

Abstract
Background: Integrity of functional brain networks is closely associated with maintained cognitive performance at old age. Consistently, both carrier status of Apolipoprotein E ε4 allele (APOE4), and age-related aggregation of Alzheimer’s disease (AD) pathology result in altered brain network connectivity. The posterior cingulate and precuneus (PCP) is a node of particular interest due to its role in crucial memory processes. Moreover, the PCP is subject to the early aggregation of AD pathology. The current study aimed at characterizing brain network properties associated with unimpaired cognition in old aged adults. To determine the effects of age-related brain change and genetic risk for AD, pathological proteins β-amyloid and tau were measured by Positron-emission tomography (PET), PCP connectivity as a proxy of cognitive network integrity, and genetic risk by APOE4 carrier status.
Methods: Fifty-seven cognitively unimpaired old-aged adults (MMSE = 29.20 ± 1.11; 73 ± 8.32 years) were administered 11C Pittsburgh Compound B and 18F Flutemetamol PET for assessing β-amyloid, and 18F AV-1451 PET for tau. Individual functional connectivity seed maps of the PCP were obtained by resting-state multiband BOLD functional MRI at 3-Tesla for increased temporal resolution. Voxelwise correlations between functional connectivity, β-amyloid- and tau-PET were explored by Biological Parametric Mapping (BPM).
Results: Local β-amyloid was associated with increased connectivity in frontal and parietal regions of the brain. Tau was linked to increased connectivity in more spatially distributed clusters in frontal, parietal, occipital, temporal, and cerebellar regions. A positive interaction was observable for APOE4 carrier status and functional connectivity with brain regions characterized by increased local β-amyloid and tau tracer retention.
Conclusions: Our data suggest an association between spatially differing connectivity systems and local β-amyloid, and tau aggregates in cognitively normal, old-aged adults, which is moderated by APOE4. Additional longitudinal studies may determine protective connectivity patterns associated with healthy aging trajectories of AD-pathology aggregation.
Keywords: MRI, functional connectivity, PET, beta-amyloid, tau, multiband fMRI, preclinical, aging
Background
Aggregation of beta-amyloid (β-amyloid) and hyperphosphorylated tau are hallmarks of brain pathology associated with Alzheimer’s Disease (AD). The age-related accumulation of both proteins precedes disease onset by decades (Price and Morris, 1999; Bateman et al., 2012). Recently published data indicate a sequential association between brain β-amyloid accumulation, subsequent tau change and resulting cognitive decline (Hanseeuw et al., 2019). However, the precise impact of β-amyloid and pathological tau on brain functionality is not yet completely understood. Several studies demonstrated the validity of positron-emission-tomography (PET) tracers for inferring the relationship between AD-pathology burden, and risk for severe cognitive impairment at old age (Klunk, 2011; Villemagne et al., 2011; Vos et al., 2013). Distinct effects on brain network connectivity have been shown for both β-amyloid and pathological tau burden (Hansson et al., 2017; Hoenig et al., 2018; Korthauer et al., 2018; Franzmeier et al., 2019; Hodgetts et al., 2019). Functional connectivity changes within the “default mode network” (DMN) relate to AD-pathology and precede the manifestation of cognitive disorder by years (Sorg et al., 2007; Sperling et al., 2009; Wang et al., 2013; Sorg and Grothe, 2015). Moreover, large clinical studies demonstrate an association between AD-pathology and brain functionality in non-demented old-aged adults, as reflected by functional network integrity (Jack et al., 2019; Maass et al., 2019; Ossenkoppele et al., 2019). This may be consistent with functional network change in other neurodegenerative diseases (Rektorova et al., 2012; Ross et al., 2014; Kronenbuerger et al., 2019).
Despite the consensus in the literature that AD-pathology is related to a breakdown in functional brain networks, its associations with functional connectivity are tenuous. The relationship between β-amyloid and decreased connectivity, mainly in the DMN, is well established for clinically manifest AD (Hedden et al., 2009; Sheline et al., 2010; Chhatwal et al., 2013; Wang et al., 2013). However, during aging and in the absence of cognitive disorder, both increased functional connectivity (Mormino et al., 2011; Lim et al., 2014), but also decreased network connectivity (Wang et al., 2013) has been observed to be associated with AD-pathology when assessing total β-amyloid load, and tau burden.
The posterior cingulate and precuneus (PCP) represents a central node of the DMN needed for memory processing (Sperling et al., 2009; Cieri and Esposito, 2018). β-amyloid load affects the functionality of the PCP in old aged adults with, and without cognitive impairment (Unschuld et al., 2012; Schreiner et al., 2016; Grothe et al., 2017; Quevenco et al., 2017, 2019). The Apolipoprotein E ε4 allele (APOE4) is the strongest known genetic risk factor for late-onset AD (Corder et al., 1993; Strittmatter et al., 1993; Beffert and Poirier, 1996). In the general population, APOE4 carrier status is associated with an increased risk for the presence of AD-pathology (Schipper, 2011; Verghese et al., 2011; Liu et al., 2013; Jack et al., 2019; Yamazaki et al., 2019). Consistently, brain network alterations involving the PCP are pronounced in carriers of the APOE4 genotype (Sperling et al., 2009; De Vogelaere et al., 2012).
PET using tracers 18F-Flutemetamol and Pittsburgh Compound B (PiB) by now is a well-established neuroimaging technique for measuring individual β-amyloid plaque burden (Vandenberghe et al., 2010; Klunk, 2011; Frisoni and Blennow, 2013). Moreover, PET tau tracers such as 18F AV-1451 have enabled in vivo assessment of pathological, hyperphosphorylated tau burden in humans (Schöll et al., 2017; Mainta et al., 2018; Villemagne et al., 2018). These advances in PET technologies allow for investigating the relationship of β-amyloid and tau, whose interactions are considered to significantly contribute to disease progression and functional decline (Selkoe and Hardy, 2016; Tosun et al., 2017; Kametani and Hasegawa, 2018) Spatial distribution patterns of both proteins diverge substantially, with β-amyloid accumulation beginning in the neocortex and progressing towards subcortical structures (Braak and Braak, 1991a), whereas tau accumulation begins in the brainstem and transentorhinal regions and spreads towards neocortical structures as the disease progresses (Braak and Braak, 1991b). Considering these earlier reports, we hypothesized that PCP functional connectivity in cognitively normal old-aged adults might reflect the interplay between β-amyloid and tau pathology on a local level. Moreover, we hypothesized, that such effects should be more pronounced in APOE4 carriers. To test these hypotheses, the relationship between local β-amyloid, local tau and PCP connectivity was investigated on a voxel-level. We used the software biological parametric mapping (BPM; Casanova et al., 2007) for generating subject-specific PET tracer retention patterns and beta-maps (Whitfield-Gabrieli and Nieto-Castanon, 2012) as a measure of individual connectivity patterns (Biswal et al., 1995), associated with AD pathology. High temporal resolution of connectivity data was achieved by using multiband functional MRI (fMRI) for the simultaneous acquisition of multiple slices (Feinberg and Setsompop, 2013; Preibisch et al., 2015).
Materials and Methods
Study Population
For the current study, 57 old-aged cognitively unimpaired (mean MMSE = 29.20 ± 1.11), and medically healthy adults (mean age = 73 ± 8.32 years) were included from an on-going neuroimaging study at our institute, using study procedures reported earlier (van Bergen et al., 2018a; Vandenberghe et al., 2010). Written informed consent was obtained from all participants before inclusion in the study. Study procedures were carried out in accordance with the Good Clinical Practice (GCP) guidelines issued by the local ethics authority (Kantonale Ethikkommision Zürich1), and with the Human Research Act of Switzerland and the declaration of Helsinki (World_Medical_Association, 1991).
Inclusion criteria were age above 50 years, pre-existing PET information on cerebral amyloid deposition, German language proficiency and capacity to consent to study procedures. Exclusion criteria were: significant medication or drug abuse that might have possible effects on cognition, a history of severe allergic reaction or known allergy against components of 11C-PiB, 18-Flutemetamol, AV-1451, general MRI exclusion criteria, MRI scans with evidence of infection or infarction and severe atrophy, clinically relevant changes in red blood cell count, serious medical or neuropsychiatric illness and significant exposure to radiation.
