

Abstract

Class I enveloped viruses share similarities in their apparent use of a hexameric coiled-coil assembly to drive the merging of virus and host cell membranes. Inhibition of coiled coil-mediated interactions using bioactive peptides that replicate an α-helical chain from the viral fusion machinery has significant antiviral potential. Here, we present the construction of a series of lipopeptides composed of a de novo heptad repeat sequence-based α-helical peptide plus a hydrocarbon tail. Promisingly, the constructs adopted stable α-helical conformations and exhibited relatively broad-spectrum antiviral activities against Middle East respiratory syndrome coronavirus (MERS-CoV) and influenza A viruses (IAVs). Together, these findings reveal a new strategy for relatively broad-spectrum antiviral drug discovery by relying on the tunability of the α-helical coiled-coil domains present in all class I fusion proteins and the amphiphilic nature of the individual helices from this multihelix motif.
Introduction
The traditional “one bug–one drug” paradigm for the development of antiviral therapeutics has yielded laudable successes to control the emerging and re-emerging threat of viral pathogens.1−3 However, a broad-spectrum antiviral strategy that affords timely and effective pharmacological agents that can respond to an increasing diversity of highly pathogenic viruses remains elusive.4 A commonality in the viral life cycle, i.e., the fusion of enveloped viruses with the host cell membrane, represents a viable target for the discovery of broad-spectrum therapeutics.5,6 In this fusion process, the triggered formation of a leucine zipper-like α-helical hexamer,7,8 either at the cell surface or within some later endosomes, is the typical structural feature of class I fusion glycoproteins used by enveloped viruses such as human immunodeficiency virus type 1 (HIV-1),9−11 influenza A viruses (IAVs),12 Middle East respiratory syndrome coronavirus (MERS-CoV),13 and Ebola virus (EboV)14 (Figure 1A).
Figure 1.

Six-helix bundle (6HB) fusion core structure and the design of a lipopeptide template based on the interaction between the NHR and CHR domains. (A) Cartoon representations of the HIV (PDB 1AIK), MERS-CoV (PDB 4NJL), and influenza H3N2 (PDB 1QU1) 6HBs, in which the NHR trimers and CHR segments are colored in gray and green, respectively. (B) Helical wheel representation of a 6HB. The residues at the a, d, and e positions (yellow) form the buried face that interacts with the NHR trimers, while those at the b, c, f, and g positions (blue) are solvent-accessible sites. (C) The de novo designed lipopeptide template, in which the critical residues at the a, d, and e positions are highlighted in red font. The dotted lines show the predicted intramolecular salt bridges formed by the acidic amino acids at the i positions and the basic residues at the i + 4 positions.
During the coiled-coil six-helix bundle (6HB) assembly, three N-terminal heptad repeat (NHR) regions of viral fusion proteins initially form a central trimeric helix scaffold that becomes temporarily exposed, creating a metastable prehairpin conformation; three C-terminal heptad repeat (CHR) regions then pack onto the periphery of the trimeric NHR inner core in an antiparallel orientation.15 Bioactive peptides derived from the CHR motif of class I viral fusion proteins, designated as C-peptides, act as decoy α-helices and are able to bind to their corresponding NHR helical trimers to form a heterologous nonfunctional 6HB structure in a virus-specific manner, thereby antagonizing the refolding of the endogenous CHR region and competitively inhibiting virus–host cell membrane fusion.16 In 1993, Jiang and colleagues discovered a highly potent HIV-1 fusion inhibitory peptides derived from the CHR region of HIV-1 gp41, designated SJ-2176; in 1994, Wild et al. reported another CHR-peptide, DP-178 (also named T20 later).17,18 T20 (brand name, Fuzeon; generic name, enfuvirtide) was finally approved by the U.S. Food and Drug Administration in 2003 for clinical use as the first fusion inhibitor-based anti-HIV drug. The discovery of these anti-HIV peptides spurred the identification of antiviral peptides against other viruses that utilize class I fusion proteins, including the recently identified MERS-CoV. After the emergence of MERS-CoV infection, Gao’s group and Jiang’s group independently solved the crystal structure of MERS-CoV’s 6HB fusion core.13,19 On the basis of the 6HB structure, Jiang’s group reported the highly effective anti-MERS-CoV peptide HR2PM2. Most recently, we identified a hydrocarbon-stapled short α-helical peptide that could inhibit MERS-CoV infection at the low micromolar level.20 Considering the universal 6HB fusion mechanism employed by class I enveloped viruses, inhibition of NHR/CHR coiled coil-mediated interactions has significant potential for the development of broad-spectrum therapeutic interventions.
Coiled coils are ubiquitous protein folding motifs found in nature, and they orchestrate the association of numerous complexes implicated in biological processes.21 A wealth of structural information has provided a relatively detailed understanding of the sequence-to-structure relationships for this supercoiled scaffold.22 In structural terms, amphipathic α-helices encode canonical coiled coils via burial of their hydrophobic faces to drive the multimerization of constituent helices.23 The hexameric coiled-coil assembly present in HIV-1 is arguably the best characterized class I viral fusion apparatus,24,25 in which the C-helices are divided into a hydrophobic buried binding interface and hydrophilic solvent-accessible sites (Figure 1B). Structural analysis has demonstrated that a high variability in the primary sequence of CHR motifs within the HIV-1 6HB is allowed as long as the driving force for coiled-coil assembly, i.e., amphiphilic characteristics of the individual helices, is maintained.26,27 Accordingly, α-helix-constrained HIV-neutralizing C-peptides, engineered by strategies such as salt bridges,28,29 helix-favoring amino acids,30 and hydrophobic mutations in buried residues,31 can significantly enhance the bundle stability compared with their corresponding wild-type ligands despite the substitution of approximately half of the parent residues.
Incorporation of an alkyl hydrocarbon tail onto a peptide represents another promising α-helix-promoting technique. Recent studies on HIV-1 fusion inhibitors have revealed that lipid-conjugated C-peptides have a greatly increased α-helicity and NHR-binding capability.32,33 Moreover, for viruses that fuse at the cell surface, lipidation also provides the critical advantage of endowing C-peptides with the membrane-tropic feature. Enrichment of the local concentration of helical peptides at the membrane level further facilitates their assembly with the intermediate-stage NHR-helical trimers.34 For intracellularly fusing viruses, lipidation allows antiviral peptides to internalize along with viruses into host cells, thereby arresting 6HB formation in the endosomes.12
Taking advantage of the adjustability and tractability of α-helix-mediated interactions in the 6HB core structure typical of class I viral fusion proteins, we herein describe a rational approach for the design of relatively broad-spectrum inhibitors against infection of MERS-CoV, of the family Coronaviridae, that primarily employs the cell surface pathway,13,35 and IAV, of the family Orthomyxoviridae, that uses the endocytic route,6,7 based on replicating the topography of CHR helices by using de novo designed amphiphilic α-helical peptides with the addition of a fatty acid tail.
Design
In terms of the sequence, coiled-coil domains share a characteristic heptad repeat, usually denoted as a-b-c-d-e-f-g, with hydrophobic residues at the core a-d positions and hydrophilic residues at the other sites.20,21 In the helix wheel model of the NHR/CHR 6HB structure, the residues at the a-d-e sites on the C-helices face the inner N-helices and are largely buried; these residues are primarily responsible for CHR–NHR interaction. Meanwhile, the residues at the b-c-f-g sites are located away from the interaction center. Therefore, we adopted the heptad repeat approach to design amphiphilic peptides based on the sequence Ac-(XaEbEcXdZeKfKg)5-βAla-K(C16)-NH2. In the repeated heptapeptide sequence, we placed hydrophobic residues at the “X” positions and polar/charged residues at the “Z” positions. With the foregrounda-d and e positions of the model sequence set, the backgroundb-c-f-g sites were populated with combinations of glutamic acid and lysine to form double Glu-Lys intrastrand salt bridges at the i to i + 4 positions to favor the overall α-helicity and solubility of the heptad repeat peptides. Seminal work investigating the structural characterization of coiled coils suggests that the stability of helical bundles is directly proportional to the number of heptad repeats. Combined with the optimal peptide length paradigm established by current peptidic fusion inhibitors, the designed sequence was made with five heptads, i.e., 35 residues, and an β-alanine (βAla) was additionally appended outside of the heptad repeat region in order to link an extra lysine residue,36,37 which was capped by a palmitoyl group (C16), thus providing its membrane-tropic feature and favorable safety profile for drug development (Figure 1C).
