

Abstract
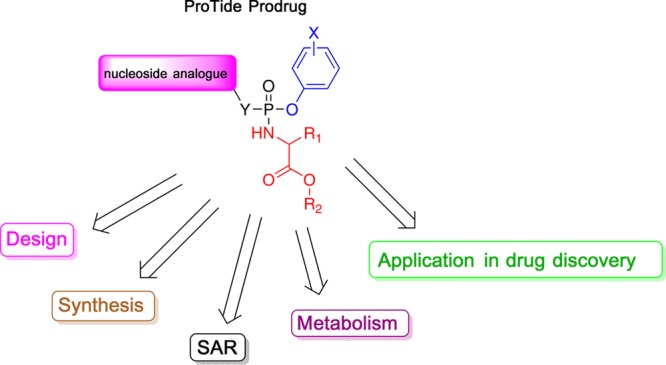
The ProTide technology is a prodrug approach developed for the efficient intracellular delivery of nucleoside analogue monophosphates and monophosphonates. In this approach, the hydroxyls of the monophosphate or monophosphonate groups are masked by an aromatic group and an amino acid ester moiety, which are enzymatically cleaved-off inside cells to release the free nucleoside monophosphate and monophosphonate species. Structurally, this represents the current end-point of an extensive medicinal chemistry endeavor that spans almost three decades. It started from the masking of nucleoside monophosphate and monophosphonate groups by simple alkyl groups and evolved into the sophisticated ProTide system as known today. This technology has been extensively employed in drug discovery, and it has already led to the discovery of two FDA-approved (antiviral) ProTides. In this work, we will review the development of the ProTide technology, its application in drug discovery, and its role in the improvement of drug delivery and efficacy.
1. Introduction
Nucleoside analogues were first introduced a number of decades ago as an effective way of treating various diseases such as cancer and viral infections like herpes simplex virus (HSV), human cytomegalovirus (HCMV), human immunodeficiency virus (HIV), hepatitis B virus (HBV), and recently also hepatitis C virus (HCV).1−6 Currently, there are more than 20 different nucleoside analogues available in the clinic to treat the viral infections and cancer.7,8 These molecules enter the cell with the aid of transporters such as concentrative nucleoside transporters as well as peptide transporters or via passive diffusion.9,10 Once inside the cell, the nucleoside analogues are activated by a number of nucleoside and nucleotide kinases which phosphorylate them in a stepwise manner resulting in the formation of the mono-, di-, and triphosphorylated nucleoside analogue metabolites (Figure 1).6,11
Figure 1.

Intracellular activation of therapeutic and experimental nucleoside analogues. NA: nucleoside analogue.
The activated (phosphorylated) antiviral nucleoside analogues exert their therapeutic effects by targeting and inhibiting intracellular enzymes (usually the virus-encoded DNA or RNA polymerases) and/or by being incorporated into the viral nucleic acid chains leading to the termination of the elongation process.7,8,12 However, there are a number of limitations associated with the administration of nucleoside analogue drugs. Nucleoside analogues are structurally different to natural nucleosides, making their phosphorylation by cellular or viral kinases often inefficient.13,14 This ultimately limits the formation of the active triphosphate metabolites, often the nucleoside analogue active metabolite.13 They also often demonstrate poor oral bioavailability due to their low intestinal permeability, as they are usually polar molecules that hinder their transport through the cell border via the paracellular route.15 Additionally, the therapeutic use of many antiviral and anticancer nucleoside analogues is further limited by resistance, which occurs via a number of mechanisms8 that include (1) down regulation of the nucleoside kinases responsible for activating nucleoside analogues,16 (2) depletion of transporters that affect their efficient trafficking into the target cells,17−19 (3) activation of phosphatases (i.e., 5′-nucleotidases) that dephosphorylate the active metabolites of nucleoside analogues,20,21 and (4) increased catabolic bioconversion of the nucleoside analogues, e.g. (deoxy)cytidine deamination, which may yield inactive versions of some of the nucleoside analogues.22,23
To overcome these limitations, it was initially suggested that delivering phosphorylated metabolites of the nucleoside analogue would address some of these shortages. This notion was further supported by numerous studies that reported the poor phosphorylation of nucleoside analogues into their nucleoside monophosphates as the rate-limiting step in their in situ activation.24−27 However, the idea of using nucleoside analogue monophosphates as therapeutics faced two main challenges. First, these compounds would have poor in vivo stability due to dephosphorylation in the bloodstream, and second, their transport into cells will be inefficient because of the charged nature of the phosphorylated nucleoside. To address the P–O− bond instability, Holý and De Clercq switched the P–O– bonds in the monophosphate group of nucleoside analogues into monophosphonate P-CH2– bonds.28 This led to improved in situ stability, which translated into longer half-lives of the nucleoside monophosphonates as compared to their monophosphates.28 This advance in the design of stable nucleotide therapeutics inspired the discovery of many nucleoside analogue monophosphonates, some of which were eventually approved for clinical use as antivirals against HCMV [i.e., cidofovir], HBV [i.e., adefovir and tenofovir], and HIV [i.e., tenofovir] (Figure 2).7
Figure 2.

Chemical structures of therapeutic nucleoside analogue monophosphonates, cidofovir, adefovir, and tenofovir.
Although the use of monophosphonates efficiently addressed the in situ and in vivo instability issue associated with monophosphate (P–O−) bonds, these compounds still had poor cellular uptake due to the negative charges on the monophosphonate group at physiological pH (<7.4). This limitation acted as a challenge in the medicinal chemistry field to develop prodrugs of nucleoside analogue monophosphates and monophosphonates that can deliver them into cells more efficiently. Numerous prodrug technologies that mask either one or two of the oxygens of the monophosphate and monophosphonate groups making the molecule more lipophilic and hence improve transport into the cells have been developed.13,29−31 Among these prodrug approaches is the ProTide prodrug technology that was pioneered by Prof. Chris McGuigan (Cardiff, UK).32 This prodrug technology has proved to be a powerful tool in the discovery of efficacious nucleoside monophosphate and monophosphonate prodrug therapeutics.33 To date, it has delivered at least 10 clinical candidates that reached, or successfully passed, phase I clinical trials.33 Although most of these nucleotide prodrugs were developed as antiviral agents against HIV, HBV, HCV, and Ebola virus, the most recent ones are also being pursued as potential anticancer agents. Moreover, the ProTide prodrug technology has so far led to the discovery of two FDA-approved (antiviral) drugs, tenofovir alafenamide (TAF)34 and sofosbuvir35 (Figure 3).13
Figure 3.

Chemical structures of the two FDA-approved antiviral ProTides, tenofovir alafenamide (TAF) and sofosbuvir.
Because of the effectiveness of the ProTide technology in the in vivo intracellular delivery of nucleoside analogue monophosphates and monophosphonates, there has been a growing interest in the application of this technology in drug discovery. The last comprehensive review on the development of the ProTide technology appeared in 2009 (Mehellou, Balzarini and McGuigan).32 However, at that time, the ProTides had just entered clinical trials and significant advances in their synthesis, metabolism, and clinical studies have been achieved since then. Thus, we felt that there is a need for an updated and comprehensive report on the ProTide technology, which this review aims to address and therefore provide a much needed updated resource for the scientific community.
2. Inception and Evolution of the ProTides
Initial work on the ProTides can be tracked to the late 1980s.36−38 The mission was to mask the oxygen atoms of phosphate groups in nucleoside monophosphate analogues so that they are neutral at physiological pH and hence have a better uptake into cells. The process started from the simple masking of the phosphate groups by alkyl groups but eventually evolved into the intelligent masking of the phosphate with aryl groups and amino acid esters, which can be cleaved off inside the intact cells. Such a development process, which took at least two decades, could be divided into six phases as discussed below.
Stage One: Alkyl and Haloalkyl Phosphate Esters
The first attempt of making nucleoside analogue monophosphate prodrugs was to mask the phosphate group of 9-β-d-arabinofuranosyladenine (AraA)39 monophosphate, which exerts antiviral activity, by simple dialkyl triesters (Stage 1, Table 1).36 Although these prodrugs led to increased stability toward adenosine/adenylate deaminases, they demonstrated a moderate biological activity.36 Similar results were found when the same principle was applied to the anticancer agent 1-β-d-arabinofuranosylcytosine (AraC).37,40 However, when the dialkyl phosphate approach was used to mask the monophosphate group of the anti-HIV agent 3′-azidothymidine (AZT),41 no significant activity was observed in vitro.42 This was attributed to several parameters, but the most likely explanation for the lack of activity was due to the high stability of the dialkyl triesters, which hindered the eventual conversion to the biologically active 5′-O-triphosphate form of AZT.43 Masking the monophosphate groups of AZT and 2′,3′-dideoxycytidine (ddC)44 with haloalkyl derivatives also yielded agents with poor biological activity.45 To enhance the intracellular cleavage of the haloalkyl masking groups, more haloalkyl phosphate masking groups with halogen atoms incorporated either on one or in both masking groups were used for making AZT monophosphate prodrugs. However, these prodrugs also displayed moderate antiviral activities.46 However, the use of haloalkyl groups in AraA and AraC monophosphates displayed significant increases in biological activity (Stage 1, Table 1).47 This increase in activity was mainly attributed to increased lipophilicity, resulting in better membrane permeability of the prodrug.47 The lack of increased AZT activity through administration of haloalkyl prodrugs of AZT could now be explained by the fact that the first phosphorylation step of AZT is not the rate-limiting step in its activation pathway while it certainly is for AraA and AraC.48 Nevertheless, the alkyl and haloalkyl phosphate masking groups, especially for their application to AraA and AraC, indicated that these masking groups showed good activity due to better membrane-crossing ability, because the biological activity correlated with lipophilicity, rather than increased intracellular levels of the phosphate species.36,37,47 Such conclusion shifted the thinking toward phosphate prodrugs to a different strategy.
Table 1. Stages of the Development of the ProTide Prodrug Technology.

Stage Two: Alkyloxy and Haloalkyloxy Phosphoramidates
Subsequently, the design of nucleoside phosphate prodrugs focused on alkyloxy and haloalkyl phosphoramidate prodrugs (Stage 2, Table 1). The move toward phosphoramidate prodrugs was provoked by the findings that the HIV protease can cleave an oligopeptide from the phosphate group of blocked nucleotide phosphoramidates.49 Initially, a selection of AZT alkyloxy phosphoramidates with methyl-esterified amino acids were synthesized.42 These compounds showed better anti-HIV activity than the dialkyl AZT prodrugs.42 Although the anti-HIV activity did not correlate with the alkyl phosphate chain, it did correlate with the amino acid side chain, with l-alanine showing the better activity.42,50 Retrospectively, this observation proved to be critical for the success of the ProTide technology in drug discovery since all of the ProTides that entered clinical trials and those that have been approved for clinical use bear an l-alanine moiety.33 Further studies into different, i.e. α, β, and γ, types of amino acids, indicated the superiority of α-amino acids.51
Encouraged by the influence of the amino acid moiety on the anti-HIV activity of AZT alkyloxy phosphoramidates, the masking of phosphate groups by the methyl-esterified amino acids and haloalkyl groups, which increased the membrane-crossing ability, were explored next. Haloalkyl phosphoramidates of AZT were synthesized with glycine, alanine, and valine amino acids as well as ethyl, trifluoroethyl, or trichloroethyl as the haloalkyl motifs.52 These, surprisingly, did not show good anti-HIV activity with the exception of the trichloroethyl alanine prodrug. Because no clear structure–activity relationship was apparent from the haloalkyl phosphoramidates, they were not pursued any further. However, the key finding of the enhancement of activity with nucleoside phosphoramidate prodrugs, especially with l-alanine, proved to be important for the future evolution of this nucleoside monophosphate prodrug strategy.
Stage Three: Phosphorodiamidates
Given that the introduction of amino acids to generate phosphoramidates led to enhanced anti-HIV activity, it was logical that the next step would be to mask both oxygen groups of the phosphate group by amino acid esters. Indeed, a series of amino acid methyl esters with different side chains were used to mask both oxygens of the phosphate group of AZT monophosphate (Stage 3, Table 1).53 Unlike previous studies, where l-alanine appeared to give the better biological activity, in these phosphorodiamidate systems amino acids with aromatic side chains, e.g., phenylalanine, showed better improvement of biological activity.53 Although this was surprising in the context of the ProTides’ development, such finding was consistent with other studies where a preference was found for amino acids with aromatic side chains in the development of unrelated phosphoramidate diesters.29,31 Although no further studies with these phosphorodiamidates were conducted since the early 1990s and the development of the ProTides moved on, phosphorodiamidates as phosphate prodrugs recently (in 2011) made a comeback.54
Stage Four: Lactyl-Derived Systems
Continuing with the probing of the phosphoramidates and particularly studies on the importance of the P–N bond, isosteric O-linked nucleoside analogues derived from lactic and glycolic acids of AZT were prepared and evaluated for their biological activity (Stage 4, Table 1).55 Although no significant improvement in the anti-HIV activity was noted for the actyl-derived AZT monophosphate prodrugs, the observed activity was found to correlate with the lengthening of the alkyl phosphate chain (R2) because longer chains resulted in poorer anti-HIV activity. As for the glycol systems (R1 = H), the prodrugs were generally more active than the lactyl systems. The poor anti-HIV activity of these phosphate prodrug systems was not very encouraging, and they were therefore not optimized or explored any further.
