

Abstract
Asymptomatic hyperuricemia affects approximately 20 percent of the general population in the USA, with variable rates in other countries. Historically asymptomatic hyperuricemia was considered a benign laboratory abnormality with little clinical significance in the absence of gout or kidney stones. Yet, there is increasing evidence that asymptomatic hyperuricemia can predict the development of hypertension, obesity, diabetes mellitus and chronic kidney disease. One potential mechanism by which asymptomatic hyperuricemia may contribute to disease is by stimulating inflammation. While urate has been classically viewed as an anti-oxidant with beneficial effects, more recent studies suggest both crystalline and soluble urate may activate various inflammatory pathways. Herein we review the role of urate in the inflammatory response. Further research is needed to define the role of asymptomatic hyperuricemia in these proinflammatory pathways.
Introduction
Hyperuricemia is classically defined as a serum urate of more than 7.0 mg/dL in men or more than 6.0 mg/dL in women. Today approximately 20 percent of the US population have hyperuricemia.1 The well accepted consequence of hyperuricemia is an increased risk for gout and kidney stones, and currently treatment of asymptomatic hyperuricemia is recommended only in people with these established conditions.2 Yet, people with asymptomatic hyperuricemia are also at risk for developing a variety of other conditions, including hypertension, acute and chronic kidney disease, obesity, metabolic syndrome, fatty liver and diabetes mellitus.3,4 While it was often viewed that the hyperuricemia develops secondary to these conditions, most studies show that the hyperuricemia precedes these diseases5–7. Indeed, a recent study in the Japanese population showed that the presence of hyperuricemia in healthy, lean, normotensive people still carries an increased risk of cardiometabolic disease8 and the treatment of asymptomatic hyperuricemia above 8 mg/dL is recommended in Japan.9 Given these findings, we herein review studies examining the role of uric acid in driving inflammation.
Throughout this paper, we use the generic term “uric acid” for the end-product of purine catabolism in humans and to describe data from experimental models in which exogenous uric acid was dissolved and administered in order to study inflammatory effects. Knowing that uric acid is predominantly found in ionized form in the circulation, we use the term “urate” when referring to circulating plasma or serum levels10 and discuss inflammatory effects of both crystalline and soluble uric acid. In line with the consensus gout labels11, hyperuricemia represents the elevated circulating levels of urate of 7 mg/dl in men and 6 mg/dl in women. Asymptomatic hyperuricemia is defined as elevated urate levels in absence of episodes of inflammation triggered by urate crystals (gout flares).
Uric acid and Susceptibility to Hyperuricemia and Gout in Humans
Uric acid is a purine degradation product generated from the breakdown of nucleic acids, purine-containing compounds such as adenosine triphosphate (ATP), and guanine and adenine. Uric acid can also be generated from ribose phosphate by a series of enzymatic reactions. In most mammals, circulating urate (the form in the blood) is relatively low (1-3 mg/dl) due to the presence of urate oxidase (uricase), an enzyme in the liver (or occasionally the kidney) that degrades uric acid to 5-hydroxyisourate, and eventually allantoin (Figure 1).12 However, a number of species lack functional uricase, including birds and most reptiles. In these species, serum urate is higher, and the excretion of uric acid is the primary means for eliminating excess nitrogen. In fact, it has been proposed that the loss of functional uricase in these species provided an evolutionary benefit for living in terrestrial versus marine environments, as the uric acid could be precipitated in the cloaca and excreted with the minimal loss of water.13
Figure 1. Regulation of serum urate.