Flutemetamol and PiB-PET for Estimation of β-amyloid Load
Individual measures of local β-amyloid burden were determined by quantification of standardized uptake values of either 18F-Flutemetamol (34 participants) or 11C-Pittsburgh Compound B (11C-PiB, 23 participants). PET was performed as reported in earlier studies of ours (Schreiner et al., 2014; Steininger et al., 2014; van Bergen et al., 2018a). A General Electric (GE) healthcare SIGNA PETMR was used for measuring 18F-Flutemetamol uptake, and a GE healthcare Discovery scanner for 11C-PiB. Average and standard deviation (SD) maps of all amyloid-PET SUVR are indicated in Supplementary Figures S1, S2. For the former, an individual dose of approximately 140 MBq of Flutemetamol was injected into the cubital vein. Time-of-flight algorithms and the required corrections were applied to reconstruct the PET images. Late-frame values (85–105 min) were referenced to the cerebellar gray matter (Vandenberghe et al., 2010) to obtain 3D volumes of Flutemetamol retention as an estimate β-amyloid load using standardized uptake value ratios (SUVR; matrix = 256 × 256 × 89, voxel size = 1.2 × 1.2 × 2.78 mm3). For the PiB protocol, approximately 350 MBq were administered intravenously and late-frame values (50–70 min) were used to obtain mean PiB uptake in all cortical VOIs and cerebellar regions using a volume-weighted averaging procedure (Gietl et al., 2015). Identical to the Flutemetamol protocol, these were normalized and referenced to the cerebellar gray matter to obtain SUVRs as estimates of the β-amyloid load.
AV1451-PET for the Estimation of Tau Load
The current study also included measures of tau load, by assessing local retention of 18F-AV1451 using an established PET procedure (Chien et al., 2013). A GE healthcare SIGNA PETMR was used for measuring 18F-AV1451 uptake. Average and SD maps of all tau-PET SUVR are indicated in Supplementary Figures S3, S4. In our study, an individual dose of approximately 200 MBq of 18F-AV1451 was administered into the cubital vein. Similar to the β-amyloid procedure, time-of-flight algorithms with the necessary corrections were applied to construct the tau-PET images based on regional uptake values. For analysis, 8 × 5 min frames were acquired. These were summed to late-frame values (80–120 min) and referenced to the cerebellar gray matter for generation of SUVRs.
MRI Acquisition
MR-Images were acquired on a 3-Tesla GE Discovery 750w MR whole-body scanner (GE Medical Systems, Milwaukee, WI, USA) equipped with a 32 Channel head coil array. A 3D T1-weighted (IR-SPGR) sequence (TI = 600 ms, TE = minimum, voxel size = 1 × 1 × 1 mm3, FOV = 256 × 256 × 256 mm3, flip angle = 8°, scan time = 5:49 min) was used to obtain structural images for anatomical referencing and automated image segmentation at the same visit the AV1451-PET was performed for tau. The average time between MRI and β-amyloid PET was 1,138 days, with a minimum of 49 days, and a maximum of 2,414 days.
Resting-state BOLD fMRI images were acquired using a 2D Gradient Echo Mux Multiband sequence (TR = 2,000 ms, TE = 30 ms, flip angle = 70°, slice thickness = 2.4 mm, number of slices = 22, number of MUXed slices (1–8) = 3, Autocalibrating Reconstruction for Cartesian imaging (ARC) acceleration factor = 2, ARC enabling = 1, scan time = 8:32 min) to obtain individual beta maps, indicating functional connectedness of each voxel to the PCP based on BOLD synchronicity (Biswal et al., 1995; Whitfield-Gabrieli and Nieto-Castanon, 2012). Average and SD maps PCC connectivity are indicated in Supplementary Figures S5, S6.
Pre-processing of Functional Images and Obtaining PCP Seed Maps
Functional images were spatially preprocessed using an in-house script implementing functions from the SPM12 toolbox2 with MATLAB 2016a (Mathworks, Natick, MA, USA). The following spatial preprocessing measures were covered in the script: (1) Functional image time-series were realigned using a 6-parameter rigid-body transformation for each image. (2) Anatomical (T1) scans were coregistered to the mean functional image. (3) Structural T1 images were segmented using SPM12’s Segment function in order to obtain tissue probability maps to warp the functional images into normalized MNI space. (4) Functional and structural images were normalized by using the forward deformations obtained in the previous step to warp the images from native into MNI space. (5) Normalized functional images were then smoothed with an 8 mm FWHM Gaussian kernel. Slice-time correction is not a necessity with multiband images due to the low TR, which reduces temporal differences between slices. In addition, as multiband acquires a high number of slices, the slight movement could have corrections done for the wrong slice. In order to avoid this complication, this step was omitted from spatial preprocessing. Normalized and smoothed functional and normalized T1 images were processed using the CONN v12 toolbox (Whitfield-Gabrieli and Nieto-Castanon, 2012). This toolbox uses the CompCor denoising method procedure, which regresses out nuisance regressors without including the global signal. After the denoising step, PCP seed maps were obtained using the CONN toolbox by calculating the correlation between the PCP and other regions of the brain.
Biological Parametric Mapping (BPM) for Voxel-Level Analysis
PET images were, based on their co-registered T1 image, aligned to the TAU PET/MR T1 image using the PMOD software, Version 3.8 (PMOD Technologies Limited, Zurich, Switzerland). T1 MRI images were normalized to MNI space and matrices were applied to the PET images, which were then scaled by cerebellum gray matter values derived from an intersected Hammers atlas derived gray matter region, which fit all normalized PET images. These were then smoothed with a 5 mm FWHM Gaussian kernel. Preprocessing of the functional images for BPM analysis required realignment, coregistration, segmentation and warping into MNI space and finally smoothed with an 8 mm FWHM Gaussian kernel using in-house scripts running on SPM12\footnotemark with MATLAB 2016a (Mathworks, Natick, MA, USA). The smoothed and normalized functional images were processed using the CONN v.12 toolbox (Whitfield-Gabrieli and Nieto-Castanon, 2012). Individual Fisher r-to-z transformed beta PCP seed connectivity maps were calculated for each participant. A second level t-test against zero was applied on the data and multiple test correction was performed at the voxel level, using FDR (p-FDR corrected, q < 0.05 for p-uncorrected = 0.001, Figure 1). To limit the analysis to the gray matter, images were masked using a multi-atlas matching approach (van Bergen et al., 2018b). Regression analysis of normalized and smoothed PET and fMRI images was performed by administering BPM3 (Casanova et al., 2007; Yang et al., 2011), a toolbox useful for exploring multimodal voxel-level correlations using the General Linear Model (GLM), running under Matlab 2010a and SPM54. The common spatial resolution of the images used in the analysis was determined by the spatial modality of PET (the lowest resolution) of approximately 3 mm. BPM was used to investigate regression models for investigating the relationship between local β-amyloid PET, tau-PET and DMN connectivity, as measured by beta maps, with a BPM cluster threshold set at 100 voxels. Additionally, the potential moderating effects of APOE4 carrier status were explored by multiple regression.
Figure 1.
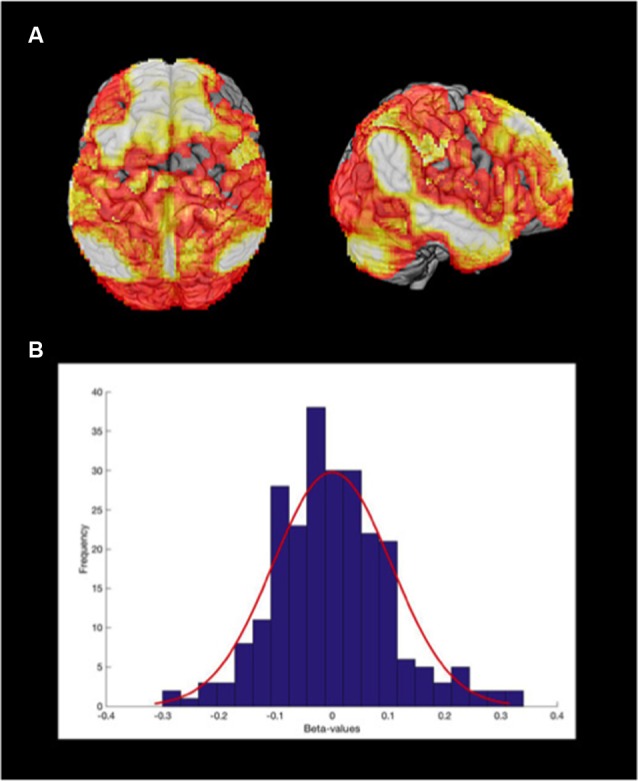
Posterior cingulate and precuneus (PCP) connectivity seed map of sample population. (A) Connectivity map of the whole sample (p-FDR < 0.05), using the PCP as a seed, projected on a 3D brain. (B) The median distribution of the Fisher r-to-z transformed beta maps.
