formed palladacycle was found to be crucial for the success of this reaction: while 6- or 7-membered palladacycles could engage in the following steps under these conditions, 5-membered palladacycles were found to be too stable to react. This represents a difference from Dong’s benzylamine system (vide supra, Scheme 108).
Scheme 112.

Meta-Arylation of Benzylamines and Protected Aldehydes with Pyridine-Type DGs
In 2017, Ferreira and co-workers used quinoline-derived DGs to realize meta C–H arylation of masked benzyl alcohols with 2-carbomethoxy-substituted NBE 115 (Scheme 113).230 Different from Yu’s system, TFA-Gly-OH performed better than the 2-pyridione-type ligands. Notably, the DG can be easily cleaved under acidic conditions to generate free benzyl alcohols.
Scheme 113.

Meta-Arylation of Benzyl Alcohol with Quinoline-Type DG
In 2017, Yu and co-workers found that nosyl-protected amines can serve as excellent DGs (Scheme 114).231 4-Acetylpyridine was identified to be the optimal ligand, and simple NBE was used in a catalytic amount. Nosyl-protected phenethylamines (210a), 2-aryl anilines (210b), and benzylamines (210c) were all suitable substrates. Similar to the Zhao and Shi’s case (vide supra, Scheme 110), aryl iodides without ortho chelating groups could be coupled but less effectively. In addition, aryl bromides with ortho chelating groups could be used. Meta C–H alkylation of nosyl-protected benzylamines was later reported by Ding and co-workers.232 Almost simultaneously, Shi reported meta-arylation using trifluoroacetyl-protected 2-aryl anilines as the DG with 4-methoxypyridine as the ligand and 2-carbomethoxy-substituted NBE 115 as the cocatalyst (211).233 Later in 2017, Yu described the use of benzylsulfonamides as the DG in the Pd/NBE-catalyzed meta C–H arylation and alkylation with isoquinoline as the ligand.234 A broad range of alkyl and aryl iodides as electrophiles has been demonstrated.
Scheme 114.

Meta-Arylations with the Amide-Type DG
In the same year, the use of carboxylic acids as the DG in the meta C–H arylation was reported by the Yu group (Scheme 115).235 Substituted 2-pyridone 214 proved to be the best ligand, which was found to accelerate the ortho C–H activation step. Note that noncoordinating ortho substituents in the aryl iodides were compatible in this transformation (213b).
Scheme 115.

Meta-Arylations of Phenylacetic Acids
In 2018, Yu reported the first example of enantioselective meta C–H arylation and alkylation using a chiral NBE cocatalyst (Scheme 116).236 With diarylmethylamines as the model substrate, chiral phosphoric acid (R)-BNDHP (1,1′-binaphth-yl-2,2′-diyl hydrogen phosphate) was used as an additive. Control experiments suggested that chiral NBE (+)-115 was responsible for the chiral induction while the chiral acid has a minor beneficial effect. A broad aryl iodide scope (215a and 215b) and good FG tolerance (215c and 215d) were observed. Asymmetric meta C–H arylation and alkylation of nosyl-protected homobenzylamines were also achieved (216), through either desymmetrization or kinetic resolution. Parallel KIEs were measured to be 1.03 and 1.33 for ortho and meta C–H bonds, respectively, indicating that neither C–H bond cleavage was the rate-determining step. The reaction rate was first order on [Ar–I], indicating that the reaction of ANP with aryl iodides was likely the rate-determining step.
3.3.2.2. Meta-Amination, Alkynylation, and Chlorination of Arenes.
Besides arylation and alkylation, other types of meta functionalizations have also been demonstrated. In 2016, Yu and co-workers achieved meta amination of aniline and phenol derivatives by using O-benzoylhydroxylamines as the electrophile (Scheme 117).237 The synthetic utility was demonstrated in the synthesis of 3-fluoro-5-morpholinoaniline, which was the synthetic intermediate of a BRAF inhibitor. In 2017, a single example of meta amination of a benzyl amine derivative was reported using similar conditions.227
Scheme 117.

Meta-Amination of Anilines and Phenols with Pyridine-Type DGs
Since the reaction is not initiated by a Pd(0)-mediated process, there is less limitation on using stronger electrophiles. Though alkynyl bromides and aryl chlorosulfates have not been used as electrophiles in the Pd(0)-initiated Pd/NBE catalysis, the corresponding meta alkynylation and chlorination have been successfully demonstrated by Yu and co-workers (Scheme 118).238,239 The meta alkynylation required bulky silyl-protected alkynyl bromides, while alkyl or aryl alkynyl bromides were not effective. For the meta chlorination, the use of less electrophilic aryl chlorosulfates was important, while N-chlorosuccinimide (NCS) only gave ortho chlorination products. In 2017, a single example of meta chlorination of a benzyl amine derivative was reported under similar conditions.227
Scheme 118.

Meta-Alkynylation and Chlorination with Pyridine-Type DGs
3.3.3. Transmetalation-Initiated Ortho Functionalization of Arylboron Species.
Besides oxidative addition of aryl halides to Pd(0) and directed C–H activation of arenes, another way to generate the aryl-Pd(II) species (38) in the general catalytic cycle (vide supra, Scheme 14) is through a transmetalation process from aryl nucleophiles to Pd(II) (Scheme 119). In 2018, Zhang240 and Zhou241 independently reported the Pd/NBE-catalyzed ortho alkylation/ipso Heck reaction using arylboron species as substrates (Scheme 120). In Zhang’s reactions, arylboronic acids were used and the Pd(0) generated from the Heck termination was oxidized back to Pd(II) using Cu(OAc)2 as the stoichiometric oxidant. Zhou and co-workers employed a phosphine ligand-free open-flask condition, in which air was used as the terminal oxidant to regenerate Pd(II) from Pd(0). Thus, both systems need stoichiometric bases during the termination step. In Zhou’s reaction, 5-NBE-2-carbonitrile 224 was identified to be more effective than simple NBE; both arylboronic acids and pinacol boronates were suitable substrates. Similar to the aforementioned C–H activation-initiated processes, aryl iodides were also tolerated in the transmetalation-based reactions. In both systems, arylboronic acids without ortho substituents gave bis-alkylation products.
Scheme 119.

Transmetalation-Initiated Pd/NBE Catalysis
Scheme 120.
Oxidative Ortho Alkylation/Ipso Heck Reaction of Arylboronic Acids
In the transmetalation-initiated process, one could imagine that, instead of terminating the aryl-Pd(II) species with an nucleophile (or an olefin) and then reforming the Pd(II) catalyst through oxidizing the resulting Pd(0) intermediate, an alternative pathway involves direct protonation of the aryl-Pd(II) species for catalyst regeneration (Scheme 121). This would prevent using stoichiometric oxidants, and further avoid stoichiometric bases, which leads to a redox-neutral ortho functionalization of aryl nucleophiles.
Scheme 121.
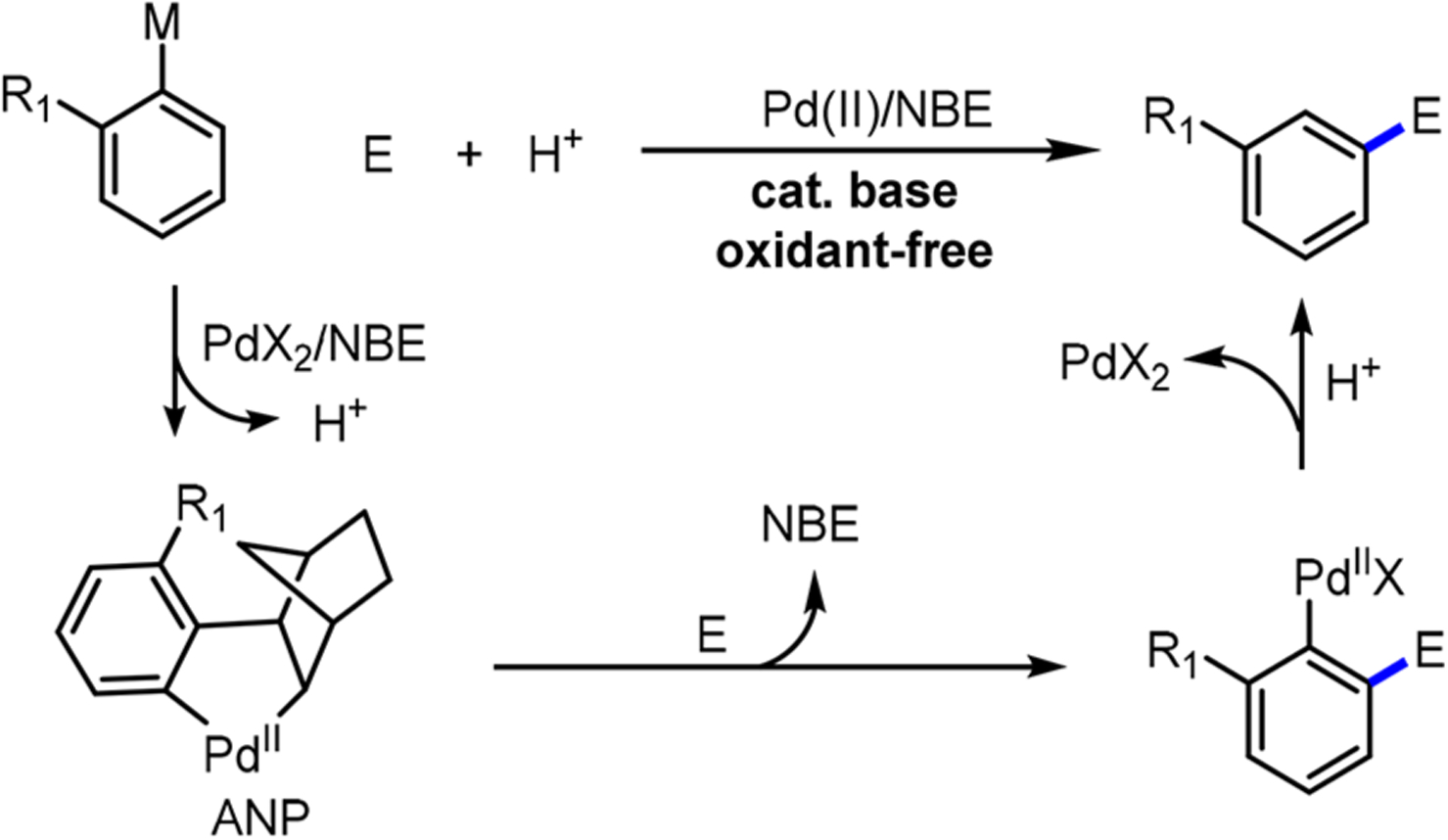
Redox-Neutral Ortho Functionalizations of Aryl Nucleophiles
Very recently, the Dong group developed a Pd/NBE-catalyzed ortho functionalization of aryl boroxines (Scheme 122).242 Different from Zhang and Zhou’s conditions, this reaction is terminated via protodepalladation, therefore not requiring stoichiometric external oxidants or bases. Both ortho acylation and amination have been demonstrated using carboxylic acid anhydrides and O-benzoylhydroxylamines as electrophiles, respectively. For the ortho acylation reaction, arsine-type ligands, i.e., AsPh3, were found to be most effective, likely through promoting both the transmetalation and protodepalladation steps. Notably, both arylboronic esters (225b) and aryl iodides (225c) were tolerated. Further deuterium labeling studies were consistent with the ipso protodepalladation pathway. For the ortho amination with aryl boroxines, phosphite-type ligands, such as P(OPh)3, were most efficient. The major side reaction in these redox-neutral transformations was the reductive elimination of the ANP intermediate to form benzocyclobutenes.
Scheme 122.

Redox-Neutral Ortho Functionalizations of Aryl Boroxines
4. SYNTHETIC APPLICATIONS
4.1. Applications in Synthesis of Bioactive Compounds
The Pd/NBE cooperative catalysis has been widely utilized in the synthesis of biologically important compounds, including natural products, drugs, and agrochemicals. In particular, the ortho/ipso bisfunctionalziations of aryl halides are highly powerful for preparing polysubstituted arenes in a site-selective and step-economical manner. In this section, these examples are summarized according to the type of the key transformations used in the syntheses.
4.1.1. Synthesis using Ortho Alkylation Reactions.
In 2013, Lautens and co-workers reported the enantioselective total synthesis of (+)-linoxepin (Scheme 123).243,244 Using the ortho alkylation/ipso Heck reaction, the tetrasubstituted arene core was efficiently constructed in a convergent way. Oxidative cleavage of the olefin to an aldehyde followed by intramolecular aldol condensation afforded 228, which was subjected to the Mizoroki–Heck reaction to furnish the synthesis of (+)-linoxepin (229).
Scheme 123.