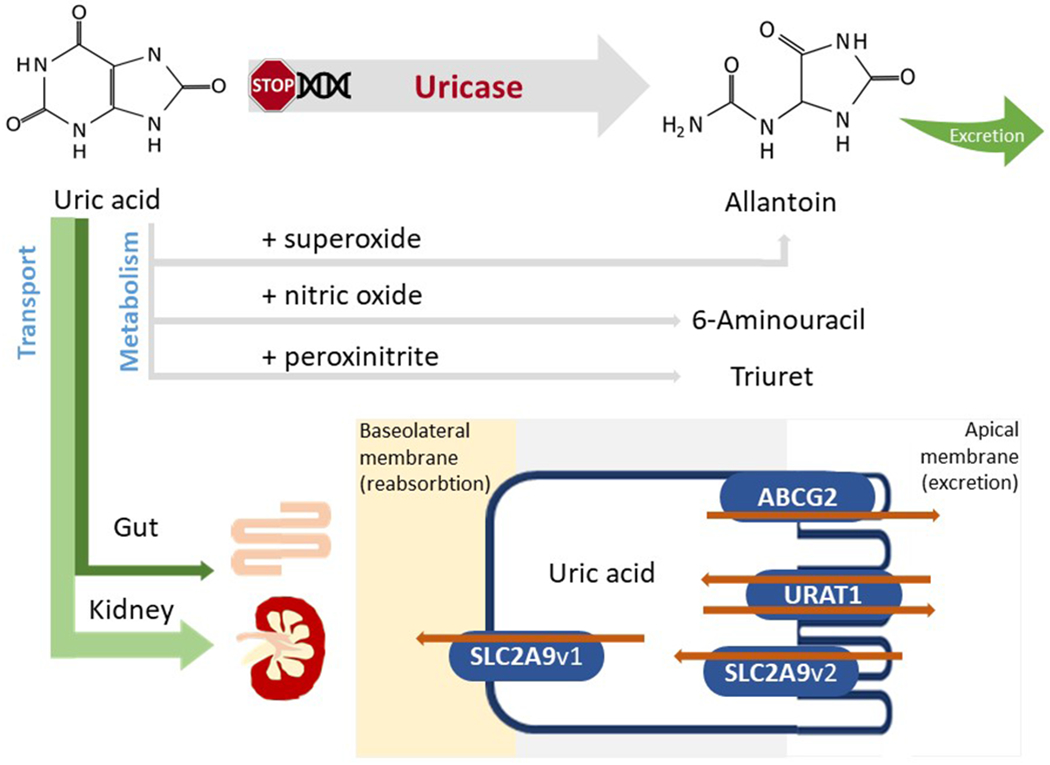
Following sequential mutations in the uricase gene, humans and higher primates do not express functional uricase, needed to metabolize uric acid to allantoin. Serum urate can interact with reactive species generating substances or radicals such as allantoin, 6-aminouracil or triuret. However, most of the serum urate will be excreted via the kidney or, alternatively, via the gut. Both proximal tubule and intestinal cells express several urate transporters (collectively known as the transportasome) that are responsible for the excretion and/or reabsorption of urate. Several main urate transporters are illustrated: URAT1, GLUT9, OAT4 and OAT10 are involved in urate reabsorption at the level of the apical membrane (brush border) of proximal renal tubular cells; GLUT9 is responsible for urate transport out of the cell via the basolateral membrane into the blood; ABCG2 is a unidirectional transporter mediating the secretion of urate via the apical membrane; OAT1 and OAT3 localized on the basolateral membrane are involved in urate excretion. OAT, organic anion transporter; URAT1, urate transporter 1; GLUT9, glucose transporter 9; ABCG2, ATP-binding cassette super-family G member 2.
Uricase is also absent in hominoids due to a series of mutations that affected the great apes and humans, as well as a parallel mutation that eliminated uricase in the lesser apes (gibbons).14,15 The parallel loss of uricase during the mid-Miocene strongly suggests that there was a natural selection advantage at that time. Moreover, the gene encoding urate transporter URAT1 was found to have coevolved with the sequential reduction of uricase enzymatic activity, enhancing the URAT1 affinity in primates and allowing for better control of higher serum urate levels.16
The observation that all mammals including humans excrete most of their nitrogen as urea suggests different benefits for why humans and apes lost uricase. Several hypotheses have been proposed. First, uric acid has a chemical structure similar to caffeine, and it has been posited that uric acid may have effects on mental performance, drive, response time, and impulsivity.17–19 Second, uric acid can function as an anti-oxidant, and it has been hypothesized that its anti-oxidant effects may have been of benefit by blocking the oxidative stress associated with aging and cancer.20 Indeed, it has been proposed that the mutation may have countered the loss of antioxidant activity associated with the mutation in L-gluconolactone oxidase that resulted in the inability to synthesize vitamin C.21 Finally, there is emerging evidence that the loss of uricase may have had survival benefits by its ability to help maintain blood pressure, stimulate fat and glycogen stores, and stimulate gluconeogenesis.14,22–24 It is also possible that the benefit of uricase may involve some aspect of each of these proposed mechanisms.