Results
Neuropsychological Performance and APOE4 Carrier Frequency
All participants underwent a battery of neuropsychological tests including the Mini-Mental State Exam (MMSE), the Boston Naming, Trail-Making and the VLMT Delayed Recall test. Mini-Mental State (MMSE) scores were within the normal range (mean MMSE = 29.20 ± 1.11), and the other neuropsychological tests indicated consistently high performance of the studied population in the investigated domains (Supplementary Table S1). Moreover, genotyping identified 11 APOE4 carriers within the 57 study participants (19.3% of the population).
Local β-amyloid and Tau Deposits Are Associated With Locally Increased Functional Connectivity
By applying voxel-wise regression, eight positively associated clusters with a size of at least 100 voxels (Total voxels = 2,030, T > 2.3, p < 0.05) could be identified, indicating relationships between local β-amyloid load and increased PCP connectivity. Clusters were mainly located in the frontal and parietal lobe (Figure 2A), including the middle frontal gyrus, the anterior orbital gyrus (right), the medial orbital gyrus (right), the lateral orbital gyrus (right), the left precentral gyrus, the inferior lateral remainder of the parietal lobe (right) and other regions such as the left anterior temporal lobe (medial part) and the left cerebellum. No significant association between local β-amyloid burden and decreased PCP connectivity could be observed. Also, when applying voxel-wise regression, no significant relationship could be observed between age and gender, and PCP connectivity.
Figure 2.
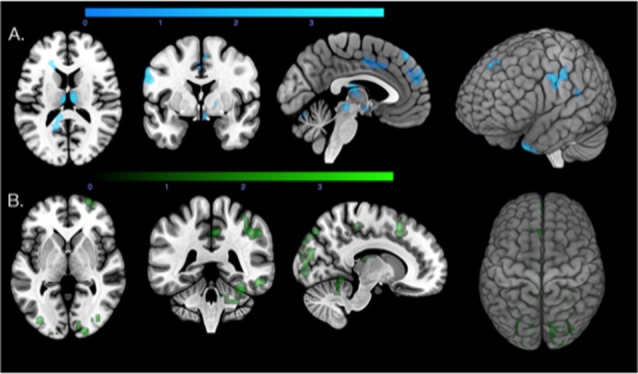
Regions that show correlations between local β-amyloid, local tau, and increased functional connectivity. (A) Axial, coronal, and sagittal slices and a 3D render of the T-map (p < 0.05, extent threshold = 100 voxels) illustrating the regions with correlations between local β-amyloid and increased connectivity (blue). (B) Axial, coronal, and sagittal slices and a 3D render of the T-map (p < 0.05, extent threshold = 100) illustrating the regions with correlations between local tau load (green) and increased functional connectivity.
For local tau associated PCP connectivity changes, the voxel-based analysis identified 14 positively associated clusters with a minimum size of 100 voxels (Total voxels = 5,662, T > 2.26, p < 0.05) widely distributed across the brain (Figure 2B). Regions primarily involved were thalamus, superior frontal gyrus, middle frontal gyrus, the inferolateral remainder of the parietal lobe (right), superior parietal gyrus, the lateral remainder of the occipital lobe, posterior temporal lobe and middle and inferior temporal gyrus. Smaller clusters included the cingulate gyrus and the right cerebellum.
There were small regions of overlap (257 voxels) between local β-amyloid associated connectivity and tau-related connectivity (Figure 3), representing ca. 12.66% of voxels included in the β-amyloid PCP connectivity map and 4.54% of the tau PCP connectivity map. The overlap between β-amyloid and tau-related connectivity was observable in particular for the left and right cerebellum, superior frontal gyrus and the inferolateral remainder of the parietal lobe (right).
Figure 3.
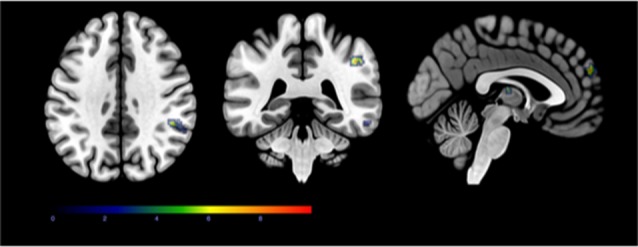
The minor regional overlap between local β-amyloid and increased PCP connectivity and local tau and increased PCP connectivity. Axial, coronal, and sagittal slices showing the small regional overlap of regions that are associated with local β-amyloid and increased PCP connectivity (p < 0.05, extent threshold = 100 voxels), and local tau-load and increased PCP connectivity (p < 0.05, extent threshold = 100 voxels).
APOE4 Carrier Status Moderates the Association Between β-amyloid and Tau Load With Functional Connectivity
APOE4 carrier status was included in the regression model as a non-imaging covariate and was found to have positive moderator effects on both β-amyloid and tau-related functional connectivity properties (Figure 4). APOE4 carrier status and local β-amyloid load were associated with increased PCP connectivity in 14 clusters of at least 100 voxels (Total voxels = 3,666, T > 2.80, p < 0.05). In contrast to the regression model with just β-amyloid and the Fisher r-to-z beta maps, which found regions with the strongest associations in the frontal lobe, APOE4 status and local β-amyloid were associated with more widely distributed PCP connectivity changes primarily in the temporal and frontal lobe (Figure 5A). These included the left anterior temporal lobe (medial and lateral part), posterior temporal lobe, right middle and inferior temporal gyrus, left middle frontal gyrus, left precentral gyrus, right medial orbital gyrus, and the right anterior orbital gyrus. Other regions included in the clusters were the right thalamus, the inferolateral remainder of the parietal lobe and the left lingual gyrus in the occipital lobe.
Figure 4.
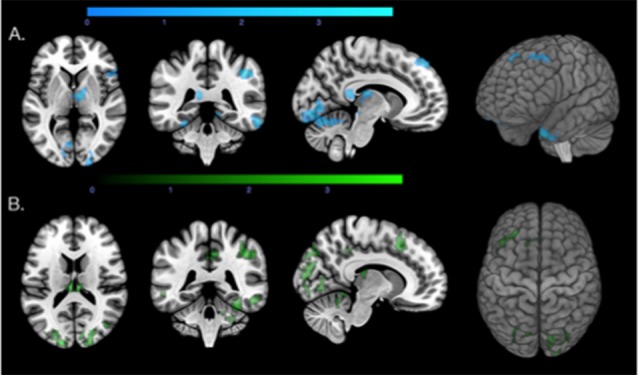
Regions with increased connectivity associated with local β-amyloid, local tau, and Apolipoprotein E ε4 allele (APOE4) carrier status. (A) Axial, coronal, and sagittal slices and a 3D view of the biological parametric mapping (BPM) T-map (p < 0.05, extent threshold = 100) illustrating regions with associations between local β-amyloid, increased functional connectivity, moderated by APOE4 carrier status (blue). (B) Axial, coronal, and sagittal slices and a 3D view of the BPM T-map (p < 0.05, extent threshold = 100) illustrating regions that showed associations between local tau, increased connectivity, moderated by APOE4 carrier status (green).
Figure 5.
