

Abstract
Ketamine, N,N-dimethyltryptamine (DMT), and other psychoplastogens possess enormous potential as neurotherapeutics due to their ability to potently promote neuronal growth. Here, we report the first-ever structure-activity relationship study with the explicit goal of identifying novel psychoplastogens. We have discovered several key features of the psychoplastogenic pharmacophore and used this information to develop isoDMT psychoplastogens that are easier to synthesize, have improved physicochemical properties, and possess reduced hallucinogenic potential as compared to their DMT counterparts.
Keywords: N,N-Dimethyltryptamine; isoDMT; Psychoplastogen; Neural Plasticity; Depression
Graphical Abstract
INTRODUCTION
Major depressive disorder and related neuropsychiatric diseases are among the leading causes of disability worldwide.1 Despite the prevalence of these illnesses, we still lack broadly efficacious treatments capable of producing both fast-acting and sustained effects. Recently, the Food and Drug Administration (FDA) approved the dissociative anesthetic esketamine for treatment-resistant depression, making it the first mechanistically distinct medicine to be introduced to psychiatry in nearly 30 years. Accumulating evidence suggests that ketamine is capable of rectifying the deleterious changes in neuronal structure that are associated with depression.2,3 Such structural alterations include the loss of dendritic spines and synapses in the prefrontal cortex (PFC), as well as reductions in dendritic arbor complexity.4,5 While the advent of ketamine represents an incredibly important milestone in the history of neuropsychiatric disease drug discovery, ketamine is an imperfect drug with potential for abuse,6 and moreover, its dissociative effects necessitate the hospitalization of patients during treatment. Therefore, the identification of safer alternatives to ketamine is an incredibly important goal.
Until recently, relatively few compounds were known to possess neural plasticity-promoting properties comparable to ketamine. Known as psychoplastogens,7 these molecules promote neuronal growth through a mechanism involving the activation of AMPA receptors, the tropomyosin receptor kinase B (TrkB), and the mammalian target of rapamycin (mTOR). In addition to ketamine, the tropane alkaloid scopolamine8 and GLYX-13 (i.e., rapastinel)9 have demonstrated psychoplastogenic properties, and this class of compounds has enormous potential for treating a variety of neuropsychiatric diseases. Our group has demonstrated that classic serotonergic psychedelics are among the most potent psychoplastogens, producing effects on neuronal structure comparable to ketamine in both cellular assays and in vivo.10 As pyramidal neurons in the PFC are known to exhibit top-down control over areas of the brain controlling motivation, fear, and reward, these results provide a potential explanation for the known antidepressant, anxiolytic, and anti-addictive effects of psychedelics in the clinic.11
Careful inspection of the molecular structures of psychedelic compounds reveals that N,N-dimethyltryptamine (DMT, 1) is a core feature of many of these molecules (Figure 1).12 As DMT produces antidepressant and anxiolytic behavioral effects in rodents13,14 and a DMT-containing tisane has demonstrated clinical efficacy for treatment-resistant depression,15,16,17 we reasoned that DMT was an excellent starting point for medicinal chemistry efforts aimed at identifying novel psychoplastogenic therapeutics. However, the synthesis of DMT derivatives from simple indoles is typically accomplished using the method of Speeter and Anthony,18 which requires multiple steps, harsh reaction conditions, and electron-rich indoles, thus limiting the number of derivatives we would be able to access rapidly for structure-activity relationship (SAR) studies. Moreover, many DMT derivatives are well known to be potent hallucinogens.
Figure 1. Structure of compounds possessing the DMT pharmacophore.
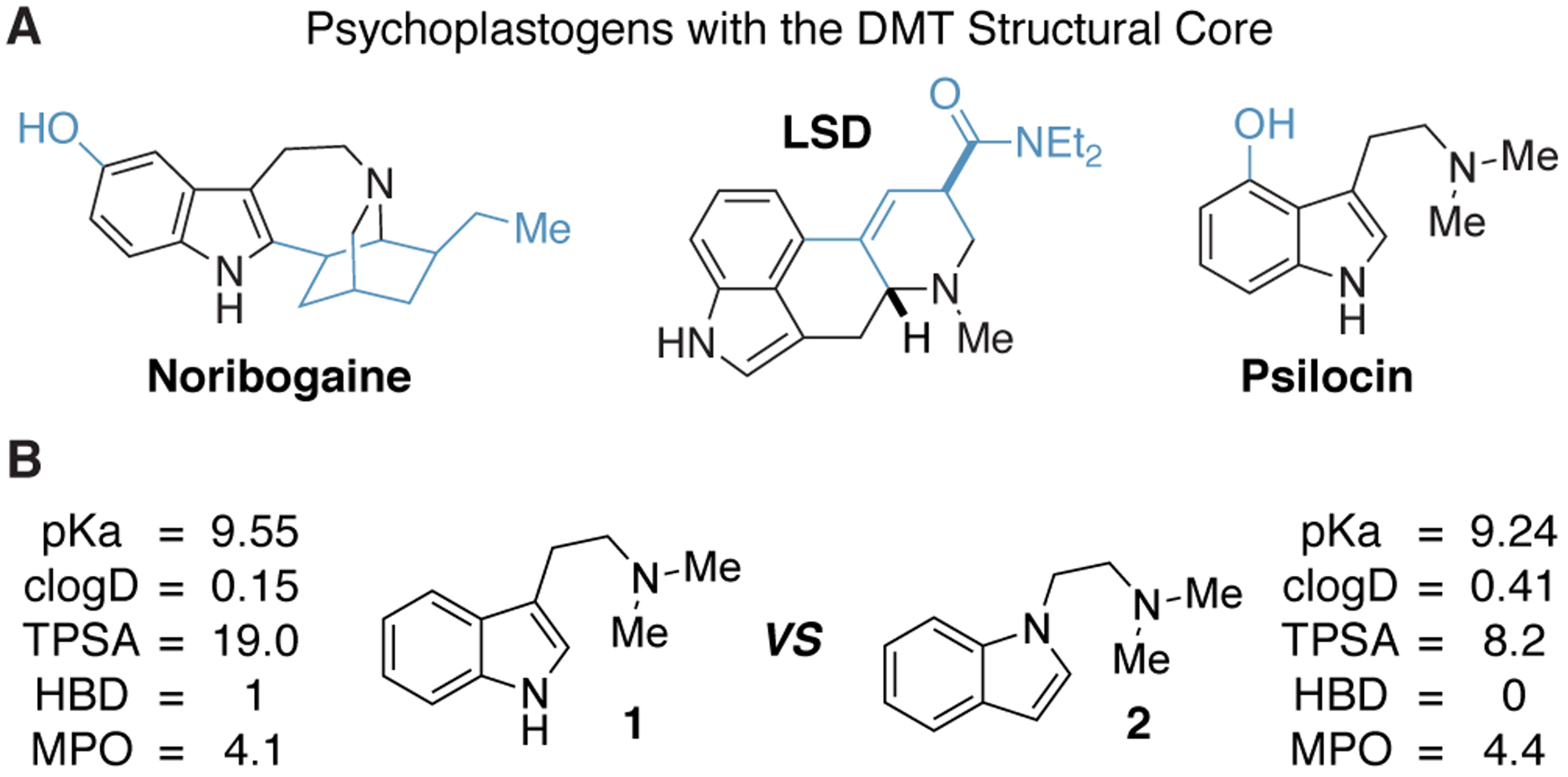
(A) The DMT structure (highlighted in black) is the core scaffold of several known psychoplastogenic compounds. (B) The only difference between the chemical structures of DMT (1) and isoDMT (2) is that the C1 and C3 atoms of the indole are transposed. Predicted chemical properties and calculated MPO scores are shown. clogD = calculated log D; TPSA = total polar surface area; HBD = hydrogen bond donor; MPO = multiparameter optimization score.
When considering how we could address these issues, we were inspired by the elegant work of Glennon and co-workers.19 By transposing the N1 and C3 atoms of DMT, they produced a small series of N,N-dimethylaminoisotryptamine (isoDMT, 2) analogs with reduced hallucinogenic potential as measured by their abilities to substitute for known hallucinogens in rodent drug discrimination assays. In principle, related analogs could be accessed in a single step through N-alkylation of the corresponding indoles or related heterocycles. Additionally, several isoDMTs have been shown to possess comparable affinity for serotonin receptors as compared to their DMT counterparts.19,20 Our group has demonstrated that the 5-HT2A receptor is necessary for the psychoplastogenic effects of DMT,10 As isoDMTs are known to bind to 5-HT2A receptors,20 we hypothesized that isoDMT analogs would still be capable of promoting neuronal growth despite lacking indole N–H bonds. Furthermore, isoDMT analogs are likely to exhibit improved physicochemical properties as the loss of a hydrogen bond donor decreases total polar surface area and improves central nervous system multiparameter optimization (MPO) scores (Figure 1).21 Here, we describe our efforts to develop an efficient method to access a variety of isoDMT derivatives as well as their subsequent evaluation in cellular neural plasticity assays.
RESULTS AND DISCUSSION
Chemistry.
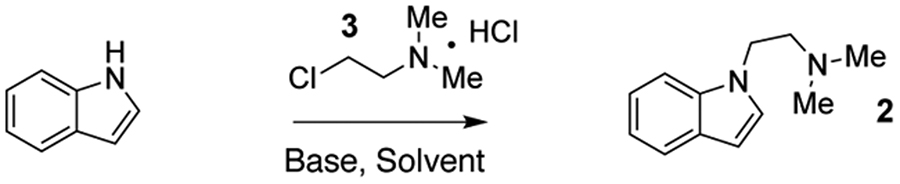
Surprisingly, there have been relatively few reports concerning the synthesis of isoDMT or related analogs, and they all require multiple steps or employ harsh reaction conditions.19,22,23 Therefore, we sought to develop an operationally simple and robust method for synthesizing a variety of isoDMTs under mild reaction conditions. We screened several conditions for performing the desired N-alkylation of indole, including the previously reported methods (Table 1), and found that alkylation could be achieved without using NaH or refluxing the reaction. The use of DMSO as the solvent proved to be critical, and ultimately, we were able to obtain 2 in good yield using KOH as the base and KI to enhance reactivity via an in situ Finkelstein reaction. Maintaining the reaction at 0.4 M proved optimal with both higher and lower concentrations resulting in a reduction in yield.
Table 1.
Optimization of Indole N-Alkylation.
 | |||||
|---|---|---|---|---|---|
| Equiv of 3 | Basea | Solventb | Additivec | Temp | Yieldd |
| 1.3 | NaH (2.6) | THF (0.5) | – | 66°C | 66% |
| 1.1 | NaH (2.6) | DMF (0.4) | – | 23°C | 67% |
| 1.1 | KOH (5) | DMF (0.4) | – | 23°C | 31% |
| 1.1 | KOH (5) | DMSO (0.4) | – | 23°C | 51% |
| 1.1 | KOH (5) | DMSO (0.4) | KI (1.1) | 23°C | 69% |
| 1.1 | KOH (5) | DMSO (0.1) | KI(1.1) | 23°C | 24% |
| 1.1 | KOH (5) | DMSO (1.0) | KI (1.1) | 23°C | 38% |
| 3 | KOH (5) | DMSO (0.4) | KI (3) | 23°C | 69% |
Number of equivalents of base are shown in parentheses.
Reaction molarities based on indole are shown in parentheses.
Number of equivalents of additive are shown in parentheses.
Yields are based on 1H NMR spectra obtained after aqueous workup with 6-flouroindole serving as an internal standard.
Previously published methods for the synthesis of isoDMT have either required purification via column chromatography or vacuum distillation followed by oxalate salt formation.19,22,23 As indole was cleanly converted to 2 using our reaction conditions, we reasoned that crystallization following a simple aqueous workup might obviate the need for further purification. Using this operationally simple method, we synthesized 20 isoDMT analogs in modest to good yields without the need for chromatography (Figure 2). Low yielding reactions could often be attributed to difficulties with crystallization, as the NMR yields of those reactions were often substantially higher than the isolated yields. An additional 4 compounds proved recalcitrant toward crystallization, and hence, were isolated following column chromatography on silica gel. The method is quite general, with electron-rich (4–11, 16) and electron-poor (12–15, 17–21) indoles performing equally well. Additionally, substitution on the indole did not have an obvious impact on reaction performance with substitution at all positions being tolerated. Lastly, related heterocycles including benzimidazole, pyrrole, and carbazole are efficiently alkylated using these same conditions (22–24).
Figure 2. Substrate scope for the N-alkylation of various indoles with 3.
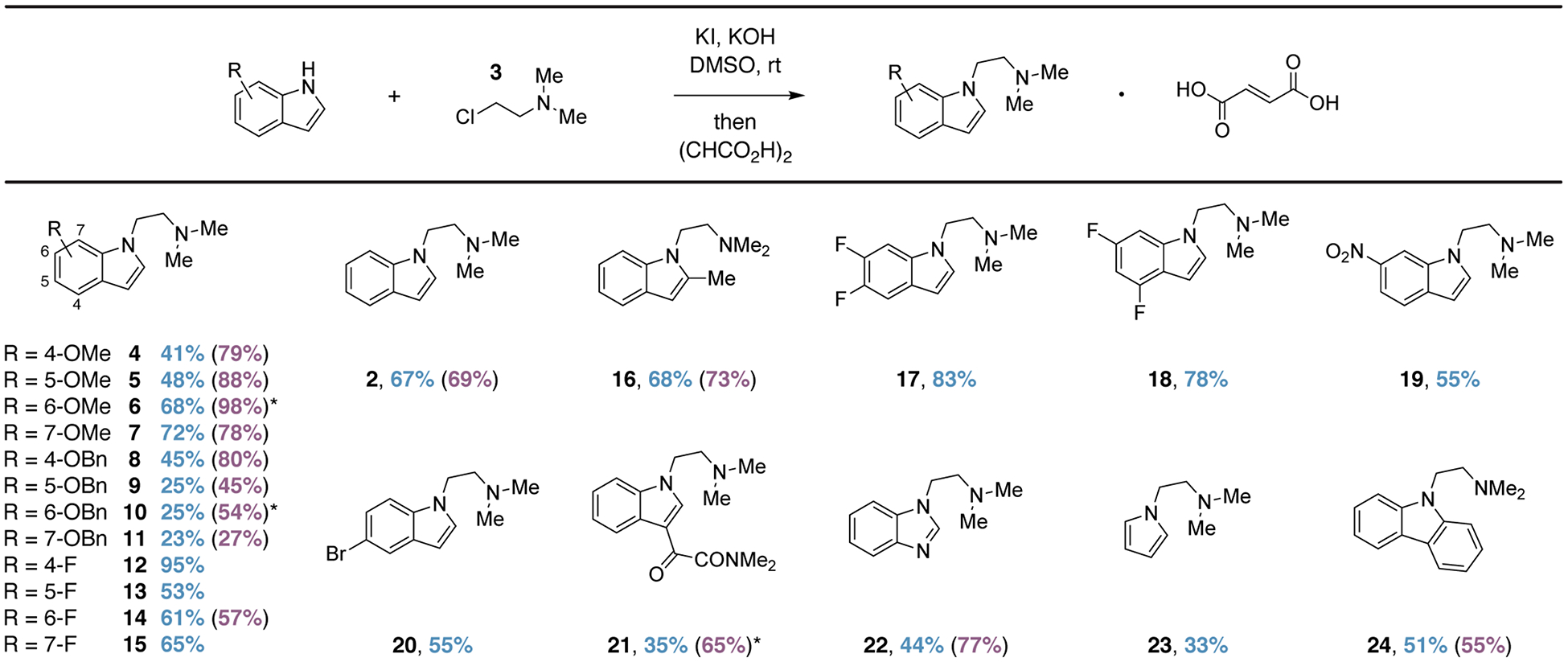
Percent yields following crystallization are indicated. Values in parentheses denote yields based on 1H NMR spectra obtained after aqueous workup with indole serving as an internal standard. Note: fluoroindole was used as the internal standard when determining the yield of 2. An asterisk indicates that a compound was purified via chromatography.
We hypothesized that the efficiency of the alkylation was due to the formation of a reactive aziridinium intermediate (Figure 3A). However, the use of 1-chloro-3-methylbutane as the alkylating agent results in a comparable yield (Figure 3B). Moreover, increasing the distance between the electrophilic carbon bearing the halide and the nucleophilic nitrogen doesn’t drastically reduce reaction performance (Figure 3B). These results suggest that the reaction is proceeding through a traditional SN2 reaction with negligible to no enhancement via neighboring group participation.