Results and Discussion
Initially, we aimed to establish combinations of foregrounda-d residues in the heptad repeat moiety. As previous experiments have indicated that alanine residues are strongly biased toward helix formation in peptides,38 the design of the novel lipopeptide began with the peptide named AAS (Table 1), in which hydrophobic alanine residues were placed at the a and d positions of the heptad, and a small helix-favoring polar residue, i.e., serine, which has been widely utilized in protein functional scanning strategies,39 was populated at the e positions. To explore the influence of hydrophobic residue contact at α-helix-mediated coiled-coil interaction sites on antiviral activity, substitutions at the a-d positions of the peptide AAS were made with different hydrophobic residues, including Val, Phe, Tyr, Leu, and Ile. We used our previously developed MERS-CoV S protein-mediated cell–cell fusion assay to test the biological activity of these peptides.40 As shown in Table 1, all of the lipopeptides potently inhibited cell–cell fusion mediated by MERS-CoV S protein, with 50% effective concentration (EC50) values ranging from 0.1 to >10.0 μM. Of the six lipopeptides we designed, both LLS and IIS were found to be potent MERS-CoV fusion inhibitors, with EC50 values of 0.24 and 0.10 μM, respectively. The possible reasons for this activity include the evolutionary preference for Leu and Ile in the heptad positions a-d for viral fusion glycoprotein sequences and the significance of creating enough hydrophobicity/amphiphilicity to yield a stable coiled-coil structure.41 Compared to LLS, IIS possessed obviously lower cytotoxicity in Huh-7 cells, which were used as target cells in the cell–cell fusion assay. Therefore, in the next round of analogues, we further mutated residues at the e position of the heptad of IIS with different types of polar residues, including cationic Lys, anionic Glu, and uncharged Gln, as well as aromatic/heterocyclic amino acids such as Tyr, Trp, and His, expecting that these optimizations would provide further improvement in potency and cytotoxicity. Strikingly, Gln-containing IIQ and Tyr-containing IIY had EC50 values of 0.11 and 0.52 μM, respectively, even reaching the potencies of the MERS-CoV-specific fusion inhibitor HR2PM2 peptide.42 In addition, both IIQ and IIY possessed lower cytotoxicity values than the selected lead IIS, with a 50% cytotoxicity concentration (CC50) value of >100 μM. Subsequently, we tested the inhibitory activity of the peptide IIQ on the entry of pseudovirus carrying the wild-type MERS-CoV S protein. As shown in Supporting Information, Figure S1, IIQ was effective against MERS-CoV pseudovirus infection, with an EC50 value of 0.13 μM; this finding was in close agreement with the cell–cell fusion assay results. These data imply that the de novo designed peptides that are nonhomologous with the naturally occurring MERS-CoV S protein sequence could effectively inhibit MERS-CoV infection by targeting virus–host cell membrane fusion. Moreover, we explored the structural properties of these lipopeptides by circular dichroism (CD) spectroscopy to determine if their propensities to adopt α-helical structure were correlated with anti-MERS-CoV activity. The relationships between the EC50s of lipopeptides and their α-helical content are shown in Figure S2 in the Supporting Information. We found that the potency was not strictly dependent on their α-helicity (r2 = 0.2439), suggesting that other factors, such as solubility and target binding affinity and kinetics, may be involved in their biological activity. This phenomena has also been observed in the studies on HIV-1 fusion inhibitors, where several C-peptides exhibited no anti-HIV-1 potency despite they have fully α-helical structure in solution.29,43
Table 1. Inhibitory Activities of Lipopeptides on MERS-CoV S Protein-Mediated Cell–Cell Fusiona.

The number of 293T/MERS/EGFP cells fused or unfused with Huh-7 cells were countered, and the percentage of inhibition was calculated as described in the experimental section. Data were derived from the results of three independent experiments and are expressed as the mean ± standard deviation.
These peptides have an acetyl group at the N-terminus and carboxyamide at the C-terminus. The letters a-g indicate the positions of the corresponding residues in a helical wheel presentation. βA, β-alanine.
Cytotoxicity to Huh-7 cells.
The ongoing threat of the emergence of resistant variants that diminish or ablate the effectiveness of the currently available anti-IAV drugs underscores the demand for new antiviral strategies targeting other proteins in the influenza virus life cycle.44 Therefore, we employed a cytopathic effect inhibition assay to evaluate the inhibition effect of compounds IIS, IIY, and IIQ, which exhibited promising potency in inhibiting MERS-CoV infection, against IAVs. Surprisingly, one of the lipopeptides, IIQ, displayed potent inhibitory activity toward both strains A/Puerto Rico/8/34 (H1N1) and A/Hong Kong/8/68 (H3N2), with EC50 values of 1.73 and 0.70 μM, respectively (Table 2). Furthermore, none of these peptides displayed a cytotoxic effect against Madin–Darby canine kidney (MDCK) cells at concentrations up to 100 μM. The selective index of IIQ was larger than 143, indicating a safe profile for further exploration. The data presented in Table S1 in the Supporting Information show that IIQ was also potent against oseltamivir-resistant strains, including LN/1109 (H1N1) and TJ/15 (H1N1), with EC50 values of 4.36 and 3.03 μM, respectively, whereas oseltamivir was much less effective (EC50 = 21.7 μM) against LN/1109 (H1N1) and inactive against TJ/15 (H1N1) at concentrations up to 100 μM. In addition, IIQ also exerted remarkable inhibitory activity against B/Lee/40 strain infection with an EC50 value of 1.87 ± 0.95 μM. As expected, MERS-CoV HR2PM2 displayed no inhibition effect against A/Puerto Rico/8/34 (H1N1) and A/Hong Kong/68 (H3N2) infection at the concentration up to 20 μM. The life cycle of influenza virus is divided into three steps, i.e., virus entry, viral genome replication, and progeny virion release.45 Currently, several anti-influenza drugs have been developed for interruption of specific processes in influenza infection. Among them, oseltamivir (Tamiflu) targets neuraminidase (NA) protein, thus preventing release of tethered progeny virus from its host cells.46 Favipiravir that selectively inhibits RNA-dependent RNA polymerase has been approved in Japan as an inhibitor of influenza virus replication for the treatment of influenza virus infection in 2014.47 We then asked if the IIQ peptide acts by inhibiting the entry step of IAV into host cells. To address this question, a viral replicon assay and a neuraminidase inhibition assay were performed to rule out any effect of IIQ on viral replication and progeny virion release, respectively. In the mini-replicon system, viral RNA polymerase protein expression plasmids mediate the expression of a reporter genome encoding firefly luciferase. Through determination of the normalized firefly luminescence/Renilla luminescence ratio, we found that favipiravir could inhibit influenza replicon activity in a dose-dependent manner, with a 50% inhibitory concentration (IC50) value of 26.2 ± 3.7 μM, whereas IIQ and the neuraminidase inhibitor oseltamivir exhibited no significant inhibition at concentrations up to 100 μM (Figure 2A). As shown in Figure 2B,C, neither IIQ nor favipiravir was able to disturb cleavage of the substrate by neuraminidase from the two IAV strains A/Puerto Rico/8/34 (H1N1) and A/Hong Kong/8/68 (H3N2) in the test range. In contrast, oseltamivir could inhibit neuraminidase of these two viruses, with IC50 values of 1.71 ± 0.2 and 0.51 ± 0.01 nM, respectively. Taken together, these results suggest IIQ may affect the entry stage of viral life cycle to block virus infection. Moreover, using a time-of-addition assay, we found that the viral load at intervals of 0–10 h (covering the whole life cycle) and 0–2 h (covering the entry step) was reduced by approximately 60%, with the addition of 10 μM IIQ, as compared with the PBS control, and no antiviral activity was observed for the remaining three time intervals (2–5, 5–8, and 8–10 h). However, favipiravir continued to exert its full effect, even at the interval of 5–8 h. These results further confirmed that IIQ targets the entry step in the IAV life cycle (Figure 2D–F). Hemagglutination inhibition (HI) assay is well-established to determine whether the sialic acid-binding site on HA1 subunit acts as potential drug target; meanwhile, hemolysis inhibition assay is commonly used to determine whether the HA2 subunit can be a possible target.48 Thus, an HI assay was first performed to determine the potential effect of IIQ peptide on the HA1 subunit sialic acid-binding site. The result showed that no apparent inhibition of influenza virus-induced aggregation of chicken erythrocytes was observed in the test range, indicating IIQ has no effect on the interaction of HA1 subunit with its sialic acid receptor (Supporting Information, Figure S3A). Moreover, the hemolysis inhibition assay showed that the lysis of erythrocytes caused by the conformational rearrangements of HA2 subunit under acidic condition was decreased compared with the hemolytic effect of virus only (Supporting Information, Figure S3B). The combined results indicate that IIQ may bind to the HA2 subunit to interrupt the conformational changes in HA2 rather than interacting with HA1 subunit via inhibiting the absorption of viruses into host cells, consistent with our design rationale.