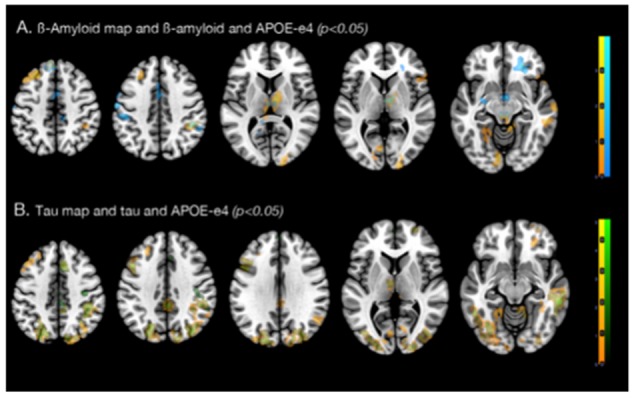
Effects of APOE4 carrier status on regional associations between increased connectivity with (A) local β-amyloid, and (B) local tau. (A) Axial slices of regions with correlations between local β-amyloid and increased connectivity (blue), and local β-amyloid, APOE4 carrier status and increased PCP connectivity (orange). (B) Axial slices of regions associated with local tau-load and the Fisher r-to-z transformed beta-maps (green), local tau-load, APOE4 carrier status and increased PCP connectivity (orange).
Local tau-tracer retention and APOE4 carrier status were associated with increased PCP connectivity in 13 clusters with at least 100 voxels in size (Total voxels = 7,045, T > 2.5, p < 0.05). Similar to the findings from the local tau and connectivity regression model, regions with strong associations between increased connectivity, local tau, and APOE4 carrier status were widely distributed across the brain (Figure 5B). Primary regions were located in the neocortex and also include other central structures, such as the cerebellum, the thalamus and caudate nucleus. These clusters enclosed brain regions within the right superior frontal gyrus, middle frontal gyrus, superior parietal gyrus, the lateral remainder of the occipital lobe, right middle and inferior temporal gyrus, and posterior temporal lobe. Similarly as with β-amyloid, no significant association between local retention of the tau-tracer and decreased PCP connectivity could be observed.
Discussion
By applying a multimodal voxel-wise regression approach to non-demented old-aged adults, we identified relationships between local β-amyloid and tau deposits with functional connectivity changes of the PCP. While local aggregation of both proteins was associated with distinct patterns of functional hyperconnectivity, the regional overlap was small. As we did find a positive moderator effect of APOE4 for both β-amyloid and tau related connectivity, we assume potential relevance for risk and progression of AD.
The majority of studies in the past have found decreased functional connectivity in the presence of AD (Sheline et al., 2010; Ouchi and Kikuchi, 2012; Song et al., 2013) and β-amyloid deposition (Sperling et al., 2010), particularly affecting the DMN and even reduction of connectivity in specific DMN nodes, such as the posterior cingulate (Weiler et al., 2014). There is however a handful of contradicting findings that observed increased connectivity and postulated that these changes may be exemplary of early-stage compensatory mechanisms in reaction to brain pathology (Mormino et al., 2011; Lim et al., 2014).
Though the reduction in connectivity is a prominent finding in the literature, our results coincide with a longitudinal PET connectivity study (Jack et al., 2013) and a recent β-amyloid and tau study (Schultz et al., 2017), that suggests that along the AD spectrum there are phases of hyper- and hypoconnectivity, with the former preceding the latter. Moreover, our findings are consistent with recent data on functional alterations associated with the presence of AD-pathology in non-demented old-aged adults (Jack et al., 2019; Maass et al., 2019; Ossenkoppele et al., 2019). Considering that the here investigated study population still had a low β-amyloid burden (Aβ−) and was cognitively relatively healthy, the observed increase in PCP connectivity may reflect the presence of pathological impairment prior to significant β-amyloid aggregation. The observed β-amyloid associated PCP connectivity was focal and significant clusters were located in the frontal and temporal lobe. Tau, however, was associated with increased activity in a wider distribution of regions across the brain. Only limited spatial overlap between PCP connectivity networks affected by β-amyloid and tau were observable. Overlap was present in the superior frontal gyrus, the cerebellum and the inferolateral remainder of the parietal lobe. While our data needs to be interpreted with caution due to the low level of PET tracer retention in an Aβ− population, possible off-target tau signal (Baker et al., 2019) and the possibility of slightly different attenuation correction due to the use of different PET scanners, our data nevertheless may accord with recent fMRI studies that suggest distinct PCP functional connectivity networks associated with β-amyloid and tau (Sepulcre et al., 2017; Franzmeier et al., 2019).
Interestingly, we found more clusters to be associated with local β-amyloid load and APOE4 carrier status than β-amyloid alone and these clusters are no longer primarily in the frontal lobe but cover many parts of the neocortex as well as other central structures, such as the cerebellum, thalamus and the caudate nucleus. While the addition of the genetic AD risk factor to the regression model better explains PCP associated increased connectivity in our data, this may be consistent with earlier reports on APOE4 associated connectivity changes in non-demented populations (Westlye et al., 2011; Wang et al., 2015; Hodgetts et al., 2019). However, this relationship was less prominent for local tau and PCP connectivity. Although adding APOE4 carrier status as an independent covariate did show some regions that were not affected with only tau alone, APOE4 effects on tau appeared less widespread. While our findings might support earlier reports on an association between APOE4 and tauopathy (Beffert and Poirier, 1996; Tiraboschi et al., 2004; Shi et al., 2017), in our study APOE4 moderated the relationship between local tau and functional connectivity to a lesser degree than β-amyloid.
Nonetheless, this observation could be a reflection of the relatively healthy status of our population, where neurodegenerative brain damage, indicated by increased tau, may still be very limited, resulting in low power for the detection of interactive effects between tau, APOE4 and functional connectivity. This may accord with the amyloid cascade hypothesis, which postulates that β-amyloid dysregulation precedes tau related neurodegeneration in preclinical stages of AD (Hardy and Selkoe, 2002). Furthermore, this study only finds relationships between β-amyloid and tau with increased PCP connectivity. Our finding of distinct functional impacts of β-amyloid and tau might be consistent with the notion of pathogenic synergism of protein aggregates in aging and neurodegenerative disease (Nelson et al., 2012). However, our findings may also reflect other non-AD related tauopathy, which might concur with very early stages of potential β-amyloid pathology in the studied population. Possible implications for therapeutic intervention strategies against age-related cognitive decline have been reviewed recently (Pini et al., 2018).
For the current study multiband fMRI was used, allowing for increased temporal resolution of resting-state data (Feinberg and Setsompop, 2013; Preibisch et al., 2015) while maintaining reproducibility of results with conventional EPI based fMRI protocols (Smitha et al., 2018). The increase in temporal resolution made possible by multiband fMRI has been suggested to allow for novel insights in brain network dynamics (Preibisch et al., 2015).
However, the cross-sectional nature and small sample size is a limitation of our study. Furthermore, due to the small sample size, further stratification of the studied population by AD-relevant properties is not compatible with the chosen statistical analysis strategy. While we did not find effects of gender and age, these might be present in a larger study population, particularly if a wider age range is included. Also, the specific investigation of APOE4 carriers, and also limiting the analysis to individuals with high cerebral β-amyloid (Aβ+ vs. Aβ−), and effects associated with tau progression beyond the entorhinal cortex, may provide additional insights on AD pathology (Braak and Braak, 1991b; Thal et al., 2002). While it is difficult to conclude whether our findings reflect pathological stages associated with either β-amyloid or tau, recent data suggest a sequential association of pathological change in AD (Hanseeuw et al., 2019). Prospective, longitudinal studies may include trajectories of gray matter integrity, as demonstrated recently for estimating neural tissue disintegration (Koini et al., 2018). Moreover, as we investigated a relatively healthy population, information on a relationship between connectivity-properties and structural change of vulnerable brain regions is limited. At present, 18F AV-1451 has become a popular choice for in vivo tau imaging (Villemagne et al., 2018; Mattsson et al., 2019; Smith et al., 2019). However, AV-1451 off-target binding unrelated to tau is a frequent finding particularly at low tracer retention, as observed in our study. Here, AV-1451 off-target signal may reflect iron deposition, neuromelanin, or vascular factors (Lockhart et al., 2017). This particularly affects brain regions such as the thalamus, putamen, superior cerebellar gray, choroid plexus, but also meninges and skull, posing challenges when studying early tau accumulation, as done in the current study (Baker et al., 2019). Functional coupling between the PCP and the PCC is well known, and our finding of an association with local β-amyloid may accord with earlier reports on altered functional brain changes during aging (Steininger et al., 2014; Li et al., 2017; Liu et al., 2017; Luo et al., 2019). However, considering that no significant cerebellar β-amyloid accumulation is to be expected in cognitively healthy individuals (Thal et al., 2002), also this finding needs further replication, and for now, needs to be interpreted with caution. Another limitation is the fact that there was significant temporal delay between β-amyloid PET and MRI, while tau PET and MRI took place at once. This difference needs to be taken into account when interpreting our findings on connectivity effects associated with β-amyloid vs. tau. However, β-amyloid effects may manifest in the long term, considering that aggregation of β-amyloid has been estimated to take about 19 years from β-amyloid positivity-threshold to mean values observed in AD dementia (Roberts et al., 2017).