Figure 3. Neighboring group participation has a minimal impact on reaction performance.
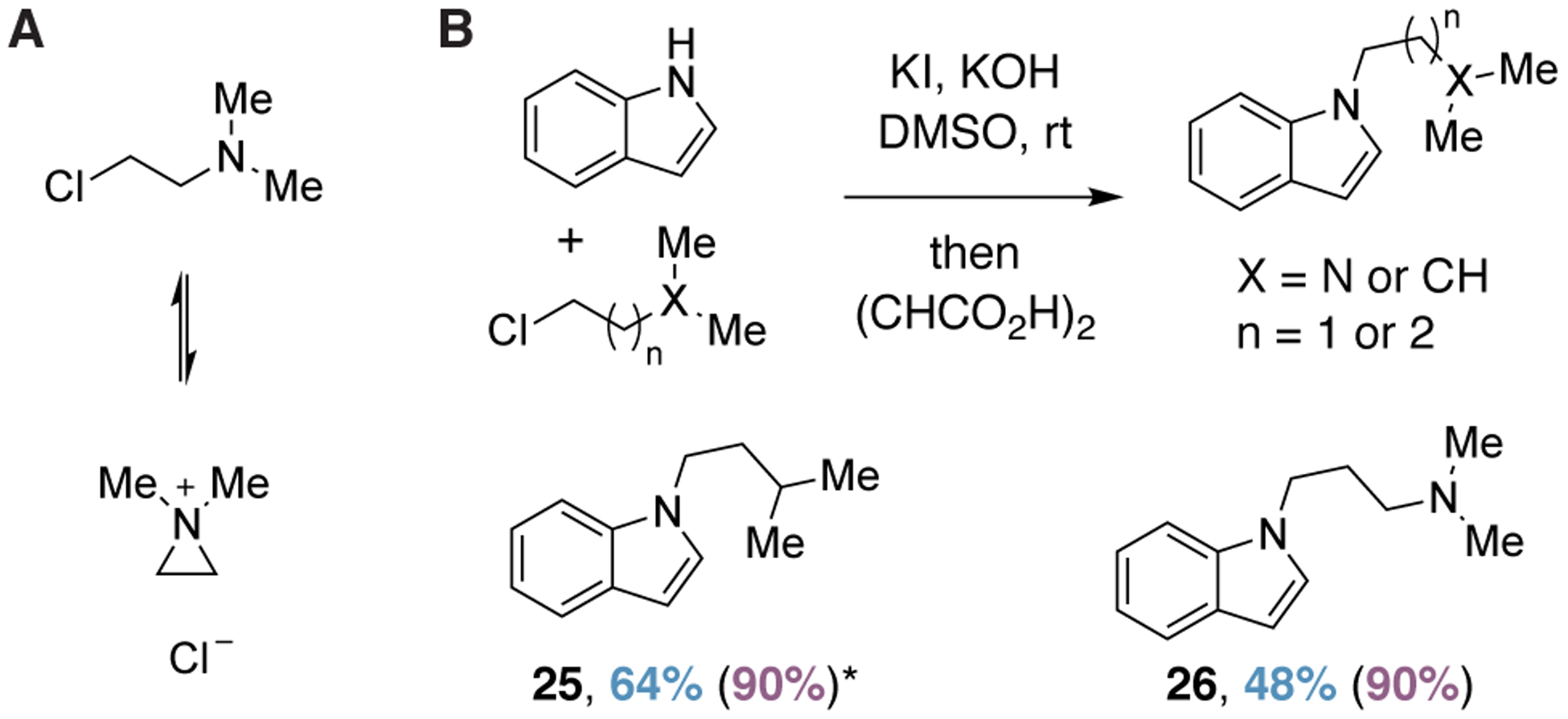
(A) Hypothesized reactive aziridinium intermediate. (B) Reaction efficiency remains high when using alkylating agents that cannot form a reactive aziridinium intermediate. Percent yields following crystallization are indicated. Values in parentheses denote yields based on 1H NMR spectra obtained after aqueous workup with 6-fluoroindole serving as an internal standard. An asterisk indicates that a compound was purified via chromatography.
Dendritogenesis Assays.
Phenotypic screening has historically proven more successful than target-based approaches for identifying drugs with novel mechanisms of action.24,25 Having established a simple and robust method for accessing isoDMT analogs, we next tested their ability to increase dendritic arbor complexity in cultures of cortical neurons using a phenotypic assay. Following treatment, neurons were fixed and visualized using an antibody against MAP2—a cytoskeletal protein localized to the somatodendritic compartment of neurons.26 Sholl analysis27 was then performed, and the maximum number of crossings (Nmax) was used as a quantitative metric of dendritic arbor complexity. For statistical comparisons between specific compounds, we compared the raw Nmax values; however, we have also calculated a percent efficacy as well as an MPO score for every compound tested in this study (Figure S1). Percent efficacies were determined by setting the Nmax values for the vehicle (DMSO) and positive (ketamine) controls equal to 0% and 100%, respectively.
We began our SAR studies by comparing the effect of DMT (1) to that of 1-Me-DMT (27) and isoDMT (2). While DMT has the potential to serve as a hydrogen bond donor when bound to its target receptors, 27 and 2 do not. Therefore, this potential hydrogen bonding interaction must not be critical for a compound to induce plasticity, as both 27 and 2 increased dendritic arbor complexity to a comparable extent as 1, despite lacking an indole N–H bond (Figure 4).
Figure 4. The indole N–H of tryptamine derivatives is not necessary to promote dendritogenesis.
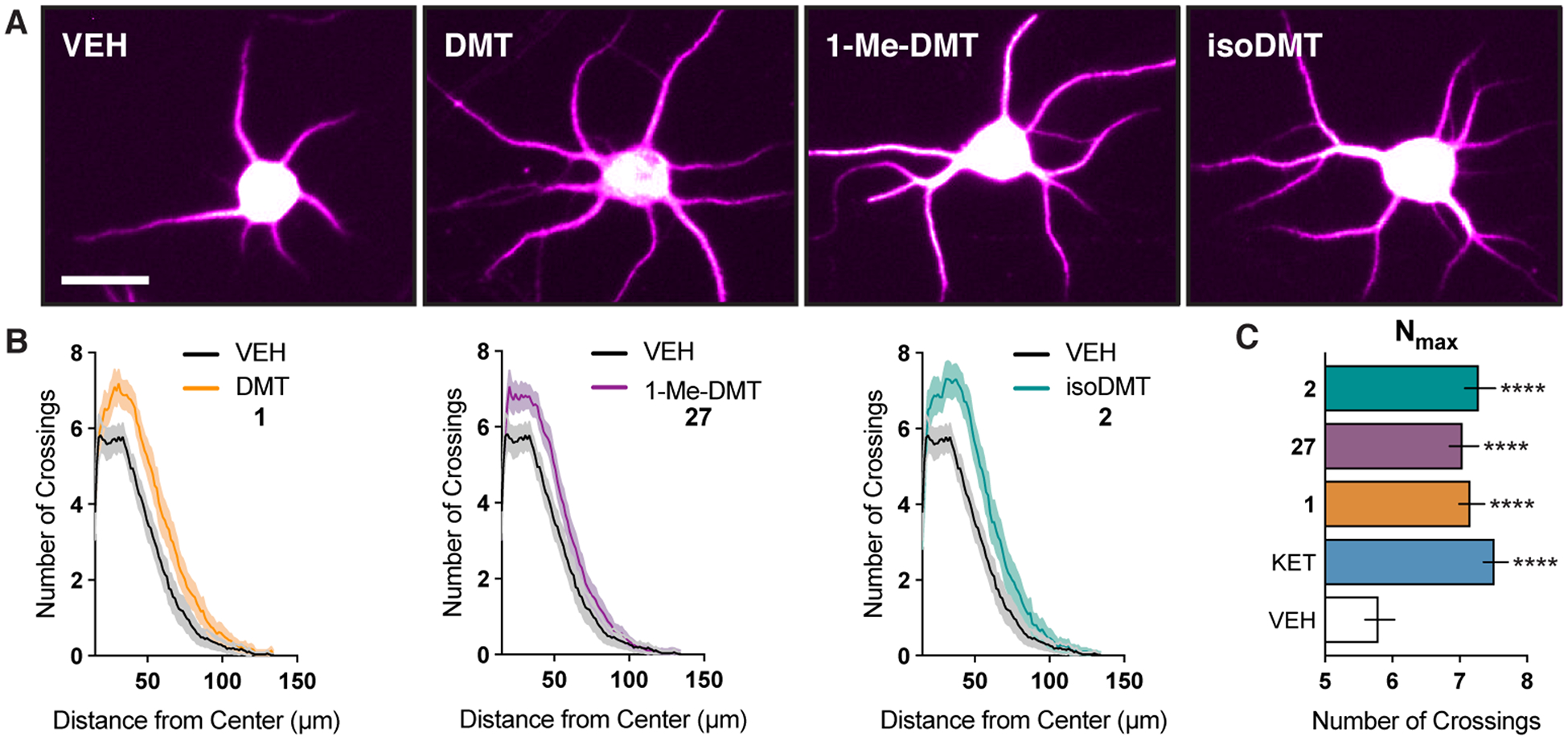
(A) Representative images of cortical neurons (DIV6) treated with compounds. See Figure S2 for the entire field of view from which these neurons were selected. (B) Sholl analysis demonstrates that 1-Me-DMT (27) and isoDMT (2) increase dendritic arbor complexity to a comparable extent as DMT (1) (n = 46–79 neurons). (C) Maximum number of crossings (Nmax) of the Sholl plots in B. Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, as compared to vehicle control following a one-way ANOVA with Dunnett’s post hoc test (F = 9.702; DFn = 4; DFd = 304; p-value < 0.0001). VEH = vehicle, KET = ketamine. Scale bar = 20 μm.
Next, we were interested in performing head-to-head comparisons between DMT analogs and their isoDMT counterparts, as Glennon and co-workers previously used serotonin receptor binding affinities and drug discrimination assays to demonstrate that these pairs of compounds can exhibit bioisosterism.19 We chose 5-MeO-DMT (28) and 6-F-DMT (29) as electron-rich and electron-poor DMT analogs, respectively. Compound 28 has been reported to promote neuritogenesis in the dentate gyrus28 and alleviate symptoms of depression and anxiety in humans.29,30 Compound 29 is predicted to be non-hallucinogenic, as fluorination of DMT analogs is known to attenuate their hallucinogenic potential.31 When compared directly, isoDMT analogs 6 and 13 performed identically to 28 and 29 (Figure 5), suggesting that SAR data related to neuronal growth obtained using derivatives of the isoDMT scaffold could be applied to derivatives of the DMT scaffold through analogy due to the isosteric nature of the two structures.
Figure 5. DMT and isoDMT analogs produce comparable effects on dendritic arbor complexity.
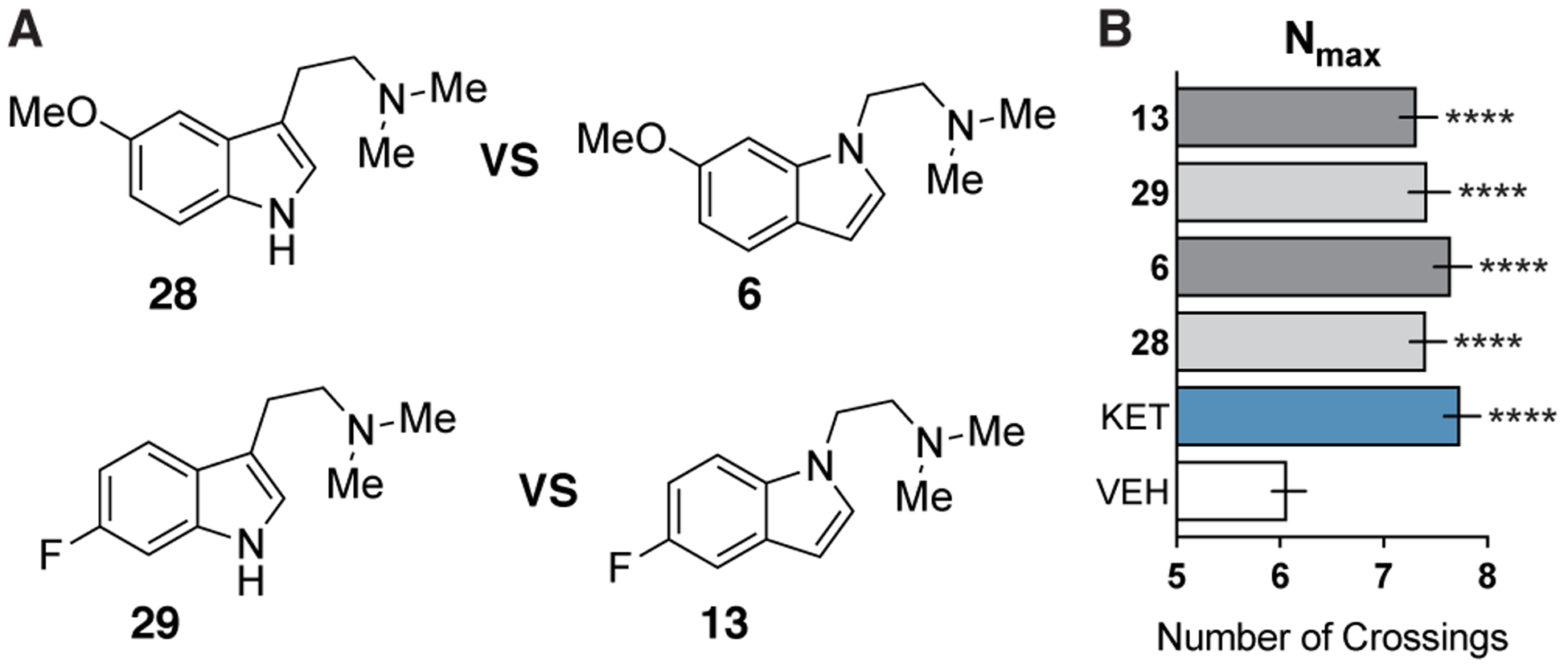
(A) Chemical structures of DMT derivatives and analogous isoDMTs. (B) Maximum number of crossings (Nmax) of the Sholl analysis for cortical neurons treated with compounds (n = 82–95 neurons). Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, as compared to vehicle control following a one-way ANOVA with Dunnett’s post hoc test (F = 11.17; DFn = 5; DFd = 524; p-value < 0.0001). VEH = vehicle, KET = ketamine.
Confident that the DMT and analogous isoDMT derivatives would behave similarly, we next attempted to use various isoDMT analogs to establish the key features of the psychoplastogen pharmacophore (Figure 6). Removing the basic amine of isoDMT to produce 25 yielded a molecule that did not promote dendritogenesis. Furthermore, compound 31—the N,N-dimethylamide analog of isoDMT—did not promote neuronal growth, confirming our hypothesis that a basic nitrogen is necessary to promote plasticity (Figure 6A and B). Extending the distance between the aromatic ring and the amine by one carbon (26) resulted in only a slight decrease in the Nmax value (Figure 6B).
Figure 6. Establishment of the essential psychoplastogen pharmacophore.
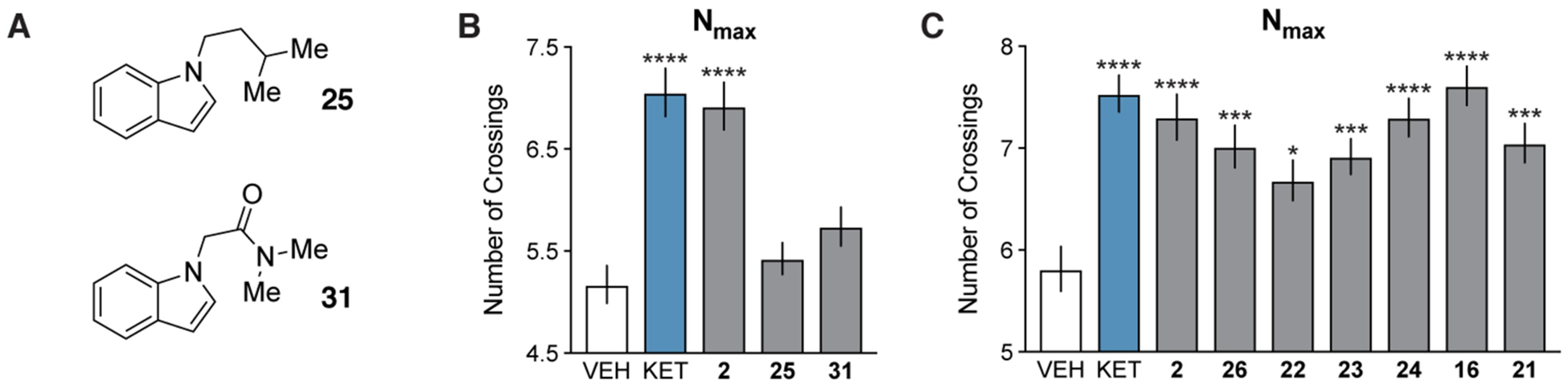
(A) Chemical structures of non-basic analogs of of isoDMT 2. (B–C) Maximum number of crossings (Nmax) of the Sholl plots for cortical neurons treated with compounds (n = 46–85 neurons). The effects of nitrogen basicity and modifications to the aromatic ring were assessed in B and C, respectively. Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, as compared to vehicle control following a one-way ANOVA with Dunnett’s post hoc test (For B: F = 19.03; DFn = 4; DFd = 273; p-value < 0.0001. For C: F = 6.933; DFn = 8; DFd = 599; p-value < 0.0001). VEH = vehicle, KET = ketamine.