Table 2. Inhibitory Activities of Lipopeptides against Influenza A Virus Strains Infection in Cell Culturea.
| EC50 (μM) for inhibiting |
|||
|---|---|---|---|
| compd | A/Puerto Rico/8/34 (H1N1) | A/Hong Kong/8/68 (H3N2) | CC50 (μM)b |
| IIS | 1.96 ± 0.28 | 6.38 ± 1.06 | >100 |
| IIY | 3.15 ± 1.79 | 12.9 ± 5.55 | >100 |
| IIQ | 1.73 ± 0.81 | 0.70 ± 0.09 | >100 |
| oseltamivir | 1.48 ± 0.05 | 0.01 ± 0.004 | >100 |
Data were derived from the results of three independent experiments and are expressed as the mean ± standard deviation.
The cytotoxicity of compounds on MDCK cells.
Figure 2.

Exploration of the viral life cycle stage in which IIQ performed its inhibitory activity. (A) The inhibitory effect of IIQ on IAV polymerase activity was tested by a mini-replicon assay. Data represent the average of three independent measurements and are shown as the mean with standard deviation (bars). Oseltamivir and favipiravir were employed as negative and positive controls, respectively. Inhibition of the neuraminidase of influenza virus strains (B) A/Puerto Rico/8/34 and (C) A/Hong Kong/8/68 by IIQ as determined by chemiluminescence-based enzyme inhibition assays. The results are from three independent experiments. Oseltamivir and favipiravir were employed as positive and negative controls, respectively. (D) Strategy for the time-of-addition assay. The life cycle of IVA is divided into three steps, i.e., viral entry (0–2 h), viral genome replication and translation (2–8 h), and progeny virion release (8–10 h). MDCK cells were treated with inhibitor at five time intervals (0–10, 0–2, 2–5, 5–8, and 8–10 h) postinfection. The blue lines indicate the exposure intervals of inhibitor during the first viral life cycle. Time-of-addition assay to analyze the life cycle step specifically targeted by (E) IIQ and (F) favipiravir. Quantitative real-time PCR was used to detect the viral load at the indicated times. Data are expressed as the mean ± standard deviation of triplicate experiments. Statistical significance was evaluated with MANOVA. *, p < 0.05 compared to PBS treatment. **, p < 0.01 compared to PBS treatment.
After identifying that the entry/fusion process phase of the life cycle of MERS-CoV and IAV was the point of interference, we explored whether the IIQ peptide would interact with the corresponding NHR region of viral fusion proteins with a mechanism of action similar to virus-specific C-peptides. The first piece of evidence came from native polyacrylamide gel electrophoresis (N-PAGE) of equimolar mixtures of IIQ plus synthetic peptides containing the NHR segments of the MERS-CoV spike (S) protein S2 subunit, designated as HR1P,13 and the postfusion structure of the H3N2 hemagglutinin (HA) HA2 subunit, namely N66,49 respectively. The mixtures of IIQ/HR1P and IIQ/N66 showed new bands at the upper positions in the gel, demonstrating that tightly associated complexes were formed (Figure 3A). These findings are consistent with the results obtained from circular dichroism (CD) spectroscopy, in which the signal of these mixtures was dramatically greater than that of the mathematical sum of the corresponding isolated peptides, suggesting induction of a large α-helical structure resulting from their interaction (Figure 3B,C).50 Moreover, data from CD analysis indicated that the IIQ/HR1P and IIQ/N66 helical bundles showed strong thermal stability, with melting temperature values of >90 and 83.1 °C, respectively (Figure 3D and Supporting Information, Table S2). In CD analysis, N36/C34 6HB,31 which has an available crystal structure and is widely used to represent the HIV-1 fusion core, was used as a positive control (Supporting Information, Figure S4). Finally, sedimentation velocity analysis demonstrated the heterogeneous 6HB states of the IIQ/HR1P complex and the IIQ/N66 complex (Supporting Information, Table S3 and Figure S5). Together, these data suggest that the active IIQ peptide does indeed associate with a site in the NHR region and forms heterogeneous 6HB structures that interfere with the fusion between virus and target cell membrane.
Figure 3.

Identification of the NHR region of viral fusion proteins as the potential target of IIQ. (A) Association of IIQ with HR1P, a synthetic peptide spanning residues 998–1039 of the MERS-CoV S2 subunit (PDB 4NJL), and N66 peptide, which corresponds to residues 40–105 of IAV H3 (X31) HA (PDB 1QU1), respectively, as determined by N-PAGE. The final peptide concentration in each preparation was 75 μM. Left panel: Peptides were electrophoresed in a 15% native polyacrylamide continuous gel at pH 3.4. HR1P alone showed no band in the gel, likely because of its tendency to aggregate when sample was prepared in PBS (pH 7.4) before analysis in the acidic electrophoresis system. Right panel: Peptides were loaded on a 15% Tris-glycine gel with Tris−glycine running buffer (pH 8.8). IIQ alone showed no band in the gel, probably because the pH of the gel buffer is only slightly higher than the isoelectric point value of IIQ, thus it carries few net charges and cannot migrate into the gel. CD spectra for (B) IIQ, HR1P, and the IIQ/HR1P complex at neutral pH (solid lines) and for (C) IIQ, N66, and the IIQ/N66 complex at pH 5.0 (the pH of endosomes, solid lines). The theoretical noninteracting spectra of the related isolated peptides ((IIQ + HR1P) and (IIQ + N66), dashed lines) are shown for comparison. All spectra were obtained with 10 μM peptide at 25 °C. (D) CD signals at 222 nm for IIQ/HR1P (pH 7.4) and IIQ/N66 (pH 5.0) mixtures as a function of temperature.
We used the palmitoyl-2-oleoyl-sn-glycero-3-phospholcholine large unilamellar vesicle (POPC LUV) liposome system to examine the ability of IIQ to interact with a lipid bilayer by measuring the heat generated during the peptide–lipid binding in an isothermal titration calorimetry assay.51 As shown in Figure 4, a huge amount of released heat was observed after the titration of POPC LUVs into solutions containing IIQ peptide, with a binding constant of 1.04 × 105 M–1. In comparison, the unconjugated peptide IIQΔ, which exhibited no significant inhibitory activity at the concentration up to 40 μM on both MERS-CoV pseudovirus and A/Puerto Rico/8/34 (H1N1) infection, did not show lipid vesicle binding. These observations indicate that IIQ can bind to a lipid bilayer. Although HIV-1-neutralizing CHR peptides provide a classic paradigm for the rational design of fusion inhibitors that interfere with the interaction between the heptad-repeat regions of class I viral fusion proteins, the development of such drugs with novel modes of action against IAV remains a challenge, mainly by the inaccessibility of exogenously added C-peptides to the endocytic compartment, where fusogenic 6HB formation takes place. Thus, we next appended the fluorescent probe nitrobenzoxadiazole (NBD) to the N-terminus of IIQ via a β-alanine spacer to generate IIQNBD and monitored its cellular uptake. IIQNBD was found to be similar to IIQ in terms of its α-helicity and inhibitory potency against IAV infection, indicating that the addition of the fluorophore did not dramatically affect the biophysical/biological properties of IIQ (Supporting Information, Table S4). To demonstrate cellular uptake, MDCK cells treated with IIQNBD were observed after incubation for 0.5, 1, and 4 h by confocal microscopy. The lysotracker probe specifically fluoresces in acidic vesicles.52 As shown in Figure 5, MDCK cells exposed to IIQNBD for 0.5 h showed that the lipopeptides were distributed on the periphery of the cell membrane. After 1 h, the accumulation of IIQNBD within the MDCK cells was detected in the acidic intracellular compartments, colocalized with LysoTracker Red. Moreover, after 4 h, an increasing amount of IIQNBD was internalized and colocalized extensively with LysoTracker Red. Taken together, these results suggest that the α-helical lipopeptide could cross the plasma membrane efficiently via endocytosis and trap IAV fusion proteins in their prehairpin intermediate state, thereby blocking membrane fusion.
Figure 4.

