

Abstract

Severe acute respiratory syndrome (SARS) is caused by a newly emerged coronavirus that infected more than 8000 individuals and resulted in more than 800 (10–15%) fatalities in 2003. The causative agent of SARS has been identified as a novel human coronavirus (SARS-CoV), and its viral protease, SARS-CoV 3CLpro, has been shown to be essential for replication and has hence been recognized as a potent drug target for SARS infection. Currently, there is no effective treatment for this epidemic despite the intensive research that has been undertaken since 2003 (over 3500 publications). This perspective focuses on the status of various efficacious anti-SARS-CoV 3CLpro chemotherapies discovered during the last 12 years (2003–2015) from all sources, including laboratory synthetic methods, natural products, and virtual screening. We describe here mainly peptidomimetic and small molecule inhibitors of SARS-CoV 3CLpro. Attempts have been made to provide a complete description of the structural features and binding modes of these inhibitors under many conditions.
1. Introduction
Coronaviruses have been known for more than five decades since the first prototype murine strain, JHM, was reported in 1947.1,2 Viruses such as porcine transmissible gastroenteritis virus (TGEV), avian infectious bronchitis virus (IBV), and bovine coronavirus (BCoV) severely infect animals. The murine coronavirus mouse hepatitis virus (MHV) was studied as a model for the human disease. Although studies of the mechanism of replication as well as the pathogenesis of several coronaviruses have been very active since 1970s, this family of coronaviruses received much attention when it was recognized that a new human coronavirus was responsible for severe acute respiratory syndrome (SARS), a contagious and fatal illness.3,4
Coronaviruses belong to one of two subfamilies of (Coronavirinae and Torovirinae) of the family Coronaviridae, which in turn comprise the order Nidovirales (Figure 1).5,6 They are classified into four genera (α, β, γ, and δ), and each genus can be further divided into lineage subgroups. SARS-CoV belongs to the Betacoronavirus group (see Figure 1).
Figure 1.

Schematic representation of the taxonomy of Coronaviridae (according to the International Committee on Taxonomy of Viruses). SARS-CoV belongs to the Betacoronavirus family but has a “b” lineage. *Coronaviridae, along with Arteriviridae, Mesoniviridae, and Roniviridae, are members of this family.
In 2003, a new human coronavirus was identified as an etiological agent of the first global pandemic of the 21st century, severe-acute respiratory syndrome (SARS), and the virus was named SARS-CoV. The first case of “an atypical pneumonia” was reported in China during November 2002.7 Its rapid and unexpected spread to another 29 countries, mostly in Asia and North America, alarmed both the public and World Health Organization (WHO). Within a few months of this outbreak in 2003, the WHO announced in a cumulative report about its emergence that it had caused 916 deaths among 8422 cases (fatality rate of 10–15%) worldwide, as shown in Table 1.8 This incidence indicates how rapidly a contagious illness can spread in this highly interconnected society.
Table 1. Summary of SARS Cases by Country or Area, November 1, 2002 to August 7, 2003.
| cumulative
number of cases |
status |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| country/areas | Fa | Ma | Ta | median age (range) | no. of cases hospitalized | no. of cases recovered | no. of deaths | CFRb (%) | no. of imported cases (%) | no. of HCW affected (%)c | date onset first probable case | date onset last probable case |
| Australia | 4 | 2 | 6 | 15 (1–45) | 0 | 6 | 0 | 0 | 6 (100) | 0 (0) | 24-Mar-03 | 1-Apr-03 |
| Brazil | 1 | 1 | 4 | 0 | 1 | 0 | 0 | 1 (100) | 0 (0) | 3-Apr-03 | 3-Apr-03 | |
| Canada | 151 | 100 | 251 | 49 (1–98) | 10 | 200 | 41 | 17 | 5 (2) | 108 (43) | 23-Feb-03 | 12-Jun-03 |
| China | P | P | 5327 | P | 29 | 4949 | 349 | 7 | NA | 1002 (19) | 16-Nov-02 | 25-Jun-03 |
| Hong Kong | 977 | 778 | 1755 | 40 (0–100) | 7 | 1448 | 300 | 17 | NA | 386 (22) | 15-Feb-03 | 31-May-03 |
| Macao | 0 | 1 | 1 | 28 | 0 | 1 | 0 | 0 | 1 (100) | 0 (0) | 5-May-03 | 5-May-03 |
| Taiwan | 349d | 319d | 665 | 46 (2–79) | 10 | 475 | 180 | 27 | 50 (8) | 86 (13) | 25-Feb-03 | 15-Jun-03 |
| Colombia | 1 | 0 | 1 | 28 | 0 | 1 | 0 | 0 | 1 (100) | 0 (0) | 2-Apr-03 | 2-Apr-03 |
| Finland | 0 | 1 | 1 | 24 | 0 | 1 | 0 | 0 | 1 (100) | 0 (0) | 30-Apr-03 | 30-Apr-03 |
| France | 1 | 6 | 7 | 49 (26–61) | 0 | 6 | 1 | 14 | 7 (100) | 2 2 (29) | 21-Mar-03 | 3-May-03 |
| Germany | 4 | 5 | 9 | 44 (4–73) | 0 | 9 | 0 | 0 | 9 (100) | 1 (11) | 9-Mar-03 | 6-May-03 |
| India | 0 | 3 | 3 | 25 (25–30) | 0 | 3 | 0 | 0 | 3 (100) | 0 (0) | 25-Apr-03 | 6-May-03 |
| Indonesia | 0 | 2 | 2 | 56 (47–65) | 0 | 2 | 0 | 0 | 2 (100) | 0 (0) | 6-Apr-03 | 17-Apr-03 |
| Italy | 1 | 3 | 4 | 30.5 (25–54) | 0 | 4 | 0 | 0 | 4 (100) | 0 (0) | 12-Mar-03 | 20-Apr-03 |
| Kuwait | 1 | 0 | 1 | 50 | 0 | 1 | 0 | 0 | 1 (100) | 0 (0) | 9-Apr-03 | 9-Apr-03 |
| Malaysia | 1 | 4 | 5 | 30 (26–84) | 0 | 3 | 2 | 40 | 5 (100) | 0 (0) | 14-Mar-03 | 22-Apr-03 |
| Mongolia | 8 | 1 | 9 | 32 (17–63) | 0 | 9 | 0 | 0 | 8 (89) | 1 (11) | 31-Mar-03 | 6-May-03 |
| New Zealand | 1 | 0 | 1 | 67 | 0 | 1 | 0 | 0 | 1 (100) | 20-Apr-03 | 20-Apr-03 | |
| Philippines | 8 | 6 | 14 | 41 (29–73) | 0 | 12 | 2 | 14 | 7 (50) | 4 (29) | 25-Feb-03 | 5-May-03 |
| Republic of Ireland | 0 | 1 | 1 | 56 | 0 | 1 | 0 | 0 | 1 (100) | 0 (0) | 27-Feb-03 | 27-Feb-03 |
| Republic of Korea | 0 | 3 | 3 | 40 (20–80) | 0 | 3 | 0 | 0 | 3 (100) | 0 (0) | 25-Apr-03 | 10-May-03 |
| Romania | 0 | 1 | 1 | 52 | 0 | 1 | 0 | 0 | 1 (100) | 0 (0) | 19-Mar-03 | 19-Mar-03 |
| Russian Federation | 0 | 1 | 1 | 25 | 1 | 0 | 0 | NA | 0 (0) | 5-May-03 | 5-May-03 | |
| Singapore | 161 | 77 | 238 | 35 (1–90) | 0 | 205 | 33 | 14 | 8 (3) | 97 (41) | 25-Feb-03 | 5-May-03 |
| South Africa | 0 | 1 | 1 | 62 | 0 | 0 | 1 | 100 | 1 (100) | 0 (0) | 3-Apr-03 | 3-Apr-03 |
| Spain | 0 | 1 | 1 | 33 | 0 | 1 | 0 | 0 | 1 (100) | 0 (0) | 26-Mar-03 | 26-Mar-03 |
| Sweden | 1 | 2 | 3 | 33 | 0 | 3 | 0 | 0 | 3 (100) | 0 (0) | ||
| Switzerland | 0 | 1 | 1 | 35 | 0 | 1 | 0 | 0 | 1 (100) | 0 (0) | 9-Mar-03 | 9-Mar-03 |
| Thailand | 5 | 4 | 9 | 42 (2–79) | 0 | 7 | 2 | 22 | 9 (100) | 1 2 (11) | 11-Mar-03 | 27-May-03 |
| United Kingdom | 2 | 2 | 4 | 59 (28–74) | 0 | 4 | 0 | 0 | 4 (100) | 0 (0) | 1-Mar-03 | 1-Apr-03 |
| United States | 16 | 17 | 33 | 36 (0–83) | 7 | 26 | 0 | 0 | 31 (94) | 1 (3) | 9-Jan-03 | 13-Jul-03 |
| Vietnam | 39 | 24 | 63 | 43 (20–76) | 0 | 58 | 5 | 8 | 1 (2) | 36 (57) | 23-Feb-0 | 14-Apr-03 |
Note: F, female; M, male; P, pending; T, total.
Case fatality based on cases with known outcome and irrespective of immediate cause of death.
Health care worker (HCW).
Discarding of three cases, new breakdown by sex pending.
SARS is mainly characterized by a high fever (>38 °C), dyspnea, lymphopenia, headache, and lower respiratory tract infections;9,10 concurrent gastrointestinal symptoms and diarrhea are also common.11−13 With the enormous efforts of the WHO and expert scientists from various countries, a novel human coronavirus was identified as the etiological agent for SARS.4,14 The sequence information on the coronavirus polymerase gene, along with all other previously characterized strains, demonstrated that this was a previously unrecognized coronavirus in humans.3,15−17 Although the SARS epidemic was successfully controlled in 2003,18,19 the identification of animal reservoirs for this virus and the recent report of a new virus related to SARS, called Middle East respiratory syndrome (MERS),20 provide strong motivation for the development of anti-SARS agents to treat this potentially fatal respiratory illness.
The recent outbreak of MERS in South Korea alarmed the public, and the number of patients under quarantine was reported to be 1600.21 After the first patient was diagnosed with MERS on May 20, 2015, within a period of two months, the total number of cases identified had increased to 186 with 36 fatalities and possible infection of 16700 individuals who were subjected to isolation.22,23 By the end of August 2015, a total of 1511 patients were infected worldwide with this virus, of which 574 (∼39%) had died after the first case was recorded in June 2012 in Saudi Arabia.24
To date, the FDA has not approved an antiviral agent for the treatment of SARS, although the clinical treatments are directed toward symptomatic relief. Therefore, the development of effective antiviral chemotherapy against SARS-CoV is important for future outbreaks. Numerous reports (over 3500 publications) have been published on SARS-CoV since 2002. Recently, a brief review on the progress of anti-SARS chemotherapy was reported.25 However; no reports have been published about the substrate selectivity, mechanism of action, and SARs of the inhibitors. Therefore, to overcome the drawbacks and to enhance the qualitative understanding of the etiology, pathology, and possible therapeutic targets against this virus, a comprehensive review is currently needed.
This perspective focuses on the status of SARS-CoV 3 chymotrypsin-like protease (3CLpro) inhibitors discovered during last 12 years from all sources, including laboratory synthetic methods, natural products, virtual screening, and structure-based molecular docking studies. Attempts have been made to provide a complete description of the structural features (SARs) and detailed mechanisms of action of inhibitors. We believe that this perspective will comprise a cumulative source of SARS-CoV 3CLpro inhibitors for researchers and further the understanding of anti-SARS chemotherapy.
2. SARS-CoV and Structure of 3CLpro
Coronaviruses are a family of positive strand, enveloped RNA viruses that can cause acute and chronic respiratory, enteric, and central nervous system diseases in many species of animals, including humans.26,27 This family features the largest viral genomes (27–31 kb) found to date.28,29 The genomic RNA is complexed with the basic nucleocapsid (N) protein to form a helical capsid within the membrane. The membrane of all coronaviruses is comprised of a minimum of three viral proteins: (i) a spike protein (S), a type of glycoprotein I, (ii) a membrane protein (M) that spans the membrane, and (iii) an envelope protein (E), a highly hydrophobic protein that covers the entire structure of the coronavirus (Figure 2).30 The SARS-CoV genome contains two open reading frames, connected by a ribosomal frame shift, which encode two large overlapping replicase polyproteins, pp1a (∼450 kDa) and pp1ab (∼750 kDa), from which the functional proteins are produced by an extensive proteolytic process.31,32 While other coronaviruses utilize three proteases for proteolytic processing, the SARS-CoV is known to encode only two proteases, which include a papain-like cysteine protease (PLpro)33 and a chymotrypsin-like cysteine protease known as 3C-like protease (3CLpro).34−39 The 3CLpro enzyme, also called Main protease (Mpro), is indispensable to the viral replication and infection process, thereby making it an ideal target for antiviral therapy.
Figure 2.
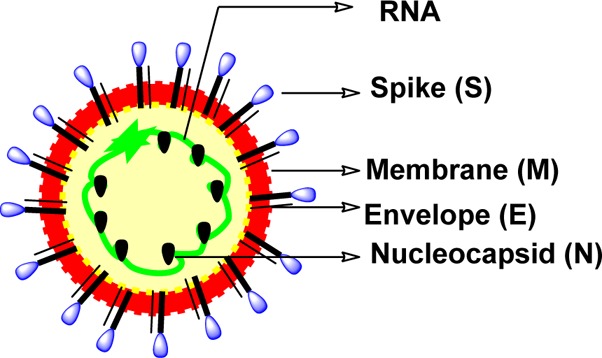
Structure of a coronavirus showing proteins used for replication.
The X-ray crystallographic structure of hexapeptidyl chlromethyl ketone (CMK) inhibitor bound to 3CLpro at different pH values was solved by Yang et al. in 2003 (see Figure 3).38 It was explained that SARS-CoV 3CLpro forms as a dimer with the two promoters (denoted as “A” and “B”) oriented almost at right angles to each other (Figure 3A,B). The crystal structure of the SARS-CoV 3CLpro, similar those of other 3CLpro, comprises three domains. Domains I (residues 8–101) and II (residues 102–184) contain β-barrels that form the chymotrypsin structure, whereas domain III (residues 201–306) consists mainly of α-helices (Figure 3).38−40 SARS-CoV 3CLpro has a Cys-His catalytic dyad, and the substrate or inhibitor binding site is located in a cleft between domain I and II. The substrate-binding subsite S1 specificity in protomer A of a CoV protease confers absolute specificity for the P1-Gln substrate residue on the enzyme. Each N-terminus residue (N-finger) squeezed between domains II and III of the parent monomer and domain II of the other monomer, plays an important role in dimerization and formation of the active site of 3CLpro. The SARS-CoV 3CLpro dimer is highly active, while the monomer is principally inactive.41
Figure 3.

SARS-CoV 3CLpro dimer structure complexed with a substrate-analogue hexapeptidyl CMK inhibitor (PDB ID 1UK4).38 (A) SARS-CoV 3CLpro dimer structure is presented as ribbons, and inhibitor molecules are shown as ball-and-stick models. Protomer A (the catalytically competent enzyme) is shown in red, protomer B (the inactive enzyme) is shown in blue, and the inhibitor molecules are shown in yellow. The N-finger residues of protomer B are shown in green. The molecular surface of the dimer is superimposed. (B) Cartoon diagram illustrating the important role of the N-finger in both the dimerization and maintenance of the active form of the enzyme is shown. Adapted from Yang, H. et al. (permission Copyright (2003) National Academy of Sciences, U.S.A.38
3. SARS-CoV 3CLpro Inhibitors
In 2004, Kua et al. reported the first preparation of the fully active dimeric SARS-CoV 3CLpro with the authentic sequence.42 To screen for inhibitors of SARS-CoV 3CLpro, they prepared a peptide substrate with a fluorescence quenching pair 4-(4-dimethylaminophenylazo)benzoic acid (Dabcyl) and 5-[(2-aminoethyl)amino]naphthalene-1-sulfonic acid (Edans) at the N- and C-termini, respectively, which resulted extremely sensitive assay and allowed many potent inhibitors of SARS-CoV 3CLpro to be identified.
3CLpro are cysteine proteases, which are analogues to the main picornavirus 3C protease, a family of viruses that also cause respiratory illness. The conservation of specificities within the 3CLpro family of coronaviruses has been reported with the amino acid sequence Leu-Gln-Ser or Leu-Gly-Ala as the preferred P2–P1–P1′ sequence (Table 2).1 Although the functional similarities of 3CLpro have “cleavage site-specificity” to that of picornavirus 3C proteases, the structural similarities between the two families are limited.43 The SARS-CoV 3CLpro cleaves polyproteins at no less than 11 conserved sites involving the Leu-Gln↓(Ser, Ala, Gly) sequence, which appears to be a conserved pattern of the 3CLpro of SARS-CoV.3,37 The active site of SARS-CoV 3CLpro contains Cys145 and His41, creating a catalytic dyad in which the cysteine functions as a common nucleophile in the proteolytic process (Figure 4).39,43,44
Table 2. Predicted Cleavage Sites by SARS-CoV 3CLpro.
| P4P3P2P1–P1′P2′P3P4′ | proteinsa |
|---|---|
| AVLQ-SGFR | TM2/3CLpro |
| VTFQ-GKFK | 3CLpro/TM3 |
| ATVQ-SKMS | TM3/? |
| ATLQ-AIAS | ? |
| VKLQ-NNEL | ? |
| VRLQ-AGNA | ?/GFL |
| PLMQ-SADA | GFL/? |
| TVLG-AVGA | ?/RdRp |
| ATLQ-AENV | RdRp/NTPase, etc. |
| TRLQ-SLEN | NTPase, etc./exonuclease |
| PKLQ-ASQA | exonuclease/2′-O-MT |
TM, Transmembrane; GFL, growth factor-like domain; RdRp, RNA-dependent RNA polymerase; 2′-O-MT, 2′-O-methyltransferase.
Figure 4.

Natural amide substrate hydrolysis by Cys145 and His41 at the active site of 3CLpro.
The initial step in the process is deprotonation of Cys-thiol (I) and followed by nucleophilic attack of resulting anionic sulfur on the substrate carbonyl carbon(II). In this step, a peptide product is released that has an amine terminus, while histidine is restored its deprotonated form (III). In the next step, the resulting thioester is hydrolyzed (IV) to release a carboxylic acid, and the free enzyme (V) is regenerated in the final step. Therefore, the functional significance of 3CLpro in the viral life cycle makes this protease an ideal target for the development of drugs against SARS and other coronavirus infections.
In 2003, the first X-ray structure of the SARS-3CLpro dimer with a peptidic CMK (1; Cbz-Val-Asn-Ser-Thr-Leu-Gln-CMK, see Figure 5) inhibitor was elucidated (Yang, H. et al.).38 The unexpected binding mode of the substrate–analogue 1 provides a structural explanation for the P1-Gln entering into the specific pocket and for the decreased P2-Leu specificity of the SARS enzyme. However, specificities for P2-Leu and P4-Ser have been observed in the structure of 1 bound to TGEV 3CLpro,43 whereas P3-Thr is orientated toward bulk solvents. In addition, compound 2 or rupintrivir (AG7088)43 shown in Figure 5 has already been clinically tested for common cold (targeting rhinovirus 3C protease) binds to human rhinovirus 3C protease in the same orientation as that observed for the CMK inhibitor of TGEV. The X-ray crystal structure of 1 with TGEV 3CLpro and superimposed 2 (AG7088) with HRV2 3Cpro is depicted in Figure 6.
Figure 5.