Moreover, although the PCP is considered to play a central role in various cognitive processes associated with the DMN (Choo et al., 2010; Teipel et al., 2015; Quevenco et al., 2017; Cieri and Esposito, 2018), PCP seed map-related effects observed in this study may nevertheless primarily reflect PCP specific pathology than general alterations of the DMN. Follow-up studies using whole-brain connectivity approaches both at rest and during cognitive tasks may thus expand findings of the current study to investigate the DMN and also other brain networks in the context of AD-risk. Here, the advent of novel blood-based biomarkers might help to further characterize processes resulting in impaired brain functionality in preclinical AD (Bacioglu et al., 2016; Mattsson et al., 2017; Zetterberg and Blennow, 2018).
In conclusion, our findings are consistent with recently published studies that describe phases of PCP hyperconnectivity during the early stages of age-related β-amyloid and tau-related aggregation (Sepulcre et al., 2017). Considering the central role of the PCP for DMN connectivity, our study suggests that APOE4 carrier status, aggregation of β-amyloid and tau closely interact regarding their effects on DMN connectivity. Additional studies are needed to clarify connectivity in the context of the sequential progression of AD pathology (Hanseeuw et al., 2019), early functional change (McLaren et al., 2012), and potential opportunities for therapeutic intervention (Pini et al., 2018). The fact that these effects are observable before the manifestation of dementia, but still are related to APOE4 carrier status, may indicate relevance for the progression of early AD-pathology (Haller et al., 2019). Thus, further longitudinal studies are needed to investigate the temporal sequence of the observed changes, and whether they are associated with accelerated disease progression. Such studies may also inform on functional connectivity in a context of resilience against the aging-related accumulation of AD pathology. Possibly protective effects of functional connectivity patterns may allow for compensating emerging brain pathology during aging, allowing for preserved cognition.
Data Availability Statement
Openly available datasets generated for this study are included in the article/Supplementary Material.
Ethics Statement
The studies involving human participants were reviewed and approved by Kantonale Ethikkommission Zürich. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
FQ: performed data processing, analysis and interpretation of neuroimaging data and contributed to writing of the manuscript together with PU, and has performed a final revision. JB: established the multiband fMRI protocol on the GE scanner in Schlieren, and installed the pipeline for performing multimodal analyses using Biological Parametric Mapping. Also, he performed data processing of MR- and PET-data for integrated, voxel-level analysis. SS, AG, RM, and VT: contacted participants, administered MRI and PET. AG: coordinated the study and genotyping of APOE, interaction with ethics committee and acquisition of PET-MR data. SK: revised MR-data analysis and wrote the manuscript. RM and VT: supervised data acquisition (MRI and PET). VT and PK: supervised the preparation of the 18F-tracer for measuring brain Aβ-plaque density, quality control of PET-data and analysis. RN and CH are the chairmen of the department and sponsors of the study, they provided critical revisions of the final manuscript. PU: proposed the research question, supervised all data analysis, wrote the manuscript and is corresponding author. Also, he supervised the acquisition, processing, analysis, interpretation of acquired data and has performed the final revision.
Conflict of Interest
RN (CEO) and CH (CMO) are members of the board of directors at Neurimmune, Schlieren, Switzerland. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank all study participants for their time, enthusiasm and unsalaried commitment to thisresearch. We thank Esmeralda Gruber (IREM) for her assistance in participant recruitment.
Funding. This work was supported by the Clinical Research Priority Program (CRPP) of the University of Zurich (UZH) on Molecular Imaging (MINZ), Zurich Neuroscience Center (ZNZ), Zurich Program for Sustainable Development of Mental Health Services, Synapsis Foundation Alzheimer Research Switzerland, the Mäxi-Foundation and with institutional support from the Institute for Regenerative Medicine (IREM), University of Zurich, Switzerland.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2020.00046/full#supplementary-material.
Amyloid average over all PET scans performed (18F Flutemetamol and 11C PiB). Scaled from 0 to 2.8 SUVR.
Amyloid standard deviation (SD) over all PET scans performed (18F Flutemetamol and 11C PiB). Scaled from 0 to 0.8 SUVR SD.
Tau average over all PET scans performed (18F AV1451). Scaled from 1.0 to 2.0 SUVR.
Tau SD over all PET scans performed (18F AV1451). Scaled from 0 to 0.6 SUVR SD.
Beta-maps average, scaled from 0 to 1.0.
Beta-maps SD, scaled from 0 to 0.3.
References
- Bacioglu M., Maia L. F., Preische O., Schelle J., Apel A., Kaeser S. A., et al. (2016). Neurofilament light chain in blood and csf as marker of disease progression in mouse models and in neurodegenerative diseases. Neuron 91, 56–66. 10.1016/j.neuron.2016.05.018 [DOI] [PubMed] [Google Scholar]
- Baker S. L., Harrison T. M., Maass A., La Joie R., Jagust W. J. (2019). Effect of off-target binding on 18F-Flortaucipir variability in healthy controls across the life span. J. Nucl. Med. 60, 1444–1451. 10.2967/jnumed.118.224113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman R. J., Xiong C., Benzinger T. L., Fagan A. M., Goate A., Fox N. C., et al. (2012). Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 367, 795–804. 10.1056/NEJMoa1202753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beffert U., Poirier J. (1996). Apolipoprotein E, plaques, tangles and cholinergic dysfunction in Alzheimer’s disease. Ann. N Y Acad. Sci. 777, 166–174. 10.1111/j.1749-6632.1996.tb34415.x [DOI] [PubMed] [Google Scholar]
- Biswal B., Yetkin F. Z., Haughton V. M., Hyde J. S. (1995). Functional connectivity in the motor cortex of resting human brain using echo-planar MRI. Magn. Reson. Med. 34, 537–541. 10.1002/mrm.1910340409 [DOI] [PubMed] [Google Scholar]
- Braak H., Braak E. (1991a). Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathol. 1, 213–216. 10.1111/j.1750-3639.1991.tb00661.x [DOI] [PubMed] [Google Scholar]
- Braak H., Braak E. (1991b). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. 10.1007/bf00308809 [DOI] [PubMed] [Google Scholar]
- Casanova R., Srikanth R., Baer A., Laurienti P. J., Burdette J. H., Hayasaka S., et al. (2007). Biological parametric mapping: a statistical toolbox for multimodality brain image analysis. NeuroImage 34, 137–143. 10.1016/j.neuroimage.2006.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhatwal J. P., Schultz A. P., Johnson K., Benzinger T. L., Jack C., Jr., Ances B. M., et al. (2013). Impaired default network functional connectivity in autosomal dominant Alzheimer disease. Neurology 81, 736–744. 10.1212/WNL.0b013e3182a1aafe [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien D. T., Bahri S., Szardenings A. K., Walsh J. C., Mu F., Su M. Y., et al. (2013). Early clinical PET imaging results with the novel PHF-tau radioligand [F-18]-T807. J. Alzheimers Dis. 34, 457–468. 10.3233/jad-122059 [DOI] [PubMed] [Google Scholar]
- Choo I. H., Lee D. Y., Oh J. S., Lee J. S., Lee D. S., Song I. C., et al. (2010). Posterior cingulate cortex atrophy and regional cingulum disruption in mild cognitive impairment and Alzheimer’s disease. Neurobiol. Aging 31, 772–779. 10.1016/j.neurobiolaging.2008.06.015 [DOI] [PubMed] [Google Scholar]
