

Abstract
α-Boryl ethers, carbonates, and acetals, readily prepared from the corresponding alcohols that are accessed through ketone diboration, react rapidly with hydrogen peroxide to release alcohols, aldehydes, and ketones through the collapse of hemiacetal intermediates. Experiments with α-boryl acetals containing a latent fluorophore clearly demonstrate that cargo can be released inside cells in the presence of exogenous or endogenous hydrogen peroxide. These experiments show that this protocol can be used for drug activation in an oxidative environment without generating toxic byproducts.
Graphical Abstract

INTRODUCTION
Reactive oxygen species (ROS), including hydrogen peroxide, are linked to a number of disparate medical conditions including neurological diseases,1 cancer,2 aging,3 and diabetes.4 ROS-rich environments are also created through exposure to ionizing radiation, as encountered in radiotherapy.5 Hydrogen peroxide’s unique reactivity properties and importance in these conditions have resulted in its utilization to initiate a number of processes in biological and materials chemistry. Initial studies from Chang’s group demonstrated that aryl boronates can be converted to fluorescent phenols by cellular H2O2.6 This result, coupled with Lo’s employment of the boronate to phenol conversion to effect benzylic leaving group departure,7 led to the development of numerous compounds that release fluorophores and other diagnostic tools in oxidatively stressed cells.8 Additional applications of oxidatively triggered self-immolative spacers9 have been developed to promote particle breakdown10 and signal amplification.11 H2O2 is an attractive agent for initiating prodrug unraveling in many cases because it is small and can access sterically hindered sites in structures that are inaccessible to enzymes, which are commonly utilized for this purpose. Cargo release from antibodies12 serves as an example of a process that can benefit from activation by a small molecule. Peroxide-mediated drug release has been explored to a limited extent.13 However, substrates for these processes employ aryl or vinyl boronates as oxidative triggers to promote release from the benzylic or allylic position. Therapeutic applications of these systems, therefore, can be complicated by the significant toxicity of the resultant quinone methide14 or acrolein15 byproducts. Thus, alternative structural motifs that release compounds in the presence of H2O2 without generating toxic byproducts would be valuable for applications in oxidative drug release.
We have initiated a program with the objective of designing readily accessible structures that have the capacity to localize toward a cellular target and decompose under oxidative conditions to release a biological effector. Our initial design for alcohol release13b (Scheme 1) employed acyl aminal substrates (1) that are available through reductive multicomponent unions of nitriles, chloroformates, and alcohols.16 Aryl or vinyl boronate oxidation with H2O2 releases a quinone methide or acrolein and CO2 to form an unstable hemiaminal (2) that collapses to release the alcohol. We reasoned that oxidation of α-boryl ethers or carbonates (3) would provide a similar unstable hemiacetal (4) that releases an alcohol directly or through carbonate breakdown with less byproduct generation. This approach allows for the selection of a non-toxic ketone byproduct to serve as a guide in substrate design.
Scheme 1.

Alcohol Release through Boronate Oxidation
This manuscript describes the realization of this approach through the release of several diverse structures via oxidative fragmentation of α-boryl ethers, carbonates, and acetals. Specific advances include (1) the development of experiment-tally facile conditions for the synthesis of α-boryl alcohols through a variant of a known ketone diboration protocol, (2) the preparation of α-boryl ethers and carbonates through conditions that avoid strong base, (3) the demonstration that α-boryl ethers decompose rapidly and efficiently in the presence of H2O2 under mildly basic conditions while α-boryl carbonates decompose more slowly, 4) the elaboration of several protocols for preparing cyclic boryl-substituted acetals, (5) the observation that the acetals can liberate aldehydes and ketones in the presence of H2O2, (6) the application of the acetal breakdown to release fluorophores at low substrate and peroxide concentrations, (7) the validation of the capacity of the acetals to release cargo in cells through stimulation with exogenous H2O2, and (8) the demonstration that cargo can be released in cells by endogenous H2O2 resulting from chemically stimulated oxidative stress. These results clearly illustrate that α-boryl ethers, carbonates, and acetals are viable substrates for releasing biological effectors in cells in response to oxidative conditions while avoiding the generation of toxic byproducts.
RESULTS AND DISCUSSION
Ether, Carbonate, and Cyclic Acetal Substrate Synthesis and Decomposition.
The success of this project was contingent upon identifying suitable approach to α-boryl alcohol formation.17 We initially employed Clark’s ketone-relevant variation18 of the Sadighi carbonyl diboration protocol19 for the conversion of 5 to boryl alcohol 6 (Scheme 2).20 These conditions (PinB–BPin, (ICy)CuCl, NaOt-Bu, PhMe, 50 °C followed by borate protodeboration on silica gel) provided 6 but were deemed unacceptable due to the low reaction rate and the technical difficulty associated with the need to initiate the reaction in a glovebox. We reasoned that the relevant copper carbene catalyst could be prepared in situ by deprotonating the imidazolium salt in the presence of CuCl, thereby obviating the need to isolate this sensitive species. Moreover, adding MeOH to the reaction mixture substantially increased the rate of the reaction, in accord with Molander’s observations.21 These changes resulted in the conversion of 5 to 6 in 82% yield within 1 h and without recourse to glovebox or Schlenk line techniques. The experimental facility of this protocol appreciably enhances access to α-boryl alcohols. This is significant because boronates and related species with α-heteroatom substitution are useful as substrates for cross-coupling21 and chain-elongation reactions22 and as surrogates of functionalized carboxylic acids for applications in medicinal chemistry.23 The hydroxy groups can be functionalized readily, as demonstrated through the formation of methoxymethyl ether 7 and phenyl carbonate 8.
Scheme 2.