Stage Five: Diaryl Phosphates
The next masking groups studied in the design of a novel phosphate prodrug strategy were aryl motifs. Indeed, the masking of AZT monophosphate by diaryl motifs was pursued (Stage 5, Table 1).56 These prodrugs showed good anti-HIV activity and, remarkably, some of them showed much better anti-HIV activity than the parent compound AZT.56 In the journey toward the ProTides, this was the first time a phosphate prodrug was more active than the parent compound. Interestingly, these phosphate prodrugs retained their anti-HIV activity in cell lines that were considered to be resistant to AZT.56 However, when these cells (T lymphocyte cell line, JM) were used for this study back in 1992, they were considered as AZT insensitive due to poor phosphorylation of AZT.57 But, it was later shown that such AZT resistance was a result of an active AZT-efflux pump rather than inefficient intracellular phosphorylation.58 Regardless of the exact AZT-insensitivity mechanism in this cell line, the AZT diaryl phosphate prodrugs were more active than their parent compound, providing a clear indication of an increased delivery of AZT monophosphate into the cells. This notion was further supported by the fact that para-nitrophenyl phosphate masking groups had ca. 100-fold better anti-HIV activity than their unsubstituted phenyl masking groups most probably due to the electron-withdrawing effect of the para-nitro group that speeds up the demasking of the prodrugs in cells.56 A further study on the effect of the nature of the phenyl substituents on the antiviral activity of diaryl phosphate prodrugs of AZT was then conducted.59,60 This study confirmed that stronger para-electron-withdrawing groups on the phenyl phosphate masking groups gave better anti-HIV activity.59,60 Intriguingly, these AZT diaryl phosphate prodrugs as well as AZT did not exhibit any anti-HIV activity in a thymidine kinase (TK)-deficient human T-lymphocyte cell line (CEM/TK–).60 McGuigan and colleagues concluded that this might have been due to poor intracellular release of AZT monophosphate. The development of diaryl monophosphate prodrugs was not progressed any further and the focus turned back to phosphoramidates, which showed good biological activity.56
Stage Six: Aryloxy Phosphoramidates (Generally Referred to as ProTides)
Inspired by the improvement of the biological activities of the earlier examples of nucleoside monophosphate prodrugs, particularly the para-substituted aryls and the amino acid esters, McGuigan and co-workers took the next logical step, which was to combine these two masking groups in the design of new monophosphate prodrugs.61 Indeed, the masking of the AZT monophosphate group with para-substituted aryls and an amino acid ester was conducted and these efforts yielded prodrugs that showed improved anti-HIV activity as compared to the parent nucleoside AZT. In particular, the combination of a phenyl and l-alanine methyl ester showed the most potent anti-HIV activity in this study, EC50 = 0.8 μM (AZT, IC50 (toxicity) ca. 100 μM).61 Interesting structure–activity relationships on these aryloxy phosphoramidate prodrugs emerged from this study. Notably, there was a preference for the l-alanine amino acid as compared to l-leucine (>10-fold) and glycine (>100-fold), while electron-withdrawing para-substituted aryls were less potent that their unsubstituted moieties.61 Further studies into these substitutions concluded that para-fluoro aryls did not largely affect the biological activity while para-nitro-substitutions had detrimental effects on the biological activity, namely a 100-fold loss in activity.62 In a separate study, it was shown that para-nitrophenyl-containing nucleoside monophosphate prodrugs were toxic even to uninfected cells.63 Remarkably, AZT aryloxy phosphoramidates retained their anti-HIV activities in thymidine kinase deficient (TK–) cells.62 These observations provided a direct proof of the ability of aryloxy phosphoramidates to deliver intact nucleoside analogue monophosphates into intact cells. Hence, the masking of nucleoside analogue monophosphates, and monophosphonates with an aryl motif and an amino acid ester, which is nowadays known as the ProTide technology, was extensively pursued in the future discovery of novel antiviral and anticancer nucleotide therapeutics.
3. Synthesis of the ProTides
Although the synthesis of the aryloxy triester phosphoramidate ProTides has recently been excellently reviewed by Pradere et al.,29 for the completion of this Perspective, we will briefly discuss the different strategies used in the synthesis of the ProTides. We will also include recent reports on the development and use of new catalysts that improve the synthesis of the ProTides, which have been published after the comprehensive review by Pradere et al.29 There are three different strategies for synthesizing ProTides: (1) the coupling of a nucleoside with a diarylphosphite followed by subsequent oxidative amination, (2) the coupling of the nucleoside with a phosphorochloridate reagent, and (3) coupling of an amino acid to a nucleoside aryl phosphate (Figure 4).
Figure 4.

Representation of the three main strategies for synthesizing ProTides. LG: leaving group. NA: nucleoside analogue. R: any ester. X: any aromatic substitution.
Among these three strategies, the second synthetic strategy, which involves the coupling of the nucleoside to a phosphorochloridate is the most common method and has been the method of choice of the initial synthesis of the ProTides. The coupling reaction takes place in the presence of either the Grignard reagent tert-butyl magnesium chloride (tBuMgCl) or N-methylimidazole (NMI), which act as a base. The choice of the base is often influenced by the nucleoside substrate, most precisely the number of hydroxyl groups that could couple to the phosphorochloridate. For instance, in ribonucleosides, the base used is often NMI and this generates mostly the 5′-O- hosphoramidates due to steric hindrance and the fact that it is a weaker base than tBuMgCl. However, in cases where the nucleoside has only one hydroxyl group, the base of choice is tBuMgCl, which often gives higher yields of the product as compared to NMI. Notably, the approach of coupling the nucleoside to a phosphorochloridate yields a mixture of diastereoisomers. In some cases, the two ProTide diastereoisomers exhibit similar potency and their development therefore is pursued as a mixture of diastereoisomers (see examples in Figure 10).64 In other cases, the two diastereoisomers may have different rates of metabolism and hence varying potency. For instance, the Sp-diastereoisomer of the clinically used ProTide sofosbuvir exhibits a 10-fold increase in antiviral activity than its Rp-diastereoisomer.35 In these instances, the separation of the two diastereoisomers becomes necessary. The separation of these by column chromatography has proved to be a stern challenge.
Figure 10.

Chemical structures of key ProTides undergoing clinical trials.
To address this, Ross et al.65 developed a system that allows for the facile generation and crystallization of single isomer phosphoramidating reagents that could be coupled to nucleosides to yield pure single ProTide isomers. From this study, phosphorochloridates with a pentafluorophenol were found to be optimal because the desired Sp-isomer phosphorochloridate was readily crystallized, stable, and allowed for the formation of the Sp-isomer ProTides, which is often the more active isomer.65
Continuing on the theme of synthetic procedures with stereocontrol of the phosphorus chirality of the ProTides, researchers from Merck recently reported the discovery of a small molecule catalyst that, upon the addition to the reaction of a nucleoside with a phosphorochloridate, leads to the generation of a single isomer with significant preference for the 5′-hydroxyl compared to the 3′-one.66 However, the reported catalyst generates the Rp-isomer and not the Sp-one, which often shows better pharmacological activity. This represents a challenge for the wide use of this catalyst. Impressively, more work from Merck researchers led to the identification of various Lewis acids as being able to improve the yield, stereoselectivity, and regioselectivity of the reaction of phosphorochlorides with nucleosides to generate ProTides.67 Indeed, a selection of Lewis acids were studied and dimethyl aluminum chloride emerged as the optimal catalyst for this reaction.
In the synthesis of nucleoside monophosphonate ProTides three different synthetic strategies are employed (Figure 5). The first strategy involves the bis-chlorination of a phosphate group to form a monophosphonate dichloridate. This is then followed by the addition of 1:1 mol equiv of the phenol and amino acid ester yielding the desired ProTides. Alternatively, in the second synthetic strategy, the monophosphonate group is initially coupled to one phenol group using N,N′-dicyclohexylcarbodiimide (DCC) and the product is then chlorinated. Finally, the chloride group is substituted by the amino acid ester.
Figure 5.
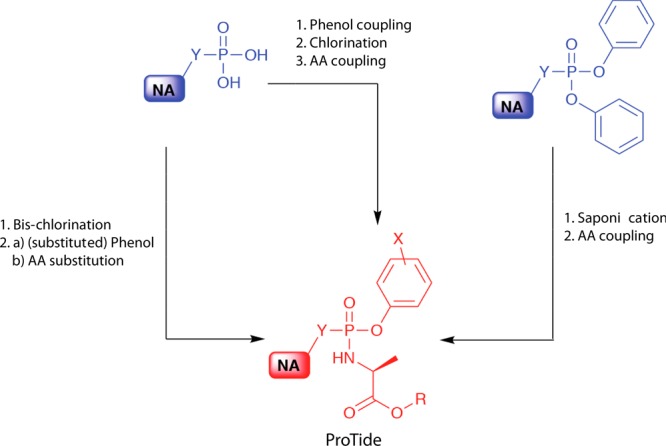
Synthetic strategies for accessing monophosphonate ProTides. AA: amino acid. NA: nucleoside analogue. R: any ester. X: any aromatic substitution. Y: O or CH2.
For the third synthetic strategy, the starting material employed is the diphenyl-masked nucleoside monophosphonate, which first undergoes selective saponification that results in the removal of one of the phenyl masking groups. The product is subsequently coupled to the amino acid ester to generate the desired nucleoside analogue monophosphonate ProTide. Further details on the synthesis of the ProTides can be found in Pradere et al.29 and Serpi et al.68
4. Metabolism of the ProTides
Once inside the cell, the ProTides require metabolism to release the nucleoside monophosphate. The metabolic conversion of the 2′,3′-didehydro-2′,3′-dideoxythymidine (d4T)69−71 monophosphate (d4T-MP) ProTide to the eventual active metabolite d4T-TP has been extensively studied in several cell systems, including human lymphocytes (Figure 6). Beside efficient formation of d4T-MP, d4T-diphosphate (d4T-DP), and d4T-triphosphate (d4T-TP), an additional metabolite markedly accumulated upon exposure of the cell culture to the d4T-MP ProTide, which was identified as being the alaninyl d4T-MP. This metabolite may act as an intracellular depot form of d4T-MP, explaining not only the superior antiviral activity of the ProTide versus the parental d4T but also the marked retainment of antiviral activity in thymidine kinase-deficient cells in which the parental d4T markedly loses antiviral efficacy.72,73 Also, the activation pathway of the ProTides of d4T-MP and AZT-MP containing different amino acids and/or ester moieties has been investigated. It was found that, overall, there was a close correlation between the anti-HIV activity of these prodrugs and their conversion rate to the amino acyl nucleotide metabolite (metabolite C, Figure 6).74,75 The metabolic conversion process is believed to be mediated by a two-step enzymatic process, which eventually releases the active monophosphate (or monophosphonate) form of the parent nucleoside as summarized in Figure 6.
Figure 6.

Postulated mechanism of ProTide metabolism illustrated using the ProTide of the anti-HIV agent d4T as an example.
The first step involves the cleavage of the amino acid ester by intracellular esterases. Researchers at Gilead Sciences identified cathepsin A as a major esterase that mediates the hydrolysis of ester motifs of ProTides.76 Such finding has made cathepsin A widely used in the field in probing the hydrolysis of ProTides in vitro.64,77−79 Notably, a second enzyme that was able to hydrolyze the ester group of the two antiviral ProTides studied, TAF and 9-(R)-4-(R)-[[[(S)-1-[(ethoxycarbonyl)ethyl]amino]phenoxy-phosphonyl]methoxy]-2-fluoro-1-furanyladenine (GS-9131)80 (structure not disclosed), was also reported in this study but its identity was not determined.76 In 2011, however, this identity of this second enzyme was established and was shown to be carboxylesterase 1 (CES1).81 Although both cathepsin A and CES1 were found to be expressed in primary hepatocytes, CES1 expression was undetectable in clone A replicon cells.81 This indicated that in primary human hepatocytes, both cathepsin A and CES1 are involved in the hydrolyzing of the carboxyl ester motif of the ProTides, whereas in clone A cells, cathepsin A is the major enzyme that mediates such process. Beyond cathepsin A and CES1, a series of cysteine and (lysosomal) serine proteases have also been identified as being able to hydrolyze the carboxyl ester motif of the ProTides in quiescent cells and peripheral blood mononuclear cells (PBMC).82
Earlier work by McGuigan and colleagues83 into the efficiency of the ester group hydrolysis in various ProTides indicated that this is dependent on the structure of the ester moiety. In particular, it was noted that ProTides with a tert-butyl ester group were more slowly hydrolyzed than those bearing other esters, e.g., methyl, isopropyl, and benzyl, which correlated with the biological activity.83 Although the lipophilicity of the ester motif seemed to give an increase in biological activity most likely due to increased cellular uptake, the rate of ester hydrolysis is another influencing factor in the ultimate biological activity of the ProTides.83 For instance, ProTides with naphthyl esters yield more active ProTides than their phenyl ester counterparts due to higher lipophilicity and faster hydrolysis (Figure 7). However, propylbenzene esters, which are more lipophilic, are not as active as phenyl esters probably due to poor in vivo hydrolysis.
Figure 7.

Effect of the ester motif on the ProTides’ biological activity. The data presented regard d4T and its ProTides and were obtained from McGuigan et al.83
In addition to the ester group’s influence on the overall lipophilicity of the ProTides and its effect on the biological activity, the aryl motif has significant impact on the lipophilicity. In McGuigan’s work, the aryl group mostly used was the phenyl group although the naphthyl group has also been explored. The biological activity between these two aryl motifs often correlated with lipophilicity, and hence the naphthyl-containing ProTides exhibit a better biological activity than their phenyl counterparts. Notably, substitutions on these aryl motifs exerts a significant impact on the biological activity (Figure 8). Electron-withdrawing substitutions were found to be associated with increased biological activity as compared to the electron-donating groups. The position on the aryl group that allows maximum electron-withdrawing effects, i.e., ortho and para, had more pronounced effects than the meta position.
Figure 8.