Chemical structures of inhibitors 1, 2, and 3.
Figure 6.

(A) The crystal structure of 1 with TGEV 3CLpro (PDB ID 1P9U) and superimposed 2 with HRV2 3Cpro (PDB ID 1CQQ). The protein binding pocket is shown in surface representation (pink color). The carbon color of compounds 1 (B), 2 (C), and the binding pocket residues of TGEV 3CLpro and HRV2 3Cpro are represented in magenta, green, and dark- and light-gray, respectively. Oxygen atoms are colored in red, nitrogen atoms in blue, sulfur atoms in yellow and hydrogen atoms in white.
Because the substrate specificity of picornavirus 3Cpro for the P1–P1′ and P4 sites is very similar to that of coronavirus 3CLpro, compounds 1 and 2 have been proposed as a starting point in the development of new SARS-CoV 3CLpro inhibitors (Figure 5).45−48 In addition, the HIV-1 protease inhibitor 3 (Figure 5)46,49 was found to have high binding affinity toward SARS-CoV 3CLpro as well. Using the above three molecules as peptidomimetics, many medicinal chemistry studies have been focused on developing a potent chemotherapy method for SARS.
Drugs designed to treat SARS-CoV 3CLpro can be broadly classified into two types: (i) peptidic inhibitors, which mimic natural peptide substrates, and (ii) small molecule-based inhibitors, obtained from modifications of existing protease inhibitors, virtual screening, structure-based molecular docking studies, and natural products. Additionally, metal-conjugated inhibitors as well as some miscellaneous SARS-CoV 3CLpro inhibitors are also discussed in this perspective.
4. Peptidomimetic Inhibitors
In principle, a good substrate can be converted to a good inhibitor by replacement of a part of the substrate sequence that binds directly to the active site of the protease (reversible or irreversible) with the chemical “warhead” targeting the catalytic mechanism. Peptidic inhibitors were designed by attaching a chemical “warhead” type agent to a peptide that mimics the natural substrate. These warhead groups include Michael acceptors, aldehydes, epoxy ketones, halomethyl ketones, and several others (for example, see Figure 7). Mechanistically, these inhibitors act through a two-step procedure, wherein they first bind and form a noncovalent complex with the enzyme such that the warhead is located in close proximity to the catalytic residue. This is followed by a nucleophilic attack by the catalytic cysteine and covalent bond formation. In this perspective, the discussion of peptidomimetics is focused on the substrate selectivity to each specific site (S1′–S1–S2–S3–S4) of 3CLpro, mode of action, and SAR studies.
Figure 7.

Proposed mechanism of cysteine protease inactivation by inhibitors containing Michael acceptor groups.
4.1. Peptides with a Michael Acceptor
Peptidyl or peptidomimetic derivatives contain Michael acceptors as warheads and are an important class of cysteine protease inhibitors. In general, inhibitor design strategies involve the replacement of a substrate’s scissile amide bond with an appropriate Michael acceptor group. The inactivation of a cysteine protease by a Michael acceptor group is depicted in Figure 7. The cysteine residue undergoes 1,4-addition to the inhibitor at the Michael acceptor warhead group, and the subsequent protonation of the α-carbanion results in the irreversible inhibition of the enzyme.
The SAR study of compound 2 indicated that the inhibitory activity was improved by replacing the following side chain residues: the P1-lactam with a phenyl group (4) and the P2-fluorobenzyl with a benzyl group (5), as shown in Figure 8.50 It was noted that compound 5 had two P1 and P2-phenylalanine groups and could fit in the S2 and S3 pockets of SARS-CoV 3CLpro, respectively. In addition, the isoxazole moiety of these analogues adopted a conformation different from that of inhibitor 2 and thus undergoes hydrogen bonding with Gln192 in the S4 pocket. However, the conjugated ester was not accessible (>4.5 Å) to Cys145 to allow a Michael addition for covalent bond (C–S bond) formation. Consequently, this process was achieved by a subsequent strategy using pseudo-C2 symmetric analogues (6–9, Figure 8),50 thus exhibiting good inhibitory activity against 3CLpro. In particular, a compound comprised of Phe–Phe dipeptide unsaturated ester and 4-(dimethylaminocinnamic acid) (8) exhibited potent inhibitory activity with an IC50 value of approximately 1.0 μM and a Ki value of 0.52 μM. The cell-based bioassay gave an EC50 = 0.18 μM. The presence of a 4-dimethylamino moiety on the phenyl ring of these cinnamic analogues was found to be an important structural functionality for activity enhancement.
Figure 8.

Structural modifications of compound 2 with a Michael acceptor to produce active compounds 4–15.
Another series of compounds were reported based on the modification of compound 2 at the P2 side chain by converting the p-fluorobenzyl group to a smaller benzyl (10) or prenyl group (11).51 These inhibitors (10 and 11) possess P1/P1′-Michael acceptor groups, which can covalently link to the Cys145 (Figure 8). The resulting analogues are not only potential inhibitors of SARS-CoV 3CLpro (Kinact values) but are effective in SARS-CoV cell-based bioassays. No toxicity was observed up to 100 μM. In addition, it was observed that compound 12, which contains a hydroxyethylene isostere (12) in place of the ketoethylene of compound 10, was inactive due to the loss of an important hydrogen bond interaction between the backbone amide nitrogen of Glu166 and the carbonyl oxygen of the inhibitor (Figure 8).51 Further replacement of the P4-isoxazole unit with a Boc-serine and a P2-benzyl, prenyl, or isobutyl (13–15: Figure 7)52 increased the inhibitory activity against 3CLpro to several times of that of the lead inhibitor (2) (IC50 = 800 μM), which confirmed both the P4-Boc-serine and P2-isopropyl groups as important structural requirements for greater potency.
Although the activity of the potent analogue 13 was improved to several times that of compound 2 against SARS-CoV 3CLpro, substrate specificity for each site in 3CLpro could not be identified because the inhibitory activity was absolutely dependent on the other residues in these peptides. Therefore, the backbone structure of compound 2 was modified in a systematic manner as reported by Yang, S. et al.53 As a result, a five-member lactam ring was found to be more specific for the P1-site, and leucine was used at the P2-site, which showed much better enzyme activity (>15-fold) than the other residues (Table 3). The strong binding of the five-member ring was evidenced by multiple hydrogen-bonds in the X-ray crystal structure (PDB ID 2GX4).53 For the P2-site, replacement of phenylalanine or 4-fluorophenylalanine with a leucine group increased the inhibitory activity of the enzyme by 4-fold. This result indicated that the rigid and planar phenyl ring is not favorable for binding to the S2 hydrophobic pocket (16 and 17). A lipophilic tert-butyl group at the P3 site further enhances the binding affinity more than 10-fold (17 and 18). Furthermore, the benzyloxy group was found to be the best replacement moiety for P4-methylisoxazole, resulting in a more than 4-fold increment in enzyme inhibitory activity (2 and 16); this group was found to be the best group for this site. On the basis of the docking study, this benzyloxy group has also been observed in a unique conformation in the X-ray crystal structure (docking study of 18 with PDB ID 2GX4; see Supporting Information (SI), Figure S1).53
Table 3. Peptidomimetics with a Michael Acceptor.

Ki, binding affinity; IC50, half-maximal inhibitory concentration.
4.2. Peptides with Keto-glutamine
A novel series of keto-glutamine analogues (19–26) with a phthalhydrazido group at the α-position were reported as reversible inhibitors against SARS-CoV 3CLpro (Figure 9).54 This discovery originated due to their inhibitory activity against the human hepatitis A virus 3C protease.55,56 These compounds feature β and β′ functionalities adjacent to the keto group as well as intramolecular hydrogen bonding to the carbonyl, which makes them more electrophilic and susceptible to hemithioacetal formation with Cys145 in the active site of the protease. Compound 25 was recognized as the most potent analogue with an inhibitory value (IC50) of 0.65 μM. SAR studies indicated that both γ-lactam and phthalhydrazide moieties are very important for good inhibition. Specifically, the introduction of the γ-lactam into the inhibitor containing a phthalhydrazide moiety greatly enhanced the inhibitory activity against SARS-CoV 3CLpro (compare inhibitors 19–22 vs 23–26). This was further supported by molecular modeling studies of the active inhibitors (24–26), which show binding via an extended β-sheet interaction with residues 163–166 of the 3CLpro and formation of hydrogen bonds between the His163 and the P1 side chain.
Figure 9.

Keto-glutamine derivatives with phthalhydrazide (19–27) and thiophene group (28).
A recent report disclosed the X-ray crystal structure of SARS-CoV 3CLpro complexed with one of the phthalhydrazide (19)-based peptide inhibitors (Figure 10, PDB ID 2Z3C).57 The inhibitor forms an unusual thiiranium ring with the nucleophilic sulfur atom of Cys145, trapping the enzyme’s catalytic residues in configurations similar to the intermediate states proposed to exist during the hydrolysis of the native substrate.57 Additionally, the data suggest that this structure resembles the proposed tetrahedral intermediate during the deacylation step of normal peptide hydrolysis cleavage.57 Furthermore, to prove the importance of P1-lactam and phthalhydrazide units in inhibitor 23, a series of analogues modified from P1-lactam to P1-phenyalanine (27) or from phthalhydrazide to thiophene (28) were reported to have only weak activity against SARS-CoV 3CLpro.54
Figure 10.

Crystal structure of phthalhydrazide-based inhibitor 19 bound to SARS-CoV 3CLpro (PDB ID 2Z3C). The protein binding pocket is shown in surface representation and colored in orange. The carbon atoms of the inhibitor 19 and the binding pocket residues are shown in stick model and colored in green and yellow, respectively. The thiiranium ring formed by amino acid Cys145 is colored in magenta.
4.3. Peptides with Nitroanilide
A diverse series of peptide anilides (29–35) were reported based on niclosamide (Figure 11).58 Unlike typical nitroanilide-based peptides, which are readily hydrolyzed by serine and cysteine protease,59 these peptides were not efficiently cleaved by SARS-CoV 3CLpro. Niclosamide showed no inhibitory activity at a concentration of 50 μM. The most potent inhibitor (29) is an anilide derived from 2-chloro-4-nitro aniline, l-phenylalanine, and 4-(dimethylamino)benzoic acid. This anilide is a competitive inhibitor of the SARS-CoV 3CLpro with a Ki value of 0.03 μM and showed high selectivity toward SARS-CoV 3CLpro (IC50 = 0.06 μM) rather than other proteases such as trypsin (IC50 = 110 μM), chymotrypsin (IC50 = 200 μM), and papain (IC50 = 220 μM). Because of the chlorine atom at the o-position, the 2-chloro-4-nitrophenyl ring and amido group cannot be in a coplanar conformation, thus making hydrolysis unfavorable.
Figure 11.

Anilide-type peptidomimetics (29–35) and (2S,2S)-aza epoxide (36) and trans-aziridine (37) inhibitors.
Modification of compound 29 to a series of analogues resulted in reduced potency (30–35).58 A docking study (Figure 12, PDB ID 1UK4) showed that the 2-chloro-4-nitroanilide unit of compound 29 occupies the second preferred pocket. Thus, the nitro group was predicted to be hydrogen bonded with Ala46 and His41, providing a possible key interaction with the catalytic dyad. The (dimethylamino)phenyl group fit into the cleft formed by Gln189–Gln192 and Met165–Pro68.58 A docking study also suggested that anilide 29 has the lowest binding energy (−9.1 kcal/mol) compared to the other derivatives. This experiment supports the observations of the enzymatic assay, which revealed the important roles of 2-chloro-4-nitroaniline and 4-(dimethylamino)benzoic acid residues in effective inhibition.58
Figure 12.
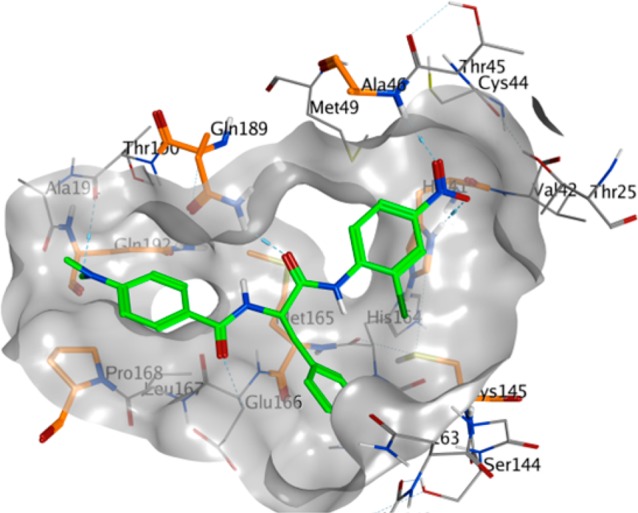
Docked pose of 29 (green, stick model) is shown with the binding pocket residues (gray, line model) and interacting residues (orange, stick model) with SARS-CoV 3CLpro (PDB ID 1UK4). The binding pocket of the protein is shown in surface representation and gray in color.
4.4. Aza-epoxide and Aziridine Peptides
It has been reported that some novel classes of aza-peptide epoxides (APEs) act as inhibitors for clan CD cysteine peptidase.60,61 In the compound library screening, compound 36 (Figure 11) showed prominent activity with irreversible inhibition of SARS-CoV 3CLpro (Kinact/Ki = 1900 (±400) M–1 s–1).62 From the kinetic data and crystal structure of APEs reported by Lee T-W. et al., the 3CLpro reacts only with the S,S-diastereomer and not its R,R-diastereomer. In addition, the epoxide C3 atom of APE must be in the S-configuration.
A comprehensive screening of various peptides with electrophilic building block-attached groups (e.g., epoxides and aziridines) identified potential 3CLpro inhibitors. The data revealed that the aziridine- and oxirane-2-carboxylates are important for the inhibition of 3CLpro. A trans-configured compound containing Gly-Gly-aziridine peptide 37 (54% inhibition at 100 μM) was selected as a modest active-site directed irreversible SARS-CoV 3CLpro inhibitor (Figure 11).63 This study also revealed that epoxide or aziridine building blocks alone, which do not contain an amino acid moiety, are not active.
4.5. Peptide Aldehydes
A series of peptide aldehyde libraries were designed to target the SARS coronavirus, based on the irreversible inhibitor CMK, and were shown to possess very weak inhibitory activity against SARS protease (IC50 > 500 μM).64 The inhibitor CMK binds in a canonical mode to TGEV 3CLpro and resulted in a binding mode with P2, P4, and P5 addressing the respective S pockets, while P3 and P6 were exposed to the solvent (Figure 13A). However, in monomer A SARS-CoV 3CLpro, the CMK inhibitor follows a different side chain orientation (noncannonical binding mode): P2, P4, and P6 residues were not positioned to the respective pockets of the enzyme but remain solvent exposed. Instead, P3-threonine associates with the S2 pocket, and the S4 pocket is occupied by P5-aspargine (Figure 13B).
Figure 13.

(A) CMK-canonical binding mode with TGEV 3CLpro (PDB code 1P9U),43 CMK-noncanonical binding mode with active monomer A of SARS CoV 3CLpro (PDB code 1UK4) (B),38 and (C) the derived inhibitors 38 and 39.
On the basis of these structural findings, it was observed that the sequential variations at the P sites of this initial structure produced potent inhibitors, especially after modifications of the P2 and P5 sites, whereas mutations of the P1 and P3 sites yielded only moderately improved inhibitors. Peptides 38 (AcNSTSQ-H) and 39 (AcESTLQ-H) were found to be more potent, with the best reversible inhibitors having IC50 values in the low micromolar range (7.5 μM) (Figure 13C). Interestingly, these inhibitors are assumed to bind in a noncanonical mode similar to that of CMK with TGEV 3CLpro (Figure 13B). In addition, the SAR suggested that the substrate specificity of SARS-CoV 3CLpro requires glutamine in the P1 position and a large hydrophobic residue in the P2 position. Moreover, X-ray crystal structures of some pentapeptide aldehydes Ac-ESTLQ-H (40, PDB ID 3SNE), Ac-NSFSQ-H (41, PDB ID 3SNA), Ac-DSFDQ-H (42, PDB ID 3SNB), and Ac-NSTSQ-H (43, PDB ID 3SNC), complexed with SARS-CoV 3CLpro, revealed that the S2 pocket of the enzyme can accommodate serine and even an aspartic acid side chain in the P2 position (see SI, Figure S2).65 However, the cleavage efficiency of serine in the P2-position was 160 times lower than the original substrate (P2-Leu), and with aspartic acid, cleavage was not observed at all. Furthermore, the same research group also determined the X-ray crystal structure of SARS-CoV 3CLpro in complex with Cm-FF-H (44, Ki = 2.24 μM, see Figure 14A). From the complex structure (see SI, Figure S3, PDB ID 3SN8), compound 44 had a P1-phenylalanine residue located in the hydrophilic S1 subsite resulted in hydrophobic interactions with Phe140, Leu141, Asn142, and the P3-cinnamoyl group of Cm-FF-H. This result suggests that the stringent specificity of SARS-CoV 3CLpro with respect to the P1 and P2 positions can be overcome by the highly electrophilic character of the aldehyde warhead.
Figure 14.

(A) Structure of aldehydes 44 and 45 and (B) substrate based inhibitors 46–48.
A novel potent SARS-CoV 3CLpro peptide–aldehyde inhibitor (45: Ki = 53 nM) was developed as an antiviral agent against SARS-CoV and human coronavirus HCoV 229E replication, which reduced the viral titer by 4.7 log (at 5 μM) for SARS-CoV and 5.2 log (at 1.25 μM) for HCoV 229E (Figure 14A).53 This inhibitor has distinct functional groups at the P1 to P4 sites compared to those of reference compound 2. This inhibitor was designed to evaluate the issues of cell viability, stability, and drug-like properties based on compound 18. Accordingly, the leucine moiety was replaced with a bulky cyclohexylalanine to improve the cell activity, and the ester group was replaced with an aldehyde to avoid hydrolysis by esterase. As a result, compound 45 (TG-0205221)53 displayed a very stable profile in mouse, rat, and human plasma (Table 4). The X-ray crystal structure of 45 (PDB ID 2GX4) revealed a unique binding mode comprising a covalent bond, hydrogen bonds, and numerous hydrophobic interactions (see SI, Figure S4).53
Table 4. In Vivo Evaluation of Compound 45 for Stabilitya.