- Cieri F., Esposito R. (2018). Neuroaging through the lens of the resting state networks. Biomed Res. Int. 2018:5080981. 10.1155/2018/5080981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder E. H., Saunders A. M., Strittmatter W. J., Schmechel D. E., Gaskell P. C., Small G. W., et al. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923. 10.1126/science.8346443 [DOI] [PubMed] [Google Scholar]
- De Vogelaere F., Santens P., Achten E., Boon P., Vingerhoets G. (2012). Altered default-mode network activation in mild cognitive impairment compared with healthy aging. Neuroradiology 54, 1195–1206. 10.1007/s00234-012-1036-6 [DOI] [PubMed] [Google Scholar]
- Feinberg D. A., Setsompop K. (2013). Ultra-fast MRI of the human brain with simultaneous multi-slice imaging. J. Magn. Reson. 229, 90–100. 10.1016/j.jmr.2013.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzmeier N., Rubinski A., Neitzel J., Kim Y., Damm A., Na D. L., et al. (2019). Functional connectivity associated with tau levels in ageing, Alzheimer’s, and small vessel disease. Brain 142, 1093–1107. 10.1093/brain/awz026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisoni G. B., Blennow K. (2013). Biomarkers for Alzheimer’s: the sequel of an original model. Lancet Neurol. 12, 126–128. 10.1016/s1474-4422(12)70305-8 [DOI] [PubMed] [Google Scholar]
- Gietl A. F., Warnock G., Riese F., Kalin A. M., Saake A., Gruber E., et al. (2015). Regional cerebral blood flow estimated by early PiB uptake is reduced in mild cognitive impairment and associated with age in an amyloid-dependent manner. Neurobiol. Aging 36, 1619–1628. 10.1016/j.neurobiolaging.2014.12.036 [DOI] [PubMed] [Google Scholar]
- Grothe M. J., Barthel H., Sepulcre J., Dyrba M., Sabri O., Teipel S. J., et al. (2017). in vivo staging of regional amyloid deposition. Neurology 89, 2031–2038. 10.1212/wnl.0000000000004643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller S., Montandon M. L., Rodriguez C., Garibotto V., Herrmann F. R., Giannakopoulos P. (2019). Hippocampal volume loss, brain amyloid accumulation and APOE status in cognitively intact elderly subjects. Neurodegener. Dis. [Epub ahead of print]. 10.1159/000504302 [DOI] [PubMed] [Google Scholar]
- Hanseeuw B. J., Betensky R. A., Jacobs H. I. L., Schultz A. P., Sepulcre J., Becker J. A., et al. (2019). Association of amyloid and tau with cognition in preclinical Alzheimer disease: a longitudinal study. JAMA Neurol. [Epub ahead of print]. 10.1001/jamaneurol.2019.1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson O., Grothe M. J., Strandberg T. O., Ohlsson T., Hagerstrom D., Jogi J., et al. (2017). Tau pathology distribution in Alzheimer’s disease corresponds differentially to cognition-relevant functional brain networks. Front. Neurosci. 11:167. 10.3389/fnins.2017.00167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J., Selkoe D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. 10.1126/science.1072994 [DOI] [PubMed] [Google Scholar]
- Hedden T., Van Dijk K. R., Becker J. A., Mehta A., Sperling R. A., Johnson K. A., et al. (2009). Disruption of functional connectivity in clinically normal older adults harboring amyloid burden. J. Neurosci. 29, 12686–12694. 10.1523/JNEUROSCI.3189-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgetts C. J., Shine J. P., Williams H., Postans M., Sims R., Williams J., et al. (2019). Increased posterior default mode network activity and structural connectivity in young adult APOE-epsilon4 carriers: a multimodal imaging investigation. Neurobiol. Aging 73, 82–91. 10.1016/j.neurobiolaging.2018.08.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoenig M. C., Bischof G. N., Seemiller J., Hammes J., Kukolja J., Onur O. A., et al. (2018). Networks of tau distribution in Alzheimer’s disease. Brain 141, 568–581. 10.1093/brain/awx353 [DOI] [PubMed] [Google Scholar]
- Jack C. R., Jr., Wiste H. J., Therneau T. M., Weigand S. D., Knopman D. S., Mielke M. M., et al. (2019). Associations of amyloid, tau, and neurodegeneration biomarker profiles with rates of memory decline among individuals without dementia. JAMA 321, 2316–2325. 10.1001/jama.2019.7437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack C. R., Jr., Wiste H. J., Weigand S. D., Knopman D. S., Lowe V., Vemuri P., et al. (2013). Amyloid-first and neurodegeneration-first profiles characterize incident amyloid PET positivity. Neurology 81, 1732–1740. 10.1212/01.wnl.0000435556.21319.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kametani F., Hasegawa M. (2018). Reconsideration of amyloid hypothesis and tau hypothesis in Alzheimer’s disease. Front. Neurosci. 12:25. 10.3389/fnins.2018.00025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk W. E. (2011). Amyloid imaging as a biomarker for cerebral β-amyloidosis and risk prediction for Alzheimer dementia. Neurobiol. Aging 32, S20–S36. 10.1016/j.neurobiolaging.2011.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koini M., Duering M., Gesierich B. G., Rombouts S., Ropele S., Wagner F., et al. (2018). Grey-matter network disintegration as predictor of cognitive and motor function with aging. Brain Struct. Funct. 223, 2475–2487. 10.1007/s00429-018-1642-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korthauer L. E., Zhan L., Ajilore O., Leow A., Driscoll I. (2018). Disrupted topology of the resting state structural connectome in middle-aged APOE ε4 carriers. NeuroImage 178, 295–305. 10.1016/j.neuroimage.2018.05.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronenbuerger M., Hua J., Bang J. Y. A., Ultz K. E., Miao X., Zhang X., et al. (2019). Differential changes in functional connectivity of striatum-prefrontal and striatum-motor circuits in premanifest Huntington’s disease. Neurodegener. Dis. 19, 78–87. 10.1159/000501616 [DOI] [PubMed] [Google Scholar]
- Li R., Yin S., Zhu X., Ren W., Yu J., Wang P., et al. (2017). Linking inter-individual variability in functional brain connectivity to cognitive ability in elderly individuals. Front. Aging Neurosci. 9:385. 10.3389/fnagi.2017.00385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim H. K., Nebes R., Snitz B., Cohen A., Mathis C., Price J., et al. (2014). Regional amyloid burden and intrinsic connectivity networks in cognitively normal elderly subjects. Brain 137, 3327–3338. 10.1093/brain/awu271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. C., Kanekiyo T., Xu H., Bu G. (2013). Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat. Rev. Neurol. 9, 106–118. 10.1038/nrneurol.2012.263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K., Yao S., Chen K., Zhang J., Yao L., Li K., et al. (2017). Structural brain network changes across the adult lifespan. Front. Aging Neurosci. 9:275. 10.3389/fnagi.2017.00275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockhart S. N., Ayakta N., Winer J. R., La Joie R., Rabinovici G. D., Jagust W. J. (2017). Elevated 18F-AV-1451 PET tracer uptake detected in incidental imaging findings. Neurology 88, 1095–1097. 10.1212/wnl.0000000000003724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo N., Sui J., Abrol A., Lin D., Chen J., Vergara V. M., et al. (2019). Age-related structural and functional variations in 5,967 individuals across the adult lifespan. Hum. Brain Mapp. [Epub ahead of print]. 10.1002/hbm.24905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maass A., Berron D., Harrison T. M., Adams J. N., La Joie R., Baker S., et al. (2019). Alzheimer’s pathology targets distinct memory networks in the ageing brain. Brain 142, 2492–2509. 10.1093/brain/awz154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainta I. C., Vargas M. I., Trombella S., Frisoni G. B., Unschuld P. G., Garibotto V. (2018). Hybrid PET-MRI in Alzheimer’s disease research. Methods Mol. Biol. 1750, 185–200. 10.1007/978-1-4939-7704-8_12 [DOI] [PubMed] [Google Scholar]