α-Boryl Alcohol Synthesis and Functionalization
The oxidative breakdown of compounds 7 and 8 was achieved by subjecting them (~25 mM) to urea·H2O2 (300 mM) in a mixture of CD3CN and aqueous (D2O) buffer (pH 8.0). The buffer was selected to mimic the experimentally determined pH of mitochondria24 in consideration of potential applications to mitigating neuronal oxidative stress. Initial experiments were conducted in a 5:1 ratio of CD3CN and buffer (Scheme 3). Reaction progress was monitored by 1H NMR through following the disappearance of the signals for diastereotopic hydrogens from the methylene group in the starting materials and the appearance of the corresponding enantiotopic hydrogens in butanone. Conversions were calculated by comparison to the internal standard 1,2-dimethoxyethane.
Scheme 3.

Oxidative Alcohol Release
Methoxymethyl ether 7 fragmented quite rapidly in the presence of hydrogen peroxide. Over 50% of the starting material was consumed in less than 2 min (Figure 1A), and complete conversion was observed within 20 min with an 89% NMR yield of butanone. Changing the solvent to a 1:1 ratio of CD3CN to buffer did not slow the reaction and resulted in a slightly increased NMR yield of 94%. Moreover lowering the pH to a cytosolic-relevant value of 7.2 had only a minimal effect on the rate despite the diminished peroxy anion concentration (Figure 1B), providing a 91% NMR yield of butanone.
Figure 1.

Oxidative breakdown of 7. (A) Reaction progress as determined by 1H NMR. (B) Reaction progress as a function of pH.
Carbonate 8, however, broke down much more slowly under the oxidative conditions.25 Consumption of 50% of the starting material required 22 min when the reaction was conducted in a 5:1 mixture of CD3CN and buffer. Changing the solvent to a 1:1 mixture of CD3CN and buffer resulted in a slightly increased rate, with 50% of the starting material being consumed within 12 min. The reactions were quite efficient, with both providing an 84% yield of the desired products.
The rate difference for alcohol release between acetal and carbonate substrates indicates that the rate-determining step in these processes is boronate oxidation rather than hemiacetal collapse. The slow breakdown of the carbonate could result from intramolecular coordination between the carbonyl oxygen and the boron, as illustrated by 9 (Figure 2) thereby inhibiting the approach of HOO− to the boron. Crystal structures show this type of coordination in α-boryl amides,26 and intramolecular coordination has been shown to confer stability to boronates.27 However, the 11B chemical shift of 8 (δ 32.2 ppm) is nearly identical to the 11B chemical shift in 7 (δ 32.1 ppm) and is significantly different from that of amido pinacolboronates, which show 11B chemical shifts of approximately 15 ppm.26,28 Alternatively the breakdown could be slowed by a diminished migratory aptitude resulting from the presence of an electron withdrawing acyl group. No evidence of a persistent peroxyboronate intermediate, such as 10, was observed upon monitoring the progress of the reaction with 11B NMR, however. Regardless of the origin of the effect, the capacity to control the breakdown rate through a simple structural modification provides kinetic versatility in drug release strategy.
Figure 2.

Structures 9 and 10 as potential origins for the slow breakdown of 8.
Several additional substrates were prepared to define the scope of the process (Table 1). Secondary alcohols such as cyclohexanol (from the breakdown of 11) and the more complex menthol (from the breakdown of 13) are released smoothly. Although the formation and fragmentation of alkoxymethyl ethers proceed rapidly and smoothly, direct release of alcohols would be desirable for avoiding the generation of toxic formaldehyde,29 particularly if the cargo is not intended to effect a cytotoxic response. Primary and secondary alcohols can be released directly, as shown in entries 3–5. The use of an aldehyde-derived boronate in entry 5 facilitated the synthesis of the ether. The oxidative cleavage of 15 and 20 (entries 3 and 6) are also significant because they show that functionalized substrates participate well in this process, providing potential handles for incorporating tissue-, cell-, or organelle-targeting functional groups. Boronate 20 releases the antioxidant pentamethyl chromanol (21),30 showing that this method could be applied to the release of radical scavengers in the presence of environments that are rich in reactive oxygen species, such as mitochondria. As previously discussed, this release was predictably somewhat slow due to the carbonate linker. The release of carboxylic acids (entry 7), while possible, is substantially slower than the release of alcohols or carbonates and is therefore not likely to be useful. Compound 22 showed a chemical shift of 27.0 ppm in the 11B NMR spectrum, indicating that coordination between the boron and the carbonyl group is likely to play a role in preventing oxidative cleavage through peroxide attack.
Table 1.
Alcohol Release Scope
 |
Reactions run with 6–12 equiv of H2O2·urea at pH 8.0 in CD3CN and D2O (5:1) at rt.
See the Supporting Information for the preparation of the substrates.
As determined by monitoring substrate consumption.
Determined by 1H NMR through comparing to the internal standard 1,2-dimethoxyethane.
The synthesis of alkyl ethers is challenging in comparison to the synthesis of alkoxymethyl ethers because direct Williamson ether syntheses with α-boryl alcohols are prone to undergo bora-Brook rearrangements31 that render the oxygen non-nucleophilic. Direct etherification requires sufficiently potent electrophiles to subvert the need for alkoxide generation. This can be achieved (Scheme 4) by activating halide leaving groups with AgOTf,32 allowing for hindered pyridines to be used as proton scavengers. This is illustrated by the ethylation of α-boryl alcohol 24 to yield 15. Alternatively, reductive etherification of α-boryl silyl ethers in the presence of BiBr333 is a versatile method for preparing these substrates under non-basic conditions. Thus, silyl ether 25, readily available from 6, can be condensed with isobutyraldehyde in the presence of Et3SiH to yield 17. An additional benefit of the reductive annulation protocol lies in the enhanced stability of α-boryl silyl ethers in comparison to α-boryl alcohols. This allows for the substrate scope to be broadened to include aldehyde-derived boronates such as 19.
Scheme 4.