The drug was added to 90% rat, mouse, or human plasma and incubated for 0, 30, and 120 min in respective wells.
In the course of studies on the SARS-CoV 3CLpro and its inhibitors,66 it was found that the mature SARS-CoV 3CLpro is very sensitive to degradation at the Arg188/Gln189 site, which causes a loss of catalytic activity. The stability of the SARS-CoV 3CLpro is dramatically increased by mutating the Arg at position 188 to Ile. The enzymatic efficiency of the R188I mutant was increased by a factor of more than 1 × 106. The potency of the mutant protease makes it possible to quantitatively evaluate substrate-based peptide–aldehyde inhibitors using conventional high-performance liquid chromatography (HPLC). A P-site pentapeptide sequence, Ac-Ser-Ala-Val-Leu-NHCH-(CH2CH2CON(CH3)2)-CHO (46: Figure 14B), inhibits the catalytic activity of the SARS-CoV 3CLpro with an IC50 value of 37 μM. The side chain structures, especially at sites P1, P2, and P4, were then optimized step by step based on X-ray crystallographic analyses of the inhibitor–protease complex to provide potent tetra peptide aldehyde inhibitors (47 and 48) (Figure 14B).67
4.6. Peptides with Halomethyl Ketone or Electrophilic Substituents
A new series of N,N′-dimethyl glutaminyl (49–53) or aspartic acid (54) inhibitors with fluoromethyl a ketone warhead were reported as SARS-CoV 3CLpro inhibitors (Table 5).68 These inhibitors were designed based on their caspase inhibitory activities.69,70 Antiviral activity assessed by cytopathic effect (CPE) inhibition in SARS-CoV infected Vero cultures revealed that compounds effectively inhibit both FFM1 and 6109 strains of SARS-CoV replication.
Table 5. Inhibitory Values of Analogues 49–54.
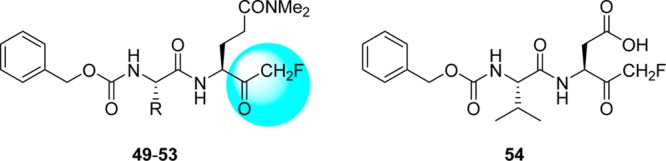

Concentration of compound inhibiting cytopathic effect to 50% of untreated cells. Values represent the mean (standard deviation) from three independent experiments.
Incubation of confluent CaCo2 or Vero cell layers with different concentrations of all the dipeptides for 3 days.
CC50, 50% cytotoxic concentration.
Among these inhibitors, compound 49 exhibited promising activity with low toxicity in cells, protecting the cells with an EC50 value of 2.5 μM and exhibiting a selectivity index >40.68 In addition, compound 49 showed low toxicity in mice. From the SAR studies, P1-glutamine, a residue that has been identified as a conservative recognition site in SARS-CoV 3CLpro, can be replaced by N,N′-dimethyl glutamine (see 49–51). However, compound 54, a potent caspase inhibitor with P1-aspartic acid, abolished activity in this series.68 Furthermore, the P2-leucine can also be replaced by isoleucine (50) and valine (51). The active compounds 49–51 were found to be inactive against rhinovirus type-2 in a cell-based assay suggested that compounds 49–51 are specific against SARS-CoV. Compound 51 was found to have low toxicity in mice after administration of a single dose at 25, 50, and 100 mg/kg. No weight loss or behavioral changes nor any gross pathology of the major organs was observed at the tested doses. This study suggested that compound 51 could be a promising candidate for animal efficacy studies68
Abeles et al. proposed that trifluoromethyl ketones (FMK)71 can also be used as protease inhibitors.72 An interesting feature of these inhibitors is the formation of thermodynamically stable hemiketal or hemithioketal that occurs upon nucleophilic attack by the Ser-hydroxyl or Cys-thiol groups present in the serine or cysteine protease, respectively. On the basis of this observation, Hayashi et al. reported Gln-derived CF3– ketones 55 and 56 as SARS-CoV 3CLpro inhibitors (Figure 15).73 Compounds 55 and 56 showed modest inhibitory activity due to the formation of typical cyclic structures that are not expected to interact effectively with the active site.69 To avoid this problem, the side chain at the P1 site was modified in order to block cyclization.74−77 As shown in Figure 15, compounds 57(74) and 58(75) showed excellent activities and further optimization provided compounds 59–60,76,77 which showed low nanomolar inhibition of SARS-CoV 3CLpro.
Figure 15.

Inhibitors with halomethyl ketones and their derivatives 55–60.
While continuing to explore the SARs based on FMK inhibitors, a series of trifluoro methyl ketones 61–68 were developed, mainly focusing on the P1 and P2–P4 positions (Table 6).78 Three different amino acids were demonstrated as variable residues at positions P1–P4. The inhibitory activities were observed to range from 10 to 50 μM. The potent inhibitor, compound 61, which possesses the same moiety as the substrate sequence of the peptide at the P1–P4 sites, exhibited comparable activity to other compounds. As shown in Table 6, replacement of the P1-benzyl (62) with a methyl group (64) or hydrogen (66) resulted in a loss of activity. Inhibitor 61 showed time-dependent inhibition, with a Ki value of 0.3 μM after a 4 h incubation.78
Table 6. Inhibitory Activity of Halomethyl Ketonesa.
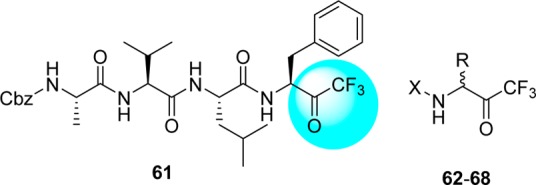
| compd | R | X | IC50 (μM) |
|---|---|---|---|
| 61 | see above structure | see above structure | 10 |
| 62 | Bn | Cbz-Leu | 15 |
| 63 | Bn | Cbz-Phe | 20 |
| 64 | Me | Boc-Leu | 40 |
| 65 | H | Boc-γ-Glu(OtBu)-Ala | 40 |
| 66 | H | γ-Glu-Ala | 50 |
| 67 | Bn | CH3(CH2)8CO-Leu | 50 |
| 68 | Bn | CH3(CH2)7CO-Leu | >50 |
IC50, half-maximal inhibitory concentration; Bn, benzyl; Me, methyl.
4.7. Symmetric Peptides
It was previously proposed that HIV protease inhibitors could serve as good starting points for the development of SARS-CoV 3CLpro inhibitors. In general, reversible inhibitors produce fewer side effects than suicide inhibitors and are thus more suitable for drug development. Recently, compound 69, a noncovalent HIV protease inhibitor (Ki = 1.5 nM), was used as a lead structure and optimized using computational analysis for the development of SARS-CoV 3CLpro inhibitors.79 As shown in the Figure 16, introduction of peripheral Val-Ala residues in place of the Cbz groups or introduction of 3-indolyl groups in place of the phenyl groups in compound 69 led to the formation of inhibitors (70 and 71) that were potent SARS-CoV 3CLpro inhibitors with Ki values of 0.34 and 0.073 μM, respectively. In addition, compound 71 is highly selective for the 3CLpro, with no inhibition observed against HIV protease at 100 μM.
Figure 16.
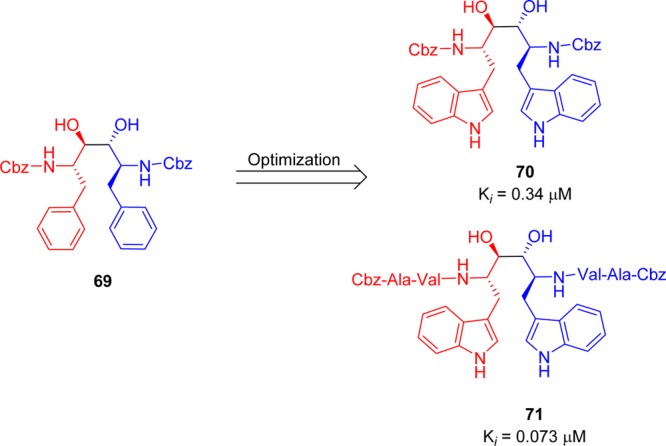
Symmetric peptide diols 69–71.
5. Small Molecule Inhibitors of SARS-CoV 3CLpro
The other category of inhibitors against SARS-CoV 3CLpro includes nonpeptidic small molecules. In general, small molecules have been found to be noncovalent or reversible covalent inhibitors, which have advantages regarding side effects and toxicity which often arise with covalent inhibitors. These inhibitors were discovered by high throughput screening of synthetic compounds and natural products.
5.1. Etacrynic Acid Derivatives
An HPLC-based screen of electrophilic compounds revealed etacrynic acid derivatives 73 (75% inhibition at 100 μM and Ki = 45.8 μM) and 74 (88% inhibition at 100 μM and Ki = 35.3 μM) as effective inhibitors of SARS-CoV 3CLpro.80 These inhibitors were obtained from the sequential modifications of an etacrynic acid (72), a well-known diuretic drug,81 and also showed activity toward the cysteine proteases such as papain protease (Ki = 375 μM).82 Ester 73 showed more potency toward papain protease (Ki = 3.2 μM) than SARS-CoV 3CLpro (Ki = 45.8 μM). However, etacrynic acid amide (74, Ki = 35.3 μM) was found to have more affinity toward SARS-CoV 3CLpro. The SAR studies revealed that chloro substituents on the phenyl moiety were necessary for SARS-CoV 3CLpro inhibition (Figure 17). Compounds with an unsubstituted phenyl ring or methyl substituent were inactive at 100 μM.80 In addition, it is quite promising that only esters or amides display 3CLpro inhibition.80
Figure 17.
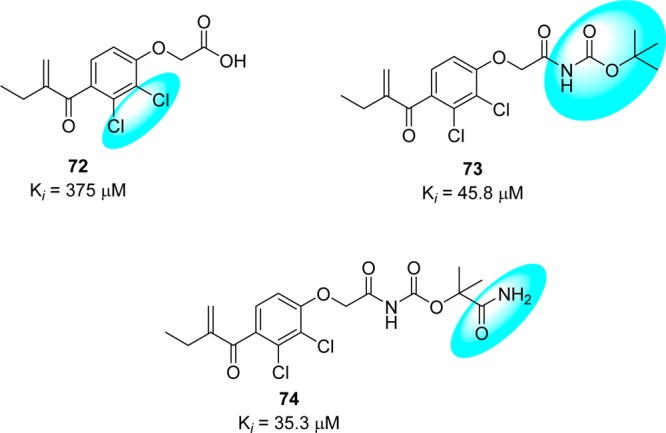
Structural features of etacrynic acids produce their inhibitory activity against SARS-CoV 3CLpro.
5.2. Isatin (2,3-Dioxindole) Inhibitors
It has been established that certain isatin (2,3-dioxindole) compounds are potent inhibitors of rhinovirus 3Cpro.83 Because the proteases of SARS-CoV and rhinovirus share similar active sites and catalytic residues,15 isatin derivatives may also be good candidates for anti-SARS drug development. Accordingly, a series of synthetic isatin derivatives (75–81) were reported as noncovalent SARS protease inhibitors,84,85 unlike rhinovirus 3Cpro, which has a covalent bond binding mode (Table 7). These isatin derivatives inhibited SARS-CoV 3CLpro in the low micromolar range, and inhibitors 78 and 80 were found to be the most potent. SAR studies revealed that the inhibitory potency heavily depended on the hydrophobicity and electron affinity of the substituents on the isatin core. Moreover, computational analysis (docking studies of 78 with PDB ID 1UK4, see SI, Figure S5) of both active compounds showed that they fit very well into the active pocket of SARS-CoV 3CLpro. The two carbonyl groups on isatin could form hydrogen bond interactions with the NH groups on Gly143, Ser144, Cys145, and the His41 side chain. In addition, compounds 78 and 80(86) exhibited better selectivity for SARS than for other proteases including papain (103, 87.24 μM), chymotrypsin (1 mM, 10.4 μM) and trypsin (362, 243 μM).
Table 7. Inhibitory Activities of Isatin Derivatives.

5.3. Flavonoid and Biflavonoid Derivatives
Chemotherapeutic agents that target viral entry are an important class of antiviral therapy as they can block the propagation of the virus at an early stage, thus minimizing the chance for the virus to evolve and acquire drug resistance. Screening of Chinese herbal medicine-based molecules resulted in the discovery of luteolin (82) as inhibitor of wild-type SARS-CoV activity with an effective concentration (EC50) of 10.6 μM (Figure 18).87 Compound 82 was identified as active using a two-step screening method consisting of frontal affinity chromatography–mass spectrometry coupled with a viral infection assay based on a human immunodeficiency virus (HIV)-luc/SARS pseudotyped virus. This flavone analogue binds with the surface spike protein of SARS-CoV and thus can interfere with the entry of the virus into the host cells. However, the related flavone quercetin (83) and its derivatives exhibited modest inhibitory activity against the SARS virus (Figure 18).
Figure 18.

Flavonoids and biflavonoid derivatives.
Quercetin-3-β-galactoside (84) was identified as a potential inhibitor of SARS-CoV and showed inhibitory activity with an IC50 of 42.79 ± 4.97 μM in a SPR/FRET-based enzymatic inhibition assay.88 The docking study of 84 with SARS-CoV 3CLpro suggested that the residue Gln189 (Q189) plays a key role in the binding interaction. To confirm this prediction, the binding mode of 84 was compared between the wild-type SARS-CoV 3CLpro and its mutated SARS-CoV 3CLpro Q189A. This comparative study was consistent with the docking prediction and the inhibitory potency of 84 on SARS-CoV 3CLpro Q189A was significantly decreased to 127.89 ± 10.06 μM. Besides, the experimental evidence showed that the enzymatic activity of SARS-CoV 3CLpro was not affected by the Q189A mutation. The l-fucose derivative (85) exhibited 2-fold potent inhibitory activity compared to 84. The SAR and molecular docking studies of these new derivatives revealed that four hydroxy groups on the quercetin moiety are key determinants for its potential biological activity.
As part of ongoing investigation of bioflavonoids from medicinal plants as potential SARS-CoV 3CLpro inhibitors, a series of inhibitors (86–90) were reported from the leaves of Torreya nucifera (Figure 18).89 Among the isolated compounds, biflavone amentoflavone (86) was recognized as a potent noncompetitive inhibitor, exhibiting an IC50 value of 8.3 μM. An SAR study demonstrated the three authentic flavones, apigenin (90), luteolin (82), and quercetin (83), showed inhibitory activities (IC50) of 280.8, 20.2, and 23.8 μM, respectively. The activity of amentoflavone (86) was consistent with the binding interactions (docking studies of 86 with PDB ID 2Z3E, see SI, Figure S6), with Val186 and Gln192 as one of the key binding modes with the target site. Moreover, the binding energy difference between apigenin (90; −7.79 kcal/mol) and amentoflavone (86; −11.42 kcal/mol) are consistent with a 30-fold lower IC50 value of 86 toward SARS-CoV 3CLpro than apigenin (90).
5.4. Terpenoid Derivatives
A series of diterpenoids (91–93) from Torreya nucifera were evaluated for their anti-SARS activity (Figure 19).89 However, these terpenoids exhibited very low activity compared to biflavonoids against SARS-CoV 3CLpro at concentrations up to 100 μM. One exception was ferruginol (91, IC50 = 49.6 μM), which exhibited significantly greater activity. Moreover, the quinone-methide triterpenoids celastrol (94), pritimererin (95), tingenone (96), and iguesterin (97) were isolated from the methanol (95%) extracts of Tripterygium regelii (Celastraceae) and showed moderate inhibitory activities with IC50 values of 2.6, 9.9, 5.5, and 10.3 μM, respectively, whereas the corresponding a semisynthetic analogue dihydrocelastrol (98: IC50 = 21.7 μM) reduced the inhibitory potency (Figure 19).90 A SAR study suggested that the quinone–methide moiety in the A ring and the more hydrophobic E-ring assist in producing the potent inhibitory activity. The compounds mentioned above (91–98) have been proven to be competitive inhibitors using kinetic analysis.
Figure 19.

Terpenoid derivatives with inhibitory activity against SARS-CoV 3CLpro.
Furthermore, abietane-type diterpenoids and lignoids exhibit a strong anti-SARS-CoV effect.91 In particular, betulinic acid 99 and savinin 100 were shown to act as competitive inhibitors against SARS-CoV 3CLpro with the Ki values of 8.2 and 9.1 μM, respectively (Figure 19).91 On the basis of molecular modeling analysis, it was observed that the competitive inhibition of 99 and 100 on SARS-CoV 3CLpro activity was consistent with the formation of multiple hydrogen bond interactions between the compound and specific amino acid residues located at the active site of the pocket of the protease enzyme.
5.5. Sulfone, Dihydroimidazole, and N-Phenyl-2-(2-pyrimidinylthio)acetamide Type Analogues
Structure-based virtual screening of a chemical database containing 58855 compounds for SARS-CoV 3CLpro inhibition produced two hits, sulfone (101) and dihydroimidazole (102) (Figure 20).92 The core structures of these two hits, defined by a molecular docking study, were used for further searches of analogues.
Figure 20.

Sulfone, dihydroimidazole, and N-phenyl-2-(2-pyrimidinylthio)acetamide-type analogues.
Accordingly, 21 analogues derived from these two hits exhibited IC50 values below 50 μM, and the two most potent compounds (103 and 104) obtained from each hit show IC50 values of 0.3 and 3 μM, respectively.92 Furthermore, a combination of structure-based virtual screening and three-dimensional quantitative structure–activity relationship (3D-QSAR) studies of compound databases of 59363 compounds led to the identification of compounds 105–110, which exhibited modest inhibition with IC50 values of 3, 10, 11, 12, 14, and 15 μM, respectively (Figure 20).93 On the basis of the structure–functional analysis, a common core structure, N-phenyl-2-(2-pyrimidinylthio)acetamide, was identified. A potential binding mode of compound 105 was predicted by the molecular modeling study (docking study of 107 with PDB ID 1UK4, see SI, Figure S7); the strong interaction of benzene and thiazole units with Glu166, Leu167, Pro168, and Gln192 at the SARS-CoV 3CLpro active site could explain its increase in potency.
5.6. Active Heterocyclic Ester Analogues
Wong and co-workers94 reported a novel class of mechanism-based irreversible inhibitors with activity in the nanomolar range, using combinatorial synthesis in microtiter plates followed by in situ screening.95−97 Instead of the expected amide reaction products, a series of benzotriazole esters (111–114) were isolated. Surprisingly, the inhibitory activity of these analogues was much higher than that of the other small molecules or peptidomimetics. Further SAR optimization yielded analogues 115–118 with nanomolar inhibitory activities (Figure 21). An interesting point was found that the esters derived from the benzoic acid-containing electron withdrawing substituents, e.g., NO2, CN and CF3 were susceptible to hydrolysis, whereas esters 111–114 and those with electron-donating substituents were relatively stable in pH 5.0–8.0 solutions over 24 h at room temperature. Compound 116 (Ki = 7.5 nM) was the most potent among the benzotriazole esters.94 The possible mode of action could be acylation of Cys145 at the active site assisted by the catalytic dyad; this irreversible enzyme acylation was verified by electrospray ionization mass spectrometry of the inhibited enzyme with the compound 112 (Figure 22).
Figure 21.

Active heterocyclic ester analogues and their inhibitory activities against SARS-CoV 3CLpro.
Figure 22.