Regulation of Serum Urate in the Absence of Uricase
In the absence of uricase, the kidney and the gut became the primary means for removing uric acid. Whereas some urate can be degraded by oxidants or nitric oxide (generating allantoin, triuret and 6-aminouracil)25,26, most urate (two-thirds) is excreted by the kidneys, largely by a series of transporters in the proximal tubules, of which SLC2A9, URAT1, and ABCG2 are especially important (Figure 1). Some urate (one-third) also passes into the gut via SLC2A9 and ABCG2 transporters where the uric acid is degraded by uricolytic bacteria. These excretory pathways are fairly effective, and in people who eat native (nonwestern) diets, serum urate tends to be low, classically in the 3 to 5 mg/dl range.27
Causes of Hyperuricemia
While the absence of uricase only results in a slight increase in serum urate (of 1-2 mg/dl), additional genetic variants in genes encoding urate transporters or enzymes involved in uric acid metabolism have been shown to modify serum urate levels, either through small effects when considering common variants28–30, or through damaging effects in the case of rare alleles31. Environmental factors can also contribute to hyperuricemia as subjects on a western diet tend to have significantly higher serum urate concentrations.32,33 This is partly driven by diets high in sugar (which contains fructose), purines (especially from animal proteins), and alcohol (especially beer).33,34 However, there is also reduced fractional excretion of urate in people with obesity, insulin resistance and hypertension 35, and there are also drugs that can impair urate excretion, especially the thiazide and loop diuretics. Hyperuricemia is also common as kidney function fails, and at the time of dialysis initiation approximately half of patients demonstrate hyperuricemia.36,37
Clinical Consequences of Hyperuricemia
One of the consequences of higher serum urate is the risk for gout, urate kidney stones, and acute kidney injury from urate crystalluria. The prevalence of gout in the US approaches 6 percent in men and 2 percent in women.1 with a worldwide range of 0.1 to 10% and increasing trend in developed countries38. People with acute gout present with severe joint pain due to the deposition of urate crystals in the synovium associated with a marked inflammatory response.33,39 Kidney stones from uric acid are also common in people with gout and hyperuricemia40,41 , and even calcium stones may occur in which the initial nidus is from urate crystal deposition42.
Another risk resulting from the uricase mutation is that the renal excretory system is not adapted for large increases in urate excretion that can occur when there is sudden production, such as following the release of DNA and RNA from tissue injury or tumor lysis.43 For example, marked uricosuria with crystal formation, intratubular obstruction and renal inflammation may result when there is a sudden rise in urinary uric acid from rapid tumor death following chemotherapy.44 Urate concentrations may also increase in the setting of dehydration and low urine volumes, leading to elevated concentrations that can result in urate crystal formation.45 High urinary urate concentrations have also been associated with tubular injury and activation in experimental models even in the absence of documented crystalluria.46–48
More recently the potential role uric acid may play with chronic inflammatory disorders has been comprehensively investigated, particularly the association of uric acid with obesity, hypertension, metabolic syndrome, diabetes mellitus, kidney disease and cardiovascular disease.4 The literature remains inconsistent, for, while epidemiological studies are generally positive, genetic Mendelian randomization studies, that are supposed to better predict whether causality is present, are often negative49–51. However, a problem with the Mendelian randomization studies is that they often of rely on single nucleotide polymorphisms (SNPs) that modulate urate transport as opposed to urate synthesis, and it is not known how these polymorphisms affect intracellular urate levels that are thought to drive the biological effects52. Moreover, only very recently the functional consequence of SNPs have been taken into account to the importance of a certain SNP in inflammation.53,54 In addition, while pilot studies with urate lowering therapies often suggest benefit, the trials usually enroll a limited numbers participants, are often nonblinded and the beneficial effects are typically modest, while other studies are negative.4 Nevertheless, meta-analyses have largely supported a potential benefit of urate lowering in subjects with hypertension and/or chronic kidney disease 55,56.
At the heart of the controversy is the question of whether a substance that is thought to be an antioxidant can actually be prooxidative and proinflammatory. In the next sections we investigate potential proinflammatory effects of both soluble and crystalline uric acid.