Effect of the aryl group substitutions on the ProTides’ biological activity. The data presented regard d4T and its ProTides and were obtained from Siddiqui et al.84
Beyond the nature of the ester group, it has been shown that the nature of the amino acid of the ProTides also affects the rate of cathepsin A-mediated hydrolysis of the ester group (Figure 9).76 Indeed, cathepsin A was unable to hydrolyze ProTides with branched amino acids such as valine and isoleucine. Subsequent studies showed that the nature of the amino acid side chain, primarily its size (bulkiness) and charge, has great influence on what enzyme hydrolyzes the ester motif.82 Indeed, an interesting structure–activity relationship emerged from the influence of the amino acid on the rate of the metabolism of ProTides. For instance, it was found that the ester motif of ProTides with alanine were efficiently processed by esterases (e.g., leukocyte elastase and proteinase 3) and serine proteases but not cysteine, aspartic, or metalloproteases.82 Amino acids with bulkier side chains, e.g., phenylalanine and leucine, were more efficiently hydrolyzed by chymotrypsin-related serine proteases such as cathepsin G and chymase.82
Figure 9.

Effect of the amino acid on the ProTides’ biological activity. The data presented regard d4T and its ProTides and were obtained from McGuigan et al.85
After the ester motif at the amino acid moiety is cleaved off, a free carboxylate, which under physiological pH carries a negative charge, is generated. Subsequently, a nucleophilic attack by the carboxylate functionality on the phosphate or phosphonate group causes the loss of the aryl-leaving group, yielding a highly unstable five-membered anhydride ring. The speed of the nucleophilic attack from the carboxylate on the phosphate or phosphonate group varies depending on the nucleoside. For instance, AZT metabolite A formation (Figure 6) was slower than that of d4T.75 Interestingly, the hydrolysis of ProTides with β-alanine did not release the nucleoside monophosphate because the hydrolysis did not proceed beyond the hydrolysis of the ester motif.75 Metabolite B formation is followed by a rapid nucleophilic attack by a water molecule on the phosphate (option 1, Figure 6) or carbon of the carbonyl group of the amino acid (option 2, Figure 6) to open up the ring and to generate the phosphoramidate metabolite (metabolite C, Figure 6). Although no compelling evidence exists to whether the water molecule attacks the phosphate or the carbon of the carbonyl group of the amino acid, which both yield the same product, the cleavage of the P–N bond at this stage has never been observed.86
The final step in the metabolism of the ProTides is the cleavage of the P–N bond by a second enzyme known as a phosphoramidase-type enzyme (histidine triad nucleotide binding protein 1,87 HINT1).81,88,89 This eventually leads to the release of the nucleoside monophosphate/phosphonate. Notably, the cleavage of the P–N bond can be resistant to HINT1 as is the case with TAF. It is hypothesized that the hydrolysis of the P–N bond in this case proceeds because of the chemical instability of the P–N bond in the acidic environment of endosomes/lysosomes.90
5. ProTides in Drug Discovery
Since the invention of the ProTide technology by the McGuigan laboratory in the early 1990s, this modality has been widely employed in the synthesis of many nucleoside monophosphate or monophosphonate ProTides. Although initially the structure–activity relationship (SAR) studies were performed on pyrimidine nucleosides such as AZT and d4T, a wide variety of other ProTide derivatives have also been synthesized and antivirally evaluated. Notably, application of the phosphoramidate ProTide technology on purine nucleoside analogues such as 2′,3′-dideoxyadenosine (ddA) and 2′,3′-didehydro-2′,3′-dideoxyadenosine(D4A),91 carbocyclic adenosine derivatives,92 and carbovir/abacavir93,94 often led to markedly enhanced antiviral potencies of such ProTides. More recently, it was even shown that ProTides of the antiherpetic acyclovir resulted in both antiherpetic and anti-HIV activity95,96 and thus, enabled broadening of the antiviral spectrum of such drug. In this respect, it should be mentioned that the ProTide technology not only enables significant optimization of existing antiviral drugs, but it also allows conversion of apparently looking inactive molecules to active antivirals by the improved uptake and direct delivery of their activated (monophosphate) species. A series of ProTides have reached clinical trials,33 with two of them already approved for clinical use.13 To highlight the usefulness of this prodrug technology in drug discovery, key ProTides that are currently reported to be undergoing clinical trials and those that have received FDA-approval will be discussed.
5.1. ProTides as Clinical Candidates
5.1.1. Benzyl ((((2R,3R,5R)-5-(4-Amino-2-oxopyrimidin-1(2H)-yl)-4,4-difluoro-3-hydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)-L-alaninate (NUC-1031)
NUC-1031 (1, Figure 10)64 is a phenyl l-alanine benzyl ester ProTide of the FDA-approved anticancer nucleoside analogue gemcitabine (Gemzar)97 and developed for clinical use by NuCana Biomed Ltd. Gemcitabine emerged as an ideal candidate for the application of the ProTide technology because of the emerging resistance against this anticancer therapeutic agent. Gemcitabine resistance mechanisms include inadequate conversion of gemcitabine into its pharmacologically active di- and triphosphate forms due to downregulation of deoxycytidine kinase, deamination of gemcitabine to its much less active uridine derivative, and mutations in the membrane transporter that mediates its active uptake into cells.98
A series of gemcitabine ProTides were synthesized and probed for their cytostatic activities.64 The lead structure from these studies was 1 as it exhibited significant antiproliferative activity, superior in vivo stability, and an excellent PK profile.64
A notable result that emerged from the ProTide was that it was able to effectively overcome gemcitabine resistance. This was attributed to three main reasons: first, cytidine deaminase is an enzyme found in both serum and cells and is known to catabolize gemcitabine. For deamination to occur, unprotected hydroxyl groups on the 3′- and 5′-position of the nucleoside analogue must be present.99 ProTide 1 demonstrated resistance to deamination because of the presence of the phosphoramidate moiety on the 5′-O-position of the ribose moiety. A second advantage of being resistant to deaminase breakdown is an increased safety profile, as lesser toxic metabolite (2′,2′-difluorodeoxyuridine) would be produced.64 Second, it was found that the cellular uptake of 1 was not dependent upon nucleoside transporters and that the ProTide was lipophilic enough to enter the cells via passive diffusion. This was deduced following an experiment involving dipyridamole, a known inhibitor of the human equilibrative nucleoside transporter 1 (hENT1), to mimic the downregulation of nucleoside transport in cancer cells. Gemcitabine was incubated with pancreatic PANC1 cancer cells in the presence of dipyridamole, and it was found that its cytotoxic activity was significantly reduced, with the cytostatic activity decreasing from 611 nM to >2000 nM (CC50). Contrastingly, even with blockage of hENT1, the activity of 1 remained fairly constant (CC50 = 162 μM), thus confirming that the cellular uptake of 1 is independent of nucleoside transporters.64 Third, the structure of the ProTide is such that it is able to overcome the rate-limiting first phosphorylation-mediated by deoxycytidine kinase which is downregulated in certain cancers. This was confirmed by little change in the activity of 1 when incubated with RT112 cancer cells in the presence of 2′-deoxycytidine being the competing natural substrate for gemcitabine activation to its 5′-monophosphate by 2′-deoxycytidine kinase (EC50 = 0.7 μM in the presence of 2′-deoxycytidine compared to 0.2 μM in the dCyd absence). Conversely, the activity of gemcitabine decreased when 2′-deoxcytidine was coadministered as the two compounds competed for the same active site on the deoxycytidine kinase.64
In vivo animal studies of compound 1 were conducted, focusing in particular on gemcitabine (partially) resistant pancreatic cancers. The initial results obtained were very promising. In mouse models with human tumor xenografts, 1 reduced the tumor size faster than gemcitabine, and significant reductions in tumor size were noticeable on day 7 post administration of the ProTide. The body weight of the animals was also recorded to assess their tolerance to the compound. Overall, ProTide 1 was associated with less than 4% reduction in body weight and it was found that over the course of the treatment, the mean body weight change was less pronounced in mice that were treated with 1 compared to those treated with gemcitabine. This indicated that the gemcitabine ProTide was better tolerated than the parent nucleoside analogue.64 The pharmacokinetics of the ProTide were also considerably better than those of gemcitabine: the half-life of 1 was 7.9 h compared to 1.5 h for gemcitabine. The ProTide also led to intracellular concentrations of the active gemcitabine triphosphate metabolite that was 13 times higher than achieved by gemcitabine.100
Phase I/II clinical trials were conducted involving 68 patients and showed that 1 was effective in a variety of different cancers but most notably in patients with ovarian, pancreatic, and biliary cancers. Phase III studies will be conducted focusing on these cancers in particular. The ProTide was found to be stable in vivo, exhibiting a plasma half-life of 8.3 h. Maximum intracellular concentrations of the active metabolite were quickly achieved, peaked at 475 μM, and were maintained for 24 h. In several patients, the tumor had shrunk by more than 30%, whereas in 33 patients, the disease had stabilized.101
5.1.2. Benzyl ((((2R,3S,5R)-5-(5-fluoro-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3-hydroxytetrahydrofuran-2-yl)methoxy)(naphthalen-1-yloxy)phosphoryl)-l-alaninate (NUC-3373)
Another ProTide currently being developed for clinical use by NuCana Biomed Ltd. is NUC-3373 (2, Figure 10),79,102 a phosphoramidate prodrug of 5-fluoro-2′-deoxyuridine (FdU). FdU is a metabolite which gets formed in vivo from the pharmacologically active anticancer agent 5-fluorouracil (5-FU). Initially discovered by McGuigan and his colleagues in 2011 after extensive SAR studies,79 ProTide 2 displayed promising preclinical results and initial studies highlighted its superiority over its parent nucleoside, 5-FU. The path toward the final structure of 2 began with McGuigan and his co-workers by synthesizing and comparing 39 different ProTides, with variations in the aryl, ester, and amino acid regions.79 There was little difference observed in the potency of the ProTides upon varying the amino acid ester region and also upon changing the nature of the amino acid. l-Alanine has long been the amino acid of choice when formulating ProTides, but there was little to discern between l-alanine and the other amino acids in these experiments. Generally, the ProTides yielding the 1-naphthyl group displayed more potency than phenyl-containing ProTides, thus leading to the deduction that the 1-naphthyl, benzyl ester-containing ProTide was the most effective.79 To determine the cytotoxic activity of 2, in vitro studies were conducted involving human tumor cell lines (colorectal, lung, ovarian, acute lymphoblastic leukemia, and cervix). Attention to thymidine kinase and the nucleoside transporter hENT1 was also given to investigate the characteristics of cancer resistance. Xenograft mouse models were also used to further analyze the anticancer activity of the ProTide.103
It was found that compound 2 was, on the whole, significantly more cytotoxic than 5-FU, displaying IC50 values 2–333-times lower in the majority of the tumor cell lines tested. The ProTide also produced 363-times more of the active metabolite (FdU) than 5-FU, thus further displaying its superiority in efficacy. Thymidine kinase inhibition only slightly affected ProTide 2 (∼4-fold) compared to FdU that showed reductions in cytotoxicity of up to 136-fold, respectively, indicating the independence of the ProTide for the FdU-activating enzyme. Significant results were obtained with the colorectal tumor xenografts: ProTide 2 inhibited tumor growth by 47% compared to 25% tumor growth inhibition demonstrated by 5-FU.103
Compound 2 was found to overcome the main mechanisms of resistance that is associated with 5-FU and other drugs within this family, such as decreased levels of the necessary phosphorylating (activating) enzyme thymidine kinase, which is required to produce the active metabolite; an overexpression of thymidylate synthase (an enzyme responsible for DNA synthesis and a target for FdUMP (the active metabolite of 5-FU) altered levels of thymidine phosphorylase and reduced entry into the cells via nucleoside transporters.79,102 ProTide 2 overcomes these pathways to resistance by preventing degradation by phosphorolytic enzymes, bypassing the need to be activated by thymidine kinase, and being able to diffuse into cells, independent of nucleoside transporters such as hENT1.104 Given the encouraging results obtained in the preclinical studies, ProTide 2 entered phase I clinical trials in the first quarter of 2016 in pursuance of determining the pharmacokinetic profile of the ProTide, the safety of the compound in human participants and clinical activity of 2 in tumor-bearing patients.103 The trial itself consists of two parts: the first part assessing the optimum dose that will maximize the risk/benefit reward to the participants when given a weekly dose of the ProTide, administered as a single intravenous infusion. The second part will assess the same as part one, except the dosing interval which will be increased to fortnightly (www.clinicaltrials.gov, trial identifier: NCT02723240).
5.1.3. 2-Ethylbutyl ((S)-(((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)-l-alaninate (GS-5734)
In 2014, there was a mass outbreak of the Ebola virus, most prevalent in Western Africa, although there were a number of reported cases in the USA and the United Kingdom. In West Africa alone, there were a total of 28616 confirmed cases in Guinea, Liberia, and Sierra Leone, leading to 11310 deaths (WHO 2016). To this date, there is no drug treatment available on the market to treat this disease. However, GS-5734 (3, Figure 10),105 developed by Gilead Sciences, has shown promising results as a potential treatment for the Ebola virus. Although most of the interest in this compound has been due to its activity against Ebola, recently, 3 was reported to be a broad-spectrum antiviral agent because it exhibited potent antiviral activity against a variety of viruses including filoviruses, coronaviruses, and paramyxoviruses.106
Compound 3 is a ProTide of an adenosine analogue which, following intracellular delivery, gets metabolized into the pharmacologically active nucleoside triphosphate form, which then inhibits viral RNA dependent RNA polymerase.105
The activity of 3 was initially tested using cell-based assays, followed by the administration of the ProTide to rhesus monkeys that were infected with the Ebola virus. Rodent studies were deemed not suitable to determine the pharmacokinetics of 3 due to the high serum concentrations of esterases present in many rodent species, which would prematurely degrade the ProTide and impact on the findings. Thus, rhesus monkeys were chosen as they closely resemble humans and they do not express a high serum esterase concentration.105 Then 10 μM 3 was incubated in a 2 h pulse or 72 h continuous incubation of Ebola virus-infected human primary macrophages, endothelial cells, and liver cells to deduce its activity. Then 10 μM nucleoside analogue was also incubated for 72 h in human monocyte-derived macrophages for comparison. ProTide 3 was found to be rapidly metabolized in the primary macrophages, with maximum intracellular concentrations of nucleoside triphosphate being achieved after 4 h post incubation, with a half-life of 16 h. It was found that 3 displayed an EC50 value against Ebola virus of 0.086 μM in the human primary macrophages, 0.14 μM in the endothelial cells, and 0.07 μM in the Huh-7 liver cells. In comparison, the nucleoside analogue displayed an EC50 value of greater than 20 μM in both the human macrophages and endothelial cells, and 1.5 μM in the liver cells. The cytotoxic concentration (CC50) of the ProTide in all cell lines was above 20 μM. The different cell lines were also infected with different variants of the Ebola virus (Makona and Kikwit variants) and other filoviruses. The cell-based assays gave strong indication that compound 3 would be effective in treating the other viruses as well.105
The pharmacokinetics of 3 were examined in rhesus monkeys. After being administered intravenously with 10 mg/kg of the ProTide, 3 displayed a very short plasma half-life of just 0.39 h. There was rapid distribution into the peripheral compartments within 2 h post administration, and a half-life in the peripheral blood mononuclear cells of 14 h was observed. Levels required to achieve a 50% inhibition of the Ebola virus were maintained for 24 h.105 Compiling the pharmacokinetics results and tissue distribution data led to the conclusion that once daily dosing would be effective to achieve a clinical effect.