Modification of the aromatic ring was generally well tolerated (Figure 6C). Converting the indole into a benzimidazole (22), pyrrole (23), or carbazole (24) had a minimal effect on the ability of these molecules to promote neuronal growth. Moreover, substitution at the 2- and 3-positions of the indole (16 and 21, respectively) was well tolerated. Taken together, the minimal psychoplastogen pharmacophore appears to involve a modifiable aromatic ring separated from a basic nitrogen by a short linker.
Substitution on the benzene ring of both DMTs and isoDMTs is known to impact hallucinogenic potential.19,32 For example, 5-MeO-DMT (28) substitutes for the hallucinogen 2,5-dimethoxy-4-methylamphetamine (DOM) in rats trained to discriminate DOM from saline, whereas 6-MeO-DMT does not.32 Similarly, 6-MeO-isoDMT (5) substitutes for a hallucinogenic training drug while 5-MeO-isoDMT (6) does not.19 Therefore, we desired to test the effects of steric and electronic perturbations at positions 4–7 of the indole. We synthesized and tested three series of analogs substituted with either methoxy (electron-donating; 4–7), benzyloxy (electron-donating, but sterically demanding; 8–11), or fluoro (electron-withdrawing; 12–15) groups (Figure 7). We found that substitution of the 5-, 6-, and 7-positions were well tolerated regardless of the substituent. However, substitution at the 4-position resulted in compounds incapable of increasing dendritic arbor complexity. This result was quite striking as the specific electronic or steric properties of the substituents were inconsequential. Even a fluorine substituent with a very small van der Waals radius (1.2 and 1.47 for H and F, respectively)33 was not tolerated. Careful inspection of the 5-HT2B crystal structure bound to LSD suggests that the 7-position of the LSD indole abuts helix V, and thus, is not likely to tolerate any substituent at that position.34 Assuming that DMTs and isoDMTs bind to 5-HT2 receptors in a similar conformation as LSD, we would predict that substitution at the 4-position of an isoDMT (corresponding to the 7-position of the LSD indole) would not be tolerated. However, this hypothesis is purely speculative at the moment, as there are no published crystal structures of DMTs or isoDMTs bound to 5-HT2 receptors.
Figure 7. The impact of indole substitution on the ability of isoDMTs to promote neuronal growth.
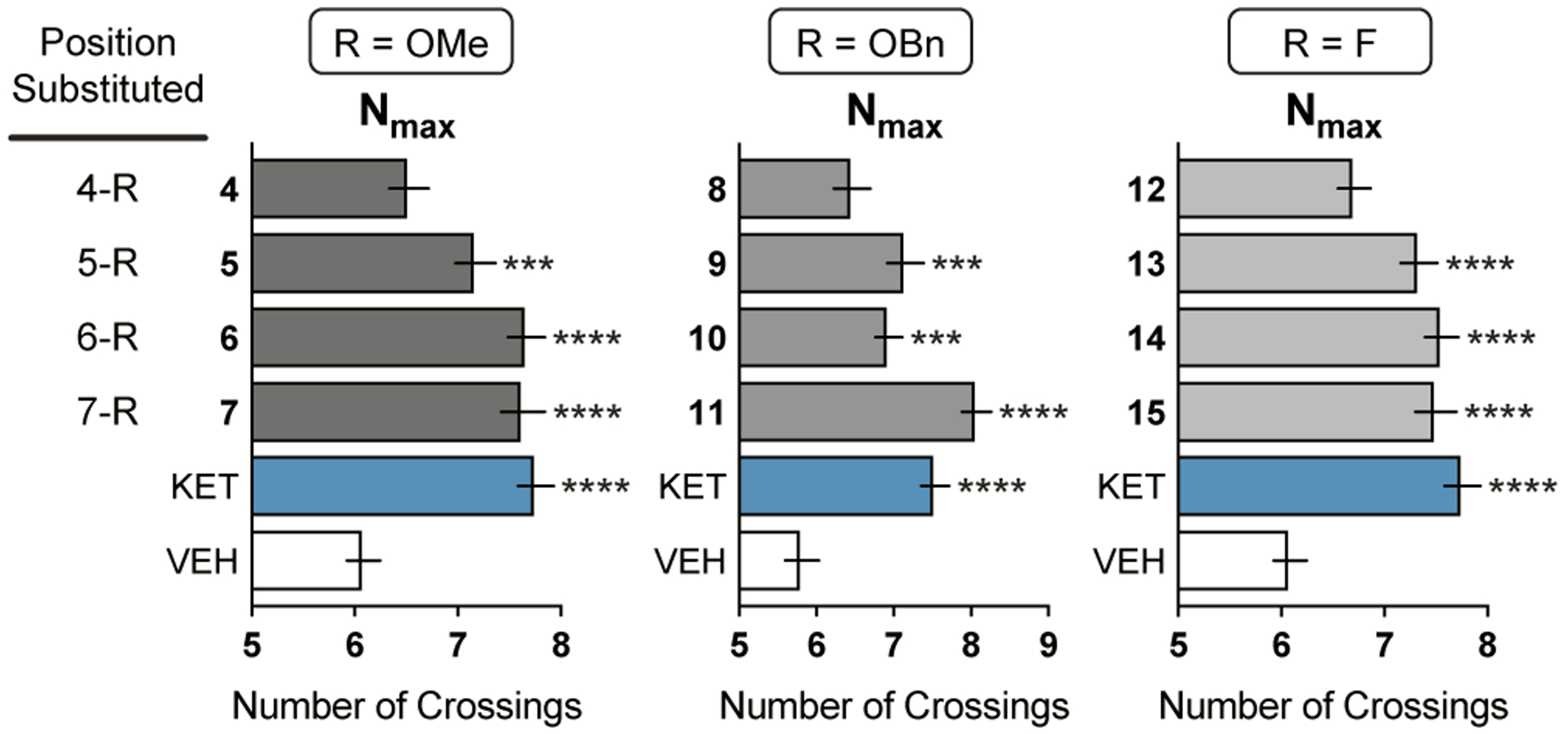
Maximum number of crossings (Nmax) of the Sholl plots for cortical neurons treated with compounds (n = 39–93 neurons). Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, as compared to vehicle control following a one-way ANOVA with Dunnett’s post hoc test (For R = OMe: F = 13.85; DFn = 5; DFd = 493; p-value < 0.0001. For R = OBn: F = 15.44; DFn = 5; DFd = 372; p-value < 0.0001. For R = F: F = 13.24; DFn = 5; DFd = 506; p-value < 0.0001). VEH = vehicle, KET = ketamine.
To determine if DMT and isoDMT derivatives exhibited differences in psychoplastogenic potency, we performed concentration–response experiments (Figure 8). We found that isoDMTs (2 and 6) produced comparable maximum efficacies and had similar potencies as isosteric DMTs (1 and 28). Moreover, they were capable of increasing dendritic arbor complexity at concentrations as low as 1 nM. We also observed that these compounds exhibited comparable efficacies and potencies to ketamine, further emphasizing their potential as antidepressants. Finally, compound 5 proved to be an exceptional psychoplastogen, which is highly significant due to its low hallucinogenic potential in both drug-discrimination19 and head-twitch response (HTR) assays (Figure 11).
Figure 8. Concentration–response experiments demonstrate that DMTs and isoDMTs have similar psychoplastogenic potencies.
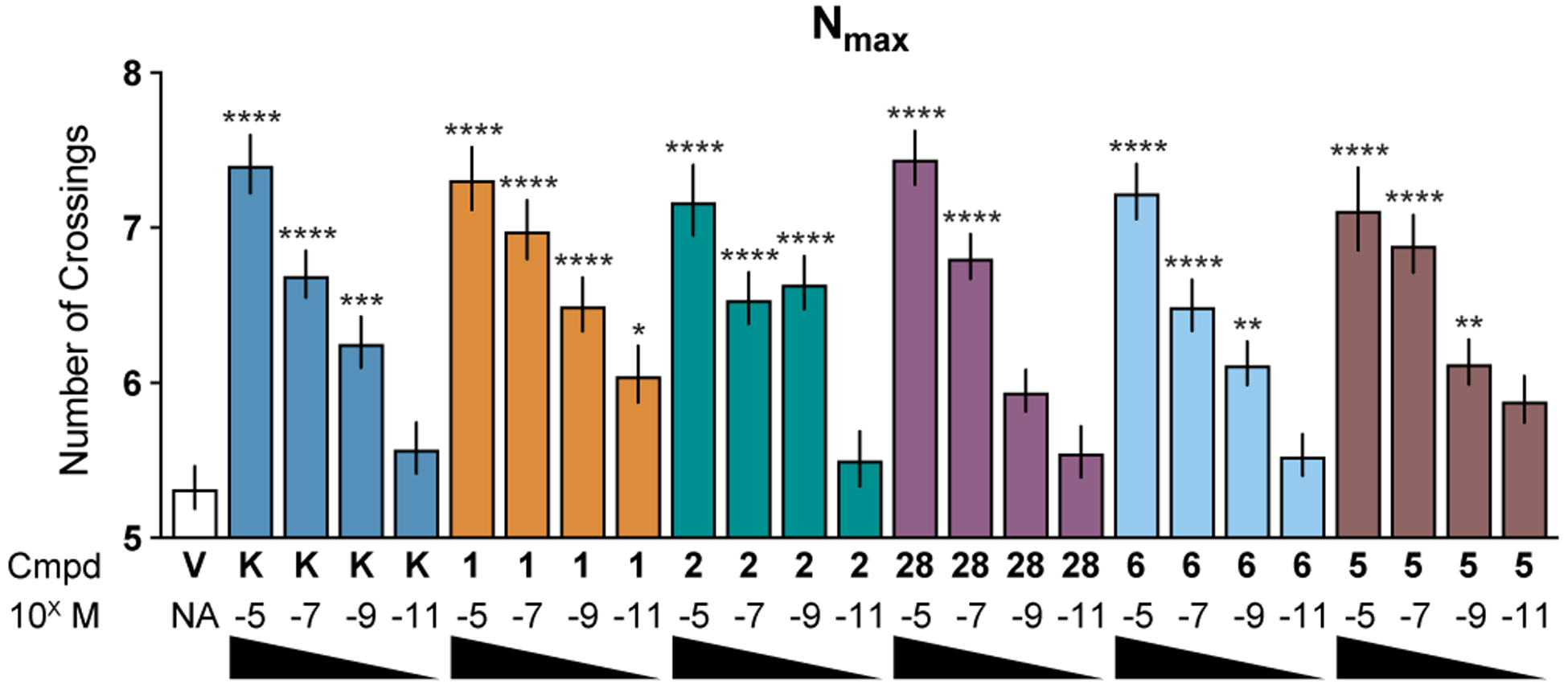
Maximum number of crossings (Nmax) of the Sholl plots for cortical neurons treated with compounds at concentrations ranging from 10 μM to 10 pM (n = 66–123 neurons). Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, as compared to vehicle control following a one-way ANOVA with Dunnett’s post hoc test (F = 15.40; DFn = 24; DFd = 2,276; p-value < 0.0001). V = vehicle, K = ketamine.
Figure 11. Mouse HTR assays demonstrate that psychoplastogenic isoDMTs exhibit reduced hallucinogenic potential.
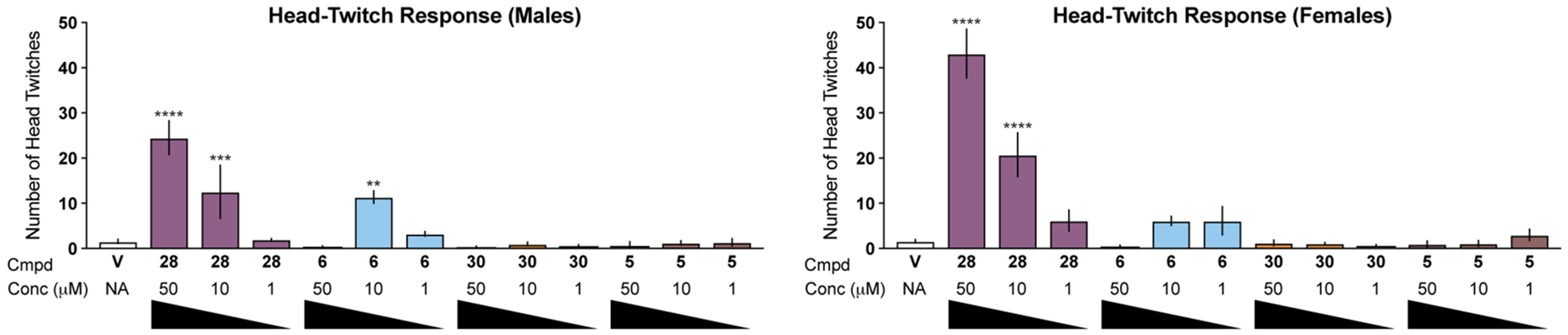
Male and female mice were administered drugs via intraperitoneal injection, and the number of head-twitches were recorded over the next 20 mins (n = 3–8 mice per condition). Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, as compared to vehicle control following a one-way ANOVA with Dunnett’s post hoc test. V = vehicle.
Previously, we demonstrated that DMT and other psychedelic compounds promote increased dendritic arbor complexity, dendritic spine density, and synaptogenesis through a 5-HT2A-dependent process.10 As expected, we found that pretreating cortical cultures with a 5-HT2A antagonist blocked the ability of 5-MeO-DMT (28) to increase dendritic growth (Figure 9). Importantly, the psychoplastogenic effects of isoDMTs were also blocked under these conditions, suggesting that 5-HT2A receptors may be involved in their mechanism of action (Figure 9). However, the involvement of other receptors cannot be ruled out at this time.
Figure 9. The psychoplastogenic effects of isoDMTs are blocked by a 5-HT2A antagonist.