Total Synthesis of (+)-Linoxepin
Fused ring structures can be efficiently generated using Type A bifunctional alkylation reagents. In 2017, the Dong group applied the annulation reaction with epoxides in the synthesis of insecticide fufenozide (Scheme 124).131 Dihydrobenzofuran 230 was efficiently constructed through the direct coupling between the aryl iodide and propylene oxide. Subsequent hydrolysis and amide condensation provided fufenozide (231) in three total steps. Later, Zhou and co-workers applied their ortho alkylation/ipso Heck annulation in a concise synthesis of eptazocine, which is a benzomorphan-type analgesic drug.96 The reaction of aryl iodide 232 with bromo-allyl alcohol 233 afforded aldehyde 234 in 53% yield, which was transformed into eptazocine (235) in three steps.
Scheme 124.

Synthesis of (±)-Fufenozide and (±)-Eptazocine
4.1.2. Synthesis Using Ortho Arylation Reactions.
Likewise, ortho arylation with Type A bifunctional reagents provides a quick access to poly fused aromatic rings. In 2008, Catellani reported direct synthesis of antibiotics carbazomycin A (236) through ortho arylation/ipso amination using o-bromo-N-acetylaniline as the reactant (Scheme 125).163 Later, Lautens achieved formal synthesis of benzo[c]phenanthridine alkaloids, nitidine (237) and NK109, taking advantage of the ortho arylation/ipso amination with aryl triflates.205 In 2014, assoanine and pratosine were synthesized from a common intermediate 238 by Takemoto, utilizing a similar ortho arylation/ipso amination reaction.167
Scheme 125.

Synthesis of Carbazomycin A, Nitidine, Assoanine, and Pratosine
On the other hand, ortho arylation with Type B bifunctional reagents leads to a different type of fused ring systems. In 2013, Gu and co-workers employed the ortho arylation/ipso Heck reaction in the total synthesis of rhazinal (242). 1-Bromo-2-nitrobenzene was found to be an effective aryl electrophile, and the EWG in the pyrrole ring was essential in this Pd/NBE-catalyzed reaction. The resulting key intermediate 241 was transformed into rhazinal in three steps (Scheme 126).176 Later, the same group achieved the enantioselective version of the synthesis using chiral ligand 243,245 which featured the first catalytic asymmetric example in the Pd/NBE catalysis. Total syntheses of related alkaloids, (+)-rhazinilam and (+)-kopsiyunnanine C1–3, have also been achieved.
Scheme 126.

Total Synthesis of (+)-Rhazinal
4.1.3. Synthesis Using Ortho Amination Reactions.
In 2015, Ritter demonstrated that the ortho amination/ipso borylation reaction could be used to quickly assemble the antipsychotic drug, Abilify (245), and the anti-inflammatory drug, Flunixin (247) (Scheme 127).187 The key arylboronic ester intermediates generated via the Pd/NBE catalysis were conveniently converted to other FGs, such as chloride in the synthesis of Abilify and a methyl group in the synthesis of Flunixin.
Scheme 127.

Syntheses of Abilify and Flunixin
4.1.4. Synthesis Using Ortho Acylation Reactions.
In 2015, the Dong group applied the ortho acylation/ipso hydrogenation in a concise synthesis of ketoprofen (249) (Scheme 128),193 which is a nonsteroidal anti-inflammatory drug for relieving arthritis-related inflammatory pains or severe toothaches. This method shows an advantage of preparing meta-substituted aryl ketones.
Scheme 128.

Formal Synthesis of Ketoprofen
In addition, utilizing an intramolecular Heck termination has also been employed recently by the same group in the syntheses of indanone-type natural products (Scheme 129).202 For example, pauciflorol F (261) were synthesized in five steps. They also reported the first total synthesis of acredinone A (266), which represents the first nonpeptidic natural product that can inhibit the voltage-gated potassium channel. The synthesis features two Pd/NBE-catalyzed ortho acylation reactions for constructing the two penta-substituted arene fragments. To be specific, the ortho acylation/ipso Heck annulation of aryl iodide 252 led to indenone 254, and the ortho acylation/ipso borylation of aryl iodide 253 afforded intermediate 255. The fragments 254 and 255 were ultimately coupled through the Suzuki–Miyaura reaction, followed by deprotection to give acredinone A (256) in eight steps in the longest linear sequence.
Scheme 129.

Syntheses of Pauciflorol F and Acredinone A
4.1.5. Synthesis Using 2-Alkylation of Indoles.
Due to the biological importance of the indole moiety, Bach’s 2-alkylation of indoles has been applied in a number of total syntheses of indole alkaloids. In 2012, Jiao and Bach reported the total syntheses of (±)-aspidospermidine and (±)-goniomitine (Scheme 130).218 In both cases, 2-alkylation of indoles was carried out at an early stage of the synthesis, demonstrating the practicability of this method. The intermediate 257 was further converted to 258 in five steps. Upon treatment with acids, annulation of 258 resulted in tetracycle 259, which was further transformed to (±)-aspidospermidine (260). Similarly, total synthesis of (±)-goniomitine started from 2-alkylation of TBS-protected tryptophol. An improved catalytic condition was identified for the synthesis of indole 261 in order to accommodate the C3-substitution. Ultimately, (±)-goniomitine (262) was synthesized in five steps from intermediate 261.
Scheme 130.

Total Syntheses of (±)-Aspidospermidine and (±)-Goniomitine
Subsequently, this 2-alkylation method has been frequently adopted in the total synthesis of other indole alkaloids. For example, Mukai,246 Yang,247 and Qin248 successfully employed such an approach in the total syntheses of (+)-kopsihainanine A (263), (−)-aspidophylline A (264), and (+)-strictamine (265), respectively (Scheme 131). In addition, this method has also been used in the synthesis of certain pharmaceutical intermediates.249–251
Scheme 131.

Total Syntheses of (+)-Kopsihainanine A, (−)-Aspidophylline A, and (+)-Strictamine
4.2. Applications in Polymer Chemistry
Besides in bioactive compounds, polysubstituted arenes are also commonly found in organic polymers. Thus, the Pd/NBE catalysis is expected to be useful in preparing aromatic polymers due to its capability to construct multiple bonds simultaneously in one single transformation.
Reductive elimination of ANP to form norbornyl benzocyclobutenes is a common side reaction in the Pd/NBE catalysis.28,252,253 However, the Xia group nicely took advantage of this unique arene/NBE annulation reaction and applied it in the synthesis of a new class of ladder polymers (Scheme 132).254 Both aryl bromides and triflates could be used as monomers to couple with norbornadienes. They found C1 and C4 substitutions in aryl bromides to be important for the efficiency of the annulation reaction. Besides the beneficial effect of the ortho substituent, the additional meta substituent suppressed side reactions, such as ortho arylation and NBE multi-insertion. The model study of 2,5-dimethyl-bromobenzene with norbornadiene gave >98% yield with excellent exo selectivity, which showed that this method was suitable for polymer synthesis. Polymers with a backbone in a bowing-ribbon conformation (267) can be conveniently synthesized from disubstituted p-dibromobenzenes and norbornadiene. Use of the biphenyl monomer afforded the polymer with a certain degree of bending freedom (268) due to the presence of the restricted yet rotatable biphenyl bond. Besides AA- and BB-type monomers, AB-type monomers could also be used (269). Ladder polymers with high molecular weights (10–50 kDa) can be obtained. These polymers exhibited excellent thermal stability, high carbonization yields, and large intrinsic porosity. Later, the same group demonstrated that spirocyclic motif could be introduced into the polymer backbone255 and different FGs could be introduced into the side chains.256 Besides using norbornadienes, benzooxanorbornadienes can also be used to construct polycyclic conjugated hydrocarbons using the same annulation reaction followed by aromatization.257–260
Scheme 132.

Synthesis of Ladder Polymers Using Catalytic Arene-NBE Annulation
In the aforementioned polymerizations, norbornadiene was used as the monomer instead of a cocatalyst. In 2018, Yoon and Dong reported the first Pd/NBE-catalyzed polymerization, which simplified the synthesis of certain classes of functional aromatic polymers (Scheme 133).261 Based on the ortho amination/ipso Sonogashira coupling, an A2B2C-type multicomponent polymerization was developed to prepare various amine-functionalized arylacetylene-containing polymers (270). Compared to the conventional “pre-functionalization” or “post-functionalization” approaches, this “in situ functionalization” strategy allows for simultaneously constructing polymer backbones and installing side chains in one catalytic cycle. Thus, this method provides a rapid and modular preparation of various amine-substituted polymers from readily available monomers. For the AA diaryl iodide monomers, both flexible and rigid linkers could be used. For the diacetylene BB monomers, both electron-rich and -poor linkers were compatible in the polymerization process. Simply through changing the amine C monomer, a range of pendant-functionalized aromatic polymers were prepared. For example, the polymer containing the ferrocene moiety showed redox response from the cyclic voltammogram (270a), and the polymer containing a tetraphenylethylene motif displayed solid-state photoluminescence (270b).
Scheme 133.
Multicomponent “In Situ Functionalization” Polymerization by Palladium/NBE Catalysis
Very recently, the same group disclosed a three-step preparation of highly water-soluble poly(para-phenylene ethynylene)s (PPEs) via the Pd/NBE-catalyzed AB-C-type polymerization (Scheme 134).262 Instead of using the AA- and BB-type monomers, an AB-type alkyne-substituted aryl iodide monomer was employed, in which two piperazine meta side chains were concurrently installed with the construction of the PPE backbone. Upon removal of the N-Boc-protecting group, the polymer was found to be highly water-soluble since the piperazine side chains can be doubly protonated (271) under aqueous acidic conditions. Interestingly, these PPEs with meta side chains were found less aggregated and more soluble with higher fluorescent quantum yields compared to the corresponding PPEs with para side chains.
Scheme 134.