Etherification in the Absence of Strong Base
The functional group tolerance of the process and the capacity for α-boryl alcohols to add into oxocarbenium ions suggested that the scope could be expanded further to promote aldehyde and ketone release. The preparation of the substrates for these studies is illustrated in Scheme 5. Cyclic acetal substrates can be prepared either through oxidative or classical exchange reactions. Ketone 26, available from commercially available 4-hydroxy-2-butanone,34 underwent copper-catalyzed borylation smoothly to yield alcohol 27. DDQ-mediated oxidative cyclization35 provided acetal 28 in 78% yield. Removing the PMB group from 27 under hydrogenolytic conditions followed by acetal exchange with the dimethyl acetal of benzophenone provided acetal 29 in 49% yield over two steps.
Scheme 5.

Synthesis of Cyclic Acetal Substrates
The boryl-substituted acetals release their cargo readily, as shown in Scheme 6. Boronate 28 reacted with H2O2 at pH 8.0 to provide hemiacetal 30, which broke down to form anisaldehyde and 1-hydroxy-3-butanone in 94% yield. Over 50% of the starting material was consumed within 90 s, and complete conversion occurred in <15 min. Similarly, boronate 29 reacted to form benzophenone quickly and efficiently. Therefore, this variation of the protocol significantly extends the range of structures that can be released in the presence of hydrogen peroxide. Moreover, this strategy illustrates a new approach to designing prodrugs for aldehydes and ketones, as previous efforts have largely centered on the use of oximes and derivatives.36,37
Scheme 6.