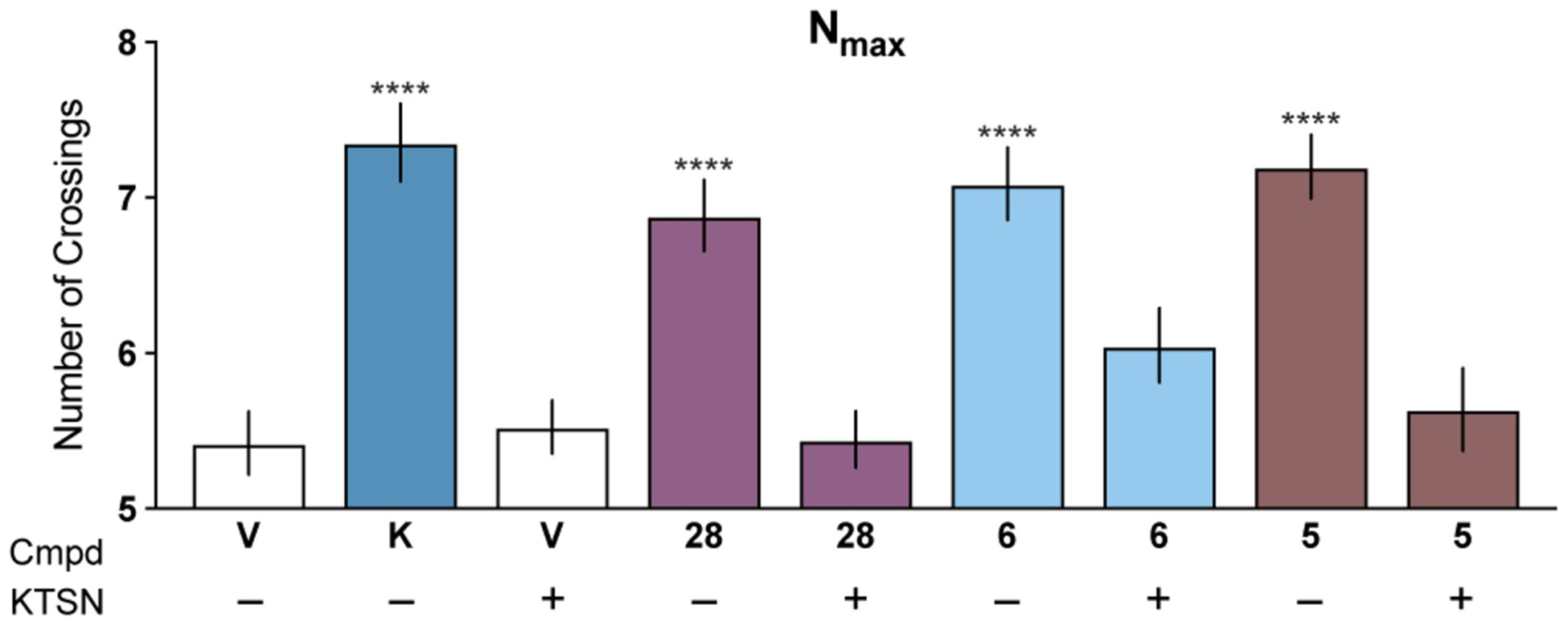
Maximum number of crossings (Nmax) of the Sholl plots for cortical neurons treated with compounds (n = 45–63 neurons) in the presence (+) or absence (–) of the 5-HT2A antagonist ketanserin. Data are represented as mean ± SEM. ****p < 0.0001, as compared to vehicle control following a one-way ANOVA with Dunnett’s post hoc test (F = 13.92; DFn = 8; DFd = 461; p-value < 0.0001). V = vehicle, K = ketamine, KTSN = ketanserin.
Zebrafish Behavioral Assays.
While our cellular dendritogenesis assays indicated that isosteric molecules from the DMT and isoDMT classes performed comparably, we wanted to demonstrate bioisosterism in a different context. To this end, we decided to employ an in vivo zebrafish behavioral assay that has been previously validated for assessing similarities between compounds.35 Due to the short timescale of the zebrafish behavioral effects (hours) relative to changes in neuronal structure (days), it is unlikely that psychoplastogenicity plays a role in modulating acute zebrafish behavior. Larval zebrafish were treated with 5-MeO-DMT (28), 6-MeO-isoDMT (6), 6-MeO-DMT (30), or 5-MeO-isoDMT (5) and video-recorded during a 17-minute battery of acoustic and visual stimuli (Figure 10A, Figure S3). Aggregate locomotion over time was determined per well. All compounds tested induced significant behavioral effects relative to the vehicle control in a concentration–dependent manner (Figure 10B) with the largest effects (ECmax) being observed at 200 μM for each compound.
Figure 10. Zebrafish behavioral assays demonstrate the similarity between isosteres.
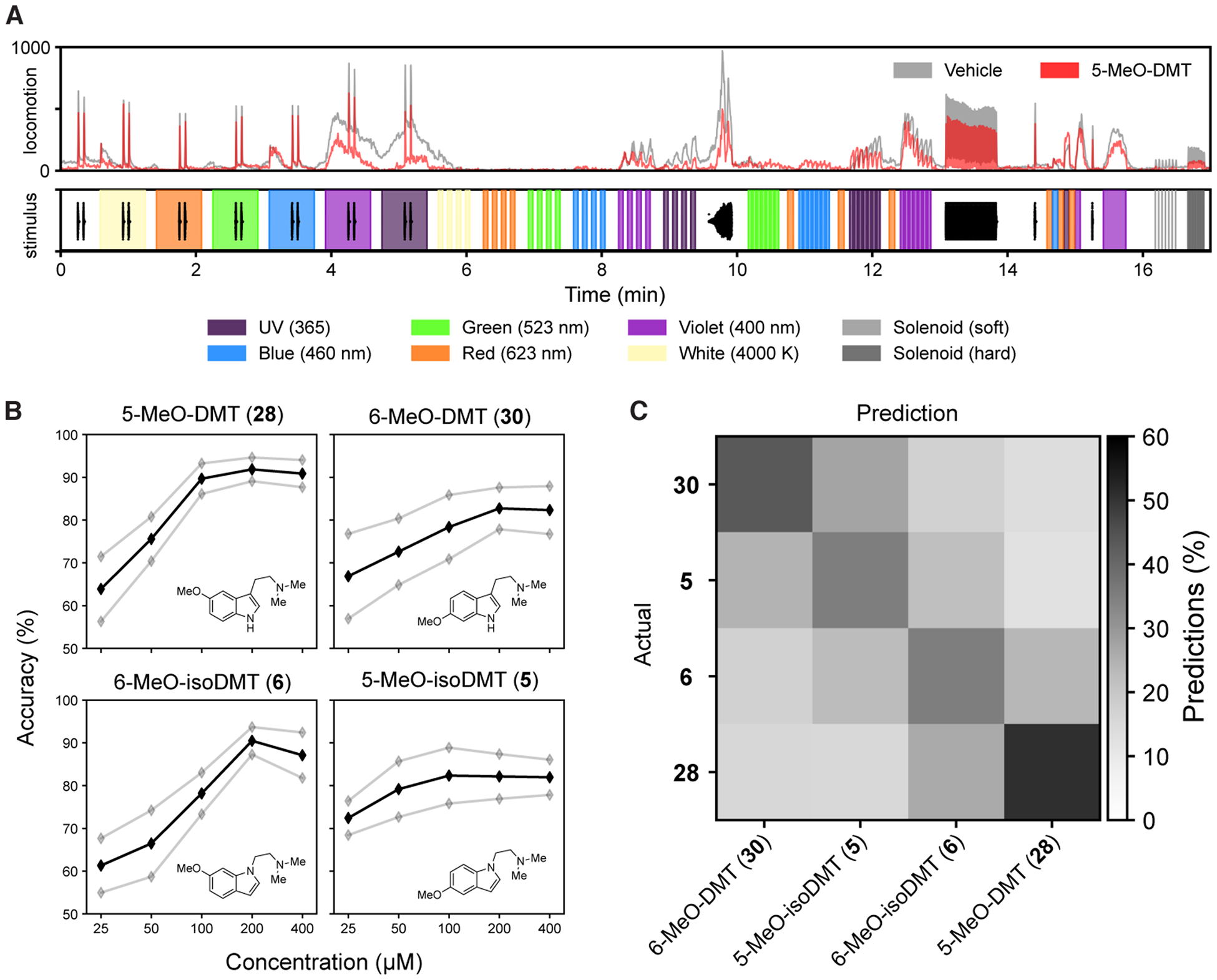
(A) An example motion trace with associated stimuli. Top: Locomotion in wells treated with vehicle (gray) or 5-MeO-DMT (28, 200 μM, red). Individual traces for the 5, 6, and 30 are shown in Figure S3. Bottom: Stimuli applied over time. Colors indicate bright LED light of respective colors. Black traces represent the waveform of acoustic stimuli, and gray vertical lines indicate physical tapping as secondary acoustic stimuli. (B) Concentration–response curves for DMT and isoDMT analogs (n = 21 wells per condition). The y-axis is the mean accuracy of classification against vehicle controls. Gray lines are the mean ± STD under bootstrap. (C) Confusion matrix showing that isosteric pairs produce similar behavioral phenotypes (n = 18 wells per condition). Darkness indicates the percentage of wells classified; the maximum is 50.7% (5-MeO-DMT as itself).
To test our hypothesis that pairs of isosteric compounds would produce behavioral phenotypes similar to each other but dissimilar from non-isosteric compounds, we trained a multiclassification model to choose between animals treated with 5-MeO-DMT (28), 6-MeO-isoDMT (6), 6-MeO-DMT (30), and 5-MeO-isoDMT (5) at 200 μM. We found that misclassification of isosteric pairs (e.g. 5-MeO-DMT (28) and 6-MeO-isoDMT (6); or 6-MeO-DMT (30) and 5-MeO-isoDMT (5)) was more likely (Figure 10C; 50.4% higher, p = 0.154 by permutation test), indicating that isosteric compounds produce more similar behavioral phenotypes. Though the acute behavioral changes in zebrafish are unlikely the result of compound-induced neural plasticity, these results suggest that the biological effects of isoDMT compounds are likely to be similar to their DMT isosteres.
Hallucinogenic Potential.
While Glennon and co-workers have shown that isoDMTs such as 5 and 6 exhibit reduced hallucinogenic potential than their DMT counterparts in drug-discrimination assays,19 we were interested in testing their abilities to elicit a mouse head-twitch response (HTR)—a well validated behavioral proxy for hallucinations.36 The known hallucinogenic compound 5-MeO-DMT (28) produces a robust, dose-dependent HTR that was greater in female mice. However, the isosteric compound 6-MeO-isoDMT (6) is significantly less potent (Figure 11). As expected based on drug-discrimination data,32 6-MeO-DMT (30) did not produce a HTR. Finally, potent plasticity-promoting compound 5-MeO-isoDMT (5) did not produce any HTR (Figure 11), demonstrating that hallucinogenic potential and psychoplastogenicity can be decoupled.
CONCLUSIONS
While the potent psychoplastogenic properties of psychedelics have been hypothesized to play a key role in their therapeutic mechanism of action, the hallucinogenic effects of these compounds have limited their potential to serve as widespread medicines. Moreover, very little is known about how the structures of these compounds impact their abilities to promote dendritic growth. Here, we address both of these issues by demonstrating that isoDMT derivatives with low hallucinogenic potential are capable of promoting dendritogenesis to a comparable extent as the psychedelic DMT and the state-of-the-art fast-acting antidepressant ketamine. Our SAR studies have defined the minimal psychoplastogen pharmacophore as an aromatic ring separated from a basic nitrogen by a short linker. Additionally, we have discovered that substitution at the 4-position of isoDMT derivatives renders them devoid of psychoplastogenic properties. Importantly, this study is the first to demonstrate that a psychedelic compound (i.e., DMT) can be engineered to lack hallucinogenic potential while retaining the ability to promote neural plasticity (e.g., 5). All told, our studies will inform future medicinal chemistry efforts to identify novel psychoplastogens with improved safety profiles for treating a variety of neuropsychiatric disorders.
EXPERIMENTAL SECTION
Chemistry (General).
All reagents were obtained from commercial sources and used without purification unless otherwise noted. DMSO was purified by passage under 12 psi N2 through activated alumina columns. Reactions were performed using glassware that was flame-dried under reduced pressure (~1 Torr). Chromatography was performed using Fisher Chemical™ Silica Gel Sorbent (230–400 Mesh, Grade 60). Compounds purified by chromatography were dissolved in a minimal amount of chloroform for loading. Thin layer chromatography (TLC) was performed on Millipore silica gel 60 F254 plates. Visualization of the developed chromatogram was accomplished by fluorescence quenching or by staining with ninhydrin.
Nuclear magnetic resonance (NMR) spectra were acquired on either a Bruker 400 operating at 400 and 100 MHz, a Varian 600 operating at 600 and 150 MHz, or a Bruker 800 operating at 800 and 200 MHz for 1H and 13C, respectively, and are referenced internally according to residual solvent signals. Data for 1H NMR are recorded as follows: chemical shift (δ, ppm), multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet), integration, coupling constant (Hz). Data for 13C NMR are reported in terms of chemical shift (δ, ppm). Infrared spectra were recorded using a Thermo Nicolet iS10 FT-IR spectrometer with a Smart iTX Accessory (diamond ATR) and are reported in frequency of absorption (ν, cm−1). Liquid chromatography-mass spectrometry (LC-MS) was performed using using a Waters LC-MS with an ACQUITY Arc QDa detector. Ketamine was purchased from Fagron. DMT (1) and 6-F-DMT (29) were synthesized using previously published methods (Purity >99% as determined by UHPLC).13,37 For cellular plasticity assays (i.e., dendritogenesis), all compounds were dissolved in DMSO and stored as 10 mM stock solutions in the dark at −20°C.
All compounds tested in cellular assays were confirmed to be of >95% purity based on UHPLC analysis (Waters ACQUITY Arc) measuring absorbance at 254 and 280 nm. Mobile phase A consisted of 0.01% formic acid in water, and mobile phase B consisted of 0.01% formic acid in acetonitrile. All samples were injected at a volume of 5 μL and the column temperature was maintained at 40 °C. One of three methods was used depending on the specific compound. Method A utilized a CORTECS C18, 2.7 μm, 4.6 × 50 mm column, a flow rate of 0.6 mL/min, and a gradient from 10% to 90% mobile phase B over 3 minutes, which was maintained for an additional 2 minutes. Method B utilized an XBridge BEH C18 2.5 μm, 2.1 × 100mm column, a flow rate of 0.6 mL/min, and a gradient from 10% to 90% mobile phase B over 0.5 minutes, which was maintained for an additional 4.5 minutes. Method C utilized a CORTECS C18, 2.7 μm, 4.6 × 50 mm column, a flow rate of 0.2 mL/min, and a gradient from 10% to 90% mobile phase B over 4 minutes, which was maintained for an additional 2 minutes. As most compounds reported in this study were isolated as the fumarate salts, peaks in UHPLC traces corresponding to fumaric acid were not included in the calculation of purity.
Synthesis of isoDMTs.
To a solution of respective indole or related heterocycle in DMSO (0.4 M) was added 2-chloro-N,N-dimethylethylamine hydrochloride (1.1 equiv), potassium iodide (1.1 equiv), and potassium hydroxide pellets (5.0 equiv). The reaction was stirred at room temperature for 24 h before being diluted with 1.0 M NaOH(aq). The aqueous phase was extracted three times with DCM. The organic extracts were combined, dried over Na2SO4, filtered, and concentrated under reduced pressure to yield an oil. The unpurified oil was dissolved in a minimal amount of acetone and added dropwise to a boiling solution of fumaric acid (1.0 equiv) in acetone. In most cases, a precipitate formed immediately, which was stored at −20°C overnight. The resulting crystals were filtered and washed with several portions of ice-cold acetone to yield the desired product. In cases where the desired product did not readily crystalize as the fumarate salt, the oil was subjected to column chromatography (9:1 CH2Cl2:MeOH:1% NH4OH(aq)) unless noted otherwise.
2-(1H-indol-1-yl)-N,N-dimethylethan-1-amine fumarate salt (1:1) (2).
Reaction performed using indole (100 mg, 0.85 mmol) and purified via crystallization. Yield = 175 mg, 67%. Purity >99%. TLC Rf (free base) = 0.50 (9:1 CH2Cl2:MeOH:1% NH4OH(aq)); 1H NMR (600 MHz, DMSO-d6) δ 7.54 (d, 1H, J = 7.6 Hz), 7.48 (d, 1H, J = 7.6 Hz), 7.38 (d, 1H, J = 3.1 Hz), 7.13 (ddd, 1H, J = 7.6, 1.0 Hz), 7.01 (dd, 1H, J = 7.6, 1.0 Hz), 6.61 (s, 2H), 6.42 (d, 1H J = 3.1 Hz), 4.29 (t, 2H, J = 6.7 Hz), 2.70 (t, 2H, J = 6.7 Hz), 2.26 (s, 6H) ppm; 13C NMR (150 MHz, DMSO-d6) δ 166.32, 135.66, 134.20, 128.82, 128.06, 120.96, 120.38, 118.88, 109.68, 100.54, 58.09, 44.83, 43.11 ppm; IR (diamond, ATR) ν 3100, 2923, 2393, 1705 cm−1; LRMS (ES+) calcd for C12H16N2+ 188.13, found 189.38 (MH+); MP = 147–149°C.
2-(4-methoxy-1H-indol-1-yl)-N,N-dimethylethan-1-amine fumarate salt (1:1) (4).