Synthesis of the Water-Soluble PPE with Meta Side Chains
5. CONCLUSION AND OUTLOOK
Since Catellani’s seminal discovery in 1997 and opening of the synthetic potential by Catellani and Lautens, the field of the Pd/NBE cooperative catalysis has expanded enormously to a number of dimensions, including new method development, natural product synthesis, and polymer chemistry. Particularly over the past decade, several new directions have been found, which have significantly increased the synthetic utilities of these methods. For example, the discovery of new classes of electrophiles allows for a variety of new ortho functionalizations, which extends the scope of the aromatic products that can be accessed. The design of new reaction pathways enables various Pd(II)-initiated reactions, including 2-alkylation of indoles, meta functionalization of arenes with ortho DGs, and ortho functionalization of arylboron species. The development of new types of substituted NBEs can overcome certain limitations in these reactions, particularly regarding the substrate scope and selectivity. In addition, an increasing number of applications have appeared in streamlined syntheses of complex target molecules and functional polymers due to the efficiency of ipso/ortho difunctionalization introduced by this unique approach. Thus, this field is now in a rapidly growing stage.
Despite the myriad recent advances, several limitations remain to be addressed in the future, in order to further improve the practicality and versatility of these methods. For example, compared to the widely adopted cross-coupling reactions, the scope of the electrophiles in the Pd/NBE catalysis is still much narrower and mainly based on carbon electrophiles. Hence, enabling broad C–X (X ≠ C) bond formation at the ortho position in the Pd(0)-initiated reactions would be highly desirable for extending the reaction scope. The solution to this problem could come from careful design of the external electrophiles and/or new NBE cocatalysts. In addition, better understanding the reaction of ANP with electrophiles should offer useful insights for reaction design, which could be achieved through a combination of experimental and computational studies. Moreover, it could be exciting to discover new classes of substrates beyond arenes or heteroarenes in the Pd/NBE catalysis, which may lead to new transformations that are difficult to achieve via conventional approaches. Regarding the Pd(II)-initiated meta C–H functionalization of arenes, the direction has clearly been moving to the use of common FGs (e.g., aldehydes, ketones, alcohols) as DGs. One promising solution could be the use of catalytic directing templates, along with more enabling ligands and/or NBE cocatalysts. Besides using ortho-DGs, one could imagine that the use of a meta-DG, pioneered by Yu263–265 and others, could afford para-substituted products. Finally, another underexplored direction could be the use of undirected arene C–H activation to initiate the Pd/NBE catalysis, in which the judicious choice of ligands/NBE cocatalysts would be important for the desired selectivity and reactivity.
ACKNOWLEDGMENTS
We thank University of Chicago and NIGMS (1R01GM124414-01A1) for research support. Dr. Zhao Wu, Mr. Renhe Li, Mr. Alexander Rago, and Mr. Ki-Young Yoon are acknowledged for checking the manuscript.
Biographies
Jianchun Wang was born in Taizhou, China. He received his B.S. degree in chemistry from Peking University in 2014, where he carried out undergraduate research of olefin polymerization in the laboratory of Professor Yuguo Ma. In the same year, he joined Professor Guangbin Dong’s research lab at the University of Texas at Austin, and he moved to University of Chicago with Professor Dong in 2016. He is currently pursuing his Ph.D. with a focus on Pd/NBE cooperative catalysis.
Guangbin Dong received his B.S. degree from Peking University and completed his Ph.D. degree in chemistry from Stanford University with Professor Barry M. Trost, where he was a Larry Yung Stanford Graduate fellow. In 2009, he began to research with Professor Robert H. Grubbs at California Institute of Technology, as a Camille and Henry Dreyfus Environmental Chemistry Fellow. In 2011, he joined the department of chemistry and biochemistry at the University of Texas at Austin as an assistant professor and a CPRIT Scholar. Since 2016, he has been a Professor of Chemistry at the University of Chicago. His research interests lie in the development of powerful chemical tools for addressing questions of biological importance.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Taylor RD; MacCoss M; Lawson ADG ″Rings in Drugs″. J. Med. Chem 2014, 57, 5845–5859. [DOI] [PubMed] [Google Scholar]
- (2).Tsuji J Palladium Reagents and Catalysts: New Perspectives for the 21st Century; John Wiley & Sons, Ltd, 2005. [Google Scholar]
- (3).Terrier F Modern Nucleophilic Aromatic Substitution; Wiley-VCH Verlag GmbH & Co. KGaA, 2013. [Google Scholar]
- (4).Negishi E.-i.; De Meijere A Handbook of Organopalladium Chemistry for Organic Synthesis; John Wiley & Sons, Inc., 2003. [Google Scholar]
- (5).Buncel E; Dust JM; Terrier F ″Rationalizing the Regioselectivity in Polynitroarene Anionic σ-Adduct Formation. Relevance to Nucleophilic Aromatic Substitution″. Chem. Rev 1995, 95, 2261–2280. [Google Scholar]
- (6).Smith MB; March J March’s Advanced Organic Chemistry; John Wiley & Sons, Inc., 2006. [Google Scholar]
- (7).Snieckus V ″Directed Ortho Metalation. Tertiary Amide and O-Carbamate Directors in Synthetic Strategies for Polysubstituted Aromatics″. Chem. Rev 1990, 90, 879–933. [Google Scholar]
- (8).Lyons TW; Sanford MS ″Palladium-Catalyzed Ligand-Directed C–H Functionalization Reactions″. Chem. Rev 2010, 110, 1147–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Engle KM; Mei T-S; Wasa M; Yu J-Q ″Weak Coordination as a Powerful Means for Developing Broadly Useful C–H Functionalization Reactions″. Acc. Chem. Res 2012, 45, 788–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Kuhl N; Hopkinson MN; Wencel-Delord J; Glorius F Beyond Directing Groups: Transition-Metal-Catalyzed C–H Activation of Simple Arenes. Angew. Chem., Int. Ed 2012, 51, 10236–10254. [DOI] [PubMed] [Google Scholar]
- (11).Yang J Transition Metal Catalyzed Meta-C–H Functionalization of Aromatic Compounds. Org. Biomol. Chem 2015, 13, 1930–1941. [DOI] [PubMed] [Google Scholar]
- (12).Li J; De Sarkar S; Ackermann L Meta- and Para-Selective C–H Functionalization by C–H Activation. Top. Organomet. Chem 2015, 55, 217–257. [Google Scholar]
- (13).Dey A; Agasti S; Maiti D Palladium catalysed Meta-C–H functionalization reactions. Org. Biomol. Chem 2016, 14, 5440–5453. [DOI] [PubMed] [Google Scholar]
- (14).Catellani M Catalytic Multistep Reactions via Palladacycles. Synlett 2003, 298–313. [Google Scholar]
- (15).Catellani M Novel Methods of Aromatic Functionalization Using Palladium and Norbornene as a Unique Catalytic System. Top. Organomet. Chem 2005, 14, 21–53. [Google Scholar]
- (16).Catellani M; Motti E; Della Ca’ N Catalytic Sequential Reactions Involving Palladacycle-Directed Aryl Coupling Steps. Acc. Chem. Res 2008, 41, 1512–1522. [DOI] [PubMed] [Google Scholar]
- (17).Martins A; Mariampillai B; Lautens M Synthesis in the Key of Catellani: Norbornene-Mediated Ortho C–H Functionalization. Top. Curr. Chem 2009, 292, 1–33. [DOI] [PubMed] [Google Scholar]
- (18).Ye J; Lautens M Palladium-Catalysed Norbornene-Mediated C–H Functionalization of Arenes. Nat. Chem 2015, 7, 863–870. [DOI] [PubMed] [Google Scholar]
- (19).Della Ca’ N; Fontana M; Motti E; Catellani M Pd/Norbornene: A Winning Combination for Selective Aromatic Functionalization via C–H Bond Activation. Acc. Chem. Res 2016, 49, 1389–1400. [DOI] [PubMed] [Google Scholar]
- (20).Wegmann M; Henkel M; Bach T C–H Alkylation Reactions of Indoles Mediated by Pd(II) and Norbornene: Applications and Recent Developments. Org. Biomol. Chem 2018, 16, 5376–5385. [DOI] [PubMed] [Google Scholar]
- (21).Zhao K; Ding L; Gu Z Development of New Electrophiles in Palladium/Norbornene-Catalyzed Ortho-Functionalization of Aryl Halides. Synlett 2019, 30, 129–140. [Google Scholar]
- (22).Cheng H-G; Chen S; Chen R; Zhou Q The Pd(II)-Initiated Catellani-Type Reactions. Angew. Chem., Int. Ed 2019, 58, 5832–5844. [DOI] [PubMed] [Google Scholar]
- (23).Mizoroki T; Mori K; Ozaki A Arylation of Olefin with Aryl Iodide Catalyzed by Palladium. Bull. Chem. Soc. Jpn 1971, 44, 580–581. [Google Scholar]
- (24).Heck R; Nolley J Jr Palladium-Catalyzed Vinylic Hydrogen Substitution Reactions with Aryl, Benzyl, and Styryl Halides. J. Org. Chem 1972, 37, 2320–2322. [Google Scholar]
- (25).Horino H; Arai M; Inoue N Reaction of Norbornenes with Phenylpalladium Chloride. Tetrahedron Lett. 1974, 15, 647–650. [Google Scholar]
- (26).Catellani M; Chiusoli GP Catalytic Activation of Aromatic C–H Bonds. J. Organomet. Chem 1982, 239, C35–C37. [Google Scholar]
- (27).Catellani M; Chiusoli GP Palladium-Catalyzed Synthesis of 1,2,3,4,4a,12b-Hexahydro-1,4-Methanotriphenylenes. J. Organomet. Chem 1985, 286, C13–C16. [Google Scholar]
- (28).Catellani M; Chiusoli GP; Ricotti S A New Palladium-Catalyzed Synthesis of 1,2,3,4,4a,8b-Hexahydro-1,4-methanobiphenylenes and 2-Phenylbicyclo[2.2.1]hept-2-enes. J. Organomet. Chem 1985, 296, C11–C15. [Google Scholar]
- (29).Catellani M; Chiusoli GP Palladium-(II) and Palladium-(IV) Complexes as Intermediates in Catalytic C–C Bond-Forming Reactions. J. Organomet. Chem 1988, 346, C27–C30. [Google Scholar]
- (30).Catellani M; Chiusoli GP Palladacycle Formation by Electrophilic Aromatic Substitution, as Monitored by 1H NMR Spectroscopy. J. Organomet. Chem 1992, 425, 151–154. [Google Scholar]
- (31).Gorelsky SI; Lapointe D; Fagnou K Analysis of the Concerted Metalation-Deprotonation Mechanism in Palladium-Catalyzed Direct Arylation Across a Broad Range of Aromatic Substrates. J. Am. Chem. Soc 2008, 130, 10848–10849. [DOI] [PubMed] [Google Scholar]
- (32).Liegault B; Lapointe D; Caron L; Vlassova A; Fagnou K Establishment of Broadly Applicable Reaction Conditions for the Palladium-Catalyzed Direct Arylation of Heteroatom-Containing Aromatic Compounds. J. Org. Chem 2009, 74, 1826–1834. [DOI] [PubMed] [Google Scholar]