Mechanism of covalent bond formation of inhibitors 112 and 120 with the active site cysteine residue of SARS-CoV 3CLpro.
In addition, the recent X-ray crystal structure of the SARS-CoV 3CLpro complex with the benzotriazole ester also confirmed that the active-site cysteine is acylated by the ester ligand which acts as a suicide inhibitor.98 It should be noted that the formation of N-hydroxybenzotriazole is a very potent inhibitor of CYP450 enzymes. Heteroaromatic ester 119 (IC50 = 0.5 μM) was also identified as a potent inhibitor of the SARS coronavirus.99 The 5-chloropyridine moiety in compound 119 proved to be the key unit for activity against SARS-CoV 3CLpro. Continuing SAR studies provided the very potent inhibitors 120–123, with inhibitory activities spanning from the micromolar to nanomolar range.
The structural biology analysis suggested, in addition to the halopyridyl unit, the other aromatic rings are also key factors for potent inhibition (Figure 21).100,101 A covalent bond formation mechanism for the enzyme–inhibitor complex (120) has been proposed on the basis of electrospray mass spectrometry investigation (Figure 22).
However, another strategy was demonstrated by combining key parts of the previously mentioned mechanism-based inhibitors (116 and 119) to produce a novel series of 5-chloropyridinyl indolecarboxylate inhibitors (124–128) with enzymatic potency in the submicromolar range (Figure 23).102 The SAR study suggested that the positions of the carboxylic acid ester and free indole hydrogen (NH) are critical for activity. Indole carboxylate 124 with carboxylate functionality at position 4 was the most potent inhibitor with an enzyme inhibitory activity (IC50) of 30 nM and an antiviral EC50 value of 6.9 μM.
Figure 23.

Active 5-chloropyridine ester analogues and their inhibitory activity against SARS 3CLpro.
5.7. Aryl Methylene Ketones and Fluoro Methylene Ketones
5-Halopyridinyl-3-aromatic esters, as described in a previous section 5.6, act as highly potent inhibitors of SARS-CoV 3CLpro with IC50 values in the low nanomolar range. They initially bind competitively and strongly to the active site but are then hydrolyzed by the enzyme as substrates and released. Despite their potent inhibition of SARS-CoV 3CLpro and relatively long half-life in buffer at neutral pH values, they are likely to be problematic as drug candidates due to their propensity to be rapidly hydrolyzed by lipase, esterase, and other enzyme in the mammalian cells. Moreover, these compounds can also potentially react nonspecifically with other thiols or nucleophiles in mammalian cells, thereby leading to toxicity. Therefore, to develop stable and noncovalent inhibitors based on pyridinyl esters, a group of methylene ketones and corresponding mono and difluorinated methylene ketones were reported as SARS-CoV 3CLpro inhibitors by Zhang. J. et al. (Figure 24).103 Compounds 129, 131, and 132 showed the best inhibition, and specifically, inhibitor 129 was the most potent among these analogues. The molecular modeling study of these active ketone analogues predicts a binding conformation similar to that of corresponding pyridinyl esters.100,101 A SAR study suggested that fluorination decreases inhibition despite enhancing the electrophilicity of the carbonyl carbon. Enzymatic analysis and ESI-MS studies indicate that these inhibitors utilize a noncovalent, reversible mechanism of action.
Figure 24.
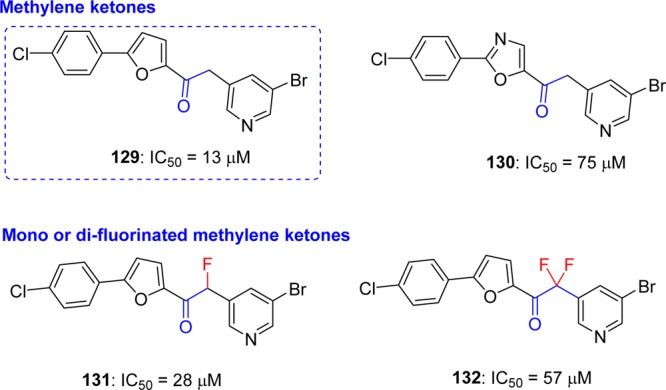
Halomethyl pyridyl ketones and their inhibition potential against SARS-CoV 3CLpro.
5.8. Pyrazolone and Pyrimidines
High throughput screening identified 3,3-dihydropyrazolidine 133(104) and tetrasubstituted pyrazole 134,105 which displayed 1,3,5-triaryl substitution patterns, as SARS-CoV 3CLpro inhibitors. Further exploration of SAR produced a series of pyrazolones that demonstrated inhibitory activities against SARS-CoV 3CLpro (Figure 25).106 Among them, compounds 135–137 exhibited potent inhibitory activities with the IC50 values of 5.5, 6.8, and 8.4 μM, respectively.
Figure 25.

Pyrazolones and pyrimidines and their inhibition potential against SARS-CoV 3CLpro.
Structure–functionality analysis indicated that the 4-carboxylbenzylidine-aryl ring attached to C4-of pyrazolone accompanied by electron withdrawing groups, such as CN, NO2, and F, favors inhibitory activity. Molecular modeling studies of the active compound 137 predicted that the N1-phenyl group located in the S1 pocket and the carboxyl benzylidene group in the S3 pocket of 3CLpro is crucial for its inhibitory activity. Pyrimidine derivatives (138–140) were designed, and their anti-SARS activity was reported (Figure 25).107,108 Compound 140 was the most potent inhibitor that showed enzyme inhibitory activity (IC50 = 6.1 μM) against SARS-CoV 3CLpro. SAR studies revealed that the presence of nitro functionality at position 4 on the benzylidene ring was more important for activity enhancement. This potent activity was consistent with a molecular docking study (docking study of 140 with PDB ID 1UK4, see SI, Figure S8);38 the oxygen of the nitro group formed a hydrogen bond with side chains of Gly143 and Cys145. In addition, the 4-chloro phenyl ring was predicted to fit into the S2 pocket due to hydrophobic inter actions.
5.9. Decahydroisoquinoline Derivatives
Starting from the peptide inhibitor 47 (see section 4.5),67 a novel nonpeptide decahydroisoquinoline inhibitor was designed and synthesized based on the cleavage site interactions at the S1, and hydrophobic interaction at the S2 sites of SARS SARS-CoV 3CLpro.109 The decahydroisoquinoline inhibitors (141–144, Figure 26) showed weak inhibitory activities for SARS-CoV 3CLpro, which confirmed that the fused ring structure of the decahydroisoquinolin scaffold can be accommodated in the active site of SARS-CoV 3CLpro. From the X-ray crystallographic studies (PDB ID 4TWW), it was confirmed that the decahydroisoquinoline inhibitors were at the active site cleft of 3CLpro, as observed in peptide–aldehyde inhibitors. The decahydroisoquinoline scaffold was inserted into a large S2 pocket and occupied most of the pocket. The P1 site imidazole was inserted into the S1 pocket as expected. These interactions were effective in holding the terminal aldehyde tightly inside the active site cleft, which resulted in the compact fitting of the novel scaffold to SARS-CoV 3CLpro.
Figure 26.

Novel decahydroisoquinoline derivatives as SARS-CoV 3CLpro inhibitors.
5.10. 3-Pyridyl and Benzotriazole-Based SARS-CoV 3CLpro Inhibitors
Jacobs et al. conducted a high-throughput screening of NIH molecular libraries (∼293000 compounds) by evaluating the inhibition of 3CLpro mediated peptide cleavage using a novel FRET-based substrate.110,111 In this screen, a dipeptide class, represented by 3-pyridyl-based hit 145 (Figure 27) was identified.
Figure 27.

Primary SAR study at hit furyl amide 145 and schematic representation of enzyme pockets occupied by 146 and 11.
Optimization study based on derivatives (Ugi library) structurally related to hit compound 145 resulted in a series of 3-pyridyl-based inhibitors among which the two compounds, 146 and 147 (Figure 27), were shown to be active against SARS-CoV 3CLpro.
The X-ray crystal structure of 146 bound to SARS-CoV 3CLpro (Figure 28) demonstrated that the binding orientation of 146 was similar to that of known covalent peptidomimetic inhibitors (for example compound 11) and preferentially occupies the S3–S1′ subpockets of SARS-CoV 3CLpro enzyme as R-enantiomer. The tert-butyl amide occupies the S3-pocket, the tert-butylanilido group occupies the deep S2-pocket, and the 3-pyridyl moiety occupies the S1; the furyl amide acts as a P1′ group. Inhibitor 146 lacks a reactive warhead.
Figure 28.
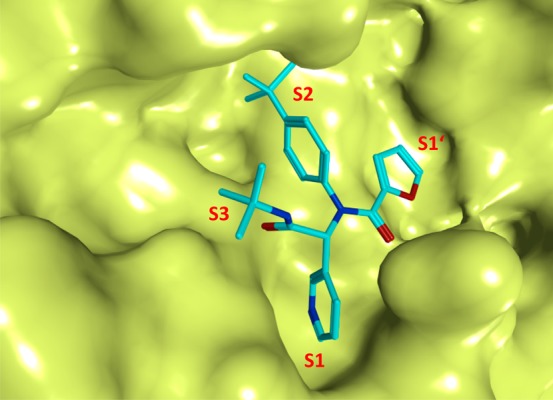
X-ray crystal structure of 146 bound to the binding pocket SARS-CoV 3CLpro (PDB ID 3V3M). The pockets S1′–S3 are highlighted, and the compound 146 is represented in stick model and colored in cyan.
On the basis of the SAR for 146 and related analogues, first a chemical library focusing exclusively on the P1′ group was synthesized while holding the P1–P3 groups constant. This resulted in a series of inhibitors.110 The SAR study around P1′ of 146 showed that the five-membered π-excessive heterocycles proved the most successful 148–153 (Figure 29A). Especially, compound bearing imidazole (150) and 5-chlorofuran (152) analogue exhibited equipotent to 146 with IC50 values of 6.0 and 5.2 μM, respectively. Next, the P1 3-pyridyl unit in 146 was replaced with its isosteres in order to identify alternate hydrogen bond acceptor groups. This effort led to identify another set of compounds (154–156, Figure 29B). Among them, only pyridazine (154) and pyrazine (155) were tolerated, although no improvement was found around the pyridyl ring over 146. Both 2-and 4-pyridyl (156) analogues were not tolerable and reduced the potency.110
Figure 29.

SAR studies at the P1′ (A) and P1 sites (B) of 146 and chiral separation of 146-(R,S) (C) to 146-(R) and 146-(S) enantiomers.
In a continuing study, the racemic compound 146 was purified by chiral supercritical fluid chromatography to separate 146-(R) (ML188)110 and 146-(S) enantiomers (Figure 29C). The evaluation of a compound 146-(R) exhibited inhibitory activity with an IC50 of 1.5 ± 0.3 μM against SARS-CoV 3CLpro, while the other enantiomer 146-(S) was inactive. The mechanism of inhibition of SARS-CoV 3CLpro by 146-(R) was determined to be competitive (Ki, 1.6 ± 0.26 μM) with noncovalent inhibition. Owing to the excellent 3CLpro inhibition and antiviral activity (12.9 ± 0.7 μM) against mock-infected and SARS-CoV infected Vero E6 Cells, 146-(R) was elected as a first in class probe candidate from the furyl amide.
Following the identification of probe compound 146-(R), the same research group continued their further efforts to develop potent, noncovalent SARS-CoV 3CLpro inhibitors based upon a chemical class of benzotriazoles from MLPCN screening.112 This resulted in a hit compound 157 (Figure 30A) demonstrating a SARS-CoV 3CLpro IC50 of 6.2 μM and good selectivity versus PLpro (IC50 > 60 μM).
Figure 30.

(A) SAR studies at the P1, (B) P2–P1′, and (C) P3-truncation of hit 157 to inhibitors (158–167).
The X-ray crystal structure of 157 bound to SARS-CoV 3CLpro shows the diamide 157 binds into an induced-fit binding site that is formed by a rearrangement of the Gln189 and Met49 residue side chains (PDB ID 4MDS, Figure 31). This induced fit site accommodates the syn-N-methyl pyrrole and anilido acetamide moieties of the inhibitors within subpockets that can be characterized as S2–S4 and S2–S1′ subpockets, respectively. Figure 30A schematically illustrates the inhibitor-active site interactions oriented in a similar manner as depicted in Figure 31.
Figure 31.
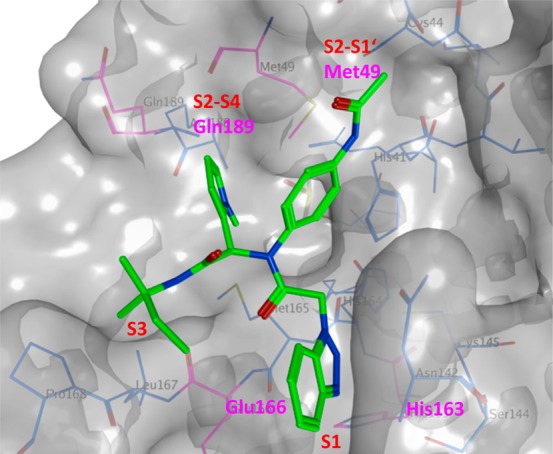
X-ray crystal structure of 157 bound to SARS-CoV 3CLpro (PDB ID: 4MDS) is represented in surface model. The compound 157 (green) is shown in stick model, and the interacting residues (magenta) and the binding pocket residues (gray) are shown in line model.
To improve the activity, first, the SAR study focusing on benzotriazole replacements in 157 for alternate hydrogen bond acceptor functionality was demonstrated. This resulted the replacement of benzotriazole with 4-phenyl 1,2,3-trizole 158 (IC50 of 11 μM, Figure 30A) was tolerable.
Second, the acetamide modification (P2–P1′ region) with a series of cyclic and acyclic congeners yielded many inhibitors which show activities below 10 μM (159–162, Figure 30B), specifically, the branched i-propyl derivative (159) and cyclobutylamide (160) having the greatest activity below 5 μM.
Third, the researchers turned to P3-truncation for minimum pharmacophore to reduce overall molecular weight. This effort led to a series of analogues and SAR proved that truncated amides (163–167, Figure 30C) have comparable activity versus the elaborated amides; for example, compare 163–167 Vs 159–162. The compound 167 represented the first sub-100 nM inhibitor for the series and one of the most potent nonwarhead based SARS-CoV 3CLpro inhibitors to date.
From the above compounds, one of the potent inhibitors, 165 (ML300)112 was selected for probe declaration.113 The biological profiles of inhibitors 146-(R), 160, and 165 are indicated in Figure 32. Relative to probe 146-(R) and the equipotent diamide 160, the compound 165 proved to offer progresses in several areas. Inhibitor 165 is ∼100 amu lower MW (MW = 431) relative to 160 with moderate ligand efficiency (LE).114 Moderate cLogP value of 165 (cLogP = 3.2) greatly improves ligand efficiency-dependent lipophilicity (LELP)114 versus 146-(R) and 160. When both probe 146-(R) and 165 tested in an in-house in vitro DMPK panel including plasma protein binding, P450 enzyme inhibition, and intrinsic clearance using liver microsomes, both 146-(R) and 165 possess good free fraction. However, intrinsic clearance indicates both 146-(R) and 165 are predicted to be highly cleared. 146-(R) and 165 possess modest P450 enzyme inhibition, with 165 maintaining 5–10 μM activity across four major CYP enzymes (see Figure 32). Probe 165 was found to be highly selective in a Eurofins lead-profiling screen,115 with only modest activity (10 μM) for melatonin MT1 receptor in a radioligand binding assay.
Figure 32.

Profiles of SARS-CoV 3CLpro inhibitors 146-(R), 160, and 165.
6. Metal Conjugated SARS-CoV 3CLpro Inhibitors
Metal ions have been shown to inhibit many viral proteases such as 3CLpro of noroviruses, papain-like protease (PLP2) of SARS-CoV, human cytomegalovirus (hCMV) protease, and hepatitis C virus (HCV) NS3 protease.116−120 The screening of 960 metal conjugated compounds allowed inhibitors with potent inhibitory activity against SARS-CoV 3CLpro to be identified. These include competitive inhibitors phenyl mercuric acetate (168, Ki = 0.7 μM), thimerosal (169, Ki = 2.4 μM), and phenyl mercuric nitrate (170, Ki = 0.3 μM) (Figure 33).121,122 However, inhibition was more pronounced using zinc-conjugated compounds (171–174), i.e., 1-hydroxypyridine-2-thione zinc (171, Ki = 0.17 μM) compared to Zn2+ ions alone (Ki = 1.1 μM).
Figure 33.

Metal-conjugated inhibitors and their inhibition potential against SARS-CoV 3CLpro.
The X-ray crystal structure of SARS-CoV 3CLpro–168 (PDB ID 1Z1I) revealed that phenyl-bound mercury occupied the S3 pocket, which is responsible for its enzymatic activity. Hg(II) ions are known to cause toxic effects because the affinity of Hg2+ ions to thiol groups in proteins leads to nonspecific inhibition of cellular enzymes.123 However, regarding the structures of zinc-centered complexes, the zinc ion plays a key role in targeting the catalytic residues via binding to the His41–Cys145 catalytic dyad to yield a zinc central tetrahedral geometry. This type of inhibition was similar to the zinc-mediated serine protease inhibitor keto-BABIM-Zn2+ for trypsin in that a zinc ion was coordinated to the two chelating nitrogen atoms of bis(5-amidino-2-benimidazilyl)methane (BABIM) and the two catalytic residues (His-Ser) of trypsin in the tetrahedral geometry.124 The safety of zinc-containing compounds for human use has been indicated by the fact that zinc acetate and zinc sulfate are added as supplements to drugs for the treatment of Wilson’s disease and Behcet’s disease, respectively.125,126 Moreover, the possibility of zinc complexes incorporated into cells through the cell membrane was also demonstrated by studies on type-2 diabetic treatment.127
Analysis of the active site cavity of this SARS–cysteine protease reveals the presence of a subsite contains a cluster of serine residues (Ser139, Ser144, and Ser147) and is an attractive target for the design of high affinity small molecule inhibitors. This cluster is conserved in all known coronavirus proteases. In particular, Ser139 and Ser147 are conserved in all known coronavirus. Because of the known potential reactivity of boronic acid compounds with the hydroxyl group of the serine residue, a series of bifunctional boronic acid-conjugated compounds (175–177) have been reported against SARS-CoV 3CLpro enzyme (Figure 33).128 The greatest improvement in affinity was achieved with an amide type compound (177) with a Ki of 40 nM. Isothermal titration microcalorimetric experiments indicated that these inhibitors bind reversibly to SARS-CoV 3CLpro in an enthalpically favorable manner, implying that they establish strong interactions with the protease molecule.
7. Miscellaneous SARS-CoV 3CLpro Inhibitors
Over the past decade, in silico virtual screening (VS), in particular structure-based virtual screening (SBVS), has emerged as a reliable, cost-effective, and time-saving technique for the discovery of lead compounds as an alternative to high throughput screening (HTPS).129 The application of VS to the discovery of new enzyme inhibitors involves docking, computational fitting of the compound structure to the active site of an enzyme, and scoring and ranking of each compound.130 On the basis of the structural information, 361413 structurally diverse small molecules were screened by a “genome-to-drug-lead” approach. Compound 178 showed modest activity against targeted human SARS-CoV 3CLpro Toronto-2-strain with an EC50 of 23 μM (Figure 34). Virtual screening of 50240 structurally diverse small molecules allowed 104 compounds with anti-SARS-CoV activities to be identified.131 Inhibitor 179 showed potent inhibitory activity with an IC50 value of 2.5 μM and an EC50 of 7 μM in a Vero cell-based SARS-CoV plaque reduction assay (Figure 34). Another group of researchers, using a quenched fluorescence resonance energy transfer assay, screened 50000 drug-like molecules, resulting in 572 hits.99 After applying a series of virtual and experimental filters, five structurally novel molecules were identified that showed potent inhibitory activity (IC50 = 0.5–7 μM) against SARS-CoV 3CLpro.
Figure 34.