To assess the efficacy of the ProTide, a blinded placebo-controlled study was conducted involving 12 Ebola-infected rhesus monkeys (six per treatment group) in two different modalities.105 The first part involved intramuscular administration of 3 either on day 0 (same day as being infected with Ebola), or day 2. Part two of the study administered a loading dose of 10 mg/kg followed by once daily doses of 3 mg/kg, commencing at various days after virus exposure or 10 mg/kg administered on day 3 post virus exposure. All of the monkeys given 3 survived until the end of the trial; 50% survival was noted with the treated group of animals given 3 mg/kg compared to no survival in the placebo group. It was noted that monkeys given the 10 mg/kg dose after day 3 had 100% survival along with reduced signs associated with the Ebola virus (thrombocytopenia, coagulopathy).105
Given the promising results obtained in the rhesus monkey study, a phase II clinical trial is due to be conducted in Liberia. Sponsored by the National Institute of Allergy and Infectious Diseases, 60–120 men infected with Ebola virus will be recruited and will be given a five-day course of 3, administered once daily, followed by monitoring over a six-month period to determine the clearance of virus from a persistent genital reservoir (www.clinicaltrials.gov, trial identifier: NCT02818582).
5.2. ProTides as Drugs
There have been two ProTide drugs approved by the FDA for clinical use. Sofosbuvir (Sovaldi), Figure 3, was approved by the FDA in December 2013 for the treatment of HCV.107 Sofosbuvir is a phosphoramidate prodrug of 2′-deoxy-2′-fluoro-2′-C-methyluridine, the parent nucleoside. Having emerged as the superior ProTide out of 50 other ProTides developed,35 sofosbuvir was acquired by Gilead Sciences, where it adopted the identity sofosbuvir and underwent further extensive clinical evaluation. Sofosbuvir was found to act by inhibiting the HCV NS5B polymerase, an enzyme responsible for the replication of the viral genome. Inhibition of this enzyme would result in RNA chain termination and ultimately stop the replication of HCV.35 Following cellular uptake by hepatocytes, sofosbuvir undergoes sequential hydrolysis by cathepsin A and CES1 to form the active nucleoside monophosphate. Further hydrolysis by HINT1 and subsequent phosphorylations yielded the pharmacologically active triphosphate metabolite, which exerts its action on NS5B polymerase.108 Initial animal studies of sofosbuvir were encouraging. The ProTide was well absorbed following oral administration in portal vein cannulated dogs, bioavailability was 9.9%, which equated to a fraction absorbed of 36%.109 In human subjects, the absolute bioavailability was not determined, although over 80% of the administered dose was absorbed. The absorption was rapid, with maximum concentrations (Cmax) of sofosbuvir achieved 0.5–2 h post oral administration and Cmax of the inactive parent nucleoside metabolite was achieved 2–4 h post administration and a half-life of 0.48–0.75 h. Ex vivo studies of the distribution of the anti-HCV agent showed a plasma protein binding of around 82% in healthy subjects and 85% in subjects with end stage renal disease.108
Studies using the diastereomeric mixture of sofosbuvir in humans showed that it was rapidly absorbed and metabolized in the liver according to the typical in vivo degradation of the ProTides (see Figure 6). Briefly, the isopropyl ester motif was hydrolyzed into the naked carboxylate group and then the P–N bond of the phosphoramidate group was cleaved off to afford parent nucleoside monophosphate.110 Notably, the parent nucleoside, an inactive metabolite, was detectable in the plasma and remained detectable for at least 24 h following the 100 and 800 mg dosing of the diastereomeric mixture of sofosbuvir.110 Although sofosbuvir showed little indication of being excreted renally (3.5% recovered in urine; renal clearance = 0.238 L/min), the inactive parent nucleoside was the predominant compound present in the urine (77.7%), indicating excretion via the kidneys. This was further confirmed when studies were conducted by observing the pharmacokinetics of sofosbuvir in patients with impaired renal function; the renal clearance of the parent nucleoside was around 2-fold higher than the glomerular filtration rate, signifying active renal excretion of the metabolites of sofosbuvir.109 Phase II studies of sofosbuvir in combination with pegylated interferon and ribavirin established the doses required to provide an optimal effect while minimizing any potential adverse effects.111 Initial studies postulated that 200 mg or 400 mg doses of sofosbuvir with pegylated interferon and ribavirin were most effective, as 100 mg dosing was found to be suboptimal due to low viral load decline than observed with the 200 mg and 400 mg doses. At these doses, the incidence of adverse effects was low, with the most frequent adverse effects being fatigue and nausea.111 Coupled with the short half-life of sofosbuvir, once daily dosing was recommended as there was little accumulation of the drug.109
The safety of sofosbuvir was later confirmed in phase II clinical trials. It was postulated that as sofosbuvir is not metabolized via the cytochrome P450–3A4 enzymes, drug–drug interactions would be minimal. However, because it seems to induce the P-glycoprotein efflux pump, caution was advised with coadministration of carbazepine and rifabutin as the concentrations of sofosbuvir can decrease significantly.112,113
Single-group, open label phase III study of treatment-naïve patients with HCV genotypes 1, 4, 5, and 6 treated with sofosbuvir plus pegylated interferon and ribavirin for 12 weeks indicated high potency of sofosbuvir.107 Indeed, 80% of cirrhotic patients versus 92% of noncirrhotic patients achieved a viral load below the lower limit of quantifications 12 weeks after completion of HCV treatment (SVR12). This indicated the efficacy of sofosbuvir in treating different HCV genotypes, confirming similar earlier preclinical observations.114 In a placebo-controlled phase III study comparing sofosbuvir and ribavirin for 12 weeks with matched interferon ineligible controls in genotype 2 and 3 patients, sustained viral response was achieved in 78% of treated patients with 93% response rate in genotype 2 patients and 61% in genotype 3 patients.115 In a study involving patients who had previously been treated with interferon who were treated with either sofosbuvir and ribavirin for 12 weeks followed by placebo for 4 weeks or sofosbuvir and ribavirin for 16 weeks, response rates showed a 73% SVR12 in the 16-week arm compared to 50% of the 12-week arm. Impressively, 94% of genotype 2 patients achieved SVR12 within 12 weeks of treatment with sofosbuvir and ribavirin, while 87% achieved it within 16 weeks. As for genotype 3 patients, 30% of patients achieved SVR12 within 12 weeks compared to 62% within 16 weeks. Notably, 46% of the patients treated with sofosbuvir and ribavirin for 12 weeks had viral relapse compared to 27% of the 16-week treatment arm. In another study, the efficacy of extended treatment of 24 weeks of sofosbuvir plus ribavirin in genotype 3 patients was evaluated.116 The results showed that 93% of the 73 patients with genotype 2 who took part achieved SVR12 within 12 weeks of sofosbuvir and ribavirin treatment, whereas 85% of the 250 patients with genotype 3 treated for 24 weeks with sofosbuvir and ribavirin achieved SVR12. This was the first large scale study showing the efficacy of prolonged interferon-free treatments in genotype 3 patients. Given the excellent safety profile and pan-genotypic efficacy of sofosbuvir, it has been licensed for clinical use in combination with pegylated interferon, ribavirin, and the NS5A inhibitors ledipasvir and velpatasvir depending on the HCV genotype to be treated.117 Collectively, this has made sofosbuvir a cornerstone in HCV therapy.
TAF, Figure 3, is the latest ProTide-based drug approved by the FDA for the treatment of HIV.118 Developed by Gilead Sciences Inc., TAF is a ProTide of the acyclic nucleoside phosphonate drug tenofovir. Another prodrug of tenofovir, tenofovir disoproxil fumarate (TDF), had long been an FDA-approved treatment for HIV, however, the ProTide TAF demonstrated increased anti-HIV activity, higher in vivo stability, and was endowed with fewer side effects than both tenofovir and TDF.118 TAF is specifically accumulating in lymphatic tissue, and in the liver, and thus, holds also great potential for the treatment of HBV infections. Markedly lower plasma levels but significantly higher intracellular tenofovir concentrations are observed with TAF as compared to TDF, explaining the lower incidence of tenofovir-associated side effects with respect to bone mineral density losses and kidney nefrotoxicity.119 The drug itself becomes phosphorylated following cellular uptake into the pharmacologically active diphosphate form and is a potent inhibitor of reverse transcriptase. Inhibition of this enzyme results in DNA chain termination and viral replication ceases to progress.120
Early studies of TAF were conducted in dogs in order to characterize its pharmacokinetic profile and understand its permeability and efficacy as a potential treatment for HIV. Initial in vitro studies using hepatic and intestinal S9 fractions from both humans and dogs yielded similar results, highlighting the suitability of using dogs for the preclinical studies. In vitro experiments showed TAF to have half-lives of 31 min in both dog intestinal and hepatic S9 fractions, a plasma half-life of 92 min, and a half-life of 28.3 min in an MT-2 cell extract.80 When 5 mg/kg of TAF were administered to the dogs, plasma concentrations peaked at 1.58 μM, achieved at 0.14 h post administration. The levels of TAF decreased rapidly (undetectable after 2 h post administration), which was accompanied by an increase in tenofovir levels. Rapid accumulation of the active metabolite tenofovir diphosphate in the peripheral blood mononuclear cells followed, with peak concentrations of 18 μM achieved at 1 h post administration. The bioavailability of TAF was found to be dependent on the administered dose, with bioavailabilities of 1.7% and 24.7% at 2 and 20 mg/kg, respectively.121 However, other studies found that oral administration of 10 mg/kg of TAF yielded a bioavailability of 17% when compared to the intravenous dose,80 with the majority of the drug metabolized in the kidneys and excreted in the urine.122
An earlier study conducted by Eisenberg et al. in 2001123 compared the activity of the ProTide with tenofovir and its prodrug TDF. Following incubation with peripheral blood mononuclear cells, it was found that the levels of tenofovir and its metabolites were 10–30 times higher compared to TDF and tenofovir, respectively.123 The in vitro anti-HIV activity of TAF was determined to be 1000-fold and 10-fold greater than tenofovir and TDF.80
Following successful in vitro assessments of TAF, the ProTide progressed into a number of phase I and II trials, where the in vivo pharmacokinetics/dynamics of the ProTide was evaluated and compared to its predecessors along with establishing the optimum dose of TAF. An initial study was conducted by Markowitz et al.,124 where either 40 or 120 mg of TAF or 300 mg of TDF were administered once daily for 14 days and the activity of both prodrugs as monotherapy for the HIV-1 infection was compared. The study showed that both 40 mg and 120 mg of TAF resulted in higher intracellular concentrations of tenofovir diphosphate in PBMC than tenofovir. TAF also produced a greater net change in HIV-1 RNA compared to TDF, thus illustrating its superiority over the existing prodrug in terms of potency.124
Phase II/III studies of TAF focused on its efficacy in different patient groups including, but not limited to, adults and adolescents. As tenofovir is eliminated via the kidneys, it was imperative to understand how renal function would affect both the accumulation of tenofovir in the body and also its excretion. TDF has been associated with effects on the bone and renal toxicity, leading to decreased glomerular filtration rates, increases in serum creatinine, and increased risk of adverse renal events. This toxicity was attributed to its effect in the proximal tubule, with efficient uptake from plasma but less efficient efflux into urine resulting in accumulation of tenofovir in the body.125 Therefore, a phase 3 double-blinded study was conducted in which TAF was administered to patients with impaired renal function. Patients were given either 10 mg of TAF or 300 mg of TDF, each coformulated with elvitegravir, cobicistat, and emtricitabine. The study found that participants on the TAF treatment regime displayed a smaller increase in creatinine, less proteinuria, and significant decreases in bone mineral density.126
TAF was demonstrated to be superior not only in its antiviral activity but also in the fact that therapeutic effects could be achieved at much lower doses than TDF, thus reducing the incidence of adverse effects. This led to the FDA approval of the ProTide in the treatment of HIV-1 in a combination therapy with other anti-HIV agents.118 Moreover, in November 2016, the FDA granted approval for a once-daily treatment of 25 mg TAF in patients suffering from chronic HBV infection (Gilead Sciences 2016). This followed extensive studies which highlighted the ProTides superiority over current therapies used to treat not only HIV but also chronic HBV infections.127
6. Conclusions and Outlook
The ProTide technology has facilitated the discovery and application of a variety of nucleoside monophosphate and monophosphonate analogues in the clinic. It took many years of trial and error, mainly by the McGuigan group, to optimize the masking groups of the phosphate to yield the successful ProTide technology as we know it today. Although this technology has largely been used in the discovery of nucleoside-based ProTides as antiviral and anticancer agents, recently it was applied in the early discovery of nucleoside-based ProTides for Parkinson’s disease.78 Additionally, the ProTide prodrug approach is receiving increasing attention as a prodrug strategy for the intracellular delivery on monophosphorylated non-nucleoside compounds such as glucosamine,128,129 Sphingosine 1-phosphate (S1P),130 4-phospho-d-erythronohydroxamic acid,131 and 5-phospho erythronohydroxamic acid.132 Together, these examples show the broad usefulness of this prodrug technology and highlight its potential in the discovery of non-nucleoside monophosphate and monophosphonate prodrugs as potential therapeutics as well. Regardless of the substrates to which the ProTide technology is applied to either nucleosides or non-nucleosides, it has already proven to be a powerful technology in drug discovery and efficient intracellular delivery of the (activated) parental drug. Given its expanding applications, there is a real possibility that this technology will deliver new (nucleoside and non-nucleoside) ProTides for the treatment of a variety of other diseases beyond microbial infections and cancer.