- (33).Li X; Pan J; Song S; Jiao N Pd-Catalyzed Dehydrogenative Annulation Approach for the Efficient Synthesis of Phenanthridinones. Chem. Sci 2016, 7, 5384–5389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Chai DI; Thansandote P; Lautens M Mechanistic Studies of Pd-Catalyzed Regioselective Aryl C–H Bond Functionalization with Strained Alkenes: Origin of Regioselectivity. Chem. - Eur. J 2011, 17, 8175–8188. [DOI] [PubMed] [Google Scholar]
- (35).Li C-S; Cheng C-H; Liao F-L; Wang S-L Insertion of Norbornadiene into the Aryl Palladium Bond; Synthesis, Structure and Dynamics of Intramolecular η2-Arene Palladium Species. J. Chem. Soc., Chem. Commun 1991, 710–712. [Google Scholar]
- (36).Catellani M; Mealli C; Motti E; Paoli P; Perez-Carreno E; Pregosin PS Palladium-Arene Interactions in Catalytic Intermediates: An Experimental and Theoretical Investigation of the Soft Rearrangement between η1 and η2 Coordination Modes. J. Am. Chem. Soc 2002, 124, 4336–4346. [DOI] [PubMed] [Google Scholar]
- (37).Liu C-H; Li C-S; Cheng C-H Ortho-Metalation of Intramolecular (η2-Arene)palladium Species and Reactivities of the Resulting Palladacycles. Organometallics 1994, 13, 18–20. [Google Scholar]
- (38).Catellani M; Mann BE Conformational Effects in Elementary Steps in Catalytic Reactions. Oxidative Addition of 3-Bromoprop-1-ene to a Palladium(II) Metallacyclic Complex. J. Organomet. Chem 1990, 390, 251–255. [Google Scholar]
- (39).Bocelli G; Catellani M; Ghelli S Regioselective Ring-Opening of a Palladium(IV) Alkylaromatic Metallacycle by Benzyl Group Migration from Palladium to the Aromatic Carbon and X-Ray Structure of the Resulting Palladium(II) Complex. J. Organomet. Chem 1993, 458, C12–C15. [Google Scholar]
- (40).Rudolph A; Rackelmann N; Lautens M Stereochemical and Mechanistic Investigations of a Palladium-Catalyzed Annulation of Secondary Alkyl Iodides. Angew. Chem., Int. Ed 2007, 46, 1485–1488. [DOI] [PubMed] [Google Scholar]
- (41).Reiser O; Weber M; de Meijere A Selective 1:3 Coupling of Norbornene and Iodobenzene: a Facile Synthesis of Benzo[E]Pyrenes with Annelated Cycloaliphatic Moieties. Angew. Chem., Int. Ed. Engl 1989, 28, 1037–1038. [Google Scholar]
- (42).Catellani M; Chiusoli GP; Castagnoli C Changing Selectivity in Palladium-Catalyzed Reactions Involving Oxidation-State IV. A New Synthesis from Iodobenzene and Norbornene. J. Organomet. Chem 1991, 407, C30–C33. [Google Scholar]
- (43).Albrecht K; Reiser O; Weber M; Knieriem B; de Meijere A Palladium-Catalyzed Domino Coupling Reactions of Aryl Halides with Norbornene and Norbornene Derivatives - A Simple Route to Polycyclic Aromatic Compounds. Tetrahedron 1994, 50, 383–401. [Google Scholar]
- (44).Catellani M; Motti E Selective Aryl Coupling via Palladacycles: A New Route to m-Alkylbiphenyls or m-Terphenyls. New J. Chem 1998, 22, 759–761. [Google Scholar]
- (45).Gallazzi MC; Porri L; Vitulli G Reversible Insertion of Norbornene into a Nickel–Methallyl Bond. J. Organomet. Chem 1975, 97, 131–138. [Google Scholar]
- (46).Catellani M; Fagnola MC Palladacycles as Intermediates for Selective Dialkylation of Arenes and Subsequent Fragmentation. Angew. Chem., Int. Ed. Engl 1994, 33, 2421–2422. [Google Scholar]
- (47).Ariafard A; Lin Z Understanding the Relative Easiness of Oxidative Addition of Aryl and Alkyl Halides to Palladium(0). Organometallics 2006, 25, 4030–4033. [Google Scholar]
- (48).Khoury PR; Goddard JD; Tam W Ring Strain Energies: Substituted Rings, Norbornanes, Norbornenes and Norbornadienes. Tetrahedron 2004, 60, 8103–8112. [Google Scholar]
- (49).Turner RB; Meador WR; Winkler RE Heats of Hydrogenation. I. ApParatus and the Heats of Hydrogenation of Bicyclo-[2,2,1]heptene, Bicyclo[2,2,1]heptadiene, Bicyclo[2,2,2]-octene and Bicyclo[2,2,2]octadiene. J. Am. Chem. Soc 1957, 79, 4116–4121. [Google Scholar]
- (50).Lopez SA; Houk KN Alkene Distortion Energies and Torsional Effects Control Reactivities, and Stereoselectivities of Azide Cycloadditions to Norbornene and Substituted Norbornenes. J. Org. Chem 2013, 78, 1778–1783. [DOI] [PubMed] [Google Scholar]
- (51).Volland WV; Davidson ER; Borden WT Effect of Carbon Atom Pyramidalization on the Bonding in Ethylene. J. Am. Chem. Soc 1979, 101, 533–537. [Google Scholar]
- (52).Williams RV; Colvin ME; Tran N; Warrener RN; Margetic D Exceptionally Pyramidalized Olefins: A Theoretical Study of the Cyclopropenyl Fused Tricycles Tricyclo[3.2.1.02,4]oct-2(4)-ene, Tricyclo[3.2.1.02,4]octa-2(4),6-diene, Tricyclo[3.2.2.02,4]non-2(4)-ene, and Tricyclo[3.2.2.02,4]nona-2(4),6-diene. J. Org. Chem 2000, 65, 562–567. [DOI] [PubMed] [Google Scholar]
- (53).Liang Y; Jiang Y-Y; Liu Y; Bi S Mechanism of Pd-Catalyzed Acylation/Alkenylation of Aryl Iodide: a DFT Study. Org. Biomol. Chem 2017, 15, 6147–6156. [DOI] [PubMed] [Google Scholar]
- (54).Mauleón P; Núñez AA; Alonso I; Carretero JC Palladium-Catalyzed Cascade Reaction of α,β-Unsaturated Sulfones with Aryl Iodides. Chem. - Eur. J 2003, 9, 1511–1520. [DOI] [PubMed] [Google Scholar]
- (55).Sickert M; Weinstabl H; Peters B; Hou X; Lautens M Intermolecular Domino Reaction of Two Aryl Iodides Involving Two C–H Functionalizations. Angew. Chem., Int. Ed 2014, 53, 5147–5151. [DOI] [PubMed] [Google Scholar]
- (56).Lv W; Wen S; Yu J; Cheng G Palladium-Catalyzed Ortho-Silylation of Aryl Iodides with Concomitant Arylsilylation of Oxanorbornadiene: Accessing Functionalized (Z)-β-Substituted Vinyl-silanes and Their Analogues. Org. Lett 2018, 20, 4984–4987. [DOI] [PubMed] [Google Scholar]
- (57).Wang X; Gensch T; Lerchen A; Daniliuc CG; Glorius F Cp*Rh(III)/Bicyclic Olefin Cocatalyzed C–H Bond Amidation by Intramolecular Amide Transfer. J. Am. Chem. Soc 2017, 139, 6506–6512. [DOI] [PubMed] [Google Scholar]
- (58).Catellani M; Frignani F; Rangoni A A Complex Catalytic Cycle Leading to a Regioselective Synthesis of o,o’-Disubstituted Vinylarenes. Angew. Chem., Int. Ed. Engl 1997, 36, 119–122. [Google Scholar]
- (59).Catellani M; Cugini F A Catalytic Process Based on Sequential Ortho-Alkylation and Vinylation of Ortho-Alkylaryl Iodides via Palladacycles. Tetrahedron 1999, 55, 6595–6602. [Google Scholar]
- (60).Gallazzi MC; Hanlon TL; Vitulli G; Porri L Stereochemistry of the Insertion Reaction of Norbornene into a Nickel–Allyl and a Palladium–Allyl Bond. J. Organomet. Chem 1971, 33, C45–C46. [Google Scholar]
- (61).Lautens M; Piguel S A New Route to Fused Aromatic Compounds by Using a Palladium-Catalyzed Alkylation-Alkenylation Sequence. Angew. Chem., Int. Ed 2000, 39, 1045–1046. [DOI] [PubMed] [Google Scholar]
- (62).Mitsudo K; Thansandote P; Wilhelm T; Mariampillai B; Lautens M Selectively Substituted Thiophenes and Indoles by a Tandem Palladium-Catalyzed Multicomponent Reaction. Org. Lett 2006, 8, 3939–3942. [DOI] [PubMed] [Google Scholar]
- (63).Jafarpour F; Jalalimanesh N Palladium/Norbornene Catalysis: A Versatile Aryl C–H Hydroxyalkylation, Alkyl Cinnamate Formation. Tetrahedron 2012, 68, 10286–10292. [Google Scholar]
- (64).Stark T; Suhartono M; Gobel MW; Lautens M A Palladium-Catalyzed Domino Reaction as Key Step for the Synthesis of Functionalized Aromatic Amino Acids. Synlett 2013, 24, 2730–2734. [Google Scholar]
- (65).Ferraccioli R; Carenzi D; Catellani M Synthesis of 1,2,3,4-Tetrahydroisoquinolines and 2,3,4,5-Tetrahydro-1H-2-benzazepines Combining Sequential Palladium-Catalysed Ortho Alkylation/Vinylation with aza-Michael Addition Reactions. Tetrahedron Lett. 2004, 45, 6903–6907. [Google Scholar]
- (66).Ferraccioli R; Giannini C; Molteni G Stereoselective Synthesis of 1,3-Substituted Tetrahydroisoquinolines Through Palladium-Catalyzed Three-Component Reaction. Tetrahedron: Asymmetry 2007, 18, 1475–1480. [Google Scholar]
- (67).Ferraccioli R; Forni A Selective Synthesis of Isoquinolin-3-one Derivatives Combining Pd-Catalysed Aromatic Alkylation/Vinylation with Addition Reactions: The Beneficial Effect of Water. Eur. J. Org. Chem 2009, 2009, 3161–3166. [Google Scholar]
- (68).Zhang H; Chen P; Liu G Palladium-Catalyzed Cascade C–H Trifluoroethylation of Aryl Iodides and Heck Reaction: Efficient Synthesis of Ortho-Trifluoroethylstyrenes. Angew. Chem., Int. Ed 2014, 53, 10174–10178. [DOI] [PubMed] [Google Scholar]
- (69).Qureshi Z; Schlundt W; Lautens M Introduction of Hindered Electrophiles via C–H Functionalization in a Palladium-Catalyzed Multicomponent Domino Reaction. Synthesis 2015, 47, 2446–2456. [Google Scholar]
- (70).Sui X; Ding L; Gu Z The Palladium/Norbornene-Catalyzed Ortho-Silylmethylation Reaction: A Practical Protocol for Ortho-Functionalized One-Carbon Homologation of Aryl Iodides. Chem. Commun 2016, 52, 13999–14002. [DOI] [PubMed] [Google Scholar]
- (71).Barreiro EJ; Kümmerle AE; Fraga CA The Methylation Effect in Medicinal Chemistry. Chem. Rev 2011, 111, 5215–5246. [DOI] [PubMed] [Google Scholar]
- (72).Mariampillai B; Alliot J; Li M; Lautens M A Convergent Synthesis of Polysubstituted Aromatic Nitriles via Palladium-Catalyzed C–H Functionalization. J. Am. Chem. Soc 2007, 129, 15372–15379. [DOI] [PubMed] [Google Scholar]
- (73).Wilson JE Palladium-Catalyzed Catellani-Type Couplings Using Methylating Reagents for the Synthesis of Highly Substituted Ortho-Methyl-Arenes and Heteroarenes. Tetrahedron Lett. 2016, 57, 5053–5056. [Google Scholar]
- (74).Catellani M; Motti E; Minari M Symmetrical and Unsymmetrical 2,6-Dialkyl-1,1’-biaryls by Combined Catalysis of Aromatic Alkylation via Palladacycles and Suzuki-Type Coupling. Chem. Commun 2000, 157–158. [Google Scholar]
- (75).Ding L; Sui X; Gu Z Enantioselective Synthesis of Biaryl Atropisomers via Pd/Norbornene-Catalyzed Three-Component Cross-Couplings. ACS Catal. 2018, 8, 5630–5635. [Google Scholar]
- (76).Motti E; Rossetti M; Bocelli G; Catellani M Palladium Catalyzed Multicomponent Reactions in Ordered Sequence: New Syntheses of o,o’-Dialkylsubstituted Diarylacetylenes and Diary-lalkylidenehexahydromethanofluorenes. J. Organomet. Chem 2004, 689, 3741–3749. [Google Scholar]
- (77).Sun F; Gu Z Decarboxylative Alkynyl Termination of Palladium-Catalyzed Catellani Reaction: A Facile Synthesis of f α-Alkynyl Anilines via Ortho C–H Amination and Alkynylation. Org. Lett 2015, 17, 2222–2225. [DOI] [PubMed] [Google Scholar]
- (78).Pan S; Ma X; Zhong D; Chen W; Liu M; Wu H Palladium-Catalyzed One-Pot Consecutive Amination and Sonogashira Coupling for Selective Synthesis of 2-Alkynylanilines. Adv. Synth. Catal 2015, 357, 3052–3056. [Google Scholar]
- (79).Sun F; Li M; Gu Z Pd/norbornene-Catalyzed Sequential Ortho-C–H Alkylation and Ipso-Alkynylation: A 1,1-Dimethyl-2-Alkynol Strategy. Org. Chem. Front 2016, 3, 309–313. [Google Scholar]