Urate as a Mechanism for Inducing an Inflammatory Response
Several studies have been performed to better understand potential mechanisms whereby urate may induce a proinflammatory response. Below we discuss several proposed mechanisms, including inflammation driven by urate crystals (including both inflammasome-independent and dependent mechanisms) as well as mechanisms mediated by soluble urate.
Urate Crystal-Dependent Inflammation.
Classically, monosodium urate crystals (MSU) activate the innate immune response by triggering inflammasome pathways57 that result in the release of interleukin-1β (IL-1β) (Figure 2). Inflammasome-independent mechanisms are also possible contributors to urate crystal induction of cytokine secretion (Figure 3).
Figure 2. Inflammasome-dependent activation of IL-1β in response to MSU crystals.
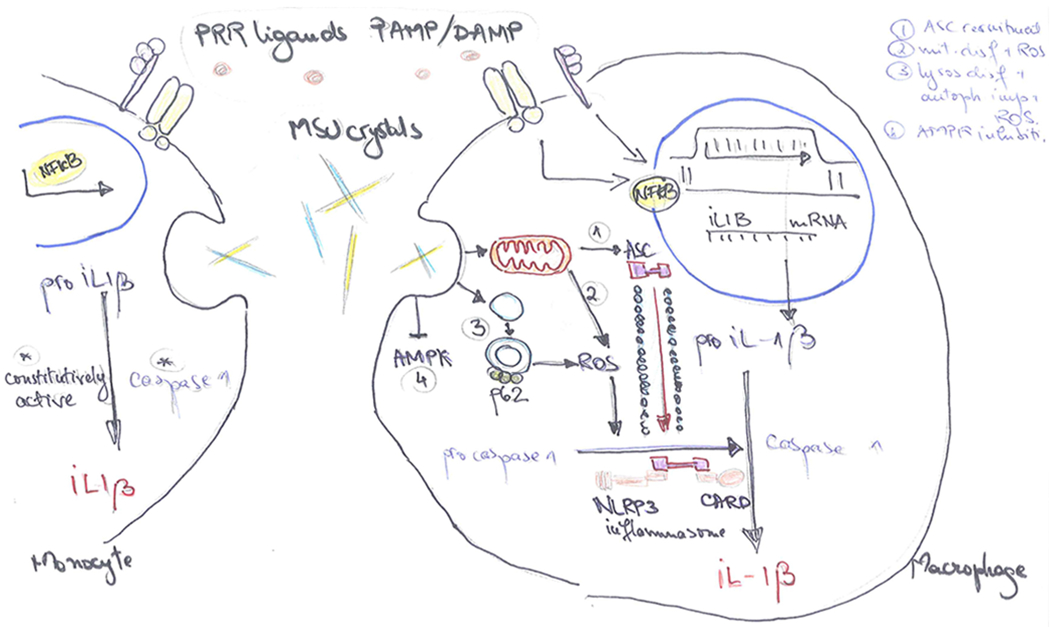
IL-1β production is initiated by exogenous or endogenous stimuli (PAMPs or DAMPs) which use the PRR machinery to induce an inflammatory response. Engagement of PRR ligands to the receptors leads to the transduction of signal to the nucleus and transcription of the inactive pro-IL-1β precursor. In the intracellular space, pro-IL-1β is proteolytically activated by specific cleavage performed by caspase-1. Caspase-1 is, in turn, present in the cytosol in a precursor form (procaspase-1). MSU crystals act as inducers of the protein platform that cleaves procaspase-1 to caspase-1, thus mediating the activation and release of biologically active IL-1β. In the monocytes (left), caspase-1 is constitutively active, thus MSU crystals potentiate IL-1β maturation. MSU crystals also upregulate mTOR gene transcription, leading to enhanced gene transcription of IL-1β and other inflammatory cytokines, and drive monocyte cell death that could, indirectly, further stimulate the inflammasome. In the macrophages (right), caspase-1 is not constitutively active, MSU crystals drive changes inside the cell that result in the assembly of the inflammasome components and caspase-1 generation. MSU crystals can do this in multiple ways: (1) induce mitochondrial ASC recruitment to the site of inflammasome assembly in a microtubule dependent manner, (2) induce mitochondrial dysfunction and ROS generation, (3) lysosomal dysfunction with impairment of autophagy, increased levels of p62 and ROS generation, (4) AMPK inhibition in an NLRP3 dependent manner. PAMP, Pathogen Associated Molecular Patterns; DAMP, Damage Associated Molecular Patterns; PRR, pattern recognition receptor; ASC, Apoptosis-Associated Speck-Like Protein Containing CARD; ROS, reactive oxygen species; AMPK, AMP-activated protein kinase.