Reaction performed using 4-methoxyindole (100 mg, 0.68 mmol) and purified via crystallization. Yield = 95 mg, 42%. Purity = 96%. TLC Rf (free base) = 0.35 (9:1 CH2Cl2:MeOH:1% NH4OH(aq)); 1H NMR (600 MHz, DMSO-d6) δ 7.26 (d, 1H, J = 3.1 Hz), 7.10 (d, 1H, J = 7.8 Hz), 6.90 (t, 1H, J = 7.8 Hz), 6.66 (d, 1H, J = 7.8 Hz), 6.60 (s, 2H), 6.35 (d, 1H, J = 3.1 Hz), 4.49 (t, 2H, J = 7.0 Hz), 3.89 (s, 3H), 2.76 (t, 2H, J = 7.0 Hz), 2.32 (s, 6H) ppm; 13C NMR (150 MHz, DMSO-d6) δ 166.30, 146.96, 134.17, 130.46, 129.79, 124.94, 119.64, 113.33, 102.42, 100.93, 59.61, 55.32, 45.63, 44.63 ppm; IR (diamond, ATR) ν 2929, 2455, 1712, 1644 cm−1. LRMS (ES+) calcd for C13H18N2O+ 219.15, found 220.33 (MH+); MP = 140–145°C.
2-(5-methoxy-1H-indol-1-yl)-N,N-dimethylethan-1-amine fumarate salt (1:1) (5).
Reaction performed using 5-methoxyindole (100 mg, 0.68 mmol) and purified via crystallization. Yield = 111 mg, 49%. Purity = 98%. TLC Rf (free base) = 0.66 (9:1 CH2Cl2:MeOH:1% NH4OH(aq)); 1H NMR (600 MHz, DMSO-d6) δ 7.38 (d, 1H, J = 8.9 Hz), 7.33 (d, 1H, J = 3.0 Hz), 7.04 (d, 1H, J = 2.4 Hz), 6.77 (dd, 1H, J = 8.9, 2.4 Hz), 6.6 (s, 2H), 6.33 (d, 1H, J = 3.0 Hz), 4.29 (t, 2H, J = 6.8 Hz), 3.74 (s, 3H), 2.79 (t, 2H, J = 6.8 Hz ), 2.30 (s, 6H) ppm; 13C NMR (150 MHz, DMSO-d6) δ 166.58, 153.42, 134.32, 130.90, 129.20, 128.48, 111.12, 110.39, 102.13, 100.34, 57.68, 55.30, 44.44, 42.91 ppm; IR (diamond, ATR) ν 3035, 2923, 2446, 1715 cm−1. LRMS (ES+) calcd for C13H18N2O+ 219.15, found 220.19 (MH+); MP = 140–142°C.
2-(−6-methoxy-1H-indol-1-yl)-N,N-dimethylethan-1-amine (6).
Reaction performed using 6-methoxyindole (147 mg, 1.0 mmol) and purified via chromatography. Yield = 148 mg, 68%. Purity >99%. TLC Rf (free base) = 0.32 (9:1 CH2Cl2:MeOH:1% NH4OH(aq)); 1H NMR (600 MHz, CDCl3) δ 7.49 (d, 1H, J = 7.8 Hz), 7.02 (d, 1H, J = 8.2 Hz), 6.82 (s, 2H), 6.78 (dd, 1H, J = 7.5 Hz), 6.42 (d, 1H, J = 7.8 Hz), 4.17 (t, 2H, J = 7.0 Hz), 2.31 (s, 3H), 2.69 (t, 2H, J = 7.0 Hz ), 2.31 (s, 6H) ppm; 13C NMR (150 MHz, CDCl3) δ 156.30, 136.77, 127.04, 123.00, 121.67, 109.22, 101.31, 93.14, 59.00, 55.91, 45.94, 44.91 ppm; IR (diamond, ATR) ν 2940, 2859, 2769, 1602 cm−1. LRMS (ES+) calcd for C13H18N2O+ 219.15, found 220.33 (MH+). The freebase was used for dendritogenesis assays.
2-(−6-methoxy-1H-indol-1-yl)-N,N-dimethylethan-1-amine oxalate (6).
Reaction performed using 6-methoxyindole (250 mg, 1.7 mmol) and purified via crystallization. Yield = 221 mg, 42%. Purity = 98%. 1H NMR (600 MHz, CD3OD) δ 7.42 (d, 1H, J = 8.5 Hz), 7.16 (d, 1H, J = 2.9 Hz), 7.02 (s, 2H), 6.74 (d, 1H, J = 8.5 Hz), 6.44 (d, 1H, J = 2.9 Hz), 4.58 (t, 2H, J = 6.8 Hz), 3.87 (s, 3H), 3.56 (t, 2H, J = 7.0 Hz ), 2.86 (s, 6H) ppm; 13C NMR (150 MHz, CD3OD) δ 166.72, 158.20, 138.02, 127.52, 124.45, 122.61, 111.06, 103.58, 93.83, 57.22, 56.19, 44.04, 42.17 ppm; IR (diamond, ATR) ν 3129, 3014, 2641, 1727 cm−1. LRMS (ES+) calcd for C13H18N2O+ 219.15, found 220.05 (MH+). MP = 165–167°C. The oxalate salt was used for HTR assays.
2-(7-methoxy-1H-indol-1-yl)-N,N-dimethylethan-1-amine fumarate salt (1:1) (7).
Reaction performed using 7-methoxyindole (100 mg, 0.68 mmol) and purified via crystallization. Yield = 162 mg, 72%. Purity >99%. TLC Rf (free base) = 0.44 (9:1 CH2Cl2:MeOH:1% NH4OH(aq)); 1H NMR (600 MHz, DMSO-d6) δ 7.26 (d, 1H, J = 3.1 Hz), 7.09–7.03 (m, 2H), 6.60 (s, 2H), 6.52 (dd, 1H, J = 6.1, 1.3 Hz), 6.41 (dd, 1H, J = 3.0, 0.7 Hz), 4.29 (t, 2H, J = 6.8 Hz), 3.86 (s, 3H), 2.75 (t, 2H, J = 6.8 Hz), 2.29 (s, 6H) ppm: 13C NMR (150 MHz, DMSO-d6) δ 166.46, 152.80, 137.02, 134.24, 127.14, 121.98, 118.38, 103.09, 99.16, 97.88, 57.83, 54.89, 44.60, 43.15 ppm; IR (diamond, ATR) ν 3435, 3034, 2653, 1705 cm−1. LRMS (ES+) calcd for C13H18N2O+ 219.15, found 220.40 (MH+); Mp 120–123°C.
Benzyloxy indoles.
The 4-, 5-, 6-, and 7-OBn indoles were synthesized using methods published previously.38
2-(4-(benzyloxy)-1H-indol-1-yl)-N,N-dimethylethan-1-amine fumarate salt (1:1) (8).
Reaction performed using 4-benzyloxyindole (200mg, 0.89 mmol) and purified via crystallization. Yield = 120 mg, 46%. Purity >99%. TLC Rf (free base) = 0.42 (9:1 CH2Cl2:MeOH:1% NH4OH(aq)); 1H NMR (400 MHz, CD3OD) δ 7.49 (d, 2H, J = 7.5 Hz), 7.37 (t, 2H, J = 7.5 Hz), 7.31 (t, 1H, J = 7.5 Hz), 7.20 (d, 1H, J = 3.26 Hz), 7.12 (m, 2H), 6.72 (s, 1H), 6.66 (m, 1H), 5.22 (s, 2H), 4.57 (t, 2H, J = 6.7 Hz), 3.50 (t, 2H, J = 6.7 Hz), 2.81 (s, 6H) ppm; 13C NMR (100 MHz, CD3OD) δ 169.90, 153.99, 139.05, 138.85, 135.75, 129.47, 128.78, 128.47, 127.21, 124.18, 121.16, 103.87, 102.67, 101.07, 70.95, 57.32, 43.90, 42.46 ppm; IR (diamond, ATR) ν 2918, 2493, 1701, 1639 cm−1. LRMS (ES+) calcd for C19H22N2O+ 294.17, found 295.24 (MH+); MP = 145–150°C.
2-(5-(benzyloxy)-1H-indol-1-yl)-N,N-dimethylethan-1-amine fumarate salt (1:1) (9).
Reaction performed using 5-benzyloxyindole (287 mg, 1.3 mmol) and purified via crystallization. Yield = 133 mg, 25%. Purity >99%. TLC Rf (free base) = 0.47 (9:1 CH2Cl2:MeOH:1% NH4OH(aq)); 1H NMR (400 MHz, CD3OD) δ 7.44 (d, 2H, J = 7.5 Hz), 7.36 (m, 3H), 7.29 (d, 2H, J = 7.5 Hz), 7.24 (d, 1H, J = 3.2Hz), 7.15 (d, 1H, J = 2.4 Hz), 6.95 (dd, 1H, J = 2.4, 8.9 Hz), 6.72 (s, 2H), 6.43 (d, 1H, J = 3.2 Hz), 5.07 (s, 2H), 4.54 (t, 2H, J = 6.8 Hz), 3.45 (t, 2H, J = 6.8 Hz), 2.78 (s, 6H) ppm; 13C NMR (200 MHz, CD3OD) δ 171.06, 154.78, 139.26, 136.11, 132.77, 130.89, 130.81, 129.44, 128.73, 128.61, 113.97, 110.95, 105.71, 103.24, 71.86, 57.66, 44.16, 42.77 ppm; IR (diamond, ATR) ν 2916, 2516, 1698, 1639 cm−1. LRMS (ES+) calcd for C19H22N2O+ 294.17, found 295.17 (MH+); MP = 133–135°C.
2-(6-(benzyloxy)-1H-indol-1-yl)-N,N-dimethylethan-1-amine (10).
Reaction performed using 6-benzyloxyindole (370 mg, 1.7 mmol) and purified via chromatogrpahy. Yield = 184 mg, 38%. Purity >97%. TLC Rf (free base) = 0.45 (9:1 CH2Cl2:MeOH:1% NH4OH(aq)); 1H NMR (600 MHz, CD3OD) δ 7.47 (d, 2H, J = 7.4 Hz), 7.40 (d, 2H, J = 8.6 Hz), 7.37 (t, 1H J = 7.4 Hz), 7.30 (t, 1H, J = 7.4 Hz), 7.08 (d, 1H, J = 3.8 Hz), 6.96 (s, 1H), 6.78 (d, 1H, J = 8.6 Hz), 6.35 (d, 1H, J = 3.8 Hz), 5.15 (s, 2H), 4.22 (t, 2H, J = 7.3 Hz), 2.66 (t, 2H, J = 7.3 Hz), 2.26 (s, 6H) ppm; 13C NMR (150 MHz, CDCl3) δ 156.39, 137.60, 136.66, 128.69, 127.97, 127.67, 127.22, 123.24, 121.68, 109.95, 101.31, 94.71, 70.87, 58.92, 45.91, 44.90 ppm; IR (diamond, ATR) ν 3030, 2952, 2768, 1621 cm−1. LRMS (ES+) calcd for C19H22N2O+ 294.17, found 295.10 (MH+).
2-(7-(benzyloxy)-1H-indol-1-yl)-N,N-dimethylethan-1-amine (11).
Reaction performed using 7-benzyloxyindole (119 mg, 0.53 mmol) and purified via chromatography. Yield = 51 mg, 23%. Purity >99%. TLC Rf (free base) = 0.48 (9:1 CH2Cl2:MeOH:1% NH4OH(aq)); 1H NMR (600 MHz, CDCl3) δ 7.49 (d, 2H, J = 7.4 Hz), 7.40 (t, 2H, J = 7.4 Hz), 7.34 (t, 1H, J = 7.4 Hz), 7.22 (d, 1H, J = 7.9 Hz), 7.01 (d, 1H, J = 3.0 Hz), 6.97 (t, 1H, J = 7.8 Hz), 6.70 (d, 1H, J = 7.8 Hz), 5.19 (s, 2H), 4.45 (t, 2H, J = 7.4 Hz), 2.62 (t, 2H, J = 7.4 Hz), 2.09 (s, 6H) ppm; 13C NMR (150 MHz, CDCl3) δ 150.59, 146.71, 137.11, 131.25, 129.45, 128.75, 128.24, 128.15, 119.82, 114.16, 103.35, 101.60, 70.55, 61.02, 47.54, 45.64 ppm; IR (diamond, ATR) ν 2940, 2821, 1575, 1439 cm−1. LRMS (ES+) calcd for C19H22N2O+ 294.17, found 295.24 (MH+).
2-(4-fluoro-1H-indol-1-yl)-N,N-dimethylethan-1-amine fumarate salt (1:1) (12).
Reaction performed using 4-fluoroindole (135 mg, 1.0 mmol) and purified via crystallization. Yield = 164 mg, 51%. Purity >99%. TLC Rf (free base) = 0.39 (9:1 CH2Cl2:MeOH:1% NH4OH(aq)); 1H NMR (400 MHz, DMSO-d6) δ 7.44 (d, 1H, J = 2.3 Hz), 7.35 (d, 2H, J = 8.3 Hz), 7.10 (dd, 2H, J = 7.4, 7.25 Hz), 6.79 (t, 2H, J = 9.5 Hz), 6.60 (s, 2H), 6.49 (d, 2H, J = 2.3 Hz), 4.32 (t, 2H, J = 6.7 Hz), 2.74 (t, 2H, J = 6.8 Hz ), 2.28 (s, 6H) ppm; 13C NMR (150 MHz, DMSO-d6) δ 166.37, 156.67, 154.34, 138.56, 138.44, 134.21, 129.35, 121.63, 121.55, 116.74, 116.51, 106.48, 106.45, 103.70, 103.51, 96.24, 57.90, 44.73, 43.40 ppm; IR (diamond, ATR) ν 3123, 2389, 1702, 1660 cm−1. LRMS (ES+) calcd for C12H16FN2+ 207.13, found 208.32 (MH+); MP = 145–149°C.
2-(5-fluoro-1H-indol-1-yl)-N,N-dimethylethan-1-amine fumarate salt (1:1) (13).
Reaction performed using 5-fluoroindole (135 mg, 1.0 mmol) and purified via crystallization. Yield = 145 mg, 45%. Purity >99%. TLC Rf (free base) = 0.35 (9:1 CH2Cl2:MeOH:1% NH4OH(aq)); 1H NMR (400 MHz, DMSO-d6) δ 7.50 (dd, 1H, J = 4.5, 4.3 Hz), 7.46 (d, 1H, J = 2.1 Hz), 7.29 (d, 1H, J = 9.5 Hz), 6.97 (t, 1H, J = 9.5 Hz), 6.60 (s, 2H), 6.41 (d, 1H, J = 2.1 Hz), 4.32 (t, 2H, J = 6.7 Hz), 2.79 (t, 2H, J = 6.7 Hz ), 2.31 (s, 6H) ppm; 13C NMR (150 MHz, DMSO-d6) δ 166.59, 159.61, 158.06, 135.77, 135.69, 134.33, 129.54, 129.51, 124.72, 121.39, 121.32, 107.48, 107.32, 100.97, 96.32, 96.15, 57.66, 44.58, 43.00 ppm; IR (diamond, ATR) ν 3036, 2049, 1723, 1663 cm−1. LRMS (ES+) calcd for C12H16FN2+ 207.13, found 207.40 (MH+); MP = 145–148°C.
2-(6-fluoro-1H-indol-1-yl)-N,N-dimethylethan-1-amine fumarate salt (1:1) (14).
Reaction performed using 6-fluoroindole (100 mg, 0.739 mmol) and purified via crystallization. Yield = 145 mg, 61%. Purity = 97%. TLC Rf (free base) = 0.45 (9:1 CH2Cl2:MeOH:1% NH4OH(aq)); 1H NMR (600 MHz, DMSO-d6) δ 7.52 (dd, 1H, J = 7.0, 3.0 Hz), 7.39–7.37 (m, 2H), 6.88–6.85 (m, 1H), 6.59 (s, 2H), 6.44 (d, 1H, J = 3.1 Hz), 4.29 (t, 2H, J = 6.8 Hz), 2.77 (t, 2H, J = 6.8 Hz ), 2.30 (s, 6H) ppm; 13C NMR (150 MHz, DMSO-d6) δ 166.59, 159.61, 158.06, 136.16, 134.75, 129.95, 125.14, 121.78, 107.91, 107.74, 101.39, 96.74, 96.57, 57.66, 44.58, 43.00 ppm; IR (diamond, ATR) ν 3058, 2385, 1698, 1634 cm−1. LRMS (ES+) calcd for C12H16FN2+ 207.13, found 208.39 (MH+); MP = 141–147°C.
2-(4-fluoro-1H-indol-1-yl)-N,N-dimethylethan-1-amine fumarate salt (1:1) (15).