- (80).Lei C; Jin X; Zhou J Palladium-Catalyzed Alkynylation and Concomitant Ortho Alkylation of Aryl Iodides. ACS Catal. 2016, 6, 1635–1639. [Google Scholar]
- (81).Wilhelm T; Lautens M Palladium-Catalyzed Alkylation-Hydride Reduction Sequence: Synthesis of Meta-Substituted Arenes. Org. Lett 2005, 7, 4053–4056. [DOI] [PubMed] [Google Scholar]
- (82).Martins A; Lautens M Aromatic Ortho-Benzylation Reveals an Unexpected Reductant. Org. Lett 2008, 10, 5095–5097. [DOI] [PubMed] [Google Scholar]
- (83).Mariampillai B; Alberico D; Bidau V; Lautens M Synthesis of Polycyclic Benzonitriles via a One-Pot Aryl Alkylation/Cyanation Reaction. J. Am. Chem. Soc 2006, 128, 14436–14437. [DOI] [PubMed] [Google Scholar]
- (84).Xia Y; Qiu D; Wang J Transition-Metal-Catalyzed Cross-Couplings through Carbene Migratory Insertion. Chem. Rev 2017, 117, 13810–13889. [DOI] [PubMed] [Google Scholar]
- (85).Barluenga J; Moriel P; Valdes C; Aznar F N-Tosylhy-drazones as Reagents for Cross-Coupling Reactions: A Route to Polysubstituted Olefins. Angew. Chem., Int. Ed 2007, 46, 5587–5590. [DOI] [PubMed] [Google Scholar]
- (86).Zhou P-X; Zheng L; Ma J-W; Ye Y-Y; Liu X-Y; Xu P-F; Liang Y-M Palladium-Catalyzed/Norbornene-Mediated C–H Activation/N-Tosylhydrazone Insertion Reaction: A Route to Highly Functionalized Vinylarenes. Chem. - Eur. J 2014, 20, 6745–6751. [DOI] [PubMed] [Google Scholar]
- (87).Bressy C; Alberico D; Lautens M A Route to Annulated Indoles via a Palladium-Catalyzed Tandem Alkylation/Direct Arylation Reaction. J. Am. Chem. Soc 2005, 127, 13148–13149. [DOI] [PubMed] [Google Scholar]
- (88).Lei C; Jin X; Zhou J Palladium-Catalyzed Heteroarylation and Concomitant Ortho-Alkylation of Aryl Iodides. Angew. Chem., Int. Ed 2015, 54, 13397–13400. [DOI] [PubMed] [Google Scholar]
- (89).Culkin DA; Hartwig JF Palladium-Catalyzed α-Arylation of Carbonyl Compounds and Nitriles. Acc. Chem. Res 2003, 36, 234–245. [DOI] [PubMed] [Google Scholar]
- (90).Maestri G; Della Ca’ N; Catellani M A Catalytic Synthesis of Selectively Substituted Biaryls through Sequential Intermolecular Coupling Involving Arene and Ketone C–H Bond Functionalization. Chem. Commun 2009, 4892–4894. [DOI] [PubMed] [Google Scholar]
- (91).Zhao Y-B; Mariampillai B; Candito DA; Laleu B; Li M; Lautens M Exploiting the Divergent Reactivity of Aryl-Palladium Intermediates for the Rapid Assembly of Fluorene and Phenanthrene Derivatives. Angew. Chem., Int. Ed 2009, 48, 1849–1852. [DOI] [PubMed] [Google Scholar]
- (92).Lei C; Cao J; Zhou J Palladium-Catalyzed Arylation of Ketones and Acetonitrile with Ortho Alkylation of Aryl Rings: De Novo Synthesis of Tetralines and Benzocycloheptenes. Org. Lett 2016, 18, 6120–6123. [DOI] [PubMed] [Google Scholar]
- (93).Lautens M; Paquin JF; Piguel S; Dahlmann M Palladium-Catalyzed Sequential Alkylation-Alkenylation Reactions and Their Application to the Synthesis of Fused Aromatic Rings. J. Org. Chem 2001, 66, 8127–8134. [DOI] [PubMed] [Google Scholar]
- (94).Lautens M; Paquin JF; Piguel S Palladium-Catalyzed Sequential Alkylation-Alkenylation Reactions. Application to the Synthesis of 2-Substituted-4-Benzoxepines and 2,5-Disubstituted-4-Benzoxepines. J. Org. Chem 2002, 67, 3972–3974. [DOI] [PubMed] [Google Scholar]
- (95).Alberico D; Paquin JF; Lautens M Palladium-Catalyzed Sequential Alkylation-Alkenylation Reactions: Application towards the Synthesis of Polyfunctionalized Fused Aromatic Rings. Tetrahedron 2005, 61, 6283–6297. [Google Scholar]
- (96).Liu Z-S; Qian G; Gao Q; Wang P; Cheng H-G; Wei Q; Liu Q; Zhou Q Palladium/Norbornene Cooperative Catalysis To Access Tetrahydronaphthalenes and Indanes with a Quaternary Center. ACS Catal. 2018, 8, 4783–4788. [Google Scholar]
- (97).Liu Z-S; Qian G; Gao Q; Wang P; Cheng H-G; Hua Y; Zhou Q 5-Norbornene-2-carboxylic acid: Another catalytic mediator for Catellani-type reactions. Tetrahedron 2019, 75, 1774–1780. [Google Scholar]
- (98).Jafarpour F; Hazrati H Direct Synthesis of Dihydrobenzo[a]-carbazoles via Palladium-Catalyzed Domino Annulation of Indoles. Adv. Synth. Catal 2010, 352, 363–367. [Google Scholar]
- (99).Jafarpour F; Otaredi-Kashani A; Behpajooh S Domino o-Alkylation and Heteroarylation of Iodoarenes: One-Pot Synthesis of Benzocyclohepta[b]indoles. Synthesis 2013, 45, 1094–1098. [Google Scholar]
- (100).Blaszykowski C; Aktoudianakis E; Bressy C; Alberico D; Lautens M Preparation of Annulated Nitrogen-Containing Heterocycles via a One-Pot Palladium-Catalyzed Alkylation/Direct Arylation Sequence. Org. Lett 2006, 8, 2043–2045. [DOI] [PubMed] [Google Scholar]
- (101).Jafarpour F; Ashtiani PT A Novel One-Step Synthesis of Imidazo[5,1-a]isoquinolines via a Tandem Pd-Catalyzed Alkylation-Direct Arylation Sequence. J. Org. Chem 2009, 74, 1364–1366. [DOI] [PubMed] [Google Scholar]
- (102).Martins A; Alberico D; Lautens M Synthesis of Polycyclic Heterocycles via a One-Pot Ortho Alkylation/Direct Heteroarylation Sequence. Org. Lett 2006, 8, 4827–4829. [DOI] [PubMed] [Google Scholar]
- (103).Martins A; Lautens M A Palladium-Catalyzed Approach to Polycyclic Sulfur Heterocycles. J. Org. Chem 2008, 73, 8705–8710. [DOI] [PubMed] [Google Scholar]
- (104).Blaszykowski C; Aktoudianakis E; Alberico D; Bressy C; Hulcoop DG; Jafarpour F; Joushaghani A; Laleu B; Lautens M A Palladium-Catalyzed Alkylation/Direct Arylation Synthesis of Nitrogen-Containing Heterocycles. J. Org. Chem 2008, 73, 1888–1897. [DOI] [PubMed] [Google Scholar]
- (105).Laleu B; Lautens M Synthesis of Annulated 2H-Indazoles and 1,2,3-and 1,2,4-Triazoles via a One-Pot Palladium-Catalyzed Alkylation/Direct Arylation Reaction. J. Org. Chem 2008, 73, 9164–9167. [DOI] [PubMed] [Google Scholar]
- (106).Wu X-X; Shen Y; Chen W-L; Chen S; Xu P-F; Liang Y-M Palladium-Catalyzed Dearomative Cyclization by A Norbornene-Mediated Sequence: A Route to Spiroindolenine Derivatives. Chem. Commun 2015, 51, 16798–16801. [DOI] [PubMed] [Google Scholar]
- (107).Thansandote P; Raemy M; Rudolph A; Lautens M Synthesis of Benzannulated N-Heterocycles by a Palladium-Catalyzed C–C/C–N Coupling of Bromoalkylamines. Org. Lett 2007, 9, 5255–5258. [DOI] [PubMed] [Google Scholar]
- (108).Zhang B-S; Hua H-L; Gao L-Y; Liu C; Qiu Y-F; Zhou P-X; Zhou Z-Z; Zhao J-H; Liang Y-M Palladium-Catalyzed Arene C–H Activation/Ketone C–H Functionalization Reaction: Route to Spirodihydroindenones. Org. Chem. Front 2017, 4, 1376–1379. [Google Scholar]
- (109).Gericke KM; Chai DI; Lautens M The Versatile Role of Norbornene in C–H Functionalization Processes: Concise Synthesis of Tetracyclic Fused Pyrroles via a Threefold Domino Reaction. Tetrahedron 2008, 64, 6002–6014. [Google Scholar]
- (110).Aureggi V; Davoust M; Gericke KM; Lautens M A Convenient Synthesis of Indolo- and Pyrrolobenzazepines via a Threefold Norbornene-Mediated Domino Reaction. Synlett 2009, 2009, 1004–1008. [Google Scholar]
- (111).Liu H; El-Salfiti M; Lautens M Expeditious Synthesis of Tetrasubstituted Helical Alkenes by a Cascade of Palladium-Catalyzed C–H Activations. Angew. Chem., Int. Ed 2012, 51, 9846–9850. [DOI] [PubMed] [Google Scholar]
- (112).Bai L; Liu J; Hu W; Li K; Wang Y; Luan X Palladium/Norbornene-Catalyzed C–H Alkylation/Alkyne Insertion/Indole Dearomatization Domino Reaction: Assembly of Spiroindolenine-Containing Pentacyclic Frameworks. Angew. Chem., Int. Ed 2018, 57, 5151–5155. [DOI] [PubMed] [Google Scholar]
- (113).Nan J; Yuan Y; Bai L; Liu J; Luan X Highly Chemoselective Construction of Spiro[4,5]decane-Embedded Polycyclic Scaffolds by a Palladium/Norbornene-Catalyzed C–H Activation/Arene Dearomatization Reaction. Org. Lett 2018, 20, 7731–7734. [DOI] [PubMed] [Google Scholar]
- (114).Gericke KM; Chai DI; Bieler N; Lautens M The Norbornene Shuttle: Multicomponent Domino Synthesis of Tetrasubstituted Helical Alkenes through Multiple C–H Functionalization. Angew. Chem., Int. Ed 2009, 48, 1447–1451. [DOI] [PubMed] [Google Scholar]
- (115).Liu H; El-Salfiti M; Chai DI; Auffret J; Lautens M Modular and Stereoselective Synthesis of Tetrasubstituted Helical Alkenes via a Palladium-Catalyzed Domino Reaction. Org. Lett 2012, 14, 3648–3651. [DOI] [PubMed] [Google Scholar]
- (116).Martins A; Marquardt U; Kasravi N; Alberico D; Lautens M Synthesis of Substituted Benzoxacycles via a Domino Ortho-Alkylation/Heck Coupling Sequence. J. Org. Chem 2006, 71, 4937–4942. [DOI] [PubMed] [Google Scholar]
- (117).Thansandote P; Chong E; Feldmann KO; Lautens M Palladium-Catalyzed Domino C–C/C–N Coupling Using a Norbornene Template: Synthesis of Substituted Benzomorpholines, Phenoxazines, and Dihydrodibenzoxazepines. J. Org. Chem 2010, 75, 3495–3498. [DOI] [PubMed] [Google Scholar]
- (118).Pache S; Lautens M Palladium-Catalyzed Sequential Alkylation-Alkenylation Reactions: New Three-Component Coupling Leading to Oxacycles. Org. Lett 2003, 5, 4827–4830. [DOI] [PubMed] [Google Scholar]
- (119).Alberico D; Rudolph A; Lautens M Synthesis of Tricyclic Heterocycles via a Tandem Aryl Alkylation/Heck Coupling Sequence. J. Org. Chem 2007, 72, 775–781. [DOI] [PubMed] [Google Scholar]
- (120).Rudolph A; Rackelmann N; Turcotte-Savard MO; Lautens M Application of Secondary Alkyl Halides to a Domino Aryl Alkylation Reaction for the Synthesis of Aromatic Heterocycles. J. Org. Chem 2009, 74, 289–297. [DOI] [PubMed] [Google Scholar]
- (121).Thansandote P; Gouliaras C; Turcotte-Savard MO; Lautens M A Rapid Approach to the Synthesis of Highly Functionalized Tetrahydroisoquinolines. J. Org. Chem 2009, 74, 1791–1793. [DOI] [PubMed] [Google Scholar]
- (122).Alberico D; Lautens M Palladium-Catalyzed Alkylation-Alkenylation Reactions: Rapid Access to Tricyclic Mescaline Analogues. Synlett 2006, 2006, 2969–2972. [Google Scholar]
- (123).Jafarpour F; Lautens M Direct, One-Step Synthesis of Condensed Heterocycles: A Palladium-Catalyzed Coupling Approach. Org. Lett 2006, 8, 3601–3604. [DOI] [PubMed] [Google Scholar]
- (124).Wu X-X; Zhou P-X; Wang L-J; Xu P-F; Liang Y-M Synthesis of Polycyclic Substituted Vinylarenes via a One-Pot Intramolecular Aryl Alkylation-N-Tosylhydrazone Insertion Reaction. Chem. Commun 2014, 50, 3882–3884. [DOI] [PubMed] [Google Scholar]
- (125).Shen Y; Wu X-X; Chen S; Xia Y; Liang Y-M Synthesis of Polyfluoroarene-Substituted Benzofuran Derivatives via Cooperative Pd/Cu Catalysis. Chem. Commun 2018, 54, 2256–2259. [DOI] [PubMed] [Google Scholar]