Figure 3. Inflammasome-independent regulation of inflammation in response to MSU crystals.
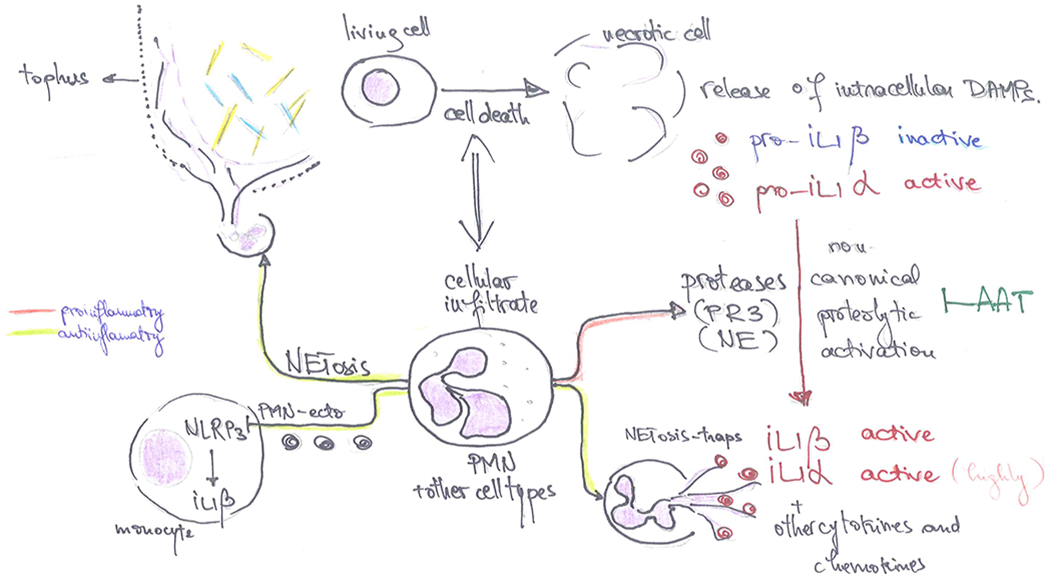
MSU crystal deposition leads to cell death and cellular infiltrate, mostly characterized by neutrophil recruitment at the site of crystal deposition, especially in the affected joints of patients with gout. Dying cells release intracellular molecules (DAMPs, alarmins) in the extracellular space, including precursors of the IL-1 cytokines. Pro-IL-1β is inactive while pro-IL-1α already has biological activity in its precursor form, therefore is able to mediate inflammatory responses in the surrounding tissue. Infiltrating cells, the majority of which are neutrophils, contain and release proteolytic enzymes (such as PR3 or NE, among others) that have the potential to cleave IL-1 precursors at alternative cleavage sites and activate IL-1β or enhance the bioactivity of IL-1α. This mechanism of protease induced activation of precursor cytokines can be limited by anti-proteases such as AAT. Conversely, neutrophils can undergo NETosis (neutrophil extracellular trap formation), which consists in the externalization of chromatin and binding of intercellular molecules. This phenomenon is considered to play an important role in the resolution of inflammation in gout: NETs can bind active cytokines or chemokines and facilitate their enzymatic digestion contributing to the limitation of inflammation; moreover, NETosis can contribute to the sequestration of MSU crystals and encapsulation of crystals within the tophus; neutrophil microvesicles (PMN-Ecto) can downregulate inflammatory pathways in paracrine manner, limiting the magnitude of the cytokine production during the acute phase by inhibiting NLRP3 or activating SOCS3. DAMP, Damage Associated Molecular Patterns; PR3, Proteinase 3; NE, Neutrophil Elastase, NLRP3, NACHT, LRR and PYD domains-containing protein 3; SOCS3, Supressor of Cytokine Signalling 3.