Glossary
Abbreviations Used
- 5-FU
5-fluorouracil
- AraA
9-β-d-arabinofuranosyladenine
- AraC
1-β-d-arabinofuranosylcytosine
- AZT
3′-azidothymidine
- d4A
2′,3′-didehydro-2′,3′-dideoxyadenosine
- CES1
carboxylesterase 1
- d4T
2′,3′-didehydro-2′,3′-dideoxythymidine
- ddC
2′,3′-dideoxyadenosine
- ddC
2′,3′-dideoxycytidine
- DCC
N,N′-dicyclohexylcarbodiimide
- FDA
Food and Drug Administration
- FdU
5-fluoro-2′-deoxyuridine
- FdUMP
5-fluoro-2′-deoxyuridine-5′-monophosphate
- HBV
hepatitis B virus
- HCMV
human cytomegalovirus
- HCV
hepatitis C virus
- hENT1
human equilibrative nucleoside transporter 1
- HINT1
histidine triad nucleotide-binding protein 1
- HIV
human immunodeficiency virus
- HSV
herpes simplex virus
- NA
nucleoside analogue
- NMI
N-methylimidazole
- PK
pharmacokinetics
- SVR12
the lower limit of quantifications 12 weeks after completion of HCV treatment
- TAF
tenofovir alafenamide
- TDF
tenofovir disoproxil fumarate
- SAR
structure–activity relationship
- tBuMgCl
tert-butyl magnesium chloride
- TK
thymidine kinase
Biographies
Youcef Mehellou obtained his M.Pharm. (Pharmacy) degree from King’s College London in 2005. This was followed by a Ph.D. on “ProTide” chemistry under the supervision of Prof. Chris McGuigan at Cardiff University, U.K. Following postdoctoral work initially with Prof. Sidney M. Hecht at Arizona State University (2009/2010) and then with Prof. Dario R. Alessi FRS at the University of Dundee, U.K. (2010/2013), Youcef moved to the University of Birmingham, U.K., in early 2013 as a Lecturer in Medicinal Chemistry. Since January 2017, Youcef has been a Lecturer in Medicinal Chemistry in Cardiff University, U.K., where his group is focusing on the discovery of small molecule modulators of signal transduction and the development of novel phosphate and phosphonate prodrug therapeutics.
Hardeep S. Rattan is currently a fourth and final year Pharmacy student at the University of Birmingham, U.K. He has a keen interest in medicinal chemistry, particularly the discovery and development of nucleoside and nucleotide therapeutics. Hardeep’s final year Masters research project focused on the ProTide prodrug technology and was carried out under the supervision of Dr. Mehellou.
Jan Balzarini graduated from the KU Leuven (Belgium) as Master in Biology (1975), Master in Bioengineering (1977), and Ph.D. in Bioengineering (1984). He is affiliated to the Rega Institute for Medical Research at the KU Leuven. In 1984, he became tenured and performed a postdoctoral stay at the National Cancer Institute of the NIH (1985/1986) with Sam Broder and David G. Johns. In 2001, he was awarded the prestigious René Descartes Research Prize of the European Commission (in collaboration with several European Research Teams), and he received several other (inter)national awards. In 2001, he became full professor at the KU Leuven; in 2006, he was appointed as Laboratory Head of Virology and Chemotherapy at the Rega Institute, and in 2014, he retired as Emeritus Professor.
The authors declare no competing financial interest.
Dedication
This review was written in memory of Prof. Christopher McGuigan (1958–2016), who provided the basis for the ProTide technology.
This article is made available for a limited time sponsored by ACS under the ACS Free to Read License, which permits copying and redistribution of the article for non-commercial scholarly purposes.
References
- Perkins E. S.; Wood R. M.; Sears M. L.; Prusoff W. H.; Welch A. D. Anti-viral activities of several iodinated pyrimidine deoxyribonucleosides. Nature 1962, 194, 985–986. 10.1038/194985a0. [DOI] [PubMed] [Google Scholar]
- Fyfe J. A.; Keller P. M.; Furman P. A.; Miller R. L.; Elion G. B. Thymidine kinase from herpes simplex virus phosphorylates the new antiviral compound, 9-(2-hydroxyethoxymethyl)guanine. J. Biol. Chem. 1978, 253, 8721–8727. [PubMed] [Google Scholar]
- De Clercq E.; Descamps J.; De Somer P.; Barr P. J.; Jones A. S.; Walker R. T. E)-5-(2-Bromovinyl)-2′-deoxyuridine: a potent and selective anti-herpes agent. Proc. Natl. Acad. Sci. U. S. A. 1979, 76, 2947–2951. 10.1073/pnas.76.6.2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E. Nucleoside analogues as antiviral agents. Acta Microbiol. Acad. Sci. Hung. 1981, 28, 289–306. [PubMed] [Google Scholar]
- Robins R. K. The potential of nucleotide analogs as inhibitors of retroviruses and tumors. Pharm. Res. 1984, 1, 11–18. 10.1023/A:1016370407633. [DOI] [PubMed] [Google Scholar]
- Jordheim L. P.; Durantel D.; Zoulim F.; Dumontet C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discovery 2013, 12, 447–464. 10.1038/nrd4010. [DOI] [PubMed] [Google Scholar]
- De Clercq E.; Li G. Approved antiviral drugs over the past 50 years. Clin. Microbiol. Rev. 2016, 29, 695–747. 10.1128/CMR.00102-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton J.; Lu X.; Hollenbaugh J. A.; Cho J. H.; Amblard F.; Schinazi R. F. Metabolism, biochemical actions, and chemical synthesis of anticancer nucleosides, nucleotides, and base analogs. Chem. Rev. 2016, 116, 14379–14455. 10.1021/acs.chemrev.6b00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loffler M.; Morote-Garcia J. C.; Eltzschig S. A.; Coe I. R.; Eltzschig H. K. Physiological roles of vascular nucleoside transporters. Arterioscler., Thromb., Vasc. Biol. 2007, 27, 1004–1013. 10.1161/ATVBAHA.106.126714. [DOI] [PubMed] [Google Scholar]
- Molina-Arcas M.; Casado F. J.; Pastor-Anglada M. Nucleoside transporter proteins. Curr. Vasc. Pharmacol. 2009, 7, 426–434. 10.2174/157016109789043892. [DOI] [PubMed] [Google Scholar]
- Anderson P. L.; Kakuda T. N.; Lichtenstein K. A. The cellular pharmacology of nucleoside- and nucleotide-analogue reverse-transcriptase inhibitors and its relationship to clinical toxicities. Clin. Infect. Dis. 2004, 38, 743–753. 10.1086/381678. [DOI] [PubMed] [Google Scholar]
- Mehellou Y.; De Clercq E. Twenty-six years of anti-HIV drug discovery: where do we stand and where do we go?. J. Med. Chem. 2010, 53, 521–538. 10.1021/jm900492g. [DOI] [PubMed] [Google Scholar]
- Thornton P. J.; Kadri H.; Miccoli A.; Mehellou Y. Nucleoside phosphate and phosphonate prodrug clinical candidates. J. Med. Chem. 2016, 59, 10400–10410. 10.1021/acs.jmedchem.6b00523. [DOI] [PubMed] [Google Scholar]
- Smal C.; Vertommen D.; Bertrand L.; Ntamashimikiro S.; Rider M. H.; Van Den Neste E.; Bontemps F. Identification of in vivo phosphorylation sites on human deoxycytidine kinase. Role of Ser-74 in the control of enzyme activity. J. Biol. Chem. 2006, 281, 4887–4893. 10.1074/jbc.M512129200. [DOI] [PubMed] [Google Scholar]
- Li F.; Maag H.; Alfredson T. Prodrugs of nucleoside analogues for improved oral absorption and tissue targeting. J. Pharm. Sci. 2008, 97, 1109–1134. 10.1002/jps.21047. [DOI] [PubMed] [Google Scholar]
- Saiki Y.; Yoshino Y.; Fujimura H.; Manabe T.; Kudo Y.; Shimada M.; Mano N.; Nakano T.; Lee Y.; Shimizu S.; Oba S.; Fujiwara S.; Shimizu H.; Chen N.; Nezhad Z. K.; Jin G.; Fukushige S.; Sunamura M.; Ishida M.; Motoi F.; Egawa S.; Unno M.; Horii A. DCK is frequently inactivated in acquired gemcitabine-resistant human cancer cells. Biochem. Biophys. Res. Commun. 2012, 421, 98–104. 10.1016/j.bbrc.2012.03.122. [DOI] [PubMed] [Google Scholar]
- Jamieson G. P.; Brocklebank A. M.; Snook M. B.; Sawyer W. H.; Buolamwini J. K.; Paterson A. R.; Wiley J. S. Flow cytometric quantitation of nucleoside transporter sites on human leukemic cells. Cytometry 1993, 14, 32–38. 10.1002/cyto.990140107. [DOI] [PubMed] [Google Scholar]
- Wiley J. S.; Jones S. P.; Sawyer W. H.; Paterson A. R. Cytosine arabinoside influx and nucleoside transport sites in acute leukemia. J. Clin. Invest. 1982, 69, 479–489. 10.1172/JCI110472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damaraju V. L.; Damaraju S.; Young J. D.; Baldwin S. A.; Mackey J.; Sawyer M. B.; Cass C. E. Nucleoside anticancer drugs: the role of nucleoside transporters in resistance to cancer chemotherapy. Oncogene 2003, 22, 7524–7536. 10.1038/sj.onc.1206952. [DOI] [PubMed] [Google Scholar]
- Eriksson S. Is the expression of deoxynucleoside kinases and 5′-nucleotidases in animal tissues related to the biological effects of nucleoside analogs?. Curr. Med. Chem. 2013, 20, 4241–4248. 10.2174/0929867311320340004. [DOI] [PubMed] [Google Scholar]
- Hunsucker S. A.; Mitchell B. S.; Spychala J. The 5′-nucleotidases as regulators of nucleotide and drug metabolism. Pharmacol. Ther. 2005, 107, 1–30. 10.1016/j.pharmthera.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Abraham A.; Varatharajan S.; Abbas S.; Zhang W.; Shaji R. V.; Ahmed R.; Abraham A.; George B.; Srivastava A.; Chandy M.; Mathews V.; Balasubramanian P. Cytidine deaminase genetic variants influence RNA expression and cytarabine cytotoxicity in acute myeloid leukemia. Pharmacogenomics 2012, 13, 269–282. 10.2217/pgs.11.149. [DOI] [PubMed] [Google Scholar]
- Steuart C. D.; Burke P. J. Cytidine deaminase and the development of resistance to arabinosyl cytosine. Nat. New Biol. 1971, 233, 109–110. 10.1038/newbio233109a0. [DOI] [PubMed] [Google Scholar]
- Balzarini J.; Herdewijn P.; De Clercq E. Differential patterns of intracellular metabolism of 2′,3′-didehydro-2′,3′-dideoxythymidine and 3′-azido-2′,3′-dideoxythymidine, two potent anti-human immunodeficiency virus compounds. J. Biol. Chem. 1989, 264, 6127–6133. [PubMed] [Google Scholar]
- Herdewijn P.; Balzarini J.; De Clercq E.; Pauwels R.; Baba M.; Broder S.; Vanderhaeghe H. 3′-substituted 2′,3′-dideoxynucleoside analogues as potential anti-HIV (HTLV-III/LAV) agents. J. Med. Chem. 1987, 30, 1270–1278. 10.1021/jm00391a003. [DOI] [PubMed] [Google Scholar]
- Balzarini J.; Kang G. J.; Dalal M.; Herdewijn P.; De Clercq E.; Broder S.; Johns D. G. The anti-HTLV-III (anti-HIV) and cytotoxic activity of 2′,3′-didehydro-2′,3′-dideoxyribonucleosides: a comparison with their parental 2′,3′-dideoxyribonucleosides. Mol. Pharmacol. 1987, 32, 162–167. [PubMed] [Google Scholar]
- Hao Z.; Cooney D. A.; Farquhar D.; Perno C. F.; Zhang K.; Masood R.; Wilson Y.; Hartman N. R.; Balzarini J.; Johns D. G. Potent DNA chain termination activity and selective inhibition of human immunodeficiency virus reverse transcriptase by 2′,3′-dideoxyuridine-5′-triphosphate. Mol. Pharmacol. 1990, 37, 157–163. [PubMed] [Google Scholar]
- De Clercq E.; Holy A. Acyclic nucleoside phosphonates: a key class of antiviral drugs. Nat. Rev. Drug Discovery 2005, 4, 928–940. 10.1038/nrd1877. [DOI] [PubMed] [Google Scholar]
- Pradere U.; Garnier-Amblard E. C.; Coats S. J.; Amblard F.; Schinazi R. F. Synthesis of nucleoside phosphate and phosphonate prodrugs. Chem. Rev. 2014, 114, 9154–9218. 10.1021/cr5002035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecker S. J.; Erion M. D. Prodrugs of phosphates and phosphonates. J. Med. Chem. 2008, 51, 2328–2345. 10.1021/jm701260b. [DOI] [PubMed] [Google Scholar]