- (126).Do HO; Khan RMK; Daugulis O A General Method for Copper-Catalyzed Arylation of Arene C–H Bonds. J. Am. Chem. Soc 2008, 130, 15185–15192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Candito DA; Lautens M Exploiting the Chemistry of Strained Rings: Synthesis of Indoles via Domino Reaction of Aryl Iodides with 2H-Azirines. Org. Lett 2010, 12, 3312–3315. [DOI] [PubMed] [Google Scholar]
- (128).Alper H; Perera CP; Ahmed FR A Novel Synthesis of β-Lactams. J. Am. Chem. Soc 1981, 103, 1289–1291. [Google Scholar]
- (129).Cheng G; Li T-J; Yu J-Q Practical Pd(II)-Catalyzed C–H Alkylation with Epoxides: One-Step Syntheses of 3,4-Dihydroisocoumarins. J. Am. Chem. Soc 2015, 137, 10950–10953. [DOI] [PubMed] [Google Scholar]
- (130).Wang Z; Kuninobu Y; Kanai M Palladium-Catalyzed Oxirane-Opening Reaction with Arenes via C–H Bond Activation. J. Am. Chem. Soc 2015, 137, 6140–6143. [DOI] [PubMed] [Google Scholar]
- (131).Li R; Dong G Direct Annulation between Aryl Iodides and Epoxides through Palladium/Norbornene Cooperative Catalysis. Angew. Chem., Int. Ed 2018, 57, 1697–1701. [DOI] [PubMed] [Google Scholar]
- (132).Li R; Liu F; Dong G Palladium-Catalyzed Asymmetric Annulation between Aryl Iodides and Racemic Epoxides using a Chiral Norbornene Cocatalyst. Org. Chem. Front 2018, 5, 3108–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Cheng H-G; Wu C; Chen H; Chen R; Qian G; Geng Z; Wei Q; Xia Y; Zhang J; Zhang Y; et al. Epoxides as Alkylating Reagents for the Catellani Reaction. Angew. Chem., Int. Ed 2018, 57, 3444–3448. [DOI] [PubMed] [Google Scholar]
- (134).Wu C; Cheng H-G; Chen R; Chen H; Liu Z-S; Zhang J; Zhang Y; Zhu Y; Geng Z; Zhou Q Convergent Syntheses of 2,3-Dihydrobenzofurans via a Catellani Strategy. Org. Chem. Front 2018, 5, 2533–2536. [Google Scholar]
- (135).Liu C; Liang Y; Zheng N; Zhang B-S; Feng Y; Bi S; Liang Y-M Synthesis of Indolines via a Palladium/Norbornene-Catalyzed Reaction of Aziridines with Aryl Iodides. Chem. Commun 2018, 54, 3407–3410. [DOI] [PubMed] [Google Scholar]
- (136).Qian G; Bai M; Gao S; Chen H; Zhou S; Cheng H-G; Yan W; Zhou Q Modular One-Step Three-Component Synthesis of Tetrahydroisoquinolines Using a Catellani Strategy. Angew. Chem., Int. Ed 2018, 57, 10980–10984. [DOI] [PubMed] [Google Scholar]
- (137).Catellani M; Motti E; Baratta S A Novel Palladium-Catalyzed Synthesis of Phenanthrenes from Ortho-Substituted Aryl Iodides and Diphenyl- or Alkylphenylacetylenes. Org. Lett 2001, 3, 3611–3614. [DOI] [PubMed] [Google Scholar]
- (138).Motti E; Ippomei G; Deledda S; Catellani M Synthesis of Selectively Substituted Ortho-Vinylbiphenyls by Palladium-Catalysed Reaction of Ortho-Substituted Aryl Iodides with Olefins. Synthesis 2003, 2671–2678. [Google Scholar]
- (139).Catellani M; Deledda S; Ganchegui B; Henin F; Motti E; Muzart J A New Catalytic Method for the Synthesis of Selectively Substituted Biphenyls Containing an Oxoalkyl Chain. J. Organomet. Chem 2003, 687, 473–482. [Google Scholar]
- (140).Motti E; Mignozzi A; Catellani M A New Type of Palladium-Catalysed Aromatic Cross-Coupling Combined with a Suzuki Reaction: Synthesis of Selectively 2, 3′-Substituted 1, 1′; 2′, 1 ″-Terphenyl Derivatives. J. Mol. Catal. A: Chem 2003, 204–205, 115–124. [Google Scholar]
- (141).Deledda S; Motti E; Catellani M Palladium-Catalysed Synthesis of Nonsymmetrically Disubstituted-1,1’-Biphenyls from o-Substituted Aryl Iodides through Aryl Coupling and Delayed Hydrogenolysis. Can. J. Chem 2005, 83, 741–747. [Google Scholar]
- (142).Della Ca’ N; Maestri G; Catellani M Palladium/Norbornene-Catalyzed Synthesis of Heteroatom-Containing o-Teraryls from Aryl Iodides and Heteroarenes through Double C–H Activation in Sequence. Chem. - Eur. J 2009, 15, 7850–7853. [DOI] [PubMed] [Google Scholar]
- (143).Faccini F; Motti E; Catellani M A New Reaction Sequence Involving Palladium-Catalyzed Unsymmetrical Aryl Coupling. J. Am. Chem. Soc 2004, 126, 78–79. [DOI] [PubMed] [Google Scholar]
- (144).Vicente J; Arcas A; Julia-Hernandez F; Bautista D Synthesis of a Palladium(IV) Complex by Oxidative Addition of an Aryl Halide to Palladium(II) and Its Use as Precatalyst in a C–C Coupling Reaction. Angew. Chem., Int. Ed 2011, 50, 6896–6899. [DOI] [PubMed] [Google Scholar]
- (145).Casnati A; Gemoets HPL; Motti E; Della Ca’ N; Noël T Homogeneous and Gas-Liquid Catellani-Type Reaction Enabled by Continuous-Flow Chemistry. Chem. - Eur. J 2018, 24, 14079–14083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Motti E; Faccini F; Ferrari I; Catellani M; Ferraccioli R Sequential Unsymmetrical Aryl Coupling of o-Substituted Aryl Iodides with o-Bromophenols and Reaction with Olefins: Palladium-Catalyzed Synthesis of 6H-Dibenzopyran Derivatives. Org. Lett 2006, 8, 3967–3970. [DOI] [PubMed] [Google Scholar]
- (147).Xu D; Dai L; Catellani M; Motti E; Della Ca’ N; Zhou Z A Novel Enantioselective Synthesis of 6H-Dibenzopyran Derivatives by Combined Palladium/Norbornene and Cinchona Alkaloid Catalysis. Org. Biomol. Chem 2015, 13, 2260–2263. [DOI] [PubMed] [Google Scholar]
- (148).Della Ca’ N; Motti E; Catellani M Palladium-Catalyzed Synthesis of Selectively Substituted Phenanthridine Derivatives. Adv. Synth. Catal 2008, 350, 2513–2516. [Google Scholar]
- (149).Della Ca’ N; Motti E; Mega A; Catellani M One-Pot Palladium-Catalyzed Synthesis of Selectively Substituted Phenanthridines by Sequential Aryl-Aryl and Heck Couplings, Aza-Michael and Retro-Mannich Reactions. Adv. Synth. Catal 2010, 352, 1451–1454. [Google Scholar]
- (150).Narbonne V; Retailleau P; Maestri G; Malacria M Diastereoselective Synthesis of Dibenzoazepines through Chelation on Palladium(IV) Intermediates. Org. Lett 2014, 16, 628–631. [DOI] [PubMed] [Google Scholar]
- (151).Martins A; Candito DA; Lautens M Palladium-Catalyzed Reductive Ortho-Arylation: Evidence for the Decomposition of 1,2-Dimethoxyethane and Subsequent Arylpalladium(II) Reduction. Org. Lett 2010, 12, 5186–5188. [DOI] [PubMed] [Google Scholar]
- (152).Motti E; Della Ca’ N; Deledda S; Fava E; Panciroli F; Catellani M Palladium-Catalyzed Unsymmetrical Aryl Couplings in Sequence Leading to o-Teraryls: Dramatic Olefin Effect on Selectivity. Chem. Commun 2010, 46, 4291–4293. [DOI] [PubMed] [Google Scholar]
- (153).Motti E; Della Ca’ N; Xu D; Piersirnoni A; Bedogni E; Zhou Z-M; Catellani M A Sequential Pd/Norbornene-Catalyzed Process Generates o-Biaryl Carbaldehydes or Ketones via a Redox Reaction or 6H-Dibenzopyrans by C–O Ring Closure. Org. Lett 2012, 14, 5792–5795. [DOI] [PubMed] [Google Scholar]
- (154).Fu WC; Wang Z; Chan WTK; Lin Z; Kwong FY Regioselective Synthesis of Polycyclic and Heptagon-Embedded Aromatic Compounds through a Versatile π-Extension of Aryl Halides. Angew. Chem., Int. Ed 2017, 56, 7166–7170. [DOI] [PubMed] [Google Scholar]
- (155).Zhao Q; Fu WC; Kwong FY Palladium-Catalyzed Regioselective Aromatic Extension of Internal Alkynes through a Norbornene-Controlled Reaction Sequence. Angew. Chem., Int. Ed 2018, 57, 3381–3385. [DOI] [PubMed] [Google Scholar]
- (156).Yang Y; Zhou B; Zhu X; Deng G; Liang Y; Yang Y Palladium-Catalyzed Synthesis of Triphenylenes via Sequential C–H Activation and Decarboxylation. Org. Lett 2018, 20, 5402–5405. [DOI] [PubMed] [Google Scholar]
- (157).Cardenas DJ; Martin-Matute B; Echavarren AM Aryl Transfer between Pd(II) Centers or Pd(IV) Intermediates in Pd-Catalyzed Domino Reactions. J. Am. Chem. Soc 2006, 128, 5033–5040. [DOI] [PubMed] [Google Scholar]
- (158).Maestri G; Motti E; Della Ca’ N; Malacria M; Derat E; Catellani M Of the Ortho Effect in Palladium/Norbornene-Catalyzed Reactions: a Theoretical Investigation. J. Am. Chem. Soc 2011, 133, 8574–8585. [DOI] [PubMed] [Google Scholar]
- (159).Shen P-X; Wang X-C; Wang P; Zhu R-Y; Yu J-Q Ligand-Enabled Meta-C–H Alkylation and Arylation using a Modified Norbornene. J. Am. Chem. Soc 2015, 137, 11574–11577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (160).Wang J; Li R; Dong Z; Liu P; Dong G Complementary Site-Selectivity in Arene Functionalization Enabled by Overcoming the Ortho Constraint in Palladium/Norbornene Catalysis. Nat. Chem 2018, 10, 866–872. [DOI] [PubMed] [Google Scholar]
- (161).Ferraccioli R; Carenzi D; Rombola O; Catellani M Synthesis of 6-Phenanthridinones and Their Heterocyclic Analogues through Palladium-Catalyzed Sequential Aryl–Aryl and N–Aryl Coupling. Org. Lett 2004, 6, 4759–4762. [DOI] [PubMed] [Google Scholar]
- (162).Ferraccioli R; Carenzi D; Motti E; Catellani M A Simple Catalytic Synthesis of Condensed Pyridones from o-Bromoarylcarboxamides Involving Ipso Substitution via Palladacycles. J. Am. Chem. Soc 2006, 128, 722–723. [DOI] [PubMed] [Google Scholar]
- (163).Della Ca’ N; Sassi G; Catellani M A Direct Palladium-Catalyzed Route to Selectively Substituted Carbazoles through Sequential C–C and C–N Bond Formation: Synthesis of Carbazomycin A. Adv. Synth. Catal 2008, 350, 2179–2182. [Google Scholar]
- (164).Candito DA; Lautens M Palladium-Catalyzed Domino Direct Arylation/N-Arylation: Convenient Synthesis of Phenanthridines. Angew. Chem., Int. Ed 2009, 48, 6713–6716. [DOI] [PubMed] [Google Scholar]
- (165).Barluenga J; Aznar F; Valdes C N-Trialkylsilylimines as Coupling Partners for Pd-Catalyzed C–N Bond-Forming Reactions: One-Step Synthesis of Imines and Azadienes from Aryl and Alkenyl Bromides. Angew. Chem., Int. Ed 2004, 43, 343–345. [DOI] [PubMed] [Google Scholar]
- (166).Maestri G; Larraufie MH; Derat E; Ollivier C; Fensterbank L; Lacote E; Malacria M Expeditious Synthesis of Phenanthridines from Benzylamines via Dual Palladium Catalysis. Org. Lett 2010, 12, 5692–5695. [DOI] [PubMed] [Google Scholar]
- (167).Tsukano C; Muto N; Enkhtaivan I; Takemoto Y Synthesis of Pyrrolophenanthridine Alkaloids Based on C(sp3)–H and C(sp2)–H Functionalization Reactions. Chem. - Asian J 2014, 9, 2628–2634. [DOI] [PubMed] [Google Scholar]
- (168).Motti E; Della Ca’ N; Xu D; Armani S; Aresta BM; Catellani M Competitive Pathways in Pd-Catalyzed Synthesis of Arylphenols. Tetrahedron 2013, 69, 4421–4428. [Google Scholar]
- (169).Zuo Z; Wang H; Fan L; Liu J; Wang Y; Luan X Modular Assembly of Spirocarbocyclic Scaffolds through Pd0 Catalyzed Intermolecular Dearomatizing [2 + 2+1] Annulation of Bromonaphthols with Aryl Iodides and Alkynes. Angew. Chem., Int. Ed 2017, 56, 2767–2771. [DOI] [PubMed] [Google Scholar]
- (170).Yamamoto Y; Murayama T; Jiang J; Yasui T; Shibuya M The Vinylogous Catellani Reaction: A Combined Computational and Experimental Study. Chem. Sci 2018, 9, 1191–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (171).Malacria M; Maestri G Palladium/Norbornene Catalytic System: Chelation as a Tool to Control Regioselectivity of Pd(IV) Reductive Elimination. J. Org. Chem 2013, 78, 1323–1328. [DOI] [PubMed] [Google Scholar]