Analysis of the binding of IIQ to a lipid bilayer. (A) The sequences IIQ and the negative control IIQΔ. (B) POPC LUVs (10 mg/mL) were injected into a chamber containing 15 μM IIQ (left) or IIQΔ (right) at 25 °C. Data acquisition and analysis were performed using MicroCal Origin software (version 7.0). The upper panels show the titration traces, and the lower panels show the binding affinity when POPC LUVs were injected into IIQ or IIQΔ solution.
Figure 5.
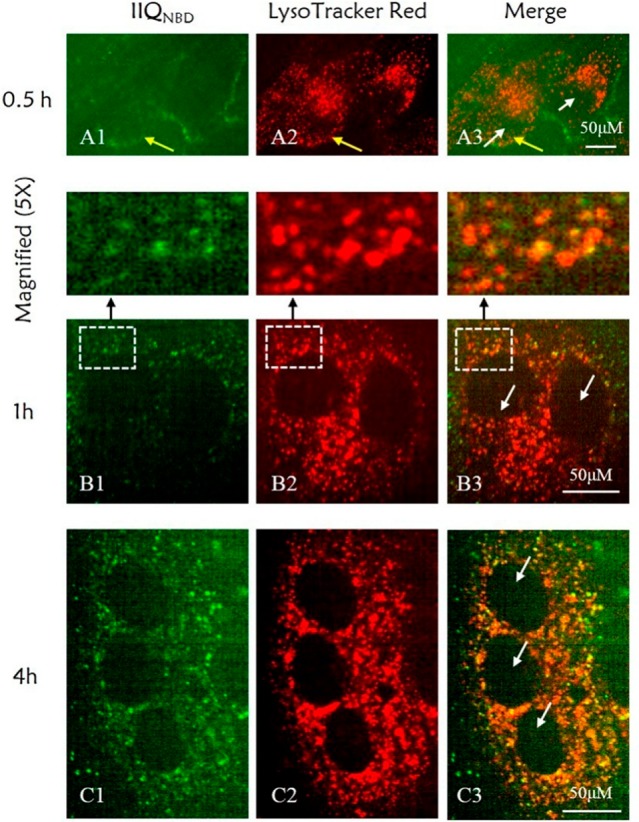
Uptake of the fluorescein-labeled peptide IIQNBD in MDCK cells. Confocal microscopy images were obtained for IIQNBD after 0.5 h (A1–A3), 1 h (B1–B3), and 4 h (C1–C3) of cell treatment. IIQ peptide was labeled with the fluorescein tag NBD (green signal, A1–C1), and intracellular acidic vesicles were stained with LysoTracker Red (red signal, A2–C2). The yellow punctate staining demonstrates the electronic merging (Merge, A3–C3) of IIQNBD and LysoTracker. The white and yellow arrows indicate the cell nuclear region and the cell membrane, respectively. Scale bar: 50 μm.
Subsequently, IIQ peptide was assessed for essential druglike properties, including water solubility and in vivo pharmacokinetic properties. IIQ showed solubility value of 8.87 ± 0.4 mg/mL in pure water. The pharmacokinetic behavior of IIQ was evaluated in rats (Figure 6 and Table 3). Sprague–Dawley rats were injected intravenously with 5 mg/kg of IIQ peptide, and blood was withdrawn after 2 min, 5 min, 15 min, 30 min, 1 h, 2 h, 4 h, 8 h 12 h, and 24 h, respectively (three animals at each time point). The LC/MS/MS method showed satisfactory results for the determination of IIQ in rat plasma and was used for the pharmacokinetic study. Inspection of the plasma concentration–time profile for IIQ revealed that the mean value of the maximum plasma concentration (Cmax) obtained 2 min after dosing was 97.6 μg/mL and the area under the plasma concentration–time curve extrapolated to the last time point (AUC0–t) was 234.7 (μg/mL)·h. The elimination kinetics of IIQ demonstrated durable plasma half-life of 6.6 h, and the concentration of IIQ in plasma remained well above the 0.1 μg/mL limit of quantitation of the analytical method at 24 h. Furthermore, IIQ peptide has a low rate of clearance of 20.7 mL·h–1·kg–1. Together, the favorable pharmacokinetic of IIQ, including the efficacious exposure level, the low clearance, and the relatively extended half-life in vivo, suggest IIQ is suitable for further study as a drug candidate.
Figure 6.

Pharmacokinetic studies of IIQ in plasma following the administration of a single intravenous dose (5 mg/kg) to Sprague–Dawley rats (n = 3).
Table 3. Pharmacokinetic Parameters of IIQ in Rats Following a Single Dose iv Administration Calculated by Noncompartmental Analysis by Using DAS, Version 3.2.8a.
| compd | AUC (0–t) ((μg/mL)·h) | MRT (0–t) (h) | t1/2 (h) | CL ((mL/h)/kg) | Cmax (μg/mL) | Vd (mL/kg) |
|---|---|---|---|---|---|---|
| IIQ | 234.7 ± 7.8 | 4.1 ± 0.1 | 6.6 ± 0.2 | 20.7 ± 0.6 | 97.6 ± 8.4 | 197.8 ± 9.3 |
MRT, mean residence time; CL, clearance; Vd, volume of distribution.
To investigate whether our engineered lipopeptides exert broad antiviral spectrum beyond Coronaviridae and Orthomyxoviridae families, IIQ peptide was further evaluated against HIV-1 envelope glycoprotein (Env)-mediated cell–cell fusion and Ebola virus (EboV) envelope glycoprotein (GP)-mediated cell entry. We found that IIQ inhibited HIV-1 Env-mediated cell fusion with an EC50 of 3.63 ± 0.54 μM and exhibited low cytotoxicity, with CC50 > 100 μM, on the TZM-b1 cell that was used for the fusion assay. Encouragingly, IIQ also inhibited the entry of pseudovirus carrying the GP of the EboV Sudan species, with EC50 value of 1.02 ± 0.54 μM. To our knowledge, the formation of a hexameric coiled-coil complex is believed to be a common element in type I fusion events. However, the fusion rates vary significantly between viruses from different families, between viruses within a family, and even between isolates of the same species, thus leading to the different window periods during which inhibitory peptides access to the target fusion protein and thereby impacting entry inhibitor efficacy.5 Furthermore, the stability of the corresponding fusogenic 6HB of different type I viral fusion proteins was also correlated with the inhibitory potency of peptides.7 Therefore, the engineered lipopeptides with simple repeating units of the heptad in our study could not be effective against all of the class I viruses entry, but first provide a proof-of-concept prototype for broad-spectrum antiviral agents design. Moreover, these lipopeptides may also be used as lead compounds for further optimization to design inhibitors with a broader antiviral spectrum.
Conclusions
On the basis of common features of the fusogenic 6HB structure formed between the CHR and NHR regions of the class I viral fusion glycoproteins, we report an effective strategy to expedite the development of relatively broad-spectrum antiviral drugs. Our study reveals that the de novo designed α-helical lipopeptides, which are nonhomologous with naturally occurring protein sequences, can interact with both MERS-CoV and IAV NHR trimeric coiled coils to prevent virus–cell membrane fusion. One of the designed peptides showed a high potency against MERS-CoV infection and effectively neutralized H1N1 A/Puerto Rico/8/34 (influenza A group 1), H3N2 A/Hong Kong/8/68 (influenza A group 2), and even the influenza B virus (B/Lee/40). The relatively broad-spectrum antiviral peptides were designed based on the secondary structure at the hexameric coiled-coil complex interface. We anticipate that this approach could also be extended to other pathogenic viruses with class I fusion proteins because they undergo fusion catalysis in a manner similar to that of MERS-CoV and IAVs.