- Wagner C. R.; Iyer V. V.; McIntee E. J. Pronucleotides: toward the in vivo delivery of antiviral and anticancer nucleotides. Med. Res. Rev. 2000, 20, 417–451. . [DOI] [PubMed] [Google Scholar]
- Mehellou Y.; Balzarini J.; McGuigan C. Aryloxy phosphoramidate triesters: a technology for delivering monophosphorylated nucleosides and sugars into cells. ChemMedChem 2009, 4, 1779–17791. 10.1002/cmdc.200900289. [DOI] [PubMed] [Google Scholar]
- Mehellou Y. The ProTides boom. ChemMedChem 2016, 11, 1114–1116. 10.1002/cmdc.201600156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman H.; Kernan M.; Prisbe E.; Rohloff J.; Sparacino M.; Terhorst T.; Yu R. Practical synthesis, separation, and stereochemical assignment of the PMPA pro-drug GS-7340. Nucleosides, Nucleotides Nucleic Acids 2001, 20, 621–628. 10.1081/NCN-100002338. [DOI] [PubMed] [Google Scholar]
- Sofia M. J.; Bao D.; Chang W.; Du J.; Nagarathnam D.; Rachakonda S.; Reddy P. G.; Ross B. S.; Wang P.; Zhang H. R.; Bansal S.; Espiritu C.; Keilman M.; Lam A. M.; Steuer H. M.; Niu C.; Otto M. J.; Furman P. A. Discovery of a beta-d-2′-deoxy-2′-alpha-fluoro-2′-beta-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J. Med. Chem. 2010, 53, 7202–7218. 10.1021/jm100863x. [DOI] [PubMed] [Google Scholar]
- McGuigan C.; Tollerfield S. M.; Riley P. A. Synthesis and biological evaluation of some phosphate triester derivatives of the anti-viral drug AraA. Nucleic Acids Res. 1989, 17, 6065–6075. 10.1093/nar/17.15.6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colin B.; Jones N. M.; McGuigan C.; Riley P. A. Synthesis and biological evaluation of some phosphate triester derivatives of the anti-cancer drug araC. Nucleic Acids Res. 1989, 17, 7195–7201. 10.1093/nar/17.18.7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuigan C.; Shackleton J. M.; Tollerfield S. M.; Riley P. A. Synthesis and evaluation of some novel phosphate and phosphinate derivatives of araA. Studies on the mechanism of action of phosphate triesters. Nucleic Acids Res. 1989, 17, 10171–10177. 10.1093/nar/17.24.10171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schabel F. M. Jr. The antiviral activity of 9-beta-D-arabinofuranosyladenine (Ara-A). Chemotherapia 1968, 13, 321–338. 10.1159/000220567. [DOI] [PubMed] [Google Scholar]
- Talley R. W.; Vaitkevicius V. K. Megaloblastosis produced by a cytosine antagonist, 1-beta-D-arabinofuranosylcytosine. Blood 1963, 21, 352–362. [PubMed] [Google Scholar]
- Mitsuya H.; Weinhold K. J.; Furman P. A.; St Clair M. H.; Lehrman S. N.; Gallo R. C.; Bolognesi D.; Barry D. W.; Broder S. 3′-Azido-3′-deoxythymidine (BW A509U): an antiviral agent that inhibits the infectivity and cytopathic effect of human T-lymphotropic virus type III/lymphadenopathy-associated virus in vitro. Proc. Natl. Acad. Sci. U. S. A. 1985, 82, 7096–7100. 10.1073/pnas.82.20.7096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine K. G.; McGuigan C.; O’Connor T. J.; Nicholls S. R.; Kinchington D. Novel phosphate derivatives of zidovudine as anti-HIV compounds. AIDS 1990, 4, 371–373. [PubMed] [Google Scholar]
- McGuigan C.; Nicholls S. R.; O’Connor T. J.; Kinchington D. Synthesis of some novel dialkyl phosphate derivatives of 3′-modified nucleosides as potential anti-AIDS drugs. Antiviral Chem. Chemother. 1990, 1, 25–33. 10.1177/095632029000100105. [DOI] [Google Scholar]
- Mitsuya H.; Broder S. Inhibition of the in vitro infectivity and cytopathic effect of human T-lymphotrophic virus type III/lymphadenopathy-associated virus (HTLV-III/LAV) by 2′,3′-dideoxynucleosides. Proc. Natl. Acad. Sci. U. S. A. 1986, 83, 1911–1915. 10.1073/pnas.83.6.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuigan C.; O’Connor T. J.; Nicholls S. R.; Nickson C.; Kinchington D. Synthesis and anti-HIV activity of some novel substituted dialkyl phosphate derivatives of AZT and ddCyd. Antiviral Chem. Chemother. 1990, 1, 355–360. 10.1177/095632029000100603. [DOI] [Google Scholar]
- McGuigan C.; Turner S.; Nicholls S. R.; O’Connor T. J.; Kinchington D. Haloalkyl phosphate derivatives of AZT as inhibitors of HIV- studies in the phosphate region. Antiviral Chem. Chemother. 1994, 5, 162–168. [Google Scholar]
- Mcguigan C.; Jones B. C. N. M.; Tollerfield S. M.; Riley P. A. Synthesis and biological evaluation of haloalkyl phosphate triester derivatives of AraA and AraC. Antiviral Chem. Chemother. 1992, 3, 79–94. 10.1177/095632029200300202. [DOI] [Google Scholar]
- Stein D. S.; Moore K. H. Phosphorylation of nucleoside analog antiretrovirals: a review for clinicians. Pharmacotherapy 2001, 21, 11–34. 10.1592/phco.21.1.11.34439. [DOI] [PubMed] [Google Scholar]
- Navia M. A.; Fitzgerald P. M.; McKeever B. M.; Leu C. T.; Heimbach J. C.; Herber W. K.; Sigal I. S.; Darke P. L.; Springer J. P. Three-dimensional structure of aspartyl protease from human immunodeficiency virus HIV-1. Nature 1989, 337, 615–620. 10.1038/337615a0. [DOI] [PubMed] [Google Scholar]
- McGuigan C.; Devine K. G.; O’Connor T. J.; Galpin S. A.; Jeffries D. J.; Kinchington D. Synthesis and evaluation of some novel phosphoramidate derivatives of 3′-azido-3′-deoxythymidine (AZT) as anti-HIV compounds. Antiviral Chem. Chemother. 1990, 1, 107–113. [Google Scholar]
- Curley D.; McGuigan C.; Devine K. G.; O’Connor T. J.; Jeffries D. J.; Kinchington D. Synthesis and anti-HIV evaluation of some phosphoramidate derivatives of AZT- studies on the effect of chain elongation on biological-activity. Antiviral Res. 1990, 14, 345–356. 10.1016/0166-3542(90)90053-A. [DOI] [PubMed] [Google Scholar]
- Mcguigan C.; Devine K. G.; Oconnor T. J.; Kinchington D. Synthesis and anti-HIV activity of some haloalkyl phosphoramidate derivatives of 3′-azido-3′-deoxythymidine (AZT)- potent activity of the trichloroethyl methoxyalaninyl compound. Antiviral Res. 1991, 15, 255–263. 10.1016/0166-3542(91)90071-X. [DOI] [PubMed] [Google Scholar]
- Jones B. C.; McGuigan C.; O’Connor J. T.; Jeffries D. J.; Kinchington D. Synthesis and anti-HIV activity of some novel phosphorodiamidate derivatives of 3′-azido-3′-deoxythymidine (AZT). Antiviral Chem. Chemother. 1991, 2, 35–39. 10.1177/095632029100200106. [DOI] [Google Scholar]
- McGuigan C.; Madela K.; Aljarah M.; Bourdin C.; Arrica M.; Barrett E.; Jones S.; Kolykhalov A.; Bleiman B.; Bryant K. D.; Ganguly B.; Gorovits E.; Henson G.; Hunley D.; Hutchins J.; Muhammad J.; Obikhod A.; Patti J.; Walters C. R.; Wang J.; Vernachio J.; Ramamurty C. V.; Battina S. K.; Chamberlain S. Phosphorodiamidates as a promising new phosphate prodrug motif for antiviral drug discovery: application to anti-HCV agents. J. Med. Chem. 2011, 54, 8632–8645. 10.1021/jm2011673. [DOI] [PubMed] [Google Scholar]
- McGuigan C.; Nickson C.; O’Connor T. J.; Kinchington D. Synthesis and anti-HIV activity of some novel lactyl and glycolyl phosphate derivatives. Antiviral Res. 1992, 17, 197–212. 10.1016/0166-3542(92)90041-3. [DOI] [PubMed] [Google Scholar]
- McGuigan C.; Pathirana R. N.; Mahmood N.; Devine K. G.; Hay A. J. Aryl phosphate derivatives of AZT retain activity against HIV1 in cell lines which are resistant to the action of AZT. Antiviral Res. 1992, 17, 311–321. 10.1016/0166-3542(92)90026-2. [DOI] [PubMed] [Google Scholar]
- Roberts N. A.; Martin J. A.; Kinchington D.; Broadhurst A. V.; Craig J. C.; Duncan I. B.; Galpin S. A.; Handa B. K.; Kay J.; Krohn A.; Lambert R. W.; Merrett J. H.; Mills J. S.; Parkes K. E. B.; Redshaw S.; Ritchie A. J.; Taylor D. L.; Thomas G. J.; Machin P. J. Rational design of peptide-based HIV proteinase inhibitors. Science 1990, 248, 358–361. 10.1126/science.2183354. [DOI] [PubMed] [Google Scholar]
- Snyder R. D.; Brennan T.; Taylor D. L.; Tyms A. S. Basis of relative insensitivity of HIV-infected JM cells to AZT. Antiviral Chem. Chemother. 1995, 6, 307–311. 10.1177/095632029500600504. [DOI] [Google Scholar]
- McGuigan C.; Davies M.; Pathirana R.; Mahmood N.; Hay A. J. Synthesis and anti-HIV activity of some novel diaryl phosphate derivatives of AZT. Antiviral Res. 1994, 24, 69–77. 10.1016/0166-3542(94)90053-1. [DOI] [PubMed] [Google Scholar]
- Mcguigan C.; Pathirana R. N.; Davies M. P. H.; Balzarini J.; Declercq E. Diaryl phosphate derivatives act as pro-drugs of AZT with reduced cytotoxicity compared to the parent nucleoside. Bioorg. Med. Chem. Lett. 1994, 4, 427–430. 10.1016/0960-894X(94)80009-X. [DOI] [Google Scholar]
- Mcguigan C.; Pathirana R. N.; Mahmood N.; Hay A. J. Aryl phosphate derivatives of AZT inhibit HIV replication in cells where the nucleoside is poorly active. Bioorg. Med. Chem. Lett. 1992, 2, 701–704. 10.1016/S0960-894X(00)80395-9. [DOI] [Google Scholar]
- McGuigan C.; Pathirana R. N.; Balzarini J.; De Clercq E. Intracellular delivery of bioactive AZT nucleotides by aryl phosphate derivatives of AZT. J. Med. Chem. 1993, 36, 1048–1052. 10.1021/jm00060a013. [DOI] [PubMed] [Google Scholar]
- Mcguigan C.; Pathirana R. N.; Mahmood N.; Devine K. G.; Hay A. J. Aryl phosphate derivatives of AZT retain activity against HIV-1 in cell-lines which are resistant to the action of AZT. Antiviral Res. 1992, 17, 311–321. 10.1016/0166-3542(92)90026-2. [DOI] [PubMed] [Google Scholar]
- Slusarczyk M.; Lopez M. H.; Balzarini J.; Mason M.; Jiang W. G.; Blagden S.; Thompson E.; Ghazaly E.; McGuigan C. Application of ProTide technology to gemcitabine: a successful approach to overcome the key cancer resistance mechanisms leads to a new agent (NUC-1031) in clinical development. J. Med. Chem. 2014, 57, 1531–1542. 10.1021/jm401853a. [DOI] [PubMed] [Google Scholar]
- Ross B. S.; Reddy P. G.; Zhang H. R.; Rachakonda S.; Sofia M. J. Synthesis of diastereomerically pure nucleotide phosphoramidates. J. Org. Chem. 2011, 76, 8311–8319. 10.1021/jo201492m. [DOI] [PubMed] [Google Scholar]
- DiRocco D. A.; Ji Y.; Sherer E. C.; Klapars A.; Reibarkh M.; Dropinski J.; Mathew R.; Maligres P.; Hyde A. M.; Limanto J.; Brunskill A.; Ruck R. T.; Campeau L. C.; Davies I. W. A multifunctional catalyst that stereoselectively assembles prodrugs. Science 2017, 356, 426–430. 10.1126/science.aam7936. [DOI] [PubMed] [Google Scholar]
- Simmons B.; Liu Z.; Klapars A.; Bellomo A.; Silverman S. M. Mechanism-based solution to the ProTide synthesis problem: selective access to Sofosbuvir, Acelarin, and INX-08189. Org. Lett. 2017, 19, 2218–2221. 10.1021/acs.orglett.7b00469. [DOI] [PubMed] [Google Scholar]
- Serpi M.; Madela K.; Pertusati F.; Slusarczyk M.. Synthesis of phosphoramidate prodrugs: ProTide approach. Current Protocols in Nucleic Acid Chemistry; Wiley: New York, 2013; Chapter 15, Unit 15.5 10.1002/0471142700.nc1505s53. [DOI] [PubMed] [Google Scholar]