- (172).Larraufie MH; Maestri G; Beaume A; Derat E; Ollivier C; Fensterbank L; Courillon C; Lacôte E; Catellani M; Malacria M Exception to the Ortho Effect in Palladium/Norbornene Catalysis. Angew. Chem., Int. Ed 2011, 50, 12253–12256. [DOI] [PubMed] [Google Scholar]
- (173).Della Ca’ N; Maestri G; Malacria M; Derat E; Catellani M Palladium-Catalyzed Reaction of Aryl Iodides with Ortho-Bromoanilines and Norbornene/Norbornadiene: Unexpected Formation of Dibenzoazepine Derivatives. Angew. Chem., Int. Ed 2011, 50, 12257–12261. [DOI] [PubMed] [Google Scholar]
- (174).Zhou H; Chen W; Chen Z Pd/Norbornene Collaborative Catalysis on the Divergent Preparation of Heterocyclic Sulfoximine Frameworks. Org. Lett 2018, 20, 2590–2594. [DOI] [PubMed] [Google Scholar]
- (175).Sole D; Fernandez I Controlling the Ambiphilic Nature of σ-Arylpalladium Intermediates in Intramolecular Cyclization Reactions. Acc. Chem. Res 2014, 47, 168–179. [DOI] [PubMed] [Google Scholar]
- (176).Sui X; Zhu R; Li G; Ma X; Gu Z Pd-Catalyzed Chemoselective Catellani Ortho-Arylation of Iodopyrroles: Rapid Total Synthesis of Rhazinal. J. Am. Chem. Soc 2013, 135, 9318–9321. [DOI] [PubMed] [Google Scholar]
- (177).Berman AM; Johnson JS Copper-Catalyzed Electrophilic Amination of Diorganozinc Reagents. J. Am. Chem. Soc 2004, 126, 5680–5681. [DOI] [PubMed] [Google Scholar]
- (178).Yoo EJ; Ma S; Mei T-S; Chan KSL; Yu J-Q Pd-Catalyzed Intermolecular C–H Amination with Alkylamines. J. Am. Chem. Soc 2011, 133, 7652–7655. [DOI] [PubMed] [Google Scholar]
- (179).Dong Z; Dong G Ortho vs Ipso: Site-Selective Pd and Norbornene-Catalyzed Arene C–H Amination Using Aryl Halides. J. Am. Chem. Soc 2013, 135, 18350–18353. [DOI] [PubMed] [Google Scholar]
- (180).Chen Z-Y; Ye C-Q; Zhu H; Zeng X-P; Yuan J-J Palladium/Norbornene-Mediated Tandem C–H Amination/C–I Alkenylation Reaction of Aryl Iodides with Secondary Cyclic O-Benzoyl Hydroxylamines and Activated Terminal Olefins. Chem. - Eur. J 2014, 20, 4237–4241. [DOI] [PubMed] [Google Scholar]
- (181).Wang J; Gu Z Synthesis of 2-(1-Alkoxyvinyl)anilines by Palladium/Norbornene-Catalyzed Amination Followed by Termination with Vinyl Ethers. Adv. Synth. Catal 2016, 358, 2990–2995. [Google Scholar]
- (182).Ye C-Q; Zhu H; Chen Z-Y Synthesis of Biaryl Tertiary Amines through Pd/Norbornene Joint Catalysis in a Remote C–H Amination/Suzuki Coupling Reaction. J. Org. Chem 2014, 79, 8900–8905. [DOI] [PubMed] [Google Scholar]
- (183).Zhou P-X; Ye Y-Y; Ma J-W; Zheng L; Tang Q; Qiu Y-F; Song B; Qiu Z-H; Xu P-F; Liang Y-M Palladium-Catalyzed/Norbornene-Mediated Ortho-Amination/N-Tosylhydrazone Insertion Reaction: An Approach to the Synthesis of Ortho-Aminated Vinylarenes. J. Org. Chem 2014, 79, 6627–6633. [DOI] [PubMed] [Google Scholar]
- (184).Luo B; Gao JM; Lautens M Palladium-Catalyzed Norbornene-Mediated Tandem Amination/Cyanation Reaction: A Method for the Synthesis of Ortho-Aminated Benzonitriles. Org. Lett 2016, 18, 4166–4169. [DOI] [PubMed] [Google Scholar]
- (185).Majhi B; Ranu BC Palladium-Catalyzed Norbornene-Mediated Tandem Ortho-C–H-Amination/Ipso-C–I-Cyanation of Iodoarenes: Regiospecific Synthesis of 2-Aminobenzonitrile. Org. Lett 2016, 18, 4162–4165. [DOI] [PubMed] [Google Scholar]
- (186).Fu WC; Zheng B; Zhao QY; Chan WTK; Kwong FY Cascade Amination and Acetone Monoarylation with Aryl Iodides by Palladium/Norbornene Cooperative Catalysis. Org. Lett 2017, 19, 4335–4338. [DOI] [PubMed] [Google Scholar]
- (187).Shi H; Babinski DJ; Ritter T Modular C–H Functionalization Cascade of Aryl Iodides. J. Am. Chem. Soc 2015, 137, 3775–3778. [DOI] [PubMed] [Google Scholar]
- (188).Fan L; Liu J; Bai L; Wang Y; Luan X Rapid Assembly of Diversely Functionalized Spiroindenes by a Three-Component Palladium-Catalyzed C–H Amination/Phenol Dearomatization Domino Reaction. Angew. Chem., Int. Ed 2017, 56, 14257–14261. [DOI] [PubMed] [Google Scholar]
- (189).Whyte A; Olson ME; Lautens M Palladium-Catalyzed, Norbornene-Mediated, Ortho-Amination Ipso-Amidation: Sequential C–N Bond Formation. Org. Lett 2018, 20, 345–348. [DOI] [PubMed] [Google Scholar]
- (190).Zhang B-S; Li Y; An Y; Zhang Z; Liu C; Wang X-G; Liang Y-M Carboxylate Ligand-Exchanged Amination/C(sp3)–H Arylation Reaction via Pd/Norbornene Cooperative Catalysis. ACS Catal. 2018, 8, 11827–11833. [Google Scholar]
- (191).Zhou P-X; Ye Y-Y; Liu C; Zhao L-B; Hou J-Y; Chen D-Q; Tang Q; Wang A-Q; Zhang J-Y; Huang Q-X; et al. Palladium-Catalyzed Acylation/Alkenylation of Aryl Iodide: A Domino Approach Based on the Catellani-Lautens Reaction. ACS Catal. 2015, 5, 4927–4931. [Google Scholar]
- (192).Huang Y; Zhu R; Zhao K; Gu Z Palladium-Catalyzed Catellani Ortho-Acylation Reaction: An Efficient and Regiospecific Synthesis of Diaryl Ketones. Angew. Chem., Int. Ed 2015, 54, 12669–12672. [DOI] [PubMed] [Google Scholar]
- (193).Dong Z; Wang J; Ren Z; Dong G Ortho C–H Acylation of Aryl Iodides by Palladium/Norbornene Catalysis. Angew. Chem., Int. Ed 2015, 54, 12664–12668. [DOI] [PubMed] [Google Scholar]
- (194).Xu S; Jiang J; Ding L; Fu Y; Gu Z Palladium/Norbornene-Catalyzed Ortho Aliphatic Acylation with Mixed Anhydride: Selectivity and Reactivity. Org. Lett 2018, 20, 325–328. [DOI] [PubMed] [Google Scholar]
- (195).Wu W; Yu S; Gu T; Fan T; Zhong Y; Li N; Tang Y; Jiang Y; Zhu X; Duan J; et al. Norbornene-Mediated Palladium-Catalysed Domino-Type Catellani Reaction: an Efficient and Regiospecific Acylation/Suzuki Coupling of Aryl Iodides. Eur. J. Org. Chem 2018, 2018, 3075–3085. [Google Scholar]
- (196).Pan S; Wu F; Yu R; Chen W Palladium-Catalyzed Sequential Acylation/Cyanation of Aryl Iodides: A Regiospecific Synthesis of 2-Cyanoaryl Ketones. J. Org. Chem 2016, 81, 1558–1564. [DOI] [PubMed] [Google Scholar]
- (197).Yu S-P; Zhong Y; Gu T; Wu W-Y; Fan T-Y; Li N-G; Shi Z-H; Tang Y-P; Duan J-A Palladium-Catalyzed One-Pot Catellani Reaction: An Efficient Synthesis of α-Alkynyl Aromatic Ketones via Ortho Acylation and Ipso Alkynylation. Tetrahedron 2018, 74, 5942–5949. [Google Scholar]
- (198).Zhang P; Pan S; Chen W; Liu M; Wu H Palladium-Catalyzed Sequential Heteroarylation/Acylation Reactions of lodobenzenes: Synthesis of Functionalized Benzo[d]oxazoles. J. Org. Chem 2018, 83, 3354–3360. [DOI] [PubMed] [Google Scholar]
- (199).Prokopcova H; Kappe CO The Liebeskind-Srogl C–C Cross-Coupling Reaction. Angew. Chem., Int. Ed 2009, 48, 2276–2286. [DOI] [PubMed] [Google Scholar]
- (200).Sun F; Li M; He C; Wang; Li B; Sui X; Gu Z Cleavage of the C(O)–S Bond of Thioesters by Palladium/Norbornene/Copper Cooperative Catalysis: An Efficient Synthesis of 2-(Arylthio)aryl Ketones. J. Am. Chem. Soc 2016, 138, 7456–7459. [DOI] [PubMed] [Google Scholar]
- (201).Fan X; Gu Z Palladium/Norbornene-Catalyzed Ortho–Acylation and Ipso-Selenation via C(O)–Se Bond Cleavage: Synthesis of α-Carbonyl Selane. Org. Lett 2018, 20, 1187–1190. [DOI] [PubMed] [Google Scholar]
- (202).Liu F; Dong Z; Wang J; Dong G Palladium/Norbornene-Catalyzed Indenone Synthesis from Simple Aryl Iodides: Concise Syntheses of Pauciflorol F and Acredinone A. Angew. Chem., Int. Ed 2019, 58, 2144–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (203).Wang Z; Li T; Zhao J; Shi X; Jiao D; Zheng H; Chen C; Zhu B Expeditious Synthesis of 6-Fluoroalkyl-Phenanthridines via Palladium-Catalyzed Norbornene-Mediated Dehydrogenative Annulation. Org. Lett 2018, 20, 6640–6645. [DOI] [PubMed] [Google Scholar]
- (204).Wang J; Zhang L; Dong Z; Dong G Reagent-Enabled Ortho-Alkoxycarbonylation of Aryl Iodides via Palladium/Norbornene Catalysis. Chem. 2016, 1, 581–591. [Google Scholar]
- (205).Blanchot M; Candito DA; Larnaud F; Lautens M Formal Synthesis of Nitidine and NK109 via Palladium-Catalyzed Domino Direct Arylation/N-Arylation of Aryl Triflates. Org. Lett 2011, 13, 1486–1489. [DOI] [PubMed] [Google Scholar]
- (206).Deyris PA; Caneque-Cobo T; Gomes F; Narbonne V; Maestri G; Malacria M Rapid and Convergent Assembly of Natural Benzo[c]phenanthridines by Palladium/Norbornene Catalysis. Heterocycles 2014, 88, 807–815. [Google Scholar]
- (207).Dong Z; Lu G; Wang J; Liu P; Dong G Modular Ipso/Ortho Difunctionalization of Aryl Bromides via Palladium/Norbornene Cooperative Catalysis. J. Am. Chem. Soc 2018, 140, 8551–8562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (208).Casnati A; Fontana M; Coruzzi G; Aresta BM; Corriero N; Maggi R; Maestri G; Motti E; Della Ca’ N Enhancing Reactivity and Selectivity of Aryl Bromides: A Complementary Approach to Dibenzo[b, f]azepine Derivatives. ChemCatChem 2018, 10, 4346–4352. [Google Scholar]
- (209).Elsayed MSA; Griggs B; Cushman M Synthesis of Benzo[1,6]naphthyridinones Using the Catellani Reaction. Org. Lett 2018, 20, 5228–5232. [DOI] [PubMed] [Google Scholar]
- (210).Tedeschi E; Dukler S; Pfeffer P; Lavie D Studies on Strychnine Derivatives and Conversion into Brucine. Tetrahedron 1968, 24, 4573–4580. [DOI] [PubMed] [Google Scholar]
- (211).Zheng H; Zhu Y; Shi Y Palladium(0)-Catalyzed Heck Reaction/C–H Activation/Amination Sequence with Diaziridinone: A Facile Approach to Indolines. Angew. Chem., Int. Ed 2014, 53, 11280–11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (212).Jafarpour F; Jalalimanesh N; Teimouri M; Shamsianpour M Palladium/Norbornene Chemistry: An Unexpected Route to Methanocarbazole Derivatives via Three Csp3–Csp2/Csp3–N/Csp2–N Bond Formations in a Single Synthetic Sequence. Chem. Commun 2015, 51, 225–228. [DOI] [PubMed] [Google Scholar]
- (213).Ma X; Lu A; Ji X; Shi G; Zhang Y Disilylation of Palladacycles that were Generated through the C–H Activation of Aryl Halides. Asian J. Org. Chem 2018, 7, 1403–1410. [Google Scholar]
- (214).Lv W; Yu J; Ge B; Wen S; Cheng G Palladium-Catalyzed Catellani-Type Bis-silylation and Bis-germanylation of Aryl Iodides and Norbornenes. J. Org. Chem 2018, 83, 12683–12693. [DOI] [PubMed] [Google Scholar]
- (215).Xu Y; Liu X; Chen W; Deng G; Liang Y; Yang Y Palladium/Norbornene Chemistry: Synthesis of Norbornene-Containing Arylsilanes Involving Double C–Si Bond Formation. J. Org. Chem 2018, 83, 13930–13939. [DOI] [PubMed] [Google Scholar]
- (216).Hao T-T; Liang H-R; Ou-Yang Y-H; Yin C-Z; Zheng X-L; Yuan M-L; Li R-X; Fu H-Y; Chen H Palladium-Catalyzed Domino Reaction for Stereoselective Synthesis of Multisubstituted Olefins: Construction of Blue Luminogens. J. Org. Chem 2018, 83, 4441–4454. [DOI] [PubMed] [Google Scholar]