Reaction performed using 7-fluoroindole (135 mg, 1.0 mmol) and purified via crystallization. Yield = 172mg, 53%. Purity = 98%. TLC Rf (free base) = 0.45 (9:1 CH2Cl2:MeOH:1% NH4OH(aq)); 1H NMR (400 MHz, DMSO-d6) δ 7.41 (s, 1H), 7.35 (d, 1H, J = 7.6 Hz), 6.98–6.88 (m, 2H), 6.61 (s, 2H), 6.48 (s, 1H,), 4.37 (t, 2H, J = 6.7 Hz), 2.69 (t, 2H, J = 6.7 Hz), 2.23 (s, 6H) ppm; 13C NMR (100 MHz, CD3OD) δ 169.37, 134.60, 133.26, 129.51, 119.90, 119.46, 116.82, 116.79, 107.06, 106.88, 102.78, 57.61, 57.61, 43.71, 43.67, 42.90 ppm; IR (diamond, ATR) ν 3040, 2429, 1718. 1661 cm−1. LRMS (ES+) calcd for C12H16FN2+ 207.13, found 207.33 (MH+); MP = 168–170°C.
N,N-dimethyl-2-(2-methyl-1H-indol-1-yl)ethan-1-amine fumarate salt (1:1) (16).
Reaction performed using 2-methylindole (100 mg, 0.76 mmol) and purified via crystallization. Yield (172 mg, 71%). Purity >99%. TLC Rf (free base) = 0.47 (9:1 CH2Cl2:MeOH:1% NH4OH(aq)); 1H NMR (400 MHz, DMSO-d6) δ 7.41 (d, 1H, J = 7.8 Hz), 7.37 (d, 1H, J = 7.8 Hz), 7.06 (t, 1H, J = 7.8 Hz), 6.96 (t, 1H, J = 7.8 Hz), 6.61 (s, 2H), 6.20 (d, J = 1.3 Hz, 1H), 4.24 (t, 2H, J = 7.0 Hz), 2.65 (t, 2H, J = 7.0 Hz), 2.42 (s, 3H), 2.31 (s, 6H) ppm; 13C NMR (150 MHz, DMSO-d6) 167.03, 137.08, 136.72, 134.75, 128.09, 120.58, 119.65, 119.62, 119.40, 109.61, 100.14, 57.82, 45.18, 12.76 ppm; IR (diamond, ATR) ν 3040, 2489, 1700, 1606 cm−1. LRMS (ES+) calcd for C13H18N2+ 203.15, found 204.43(MH+); MP = 131–133°C.
2-(5,6-difluoro-1H-indol-1-yl)-N,N-dimethylethan-1-amine fumarate salt (1:1) (17).
Reaction performed using 5,6-difluoroindole (153 mg, 1.0 mmol) and purified via crystallization. Yield = 147 mg, 43%. Purity = 98%. TLC Rf (free base) = 0.35 (9:1 CH2Cl2:MeOH:1% NH4OH(aq)); 1H NMR (400 MHz, DMSO-d6) δ 7.64 (dd, 1H, J = 7.0, 4.7 Hz), 7.51 (dd, 1H, J = 8.5, 2.1 Hz), 7.45 (d, 1H, J = 2.1 Hz), 6.60 (s, 2H), 6.43 (d, 1H, J = 2.1 Hz), 4.28 (t, 2H, J = 6.5 Hz), 2.73 (t, 2H, J = 6.5 Hz ), 2.27 (s, 6H) ppm; 13C NMR (150 MHz, DMSO-d6) δ 166.37, 134.21, 131.09, 130.99, 134.70, 130.67, 123.21, 123.12, 106.86, 106.67, 100.98, 100.94, 98.30, 98.08, 57.86, 44.71, 43.36 ppm; IR (diamond, ATR) ν 3051, 2392, 1712, 1658 cm−1. LRMS (ES+) calcd for C12H16F2N2+ 224.11, found 225.28 (MH+); MP = 162–165°C.
2-(4,6-difluoro-1H-indol-1-yl)-N,N-dimethylethan-1-amine fumarate salt (1:1) (18).
Reaction performed using 4,6-difluoroindole (153 mg, 1.0 mmol) and purified via crystallization. Yield = 265 mg, 78%. Purity = >99%. TLC Rf (free base) = 0.35 (9:1 CH2Cl2:MeOH:1% NH4OH(aq)); 1H NMR (400 MHz, DMSO-d6) δ 7.44 (d, 1H, J = 2.8 Hz), 7.32 (d, 1H, J = 10.1 Hz), 6.83 (t, 1H, J = 10.1 Hz), 6.60 (s, 2H), 6.49 (d, 1H, J = 2.8 Hz), 4.31 (t, 2H, J = 6.6 Hz), 2.78 (t, 2H, J = 6.6 Hz ), 2.31 (s, 6H) ppm; 13C NMR (150 MHz, DMSO-d6) δ 166.55, 159.33, 159.21, 156.99, 156.87, 156.03, 155.88, 153.58, 153.43, 137.50, 137.36, 137.22, 134.29, 129.82, 129.79, 113.42, 113.20, 96.61, 94.53, 94.30, 94.24, 94.00, 93.32, 93.28, 93.06, 93.02, 57.49, 44.49, 43.23 ppm; IR (diamond, ATR) ν 3026, 2398, 1706, 1640 cm−1. LRMS (ES+) calcd for C12H16F2N2+ 224.11, found 225.28 (MH+); MP = 141–145°C.
N,N-dimethyl-2-(6-nitro-1H-indol-1-yl)ethan-1-amine fumarate salt (1:1) (19).
Reaction performed using 6-nitroindole (43.6 mg, 0.269 mmol) and purified via crystallization. Yield = 52 mg, 55%. Purity >96%. TLC Rf (free base) = 0.48 (9:1 CH2Cl2:MeOH:1% NH4OH(aq)); 1H NMR (400 MHz, DMSO-d6) δ 8.54 (d, 1H, J = 2.0 Hz), 7.90 (dd, 1H, J = 8.8, 2.0 Hz), 7.82 (d, 1H, J = 3.0 Hz), 7.72 (d, 1H, J = 8.8 Hz), 7.74 (s, 1H), 6.65 (d, 1H, J = 3.0 Hz), 6.60 (s, 2H), 4.44 (t, 2H, J = 6.3 Hz), 2.68 (t, 2H, J = 6.3 Hz), 2.23 (s, 6H) ppm; 13C NMR (600 MHz, DMSO-d6) δ 166.66, 136.18, 134.76, 134.59, 133.41, 121.04, 114.52, 107.53, 102.25, 58.98, 45.50, 44.18 ppm; IR (diamond, ATR) ν 3048, 2922, 1704, 1607 cm−1. LRMS (ES+) calcd for C12H16N3O2+ 233.12, found 234.25 (MH+); MP = 159–164°C.
2-(5-bromo-1H-indol-1-yl)-N,N-dimethylethan-1-amine fumarate salt (1:1) (20).
Reaction performed using 5-bromoindole (56.1 mg, 0.281 mmol) and purified via crystallization. Yield = 60 mg, 55%. Purity >99%. TLC Rf (free base) = 0.49 (9:1 CH2Cl2:MeOH:1% NH4OH(aq)); 1H NMR (600 MHz, DMSO-d6) δ 7.72 (d, J = 1.9 Hz, 1H), 7.48 (d, 1H, J = 8.7 Hz), 7.44 (d, 1H, J = 3.1 Hz), 7.23 (dd, 1H, J = 8.7, 1.9 Hz), 6.60 (s, 3H), 6.41 (d, 1H, J = 3.1 Hz), 4.27 (t, 2H, J = 6.6 Hz), 2.67 (t, 2H, J = 6.6 Hz), 2.22 (s, 6H) ppm; 13C NMR (600 MHz, DMSO-d6) δ 166.24, 134.47, 134.15, 130.46, 129.89, 123.81, 122.54, 111.87, 111.56, 100.28, 58.09, 44.86, 43.32 ppm; IR (diamond, ATR) ν 2959, 2443, 1705, 1661 cm−1. LRMS (ES+) calcd for C12H16BrN2+ 266.04, found 267.26 (MH+); MP = 140–142°C.
2-(1-(2-(dimethylamino)ethyl)-1H-indol-3-yl)-N,N-dimethyl-2-oxoacetamide (21).
Reaction performed using 2-(1H-indol-3-yl)-N,N-dimethyl-2-oxoacetamide (synthesized using the method developed by Speeter et. al.)18 (200 mg, 0.92 mmol) and purified via chromatography. Yield = 92 mg, 35%. Purity = 98%. TLC Rf (free base) = 0.38 (9:1 CH2Cl2:MeOH:1% NH4OH(aq)); 1H NMR (600 MHz, CDCl3) δ 8.32 (s, 1H), 7.92 (s, 1H), 7.37–7.29 (m, 3H), 4.21 (t, 2H, J = 7.0 Hz), 3.08 (s, 3H), 3.04 (s, 3H), 2.71 (t, 2H, J = 7.0 Hz ), 2.27 (s, 6H) ppm; 13C NMR (150 MHz, CDCl3) δ 185.64, 167.70, 138.82, 137.01, 126.36, 124.00, 123.28, 122.48, 113.45, 110.06, 58.40, 45.68 45.42, 37.59, 34.51 ppm; IR (diamond, ATR) ν 2981, 1734 1631, 1525 cm−1. LRMS (ES+) calcd for C16H21N3O2+ 287.16, found 288.25 (MH+).
2-(1H-benzo[d]imidazol-1-yl)-N,N-dimethylethan-1-amine fumarate salt (1:1) (22).
Reaction performed using benzimidazole (200mg, 1.6 mmol) and purified via crystallization. Yield = 218 mg, 45%. Purity = 98%. TLC Rf (free base) = 0.42 (9:1 CH2Cl2:MeOH:1% NH4OH(aq)); 1H NMR (400 MHz, DMSO-d6) δ 8.23 (s, 1H), 7.71–7.56 (m, 2H), 7.34–7.10 (m, 2H), 6.61 (s, 2H), 4.41 (t, 2H, J = 6.5 Hz), 2.83 (dd, 2H, J = 7.0, 6.0 Hz), 2.32 (s, 6H) ppm; 13C NMR (100 MHz, CD3OD) δ 170.14, 144.78, 143.90, 135.81, 134.62, 124.80, 124.04, 120.36, 111.29, 57.46, 44.39, 41.80 ppm; IR (diamond, ATR) ν 3054, 2384, 1707, 1654 cm−1. LRMS (ES+) calcd for C11H15N3+ 189.13, found 190.23 (MH+); MP = 171–178°C.
N,N-dimethyl-2-(1H-pyrrol-1-yl)ethan-1-amine fumarate salt (1:1) (23).
Reaction performed using pyrrole (0.103 ml, 1.5 mmol) and purified via crystallization. Yield = 126 mg, 33%. Purity >99%. TLC Rf (free base) = 0.45 (9:1 CH2Cl2:MeOH:1% NH4OH(aq)); 1H NMR (600 MHz, CD3OD) δ 6.80 (t, 2H, J = 2.2, Hz), 6.72 (s, 2H), 6.14 (t, 2H. J = 2.2, Hz), 4.34 (t, 2H, J = 6.4 Hz), 3.50 (dd, 2H, J = 6.4 Hz), 2.78 (s, 6H) ppm; 13C NMR (100 MHz, CD3OD) δ 170.26, 136.21, 122.18, 110.89, 59.35, 45.83, 44.45 ppm; IR (diamond, ATR) ν 2998, 2532, 1662, 1421 cm−1. LRMS (ES+) calcd for C8H14N2+ 138.12, found 139.29 (MH+); MP = 174–180°C.
2-(9H-carbazol-9-yl)-N,N-dimethylethan-1-amine fumarate salt (1:1) (24).
Reaction performed using carbazole (100 mg, 0.57 mmol) and purified via crystallization. Yield = 102 mg, 51%. Purity >99%. TLC Rf (free base) = 0.42 (9:1 CH2Cl2:MeOH:1% NH4OH(aq)); 1H NMR (600 MHz, DMSO-d6) δ 8.15 (d, 2H, J = 7.8 Hz), 7.61 (d, 2H, J = 8.2 Hz), 7.48–7.44 (m, 2H), 7.23–7.19 (m, 2H), 6.61 (s, 2H), 4.52 (t, 2H, J = 7.0 Hz), 2.73 (t, 2H, J = 7.0 Hz ), 2.31 (s, 6H) ppm; 13C NMR (100 MHz, DMSO-d6) δ 169.78, 139.86, 134.76, 125.83, 123.17, 119.99, 119.39, 108.34, 54.39, 42.61, 37.85 ppm; IR (diamond, ATR) ν 3053, 2405, 1720, 1660 cm−1. LRMS (ES+) calcd for C16H18N2+ 238.15, found 239.34 (MH+); MP = 182–184°C.
1-isopentyl-1H-indole (25).
Reaction performed using indole (100 mg, 0.85 mmol) and 1-chloro-3-methylbutane (0.11 mL mg, 0.94 mmol, 1.1 equiv) and purified via chromatography (4:1 hexanes:EtOAc). Yield = 85 mg, 53%. Purity = 97%. TLC Rf = 0.70 (7:3 hexanes:EtOAc); 1H NMR (600 MHz, CDCl3) δ 7.65 (d, 1H, J = 8.0 Hz), 7.36 (d, 1H, J = 8.0 Hz), 7.22 (t, 1H, J = 6.9 Hz), 7.11 (m, 2H), 6.49 (d, 1H, J = 3.1 Hz), 4.15 (t, 2H, J = 7.5 Hz), 1.74 (dd, 2H, J = 6.7 Hz), 1.62 (quint, 2H, J = 6.7 Hz ), 0.98 (d, 1H, J = 6.7 Hz) ppm; 13C NMR (150 MHz, CDCl3) δ 135.78, 128.46, 127.58, 121.20, 120.83, 119.05, 109.28, 100.78, 44.44, 40.72, 38.92, 25.61, 22.37 ppm; IR (diamond, ATR) ν 3054, 2955, 2927, 2869 cm−1. LRMS (ES+) calcd for C13H17N+ 187.14 found 188.39 (MH+).
3-(1H-indol-1-yl)-N,N-dimethylpropan-1-amine fumarate salt (1:1) (26).
Reaction performed using indole (100 mg, 0.85 mmol) and 3-chloro-N,N-dimethylpropan-1-amine (160 mg, 0.98 mmol, 1.1 equiv) and purified via crystallization Yield = 107 mg, 48%. Purity = 98%. TLC Rf (free base) = 0.38 (9:1 CH2Cl2:MeOH:1% NH4OH(aq)); 1H NMR (600 MHz, DMSO-d6) δ 7.54 (d, 1H, J = 7.8 Hz), 7.48 (d, 1H, J = 7.8 Hz), 7.36 (d, 1H, J = 3.1 Hz), 7.13 (td, 1H, J = 7.4, 1.0 Hz), 7.01 (td, 1H, J = 7.4, 1.0 Hz), 6.55 (s, 2H), 6.43 (dd, 1H, J = 3.1, 1.0 Hz), 4.21 (t, 2H, J = 6.8 Hz), 2.56 (t, 2H, J = 7.4 Hz), 2.40 (s, 6H), 2.00 (tt, 2H, J = 7.4, 6.8 Hz) ppm; 13C NMR (150 MHz, DMSO-d6) δ 167.19, 135.60, 134.62, 128.49, 128.07, 120.99, 120.41, 118.90, 109.67, 100.61, 54.88, 43.41, 43.07, 26.26 ppm; IR (diamond, ATR) ν 3435, 3034, 2653, 1705 cm−1. LRMS (ES+) calcd for C13H18N2+ 203.15, found 204.36 (MH+); MP = 129–131°C.
Tert-butyl (2-(1H-indol-3-yl)ethyl)carbamate.
To an ice-cold solution of tryptamine (0.50 g, 3.1 mmol) and triethylamine (0.68 mL, 9.4 mmol, 3 equiv) in CH2Cl2 (44 mL) was added Boc2O (0.77 g, 3.7 mmol, 1.2 equiv). The reaction was warmed to room temperature, stirred overnight, and then quenched with H2O (200 mL). The organic phase was separated and the aqueous phase extracted with CH2Cl2 (2 × 50 mL). The organic extracts were combined, dried over Na2SO4, filtered, and concentrated under reduced pressure to afford an oil that was purified by chromatography on silica gel (8:2 hexanes:EtOAc); white solid (0.69 g, 85%).