Miscellaneous SAR–CoV 3CLpro inhibitors.
Among them, compounds 180 (IC50 = 4.3 μM) and 181 (IC50 = 4.3 μM) (Figure 34) showed good inhibitory activity of SARS-CoV 3CLpro and exhibited interesting selectivity with no inhibition against other proteases tested (HAV 3Cpro, NS3pro, chymotrypsin, and papain).99
The elucidation of the crystal structure of SARS-CoV 3CLpro provided enormous opportunities for the discovery of inhibitors through rational drug design. As part of an effort to discover small molecule inhibitors of SARS-CoV 3CLpro, structure-based virtual screening of 32000 small molecules was screened against the SARS-CoV 3CLpro enzyme.47 Use of knowledge-based filters yielded 27 molecules for follow-up. A biological evaluation of the inhibitors in the low micromolar range found two compounds, 182 and 183, with IC50 values of 18.2 and 17.2, respectively (Figure 34). It has been reported that several nucleoside derivatives have 6-chloropurine as a nucleobase showed potent antiviral activity against some types of viruses.132,133 Because 6-chloropurine analogues are known to inhibit bacterial RNA polymerases, a series of nucleoside analogues with 6-chloropurines were evaluated for anti-SARS-CoV activity by a plaque reduction activity.134 Among them, two compounds, 184 and 185, exhibited modest anti-SARS-CoV activity (IC50 values of 48.7 and 14.5 μM, respectively) that was comparable to those of mizoribine and ribavirin (Figure 34). This study revealed several SAR trends such as a 6-chloropurine moiety, 5′-hydroxy, and protected (benzylated)-5′-hydroxy group are responsible for the potent inhibitory activity.
Ribavirin, a broad-spectrum of inhibitor of RNA and DNA viruses, was used for the treatment of SARS affected patients135 but it does not inhibit viral growth at concentrations attainable in human serum. In contrast, interferon (IFN)-α showed an in vitro inhibitory effect at concentrations of 1000 IU/mL.136 Interestingly, the combination of ribavirin and IFN-β synergistically inhibited SARS-CoV replication. The HIV protease inhibitor nelfinavir137 and the antimalarial agent chloroquine138 showed strong inhibitory activity against SARS-CoV replication. However, no cytoprotective effect was found for nelfinavir in an independent study.139,140 Structure-based virtual screening of compounds was conducted to identify novel SARS-CoV 3CLpro inhibitors.141 The top-ranked 1468 compounds with free binding energy ranging from −14.0 to −17.09 kcal mol–1 were selected to evaluate the hydrogen bond interactions in the active site of SARS-CoV 3CLpro. Among them, 53 compounds were selected for their inhibitory activity toward SARS-CoV 3CLpro from Escherichia coli. Two of the compounds (186 and 187) were demonstrated to be competitive inhibitors of 3CLpro with Ki values of 9.11 and 9.93 μM, respectively (Figure 34).141 A detailed docking simulation analyses suggested that these inhibitors could be stabilized by the formation of hydrogen bonds with catalytic residues and the establishment of hydrophobic contacts at the opposite region of the active site. In particular, for the potent compound 187, the nitrophenyl group was likely to be very crucial in the SARS-CoV 3CLpro inhibitory activity through its formation of H-bonds with Cys145 and Gly143, as well as its hydrophobic interactions with His41 and Cys145.
Recently, the combination of virtual screening (VS) and high-throughput screening (HTS) techniques were applied to screen 41000 compounds from structurally diverse libraries have allowed novel, nonpeptidic small molecule inhibitors (188, IC50 = 13.9 μM) and (189, IC50 = 18.2 μM) against human SARS-CoV 3CLpro to be identified (Figure 34).142 Because the newly identified compounds are of low molecular weight, they were examined for selectivity against three proteases, namely SARS-CoV PLpro (a cysteine protease), human UCH-L1 (a cysteine protease), and hepatitis C virus NS3/4A (a serine protease), and two nonproteolytic enzymes, Bacillus anthracis dihydroorotase and Streptococcus pneumoniae PurC. Compound 189 displayed good selectivity for SARS-CoV 3CLpro and did not show inhibitory activity (>200 μM) against other five enzymes, whereas compound 188 showed 20-fold selectivity against the two SARS cysteine proteases, 3CLpro and PLpro, over other enzymes. Because low molecular weight compounds typically lack high specificity, lack of inhibition of compound 188 for other enzymes, especially the UCH-L1 cysteine protease, is particularly noteworthy.
8. Conclusion and Perspectives
The emergence of SARS and the identification of a coronavirus as the causative agent of the disease astounded the coronavirus community, as it was the first definitive association of a coronavirus with a severe disease in humans. Because the first crystal structure of the SARS-CoV 3CLpro dimer with a peptidic CMK inhibitor covalently bound was elucidated in 2003, over 20 crystal structures of the enzyme have been reported. Structure-based design and virtual screens have provided both peptidomimetic and nonpeptidomimetic inhibitors with potency in the micromolar to nanomolar range. Yet, to date, there is no effective therapy for the treatment of SARS in humans, and to our knowledge, no CoV 3CLpro inhibitor has been taken into clinical development. In this perspective, we have described the SAR for several classes of inhibitors, highlighting their structural features and binding modes. Both peptidomimetic and small molecule SARS-CoV 3CLpro are largely based on a warhead-based design strategy. So far, only a few inhibitors have been described that exhibit good enzymatic and cellular potency, and the majority of these inhibitors have not been followed up with additional studies (such as antiviral activity or in vivo evaluation), likely due to their unattractive structures and/or their nonideal physiochemical properties.
The reactive warhead groups used in peptidomimetic inhibitors for SARS-CoV 3CLpro include Michael acceptors, aldehydes, epoxy ketones, electrophilic ketones such as halomethyl ketones, and trifluoromethyl ketones. Although these peptidomimetics are covalent inhibitors with the potential for toxicity, significant improvements have been made in enzymatic and cellular potency.
Of the many peptidomimetics inhibitors described in the literature, those highlighted in Figure 35 appear to be the most promising for further optimization efforts. Compound 2 (Figure 5) is an example of an inhibitor incorporating a Michael acceptor. It was developed by Pfizer as an inhibitor of human rhinovirus 3C protease for common cold (targeted rhinovirus 3C-protease). Although 2 was not active against SARS-CoV in cell culture, it served as a good starting point for anti-SARS drug design, leading to inhibitors 8 and 18 (see section 4.1), which are the two most potent inhibitors against SARS-CoV 3CLpro incorporating a Michael acceptor warhead. Specifically, compound 8 exhibited excellent cellular potency with an EC50 value of 0.18 μM and it is a nontoxic anti-SARS agent. However, further in vivo studies for compound 8 have not been reported in the literature.
Figure 35.

Profile of representative peptidic SARS-CoV 3CLpro inhibitors highlighting reactive warhead groups (red).
Peptidic aldehydes are promising enzymatic inhibitors, but they are unlikely to be effective as therapeutic agents due to their rapid in vivo metabolism and low oral bioavailability. In contrast, the peptide aldehyde thrombin inhibitor efegatran was well tolerated in a phase I clinical trial.143,144 Inhibitor 45, a potent peptide aldehyde, showed remarkable activity against SARS-CoV and human coronavirus (HCoV) 229E replications, reducing the viral titer by 4.7 log (at 5 μM) for SARS-CoV and 5.2 log (at 1.25 μM) for HCoV 229E. This inhibitor also displayed a stable profile in mouse, rat, and human plasma (see section 4.5) and may represent a starting point for the development of an anti-SARS agent.
Inhibitor 51 is one of the potent inhibitors in the halomethyl series, exhibiting low toxicity in mice after a single ip dose at 25, 50, and 100 mg/kg, no weight loss, behavioral changes, or gross pathology of major organs was observed at the tested doses (see section 4.6). The low molecular weight of 51 is a potential advantage. Because peptidyl monofluoromethyl ketones have been shown to be effective in vivo,145−147 the inhibitor 51 may be a suitable candidate for further in vivo efficacy and toxicology studies.
Numerous small molecules were also discussed in this perspective. The majority of efforts to develop nonpeptide SARS-CoV 3CLpro inhibitors have also relied on warhead-based design strategy, and several of these nonpeptide inhibitors achieved nanomolar potency. The most interesting inhibitors (78, 116, 119, 124, 129, 146, 160, 165, and 186) are illustrated in Figure 36. In the case of pyridyl esters, the potent mechanism-based enzyme inactivator 124 (see section 5.6) achieved cell-based inhibition below 10 μM in SARS-CoV infected Vero E6 cells. Compounds 146-(R), 160, and 165 are promising examples of noncovalent SARS-CoV 3CLpro inhibitors of moderate molecular weights and good enzymatic and antiviral activity (see section 5.10). These inhibitors are potential starting points for the design of more potent 3CLpro inhibitors with a noncovalent mechanism of action. However, further in vivo studies for above-mentioned small molecules have not reported so far.
Figure 36.