- (217).Jiao L; Bach T Palladium-Catalyzed Direct 2-Alkylation of Indoles by Norbornene-Mediated Regioselective Cascade C–H Activation. J. Am. Chem. Soc 2011, 133, 12990–12993. [DOI] [PubMed] [Google Scholar]
- (218).Jiao L; Herdtweck E; Bach T Pd(II)-Catalyzed Regioselective 2-Alkylation of Indoles via a Norbornene-Mediated C–H Activation: Mechanism and Applications. J. Am. Chem. Soc 2012, 134, 14563–14572. [DOI] [PubMed] [Google Scholar]
- (219).Jiao L; Bach T Palladium-Catalyzed Direct C–H Alkylation of Electron-Deficient Pyrrole Derivatives. Angew. Chem., Int. Ed 2013, 52, 6080–6083. [DOI] [PubMed] [Google Scholar]
- (220).Potukuchi HK; Bach T Selective C-2 Alkylation of Tryptophan by a Pd(II)/Norbornene-Promoted C–H Activation Reaction. J. Org. Chem 2013, 78, 12263–12267. [DOI] [PubMed] [Google Scholar]
- (221).Gao Y; Zhu W; Yin L; Dong B; Fu J; Ye Z; Xue F; Jiang C Palladium-Catalyzed Direct C2-Arylation of Free (N–H) Indoles via Norbornene-Mediated Regioselective C–H Activation. Tetrahedron Lett. 2017, 58, 2213–2216. [Google Scholar]
- (222).Zhang H; Wang H-Y; Luo Y; Chen C; Cao Y; Chen P; Guo Y-L; Lan Y; Liu G Regioselective Palladium-Catalyzed C–H Bond Trifluoroethylation of Indoles: Exploration and Mechanistic Insight. ACS Catal. 2018, 8, 2173–2180. [Google Scholar]
- (223).Wang X-C; Gong W; Fang L-Z; Zhu R-Y; Li SH; Engle KM; Yu J-Q Ligand-Enabled Meta-C–H Activation Using a Transient Mediator. Nature 2015, 519, 334–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (224).Dong Z; Wang J; Dong G Simple Amine-Directed Meta-Selective C–H Arylation via Pd/Norbornene Catalysis. J. Am. Chem. Soc 2015, 137, 5887–5890. [DOI] [PubMed] [Google Scholar]
- (225).Han J; Zhang L; Zhu Y; Zheng Y; Chen X; Huang Z-B; Shi D-Q; Zhao Y Highly Regioselective Meta Arylation of Oxalyl Amide-Protected β-Arylethylamine via the Catellani Reaction. Chem. Commun 2016, 52, 6903–6906. [DOI] [PubMed] [Google Scholar]
- (226).Wang P; Farmer ME; Huo X; Jain P; Shen P-X; Ishoey M; Bradner JE; Wisniewski SR; Eastgate MD; Yu J-Q Ligand-Promoted Meta-C–H Arylation of Anilines, Phenols, and Heterocycles. J. Am. Chem. Soc 2016, 138, 9269–9276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (227).Wang P; Farmer ME; Yu J-Q Ligand-Promoted Meta-C–H Functionalization of Benzylamines. Angew. Chem., Int. Ed 2017, 56, 5125–5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (228).Farmer ME; Wang P; Shi H; Yu J-Q Palladium-Catalyzed Meta-C–H Functionalization of Masked Aromatic Aldehydes. ACS Catal. 2018, 8, 7362–7367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (229).Wang P; Verma P; Xia G; Shi J; Qiao JX; Tao S; Cheng PT; Poss MA; Farmer ME; Yeung K-S; et al. Ligand-Accelerated Non-directed C–H Functionalization of Arenes. Nature 2017, 551, 489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (230).Li Q; Ferreira EM Meta-Selective C–H Arylation of Aromatic Alcohols with a Readily Attachable and Cleavable Molecular Scaffold. Chem. - Eur. J 2017, 23, 11519–11523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (231).Ding Q; Ye S; Cheng G; Wang P; Farmer ME; Yu J-Q Ligand-Enabled Meta-Selective C–H Arylation of Nosyl-Protected Phenethylamines, Benzylamines, and 2-Aryl Anilines. J. Am. Chem. Soc 2017, 139, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (232).Liu J; Ding Q; Fang W; Wu W; Zhang Y; Peng Y Pd(II)/Norbornene-Catalyzed Meta-C–H Alkylation of Nosyl-Protected Phenylalanines. J. Org. Chem 2018, 83, 13211–13216. [DOI] [PubMed] [Google Scholar]
- (233).Ling P-X; Chen K; Shi B-F Palladium-Catalyzed Interannular Meta-C–H Arylation. Chem. Commun 2017, 53, 2166–2169. [DOI] [PubMed] [Google Scholar]
- (234).Cheng G; Wang P; Yu J-Q Meta-C–H Arylation and Alkylation of Benzylsulfonamide Enabled by a Palladium(II)/Isoquinoline Catalyst. Angew. Chem., Int. Ed 2017, 56, 8183–8186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (235).Li G-C; Wang P; Farmer ME; Yu J-Q Ligand-Enabled Auxiliary-Free Meta-C–H Arylation of Phenylacetic Acids. Angew. Chem., Int. Ed 2017, 56, 6874–6877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (236).Shi H; Herron AN; Shao Y; Shao Q; Yu JQ Enantioselective Remote Meta-C–H Arylation and Alkylation via A Chiral Transient Mediator. Nature 2018, 558, 581–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (237).Wang P; Li G-C; Jain P; Farmer ME; He J; Shen P-X; Yu J-Q Ligand-Promoted Meta-C–H Amination and Alkynylation. J. Am. Chem. Soc 2016, 138, 14092–14099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (238).Wang P; Li G-C; Jain P; Farmer ME; He J; Shen P-X; Yu J-Q Ligand-Promoted Meta-C–H Amination and Alkynylation. J. Am. Chem. Soc 2016, 138, 14092–14099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (239).Shi H; Wang P; Suzuki S; Farmer ME; Yu J-Q Ligand Promoted Meta-C–H Chlorination of Anilines and Phenols. J. Am. Chem. Soc 2016, 138, 14876–14879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (240).Shi G; Shao C; Ma X; Gu Y; Zhang Y Pd(II)-Catalyzed Catellani-Type Domino Reaction Utilizing Arylboronic Acids as Substrates. ACS Catal. 2018, 8, 3775–3779. [Google Scholar]
- (241).Chen S; Liu Z-S; Yang T; Hua Y; Zhou Z; Cheng H-G; Zhou Q The Discovery of a Palladium(II)-Initiated Borono-Catellani Reaction. Angew. Chem., Int. Ed 2018, 57, 7161–7165. [DOI] [PubMed] [Google Scholar]
- (242).Li R; Liu F; Dong G Redox-Neutral Ortho Functionalization of Aryl Boroxines via Palladium/Norbornene Cooperative Catalysis. Chem. 2019, 5, 929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (243).Weinstabl H; Suhartono M; Qureshi Z; Lautens M Total Synthesis of (+)-Linoxepin by Utilizing the Catellani Reaction. Angew. Chem., Int. Ed 2013, 52, 5305–5308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (244).Qureshi Z; Weinstabl H; Suhartono M; Liu H; Thesmar P; Lautens M Application of the Palladium-Catalysed Norbornene-Assisted Catellani Reaction Towards the Total Synthesis of (+)-Linoxepin and Isolinoxepin. Eur. J. Org. Chem 2014, 2014, 4053–4069. [Google Scholar]
- (245).Zhao K; Xu S; Pan C; Sui X; Gu Z Catalytically Asymmetric Pd/Norbornene Catalysis: Enantioselective Synthesis of (+)-Rhazinal, (+)-Rhazinilam, and (+)-Kopsiyunnanine C1–3. Org. Lett 2016, 18, 3782–3785. [DOI] [PubMed] [Google Scholar]
- (246).Mizutani M; Yasuda S; Mukai C Total synthesis of (+)-kopsihainanine A. Chem. Commun 2014, 50, 5782–5785. [DOI] [PubMed] [Google Scholar]
- (247).Jiang S-Z; Zeng X-Y; Liang X; Lei T; Wei K; Yang Y-R Iridium-Catalyzed Enantioselective Indole Cyclization: Application to the Total Synthesis and Absolute Stereochemical Assignment of (−)-Aspidophylline A. Angew. Chem., Int. Ed 2016, 55, 4044–4048. [DOI] [PubMed] [Google Scholar]
- (248).Xiao T; Chen Z-T; Deng L-F; Zhang D; Liu X-Y; Song H; Qin Y Formal Total Synthesis of the Akuammiline Alkaloid (+)-Strictamine. Chem. Commun 2017, 53, 12665–12667. [DOI] [PubMed] [Google Scholar]
- (249).Nuth M; Guan HC; Zhukovskaya N; Saw YL; Ricciardi RP Design of Potent Poxvirus Inhibitors of the Heterodimeric Processivity Factor Required for Viral Replication. J. Med. Chem 2013, 56, 3235–3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (250).Alverez C; Bulfer SL; Chakrasali R; Chimenti MS; Deshaies RJ; Green N; Kelly M; LaPorte MG; Lewis TS; Liang M; et al. Allosteric Indole Amide Inhibitors of p97: Identification of a Novel Probe of the Ubiquitin Pathway. ACS Med. Chem. Lett 2016, 7, 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (251).Denny RA; Flick AC; Coe J; Langille J; Basak A; Liu S; Stock I; Sahasrabudhe P; Bonin P; Hay DA; et al. Structure-Based Design of Highly Selective Inhibitors of the CREB Binding Protein Bromodomain. J. Med. Chem 2017, 60, 5349–5363. [DOI] [PubMed] [Google Scholar]
- (252).Catellani M; Ferioli L An Improved Synthesis of 1,4-cis,exo-Hexa- or Tetrahydromethano- or -ethanobiphenylene Derivatives Catalyzed by Palladium Complexes. Synthesis 1996, 1996, 769–772. [Google Scholar]
- (253).Wu X; Zhou J An Efficient Method for the Heck-Catellani Reaction of Aryl Halides. Chem. Commun 2013, 49, 11035–11037. [DOI] [PubMed] [Google Scholar]
- (254).Liu S; Jin ZX; Teo YC; Xia Y Efficient Synthesis of Rigid Ladder Polymers via Palladium Catalyzed Annulation. J. Am. Chem. Soc 2014, 136, 17434–17437. [DOI] [PubMed] [Google Scholar]
- (255).Lai HWH; Liu S; Xia Y Norbornyl Benzocyclobutene Ladder Polymers: Conformation and Microporosity. J. Polym. Sci., Part A: Polym. Chem 2017, 55, 3075–3081. [Google Scholar]
- (256).Lai HWH; Teo YC; Xia Y Functionalized Rigid Ladder Polymers from Catalytic Arene-Norbornene Annulation Polymerization. ACS Macro Lett. 2017, 6, 1357–1361. [DOI] [PubMed] [Google Scholar]
- (257).Jin Z; Teo YC; Zulaybar NG; Smith MD; Xia Y Streamlined Synthesis of Polycyclic Conjugated Hydrocarbons Containing Cyclobutadienoids via C–H Activated Annulation and Aromatization. J. Am. Chem. Soc 2017, 139, 1806–1809. [DOI] [PubMed] [Google Scholar]
- (258).Jin Z; Teo YC; Teat SJ; Xia Y Regioselective Synthesis of [3]Naphthylenes and Tuning of Their Antiaromaticity. J. Am. Chem. Soc 2017, 139, 15933–15939. [DOI] [PubMed] [Google Scholar]
- (259).Jin Z; Teo YC; Teat SJ; Xia Y Iterative Synthesis of Edge-Bent [3]Naphthylene. Synlett 2018, 29, 2547–2551. [Google Scholar]
- (260).Teo YC; Jin Z; Xia Y Synthesis of Cyclobutadienoid-Fused Phenazines with Strongly Modulated Degrees of Antiaromaticity. Org. Lett 2018, 20, 3300–3304. [DOI] [PubMed] [Google Scholar]
- (261).Yoon K-Y; Dong G Modular In Situ Functionalization Strategy: Multicomponent Polymerization by Palladium/Norbornene Cooperative Catalysis. Angew. Chem., Int. Ed 2018, 57, 8592–8596. [DOI] [PubMed] [Google Scholar]
- (262).Yoon K-Y; Xue Y; Dong G Three-Step Synthesis of A Less-Aggregated Water-Soluble Poly(Para-Phenylene Ethynylene) with Meta-Side-Chains by Palladium/Norbornene Cooperative Catalysis. Macromolecules 2019, 52, 1663–1670. [Google Scholar]
- (263).Leow D; Li G; Mei T-S; Yu J-Q Activation of Remote Meta-C–H Bonds Assisted by an End-on Template. Nature 2012, 486, 518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (264).Lee S; Lee H; Tan KL Meta-Selective C–H Functionalization Using a Nitrile-Based Directing Group and Cleavable Si-Tether. J. Am. Chem. Soc 2013, 135, 18778–18781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (265).Tang R-Y; Li G; Yu J-Q Conformation-induced Remote Meta-C–H Activation of Amines. Nature 2014, 507, 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]