Experimental Section
General Peptide Synthesis
Peptides were synthesized by using standard Fmoc solid-phase synthesis techniques with a CS Bio polypeptide synthesizer. Rink amide resin, with a resin loading of 0.44 mmol/g, was selected as the solid support. N,N-Dimethylformamide (DMF), dichloromethane (DCM), N-methyl-2-pyrrolidone (NMP), methanol, piperidine, and other reagents used in the reaction process were anhydrous reagents or dried prior to use. The synthetic steps were as follows: (1) The resin was first swelled in a reaction vessel by the addition of 5 mL of DMF and 5 mL of DCM, followed by stirring for 20 min. (2) The Fmoc protecting group was removed using 20% piperidine/DMF. The deprotection reaction was performed twice; the first reaction was 5 min, and the second reaction was 2 min. (3) The next amino acid was coupled with the addition of 5 mL of amino acid solution (0.25 M), 5 mL of condensing reagents [0.2 M O-benzotriazole-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU)/DMF and 0.2 M 1-hydroxybenzotriazole (HOBt)/DMF], and 5 mL of active base solution [0.4 M N,N-diisopropylethylamine (DIEA)/DMF] to the reactor; the reaction was stirred at room temperature for 60 min. After completion of the coupling reaction or Fmoc removal, the resin was washed with DMF (5 × 1 min) and DCM (3 × 1 min). For lipopeptides, the template peptides containing a Dde-protected lysine residue at their C-terminus required a special deprotection step (four 3 min washes of 2% hydrazinehydrate in DMF). This enabled the conjugation of a palmityl moiety, which was performed by the addition of 3 equiv of palmic acid, 3 equiv of HBTU, and 6 equiv of DIEA in DMF to the resin, followed by stirring for 2 h. Conjugation of a fluoride [4-fluoro-7- nitrobenzofurazan (NBD)] fluorescent probe to the N-terminus of the peptide was performed by the addition of 6 equiv of NBD-Cl dissolved in DMF together with 6 equiv of DIEA to the peptide resin, followed by stirring for 12 h. The peptides were cleaved from the resin and deprotected with reagent K, which contained 85% trifluoroacetic acid, 5% thioanisole, 5% m-cresol, and 5% water. The carboxyl termini were amidated upon cleavage from the resin, and the amino termini were capped with acetic acid anhydride, except NBD-conjugated peptides. All lyophilized crude peptides were purified by reversed-phase high-performance liquid chromatography (RP-HPLC; Shimadzu preparative HPLC system), and the purity of each peptide was confirmed to be ≥95% by analytical RP-HPLC (Shimadzu analytical HPLC system). Such information is provided in the Supporting Information (Table S5–S6). The molecular weight of the peptides was characterized by matrix-assisted laser desorption ionization–time-of-flight mass spectrometry (Autoflex III, Bruker Daltonics Inc., Billerica, MA).
MERS-CoV S Protein-Mediated Cell–Cell Fusion Assay
The target cells were Huh-7 cells expressing the MERS-CoV receptor dipeptidyl peptidase 4. The effector cells were 293T/MERS/enhanced GFP protein (EGFP) cells.13 The 293T/MERS/EGPF cells contained the MERS-CoV S protein gene and the EGFP gene transfected with plasmid. The 293T/EGFP cells expressing only EGFP were employed as negative control cells. Huh-7 cells were plated in 96-well plates (5 × 104 cells/well) at 37 °C for 5 h. Then, serially diluted peptide samples were added, followed by the addition of 293T/MERS/EGPF cells or 293T/EGFP cells (1 × 104 cells/well). After coculturing at 37 °C for 4 h, the 293T/MERS/EGFP cells and 293T/EGFP cells, either fused or unfused, with Huh-7 cells were counted under an inverted fluorescence microscope (Nikon Eclipse Ti-S).
Inhibition of Pseudotyped MERS-CoV Infection
Inhibition of MERS-CoV pseudovirus infection was assessed using a previously described method.42 Briefly, the serially diluted tested peptides were added to a 96-well plate and incubated with MERS-CoV pseudovirus for 30 min at 37 °C. Then, the pseudovirus/peptide mixture was added to the Huh-7 cells. Cultures were refed with fresh medium at 12 h postinfection and incubated for an additional 48 h at 37 °C. Fluorescence was determined using a luciferase kit (Promega) and an Ultra 384 luminometer (Tecan).
Cytopathic Effect Reduction Assay
Madin–Darby canine kidney (MDCK) cells were seeded in a 96-well plate (Nunc MicroWell) at a density of 1.5 × 104 cells/well in Dulbecco’s Modified Eagle’s Medium/Ham’s F-12 medium (DF-12) and were incubated overnight to adhere to the plate.53 A 3-fold dilution series of tested compounds in DF-12 with tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-trypsin was added to cells infected with influenza virus A/Puerto Rico/8/1934 (H1N1) or A/Hong Kong/8/68 (H3N2) at a final multiplicity of infection (MOI) of 0.003 Pfu/cell suspended in DF-12 medium with TPCK-trypsin. After incubation at 37 °C for 72 h, the antiviral effect of the tested compounds was measured using a CellTiter-Glo cell viability assay (Promega, USA), as described by the manufacturer. The plates were read by a SpectraMax M5 microplate reader (Molecular Devices, USA).
Cytotoxicity Assays
Cytotoxicity of Compounds on Huh-7 Cells
Briefly, 100 μL of a Huh-7 cell suspension (1 × 105 cells/mL) were added to each well of a 96-well culture plate. The plate was incubated at 37 °C in 5% CO2 for 12 h. Next, 5 μL of serially diluted peptide solution were added. At the same time, a blank control group without peptide and a positive control group with 5 μL of 10% Triton X-100 were cultured for 48 h under the conditions of 5% CO2 at 37 °C. To each well were added 10 μL of Cell Counting Kit 8 solution, and the plate was incubated for an additional 2 h. The absorbance at 450 nm was determined by a microplate reader.
Cytotoxicity of Compounds on MDCK Cells
MDCK cells were seeded in a 96-well plate at a density of 1.5 × 104 cells/well in DF-12 medium and incubated overnight before a 3-fold dilution series of tested compounds in DF-12 medium with TPCK-trypsin was added to the cells. At 72 h after treatment, the cytotoxicity was measured by a CellTiter-Glo cell viability assay (Promega, USA). The 50% cytotoxicity concentration data were fit and determined by Origin 8 software.
Influenza Virus Replication Assay
Human embryonic kidney 293T cells were seeded in a 96-well plate (Nunc MicroWell) at a density of 2.5 × 104 cells/well in DF-12 medium and were incubated overnight. The plasmid encoding the influenza replication complex component (NP, PB1, PB2, and PA) and that encoding the luciferase reporter (polI-NS-Luc and pRLSV40) were transfected into the cells using Lipofectamine 3000 (Invitrogen, USA). At 6 h post-transfection, serially diluted oseltamivir or favipiravir or IIQ was added at the indicated concentration. After 30 h, the luciferase activities were measured using a SpectraMax M5 microplate reader (Molecular Devices, USA) and a Dual-Glo luciferase assay kit (Promega, USA), according to the standard protocol. The replicon activities were represented by the ratio of firefly luciferase activity relative to Renilla luciferase activity. The replicon activity inhibition of the tested compounds was calculated using the following formula: Inhibition of replicon activity (%) = (experimental sample ratio – negative control ratio)/(positive control ratio – negative control ratio) × 100%. Curve fitting was performed by Origin 8.0 software.
Neuraminidase Inhibition Assay
The NA-Star Influenza Neuraminidase Inhibitor Resistance Detection Kit (Applied Biosystems, USA) was used to measure the inhibition of neuraminidase activity according to the manufacturer’s instructions. Neuraminidase from influenza A/Hong Kong/8/68 (H3N2) and neuraminidase from influenza A/Puerto Rico/8/1934 (H1N1) virus were used for this assay. The chemiluminescent signal intensity of the assay plate was measured immediately after accelerator injection, and the read time was set to 1 s. The inhibitory activity of oseltamivir or favipiravir or IIQ was calculated using the following formula: inhibitory activity (%) = (fluorescence of virus control – fluorescence of sample)/(fluorescence of virus control – fluorescence of substrate control) × 100%. Curve fitting and IC50 calculation were accomplished by Origin 8.0 software.
Time of Addition Experiment and Real-Time Reverse Transcription–Polymerase Chain Reaction (RT-qPCR)
MDCK cells were seeded in a 12-well plate at a density of 2.0 × 105 cells/well and incubated overnight. The cells were washed three times with phosphate-buffered saline (PBS) prior to virus (MOI of 0.05 PFU/mL) adsorption at 4 °C for 60 min. After adsorption, the cells were washed with PBS, treated with favipiravir or IIQ at different time intervals (0–10 h, 0–2 h, 2–5 h, 5–8 h, 8–10 h postinfection, respectively), harvested, and then applied to qPCR for viral load analysis. Cells were continuously cultured in fresh medium after removal of test compounds at 37 °C until 10 h postinfection, and then viral load was analyzed. Total RNA of the cell samples was extracted using an RNeasy Mini Kit (QIAGEN). Absolute RT-qPCR was performed using the ABI Step One Plus platform by using a One-Step PrimeScript RT-PCR Kit (Takara). All samples were run in triplicate. The details of the primers, probe sequences, and reaction system can be found in the Supporting Information. The mRNA expression profiles at different time intervals of IIQ or favipiravir or PBS treatment were analyzed, and the mRNA expression profiles under IIQ or favipiravir treatment were presented relative to the PBS control. Statistical significance of the data was determined by MANOVA method using SPSS 20.0 software.