- Mattsson N., Andreasson U., Zetterberg H., Blennow K., Alzheimer’s Disease Neuroimaging I. (2017). Association of plasma neurofilament light with neurodegeneration in patients with Alzheimer disease. JAMA Neurol. 74, 557–566. 10.1001/jamaneurol.2016.6117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattsson N., Insel P. S., Donohue M., Jogi J., Ossenkoppele R., Olsson T., et al. (2019). Predicting diagnosis and cognition with 18F-AV-1451 tau PET and structural MRI in Alzheimer’s disease. Alzheimers Dement. 15, 570–580. 10.1016/j.jalz.2018.12.001 [DOI] [PubMed] [Google Scholar]
- McLaren D. G., Sreenivasan A., Diamond E. L., Mitchell M. B., Van Dijk K. R., Deluca A. N., et al. (2012). Tracking cognitive change over 24 weeks with longitudinal functional magnetic resonance imaging in Alzheimer’s disease. Neurodegener. Dis. 9, 176–186. 10.1159/000335876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mormino E. C., Smiljic A., Hayenga A. O., Onami S. H., Greicius M. D., Rabinovici G. D., et al. (2011). Relationships between β-amyloid and functional connectivity in different components of the default mode network in aging. Cereb. Cortex 21, 2399–2407. 10.1093/cercor/bhr025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson P. T., Alafuzoff I., Bigio E. H., Bouras C., Braak H., Cairns N. J., et al. (2012). Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J. Neuropathol. Exp. Neurol. 71, 362–381. 10.1097/NEN.0b013e31825018f7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossenkoppele R., Iaccarino L., Schonhaut D. R., Brown J. A., La Joie R., O’Neil J. P., et al. (2019). Tau covariance patterns in Alzheimer’s disease patients match intrinsic connectivity networks in the healthy brain. NeuroImage Clin. 23:101848. 10.1016/j.nicl.2019.101848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouchi Y., Kikuchi M. (2012). A review of the default mode network in aging and dementia based on molecular imaging. Rev. Neurosci. 23, 263–268. 10.1515/revneuro-2012-0029 [DOI] [PubMed] [Google Scholar]
- Pini L., Manenti R., Cotelli M., Pizzini F. B., Frisoni G. B., Pievani M. (2018). Non-invasive brain stimulation in dementia: a complex network story. Neurodegener. Dis. 18, 281–301. 10.1159/000495945 [DOI] [PubMed] [Google Scholar]
- Preibisch C., Castrillon G. J., Buhrer M., Riedl V. (2015). Evaluation of multiband EPI acquisitions for resting state fMRI. PLoS One 10:e0136961. 10.1371/journal.pone.0136961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price J. L., Morris J. C. (1999). Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann. Neurol. 45, 358–368. [DOI] [PubMed] [Google Scholar]
- Quevenco F. C., Preti M. G., van Bergen J. M., Hua J., Wyss M., Li X., et al. (2017). Memory performance-related dynamic brain connectivity indicates pathological burden and genetic risk for Alzheimer’s disease. Alzheimers Res. Ther. 9:24. 10.1186/s13195-017-0249-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quevenco F. C., Schreiner S. J., Preti M. G., van Bergen J. M. G., Kirchner T., Wyss M., et al. (2019). GABA and glutamate moderate β-amyloid related functional connectivity in cognitively unimpaired old-aged adults. NeuroImage Clin. 22:101776. 10.1016/j.nicl.2019.101776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rektorova I., Krajcovicova L., Marecek R., Mikl M. (2012). Default mode network and extrastriate visual resting state network in patients with Parkinson’s disease dementia. Neurodegener. Dis. 10, 232–237. 10.1159/000334765 [DOI] [PubMed] [Google Scholar]
- Roberts B. R., Lind M., Wagen A. Z., Rembach A., Frugier T., Li Q. X., et al. (2017). Biochemically-defined pools of amyloid-β in sporadic Alzheimer’s disease: correlation with amyloid PET. Brain 140, 1486–1498. 10.1093/brain/awx057 [DOI] [PubMed] [Google Scholar]
- Ross C. A., Aylward E. H., Wild E. J., Langbehn D. R., Long J. D., Warner J. H., et al. (2014). Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat. Rev. Neurol. 10, 204–216. 10.1038/nrneurol.2014.24 [DOI] [PubMed] [Google Scholar]
- Schipper H. M. (2011). Presymptomatic apolipoprotein E genotyping for Alzheimer’s disease risk assessment and prevention. Alzheimers Dement. 7, e118–e123. 10.1016/j.jalz.2010.06.003 [DOI] [PubMed] [Google Scholar]
- Schöll M., Ossenkoppele R., Strandberg O., Palmqvist S., Swedish BioFINDER study. Jögi J., et al. (2017). Distinct 18F-AV-1451 tau PET retention patterns in early- and late-onset Alzheimer’s disease. Brain 140, 2286–2294. 10.1093/brain/awx171 [DOI] [PubMed] [Google Scholar]
- Schreiner S. J., Kirchner T., Wyss M., Van Bergen J. M., Quevenco F. C., Steininger S. C., et al. (2016). Low episodic memory performance in cognitively normal elderly subjects is associated with increased posterior cingulate gray matter N-acetylaspartate: a 1H MRSI study at 7 Tesla. Neurobiol. Aging 48, 195–203. 10.1016/j.neurobiolaging.2016.08.022 [DOI] [PubMed] [Google Scholar]
- Schreiner S. J., Liu X., Gietl A. F., Wyss M., Steininger S. C., Gruber E., et al. (2014). Regional fluid-attenuated inversion recovery (FLAIR) at 7 Tesla correlates with amyloid-β in hippocampus and brainstem of cognitively normal elderly subjects. Front. Aging Neurosci. 6:240. 10.3389/fnagi.2014.00240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz A. P., Chhatwal J. P., Hedden T., Mormino E. C., Hanseeuw B. J., Sepulcre J., et al. (2017). Phases of hyperconnectivity and hypoconnectivity in the default mode and salience networks track with amyloid and tau in clinically normal individuals. J. Neurosci. 37, 4323–4331. 10.1523/JNEUROSCI.3263-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe D. J., Hardy J. (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8, 595–608. 10.15252/emmm.201606210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepulcre J., Sabuncu M. R., Li Q., El Fakhri G., Sperling R., Johnson K. A. (2017). Tau and amyloid-β proteins distinctively associate to functional network changes in the aging brain. Alzheimers Dement. 13, 1261–1269. 10.1016/j.jalz.2017.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheline Y. I., Raichle M. E., Snyder A. Z., Morris J. C., Head D., Wang S., et al. (2010). Amyloid plaques disrupt resting state default mode network connectivity in cognitively normal elderly. Biol. Psychiatry 67, 584–587. 10.1016/j.biopsych.2009.08.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y., Yamada K., Liddelow S. A., Smith S. T., Zhao L., Luo W., et al. (2017). ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527. 10.1038/nature24016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R., Wibom M., Pawlik D., Englund E., Hansson O. (2019). Correlation of in vivo [18F]Flortaucipir with postmortem alzheimer disease tau pathology. JAMA Neurol. 76, 310–317. 10.1001/jamaneurol.2018.3692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smitha K. A., Arun K. M., Rajesh P. G., Joel S. E., Venkatesan R., Thomas B., et al. (2018). Multiband fMRI as a plausible, time-saving technique for resting-state data acquisition: study on functional connectivity mapping using graph theoretical measures. Magn. Reson. Imaging 53, 1–6. 10.1016/j.mri.2018.06.013 [DOI] [PubMed] [Google Scholar]
- Song X., Mitnitski A., Zhang N., Chen W., Rockwood K., Alzheimer’s Disease Neuroimaging I. (2013). Dynamics of brain structure and cognitive function in the Alzheimer’s disease neuroimaging initiative. J. Neurol. Neurosurg. Psychiatry 84, 71–78. 10.1136/jnnp-2012-303579 [DOI] [PubMed] [Google Scholar]