N-methyl-2-(1-methyl-1H-indol-3-yl)ethan-1-amine.
To an ice-cold solution of sodium hydride (0.23 g, 5.8 mmol, 2.2 equiv) in DMF (3 mL) was added tert-butyl (2-(1H-indol-3-yl)ethyl)carbamate (0.69 g, 2.6 mmol) in DMF (3 mL). The reaction mixture was allowed to stir at room temperature before being cooled to 0 °C. Methyl iodide (0.4 mL, 5.8 mmol, 2.2 equiv) was added dropwise. The reaction was stirred at room temperature for 20 h. Next, the reaction was cooled to 0°C, quenched with TFA (2 mL), and stirred for 30 min. The mixture was diluted with 1.0 M NaOH(aq) (600 mL) and extracted with CH2Cl2 (3 × 75 mL). The organic phases were combined, dried over Na2SO4, filtered, and concentrated under reduced pressure to afford an oil that was used without further purification (0.45 g, 90%).
N,N-dimethyl-2-(1-methyl-1H-indol-3-yl)ethan-1-amine fumarate salt (1:1) (1-Me-DMT, 27).
To an ice-cold solution of N-methyl-2-(1-methyl-1H-indol-3-yl)ethan-1-amine (0.14 g, 0.70 mmol) and glacial acetic acid (0.22 mL, 11 mmol, 5.0 equiv) in MeOH (12 mL) was added sodium cyanoborohydride (0.10 g, 1.6 mmol, 2.1 equiv) followed by 37% formaldehyde(aq) (0.16 mL, 1.9 mmol, 2.6 equiv). The reaction was stirred at room temperature for 5 h before being concentrated under reduced pressure. The unpurified material was then diluted with CH2Cl2 (50 mL) and 1 M NaOH(aq) (100 mL). The phases were separated, and the aqueous phase was extracted with CH2Cl2 (2 × 50 mL). The organic extracts were combined, dried over Na2SO4, filtered, and concentrated under reduced pressure. The unpurified material was dissolved in acetone (5 mL) and added to a boiling solution of fumaric acid (0.088 g, 1 mmol, 1 equiv) in acetone (20 mL). A precipitate formed immediately, and the solution was cooled to room temperature prior to being filtered. The resulting white solid was dried under reduced pressure to yield the pure compound as the fumarate salt (1:1). Yield = 0.108 g, 65%. Purity >99%. TLC Rf (free base) = 0.19 (9:1 CH2Cl2:MeOH:1% NH4OH(aq)); 1H NMR (600 MHz, CD3OD) δ 7.60 (d, 1H, J = 8.0 Hz), 7.37 (d, 1H, J = 8.0 Hz), 7.20 (t, 1H, J = 8.0 Hz), 7.14 (s, 1H), 7.09 (t, 1H, J = 8.0 Hz), 6.69 (s, 2H), 3.78 (s, 3H), 3.42 (t, 2H, J = 7.8 Hz), 3.20 (t, 2H, J = 7.8 Hz), 2.91 (s, 6H) ppm; 13C NMR (150 MHz, CD3OD) δ 171.44, 138.80, 136.23, 128.67, 128.59, 122.95, 120.20, 119.27, 110.53, 109.11, 59.12, 43.41, 32.77, 21.72 ppm; IR (diamond, ATR) ν 3435, 3034, 2653, 1705 cm−1 LRMS (ES+) m/z calcd for C13H18N2+ 202.15, found 203.37 (MH+); MP = 167−170°C.
2-(5-methoxy-1H-indol-3-yl)-N,N-dimethylethan-1-amine fumarate salt (2:1) (5-MeO-DMT, 28).
To an ice-cold solution of 5-methoxytryptamine (0.50 g, 2.2 mmol) and glacial acetic acid (0.60 mL, 11 mmol, 5.0 equiv) in MeOH (44 mL) was added sodium cyanoborohydride (0.305 g, 4.8 mmol, 2.2 equiv) followed by 37% formaldehyde(aq) (0.46 mL, 5.7mmol, 2.6 equiv). The reaction was stirred at room temperature for 5 h before being concentrated under reduced pressure. The residue was diluted with CH2Cl2 (50 mL) and 1 M NaOH(aq) (100 mL). The phases were separated and the aqueous phase was extracted with CH2Cl2 (2 × 50 mL). The organic extracts were combined, dried over Na2SO4, filtered, and concentrated under reduced pressure. The unpurified material was dissolved in acetone (5 mL) and added to a boiling solution of fumaric acid (0.26 g, 2.2 mmol, 0.7 equiv) in acetone (35 mL). A precipitate formed immediately, and the solution was cooled to room temperature prior to being filtered. The resulting white solid was dried under reduced pressure to yield the pure compound as the fumarate salt (2:1). Yield = 0.49 g, 80%. Purity = 98%. TLC Rf (free base) = 0.20 (9:1 CH2Cl2:MeOH:1% NH4OH(aq)); 1H NMR (600 MHz, CD3OD) δ 7.24 (d, 1H, J = 8.8 Hz), 7.14 (s, 1H), 7.07 (s, 1H), 6.78 (d, 1H, J = 8.8 Hz), 6.70 (s, 1H), 3.83 (s, 3H), 3.29 (m, 2H), 3.13 (t, 2H, J = 7.9 Hz), 2.83 (s, 6H) ppm; 13C NMR (150 MHz, CD3OD) δ 174.36, 155.27, 137.09, 133.44, 128.49, 124.79, 113.20, 112.96, 110.05, 101.05, 59.25, 56.37, 43.56, 22.12 ppm; IR (diamond, ATR) ν 3436, 3034 2654, 1705 cm−1. LRMS (ES+) m/z calcd for C13H18N2O+ 218.14, found 219.34 (MH+); MP = 175−177°C.
2-(6-methoxy-1H-indol-3-yl)-N,N-dimethylethan-1-amine fumarate salt (2:1) (6-MeO-DMT, 30).
To an ice-cold solution of 6-methoxytryptamine (0.40 g, 2.1 mmol) and glacial acetic acid (0.60 mL, 10 mmol, 5.0 equiv) in MeOH (42 mL) was added sodium cyanoborohydride (0.29 g, 4.6 mmol, 2.2 equiv) followed by 37% formaldehyde(aq) (0.44 mL, 5.5mmol, 2.6 equiv). The reaction was stirred at room temperature for 5 h before being concentrated under reduced pressure. The residue was diluted with CH2Cl2 (45 mL) and 1 M NaOH(aq) (100 mL). The phases were separated and the aqueous phase was extracted with CH2Cl2 (2 × 45 mL). The organic extracts were combined, dried over Na2SO4, filtered, and concentrated under reduced pressure. The unpurified material was dissolved in acetone (5 mL) and added to a boiling solution of fumaric acid (0.26 g, 2.2 mmol, 0.7 equiv) in acetone (35 mL). A precipitate formed immediately, and the solution was cooled to room temperature prior to being filtered. The resulting white solid was dried under reduced pressure to yield the pure compound as the fumarate salt (2:1). Yield = 0.320 g, 55%. Purity = 95%. TLC Rf (free base) = 0.31 (9:1 CH2Cl2:MeOH:1% NH4OH(aq)); 1H NMR (600MHz, CD3OD) δ 7.44 (d, 1H, J = 8.6 Hz), 7.04 (s, 1H), 6.88 (s, 1H), 6.70 (m, 2H), 3.78 (s, 3H), 3.32 (t, 2H, J = 7.5 Hz), 3.12 (t, 2H, J = 7.5 Hz), 2.84 (s, 6H) ppm; 13C NMR (150 MHz, CD3OD) δ 173.79, 157.87, 138.97, 136.93, 122.89, 122.53, 119.64, 110.40, 109.99, 95.62, 59.12, 55.94, 43.36, 21.96 ppm; IR (diamond, ATR) ν 2915, 2836, 1691, 1559 cm−1. LRMS (ES+) m/z calcd for C13H18N2O+ 218.14, found 219.29 (MH+); MP = 173−176°C.
2-(1H-indol-1-yl)-N,N-dimethylacetamide (31).
To a solution of indole (117 mg, 1.0 mmol) in DMSO (2.5 mL, 0.4 M) was added 2-chloro-N,N-dimethylacetamide (0.11 mL, 1.1 mmol, 1.1 equiv), potassium iodide (182 mg, 1.1 mmol, 1.1 equiv), and potassium hydroxide pellets (280 mg, 5 mmol, 5.0 equiv). The reaction was stirred at room temperature for 24 h before being diluted with 1.0 M NaOH(aq). The aqueous phase was extracted three times with DCM. The organic extracts were combined, dried over Na2SO4, filtered, and concentrated under reduced pressure to yield an oil. The oil was purified via chromatography (3:2 hexanes:EtOAc). Yield = 175 mg, 57%. Purity = 99%. TLC Rf = 0.15 (3:2 hexanes:EtOAc); 1H NMR (600 MHz, CD3OD) δ 7.53 (d, 1H, J = 7.9 Hz), 7.27 (d, 1H, J = 7.9 Hz), 7.12 (m, 2H), 7.01 (t, 1H, J = 7.5, Hz), 6.46 (d, 1H, J = 2.9 Hz), 5.01 (s, 2H), 3.15 (s, 3H), 2.96 (s, 3H) ppm; 13C NMR (150 MHz, CDCl3) δ 167.4, 136.7, 128.7, 128.5, 122.0, 121.2, 119.8, 109.1, 102.4, 48.2, 36.7, 36.1 ppm; IR (diamond, ATR) ν 3021, 2922, 2877, 1648 cm−1. LRMS (ES+) calcd for C12H14N2O+ 202.11 found 203.17 (MH+); MP = 58–61°C.
Animals.
For the dendritogenesis experiments, timed pregnant Sprague Dawley rats were obtained from Charles River Laboratories (Wilmington, MA). For the head-twitch response assay, male and female C57BL/6J mice were obtained from Jackson Laboratory (Sacramento, C.A.). Mice were housed in a temperature and humidity-controlled room maintained on a 12-h light/dark cycle in groups of 4–5 (same sex). Animals weighed between 17 and 30 g at the time of the experiments. All experimental procedures involving rodents were approved by the UC Davis Institutional Animal Care and Use Committee (IACUC) and adhered to principles described in the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Zebrafish experiments were performed in accordance with established protocols approved by the UCSF IACUC and adhered to principles described in the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The University of California, Davis and the University of California, San Francisco are accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC).
Dendritogenesis Experiments.
Dendritogenesis experiments were performed following a previously published method with slight modifications.10 Neurons were plated in 96-well format (200 μL of media per well) at a density of approximately 15,000 cells/well in Neurobasal (Life Technologies) containing 1% penicillin-streptomycin, 10% heat-inactivated fetal bovine serum, and 0.5 mM glutamine. After 24 h, the medium was replaced with Neurobasal containing 1x B27 supplement (Life Technologies), 1% penicillin-streptomycin, 0.5 mM glutamine, and 12.5 μM glutamate. After 3 days in vitro (DIV3), the cells were treated with compounds. All compounds tested in the dendritogenesis assays were treated at 10 μM unless noted otherwise. Stock solutions of the compounds in DMSO were first diluted 100-fold in Neurobasal before an additional 10-fold dilution into each well (total dilution = 1:1000; 0.1% DMSO concentration). Treatments were randomized. After 1 h, the media was removed and replaced with new Neurobasal media containing 1x B27 supplement, 1% penicillin-streptomycin, 0.5 mM glutamine, and 12.5 μM glutamate. The cells were allowed to grow for an additional 71 h. At that time, neurons were fixed by removing 80% of the media and replacing it with a volume of 4% aqueous paraformaldehyde (Alfa Aesar) equal to 50% of the working volume of the well. Then, the cells were incubated at room temperature for 20 min before the fixative was aspirated and each well washed twice with DPBS. Cells were permeabilized using 0.2% Triton X-100 (ThermoFisher) in DPBS for 20 minutes at room temperature without shaking. Plates were blocked with antibody diluting buffer (ADB) containing 2% bovine serum albumin (BSA) in DPBS for 1 h at room temperature. Then, plates were incubated overnight at 4°C with gentle shaking in ADB containing a chicken anti-MAP2 antibody (1:10,000; EnCor, CPCA-MAP2). The next day, plates were washed three times with DPBS and once with 2% ADB in DPBS. Plates were incubated for 1 h at room temperature in ADB containing an anti-chicken IgG secondary antibody conjugated to Alexa Fluor 488 (Life Technologies, 1:500) and washed five times with DPBS. After the final wash, 100 μL of DPBS was added per well and imaged on an ImageXpress Micro XL High-Content Screening System (Molecular Devices, Sunnyvale,CA) with a 20x objective. Images were analyzed using ImageJ Fiji (version 1.51W). First, images corresponding to each treatment are sorted into individual folders that are then blinded for data analysis. Plate controls (both positive and negative) were used to ensure that the assay is working properly as well as to visually determine appropriate numerical values for brightness/contrast and thresholding to be applied universally to the remainder of the randomized images. Next, the brightness/contrast settings were applied, and approximately 1–2 individual pyramidal-like neurons per image (i.e., no bipolar neurons) were selected using the rectangular selection tool and saved as separate files. Neurons were selected that did not overlap extensively with other cells or extend far beyond the field of view. The threshold settings were then applied to the individual images. The paintbrush tool was used to eliminate artifacts and dendritic processes originating from adjacent neurons (cleanup phase). See Figure S2 for a visual explanation of how neurons were selected and processed for data analysis. Next, the point tool was used to select the center of the neuron, and the images were saved and processed using the following Sholl analysis batch macro:
run(“Sholl Analysis…”, “starting=0 ending=NaN radius_step=2 #_samples=1 integration=Mean enclosing=1 #_primary=4 infer fit linear polynomial=[Best fitting degree] most semi-log normalizer=Area create background=228 save do”);
Sholl analysis circle radii = 2 pixel increments = 0.67 μm. All images were taken and analyzed by an experimenter blinded to treatment conditions. The number of crossings for each neuron at each distinct radius was averaged to produce an average Sholl plot for each treatment. The Nmax values were simply determined by identifying the maximum of each plot. For each treatment, neurons were selected from at least 6 wells spread across 2 plates (9 sites/well × 3 wells/plate × 2 plates). Each plate was prepared using neurons obtained from independent pregnant dams).
Ketanserin Blocking Experiments.
For the ketanserin blocking experiments (Figure 9), a slightly modified method was employed. On DIV 3, neurons were first treated with ketanserin (10 μM) for 1 h followed by a 1 h incubation with drug (1 μM) and ketanserin (10 μM) (final concentration of DMSO = 0.2%). After 1 h, the media was removed and replaced with new Neurobasal media containing 1x B27 supplement, 1% penicillin-streptomycin, 0.5 mM glutamine, and 12.5 μM glutamate. The cells were allowed to grow for an additional 71 h before being fixed, stained, and imaged.
Zebrafish behavioral experiments.
At 7 days post-fertilization, healthy zebrafish larvae (wild-type Singapore strain) were sorted from unhealthy fish, and their mobility was reduced by transferring approximately 500 fish to a plate containing a 2:1 ratio of 4°C to room temperature egg water.39 Using a pipette, 8 larvae were carefully distributed into each well of a 96-well plate (GE Healthcare Life Sciences) in 300 μL aliquots. Plates were incubated at room temperature for 1 h, at which time the animals were mobile. Stock solutions of the compounds in DMSO (1.2 μL) were mixed into the well (0.4% DMSO concentration) and plates were incubated at room temperature for 1 h prior to behavioral experiments. DMSO (Sigma Aldrich 472301) and eugenol (100 μM; Tokyo Chemical Industries A0232) were used as vehicle and lethal controls, respectively. Treatments were spread across 7 plates (3 replicate wells per treatment condition, 3 lethal control wells, and 21 DMSO wells on each plate) and were randomized on the plates using a Biomek FXP liquid handler. Plates were recorded at approximately 30 min intervals between the hours of 16:00 and 20:00 on a single day.