Inflammasome activation.
IL-1 is a potent proinflammatory cytokine, with a tightly regulated production mechanism and low circulating levels. There are two major IL-1 proteins: IL-1α and IL-1β. Whereas IL-1α is present primarily intracellularly and membrane bound, IL-1β can be secreted by stimulated cells 58,59. This secretion of IL-1β is differentially regulated in myeloid cells: in macrophages it requires a two-step activation, whereas in monocytes it can be directly processed into its bioactive form 60 (Figure 2). IL-1β is translated as a precursor protein (pro-IL-1 β) and needs to be activated through protein cleavage by caspase-1. Caspase-1, in turn, needs to be activated in protein platforms called inflammasomes61.
The NACHT, LRR and PYD domains-containing protein 3 (NLRP3) inflammasome57 and subsequent procaspase-1 cleavage is the most commonly known mechanism to complete the activation and release of active IL-1β upon interaction with MSU. The mechanism of MSU activation of the NLRP3 inflammasome was first described in THP1 cells62. Furthermore, MSU crystals have been shown to contribute to NLRP3 inflammasome formation through a mechanism dependent on tubulin polymerization and mitochondrial ASC recruitment at the site of inflammasome component colocalization 63. Interestingly, however, in an in vivo murine model of gouty arthritis induced by intra-articular injection of a combination of MSU crystals and stearate fatty acids, NLRP3 involvement was not observed, despite the inflammatory reaction being dependent on ASC and caspase-164. This suggests (as will be discussed below) that the in vivo effect of NLRP3 can be bypassed or overridden by other proinflammatory systems64.
As summarized in Figure 2, the inflammasome protein platform also shows interactions with organelle clearance and autophagy. MSU was found to damage lysosomal integrity and to impair autophagy, leading to p62 accumulation. Autophagy related p62 excess led to Nrf2 activation and translocation to the nucleus, where Nrf2 induced transcription of heme oxygenase-1 and superoxide dismutase65, contributing to altered intracellular redox status. In line with this, another report linked p62 expression to enhanced ROS production and, subsequent caspase-dependent apoptosis and IL-1β production66. Moreover, MSU dependent IL-1β and CXCL1 production was shown to be mediated by inhibition of AMP-activated protein kinase (AMPK) in a mechanism dependent on both NFκB and NLRP367. These findings suggest that the intracellular homeostasis is disrupted at several different networks by the sterile MSU crystals (autophagy dysfunction, diminished clearance of damaged organelles such as mitochondria, in addition to oxidative stress and, at least in part, AMPK inhibition) culminating in the activation of the inflammasomes (Figure 2, right). A recent study performed in monocytes also showed that MSU crystals lead to inflammatory cytokine production through an mTOR dependent mechanism involving pyroptosis which could further, indirectly, lead to inflammasome activation68 (Figure 2, left).
Inflammasome-independent urate crystal induced inflammation.
While inflammasome-dependent maturation and secretion of IL-1β is paramount in monocyte and macrophage mediated innate immune responses, inflammatory diseases which are characterized by polymorphonuclear leukocyte (neutrophil) infiltrates are commonly independent of inflammasome activation69. In gout, the inflammatory reaction has been shown to be mediated by inflammasome activation in response to MSU, although this is likely to be short-lived due to the subsequent clinical picture determined by rampant neutrophil infiltrate. While neutrophils are capable of transcribing IL-1β 70, an even greater inflammatory process is mediated by intracellular serine-proteases (such as neutrophil elastase (NE), cathepsin G (CG), proteinase 3 (PR3)) which are released as a consequence of cell influx, MSU mediated damage and cell death. Neutrophil-derived serine proteases, such as proteinase-3 (PR3) and elastase (NE) can process the inactive IL-1β precursor at alternative proteolytic cleavage sites, leading to bio-active IL-1β 71. Moreover, due to cell damage, IL-1α is also released from dying cells in the extracellular space and functions as an alarmin (Figure 3). 58,59. This can explain why in many in vivo scenarios, including but not restricted to gout, the inflammatory outcomes remain IL-1 driven, while the inflammasome involvement is limited or bypassed64,69. In line with this, a promising therapeutic option for gout and MSU dependent inflammation is represented by alpha-1-antytripsin (AAT), the natural plasma inhibitor of serine-proteases, which has been shown to limit inflammation through upregulation of IL-1 receptor antagonist (IL-1Ra) and through inhibition of PR3 dependent IL-1 activation72 (Figure 3).