Aldehyde and Ketone Release through Oxidative Acetal Cleavage
Latent Fluorophore Synthesis and Release.
All studies to this point were conducted at relatively high concentrations of substrate and peroxide. Determining whether these processes can proceed at biologically relevant concentrations requires an analytical technique that is more sensitive than 1H NMR. Therefore, we explored the potential for the release of a fluorophore at low substrate and peroxide concentrations. The synthesis of a latent fluorophore is shown in Scheme 7. Silyl ether 31, which was prepared from the TBS ether of 4-hydroxy-2-butanone, coupled with aldehyde 32 (prepared from commercially available materials in two steps)38 in the presence of TMSOTf39 to yield acetal 33. This acetal was formed as a single stereoisomer, with the relative configuration being determined through a NOESY experiment. The Noyori acetalization conditions were significantly superior to those of Brønsted acid mediated protocols due to the absence of protodeboration as a prominent competing reaction. Acetalization induces significantly different fluorescence properties relative to the aldehyde, with λex values of 448 and 402 nm and λem values of 510 and 452 nm for 32 and 33, respectively, thereby facilitating the monitoring of oxidative breakdown. Acetal 34 was prepared through a similar protocol to serve as a control compound in evaluating the importance of the oxidative trigger in peroxide-mediated decomposition. We also prepared benzylic carbonate 35. This compound releases its fluorophore through the common oxidative 1,6-elimination pathway and was synthesized to compare the background stability of 33 with a well-vetted latent fluorophore motif.
Scheme 7.
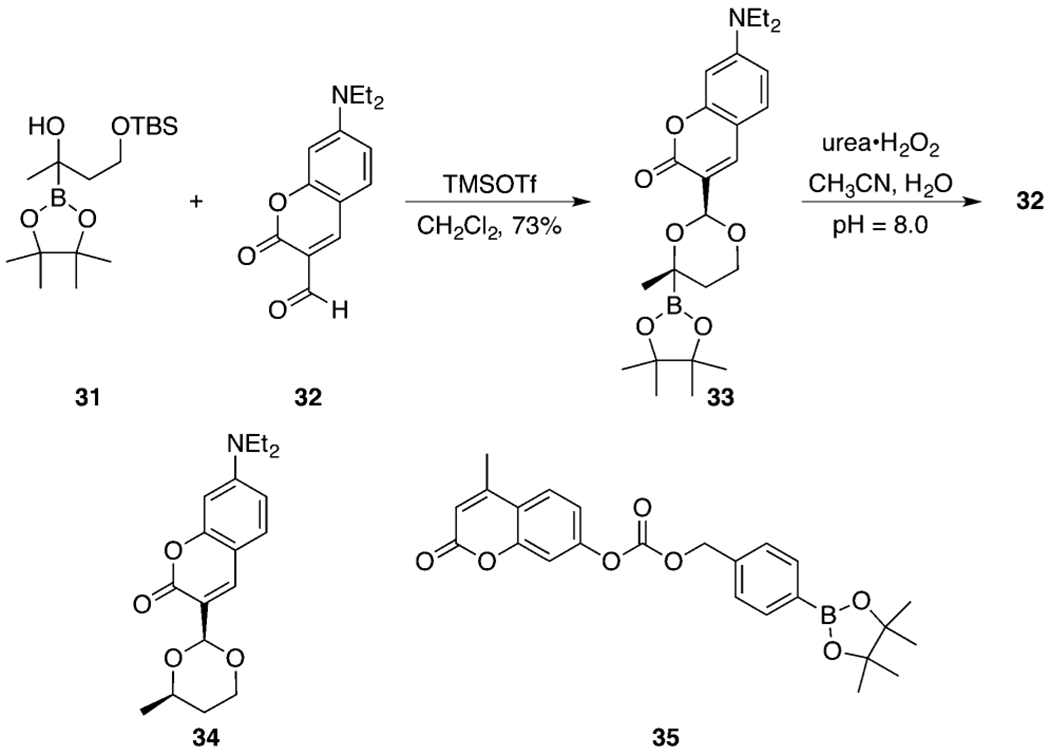
Synthesis of a Latent Fluorophore
Fluorophore release was studied at a concentration of 25 μM for 33 at pH 7.4 with H2O2 concentrations of 100 and 200 μM. The concentration of 32 was monitored by excitation at 448 nm and emission at 499 nm (a wavelength where 33 shows only slight emission), with product release being quantitated by comparison to a standard curve. The fluorophore release experiments are summarized in Figure 3. The breakdown of 33 was conducted in 1% DMSO in aqueous phosphate buffer. Fluorophore concentration increased steadily with time. The rate and extent of fluorophore release showed the expected dependency upon H2O2 concentration. Lowering the H2O2 concentration from 200 to 100 μM slowed fluorophore release to a small but noteworthy extent. The yield of 32 was 88% with 200 μM H2O2 and 78% with 100 μM H2O2. Fluorophore release was minimal in the absence of H2O2. A similar study of carbonate 35 showed that ratio of fluorophore release in the presence and absence of H2O2 was nearly equivalent to the ratio that was observed for 33.25 This indicates that the inherent stability of the α-boryl ether moiety is comparable to a well established oxidative trigger. Fluorophore release in the absence of H2O2 was studied at pH 6.0 and 4.5 to determine the stability of the acetal toward acid. Fluorophore release was slightly inhibited at lower pH values (see the Supporting Information for a graphic with an expanded y-axis), which is significant for biological applications in consideration of the capacity of endosomes to achieve pH values as low as 4.9.40
Figure 3.

Fluorophore release at low substrate and peroxide concentrations and pH stability studies.
Acetal 34 did not release 32 at any H2O2 concentration over the time span of the experiment,25 thereby validating the importance of boronate oxidation in cargo release. Separate studies in the presence of a large excess of H2O2 (10 mM) allowed for the determination of a pseudo-first-order rate constant of 1.47 × 10−3 s−1 for the decomposition of 33.25 This rate compares favorably to the peroxide-mediated decomposition of boryl-substituted benzylic carbamates to generate quinone methides via 1,6-elimination.8a The 1,6-elimination protocol is likely to be significantly slower for releasing aliphatic alcohols, however, in consideration of their lower nucleofugacity and our prior observation13b that the rates of these processes are strongly correlated with the rate of benzylic C–O bond cleavage.41
Exposing 33 to a number of reactive oxygen species showed that the breakdown is selective for H2O2 (Figure 4). Solutions of H2O2, NaOCl, KO2, and t-BuOOH were prepared by diluting commercially available material. Hydroxyl and t-butoxyl radicals were prepared by mixing the corresponding peroxide with FeSO4·5H2O and adding catalase to consume residual peroxide.8a The chart shows the ratio of fluorescence intensity after 30 min to the initial value. Aside from H2O2, only hydroxyl radical showed a notable fluorophore release, albeit significantly lower in magnitude compared to that of H2O2-mediated release.
Figure 4.

Comparison of fluorophore release by different oxidants. [33]0 = 40 μM, [oxidant]0 = 200 μM, pH 7.4.
Cellular Fluorophore Release.
These results led us to study the release of the fluorophore in cells to provide an easily visualized demonstration of these compounds’ capacity to release cargo in a biologically relevant environment. This was demonstrated in accord with Chang’s protocol,42 whereby HeLa cells were incubated with 33 (10 μM) for 45 min and fluorophore release was imaged in the absence and presence of exogenous H2O2 (100 μM). The results are shown in Figure 5. Very little fluorophore release occurred within 30 min in the absence of external H2O2. Significant fluorophore release was observed in the presence of H2O2, however. This demonstrates that α-boryl acetals are cell-permeable and can release cargo within cells. Conducting these studies with control acetal 34 resulted in no fluorophore release,25 thereby providing further evidence for the proposed release mechanism.
Figure 5.