- Sorg C., Grothe M. J. (2015). The complex link between amyloid and neuronal dysfunction in Alzheimer’s disease. Brain 138, 3472–3475. 10.1093/brain/awv302 [DOI] [PubMed] [Google Scholar]
- Sorg C., Riedl V., Muhlau M., Calhoun V. D., Eichele T., Laer L., et al. (2007). Selective changes of resting-state networks in individuals at risk for Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 104, 18760–18765. 10.1073/pnas.0708803104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling R. A., Dickerson B. C., Pihlajamaki M., Vannini P., LaViolette P. S., Vitolo O. V., et al. (2010). Functional alterations in memory networks in early Alzheimer’s disease. Neuromolecul. Med. 12, 27–43. 10.1007/s12017-009-8109-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling R. A., Laviolette P. S., O’Keefe K., O’Brien J., Rentz D. M., Pihlajamaki M., et al. (2009). Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron 63, 178–188. 10.1016/j.neuron.2009.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steininger S. C., Liu X., Gietl A., Wyss M., Schreiner S., Gruber E., et al. (2014). Cortical amyloid β in cognitively normal elderly adults is associated with decreased network efficiency within the cerebro-cerebellar system. Front. Aging Neurosci. 6:52. 10.3389/fnagi.2014.00052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter W. J., Saunders A. M., Schmechel D., Pericak-Vance M., Enghild J., Salvesen G. S., et al. (1993). Apolipoprotein E: high-avidity binding to β-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. U S A 90, 1977–1981. 10.1073/pnas.90.5.1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teipel S., Grothe M. J., Alzheimer’s Disease Neuroimaging I. (2015). Does posterior cingulate hypometabolism result from disconnection or local pathology across preclinical and clinical stages of Alzheimer’s disease? Eur. J. Nucl. Med. Mol. Imaging 43, 526–536. 10.1007/s00259-015-3222-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal D. R., Rüb U., Orantes M., Braak H. (2002). Phases of A β-deposition in the human brain and its relevance for the development of AD. Neurology 58, 1791–1800. 10.1212/wnl.58.12.1791 [DOI] [PubMed] [Google Scholar]
- Tiraboschi P., Hansen L. A., Masliah E., Alford M., Thal L. J., Corey-Bloom J. (2004). Impact of APOE genotype on neuropathologic and neurochemical markers of Alzheimer disease. Neurology 62, 1977–1983. 10.1212/01.wnl.0000128091.92139.0f [DOI] [PubMed] [Google Scholar]
- Tosun D., Landau S., Aisen P. S., Petersen R. C., Mintun M., Jagust W., et al. (2017). Association between tau deposition and antecedent amyloid-β accumulation rates in normal and early symptomatic individuals. Brain 140, 1499–1512. 10.1093/brain/awx046 [DOI] [PubMed] [Google Scholar]
- Unschuld P. G., Edden R. A., Carass A., Liu X., Shanahan M., Wang X., et al. (2012). Brain metabolite alterations and cognitive dysfunction in early Huntington’s disease. Mov. Disord. 27, 895–902. 10.1002/mds.25010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bergen J. M. G., Li X., Quevenco F. C., Gietl A. F., Treyer V., Leh S. E., et al. (2018a). Low cortical iron and high entorhinal cortex volume promote cognitive functioning in the oldest-old. Neurobiol. Aging 64, 68–75. 10.1016/j.neurobiolaging.2017.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bergen J. M. G., Li X., Quevenco F. C., Gietl A. F., Treyer V., Meyer R., et al. (2018b). Simultaneous quantitative susceptibility mapping and Flutemetamol-PET suggests local correlation of iron and β-amyloid as an indicator of cognitive performance at high age. NeuroImage 174, 308–316. 10.1016/j.neuroimage.2018.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberghe R., Van Laere K., Ivanoiu A., Salmon E., Bastin C., Triau E., et al. (2010). 18F-flutemetamol amyloid imaging in Alzheimer disease and mild cognitive impairment: a phase 2 trial. Ann. Neurol. 68, 319–329. 10.1002/ana.22068 [DOI] [PubMed] [Google Scholar]
- Verghese P. B., Castellano J. M., Holtzman D. M. (2011). Apolipoprotein E in Alzheimer’s disease and other neurological disorders. Lancet Neurol. 10, 241–252. 10.1016/S1474-4422(10)70325-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villemagne V. L., Dore V., Burnham S. C., Masters C. L., Rowe C. C. (2018). Imaging tau and amyloid-β proteinopathies in Alzheimer disease and other conditions. Nat. Rev. Neurol. 14:446. 10.1038/s41582-018-0021-z [DOI] [PubMed] [Google Scholar]
- Villemagne V. L., Pike K. E., Chetelat G., Ellis K. A., Mulligan R. S., Bourgeat P., et al. (2011). Longitudinal assessment of Aβ and cognition in aging and Alzheimer disease. Ann. Neurol. 69, 181–192. 10.1002/ana.22248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos S. J., Xiong C., Visser P. J., Jasielec M. S., Hassenstab J., Grant E. A., et al. (2013). Preclinical Alzheimer’s disease and its outcome: a longitudinal cohort study. Lancet Neurol. 12, 957–965. 10.1016/S1474-4422(13)70194-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Brier M. R., Snyder A. Z., Thomas J. B., Fagan A. M., Xiong C., et al. (2013). Cerebrospinal fluid Aβ42, phosphorylated Tau181, and resting-state functional connectivity. JAMA Neurol. 70, 1242–1248. 10.1001/jamaneurol.2013.3253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Wang X., He Y., Yu X., Wang H., He Y. (2015). Apolipoprotein E ε4 modulates functional brain connectome in Alzheimer’s disease. Hum. Brain Mapp. 36, 1828–1846. 10.1002/hbm.22740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiler M., Teixeira C. V., Nogueira M. H., de Campos B. M., Damasceno B. P., Cendes F., et al. (2014). Differences and the relationship in default mode network intrinsic activity and functional connectivity in mild Alzheimer’s disease and amnestic mild cognitive impairment. Brain Connect. 4, 567–574. 10.1089/brain.2014.0234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westlye E. T., Lundervold A., Rootwelt H., Lundervold A. J., Westlye L. T. (2011). Increased hippocampal default mode synchronization during rest in middle-aged and elderly APOE ε4 carriers: relationships with memory performance. J. Neurosci. 31, 7775–7783. 10.1523/jneurosci.1230-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield-Gabrieli S., Nieto-Castanon A. (2012). Conn: a functional connectivity toolbox for correlated and anticorrelated brain networks. Brain Connect. 2, 125–141. 10.1089/brain.2012.0073 [DOI] [PubMed] [Google Scholar]
- World_Medical_Association (1991). Declaration of Helsinki. Law Med Health Care 19, 264–265. [PubMed] [Google Scholar]
- Yamazaki Y., Zhao N., Caulfield T. R., Liu C. C., Bu G. (2019). Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat. Rev. Neurol. 15, 501–518. 10.1038/s41582-019-0228-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Beason-Held L., Resnick S. M., Landman B. A. (2011). Biological parametric mapping with robust and non-parametric statistics. NeuroImage 57, 423–430. 10.1016/j.neuroimage.2011.04.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetterberg H., Blennow K. (2018). From cerebrospinal fluid to blood: the third wave of fluid biomarkers for Alzheimer’s disease. J. Alzheimers Dis. 64, S271–S279. 10.3233/JAD-179926 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Amyloid average over all PET scans performed (18F Flutemetamol and 11C PiB). Scaled from 0 to 2.8 SUVR.
Amyloid standard deviation (SD) over all PET scans performed (18F Flutemetamol and 11C PiB). Scaled from 0 to 0.8 SUVR SD.
Tau average over all PET scans performed (18F AV1451). Scaled from 1.0 to 2.0 SUVR.
Tau SD over all PET scans performed (18F AV1451). Scaled from 0 to 0.6 SUVR SD.
Beta-maps average, scaled from 0 to 1.0.
Beta-maps SD, scaled from 0 to 0.3.
Data Availability Statement
Openly available datasets generated for this study are included in the article/Supplementary Material.