Circular Dichroism (CD) Spectroscopy
CD spectra were acquired on a MOS-450 system (BioLogic, Claix, France) with the following parameters: bandwidth, 4.0 nm; resolution, 0.1 nm; path length, 0.1 cm; response time, 4.0 s; and scanning speed, 50 nm/min. HR1P was incubated with IIQ at 25 °C for 30 min in 10 mM phosphate buffer (pH 7.4). N66 was incubated with IIQ at 30 °C for 30 min in 10 mM sodium acetate buffer (containing 10 mM sodium phosphate and 150 mM sodium chloride, pH 5.0). All samples were prepared with the buffer solution at a final concentration of 10 μM and cooled to 25 °C for measurement. The CD data were presented as the mean residue ellipticity. The α-helical content for these peptides was calculated by assuming that 100% helicity corresponds to −33000 degrees cm2 dmol–1. For the thermal unfolding experiments, the CD absorbance was monitored at 222 nm with the temperature for the peptide solutions ranging from 10 to 90 °C at a scan speed of 2 °C/min. Samples at pH 5.0 contained 10 μM peptide in 10 mM sodium acetate buffer (containing 10 mM sodium phosphate and 150 mM sodium chloride). Samples at pH 7.4 contained 10 μM peptide in 10 mM phosphate buffer.
Native Polyacrylamide Gel Electrophoresis
An equimolar mixture of IIQ and HR1P in 10 mM phosphate buffer (pH 7.4) was incubated at 25 °C for 30 min (final concentration of each peptide: 150 μM). After the addition of 2× β-alanine–formic acid native sample buffer to samples at a ratio of 1:1, all samples were loaded on a 15% β-alanine–formic acid gel with a β-alanine–formic acid running buffer (pH 3.4). After sample loading, the samples were concentrated at a constant voltage of 90 V. When the samples reached the boundary of the two gels, the voltage was raised to 150 V, and the samples were electrophoresed for 2.5 h until the indicator was within 1–2 cm of the leading edge. The gel was subsequently stained with Coomassie Blue R250. An equimolar mixture of IIQ and N66 in 10 mM sodium acetate buffer (containing 10 mM sodium phosphate and 150 mM sodium chloride, pH 5.0) was incubated at 30 °C for 30 min (final concentration of each peptide: 150 μM). After incubation, the solutions were equilibrated at room temperature, and the pH was neutralized by the addition of 200 mM Tris buffer (pH 8.5). After the addition of 2× Tris-glycine native sample buffer (Invitrogen, USA) to the samples at a ratio of 1:1, all samples were loaded onto a 15% Tris-glycine gel with Tris−glycine running buffer (pH 8.8). After sample loading, the samples were concentrated at a constant voltage of 90 V. When the samples reached the boundary of the two gels, the voltage was raised to 150 V and the samples were electrophoresed for 2.5 h until the indicator was within 1–2 cm of the leading edge. The gel was subsequently stained with Coomassie Blue R250.
Sedimentation Velocity Analysis (SVA)
A Beckman XL-A-type ultracentrifuge was used for the SVA experiments. HR1P was incubated with IIQ at 25 °C for 30 min in 10 mM phosphate buffer (pH 7.4). N66 was incubated with IIQ at 30 °C for 30 min in 10 mM sodium acetate buffer (containing 10 mM phosphate and 150 mM sodium chloride, pH 5.0). All samples were prepared at a final concentration of 150 μM, which was measured by a UV absorption experiment. Data were collected at 280 nm at a rotor speed of 3000 rpm initially and then 60000 rpm in continuous mode at 25 °C. The sedimentation coefficient distribution, c(s), was measured, and the molecular weight distribution was calculated by SEDFIT software.
Isothermal Titration Calorimetry (ITC)
ITC was performed to detect the IIQ and lipid-binding activity, as described previously.51 The solutions were degassed under vacuum prior to use. To detect the IIQ and lipid-binding activity, large unilamellar vesicles (LUVs) of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) liposomes were used. LUVs of POPC (13 mM) were injected into the cell containing peptide solution (15 μM, 300 μL). The experiments used a MicroCal ITC200 system (GE, Alpharetta, GA USA) for titration, with the following experimental parameters: total injection, 20 drops; drop volume, 2 μL; syringe concentration, 13 mM; cell concentration, 15 μM; cell temperature, 25 °C; energy reference, 3 μCal/s; titration delay, 60 s; stirring speed, 750 rpm; drop volume, 2 μL; titration time, 4 s; two-drop interval, 120 s; data collection interval, 5 s. Data acquisition and analyses were performed using Origin software (Version 8.5, MicroCal).
Immunofluorescence Assay
MDCK cells were pretreated with LysoTracker Red DND-99 (L7528), which was purchased from Thermo Fisher Scientific (Shanghai, China). LysoTracker solution (1 μL, 1 mM) was added to 10 mL of growth medium to obtain a working solution of 100 nM. Cells in a 96-well plate were incubated with 50 μL of 100 nM LysoTracker working solution for 1.5 h, and then they were washed twice with PBS. DF-12 medium without phenol red containing 5 μM IIQNBD was added to the cells (time point of 0 h), and then the cells were cultured at 37 °C under 5% CO2. At time points of 10 min, 30 min, 1 h, 2 h, and 4 h, the cells were scanned with the Operetta confocal imaging system (provided by PerkinElmer) with a 20× objective lens. The following filters were used: Alexa Fluor 548 for acidic intracellular compartments and FITC for IIQNBD.
Pharmacokinetic Assessments
Sprague–Dawley rats weighing 210 ± 10 g each were obtained from the Animal Center of Beijing Institute of Pharmacology and Toxicology and were used for pharmacokinetic assessments. Animals were treated in accordance with the Animal Welfare Act and the “Guide for the Care and Use of Laboratory Animals” (NIH Publication 86-23, revised 1985). Complete pharmacokinetic experimental procedures are provided in the Supporting Information.
Acknowledgments
This research was supported, in part, by grants from the National Science Foundation of China (81573266 and 81630090), Beijing Nova Program (Z181100006218115), National Science and Technology Major Project of China (2018ZX09711003), and the National Key Research and Development Program of China (2016YFC1201000 and 2016YFC1200400).
Glossary
Abbreviations Used
- CHR
C-terminal heptad repeat
- NHR
N-terminal heptad repeat
- 6HB
six-helix bundle
- Da
Dalton
- SVA
sedimentation velocity analysis.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.8b00890.
Inhibitory activity of compounds on oseltamivir-resistant strains; biophysical properties of the IIQ peptide; summary of the SVA results of IIQ/HR1P and IIQ/N66 complexes; biological and biophysical properties of IIQNBD; HPLC method used for the purification of peptide compounds; HPLC method used for the analysis of peptide compounds; IIQ peptide as inhibitor of MERS-CoV infection; correlation between α-helical contents of lipopeptides with their observed EC50s; identification of HA2 subunit as the potential target of IIQ compound; interaction between C34 and N36; the molecular mass of IIQ/HR1P and IIQ/N66 as determined by SVA; pharmacokinetic assessments; aqueous solubility determination; HIV-1 Env-mediated cell–cell fusion assay; inhibition of pseudotyped Ebola virus infection; hemagglutination inhibition assay; hemolysis inhibition assay; as well as MALDI-TOF-MS and analytical HPLC of designed peptides (PDF)
Author Contributions
¶ C.W., L.Z., S.X., and T.Z. contributed equally to this work. Dr. Chao Wang, Prof. Keliang Liu, Prof. Shibo Jiang, and Prof. Wu Zhong conceived and designed the study. Guodong Liang, Guangpeng Meng, and Yue Li performed synthesis. Dr. Lei Zhao, Dr. Shuai Xia, and Dr. Ruiyuan Cao performed biological evaluation. Dr. Weiguo Shi and Guodong Liang performed biophysical analysis. Dr. Tianhong Zhang and Weicong Wang performed pharmacokinetics study. Dr. Chao Wang, Prof. Keliang Liu, Prof. Shibo Jiang, and Prof. Wu Zhong analyzed the data and wrote the manuscript.
The authors declare no competing financial interest.
This article is made available for a limited time sponsored by ACS under the ACS Free to Read License, which permits copying and redistribution of the article for non-commercial scholarly purposes.