Fluorophore release in HeLa cells treated with exogenous H2O2. Cells were incubated with 33 (10 μM) in DPBS buffer for 45 min at 37 °C, followed by replacement with fresh DPBS containing (A) vehicle or (B) H2O2 (100 μM). After 30 min, fluorescence was imaged (Zeiss Axio Observer Z1, 20× objective, GFP filter (Set 38 HE; ex = 470 nm; em = 525 nm)). (C) Bright-field image of cells in panel B stained with Hoechst 33258 (1 μM) and imaged using a DAPI filter (Set 68; ex = 377 nm; em = 464 nm). (D) Mean fluorescence intensities were calculated from three individual HeLa cells and set relative to the mean fluorescence intensity prior to treatments (F/Fi). Error bars denote standard deviations; ***, P < 0.001.
HeLa cells, directly derived from cancerous cervical tissue, are exprected to contain slightly elevated levels of endogenous H2O243 and therefore show higher background emission. We repeated this experiment with 33 and HEK293T cells, derived from the transformation of noncancerous embryonic kidney tissue,44 to test whether differentiation between cell lines is possible.
Quantitation of fluorophore release in the absence and presence of exogenous H2O2 (Figure 6) indeed showed that the background signal was significantly reduced in the absence of exogenous H2O2. These results further validate the stability of α-boryl acetals in the absence of oxidants, as required for selective applications to drug release in oxidatively stressed environments. Conducting these studies with the control acetal 34 again resulted in no fluorophore release.25
Figure 6.

Fluorophore release in HEK293T cells through treatment with exogenous H2O2. Cells were treated with 33 (10 μM) in DPBS buffer for 45 min at 37 °C, followed by replacement with fresh DPBS containing (A) vehicle or (B) H2O2 (100 μM). After 30 min incubation, cellular fluorescence was imaged on a Zeiss Axio Observer Z1 microscope using a 20× objective and GFP (Set 38 HE) filter (ex = 470 nm, em = 525 nm). (C) Bright-field image of cells in panel B stained with Hoechst 33258 (1 μM) and imaged using a DAPI (Set 68) filter (ex = 377 nm, em = 464 nm). (D) Mean fluorescence intensities were calculated from individual ROIs (n = 3) and set relative to the mean fluorescence intensity prior to treatments (F/Fi). Error bars denote standard deviations.
While these studies provide compelling evidence for the capacity of α-boryl acetals to release cargo in cells, the results would have significantly more impact if fluorophore release could be achieved through endogenous H2O2 generation. Phorbol myristate acetate (PMA) promotes intracellular H2O2 generation.45 Therefore, HeLa cells were incubated with PMA (1μM) for 60 min followed by the addition of 33 (10 μM). Fluorophore release in cells that were treated with PMA was evidenced by a significant increase of fluorescence (Figure 7), in contrast to the lack of fluorophore release in cells that were not treated with PMA. These results clearly show the capacity of α-boryl acetals to release compounds inside of cells in response to endogenous concentrations of H2O2.
Figure 7.