- Hamamoto Y.; Nakashima H.; Matsui T.; Matsuda A.; Ueda T.; Yamamoto N. Inhibitory effect of 2′,3′-didehydro-2′,3′-dideoxynucleosides on infectivity, cytopathic effects, and replication of human immunodeficiency virus. Antimicrob. Agents Chemother. 1987, 31, 907–910. 10.1128/AAC.31.6.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba M.; Pauwels R.; Herdewijn P.; De Clercq E.; Desmyter J.; Vandeputte M. Both 2′,3′-dideoxythymidine and its 2′,3′-unsaturated derivative (2′,3′-dideoxythymidinene) are potent and selective inhibitors of human immunodeficiency virus replication in vitro. Biochem. Biophys. Res. Commun. 1987, 142, 128–134. 10.1016/0006-291X(87)90460-8. [DOI] [PubMed] [Google Scholar]
- Lin T. S.; Schinazi R. F.; Prusoff W. H. Potent and selective in vitro activity of 3′-deoxythymidin-2′-ene (3′-deoxy-2′,3′-didehydrothymidine) against human immunodeficiency virus. Biochem. Pharmacol. 1987, 36, 2713–2718. 10.1016/0006-2952(87)90253-X. [DOI] [PubMed] [Google Scholar]
- Balzarini J.; Karlsson A.; Aquaro S.; Perno C. F.; Cahard D.; Naesens L.; De Clercq E.; McGuigan C. Mechanism of anti-HIV action of masked alaninyl d4T-MP derivatives. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 7295–7299. 10.1073/pnas.93.14.7295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balzarini J.; Egberink H.; Hartmann K.; Cahard D.; Vahlenkamp T.; Thormar H.; De Clercq E.; McGuigan C. Antiretrovirus specificity and intracellular metabolism of 2′,3′ -didehydro-2′,3′-dideoxythymidine (stavudine) and its 5′-monophosphate triester prodrug So324. Mol. Pharmacol. 1996, 50, 1207–1213. [PubMed] [Google Scholar]
- Naesens L.; Cahard D.; Salgado A.; Bidois L.; De Clercq E.; McGuigan C.; Balzarini J. Metabolism and anti-HIV activity of phosphoramidate derivatives of d4T-MP with variations in the amino acid moiety. Adv. Exp. Med. Biol. 1998, 431, 753–757. [DOI] [PubMed] [Google Scholar]
- Saboulard D.; Naesens L.; Cahard D.; Salgado A.; Pathirana R.; Velazquez S.; McGuigan C.; De Clercq E.; Balzarini J. Characterization of the activation pathway of phosphoramidate triester prodrugs of stavudine and zidovudine. Mol. Pharmacol. 1999, 56, 693–704. [PubMed] [Google Scholar]
- Birkus G.; Wang R.; Liu X.; Kutty N.; MacArthur H.; Cihlar T.; Gibbs C.; Swaminathan S.; Lee W.; McDermott M. Cathepsin A is the major hydrolase catalyzing the intracellular hydrolysis of the antiretroviral nucleotide phosphonoamidate prodrugs GS-7340 and GS-9131. Antimicrob. Agents Chemother. 2007, 51, 543–550. 10.1128/AAC.00968-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehellou Y.; Valente R.; Mottram H.; Walsby E.; Mills K. I.; Balzarini J.; McGuigan C. Phosphoramidates of 2′-beta-D-arabinouridine (AraU) as phosphate prodrugs; design, synthesis, in vitro activity and metabolism. Bioorg. Med. Chem. 2010, 18, 2439–2446. 10.1016/j.bmc.2010.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osgerby L.; Lai Y. C.; Thornton P. J.; Amalfitano J.; Le Duff C. S.; Jabeen I.; Kadri H.; Miccoli A.; Tucker J. H.; Muqit M. M.; Mehellou Y. Kinetin riboside and its ProTides activate the Parkinson’s disease associated PTEN-induced putative kinase 1 (PINK1) independent of mitochondrial depolarization. J. Med. Chem. 2017, 60, 3518–3524. 10.1021/acs.jmedchem.6b01897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuigan C.; Murziani P.; Slusarczyk M.; Gonczy B.; Vande Voorde J.; Liekens S.; Balzarini J. Phosphoramidate ProTides of the anticancer agent FUDR successfully deliver the preformed bioactive monophosphate in cells and confer advantage over the parent nucleoside. J. Med. Chem. 2011, 54, 7247–7258. 10.1021/jm200815w. [DOI] [PubMed] [Google Scholar]
- Lee W. A.; He G. X.; Eisenberg E.; Cihlar T.; Swaminathan S.; Mulato A.; Cundy K. C. Selective intracellular activation of a novel prodrug of the human immunodeficiency virus reverse transcriptase inhibitor tenofovir leads to preferential distribution and accumulation in lymphatic tissue. Antimicrob. Agents Chemother. 2005, 49, 1898–1906. 10.1128/AAC.49.5.1898-1906.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami E.; Tolstykh T.; Bao H.; Niu C.; Steuer H. M.; Bao D.; Chang W.; Espiritu C.; Bansal S.; Lam A. M.; Otto M. J.; Sofia M. J.; Furman P. A. Mechanism of activation of PSI-7851 and its diastereoisomer PSI-7977. J. Biol. Chem. 2010, 285, 34337–34347. 10.1074/jbc.M110.161802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkus G.; Kutty N.; He G. X.; Mulato A.; Lee W.; McDermott M.; Cihlar T. Activation of 9-[(R)-2-[[(S)-[[(S)-1-(Isopropoxycarbonyl)ethyl]amino] phenoxyphosphinyl]-methoxy]propyl]adenine (GS-7340) and other tenofovir phosphonoamidate prodrugs by human proteases. Mol. Pharmacol. 2008, 74, 92–100. 10.1124/mol.108.045526. [DOI] [PubMed] [Google Scholar]
- McGuigan C.; Sutton P. W.; Cahard D.; Turner K.; O’Leary G.; Wang Y.; Gumbleton M.; De Clercq E.; Balzarini J. Synthesis, anti-human immunodeficiency virus activity and esterase lability of some novel carboxylic ester-modified phosphoramidate derivatives of stavudine (d4T). Antiviral Chem. Chemother. 1998, 9, 473–479. 10.1177/095632029800900603. [DOI] [PubMed] [Google Scholar]
- Siddiqui A. Q.; McGuigan C.; Ballatore C.; Zuccotto F.; Gilbert I. H.; De Clercq E.; Balzarini J. Design and synthesis of lipophilic phosphoramidate d4T-MP prodrugs expressing high potency against HIV in cell culture: structural determinants for in vitro activity and QSAR. J. Med. Chem. 1999, 42, 4122–4128. 10.1021/jm9807104. [DOI] [PubMed] [Google Scholar]
- McGuigan C.; Tsang H. W.; Cahard D.; Turner K.; Velazquez S.; Salgado A.; Bidois L.; Naesens L.; De Clercq E.; Balzarini J. Phosphoramidate derivatives of d4T as inhibitors of HIV: the effect of amino acid variation. Antiviral Res. 1997, 35, 195–204. 10.1016/S0166-3542(97)00029-6. [DOI] [PubMed] [Google Scholar]
- Anderson B. M.; Cordes E. H.; Jencks W. P. Reactivity and catalysis in reactions of the serine hydroxyl group and of O-acyl serines. J. Biol. Chem. 1961, 236, 455–463. [PubMed] [Google Scholar]
- Brenner C. Hint, Fhit, and GalT: function, structure, evolution, and mechanism of three branches of the histidine triad superfamily of nucleotide hydrolases and transferases. Biochemistry 2002, 41, 9003–9014. 10.1021/bi025942q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntee E. J.; Remmel R. P.; Schinazi R. F.; Abraham T. W.; Wagner C. R. Probing the mechanism of action and decomposition of amino acid phosphomonoester amidates of antiviral nucleoside prodrugs. J. Med. Chem. 1997, 40, 3323–3331. 10.1021/jm960694f. [DOI] [PubMed] [Google Scholar]
- Cheng J.; Zhou X.; Chou T. F.; Ghosh B.; Liu B.; Wagner C. R. Identification of the amino acid-AZT-phosphoramidase by affinity T7 phage display selection. Bioorg. Med. Chem. Lett. 2009, 19, 6379–6381. 10.1016/j.bmcl.2009.09.067. [DOI] [PubMed] [Google Scholar]
- Birkus G.; Kutty N.; Frey C. R.; Shribata R.; Chou T.; Wagner C.; McDermott M.; Cihlar T. Role of cathepsin A and lysosomes in the intracellular activation of novel antipapillomavirus agent GS-9191. Antimicrob. Agents Chemother. 2011, 55, 2166–2173. 10.1128/AAC.01603-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balzarini J.; Kruining J.; Wedgwood O.; Pannecouque C.; Aquaro S.; Perno C. F.; Naesens L.; Witvrouw M.; Heijtink R.; De Clercq E.; McGuigan C. Conversion of 2′,3′-dideoxyadenosine (ddA) and 2′,3′-didehydro-2′,3′-dideoxyadenosine (d4A) to their corresponding aryloxyphosphoramidate derivatives markedly potentiates their activity against human immunodeficiency virus and hepatitis B virus. FEBS Lett. 1997, 410, 324–328. 10.1016/S0014-5793(97)00616-9. [DOI] [PubMed] [Google Scholar]
- McGuigan C.; Hassan-Abdallah A.; Srinivasan S.; Wang Y.; Siddiqui A.; Daluge S. M.; Gudmundsson K. S.; Zhou H.; McLean E. W.; Peckham J. P.; Burnette T. C.; Marr H.; Hazen R.; Condreay L. D.; Johnson L.; Balzarini J. Application of phosphoramidate ProTide technology significantly improves antiviral potency of carbocyclic adenosine derivatives. J. Med. Chem. 2006, 49, 7215–7226. 10.1021/jm060776w. [DOI] [PubMed] [Google Scholar]
- Balzarini J.; Aquaro S.; Hassan-Abdallah A.; Daluge S. M.; Perno C. F.; McGuigan C. Improved antiviral activity of the aryloxymethoxyalaninyl phosphoramidate (APA) prodrug of abacavir (ABC) is due to the formation of markedly increased carbovir 5′-triphosphate metabolite levels. FEBS Lett. 2004, 573, 38–44. 10.1016/j.febslet.2004.07.049. [DOI] [PubMed] [Google Scholar]
- McGuigan C.; Harris S. A.; Daluge S. M.; Gudmundsson K. S.; McLean E. W.; Burnette T. C.; Marr H.; Hazen R.; Condreay L. D.; Johnson L.; De Clercq E.; Balzarini J. Application of phosphoramidate pronucleotide technology to abacavir leads to a significant enhancement of antiviral potency. J. Med. Chem. 2005, 48, 3504–3515. 10.1021/jm0491400. [DOI] [PubMed] [Google Scholar]
- Lisco A.; Vanpouille C.; Tchesnokov E. P.; Grivel J. C.; Biancotto A.; Brichacek B.; Elliott J.; Fromentin E.; Shattock R.; Anton P.; Gorelick R.; Balzarini J.; McGuigan C.; Derudas M.; Gotte M.; Schinazi R. F.; Margolis L. Acyclovir is activated into a HIV-1 reverse transcriptase inhibitor in herpesvirus-infected human tissues. Cell Host Microbe 2008, 4, 260–270. 10.1016/j.chom.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanpouille C.; Lisco A.; Derudas M.; Saba E.; Grivel J. C.; Brichacek B.; Scrimieri F.; Schinazi R.; Schols D.; McGuigan C.; Balzarini J.; Margolis L. A new class of dual-targeted antivirals: monophosphorylated acyclovir prodrug derivatives suppress both human immunodeficiency virus type 1 and herpes simplex virus type 2. J. Infect. Dis. 2010, 201, 635–643. 10.1086/650343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertel L. W.; Boder G. B.; Kroin J. S.; Rinzel S. M.; Poore G. A.; Todd G. C.; Grindey G. B. Evaluation of the antitumor activity of gemcitabine (2′,2′-difluoro-2′-deoxycytidine). Cancer Res. 1990, 50, 4417–4422. [PubMed] [Google Scholar]
- Saif M. W.; Lee Y.; Kim R. Harnessing gemcitabine metabolism: a step towards personalized medicine for pancreatic cancer. Ther. Adv. Med. Oncol. 2012, 4, 341–346. 10.1177/1758834012453755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camiener G. W.; Smith C. G. Studies of enzymatic deamination of cytosine arabinoside 0.1. Enzyme distribution and species specificity. Biochem. Pharmacol. 1965, 14, 1405–1416. 10.1016/0006-2952(65)90175-9. [DOI] [PubMed] [Google Scholar]
- Ghazaly E. A.; Slusarczyk M.; Mason M.; Gribben J.; McGuigan C.; Blagden S. NUC-1031: a novel ProTide that overcomes the key cancer resistance mechanisms associated with poor survival. Cancer Res. 2014, 74, CT401. 10.1158/1538-7445.AM2014-CT401. [DOI] [Google Scholar]