Profile of representative nonpeptidic SARS-CoV 3CLpro inhibitors highlighting reactive warhead groups (red).
Although many structural and nonstructural proteins are known to be potential targets for anticoronavirus therapy, none of them are well-conserved due to their possible role in the viral life cycle, thus limiting the potential success of wide-spectrum inhibitors. In contrast, the coronavirus 3CLpro is highly conserved among coronaviruses, making it an attractive target for broad-spectrum inhibitors (see SI, Table S1).148 The proteases share 40–60% sequence identity and 60–100% sequence similarity. Therefore, targeting SARS-CoV 3CLpro is an important approach for the development of antiviral therapy that can be applied for broad viral infections. Recent reports have revealed that many SARS-CoV 3CLpro inhibitors showed potential activity against the recent outbreak of MERS-CoV.149
A feasible and rapid advancement in the drug discovery for the development of effective chemotherapeutics against SARS-CoV might be achieved by repurposing existing and clinically approved drugs. It was recently reported that screening a library of drugs either clinically developed or with a well-defined cellular pathway from different classes of therapeutics produced a series of compounds with good activity against SARS-CoV.149 Drugs that inhibit CoV included neurotransmitter inhibitors, estrogen receptor antagonists, kinase signaling inhibitors, protein-processing inhibitors, inhibitors of lipid or sterol metabolism, and inhibitors of DNA synthesis or pair. However, the inhibitors (peptidomimetics or nonpeptidomimetics) that target other serine proteases (e.g., HCV protease, thrombin) and cysteine proteases (e.g., calpain, cathepsin K, caspases) have not been tested against 3CLpro. For examples, ketoamides (such as A-705253 for calpain),150 nitriles (such as odanacatib/MK-0822 and vildagliptin/LAF237 for cathepsin K and dipeptidyl peptidase-4 (DPP4)),151,152 phenyloxymethyl ketones (such as VX-166 for caspases),153 fused triazole derivatives (such as sitagliptin/MK-0431 for DPP4),154 nonpeptides (such as apixaban/BMS-562247-01 for factor Xa),155 and beta lactams for penicillin binding proteins such as penicillin.156 Therefore, these structural types should be considered in future for the development of anti-SARS therapy.
N-Finger residues (N-finger) of SARS 3CLpro play an important role in enzyme dimerization, and therefore peptides with N-terminal amino acid sequences may act as inhibitors of 3CLpro dimerization, similar HIV protease, and other viral enzymes.157−163 In 2006, Wei et al. reported that N-terminal octapeptide N8 (Ki of 2.20 mM) was the first example of inhibitor targeting the dimeric interface of SARS 3CLpro,164 providing a novel strategy for drug design against SARS and other coronaviruses. However, no peptidomimetic or small molecule inhibitor has yet been reported in the literature. Although it would be a great challenge to explore new inhibitors of dimerization, with the current development of computational approaches, the structure-based design of novel inhibitors may be successful.
In conclusion, although huge efforts have been taken by both academia and pharmaceutical industries, no coronavirus protease inhibitor has yet successfully completed a preclinical development program. We hope that this perspective will be useful to medicinal chemists targeting 3CLpro to identify novel anti-SARS CoV inhibitors with drug-like properties and that effective therapy for coronaviruses will be discovered.
Acknowledgments
T.P. thanks the Japanese Society for the Promotion of Science (JSPS) foundation for a support for postdoctoral study in Japan. We thank Proceedings of the National Academy of Sciences (PNAS) for the permission to use Figure 3.
Glossary
Abbreviations Used
- hCoV
human coronavirus
- SARS
severe acute respiratory syndrome
- Pros
proteases
- TGEV
transmissible gastroenteritis virus
- IBV
infectious bronchitis virus
- BCoV
bovine coronavirus
- hCMV
human cytomegalovirus
- HCV
hepatitis C virus
- MHV
murine coronavirus mouse hepatitis virus
- WHO
World Health Organization
- MERS
Middle East respiratory syndrome
- FDA
Food and Drug Administration
- RNA
ribonucleic acid
- DNA
DNA
- PLP
papain-like cysteine protease
- 3CLpro
chymotrypsin-like cysteine protease
- Mpro
main protease
- APEs
aza-peptide epoxides
- HPLC
high performance liquid chromatography
- HIV
human immunodeficiency virus
- HCoV-229E
human coronavirus 229E
- SAR
structure–activity relationship
- QSAR
quantitative structure–activity relationship
- Cys
cysteine
- His
histidine
- Ser
serine
- S
spike protein
- M
membrane protein
- N
nuleocapsid
- E
envelope
- BABIM
bis(5-amidino-2-benimidazilyl)methane
- SBVS
structure-based virtual screening
- HTPS
high throughput screening
- IFN
interferon
- ORF
open reading frame
- MW
molecular weight
- LELP
ligand efficiency-dependent lipophilicity
- LE
ligand efficiency
- DPP4
dipeptidyl peptidase-4
Biographies
Thanigaimalai Pillaiyar received his Master’s degree in Chemistry in 2006 from Bharathiar University, India. Prior to his doctoral study, he worked as a Research Executive at Orchid Chemicals and Pharmaceuticals Limited, India. He received his Doctoral degree in Medicinal Chemistry in 2011 under the supervision of Prof. Dr. Sang-Hun Jung at Chungnam National University, South Korea. In 2011, he won a “Japanese Society for the Promotion of Science Postdoctoral fellowship” for two years with Prof. Dr. Yoshio Hayashi at Tokyo University of Pharmacy and Life sciences, Japan. He was awarded an Alexander von Humboldt Postdoctoral fellowship” in 2013 for two years with Prof. Dr. Christa E. Müller at University of Bonn, Germany. He has been working on various therapeutic targets, focusing on infective and inflammatory diseases.
Manoj Manickam received his Ph.D. in 2010 from Bharathiar University under the supervision of Prof. Dr. K. J. Rajendra Prasad, Coimbatore, India. He continued to work as a Research Associate at Orchid Chemicals and Pharmaceuticals Ltd. Then he moved to Chungam National University, South Korea, for continuing his research. Currently, he is a Senior Research Scientist at the Department of Pharmacy and Institute of Drug Research and Development, Chungnam National University, working with Professor Sang-Hun Jung.
Vigneshwaran Namasivayam is a Scientific Staff at Pharmaceutical Institute, University of Bonn, Germany (since 2010), and involved in the field of cheminformatics, computational chemistry, and molecular modelling. He gained his Master of Technology in Bioinformatics from SASTRA University, India (2004), and Doctoral degree under the supervision of Prof. Dr. Hans-Jörg Hofmann from Leipzig University, Germany (2009). He carried out his postdoctoral research at Technical University of Munich, Germany (2010). Prior to his doctoral studies in Germany, he worked as a Research Executive (2004–2006) at Orchid Chemical and Pharmaceutical Limited, Chennai, India.
Yoshio Hayashi earned his Ph.D. in 1990 in the Faculty of Pharmaceutical Science, Kyoto University, under the guidance of Emeritus Prof. Haruaki Yajima and Prof. Nobutaka Fujii. After spending two years at Calpis Food Industry Co., Ltd. and three years at Nippon Steel Corporation (NSC) as a researcher, he was promoted to senior researcher at the Life Science Research Center of the NSC, where he stayed for another eight years. In 1999, he joined Prof. Yoshiaki Kiso’s group in the Department of Medicinal Chemistry of Kyoto Pharmaceutical University as a lecturer and, in 2001, was appointed as an associate professor. In 2007, he moved to Tokyo University of Pharmacy and Life Sciences as a full professor. His research interests include peptide chemistry, peptidomimetics, and medicinal chemistry.
Sang-Hun Jung received his M.S. degree from the College of Pharmacy of the Seoul National University in 1976. He received his Ph.D. from the Chemistry Department at the University of Houston, USA, in 1984. He served as a postdoctoral fellow at the University of Pittsburgh until 1985 and as a Principle investigator of LG life Science from 1985 to 1989. He has been a professor at the College of Pharmacy, Chungnam National University, South Korea, since 1989. He has served as a Department Chairman (1993–2000), Dean of the College of Pharmacy (2003–2004), and President of Institute of Drug Research and Development of Chungnam National University (2007–2009). His research interests include antimicrotubule-based anticancer agents, novel inotropes with selective activation of cardiac myosin, and melanogenesis inhibitors.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.5b01461.
Docking figures of compounds 18, 41–44, 45, 46, 83, 92, 112, and 155; sequence comparison analysis of 3CLpro of coronaviruses to the SARS-CoV 3CLpro; list of X-ray structures of ligands with 3Cpro and 3CLpro (PDF)
The authors declare no competing financial interest.
This article is made available for a limited time sponsored by ACS under the ACS Free to Read License, which permits copying and redistribution of the article for non-commercial scholarly purposes.
Supplementary Material
References
- Cheever F. S.; Daniels J. B.; Pappenheimer A. M.; Baily O. T. A murine virus (JHM) causing disseminated encephalomyelitis with extensive destruction of myelin: I. Isolation and biological properties of the virus. J. Exp. Med. 1949, 90, 181–210. 10.1084/jem.90.3.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey O. T.; Pappenheimer A. M.; Sargent F.; Cheever M. D.; Daniels J. B. A murine virus (jhm) causing disseminated encephalomyelitis with extensive destruction of myelin: II. Pathology. J. Exp. Med. 1949, 90, 195–212. 10.1084/jem.90.3.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rota P. A.; Oberste M. S.; Monroe S. S.; Nix W. A.; Campagnoli R.; Icenogle J. P.; Penaranda S.; Bankamp B.; Maher K.; Chen M. H.; Tong S.; Tamin A.; Lowe L.; Frace M.; DeRisi J. L.; Chen Q.; Wang D.; Erdman D. D.; Peret T. C.; Burns C.; Ksiazek T. G.; Rollin P. E.; Sanchez A.; Liffick S.; Holloway B.; Limor J.; McCaustland K.; Olsen-Rasmussen M.; Fouchier R.; Gunther S.; Osterhaus A. D.; Drosten C.; Pallansch M. A.; Anderson L. J.; Bellini W. J. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 2003, 300, 1394–1399. 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- Drosten C.; Gunther S.; Preiser W.; van der Werf S.; Brodt H. R.; Becker S.; Rabenau H.; Panning M.; Kolesnikova L.; Fouchier R. A.; Berger A.; Burguiere A. M.; Cinatl J.; Eickmann M.; Escriou N.; Grywna K.; Kramme S.; Manuguerra J. C.; Muller S.; Rickerts V.; Sturmer M.; Vieth S.; Klenk H. D.; Osterhaus A. D.; Schmitz H.; Doerr H. W. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- de Groot R. J.; Baker S. C.; Baric R.; Enjuanes L.; Gorbalenya A. E.; Holmes K. V.; Perlman S.; Poon L.; Rottier P. J. M.; Talbot P. J.; Woo P. C. Y.; Ziebuhr J.. Family Coronaviridae. In Ninth Report of the International Committee on Taxonomy of Viruses; King A. M. Q., Lefkowitz E., Adams M. J., Carstens E. B., Eds.; Elsevier: Oxford, 2011; pp 806–828. [Google Scholar]
- ICTV Master Species List 2009; International Committee on Taxonomy of Viruses, August24, 2010; vol. 10.
- Parry J. WHO investigates China’s fall in SARS cases. Br. Med. J. 2003, 326, 1285-c. 10.1136/bmj.326.7402.1285-c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Communicable Disease Surveillance and Response; World Health Organization: Geneva, May7, 2003; http://www.who.int/csr/sars/archive/2003_05_07a/en and http://www.who.int/csr/sars/country/en/country2003_08_15.pdf (August 15, 2003).
- Nie Q. H.; Luo X. D.; Zhang J. Z.; Su Q. Current status of severe acute respiratory syndrome in China. World J. Gastroenterol. 2003, 9, 1635–1645. 10.3748/wjg.v9.i8.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui P. T.; Kwok M. L.; Yuen H.; Lai S. T. Severe acute respiratory syndrome: clinical outcome and prognostic correlates. Emerging Infect. Dis. 2003, 9, 1064–1069. 10.3201/eid0909.030362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung W. K.; To K. F.; Chan P. K.; Chan H. L.; Wu A. K.; Lee N.; Yuen K. Y.; Sung J. J. Enteric involvement of severe acute respiratory syndrome-associated coronavirus infection. Gastroenterology 2003, 125, 1011–1017. 10.1016/S0016-5085(03)01215-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee N.; Hui D.; Wu A.; Chan P.; Cameron P.; Joynt G. M.; Ahuja A.; Yung M. Y.; Leung C. B.; To K. F.; Lui S. F.; Szeto C. C.; Chung S.; Sung J. J. A major outbreak of severe acute respiratory syndrome in Hong Kong. N. Engl. J. Med. 2003, 348, 1986–1994. 10.1056/NEJMoa030685. [DOI] [PubMed] [Google Scholar]
- Booth C. M.; Matukas L. M.; Tomlinson G. A.; Rachlis A. R.; Rose D. B.; Dwosh H. A.; Walmsley S. L.; Mazzulli T.; Avendano M.; Derkach P.; Ephtimios I. E.; Kitai I.; Mederski B. D.; Shadowitz S. B.; Gold W. L.; Hawryluck L. A.; Rea E.; Chenkin J. S.; Cescon D. W.; Poutanen S. M.; Detsky A. S. Clinical features and short-term outcomes of 144 patients with SARS in the greater Toronto area. JAMA 2003, 289, 2801–2809. 10.1001/jama.289.21.JOC30885. [DOI] [PubMed] [Google Scholar]
- Ksiazek T. G.; Erdman D.; Goldsmith C. S.; Zaki S. R.; Peret T.; Emery S.; Tong S.; Urbani C.; Comer J. A.; Lim W.; Rollin P. E.; Dowell S. F.; Ling A. E.; Humphrey C. D.; Shieh W. J.; Guarner J.; Paddock C. D.; Rota P.; Fields B.; DeRisi J.; Yang J. Y.; Cox N.; Hughes J. M.; LeDuc J. W.; Bellini W. J.; Anderson L. J. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- Snijder E. J.; Bredenbeek P. J.; Dobbe J. C.; Thiel V.; Ziebuhr J.; Poon L. L.; Guan Y.; Rozanov M.; Spaan W. J.; Gorbalenya A. E. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 2003, 331, 991–1004. 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuiken T.; Fouchier R. A. M.; Schutten M.; Rimmelzwaan G. F.; van Amerongen G.; Van Riel D.; Laman J. D.; de Jong T.; Van Doornum G.; Lim W.; Ling A. E.; Chan P. K. S.; Tam J. S.; Zambon M. C.; Gopal R.; Drosten C.; Van der Werf S.; Escriou N.; Manuguerra J. C.; Stohr K.; Peiris J. S. M.; Osterhaus A. D. M. E. Newly discovered coronavirus as the primary cause of severe acute respiratory syndrome. Lancet 2003, 362, 263–270. 10.1016/S0140-6736(03)13967-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiris J. S. M.; Lai T. L.; Poon L. M.; Guan Y.; Yam L. Y. C.; Lim W.; Nicholls J.; Yee W. K. S.; Yan W. W.; Cheung M. T.; Cheng V. C. C.; Chan K. H.; Tsang D. N. C.; Yung R. W. H.; Ng T. K.; Yuen K. Y. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 2003, 361, 1319–1325. 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleck F. WHO says SARS outbreak is over, but fight should go on. Br. Med. J. 2003, 327, 70-c. 10.1136/bmj.327.7406.70-c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashraf H. WHO declares Beijing to be free of SARS. Lancet 2003, 361, 2212. 10.1016/S0140-6736(03)13790-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- New 'Sars-like' Coronavirus Identified by UK Officials. BBC News, September24, 2012; http://www.bbc.co.uk/news/health-19698335.
- South Korea Declares 'DDe Facto End' to MERS Virus. BBC News, July28, 2015; http://www.bbc.com/news/world-asia-33684981.
- S. Korea Reports 23 New Cases of MERS, Bringing Total to 87. Yonhap News Agency, June8, 2015. [Google Scholar]
- South Korea MERS Outbreak Began with a Cough; The Wall Street Journal 8June2015. [Google Scholar]
- Rapid Risk Assessment: Severe Respiratory Disease Associated with MiddleEast Respiratory Syndrome Coronavirus (MERS-CoV); European Centre for Disease Prevention and Control, August28, 2015; http://ecdc.europa.eu/en/publications/Publications/MERS-CoV-rapid-risk-assessment-August-2015.pdf.
- Ghosh A. K.; Xi K.; Johnson M. E.; Baker S. C.; Mesecar A. D. Progress in anti-sars coronavirus chemistry, biology and chemotherapy. Annu. Rep. Med. Chem. 2006, 41, 183–196. 10.1016/S0065-7743(06)41011-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh K. Coronaviruses: A comparative review. Curr. Top. Microbiol. Immunol. 1974, 63, 85–129. 10.1007/978-3-642-65775-7_3. [DOI] [Google Scholar]
- Marra M. A.; Jones S. J.; Astell C. R.; Holt R. A.; Brooks-Wilson A.; Butterfield Y. S.; Khattra J.; Asano J. K.; Barber S. A.; Chan S. Y.; Cloutier A.; Coughlin S. M.; Freeman D.; Girn N.; Griffith O. L.; Leach S. R.; Mayo M.; McDonald H.; Montgomery S. B.; Pandoh P. K.; Petrescu A. S.; Robertson A. G.; Schein J. E.; Siddiqui A.; Smailus D. E.; Stott J. M.; Yang G. S.; Plummer F.; Andonov A.; Artsob H.; Bastien N.; Bernard K.; Booth T. F.; Bowness D.; Czub M.; Drebot M.; Fernando L.; Flick R.; Garbutt M.; Gray M.; Grolla A.; Jones S.; Feldmann H.; Meyers A.; Kabani A.; Li Y.; Normand S.; Stroher U.; Tipples G. A.; Tyler S.; Vogrig R.; Ward D.; Watson B.; Brunham R. C.; Krajden M.; Petric M.; Skowronski D. M.; Upton C.; Roper R. L. The genome sequence of the SARS-associated coronavirus. Science 2003, 300, 1399–1404. 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- Lomniczi B. J. Biological properties of avian coronavirus RNA. J. Gen. Virol. 1977, 36, 531–533. 10.1099/0022-1317-36-3-531. [DOI] [PubMed] [Google Scholar]
- Lee H. J.; Shieh C. K.; Gorbalenya A. E.; Koonin E. V.; La Monica N.; Tuler J.; Bagdzhadzhyan A.; Lai M. M. The complete sequence (22 kilobases) of murine coronavirus gene 1 encoding the putative proteases and RNA polymerase. Virology 1991, 180, 567–582. 10.1016/0042-6822(91)90071-I. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond C. W.; Leibowitz J. L.; Robb J. A. Pathogenic murine coronaviruses. II. Characterization of virus-specific proteins of murine coronaviruses JHMV and A59V. Virology 1979, 94, 371–384. 10.1016/0042-6822(79)90468-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebuhr J.; Heusipp G.; Siddell S. G. Biosynthesis, purification, and characterization of the human coronavirus 229E 3C-like proteinase. J. Virol. 1997, 71, 3992–3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty W. G.; Semler B. L. Expression of virus-encoded proteinases: functional and structural similarities with cellular enzymes. Microbiol. Rev. 1993, 57, 781–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratia K.; Saikatendu K. S.; Santarsiero B. D.; Barretto N.; Baker S. C.; Stevens R. C.; Mesecar A. D. Severe acute respiratory syndrome coronavirus papain-like protease: structure of a viral deubiquitinating enzyme. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 5717–5722. 10.1073/pnas.0510851103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.; Chen L.; Tan J.; Chen J.; Du L.; Sun T.; Shen J.; Chen K.; Jiang H.; Shen X. Severe acute respiratory syndrome coronavirus 3C-like proteinase N terminus is indispensable for proteolytic activity but not for enzyme dimerization. Biochemical and thermodynamic investigation in conjunction with molecular dynamics simulations. J. Biol. Chem. 2005, 280, 164–173. 10.1074/jbc.M408211200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C.; Wei P.; Fan K.; Liu Y.; Lai L. 3C-like proteinase from SARS coronavirus catalyzes substrate hydrolysis by a general base mechanism. Biochemistry 2004, 43, 4568–4574. 10.1021/bi036022q. [DOI] [PubMed] [Google Scholar]