Cellular fluorophore release in HeLa cells by endogenous, PMA-stimulated H2O2 generation. Cells were pretreated in DMEM containing (A) DMSO or (B) PMA (1 uM) and incubated at 37 °C for 60 min. Media was replaced with fresh DPBS containing 33 (10 uM) and cells were incubated for an additional 60 min at 37 °C before fluorescence was imaged (Zeiss Axio Observer Z1, 20× objective, GFP filter (Set 38 HE; ex = 470 nm; em = 525 nm)). (C) Bright-field image of cells in panel B stained with Hoechst 33258 (1 μM) and imaged using a DAPI filter (Set 68; ex = 377 nm; em = 464 nm). (D) Mean fluorescence intensities were calculated from three individual HeLa cells and set relative to the mean fluorescence intensity prior to treatments (F/Fi). Error bars denote standard deviations; **, P < 0.01.
CONCLUSIONS
We have shown that α-boryl ethers and related structures are excellent vehicles for releasing molecular cargo in an oxidative environment. These compounds are accessed from α-boryl alcohols that can be prepared by operationally facile ketone or aldehyde borylation reactions. Although these alcohols cannot be functionalized via their alkoxides, they can be alkylated or acylated in the presence of weak amine bases. Reductive alkylation provides an attractive alternative to boryl ether formation under acidic conditions. α-Boryl ethers release alcohols extremely rapidly in the presence of H2O2, while α-boryl carbonates decompose somewhat more slowly, providing a predictable mechanism for controlling the rate of alcohol release. The capacity to functionalize α-boryl alcohols under acidic conditions provides a pathway to generate α-boryl acetals. These acid-stable structures readily release aldehydes and ketones upon exposure to H2O2. The ability to liberate cargo at low substrate and peroxide concentrations was validated through the release of a fluorescent aldehyde. Fluorophore release can also be achieved inside cells with exogenous H2O2 or with endogenous, chemically stimulated H2O2 generation. The presence of the boronate group is essential to these processes, in support of the proposed pathway for the breakdown. The capacity to release molecules inside cells with a sterically non-demanding oxidant while generating non-toxic byproducts indicates that these compounds will be valuable for drug release in oxidatively stressed cells.
Supplementary Material
ACKNOWLEDGMENTS
We thank the National Institutes of Health (AI068021 for P.E.F. and GM112728 for A.D.) and the National Science Foundation (CHE-1362396 for P.E.F.) for generous support of this work. Y.N. was supported in part by a Mellon Graduate Fellowship. We thank Viswanathan Elumalai for assistance with the NMR experiments, and Dr. Detcho Stoyanovsky for valuable discussions.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b07890.
Schemes for compound synthesis, characterization of all compounds, general protocols for compound release, and procedures for fluorophore release in cells (PDF)
The authors declare the following competing financial interest(s): The authors have applied for a provisional patent covering the drug release applications of this work.
REFERENCES
- (1).(a) DiMauro S; Schon EA Annu. Rev. Neurosci 2008, 31, 91. [DOI] [PubMed] [Google Scholar]; (b) Lin MT; Beal MF Nature 2006, 443, 787. [DOI] [PubMed] [Google Scholar]; (c) Mattson MP Nature 2004, 430, 631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).(a) Fruehauf JP; Meyskens FL Jr. Clin. Cancer Res 2007, 13, 789. [DOI] [PubMed] [Google Scholar]; (b) Wilson WR; Hay MP Nat. Rev. Cancer 2011, 11, 393. [DOI] [PubMed] [Google Scholar]; (c) Ishikawa K; Takenaga K; Akimoto M; Koshikawa N; Yamaguchi A; Imanishi H; Nakada K; Honma Y; Hayashi J Science 2008, 320, 661. [DOI] [PubMed] [Google Scholar]
- (3).(a) Brewer TF; Garcia FJ; Onak CS; Carroll KS; Chang CJ Annu. Rev. Biochem 2015, 84, 765. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Balaban RS; Nemoto S; Finkel T Cell 2005, 120, 483. [DOI] [PubMed] [Google Scholar]; (c) Finkel T; Holbrook NJ Nature 2000, 408, 239. [DOI] [PubMed] [Google Scholar]
- (4).(a) Houstis N; Rosen ED; Lander ES Nature 2006, 440, 944. [DOI] [PubMed] [Google Scholar]; (b) Pop-Busui R; Sima A; Stevens M Diabetes/Metab. Res. Rev 2006, 22, 257. [DOI] [PubMed] [Google Scholar]
- (5).Azzam EI; Jay-Gerin J-P; Pain D Cancer Lett. 2012, 327, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Chang MCY; Pralle A; Isacoff EY; Chang CJ J. Am. Chem. Soc 2004, 126, 15392. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lippert AR; Van De Bittner GC; Chang CJ Acc. Chem. Res 2011, 44, 793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Lo L-C; Chu C-Y Chem. Commun 2003, 39, 2728. [DOI] [PubMed] [Google Scholar]