After 1 h of incubation, treated plates were positioned in an automated behavioral instrument and acclimated in darkness for 5 min. The plates were illuminated from below with 760 nm light through an acrylic diffuser and recorded with an overhead PointGrey Grasshopper GS3-U3-41C6M-C camera (FLIR Integrated Imaging Solutions Inc, Richmond, BC, Canada) mounted to a telecentric lens (Opto Engineering) through an infrared filter (LEE Filters LE8744 polyester #87). The camera captured 1600×1068 8-bit-deep images at 100 Hz. Stimulus lights included 623 nm (DigiKey 1537–1041-ND), 525 nm (DigiKey 1537–1039-ND), 460 nm (DigiKey 1537–1037-ND), 400 nm (Mouser LZ4–40UB00–00U7), 355 nm (Mouser 416-LST101G01UV01), and 4000 K white (Mouser 416–0D0BN240E-SB01) LEDs, which were positioned overhead. Audio stimuli were presented through two 5 W transducers (Generic) with an APA150 150 W Power Amplifier. Two 36 V SparkFun Electronics push–pull solenoids were used to deliver tapping stimuli. All stimuli were verified with sensors. A pixel intensity threshold was set such that values below the threshold represented noise. Animal locomotion was calculated as the number of pixels that changed with intensity greater than or equal to that threshold. Vectors were interpolated using ns-resolved timestamps from the image sensor and aligned to the stimuli.
Distances from vehicle controls were estimated from the out-of-bag accuracy of random forest models trained to distinguish between the motion vectors of compound-treated and vehicle-treated wells. Models (12 total, 2,000 trees each) were trained and averaged per problem (compound–concentration pair), sampling without replacement to avoid class imbalance, but maintaining the same number per plate. One plate was removed from further analysis when an independent visualization showed that all its vehicle-treated, compound-treated, and eugenol-treated wells clustered together. For multiclassification, a random forest model with 10,000 trees was trained on all 18 replicates per compound at 200 μM. For all models, scikit-learn 0.21.3 defaults were used for all remaining hyperparameters.
Head twitch response experiments.
Mice (9–10 weeks old) were injected intraperitoneally with compound (injection volume 5 ml/kg), placed in an empty cage, and filmed for 20 minutes. Cages were cleaned with 70% ethanol between experiments. Each video was scored for the number of head-twitches by two trained observers blinded to treatment condition (Pearson correlation coefficients = 0.91 and 0.99 for males and females, respectively), and these results were averaged.
Statistical analysis.
Treatments were randomized, and data were analyzed by experimenters blinded to treatment conditions. Statistical analyses were performed using GraphPad Prism (version 8.1.2). The specific tests used, F-statistics, degrees of freedom, and main effect p-values are indicated in the figure legends where appropriate. All comparisons were planned prior to performing each experiment. For dendritogenesis experiments a one-way ANOVA with Dunnett’s post hoc test was deemed most appropriate, as our research question focused on determining whether or not a particular treatment promoted neuronal growth to a greater extent than the vehicle control. Ketamine was included as a positive control to ensure that the assay was working properly.
Supplementary Material
ACKNOWLEDGMENT
We thank members of the Olson Laboratory for helpful discussions, and Steven Chen and Michelle Arkin for access to the Biomek liquid handler.
Funding
This work was supported by funds from the National Institutes of Health (R01GM128997 to D.E.O. and R01AA022583 to D.K.), a UC Davis Miller Symposium Graduate Fellowship (L.E.D), a UC Davis Provost Undergraduate Fellowship (A.A.), an NIH Chemical Biology T32 Training Grant (T32GM113770 to C.L. and R.J.T.), a UC Davis Science Translation and Innovative Research (STAIR) Grant (D.E.O.), a Hellman Fellowship (D.E.O.), The Paul G. Allen Family Foundation (D.K.), the Genentech Foundation (D.K.), and the National Institute of Child Health and Human Development (U54 HD079125).
ABBREVIATIONS USED
- DMT
N,N-dimethyltryptamine
- PFC
prefrontal cortex
- 5-HT2A
serotonin 2A
- MPO
multiparameter optimization
- LSD
lysergic acid diethylamide
- TPSA
total polar surface area
- MAP2
microtubule-associated protein 2
- Nmax
maximum number of crossings
- 5-HT2B
serotonin 2B
- DIV
days in vitro
- VEH
vehicle
- KET
ketamine
- SEM
standard error of the mean
- ANOVA
analysis of variance
- DOM
2,5-dimethoxy-4-methylamphetamine
- OMe
methoxy
- OBn
benzyloxy
- F
fluoro
- μM
micromolar
- nM
nanomolar
- pM
picomolar
- V
vehicle
- K
ketamine
- ATR
attenuated total reflectance
- FT-IR
fourier transform infrared spectroscopy
- UHPLC
ultra-high performance liquid chromatography
- LRMS
low-resolution mass spectrometry
- IACUC
institutional animal care and use committee
- AAALAC
Association for Assessment and Accreditation of Laboratory Animal Care
- BSA
bovine serum albumin
- DPBS
Dulbecco’s phosphate-buffered saline
- mTOR
mammalian target of rapamycin
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- TrkB
tropomyosin receptor kinase B
- HTR
head-twitch response
Footnotes
Supporting Information
Figure S1; Figure S2; Figure S3; 1H and 13C NMR spectra; molecular formula strings for all compounds
REFERENCES
- 1.Whiteford HA; Degenhardt L; Rehm J; Baxter AJ; Ferrari AJ; Erskine HE; Charlson FJ; Norman RE; Flaxman AD; Johns N, Murray CJL; Vos Theo. Global Burden of Disease Attributable to Mental and Substance Use Disorders: Findings from the Global Burden of Disease Study. Lancet 2013, 382, 1575–1586. [DOI] [PubMed] [Google Scholar]
- 2.Li N; Lee B; Liu RJ; Banasr M; Dwyer JM; Iwata M; Li XY; Aghaja- nian G; Duman RS mTOR-Dependent Synapse Formation Underlies the Rapid Antidepressant Effects of NMDA Antagonists. Science 2010, 329, 959–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moda-Sava RN; Murdock MH; Parekh PK; Fetcho RN; Huang BS; Huynh TN; Witztum J; Shaver DC; Rosenthal DL; Alway EJ; Lopez K; Meng Y; Nellissen L; Grosenick L; Milner TA; Deisseroth K; Bito H; Kasai H; Liston C6. Sustained Rescue of Prefrontal Circuit Dysfunction by Antidepressant-Induced Spine Formation. Science 2019, 364 DOI: 10.1126/science.aat8078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duman RS; Aghajanian GK; Sanacora G; Krystal JH Synaptic Plasticity and Depression: New Insights from Stress and Rapid-Acting Antidepressants. Nat. Med 2016, 22, 238–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duman RS; Aghajanian GK Synaptic Dysfunction in Depression: Potential Therapeutic Targets. Science 2012, 338, 68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morgan CJ; Curran HV; Independent Scientific Committee on Drugs. Ketamine Use: A Review. Addiction 2012, 107, 27–38. [DOI] [PubMed] [Google Scholar]
- 7.Olson DE Psychoplastogens: A Promising Class of Plasticity-Promoting Neurotherapeutics. J. of Exp. Neurosci 2018, 12, 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Voleti B; Navarria A; Liu RJ; Banasr M; Li N; Terwilliger R; Sanacora G; Eid T; Aghajanian G; Duman RS Scopolamine Rapidly Increases Mammalian Target of Rapamycin Complex 1 Signaling, Synaptogenesis, and Antidepressant Behavioral Responses. Biol. Psychiatry 2013, 74, 742–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu RJ; Duman C; Kato T, Hare B; Lopresto D; Bang E; Burgdorf J; Moskal J; Taylor J; Aghajanian G; Duman RS GLYX-13 Produces Rapid Antidepressant Responses with Key Synaptic and Behavioral Effects Distinct from Ketamine. Neuropsychopharmacology. 2017, 42, 1231–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ly C; Greb AC; Cameron LP; Wong JM; Barragan EV; Wilson PC; Burbach KF; Soltanzadeh Zarandi S; Sood A; Paddy MR; Duim WC; Dennis MY; McAllister AK; Ori-McKenney KM; Gray JA; Olson DE Psychedelics Promote Structural and Functional Neural Plasticity. Cell Rep. 2018, 23, 3170–3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.dos Santos RG; Osório FL; Crippa JA; Riba J; Zuardi AW; Hallak JE Antidepressive, Anxiolytic, and Antiaddictive Effects of Ayahuasca, Psilocybin and Lysergic Acid Diethylamide (LSD): A Systematic Review of Clinical Trials Published in the Last 25 Years. Ther. Adv. Psychopharmacol 2016, 6, 193–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cameron LP; Olson DE Dark Classics in Chemical Neuroscience: N,N-Dimethyltryptamine (DMT). ACS Chem. Neurosci 2018, 9, 2344–2357. [DOI] [PubMed] [Google Scholar]
- 13.Cameron LP; Benson CJ; Dunlap LE; Olson DE Effects of N,N-Dimethyltryptamine (DMT) on Rodent Behaviors Relevant to Anxiety and Depression. ACS Chem. Neurosci 2018, 97, 1582–1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cameron LP; Benson CJ; DeFelice BC; Fiehn O; Olson DE Chronic, Intermittent Microdoses of the Psychedelic N, N-Dimethyltryptamine (DMT) Produce Positive Effects on Mood and Anxiety in Rodents. ACS Chem. Neurosci 2019, 10, 3261–3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palhano-Fontes F; Barreto D; Onias H; Andrade KC; Novaes MM; Pessoa JA; Mota-Rolim SA; Osorŕio FL; Sanches R; Dos Santos RG; Tofŕoli LF; de Oliveira Silveira G; Yonamine M; Riba J; Santos FR; Silva-Junior AA; Alchieri JC; Galvaoõ-Coelho NL; Lobaoõ-Soares B; Hallak JEC; Arcoverde E, Maia-de-Oliveira JP; Arauŕjo DB Rapid Antidepressant Effects of the Psychedelic Ayahuasca in Treatment-Resistant Depression: A Randomized Placebo-Controlled Trial. Psychol. Med 2018. 49, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Osorŕio FL; Sanches RF; Macedo LR; dos Santos RG; Maia-de-Oliveira JP; Wichert-Ana L; de Araujo DB; Riba J; Crippa JA; Hallak JE Antidepressant Effects of a Single Dose of Ayahuasca in Patients with Recurrent Depression: A Preliminary Report. Rev. Bras. Psiquiatr 2015, 37, 13–20. [DOI] [PubMed] [Google Scholar]
- 17.Sanches RF; de Lima Osorŕio F; Dos Santos RG; Macedo LR; Maia-de-Oliveira JP; Wichert-Ana L; de Araujo DB; Riba J; Crippa JA; Hallak JE Antidepressant Effects of a Single Dose of Ayahuasca in Patients With Recurrent Depression: A SPECT Study. J. Clin. Psychopharmacol 2016, 36, 77–81. [DOI] [PubMed] [Google Scholar]
- 18.Speeter ME; Anthony WC The Action of Oxalyl Chloride on Indoles: A New Approach to Tryptamines. J. Am. Chem. Soc 1954, 76, 6208–6210. [Google Scholar]
- 19.Glennon RA; Jacyno JM; Young R; McKenney JD; Nelson D Synthesis and Evaluation of a Novel Series of N,N-dimethylisotryptamines. J. Med. Chem 1984, 27, 41–45. [DOI] [PubMed] [Google Scholar]
- 20.Chang-Fong J; Addo J; Dukat M; Smith C; Mitchell NA; Herrick-Davis K; Teitler M; Glennon RA Evaluation of Isotryptamine Derivatives at 5-HT(2) Serotonin Receptors. Bio. Org. Med. Chem. Lett 2002, 12, 155–158. [DOI] [PubMed] [Google Scholar]
- 21.Wager TT; Hou X; Verhoest PR; Villalobos A Central Nervous System Multiparameter Optimization Desirability: Application in Drug Discovery. ACS Chem. Neurosci 2016, 6, 767–775. [DOI] [PubMed] [Google Scholar]
- 22.Kurkin AV; Karchava AV; Yurovskaya MA Reaction of 1-(R-Sulfonyl)indoles with N,N-Dibenzyl-β-amino Alcohols. Chem. Heterocycl. Compd 2000, 36, 1141–1148. [Google Scholar]
- 23.Hill TA; Gordon CP; McGeachie AB; Venn-Brown B; Odell LR; Chau N; Quan A; Mariana A; Sakoff JA; Chircop M; Robinson PJ; McCluskey A Inhibition of Dynamin Mediated Endocytosis by the Dynoles--Synthesis and Functional Activity of a Family of Indoles. J. Med. Chem 2009, 52, 3762–3773. [DOI] [PubMed] [Google Scholar]
- 24.Swinney DC; Anthony J How Were New Medicines Discovered? Nat. Rev. Drug Discov 2011, 10, 507–519. [DOI] [PubMed] [Google Scholar]
- 25.Sundstrom LE Thinking Inside the Box. To Cope with an Increasing Disease Burden, Drug Discovery Needs Biologically Relevant and Predictive Testing Systems. EMBO Rep. 2007, 8, S40–S43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Caceres A; Banker G; Steward O; Binder L; Payne M MAP2 is Localized to the Dendrites of Hippocampal Neurons which Develop in Culture. Brain Res. 1984, 315, 314–318. [DOI] [PubMed] [Google Scholar]
- 27.Ristanovic D; Milosevic NT; Stulic V Application of Modified Sholl Analysis to Neuronal Dendritic Arborization of the Cat Spinal Cord. J. Neurosci. Methods 2006, 158, 212–218. [DOI] [PubMed] [Google Scholar]
- 28.Lima da Cruz RA; Moulin TC; Petiz LL; Leão R, A N Single Dose of 5-MeO-DMT Stimulates Cell Proliferation, Neuronal Survivability, Morphological and Functional Changes in Adult Mice Ventral Dentate Gyrus. Front. Mol. Neurosci 2018, 11, 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davis AK; Barsuglia JP; Lancelotta R; Grant RM; Renn E The Epidemiology of 5-Methoxy-N,N-dimethyltryptamine (5-MeO-DMT) Use: Benefits, Consequences, Patterns of Use, Subjective Effects, and Reasons for Consumption. J. Psychopharmacol 2018, 32, 779–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davis AK; So S; Lancelotta R; Barsuglia JP; Griffiths RR 5-Methoxy-N,N-dimethyltryptamine (5-MeO-DMT) Used in a Naturalistic Group Setting is Associated with Unintended Improvements in Depression and Anxiety. Am. J. Drug Alcohol Abuse 2019, 45, 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blair JB; Kurrasch-Orbaugh D; Marona-Lewicka D; Cumbay MG; Watts VJ; Barker EL; Nichols DE Effect of Ring Fluorination on the Pharmacology of Hallucinogenic Tryptamines. J. Med. Chem 2000, 43, 4701–4710. [DOI] [PubMed] [Google Scholar]
- 32.Glennon RA; Young R. l; Jacyno JM; Slusher M; Rosecrans JA DOM-Stimulus Generalization to LSD and other Hallucinogenic Indolealkylamines. Eur. J. Pharmacol 1983, 86, 453–459. [DOI] [PubMed] [Google Scholar]
- 33.Meanwell NA Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem 2018, 61, 5822–5880. [DOI] [PubMed] [Google Scholar]
- 34.Wacker D; Wang S; McCorvy JD; Betz RM; Venkatakrishnan AJ; Levit A; Lansu K; Schools ZL; Che T; Nichols DE; Shoichet BK; Dror RO; Roth BL Crystal Structure of an LSD-Bound Human Serotonin Receptor. Cell 2017, 168, 377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCarroll M; Gendelev L; Kinser R; Taylor J; Bruni G; Myers-Turnbull D; Helsell C; Carbajal A; Rinaldi C; Kang HJ; Gong JH; Sello J; Tomita S; Peterson R; Keiser M; Kokel D Zebrafish Behavioural Profiling Identifies GABA and Serotonin Receptor Ligands Related to Sedation and Paradoxical Excitation. Nat. Comm, 2019, 10, 4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hanks JB; González-Maeso J Animal Models of Serotonergic Psychedelics. ACS Chem. Neurosci, 2013, 4, 33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tombari RJ; Saunders CM; Wu CY; Dunlap LE; Tantillo DJ; Olson DE Ex Vivo Analysis of Tryptophan Metabolism Using 19F NMR, ACS Chem. Biol, 2019, 14, 1866–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buemi MR; De Luca L; Chimirri A; Ferro S; Gitto R; Alvarez-Builla J; Alajarin R Indole Derivatives as Dual-Effective Agents for the Treatment of Neurodegenerative Diseases: Synthesis, Biological Evaluation, and Molecular Modeling Studies. Bioorg. Med. Chem 2013, 21, 4575–4580. [DOI] [PubMed] [Google Scholar]
- 39.Westerfield Monte. The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish (Danio Rerio). University of Oregon Press; 2000. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.