- Shi J.; Wei Z.; Song J. Dissection study on the severe acute respiratory syndrome 3C-like protease reveals the critical role of the extra domain in dimerization of the enzyme: defining the extra domain as a new target for design of highly specific protease inhibitors. J. Biol. Chem. 2004, 279, 24765–24773. 10.1074/jbc.M311744200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan K.; Wei P.; Feng Q.; Chen S.; Huang C.; Ma L.; Lai B.; Pei J.; Liu Y.; Chen J.; Lai L. Biosynthesis, purification, and substrate specificity of severe acute respiratory syndrome coronavirus 3C-like proteinase. J. Biol. Chem. 2004, 279, 1637–1642. 10.1074/jbc.M310875200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.; Yang M.; Ding Y.; Liu Y.; Lou Z.; Zhou Z.; Sun L.; Mo L.; Ye S.; Pang H.; Gao G. F.; Anand K.; Bartlam M.; Hilgenfeld R.; Rao Z. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 13190–13195. 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu M. F.; Kuo C. J.; Chang K. T.; Chang H. C.; Chou C. C.; Ko T. P.; Shr H. L.; Chang G. G.; Wang H.; Liang P. H. Mechanism of the maturation process of SARS-CoV 3CL protease. J. Biol. Chem. 2005, 280, 31257–31266. 10.1074/jbc.M502577200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegyi A.; Ziebuhr J. Conservation of substrate specificities among coronavirus main proteases. J. Gen. Virol. 2002, 83, 595–599. 10.1099/0022-1317-83-3-595. [DOI] [PubMed] [Google Scholar]
- Grum-Tokars V.; Ratia K.; Begaye A.; Baker S. C.; Mesecar A. D. Evaluating the 3C-like protease activity of SARS-coronavirus: Recommendations for standardized assays for drug discovery. Virus Res. 2008, 133, 63–73. 10.1016/j.virusres.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo C. J.; Chi Y. H.; Hsu J. T.; Liang P. H. Characterization of SARS main protease and inhibitor assay using a fluorogenic substrate. Biochem. Biophys. Res. Commun. 2004, 318, 862–867. 10.1016/j.bbrc.2004.04.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand K.; Ziebuhr J.; Wadhwani P.; Mesters J. R.; Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- Chou K.; Wei D.; Zhong W. Binding mechanism of coronavirus main proteinase with ligands and its implication to drug design against SARS. Biochem. Biophys. Res. Commun. 2003, 308, 148–151. 10.1016/S0006-291X(03)01342-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke T. Nature (Science Update): SARS’ Achilles’ Heel Revealed. Science, May15, 2003; DOI: 10.1038/news030512-11. [DOI]
- Jenwitheesuk E.; Samudrala R. Identifying inhibitors of the SARS coronavirus proteinase. Bioorg. Med. Chem. Lett. 2003, 13, 3989–3992. 10.1016/j.bmcl.2003.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee P.; Desai P.; Ross L.; White E. L.; Avery M. A. Structure-based virtual screening against SARS-3CL(pro) to identify novel non-peptidic hits. Bioorg. Med. Chem. 2008, 16, 4138–4149. 10.1016/j.bmc.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A. K.; Xi K.; Ratia K.; Santarsiero B. D.; Fu W.; Harcourt B. H.; Rota P. A.; Baker S. C.; Johnson M. E.; Mesecar A. D. Design and synthesis of peptidomimetic severe acute respiratory syndrome chymotrypsin-like protease inhibitors. J. Med. Chem. 2005, 48, 6767–6771. 10.1021/jm050548m. [DOI] [PubMed] [Google Scholar]
- Bone R.; Vacca J. P.; Anderson P. S.; Holloway M. K. X-ray crystal structure of the hiv protease complex with l-700,417, an inhibitor with pseudo C2 symmetry. J. Am. Chem. Soc. 1991, 113, 9382–9384. 10.1021/ja00024a061. [DOI] [Google Scholar]
- Shie J.-J.; Fang J.-M.; Kuo T.-H.; Kuo C.-J.; Liang P.-H.; Huang H.-J.; Wu Y.-T.; Jan J.-T.; Cheng Y.-S. E.; Wong C.-H. Inhibition of the severe acute respiratory syndrome 3CL protease by peptidomimetic alpha, beta-unsaturated esters. Bioorg. Med. Chem. 2005, 13, 5240–5252. 10.1016/j.bmc.2005.05.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A. K.; Xi K.; Ratia K.; Santarsiero B. D.; Fu W.; Harcourt B. H.; Rota P. A.; Baker S. C.; Johnson M. E.; Mesecar A. D. Design and synthesis of peptidomimetic severe acute respiratory syndrome chymotrypsin-like protease inhibitors. J. Med. Chem. 2005, 48, 6767–6770. 10.1021/jm050548m. [DOI] [PubMed] [Google Scholar]
- Ghosh A. K.; Xi K.; Grum-Tokars V.; Xu X.; Ratia K.; Fu W.; Houser K. V.; Baker S. C.; Johnson M. E.; Mesecar A. D. Structure-based design, synthesis, and biological evaluation of peptidomimetic SARS-CoV 3CLpro inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 5876–5880. 10.1016/j.bmcl.2007.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S.; Chen S.- J.; Hsu M.-F.; Wu J.-D.; Tseng C.-T. K.; Liu Y.-F.; Chen H.-C.; Kuo C.-W.; Wu C.-S.; Chang L.-W.; Chen W.-C.; Liao S.-Y.; Chang T.-Y.; Hung H.-H.; Shr H.-L.; Liu C.-Y.; Huang Y.-A.; Chang L.-Y.; Hsu J.-C.; Peters C. J.; Wang A. H.-J.; Hsu M.-C. Synthesis, crystal structure, structure-activity relationships, and antiviral activity of a potent SARS coronavirus 3CL protease inhibitor. J. Med. Chem. 2006, 49, 4971–4980. 10.1021/jm0603926. [DOI] [PubMed] [Google Scholar]
- Jain R. P.; Pettersson H. I.; Zhang J.; Aull K. D.; Fortin P. D.; Huitema C.; Eltis L. D.; Parrish J. C.; James M. N. G.; Wishart D. S.; Vederas J. C. Synthesis and evaluation of keto-glutamine analogues as potent inhibitors of severe acute respiratory syndrome 3CLpro. J. Med. Chem. 2004, 47, 6113–6116. 10.1021/jm0494873. [DOI] [PubMed] [Google Scholar]
- Jain R. P.; Vederas J. C. Structural variations in keto-glutamines for improved inhibition against hepatitis A virus 3C proteinase. Bioorg. Med. Chem. Lett. 2004, 14, 3655–3658. 10.1016/j.bmcl.2004.05.021. [DOI] [PubMed] [Google Scholar]
- Ramtohul Y. K.; James M. N. G.; Vederas J. C. Synthesis and evaluation of keto-glutamine analogues as inhibitors of hepatitis A virus 3C proteinase. J. Org. Chem. 2002, 67, 3169–3178. 10.1021/jo0157831. [DOI] [PubMed] [Google Scholar]
- Yin J.; Niu C.; Cherney M. M.; Zhang J.; Huitema C.; Eltis L. D.; Vederas J. C.; James M. N. G. A mechanistic view of enzyme inhibition and peptide hydrolysis in the active site of the SARS-CoV 3C-like peptidase. J. Mol. Biol. 2007, 371, 1060–1074. 10.1016/j.jmb.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shie J. J.; Fang J.-M.; Kuo C.-J.; Kuo T.-H.; Liang P.-H.; Huang H.-J.; Yang W.-B.; Lin C.-H.; Chen J.-L.; Wu Y.-T.; Wong C.-H. Discovery of potent anilide inhibitors against the severe acute respiratory syndrome 3CL protease. J. Med. Chem. 2005, 48, 4469–4473. 10.1021/jm050184y. [DOI] [PubMed] [Google Scholar]
- Zhang R.; Malcolm B. A.; Beyer B. M.; Njoroge F. G.; Durkin J. P.; Windsor W. T.. Peptide Substrates for Hepatitis C Virus NS3 Protease Assays. U.S. Patent, US 6,251,583 B1, 2001; 21 pp.
- Asgian J. L.; James K. E.; Li Z. Z.; Carter W.; Barrett A. J.; Mikolajczyk J.; Salvesen G. S.; Powers J. C. Aza-peptide epoxides: A new class of inhibitors selective for clan CD cysteine proteases. J. Med. Chem. 2002, 45, 4958–4960. 10.1021/jm025581c. [DOI] [PubMed] [Google Scholar]
- Lee T. W.; Cherney M. M.; Huitema C.; Liu J.; James K. E.; Powers J. C.; Eltis L. D.; James M. N. G. Crystal structures of the main peptidase from the SARS coronavirus inhibited by a substrate-like aza-peptide epoxide. J. Mol. Biol. 2005, 353, 1137–1151. 10.1016/j.jmb.2005.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T. W.; Cherney M. M.; Liu J.; James K. E.; Powers J. C.; Eltis L. D.; James M. N. G. Crystal structures reveal an induced-fit binding of a substrate-like aza-peptide epoxide to SARS coronavirus main peptidase. J. Mol. Biol. 2007, 366, 916–932. 10.1016/j.jmb.2006.11.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina E.; Stiefl N.; Degel B.; Schulz F.; Breuning A.; Schiller M.; Vicik R.; Baumann K.; Ziebuhr J.; Schirmeister T. Screening of electrophilic compounds yields an aziridinyl peptide as new active-site directed SARS-CoV main protease inhibitor. Bioorg. Med. Chem. Lett. 2005, 15, 5365–5369. 10.1016/j.bmcl.2005.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Gharabli S. I.; Shah S. T.; Weik S.; Schmidt M. F.; Mesters J. R.; Kuhn D.; Klebe G.; Hilgenfeld R.; Rademann J. An efficient method for the synthesis of peptide aldehyde libraries employed in the discovery of reversible SARS coronavirus main protease (SARS-CoV Mpro) inhibitors. ChemBioChem 2006, 7, 1048–1055. 10.1002/cbic.200500533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L.; George S.; Schmidt M. F.; Al-Gharabli S. I.; Rademann J.; Hilgenfeld R. Peptide aldehyde inhibitors challenge the substrate specificity of the SARS-coronavirus main protease. Antiviral Res. 2011, 92, 204–212. 10.1016/j.antiviral.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akaji K.; Konno H.; Onozuka M.; Makino A.; Saito H.; Nosaka K. Evaluation of peptide-aldehyde inhibitors using R188I mutant of SARS 3CL protease as a proteolysis-resistant mutant. Bioorg. Med. Chem. 2008, 16, 9400–9408. 10.1016/j.bmc.2008.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akaji K.; Konno H.; Mitsui H.; Teruya K.; Shimamoto Y.; Hattori Y.; Ozaki T.; Kusunoki M.; Sanjoh A. Structure-based design, synthesis, and evaluation of peptide-mimetic SARS 3CL protease inhibitors. J. Med. Chem. 2011, 54, 7962–73. 10.1021/jm200870n. [DOI] [PubMed] [Google Scholar]
- Zhang H.- Z.; Zhang H.; Kemnitzer W.; Tseng B.; Cinatl J. Jr.; Michaelis M.; Doerr H. W.; Cai S. X. Design and synthesis of dipeptidyl glutaminyl fluoromethyl ketones as potent severe acute respiratory syndrome coronavirus (SARS-CoV) inhibitors. J. Med. Chem. 2006, 49, 1198–1201. 10.1021/jm0507678. [DOI] [PubMed] [Google Scholar]
- Yang W.; Guastella J.; Huang J.-C.; Wang Y.; Zhang L.; Xue D.; Tran M.; Woodward R.; Kasibhatla S.; Tseng B.; Drewe J.; Cai S. X. MX1013, a dipeptide caspase inhibitor with potent in vivo antiapoptotic activity. Br. J. Pharmacol. 2003, 140, 402–412. 10.1038/sj.bjp.0705450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Huang J.-C.; Zhou Z.-L.; Yang W.; Guastella J.; Drewe J.; Cai S. X. Dipeptidyl aspartyl fluoromethylketones as potent caspase-3 inhibitors: SAR of the P2 amino acid. Bioorg. Med. Chem. Lett. 2004, 14, 1269–1272. 10.1016/j.bmcl.2003.12.065. [DOI] [PubMed] [Google Scholar]
- Begue J.-P.; Bonnet-Delpon D. Preparation of trifluoromethyl ketones and related fluorinated ketones. Tetrahedron 1991, 47, 3207–3258. 10.1016/S0040-4020(01)86391-2. [DOI] [Google Scholar]
- Gelb M. H.; Svaren J. P.; Abeles R. H. Fluoro ketone inhibitors of hydrolytic enzymes. Biochemistry 1985, 24, 1813–1817. 10.1021/bi00329a001. [DOI] [PubMed] [Google Scholar]
- Sydnes M. O.; Hayashi Y.; Sharma V. K.; Hamada T.; Bacha U.; Barrila J.; Freire E.; Kiso Y. Synthesis of glutamic acid and glutamine peptides possessing a trifluoromethyl ketone group as SARS-CoV 3CL protease inhibitors. Tetrahedron 2006, 62, 8601–8609. 10.1016/j.tet.2006.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regnier T.; Sarma D.; Hidaka K.; Bacha U.; Freire E.; Hayashi Y.; Kiso Y. New developments for the design, synthesis and biological evaluation of potent SARS-CoV 3CL(pro) inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 2722–2727. 10.1016/j.bmcl.2009.03.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno S.; Thanigaimalai P.; Yamamoto T.; Nakada K.; Kakiuchi R.; Takayama K.; Yamazaki Y.; Yakushiji F.; Akaji K.; Kiso Y.; Kawasaki Y.; Chen S. E.; Freire E.; Hayashi Y. Design and synthesis of new tripeptide-type SARS-CoV 3CL protease inhibitors containing an electrophilic arylketone moiety. Bioorg. Med. Chem. 2013, 21, 412–424. 10.1016/j.bmc.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanigaimalai P.; Konno S.; Yamamoto T.; Koiwai Y.; Taguchi A.; Takayama K.; Yakushiji F.; Akaji K.; Chen S. E.; Naser-Tavakolian A.; Schön A.; Freire E.; Hayashi Y. Development of potent dipeptide-type SARS-CoV 3CL protease inhibitors with novel P3 scaffolds: design, synthesis, biological evaluation, and docking studies. Eur. J. Med. Chem. 2013, 68, 372–384. 10.1016/j.ejmech.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanigaimalai P.; Konno S.; Yamamoto T.; Koiwai Y.; Taguchi A.; Takayama K.; Yakushiji F.; Akaji K.; Kiso Y.; Kawasaki Y.; Chen S. E.; Naser-Tavakolian A.; Schön A.; Freire E.; Hayashi Y. Design, synthesis, and biological evaluation of novel dipeptide-type SARS-CoV 3CL protease inhibitors: structure-activity relationship study. Eur. J. Med. Chem. 2013, 65, 436–447. 10.1016/j.ejmech.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Y.-M.; Yang W.-B.; Kuo T.-H.; Tsai K.-C.; Lin C.-H.; Yang A.-S.; Liang P.-H.; Wong C.-H. Design, synthesis, and evaluation of trifluoromethyl ketones as inhibitors of SARS-CoV 3CL protease. Bioorg. Med. Chem. 2008, 16, 4652–4660. 10.1016/j.bmc.2008.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Y.-M.; Yang W.-B.; Peng H.-P.; Hsu M.-F.; Tsai K.-C.; Kuo T.-H.; Wang A. H.-J.; Liang P.-H.; Lin C.-H.; Yang A.-S.; Wong C.-H. Structure-based design and synthesis of highly potent SARS-CoV 3CL protease inhibitors. ChemBioChem 2007, 8, 1654–1657. 10.1002/cbic.200700254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeppler U.; Stiefl N.; Schiller M.; Vicik R.; Breuning A.; Schmitz W.; Rupprecht D.; Schmuck C.; Baumann K.; Ziebuhr J.; Schirmeister T. A new lead for nonpeptidic active-site-directed inhibitors of the severe acute respiratory syndrome coronavirus main protease discovered by a combination of screening and docking methods. J. Med. Chem. 2005, 48, 6832–6842. 10.1021/jm0501782. [DOI] [PubMed] [Google Scholar]
- Sprague J. M. Diuretics. Top. Med. Chem. 1986, 2, 1–63. [Google Scholar]
- Kaeppler U.; Schirmeister T. New non-peptidic inhibitors of papain derived from etacrynic acid. Med. Chem. 2005, 1, 361–370. 10.2174/1573406054368701. [DOI] [PubMed] [Google Scholar]
- Webber S. E.; Tikhe J.; Worland S. T.; Fuhrman S. A.; Hendrickson T. F.; Matthews D. A.; Love R. A.; Patick A. K.; Meador J. W.; Ferre R. A.; Brown E. L.; DeLisle D. M.; Ford C. E.; Binford S. L. Design, synthesis, and evaluation of nonpeptidic inhibitors of human rhinovirus 3C protease. J. Med. Chem. 1996, 39, 5072–5082. 10.1021/jm960603e. [DOI] [PubMed] [Google Scholar]
- Chen L. R.; Wang Y. C.; Lin Y. W.; Chou S. Y.; Chen S. F.; Liu L. T.; Wu Y. T.; Kuo C. J.; Chen T. S. S.; Juang S. H. Synthesis and evaluation of isatin derivatives as effective SARS coronavirus 3CL protease inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 3058–3062. 10.1016/j.bmcl.2005.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.; Zhu H.-M.; Niu G.-J.; Shi E.-Z.; Chen J.; Sun B.; Chen W.-Q.; Zhou H.-G.; Yang C. Synthesis, modification and docking studies of 5-sulfonyl isatin derivatives as SARS-CoV 3C-like protease inhibitors. Bioorg. Med. Chem. 2014, 22, 292–302. 10.1016/j.bmc.2013.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Güngör T.; Chen Y.; Golla R.; Ma Z.; Corte J. R.; Northrop J. P.; Bin B.; Disckson J. K.; Stouch T.; Zhou R.; Johnson S. E.; Seethala R.; Feyen J. H. M. Synthesis and characterization of 3-arylquinazolinone and 3-arylquinazolinethione derivatives as selective estrogen receptor beta modulators. J. Med. Chem. 2006, 49, 2440–2455. 10.1021/jm0509389. [DOI] [PubMed] [Google Scholar]
- Yi L.; Li Z.; Yuan K.; Qu X.; Chen J.; Wang G.; Zhang H.; Luo H.; Zhu L.; Jiang P.; Chen L.; Shen Y.; Luo M.; Zuo G.; Hu J.; Duan D.; Nie Y.; Shi X.; Wang W.; Han Y.; Li T.; Liu Y.; Ding M.; Deng H.; Xu X. Small molecules blocking the entry of severe acute respiratory syndrome coronavirus into host cells. J. Virol. 2004, 78, 11334–11339. 10.1128/JVI.78.20.11334-11339.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.; Li J.; Luo C.; Liu H.; Xu W.; Chen G.; Liew O. W.; Zhu W.; Puah C. M.; Shen X.; Jiang H. Binding interaction of quercetin-3-beta-galactoside and its synthetic derivatives with SARS-CoV 3CL(pro): Structure-activity relationship studies reveal salient pharmacophore features. Bioorg. Med. Chem. 2006, 14, 8295–8306. 10.1016/j.bmc.2006.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu Y. B.; Jeong H. J.; Kim J. H.; Kim Y. M.; Park J.-Y.; Kim D.; Naguyen T. T. H.; Park S.-J.; Chang J. S.; Park K. H. Biflavonoids from Torreya nucifera displaying SARS-CoV 3CLpro inhibition. Bioorg. Med. Chem. 2010, 18, 7940–7947. 10.1016/j.bmc.2010.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu Y. B.; Park S.-J.; Kim Y. M.; Lee J.-Y.; Seo W. D.; Chang J. S.; Park K. H.; Rho M.-C.; Lee W. S. SARS-CoV 3CLpro inhibitory effects of quinone-methide triterpenes from Tripterygium regelii. Bioorg. Med. Chem. Lett. 2010, 20, 1873–1876. 10.1016/j.bmcl.2010.01.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen C.-C.; Kuo Y.-H.; Jan J.-T.; Liang P.-H.; Wang S.-Y.; Liu H.-G.; Lee C.-K.; Chang S.-T.; Kuo C.-J.; Lee S.-S.; Hou C.-C.; Hsiao P.-W.; Chien S.-C.; Shyur L.-F.; Yang N.-S. Specific plant terpenoids and lignoids possess potent antiviral activities against severe acute respiratory syndrome coronavirus. J. Med. Chem. 2007, 50, 4087–4095. 10.1021/jm070295s. [DOI] [PubMed] [Google Scholar]
- Lu I.-L.; Mahindroo N.; Liang P.-H.; Peng Y.-H.; Kuo C.-J.; Tsai K.-C.; Hsieh H.-P.; Chao Y.-S.; Wu S.-Y. Structure-based drug design and structural biology study of novel nonpeptide inhibitors of severe acute respiratory syndrome coronavirus main protease. J. Med. Chem. 2006, 49, 5154–5161. 10.1021/jm060207o. [DOI] [PubMed] [Google Scholar]
- Tsai K.-C.; Chen S.-Y.; Liang P.-H.; Lu I.-L.; Mahindroo N.; Hsieh H.-P.; Chao Y.-S.; Liu L.; Liu D.; Lien W.; Lin T.-H.; Wu S.-Y. Discovery of a novel family of SARS-CoV protease inhibitors by virtual screening and 3D-QSAR studies. J. Med. Chem. 2006, 49, 3485–3495. 10.1021/jm050852f. [DOI] [PubMed] [Google Scholar]
- Wu C.-Y.; King K.-Y.; Kuo C.-J.; Fang J.-M.; Wu Y.-T.; Ho M.-Y.; Liao C.-L.; Shie J.-J.; Liang P.-H.; Wong C.-H. Stable benzotriazole esters as mechanism-based inactivators of the severe acute respiratory syndrome 3CL protease. Chem. Biol. 2006, 13, 261–268. 10.1016/j.chembiol.2005.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brik A.; Lin Y.-C.; Elder J.; Wong C.-H. A quick diversity-oriented amide-forming reaction to optimize P-subsite residues of HIV protease inhibitors. Chem. Biol. 2002, 9, 891–896. 10.1016/S1074-5521(02)00184-9. [DOI] [PubMed] [Google Scholar]
- Wu C.-Y.; Chang C.-F.; Chen J. S.-Y.; Wong C.-H.; Lin C.- H. Rapid diversity-oriented synthesis in microtiter plates for in situ screening: discovery of potent and selective alpha-fucosidase inhibitors. Angew. Chem., Int. Ed. 2003, 42, 4661–4664. 10.1002/anie.200351823. [DOI] [PubMed] [Google Scholar]
- Chang C.-F.; Ho C.-W.; Wu C.-Y.; Chao T.-A.; Wong C.-H.; Lin C.-H. Discovery of picomolar slow tight-binding inhibitors of alpha-fucosidase. Chem. Biol. 2004, 11, 1301–1306. 10.1016/j.chembiol.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Verschueren K. H. G.; Pumpor K.; Anemüller S.; Chen S.; Mesters J. R.; Hilgenfeld R. A structural view of the inactivation of the SARS coronavirus main proteinase by benzotriazole esters. Chem. Biol. 2008, 15, 597–606. 10.1016/j.chembiol.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard J. E.; Elowe N. H.; Huitema C.; Fortin P. D.; Cechetto J. D.; Eltis L. D.; Brown E. D. High-throughput screening identifies inhibitors of the SARS coronavirus main proteinase. Chem. Biol. 2004, 11, 1445–1453. 10.1016/j.chembiol.2004.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Pettersson H. I.; Huitema C.; Niu C.; Yin J.; James M. N.; Eltis L. D.; Vederas J. C. Design, synthesis, and evaluation of inhibitors for severe acute respiratory syndrome 3C-like protease based on phthalhydrazide ketones or heteroaromatic esters. J. Med. Chem. 2007, 50, 1850–1864. 10.1021/jm061425k. [DOI] [PubMed] [Google Scholar]