- (8).(a) Srikun D; Miller EW; Domaille DW; Chang CJ J. Am. Chem. Soc 2008, 130, 4596. [DOI] [PubMed] [Google Scholar]; (b) Lippert AR; Gschneidtner T; Chang CJ Chem. Commun 2010, 46, 7510. [DOI] [PubMed] [Google Scholar]; (c) Chung C; Srikun D; Lim CS; Chang CJ; Cho BR Chem. Commun 2011, 47, 9618. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Van de Bittner GC; Dubikovskaya EA; Bertozzi CR; Chang CJ Proc. Natl. Acad. Sci. U. S. A 2010, 107, 21316. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Karton-Lifshin N; Segal E; Omer L; Portnoy M; Satchi-Fainaro R; Shabat D J. Am. Chem. Soc 2011, 133, 10960. [DOI] [PubMed] [Google Scholar]; (f) Yuan L; Lin W; Xie Y; Chen B; Zhu S J. Am. Chem. Soc 2012, 134, 1305. [DOI] [PubMed] [Google Scholar]; (g) Van de Bittner GC; Bertozzi CR; Chang CJ J. Am. Chem. Soc 2013, 135, 1783. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Carroll V; Michel BW; Blecha J; VanBrocklin H; Keshari K; Wilson D; Chang CJ J. Am. Chem. Soc 2014, 136, 14742. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Kim J; Park J; Lee H; Choi Y; Kim Y Chem. Commun 2014, 50, 9353. [DOI] [PubMed] [Google Scholar]; (j) Brooks AD; Mohapatra H; Phillips ST J. Org. Chem 2015, 80, 10437. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Wang P; Wang K; Chen D; Mao Y; Gu Y RSC Adv. 2015, 5, 85957. [Google Scholar]; (l) Xu J; Zhang Y; Yu H; Gao X; Shao S Anal. Chem 2016, 88, 1455. [DOI] [PubMed] [Google Scholar]; (m) Narayanaswamy N; Narra S; Nair RR; Saini DK; Kondaiah P; Govindaraju T Chem. Sci 2016, 7, 2832. [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Xiao H; Li P; Hu X; Shi X; Zhang W; Tang B Chem. Sci 2016, 7, 6153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).For a review on this concept, see; Alouane A; Labruère R; Le Saux T; Schmidt F; Jullien L Angew. Chem., Int. Ed 2015, 54, 7492. [DOI] [PubMed] [Google Scholar]
- (10).(a) Broaders KE; Grandhe S; Fréchet JMJ J. Am. Chem. Soc 2011, 133, 756. [DOI] [PubMed] [Google Scholar]; (b) Lewis GG; Robbins JS; Phillips ST Macromolecules 2013, 46, 5177. [Google Scholar]; (c) Robbins JS; Schmid KM; Phillips ST J. Org. Chem 2013, 78, 3159. [DOI] [PubMed] [Google Scholar]; (d) Chen C-Y; Chen C-T Chem. - Eur. J 2013, 19, 16050. [DOI] [PubMed] [Google Scholar]; (e) Zhao W; Li Y; Yang S; Chen Y; Zheng J; Liu C; Qing Z; Li J; Yang R Anal. Chem 2016, 88, 4833. [DOI] [PubMed] [Google Scholar]
- (11).(a) Roth ME; Green O; Gnaim S; Shabat D Chem. Rev 2016, 116, 1309. [DOI] [PubMed] [Google Scholar]; (b) Sella E; Shabat D Chem. Commun 2008, 5701. [DOI] [PubMed] [Google Scholar]; (c) Sella E; Shabat DJ J. Am. Chem. Soc 2009, 131, 9934. [DOI] [PubMed] [Google Scholar]; (d) Nuñez SA; Yeung K; Fox NS; Phillips ST J. Org. Chem 2011, 76, 10099. [DOI] [PubMed] [Google Scholar]; (e) Yeung K; Schmid KM; Phillips ST Chem. Commun 2013, 49, 394. [DOI] [PubMed] [Google Scholar]; (f) Brooks AD; Yeung K; Lewis GG; Phillips ST Anal. Methods 2015, 7, 7186. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Brooks AD; Mohapatra H; Phillips ST J. Org. Chem 2015, 80, 10437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).For a review, see; Bo.hme D; Beck-Sickinger AG J. Pept. Sci 2015, 21, 186. [DOI] [PubMed] [Google Scholar]
- (13).(a) Major Jourden JL; Cohen SM Angew. Chem., Int. Ed 2010, 49, 6795. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mosey RA; Floreancig PE Org. Biomol. Chem 2012, 10, 7980. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kim E-J; Bhuniya S; Lee H; Kim HM; Cheong C; Maiti S; Hong KS; Kim JS J. Am. Chem. Soc 2014, 136, 13888. [DOI] [PubMed] [Google Scholar]
- (14).(a) Bolton JL Curr. Org. Chem 2014, 18, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dufrasne F; Gelbcke M; Neve J; Kiss R; Kraus J-L Curr. Med. Chem 2011, 18, 3995. [DOI] [PubMed] [Google Scholar]
- (15).Kehrer JP; Biswal SS Toxicol. Sci 2000, 57, 6. [DOI] [PubMed] [Google Scholar]
- (16).Wan S; Green ME; Park J-H; Floreancig PE Org. Lett 2007, 9, 5385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).For a review, see; Clark TB Asian J. Org. Chem 2016, 5, 31. [Google Scholar]
- (18).McIntosh ML; Moore CM; Clark TB Org. Lett 2010, 12, 1996. [DOI] [PubMed] [Google Scholar]
- (19).Laitar DS; Tsui EY; Sadighi JP J. Am. Chem. Soc 2006, 128, 11036. [DOI] [PubMed] [Google Scholar]
- (20).For an enantioselective variant on this protocol, see; Kubota K; Yamamoto E; Ito HH J. Am. Chem. Soc 2015, 137, 420. [DOI] [PubMed] [Google Scholar]