Supplementary Material
References
- De Clercq E. Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV. Int. J. Antimicrob. Agents 2009, 33, 307–320. 10.1016/j.ijantimicag.2008.10.010. [DOI] [PubMed] [Google Scholar]
- Mendes-Correa M.; Nunez M. Management of HIV and hepatitis virus coinfection. Expert Opin. Pharmacother. 2010, 11, 2497–2516. 10.1517/14656566.2010.500615. [DOI] [PubMed] [Google Scholar]
- Yen H. L. Current and novel antiviral strategies for influenza infection. Curr. Opin. Virol. 2016, 18, 126–134. 10.1016/j.coviro.2016.05.004. [DOI] [PubMed] [Google Scholar]
- Zhu J. D.; Meng W.; Wang X. J.; Wang H. C. Broad-spectrum antiviral agents. Front. Microbiol. 2015, 6, 517–531. 10.3389/fmicb.2015.00517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrov D. S. Virus entry: Molecular mechanism and biomedical applications. Nat. Rev. Microbiol. 2004, 2, 109–122. 10.1038/nrmicro817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison S. C. Viral membrane fusion. Nat. Struct. Mol. Biol. 2008, 15, 690–698. 10.1038/nsmb.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colman P. M.; Lawrence M. C. The structural biology of type I viral membrane fusion. Nat. Rev. Mol. Cell Biol. 2003, 4, 309–319. 10.1038/nrm1076. [DOI] [PubMed] [Google Scholar]
- Wang C.; Shi W.; Cai L.; Lu L.; Yu F.; Wang Q.; Jiang X.; Xu X.; Wang K.; Xu L.; Jiang S.; Liu K. Artificial peptides conjugated with cholesterol and pocket-specific small molecules potently inhibit infection by laboratory-adapted and primary HIV-1 isolates and enfuvirtide-resistant HIV-1 strains. J. Antimicrob. Chemother. 2014, 69, 1537–1545. 10.1093/jac/dku010. [DOI] [PubMed] [Google Scholar]
- Wang C.; Lai W.; Yu F.; Zhang T.; Lu L.; Jiang X.; Zhang Z.; Xu X.; Bai Y.; Jiang S.; Liu K. De novo Design of isopeptide bond-tethered triple-stranded coiled coils with exceptional resistance to unfolding and proteolysis: implication for developing antiviral therapeutics. Chem. Sci. 2015, 6, 6505–6509. 10.1039/C5SC02220G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Shi W.; Cai L.; Lu L.; Wang Q.; Zhang T.; Li J.; Zhang Z.; Wang K.; Xu L.; Jiang X.; Jiang S.; Liu K. Design, synthesis, and biological evaluation of highly potent small molecule–peptide conjugates as new HIV-1 fusion inhibitors. J. Med. Chem. 2013, 56, 2527–2539. 10.1021/jm3018964. [DOI] [PubMed] [Google Scholar]
- Qi Q.; Wang Q.; Chen W.; Du L.; Dimitrov D. S.; Lu L.; Jiang S. HIV-1 gp41-targeting fusion inhibitory peptides enhance the gp120-targeting protein-mediated inactivation of HIV-1 virions. Emerging Microbes Infect. 2017, 6, e59. 10.1038/emi.2017.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K. K.; Pessi A.; Gui L.; Santoprete A.; Talekar A.; Moscona A.; Porotto M. Capturing a fusion intermediate of influenza hemagglutinin with a cholesterol-conjugated peptide, a new antiviral strategy for influenza virus. J. Biol. Chem. 2011, 286, 42141–42149. 10.1074/jbc.M111.254243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L.; Liu Q.; Zhu Y.; Chan K. H.; Qin L.; Li Y.; Wang Q.; Chan J. F.; Du L.; Yu F.; Ma C.; Ye S.; Yuen K. Y.; Zhang R.; Jiang S. Structure-based discovery of Middle East respiratory syndrome coronavirus fusion inhibitor. Nat. Commun. 2014, 5, 3067–3078. 10.1038/ncomms4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller E. H.; Harrison J. S.; Radoshitzky S. R.; Higgins C. D.; Chi X.; Dong L.; Kuhn J. H.; Bavari S.; Lai J. R.; Chandran K. Inhibition of Ebola virus entry by a C-peptide targeted to endosomes. J. Biol. Chem. 2011, 286, 15854–15861. 10.1074/jbc.M110.207084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porotto M.; Yokoyama C. C.; Palermo L. M.; Mungall B.; Aljofan M.; Cortese R.; Pessi A.; Moscona A. Viral entry inhibitors targeted to the membrane site of action. J. Virol. 2010, 84, 6760–6768. 10.1128/JVI.00135-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L.; Jiang S. Development of peptide and small-molecule HIV-1 fusion inhibitors that target gp41. ChemMedChem 2010, 5, 1813–1824. 10.1002/cmdc.201000289. [DOI] [PubMed] [Google Scholar]
- Jiang S.; Lin K.; Strick N.; Neurath A. R. HIV-1 inhibition by a peptide. Nature 1993, 365, 113. 10.1038/365113a0. [DOI] [PubMed] [Google Scholar]
- Wild C.; Shugars D. C.; Greenwell T. K.; McDanal C. B.; Matthews T. J. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 9770–9774. 10.1073/pnas.91.21.9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J.; Lu G.; Qi J.; Li Y.; Wu Y.; Deng Y.; Geng H.; Li H.; Wang Q.; Xiao H.; Tan W.; Yan J.; Gao G. F. Structure of the fusion core and inhibition of fusion by a heptad repeat peptide derived from the S protein of Middle East respiratory syndrome coronavirus. J. Virol. 2013, 87, 13134–13140. 10.1128/JVI.02433-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Xia S.; Zhang P.; Zhang T.; Wang W.; Tian Y.; Meng G.; Jiang S.; Liu K. Discovery of hydrocarbon-stapled short α-helical peptides as promising Middle East respiratory syndrome coronavirus (MERS-CoV) fusion inhibitors. J. Med. Chem. 2018, 61, 2018–2026. 10.1021/acs.jmedchem.7b01732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolovic B.; Danial M.; Klok H. A. Coiled coils: attractive protein folding motifs for the fabrication of self-assembled, responsive and bioactive materials. Chem. Soc. Rev. 2010, 39, 3541–3575. 10.1039/b914339b. [DOI] [PubMed] [Google Scholar]
- Fletcher J. M.; Boyle A. L.; Bruning M.; Bartlett G. J.; Vincent T. L.; Zaccai N. R.; Armstrong C. T.; Bromley E. H.; Booth P. J.; Brady R. L.; Thomson A. R.; Woolfson D. N. A basis set of de novo coiled-coil peptide oligomers for rational protein design and synthetic biology. ACS Synth. Biol. 2012, 1, 240–250. 10.1021/sb300028q. [DOI] [PubMed] [Google Scholar]
- Gochin M.; Zhou G. Amphipathic properties of HIV-1 gp41 fusion inhibitors. Curr. Top. Med. Chem. 2011, 11, 3022–3032. 10.2174/156802611798808488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M.; Blacklow S. C.; Kim P. S. A trimeric structural domain of the HIV-1 transmembrane glycoprotein. Nat. Struct. Mol. Biol. 1995, 2, 1075–1082. 10.1038/nsb1295-1075. [DOI] [PubMed] [Google Scholar]
- Lai W.; Wang C.; Yu F.; Lu L.; Wang Q.; Jiang X.; Xu X.; Zhang T.; Wu S.; Zheng X.; Zhang Z.; Dong F.; Jiang S.; Liu K. An effective strategy for recapitulating N-terminal heptad repeat trimers in enveloped virus surface glycoproteins for therapeutic applications. Chem. Sci. 2016, 7, 2145–2150. 10.1039/C5SC04046A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su S.; Wang Q.; Xu W.; Yu F.; Hua C.; Zhu Y.; Jiang S.; Lu L. A novel HIV-1 gp41 tripartite model for rational design of HIV-1 fusion inhibitors with improved antiviral activity. AIDS 2017, 31, 885–894. 10.1097/QAD.0000000000001415. [DOI] [PubMed] [Google Scholar]
- Crespillo S.; Camara-Artigas A.; Casares S.; Morel B.; Cobos E. S.; Mateo P. L.; Mouz N.; Martin C. E.; Roger M. G.; El Habib R.; Su B.; Moog C.; Conejero-Lara F. Single-chain protein mimetics of the N-terminal heptad-repeat region of gp41 with potential as anti-HIV-1 drugs. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 18207–18212. 10.1073/pnas.1413592112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa H.; Nakamura S.; Kodama E.; Ito S.; Kajiwara K.; Izumi K.; Sakagami Y.; Oishi S.; Ohkubo T.; Kobayashi Y.; Otaka A.; Fujii N.; Matsuoka M. Electrostatically constrained alpha-helical peptide inhibits replication of HIV-1 resistant to enfuvirtide. Int. J. Biochem. Cell Biol. 2009, 41, 891–899. 10.1016/j.biocel.2008.08.039. [DOI] [PubMed] [Google Scholar]