- Blagden S. P.; Rizzuto I.; Stavraka C.; O’Shea D.; Suppiah P.; Patel M. K.; Sukumaran A.; Loyse N.; Bharwani N.; Rockall A.; Gabra H.; El-Bahrawy M.; Wasan H. S.; Leonard R. C.; Habib N. A.; Gribben J. G.; Ghazaly E. A.; McGuigan C. Final results of ProGem1, the first in-human phase I/II study of NUC-1031 in patients with solid malignancies. J. Clin. Oncol. 2015, 33, 2514. 10.1200/jco.2015.33.15_suppl.2514. [DOI] [Google Scholar]
- Vande Voorde J.; Liekens S.; McGuigan C.; Murziani P. G.; Slusarczyk M.; Balzarini J. The cytostatic activity of NUC-3073, a phosphoramidate prodrug of 5-fluoro-2′-deoxyuridine, is independent of activation by thymidine kinase and insensitive to degradation by phosphorolytic enzymes. Biochem. Pharmacol. 2011, 82, 441–452. 10.1016/j.bcp.2011.05.024. [DOI] [PubMed] [Google Scholar]
- Ghazaly E. A.; Slusarczyk M.; McGuigan C.; Harrison D.; Blagden S. P. NUC-3373: A novel pyrimidine nucleotide analogue that overcomes key cancer drug resistance limiting patient survival. Mol. Cancer Ther. 2015, 14, B46. 10.1158/1535-7163.TARG-15-B46. [DOI] [Google Scholar]
- Alexander P.; Kucera G.; Pardee T. S. Improving nucleoside analogs via lipid conjugation: Is fatter any better?. Crit. Rev. Oncol. Hematol. 2016, 100, 46–56. 10.1016/j.critrevonc.2016.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren T. K.; Jordan R.; Lo M. K.; Ray A. S.; Mackman R. L.; Soloveva V.; Siegel D.; Perron M.; Bannister R.; Hui H. C.; Larson N.; Strickley R.; Wells J.; Stuthman K. S.; Van Tongeren S. A.; Garza N. L.; Donnelly G.; Shurtleff A. C.; Retterer C. J.; Gharaibeh D.; Zamani R.; Kenny T.; Eaton B. P.; Grimes E.; Welch L. S.; Gomba L.; Wilhelmsen C. L.; Nichols D. K.; Nuss J. E.; Nagle E. R.; Kugelman J. R.; Palacios G.; Doerffler E.; Neville S.; Carra E.; Clarke M. O.; Zhang L.; Lew W.; Ross B.; Wang Q.; Chun K.; Wolfe L.; Babusis D.; Park Y.; Stray K. M.; Trancheva I.; Feng J. Y.; Barauskas O.; Xu Y.; Wong P.; Braun M. R.; Flint M.; McMullan L. K.; Chen S. S.; Fearns R.; Swaminathan S.; Mayers D. L.; Spiropoulou C. F.; Lee W. A.; Nichol S. T.; Cihlar T.; Bavari S. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature 2016, 531, 381–385. 10.1038/nature17180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo M. K.; Jordan R.; Arvey A.; Sudhamsu J.; Shrivastava-Ranjan P.; Hotard A. L.; Flint M.; McMullan L. K.; Siegel D.; Clarke M. O.; Mackman R. L.; Hui H. C.; Perron M.; Ray A. S.; Cihlar T.; Nichol S. T.; Spiropoulou C. F. GS-5734 and its parent nucleoside analog inhibit Filo-, Pneumo-, and Paramyxoviruses. Sci. Rep. 2017, 7, 43395. 10.1038/srep43395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawitz E.; Mangia A.; Wyles D.; Rodriguez-Torres M.; Hassanein T.; Gordon S. C.; Schultz M.; Davis M. N.; Kayali Z.; Reddy K. R.; Jacobson I. M.; Kowdley K. V.; Nyberg L.; Subramanian G. M.; Hyland R. H.; Arterburn S.; Jiang D.; McNally J.; Brainard D.; Symonds W. T.; McHutchison J. G.; Sheikh A. M.; Younossi Z.; Gane E. J. Sofosbuvir for previously untreated chronic hepatitis C infection. N. Engl. J. Med. 2013, 368, 1878–1887. 10.1056/NEJMoa1214853. [DOI] [PubMed] [Google Scholar]
- Kattakuzhy S.; Levy R.; Kottilil S. Sofosbuvir for treatment of chronic hepatitis C. Hepatol. Int. 2015, 9, 161–173. 10.1007/s12072-014-9606-9. [DOI] [PubMed] [Google Scholar]
- Kirby B. J.; Symonds W. T.; Kearney B. P.; Mathias A. A. Pharmacokinetic, pharmacodynamic, and drug-interaction profile of the hepatitis C virus NS5B polymerase inhibitor Sofosbuvir. Clin. Pharmacokinet. 2015, 54, 677–690. 10.1007/s40262-015-0261-7. [DOI] [PubMed] [Google Scholar]
- Denning J.; Cornpropst M.; Flach S. D.; Berrey M. M.; Symonds W. T. Pharmacokinetics, safety, and tolerability of GS-9851, a nucleotide analog polymerase inhibitor for hepatitis C virus, following single ascending doses in healthy subjects. Antimicrob. Agents Chemother. 2013, 57, 1201–1208. 10.1128/AAC.01262-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Torres M.; Lawitz E.; Kowdley K. V.; Nelson D. R.; Dejesus E.; McHutchison J. G.; Cornpropst M. T.; Mader M.; Albanis E.; Jiang D.; Hebner C. M.; Symonds W. T.; Berrey M. M.; Lalezari J. Sofosbuvir (GS-7977) plus peginterferon/ribavirin in treatment-naive patients with HCV genotype 1: a randomized, 28-day, dose-ranging trial. J. Hepatol. 2013, 58, 663–668. 10.1016/j.jhep.2012.11.018. [DOI] [PubMed] [Google Scholar]
- Cholongitas E.; Papatheodoridis G. V. Sofosbuvir: a novel oral agent for chronic hepatitis C. Ann. Gastroenterol. 2014, 27, 331–337. [PMC free article] [PubMed] [Google Scholar]
- Papatheodoridis G. V.; Dimou E.; Papadimitropoulos V. Nucleoside analogues for chronic hepatitis B: antiviral efficacy and viral resistance. Am. J. Gastroenterol. 2002, 97, 1618–1628. 10.1111/j.1572-0241.2002.05819.x. [DOI] [PubMed] [Google Scholar]
- Lam A. M.; Espiritu C.; Bansal S.; Micolochick Steuer H. M.; Niu C.; Zennou V.; Keilman M.; Zhu Y.; Lan S.; Otto M. J.; Furman P. A. Genotype and subtype profiling of PSI-7977 as a nucleotide inhibitor of hepatitis C virus. Antimicrob. Agents Chemother. 2012, 56, 3359–3368. 10.1128/AAC.00054-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson I. M.; Gordon S. C.; Kowdley K. V.; Yoshida E. M.; Rodriguez-Torres M.; Sulkowski M. S.; Shiffman M. L.; Lawitz E.; Everson G.; Bennett M.; Schiff E.; Al-Assi M. T.; Subramanian G. M.; An D.; Lin M.; McNally J.; Brainard D.; Symonds W. T.; McHutchison J. G.; Patel K.; Feld J.; Pianko S.; Nelson D. R. Sofosbuvir for hepatitis C genotype 2 or 3 in patients without treatment options. N. Engl. J. Med. 2013, 368, 1867–1877. 10.1056/NEJMoa1214854. [DOI] [PubMed] [Google Scholar]
- Zeuzem S.; Dusheiko G. M.; Salupere R.; Mangia A.; Flisiak R.; Hyland R. H.; Illeperuma A.; Svarovskaia E.; Brainard D. M.; Symonds W. T.; Subramanian G. M.; McHutchison J. G.; Weiland O.; Reesink H. W.; Ferenci P.; Hezode C.; Esteban R. Sofosbuvir and ribavirin in HCV genotypes 2 and 3. N. Engl. J. Med. 2014, 370, 1993–2001. 10.1056/NEJMoa1316145. [DOI] [PubMed] [Google Scholar]
- Lynch S. M.; Wu G. Y. Hepatitis C Virus: A Review of treatment guidelines, cost-effectiveness, and access to therapy. J. Clin. Transl. Hepatol. 2016, 4, 310–319. 10.14218/JCTH.2016.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E. Tenofovir alafenamide (TAF) as the successor of tenofovir disoproxil fumarate (TDF). Biochem. Pharmacol. 2016, 119, 1–7. 10.1016/j.bcp.2016.04.015. [DOI] [PubMed] [Google Scholar]
- Gibson A. K.; Shah B. M.; Nambiar P. H.; Schafer J. J. Tenofovir alafenamide. Ann. Pharmacother. 2016, 50, 942–952. 10.1177/1060028016660812. [DOI] [PubMed] [Google Scholar]
- Menendez-Arias L.; Alvarez M.; Pacheco B. Nucleoside/nucleotide analog inhibitors of hepatitis B virus polymerase: mechanism of action and resistance. Curr. Opin. Virol. 2014, 8, 1–9. 10.1016/j.coviro.2014.04.005. [DOI] [PubMed] [Google Scholar]
- Babusis D.; Phan T. K.; Lee W. A.; Watkins W. J.; Ray A. S. Mechanism for effective lymphoid cell and tissue loading following oral administration of nucleotide prodrug GS-7340. Mol. Pharmaceutics 2013, 10, 459–466. 10.1021/mp3002045. [DOI] [PubMed] [Google Scholar]
- Ray A. S.; Fordyce M. W.; Hitchcock M. J. Tenofovir alafenamide: a novel prodrug of tenofovir for the treatment of human immunodeficiency virus. Antiviral Res. 2016, 125, 63–70. 10.1016/j.antiviral.2015.11.009. [DOI] [PubMed] [Google Scholar]
- Eisenberg E. J.; He G. X.; Lee W. A. Metabolism of GS-7340, a novel phenyl monophosphoramidate intracellular prodrug of PMPA, in blood. Nucleosides, Nucleotides Nucleic Acids 2001, 20, 1091–1098. 10.1081/NCN-100002496. [DOI] [PubMed] [Google Scholar]
- Markowitz M.; Zolopa A.; Squires K.; Ruane P.; Coakley D.; Kearney B.; Zhong L.; Wulfsohn M.; Miller M. D.; Lee W. A. Phase I/II study of the pharmacokinetics, safety and antiretroviral activity of tenofovir alafenamide, a new prodrug of the HIV reverse transcriptase inhibitor tenofovir, in HIV-infected adults. J. Antimicrob. Chemother. 2014, 69, 1362–1369. 10.1093/jac/dkt532. [DOI] [PubMed] [Google Scholar]
- Ray A. S.; Fordyce M. W.; Hitchcock M. J. Tenofovir alafenamide: a novel prodrug of tenofovir for the treatment of human immunodeficiency virus. Antiviral Res. 2016, 125, 63–70. 10.1016/j.antiviral.2015.11.009. [DOI] [PubMed] [Google Scholar]
- Sax P. E.; Wohl D.; Yin M. T.; Post F.; DeJesus E.; Saag M.; Pozniak A.; Thompson M.; Podzamczer D.; Molina J. M.; Oka S.; Koenig E.; Trottier B.; Andrade-Villanueva J.; Crofoot G.; Custodio J. M.; Plummer A.; Zhong L.; Cao H.; Martin H.; Callebaut C.; Cheng A. K.; Fordyce M. W.; McCallister S.; Tenofovir alafenamide versus tenofovir disoproxil fumarate, coformulated with elvitegravir, cobicistat, and emtricitabine, for initial treatment of HIV-1 infection: two randomised, double-blind, phase 3, non-inferiority trials. Lancet 2015, 385, 2606–2615. 10.1016/S0140-6736(15)60616-X. [DOI] [PubMed] [Google Scholar]
- Agarwal K.; Fung S. K.; Nguyen T. T.; Cheng W.; Sicard E.; Ryder S. D.; Flaherty J. F.; Lawson E.; Zhao S.; Subramanian G. M.; McHutchison J. G.; Gane E. J.; Foster G. R. Twenty-eight day safety, antiviral activity, and pharmacokinetics of tenofovir alafenamide for treatment of chronic hepatitis B infection. J. Hepatol. 2015, 62, 533–540. 10.1016/j.jhep.2014.10.035. [DOI] [PubMed] [Google Scholar]
- McGuigan C.; Serpi M.; Bibbo R.; Roberts H.; Hughes C.; Caterson B.; Gibert A. T.; Verson C. R. Phosphate prodrugs derived from N-acetylglucosamine have enhanced chondroprotective activity in explant cultures and represent a new lead in antiosteoarthritis drug discovery. J. Med. Chem. 2008, 51, 5807–5812. 10.1021/jm800594c. [DOI] [PubMed] [Google Scholar]
- Serpi M.; Bibbo R.; Rat S.; Roberts H.; Hughes C.; Caterson B.; Alcaraz M. J.; Gibert A. T.; Verson C. R.; McGuigan C. Novel phosphoramidate prodrugs of N-acetyl-(D)-glucosamine with antidegenerative activity on bovine and human cartilage explants. J. Med. Chem. 2012, 55, 4629–4639. 10.1021/jm300074y. [DOI] [PubMed] [Google Scholar]
- James E.; Pertusati F.; Brancale A.; McGuigan C. Kinase-independent phosphoramidate S1P1 receptor agonist benzyl ether derivatives. Bioorg. Med. Chem. Lett. 2017, 27, 1371–1378. 10.1016/j.bmcl.2017.02.011. [DOI] [PubMed] [Google Scholar]
- Ruda G. F.; Alibu V. P.; Mitsos C.; Bidet O.; Kaiser M.; Brun R.; Barrett M. P.; Gilbert I. H. Synthesis and biological evaluation of phosphate prodrugs of 4-phospho-D-erythronohydroxamic acid, an inhibitor of 6-phosphogluconate dehydrogenase. ChemMedChem 2007, 2, 1169–1180. 10.1002/cmdc.200700040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruda G. F.; Wong P. E.; Alibu V. P.; Norval S.; Read K. D.; Barrett M. P.; Gilbert I. H. Aryl phosphoramidates of 5-phospho erythronohydroxamic acid, a new class of potent trypanocidal compounds. J. Med. Chem. 2010, 53, 6071–6078. 10.1021/jm1004754. [DOI] [PMC free article] [PubMed] [Google Scholar]