- (21).Molander GA; Wisniewski SR J. Am. Chem. Soc 2012, 134, 16856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Moore CM; Medina CR; Cannamela PC; McIntosh ML; Ferber CJ; Roering AJ; Clark TB Org. Lett 2014, 16, 6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).(a) Beenen MA; An C; Ellman JA J. Am. Chem. Soc 2008, 130, 6910. [DOI] [PubMed] [Google Scholar]; (b) Snow RJ; Bachovchin WW; Barton RW; Campbell SJ; Coutts SJ; Freeman DM; Gutheil WG; Kelly TA; Kennedy CA J. Am. Chem. Soc 1994, 116, 10860. [Google Scholar]; (c) Kettner C; Mersinger L; Knabb RJ Biol. Chem 1990, 265, 18289. [PubMed] [Google Scholar]; (d) Adams J; Stein R Annu. Rep. Med. Chem 1996, 31, 279. [Google Scholar]
- (24).(a) Chacon E; Reece JM; Nieminen A-L; Zahrebelski G; Herman B; Lemasters JJ Biophys. J 1994, 66, 942. [DOI] [PMC free article] [PubMed] [Google Scholar]; Llopis J; McCaffery JM; Miyawaki A; Farquhar MG; Tsien RY Proc. Natl. Acad. Sci. U. S. A 1998, 95, 6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Please see the Supporting Information for details and primary data.
- (26).Hu N; Zhao G; Zhang Y; Liu X; Li G; Tang WJ Am. Chem. Soc 2015, 137, 6746. [DOI] [PubMed] [Google Scholar]
- (27).(a) Carry B; Zhang L; Nishiura M; Hou Z Angew. Chem., Int. Ed 2016, 55, 6257. [DOI] [PubMed] [Google Scholar]; (b) Gillis EP; Burke MD J. Am. Chem. Soc 2008, 130, 14084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Biedrzycki M; Scouten WH; Biedrzycka Z J. Organomet. Chem 1992, 431, 255. [Google Scholar]
- (29).For a report on the carcinogenicity of formaldehyde, see; ntp.niehs.nih.gov/ntp/roc/content/profiles/formaldehyde.pdf.
- (30).(a) Samhan-Arias AK; Tyurina YY; Kagan VE J. Clin. Biochem. Nutr 2010, 48, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Staniek K; Rosenau T; Gregor W; Nohl H; Gille L Biochem. Pharmacol 2005, 70, 1361. [DOI] [PubMed] [Google Scholar]
- (31).Kisu H; Sakaino H; Ito F; Yamashita M; Nozaki K J. Am. Chem. Soc 2016, 138, 3548. [DOI] [PubMed] [Google Scholar]
- (32).Burk RM; Gac TS; Roof MB Tetrahedron Lett. 1994, 35, 8111. [Google Scholar]
- (33).Bajwa JS; Jiang X; Slade J; Prasad K; Repič O; Blacklock TJ Tetrahedron Lett. 2002, 43, 6709. [Google Scholar]
- (34).Gathirwa JW; Maki T Tetrahedron 2012, 68, 370. [Google Scholar]
- (35).Oikaw Y; Yoshioka T; Yonemitsu O Tetrahedron Lett. 1982, 23, 889. [Google Scholar]
- (36).(a) Kumpulainen H; Mähönen N; Laitinen M; Jaurakkajärvi M; Raunio H; Juvonen RO; Vepsäläinen J; Järvinen T; Rautio J J. Med. Chem 2006, 49, 1207. [DOI] [PubMed] [Google Scholar]; (b) Foley D; Bailey P; Pieri M; Meredith D Org. Biomol. Chem 2009, 7, 1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Although ketone and aldehyde release under endogenous conditions is limited to oximes and derivatives, light-mediated ketone and aldehyde release from acetals has been reported. See; (a) Lu M; Fedoryak OD; Moister BR; Dore TM Org. Lett 2003, 5, 2119. [DOI] [PubMed] [Google Scholar]; (b) Kilic F; Kashikar ND; Schmidt R; Alvarez L; Dai L; Weyand I; Wiesner B; Goodwin N; Hagen V; Kaupp UB J. Am. Chem. Soc 2009, 131, 4027. [DOI] [PubMed] [Google Scholar]; (c) Kostikov AP; Malashikhina N; Popik VV J. Org. Chem 2009, 74, 1802. [DOI] [PubMed] [Google Scholar]; (d) Wang P; Hu H; Wang Y Org. Lett 2007, 9, 1533. [DOI] [PubMed] [Google Scholar]; (e) Blanc A; Bochet CG J. Org. Chem 2003, 68, 1138. [DOI] [PubMed] [Google Scholar]
- (38).Wu J-S; Liu W-M; Zhuang X-Q; Wang F; Wang P-F; Tao S-L; Zhang X-H; Wu S-K; Lee S-T Org. Lett 2007, 9, 33. [DOI] [PubMed] [Google Scholar]
- (39).Tsunoda T; Suzuki M; Noyori R Tetrahedron Lett. 1980, 21, 1357. [Google Scholar]
- (40).Huotari J; Helenius A EMBO J. 2011, 30, 3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).A recent report has detailed the influence of leaving groups on the rate of 1,6-elimination. See; Dȩbowska K; Dȩbski D; Michałowski B; Dybala-Defratyka A; Wójcik T; Michalski R; Jakubowska M; Selmi A; Smulik R; Piotrowski L; Adamus J; Marcinek A; Chlopicki S; Sikora A Chem. Res. Toxicol 2016, 29, 735. [DOI] [PubMed] [Google Scholar]
- (42).Dickinson BC; Lin VS; Chang CJ Nat. Protoc 2013, 8, 1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).(a) Verschoor ML; Wilson LA; Singh G Can. J. Physiol. Pharmacol 2010, 88, 204. [DOI] [PubMed] [Google Scholar]; (b) Trush MA; Kensler TW Free Radical Biol. Med 1991, 10, 201. [DOI] [PubMed] [Google Scholar]; (c) Reuter S; Gupta SC; Chaturvedi MM; Aggarwal BB Free Radical Biol. Med 2010, 49, 1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Lin Y-C; Boone M; Meuris L; Lemmens I; Van Roy N; Soete A; Reumers J; Moisse M; Plaisance S; Drmanac R; Chen J; Speleman F; Lambrechts D; Van de Peer Y; Tavernier J; Callewaert N Nat. Commun 2014, 5, 4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Bellavite P Free Radical Biol. Med 1988, 4, 225. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.