- Dwyer J. J.; Wilson K. L.; Davison D. K.; Freel S. A.; Seedorff J. E.; Wring S. A.; Tvermoes N. A.; Matthews T. J.; Greenberg M. L.; Delmedico M. K. Design of helical, oligomeric HIV-1 fusion inhibitor peptides with potent activity against enfuvirtide-resistant virus. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 12772–12777. 10.1073/pnas.0701478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sia S. K.; Carr P. A.; Cochran A. G.; Malashkevich V. N.; Kim P. S. Short constrained peptides that inhibit HIV-1 entry. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 14664–14669. 10.1073/pnas.232566599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B.; Wang K.; Lu L.; Yu F.; Cheng M.; Jiang S.; Liu K.; Cai L. Hydrophobic mutations in buried polar residues enhance HIV-1 gp41 N-terminal heptad repeat-C-terminal heptad repeat interactions and C-peptides’ anti-HIV activity. AIDS 2014, 28, 1251–1260. 10.1097/QAD.0000000000000255. [DOI] [PubMed] [Google Scholar]
- Chong H.; Wu X.; Su Y.; He Y. Development of potent and long-acting HIV-1 fusion inhibitors. AIDS 2016, 30, 1187–1196. 10.1097/QAD.0000000000001073. [DOI] [PubMed] [Google Scholar]
- Ding X.; Zhang X.; Chong H.; Zhu Y.; Wei H.; Wu X.; He J.; Wang X.; He Y. Enfuvirtide (T20)-based lipopeptide is a potent HIV-1 cell fusion inhibitor: implication for viral entry and inhibition. J. Virol. 2017, 91, e00831-17. 10.1128/JVI.00831-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.; Tan J.; Mehryar M. M.; Teng Z.; Zeng Y. Peptide HIV fusion inhibitors: modifications and conjugations. MedChemComm 2014, 5, 1472–1482. 10.1039/C4MD00214H. [DOI] [Google Scholar]
- Park J. E.; Gallagher T. Lipidation increases antiviral activities of coronavirus fusion-inhibiting peptides. Virology 2017, 511, 9–18. 10.1016/j.virol.2017.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Lu L.; Na H.; Li X.; Wang Q.; Jiang X.; Xu X.; Yu F.; Zhang T.; Li J.; Zhang Z.; Zheng B.; Liang G.; Cai L.; Jiang S.; Liu K. Conjugation of a nonspecific antiviral sapogenin with a specific HIV fusion inhibitor: a promising strategy for discovering new antiviral therapeutics. J. Med. Chem. 2014, 57, 7342–7354. 10.1021/jm500763m. [DOI] [PubMed] [Google Scholar]
- Wexler-Cohen Y.; Shai Y. Demonstrating the C-terminal boundary of the HIV 1 fusion conformation in a dynamic ongoing fusion process and implication for fusion inhibition. FASEB J. 2007, 21, 3677–3684. 10.1096/fj.07-8582com. [DOI] [PubMed] [Google Scholar]
- Spek E. J.; Bui A. H.; Lu M.; Kallenbach N. R. Surface salt bridges stabilize the GCN4 leucine zipper. Protein Sci. 1998, 7, 2431–2437. 10.1002/pro.5560071121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal G.; Fong S. Y.; Kossiakoff A. A.; Sidhu S. S. Alternative views of functional protein binding epitopes obtained by combinatorial shotgun scanning mutagenesis. Protein Sci. 2005, 14, 2405–2413. 10.1110/ps.051519805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia S.; Liu Q.; Wang Q.; Sun Z.; Su S.; Du L.; Ying T.; Lu L.; Jiang S. Middle East respiratory syndrome coronavirus (MERS-CoV) entry inhibitors targeting spike protein. Virus Res. 2014, 194, 200–210. 10.1016/j.virusres.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou X.; Zhu X.; Wang J.; Dong N.; Shan A. Novel design of heptad amphiphiles to enhance cell selectivity, salt resistance, antibiofilm properties and their membrane-disruptive mechanism. J. Med. Chem. 2017, 60, 2257–2270. 10.1021/acs.jmedchem.6b01457. [DOI] [PubMed] [Google Scholar]
- Channappanavar R.; Lu L.; Xia S.; Du L.; Meyerholz D. K.; Perlman S.; Jiang S. Protective effect of intranasal regimens containing peptidic Middle East respiratory syndrome coronavirus fusion inhibitor against MERS-CoV infection. J. Infect. Dis. 2015, 212, 1894–1903. 10.1093/infdis/jiv325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L. F.; Pan C. G.; Xu L.; Shui Y.; Liu K. L.; Jiang S. B. Interactions between different generation HIV-1 fusion inhibitors and the putative mechanism underlying the synergistic anti-HIV-1 effect resulting from their combination. FASEB J. 2012, 26, 1018–1026. 10.1096/fj.11-195289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M.; Si L.; Wang Y.; Wu Y.; Yu F.; Jiao P.; Shi Y.; Wang H.; Xiao S.; Fu G.; Tian K.; Wang Y.; Guo Z.; Ye X.; Zhang L.; Zhou D. Discovery of pentacyclic triterpenoids as potential entry inhibitors of influenza viruses. J. Med. Chem. 2014, 57, 10058–10071. 10.1021/jm5014067. [DOI] [PubMed] [Google Scholar]
- Li R.; Liu T.; Liu M.; Chen F.; Liu S.; Yang J. Anti-influenza A virus activity of dendrobine and its mechanism of action. J. Agric. Food Chem. 2017, 65, 3665–3674. 10.1021/acs.jafc.7b00276. [DOI] [PubMed] [Google Scholar]
- Kadam R. U.; Juraszek J.; Brandenburg B.; Buyck C.; Schepens W. B. G.; Kesteleyn B.; Stoops B.; Vreeken R. J.; Vermond J.; Goutier W.; Tang C.; Vogels R.; Friesen R. H. E.; Goudsmit J.; van Dongen M. J. P.; Wilson I. A. Potent peptidic fusion inhibitors of influenza virus. Science 2017, 358, 496–502. 10.1126/science.aan0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawes B. E.; Kalveram B.; Ikegami T.; Juelich T.; Smith J. K.; Zhang L.; Park A.; Lee B.; Komeno T.; Furuta Y.; Freiberg A. N. Favipiravir (T-705) protects against Nipah virus infection in the hamster model. Sci. Rep. 2018, 8, 7604. 10.1038/s41598-018-25780-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D.; Luo Y.; Yang G.; Li F.; Xie X.; Chen D.; He L.; Wang J.; Ye C.; Lu S.; Lv L.; Liu S.; He J. Potent influenza A virus entry inhibitors targeting a conserved region of hemagglutinin. Biochem. Pharmacol. 2017, 144, 35–51. 10.1016/j.bcp.2017.07.023. [DOI] [PubMed] [Google Scholar]
- Bullough P. A.; Hughson F. M.; Skehel J. J.; Wiley D. C. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 1994, 371, 37–43. 10.1038/371037a0. [DOI] [PubMed] [Google Scholar]
- Lawless M. K.; Barney S.; Guthrie K. I.; Bucy T. B.; Petteway S. R. Jr.; Merutka G. HIV-1 membrane fusion mechanism: structural studies of the interactions between biologically-active peptides from gp41. Biochemistry 1996, 35, 13697–13708. 10.1021/bi9606962. [DOI] [PubMed] [Google Scholar]
- Qi Z.; Shi W. G.; Xue N.; Pan C. G.; Jing W. G.; Liu K. L.; Jiang S. B. Rationally designed anti-HIV peptides containing multifunctional domains as molecule probes for studying the mechanisms of action of the first and second generation HIV fusion inhibitors. J. Biol. Chem. 2008, 283, 30376–30384. 10.1074/jbc.M804672200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan L.; Meng Q.; Xu L.; Meng Z.; Yan H.; Liu K. Peptide amphiphiles with multifunctional fragments promoting cellular uptake and endosomal escape as efficient gene vectors. J. Mater. Chem. B 2015, 3, 1068–1078. 10.1039/C4TB01353K. [DOI] [PubMed] [Google Scholar]
- He B.; Fu Y.; Xia S.; Yu F.; Wang Q.; Lu L.; Jiang S. Intranasal application of polyethyleneimine suppresses influenza virus infection in mice. Emerging Microbes Infect. 2016, 5, e41. 10.1038/emi.2016.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.