- Niu C.; Yin J.; Zhang J.; Vederas J. C.; James M. N. Molecular docking identifies the binding of 3-chloropyridine moieties specifically to the S1 pocket of SARS-CoV Mpro. Bioorg. Med. Chem. 2008, 16, 293–302. 10.1016/j.bmc.2007.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A. K.; Gong G.; Grum-Tokars V.; Mulhearn D. C.; Baker S. C.; Coughlin M.; Prabhakar B. S.; Sleeman K.; Johnson M. E.; Mesecar A. D. Design, synthesis and antiviral efficacy of a series of potent chloropyridyl ester-derived SARS-CoV 3CLpro inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 5684–5688. 10.1016/j.bmcl.2008.08.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Huitema C.; Niu C.; Yin J.; James M. N.G.; Eltis L. D.; Vederas J. C. Aryl methylene ketones and fluorinated methylene ketones as reversible inhibitors for severe acute respiratory syndrome (SARS) 3C-like proteinase. Bioorg. Chem. 2008, 36, 229–240. 10.1016/j.bioorg.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.; Chen S.; Gui C.; Shen J.; Shen X.; Jiang H. Discovering severe acute respiratory syndrome coronavirus 3CL protease inhibitors: virtual screening, surface plasmon resonance, and fluorescence resonance energy transfer assays. J. Biomol. Screening 2006, 11, 915–921. 10.1177/1087057106293295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo C. J.; Liu H. G.; Lo Y. K.; Seong C. M.; Lee K. I.; Jung Y. S.; Liang P. H. Individual and common inhibitors of coronavirus and picornavirus main proteases. FEBS Lett. 2009, 583, 549–555. 10.1016/j.febslet.2008.12.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramajayam R.; Tan K.-P.; Liu H.-G.; Liang P.-H. Synthesis and evaluation of pyrazolone compounds as SARS-coronavirus 3C-like protease inhibitors. Bioorg. Med. Chem. 2010, 18, 7849–7854. 10.1016/j.bmc.2010.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn T. Y.; Kuo C. J.; Liu H. G.; Ha D. C.; Liang P. H.; Jung Y. S. Synthesis and evaluation of benzoquinolinone derivatives as sars-cov 3cl protease inhibitors. Bull. Korean Chem. Soc. 2010, 31, 87–91. 10.5012/bkcs.2010.31.01.087. [DOI] [Google Scholar]
- Ramajayam R.; Tan K.-P.; Liu H.-G.; Liang P.-H. Synthesis, docking studies, and evaluation of pyrimidines as inhibitors of SARS-CoV 3CL protease. Bioorg. Med. Chem. Lett. 2010, 20, 3569–3572. 10.1016/j.bmcl.2010.04.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamoto Y.; Hattori Y.; Kobayashi K.; Teruya K.; Sanjoh A.; Nakagawa A.; Yamashita K.; Akaji K. Fused-ring structure of decahydroisoquinolin as a novel scaffold for SARS 3CL protease inhibitors. Bioorg. Med. Chem. 2015, 23, 876–890. 10.1016/j.bmc.2014.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs J.; Grum-Tokars V.; Zhou Y.; Turlington M.; Saldanha S. A.; Chase P.; Eggler A.; Dawson E. S.; Baez-Santos Y. M.; Tomar S.; Mielech A. M.; Baker S. C.; Lindsley C. W.; Hodder P.; Mesecar A.; Stauffer S. R. Discovery, synthesis, and structure-based optimization of a series of N-(tert-butyl)-2-(N-arylamido)-2-(pyridin-3-yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS-CoV) 3CL protease. J. Med. Chem. 2013, 56, 534–546. 10.1021/jm301580n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretto N.; Jukneliene D.; Ratia K.; Chen Z.; Mesecar A. D.; Baker S. C. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J. Virol. 2005, 79, 15189–15198. 10.1128/JVI.79.24.15189-15198.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turlington M.; Chun A.; Tomar S.; Eggler A.; Grum-Tokars V.; Jacobs J.; Daniels J. S.; Dawson E.; Saldanha A.; Chase P.; Baez-Santos Y. M.; Lindsley C. W.; Hodder P.; Mesecar A. D.; Stauffer S. R. Discovery of N-(benzo[1,2,3]triazol-1-yl)-N-(benzyl)acetamido)phenyl) carboxamides as severe acute respiratory syndrome coronavirus (SARS-CoV) 3CLpro inhibitors: identification of ML300 and noncovalent nanomolar inhibitors with an induced-fit binding. Bioorg. Med. Chem. Lett. 2013, 23, 6172–6177. 10.1016/j.bmcl.2013.08.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turlington M.; Chun A.; Jacobs J.; Dawson E.; Daniels J. S.; Saldanha A.; Chase P.; Hodder P.; Eggler A.; Tokars V.; Mesecar A.; Lindsley C. W.; Stauffer S. R.. Noncovalent triazole-based inhibitors of the SARS main proteinase 3CLpro. Probe Reports from the NIH Molecular Libraries Program; National Center for Biotechnology Information: Bethesda, MD, 2012; http://www.ncbi.nlm.nih.gov/books/NBK143547/. [PubMed]
- Hopkins A. L.; Groom C. R.; Alex A. Drug Discovery Today 2004, 9, 430–431. 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- For information on MLPCN’s probe compound ancillary screen, see Eurofins LeadProfilingScreen
- Someya Y.; Takeda N.; Miyamura T. Characterization of the norovirus 3C-like protease. Virus Res. 2005, 110, 91–97. 10.1016/j.virusres.2005.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y. S.; Chang G. G.; Juo C. G.; Lee H. J.; Yeh S. H.; Hsu J. T.; Chen X. Papain-like protease 2 (PLP2) from severe acute respiratory syndrome coronavirus (SARS-CoV): expression, purification, characterization, and inhibition. Biochemistry 2005, 44, 10349–10359. 10.1021/bi0504761. [DOI] [PubMed] [Google Scholar]
- Dhanak D.; Burton G.; Christmann L. T.; Darcy M. G.; Elrod K. C.; Kaura A.; Keenan R. M.; Link J. O.; Peishoff C. E.; Shah D. H. Metal mediated protease inhibition: design and synthesis of inhibitors of the human cytomegalovirus (hCMV) protease. Bioorg. Med. Chem. Lett. 2000, 10, 2279–2282. 10.1016/S0960-894X(00)00462-5. [DOI] [PubMed] [Google Scholar]
- Tong L.; Qian C.; Massariol M.-J.; Bonneau P. R.; Cordingley M. G.; Lagace L. A new serine-protease fold revealed by the crystal structure of human cytomegalovirus protease. Nature 1996, 383, 272–275. 10.1038/383272a0. [DOI] [PubMed] [Google Scholar]
- Yeung K. S.; Meanwell N. A.; Qiu Z.; Hernandez D.; Zhang S.; McPhee F.; Weinheimer S.; Clark J. M.; Janc J. W. Structure-activity relationship studies of a bisbenzimidazole-based, Zn(2+)-dependent inhibitor of HCV NS3 serine protease. Bioorg. Med. Chem. Lett. 2001, 11, 2355–2359. 10.1016/S0960-894X(01)00457-7. [DOI] [PubMed] [Google Scholar]
- Hsu J. T.-A.; Kuo C.-J.; Hsieh H.-P.; Wang Y.-C.; Huang K.-K.; Lin C. P.-C.; Huang P.-F.; Chen X.; Liang P.-H. Evaluation of metal-conjugated compounds as inhibitors of 3CL protease of SARS-CoV. FEBS Lett. 2004, 574, 116–120. 10.1016/j.febslet.2004.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.-C.; Kuo C.-J.; Hsu M.-F.; Liang P.-H.; Fang J.-M.; Shie J.-J.; Wang A. H.-J. Structural basis of mercury- and zinc-conjugated complexes as SARS-CoV 3C-like protease inhibitors. FEBS Lett. 2007, 581, 5454–5458. 10.1016/j.febslet.2007.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz B. A.; Clark J. M.; Finer-Moore J. S.; Jenkins T. E.; Johnson C. R.; Ross M. J.; Luong C.; Moore W. R.; Stroud R. M. Design of potent selective zinc-mediated serine protease inhibitors. Nature 1998, 391, 608–612. 10.1038/35422. [DOI] [PubMed] [Google Scholar]
- Christianson D. W.; Lipscomb W. N. X-ray crystallographic investigation of substrate binding to carboxypeptidase A at subzero temperature. Proc. Natl. Acad. Sci. U. S. A. 1986, 83, 7568–7572. 10.1073/pnas.83.20.7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer G. J.; Johnson V. D.; Dick R. D.; Hedera P.; Fink J. K.; Kluin K. J. Treatment of Wilson’s disease with zinc. XVII: Treatment during pregnancy. Hepatology 2000, 31, 364–370. 10.1002/hep.510310216. [DOI] [PubMed] [Google Scholar]
- Sharquie K. E.; Najim R. A.; Al-Dori W. S.; Al-Hayani R. K. Oral zinc sulfate in the treatment of Behcet’s disease: a double blind cross-over study. J. Dermatol. 2006, 33, 541–546. 10.1111/j.1346-8138.2006.00128.x. [DOI] [PubMed] [Google Scholar]
- Sakurai H.; Adachi Y. The pharmacology of the insulinomimetic effect of zinc complexes. BioMetals 2005, 18, 319–323. 10.1007/s10534-005-3688-8. [DOI] [PubMed] [Google Scholar]
- Bacha U.; Barrila J.; Velazquez-Campoy A.; Leavitt S. A.; Freire E. Identification of novel inhibitors of the SARS coronavirus main protease 3CLpro. Biochemistry 2004, 43, 4906–4912. 10.1021/bi0361766. [DOI] [PubMed] [Google Scholar]
- Hou T. J.; Xu X. J. Recent development and application of virtual screening in drug discovery: an overview. Curr. Pharm. Des. 2004, 10, 1011–1033. 10.2174/1381612043452721. [DOI] [PubMed] [Google Scholar]
- Andricopulo A. D.; Guido R. V. C.; Oliva G. Virtual screening and its integration with modern drug design technologies. Curr. Med. Chem. 2008, 15, 37–46. 10.2174/092986708783330683. [DOI] [PubMed] [Google Scholar]
- Kao R. Y.; Tsui W. H. W.; Lee T. S. W.; Tanner J. A.; Watt R. M.; Huang J. D.; Hu L. H.; Chen G. H.; Chen Z. W.; Zhang L. Q.; He T.; Chan K. H.; Tse H.; To A. P. C.; Ng L. W. Y.; Wong B. C. W.; Tsoi H. W.; Yang D.; Ho D. D.; Yuen K. Y. Identification of novel small-molecule inhibitors of severe acute respiratory syndrome-associated coronavirus by chemical genetics. Chem. Biol. 2004, 11, 1293–1299. 10.1016/j.chembiol.2004.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama T.; Sato Y.; Oto Y.; Takahashi Y.; Snoeck R.; Andrei G.; Witvrouw M.; De Clercq E. Synthesis and antiviral activity of 6-chloropurine arabinoside and its 2′-deoxy-2′-fluoro derivative. Chem. Pharm. Bull. 1996, 44, 2331–2334. 10.1248/cpb.44.2331. [DOI] [PubMed] [Google Scholar]
- Honjo M.; Maruyama T.; Horikawa M.; Balzarini J.; De Clercq E. Synthesis and biological evaluation of phosphonopyrimidine and phosphonopurine ribonucleosides. Chem. Pharm. Bull. 1987, 35, 3227–3234. 10.1248/cpb.35.3227. [DOI] [PubMed] [Google Scholar]
- Ikejiri M.; Saijo M.; Morikawa S.; Fukushi S.; Mizutani T.; Kurane I.; Maruyama T. Synthesis and biological evaluation of nucleoside analogues having 6-chloropurine as anti-SARS-CoV agents. Bioorg. Med. Chem. Lett. 2007, 17, 2470–2473. 10.1016/j.bmcl.2007.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren G.; King S.; Knowles S.; Phillips E. Ribavirin in the treatment of SARS: A new trick for an old drug?. Can. Med. Assoc. J. 2003, 168, 1289–1292. [PMC free article] [PubMed] [Google Scholar]
- Stroher U.; DiCaro A.; Li Y.; Strong J. E.; Aoki F.; Plummer F.; Jones S. M.; Feldmann H. Severe acute respiratory syndrome-related coronavirus is inhibited by interferon- alpha. J. Infect. Dis. 2004, 189, 1164–1167. 10.1086/382597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto N.; Yang R.; Yoshinaka Y.; Amari S.; Nakano T.; Cinatl J.; Rabenau H.; Doerr H. W.; Hunsmann G.; Otaka A.; Tamamura H.; Fujii N.; Yamamoto N. HIV protease inhibitor nelfinavir inhibits replication of SARS-associated coronavirus. Biochem. Biophys. Res. Commun. 2004, 318, 719–725. 10.1016/j.bbrc.2004.04.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyaerts E.; Vijgen L.; Maes P.; Neyts J.; Ranst M. V. In vitro inhibition of severe acute respiratory syndrome coronavirus by chloroquine. Biochem. Biophys. Res. Commun. 2004, 323, 264–268. 10.1016/j.bbrc.2004.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan E. L. C.; Ooi E. E.; Lin C. Y.; Tan H. C.; Ling A. E.; Lim B.; Stanton L. W. Inhibition of SARS coronavirus infection in vitro with clinically approved antiviral drugs. Emerging Infect. Dis. 2004, 10, 581–586. 10.3201/eid1004.030458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. C.; Huang V.; Chao T. C.; Hsiao C. D.; Lin A.; Chang M. F.; Chow L. P. Screening of drugs by FRET analysis identifies inhibitors of SARS-CoV 3CL protease. Biochem. Biophys. Res. Commun. 2005, 333, 194–199. 10.1016/j.bbrc.2005.05.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T. T. H.; Ryu H.-J.; Lee S.-H.; Hwang S. W.; Breton V.; Rhee J. H.; Kim D. Virtual screening identification of novel severe acute respiratory syndrome 3C-like protease inhibitors and in vitro confirmation. Bioorg. Med. Chem. Lett. 2011, 21, 3088–3091. 10.1016/j.bmcl.2011.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.; Mittal A.; Patel K.; Gatuz J. L.; Truong L.; Torres J.; Mulhearn D. C.; Johnson M. E. Identification of novel drug scaffolds for inhibition of SARS-CoV 3-Chymotrypsin-like protease using virtual and high-throughput screenings. Bioorg. Med. Chem. 2014, 22, 167–177. 10.1016/j.bmc.2013.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetzer T.; Hauptmann J.; Sturzebecher J. Advances in the development of thrombin inhibitors. Expert Opin. Invest. Drugs 2001, 10, 845–864. 10.1517/13543784.10.5.845. [DOI] [PubMed] [Google Scholar]
- Klootwijk P.; Lenderink T.; Meij S.; Boersma H.; Melkert R.; Umans V. A.; Stibbe J.; Müller E. J.; Poortermans K. J.; Deckers J. W.; Simoons M. L. Anticoagulant properties, clinical efficacy and safety of efegatran, a direct thrombin inhibitor, in patients with unstable angina. Eur. Heart J. 1999, 20, 1101–1111. 10.1053/euhj.1999.1477. [DOI] [PubMed] [Google Scholar]
- Esser R. E.; Angelo R. A.; Murphey M. D.; Watts L. M.; Thornburg L. P.; Palmer J. T.; Talhouk J. W.; Smith R. E. Cysteine proteinase inhibitors decrease articular cartilage and bone destruction in chronic inflammatory arthritis. Arthritis Rheum. 1994, 37, 236–247. 10.1002/art.1780370213. [DOI] [PubMed] [Google Scholar]
- McGrath M. E.; Eakin A. E.; Engel J. C.; McKerrow J. H.; Craik C. S.; Fletterick R. J. The crystal structure of cruzain: A therapeutic target for Chagas’ disease. J. Mol. Biol. 1995, 247, 251–259. 10.1006/jmbi.1994.0137. [DOI] [PubMed] [Google Scholar]
- Richer J. K.; Hunt W. G.; Sakanari J. A.; Grieve R. B. Dirofilaria immitis: Effect of fluoromethyl ketone cysteine protease inhibitors on the third- to fourth-stage molt. Exp. Parasitol. 1993, 76, 221–231. 10.1006/expr.1993.1027. [DOI] [PubMed] [Google Scholar]
- Barrila J. A.Dimerization of the SARS coronavirus 3CL protease is controlled through long-range interactions. The Johns Hopkins University, ProQuest, UMI Dissertations Publishing, 2009, 3339678. [Google Scholar]
- Dyall J.; Coleman C. M.; Hart B. J.; Venkataraman T.; Holbrook M. R.; Kindrachuk J.; Johnson R. F.; Olinger G. G. Jr.; Jahrling P. B.; Laidlaw M.; Johansen L. M.; Lear-Rooney C. M.; Glass P. J.; Hensley L. E.; Frieman M. B. Repurposing of clinically developed drugs for treatment of Middle East respiratory syndrome coronavirus infection. Antimicrob. Agents Chemother. 2014, 58, 4885–4893. 10.1128/AAC.03036-14. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Pillaiyar T.; Manickam M.; Jung S. H. Middle East respiratory syndrome coronavirus (MERS-CoV): An updated overview and pharmacotherapeutics. Med. Chem. 2015, 5, 361–372. 10.4172/2161-0444.1000287. [DOI] [Google Scholar]
- Medeiros R.; Kitazawa M.; Chabrier M. A.; Cheng D.; Baglietto-Vargas D.; Kling A.; Moeller A.; Green K. N.; LaFerla F. M. Calpain inhibitor A-705253 mitigates Alzheimer’s disease-like pathology and cognitive decline in aged 3xTgAD mice. Am. J. Pathol. 2012, 181, 616–625. 10.1016/j.ajpath.2012.04.020. [DOI] [PubMed] [Google Scholar]
- Gauthier J. Y.; Chauret N.; Cromlish W.; Desmarais S.; Duong L. T.; Falgueyret J. P.; Kimmel D. B.; Lamontagne S.; Léger S.; LeRiche T.; Li C. S.; Massé F.; McKay D. J.; Nicoll-Griffith D. A.; Oballa R. M.; Palmer J. T.; Percival M. D.; Riendeau D.; Robichaud J.; Rodan G. A.; Rodan S. B.; Seto C.; Thérien M.; Truong V. L.; Venuti M. C.; Wesolowski G.; Young R. N.; Zamboni R.; Black W. C. The discovery of odanacatib (MK-0822), a selective inhibitor of cathepsin K. Bioorg. Med. Chem. Lett. 2008, 18, 923–928. 10.1016/j.bmcl.2007.12.047. [DOI] [PubMed] [Google Scholar]
- Ahrén B.; Landin-Olsson M.; Jansson P. A.; Svensson M.; Holmes D.; Schweizer A. Inhibition of dipeptidyl peptidase-4 reduces glycemia, sustains insulin levels, and reduces glucagon levels in type 2 diabetes. J. Clin. Endocrinol. Metab. 2004, 89, 2078–2084. 10.1210/jc.2003-031907. [DOI] [PubMed] [Google Scholar]
- Weber P.; Wang P.; Maddens S.; Wang P. S. H.; Wu R.; Miksa M.; Dong W.; Mortimore M.; Golec J. M. C.; Charlton P. VX-166: a novel potent small molecule caspase inhibitor as a potential therapy for sepsis. Crit. Care 2009, 13, R146. 10.1186/cc8041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman G. A.; Stevens C.; Van Dyck K.; Bergman A.; Yi B.; De Smet M.; Snyder K.; Hilliard D.; Tanen M.; Tanaka W.; Wang A. Q.; Zeng W.; Musson D.; Winchell G.; Davies M. J.; Ramael S.; Gottesdiener K. M.; Wagner J. A. Pharmacokinetics and pharmacodynamics of sitagliptin, an inhibitor of dipeptidyl peptidase IV, in healthy subjects: results from two randomized, double-blind, placebo-controlled studies with single oral doses. Clin. Pharmacol. Ther. 2005, 78, 675–688. 10.1016/j.clpt.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Nutescu E. Apixaban: A novel oral inhibitor of factor Xa. Am. J. Health-Syst. Pharm. 2012, 69, 1113–1126. 10.2146/ajhp110418. [DOI] [PubMed] [Google Scholar]
- Zervosen A.; Sauvage E.; Frère J. M.; Charlier P.; Luxen A. Development of new drugs for an old target: the penicillin binding proteins. Molecules 2012, 17, 12478–12505. 10.3390/molecules171112478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm H. J.; Boetzel J.; Büttner J.; Fritsche E.; Göhring W.; Jaeger E.; König S.; Thumfart O.; Wenger T.; Nagel N. E.; Schramm W. The inhibition of human immunodeficiency virus proteases by ’interface peptides’. Antiviral Res. 1996, 30, 155–170. 10.1016/0166-3542(96)00940-0. [DOI] [PubMed] [Google Scholar]
- Schramm H. J.; Nakashima H.; Schramm W.; Wakayama H.; Yamamoto N. HIV-1 reproduction is inhibited by peptides derived from the N- and C-termini of HIV-1 protease. Biochem. Biophys. Res. Commun. 1991, 179, 847–851. 10.1016/0006-291X(91)91895-J. [DOI] [PubMed] [Google Scholar]
- Boggetto N.; Reboud-Ravaux M. Dimerization inhibitors of HIV-1 protease. Biol. Chem. 2002, 383, 1321–1324. 10.1515/BC.2002.150. [DOI] [PubMed] [Google Scholar]
- Zutshi R.; Chmielewski J. Targeting the dimerization interface for irreversible inhibition of HIV-1 protease. Bioorg. Med. Chem. Lett. 2000, 10, 1901–1903. 10.1016/S0960-894X(00)00369-3. [DOI] [PubMed] [Google Scholar]
- Zutshi R.; Brickner M.; Chmielewski J. Inhibiting the assembly of protein-protein interfaces. Curr. Opin. Chem. Biol. 1998, 2, 62–66. 10.1016/S1367-5931(98)80036-7. [DOI] [PubMed] [Google Scholar]
- Weber I. T. Comparison of the crystal structures and intersubunit interactions of human immunodeficiency and Rous sarcoma virus proteases. J. Biol. Chem. 1990, 265, 10492–10496. [PubMed] [Google Scholar]
- Zhang Z. Y.; Poorman R. A.; Maggiora L. L.; Heinrikson R. L.; Kézdy F. J. Dissociative inhibition of dimeric enzymes. Kinetic characterization of the inhibition of HIV-1 protease by its COOH-terminal tetrapeptide. J. Biol. Chem. 1991, 266, 15591–15594. [PubMed] [Google Scholar]
- Wei P.; Fan K.; Chen H.; Ma L.; Huang C.; Tan L.; Xi D.; Li C.; Liu Y.; Cao A.; Lai L. The N-terminal octapeptide acts as a dimerization inhibitor of SARS coronavirus 3C-like proteinase. Biochem. Biophys. Res. Commun. 2006, 339, 865–872. 10.1016/j.bbrc.2005.11.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.