Conversely, neutrophils have been shown to be involved in the spontaneous resolution of acute attacks of gout. Neutrophil-derived micro vesicles (PMN-Ecto) have been reported to be released in the peritoneum of mice injected with MSU and to downregulate IL-1β production in response to complement 5a via upregulation of SOCS373. Moreover, neutrophils can undergo oxidative burst and form extracellular traps (NETs) which contribute to resolution of inflammation through several mechanisms. These include the immobilization of MSU crystals and proteolytic degradation of cytokines or chemokines74,75, which are protected from the activity of antiproteases while associated with aggregated NETs 76. Nevertheless, other proinflammatory cell types are likely to be entrapped in the NETs, contributing to the variability of the anti-inflammatory outcomes in NETosis77,78. Consequently, NETs are presumed to contribute to the local tolerance towards the remaining urate crystals, as aggregated NETs and subsequent granuloma formation drive the development of tophaceous deposits and tissue remodeling79,80(Figure 3).
Soluble Urate Inflammatory Pathways.
While early studies focused on the effects of crystalline uric acid on inflammatory cell activation, more recent studies show that soluble uric acid has many proinflammatory and prooxidative effects in the intracellular environment. Some of these pathways are discussed below, including oxidative stress, inflammatory signaling, autophagy and intracellular immunometabolic sensors (Figure 4).
Figure 4. Intracellular pathways involved in proinflammatory effects of soluble uric acid.
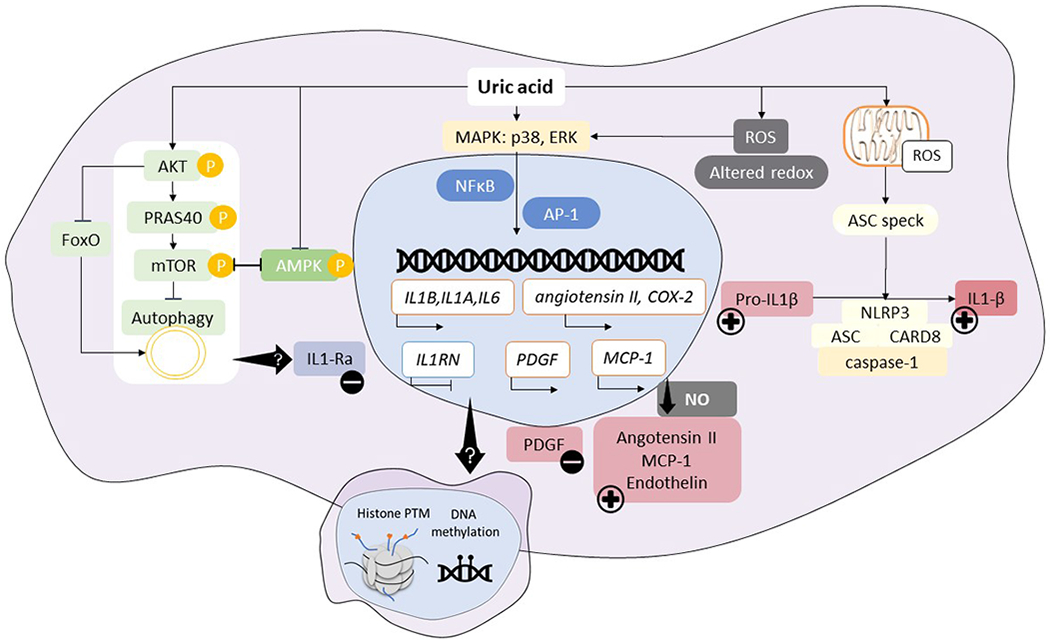
Soluble uric acid has been shown to modulate cytokine production and inflammatory outcomes via several pathways. Uric acid enhances AKT phosphorylation, leading to PRAS40 phosphorylation, and resulting in autophagy inhibition; this is in line with downregulation of expression in genes involved in FOXO pathway. Uric acid was shown to inhibit AMPK phosphorylation, which is consistent with mTOR activation. Several transcription factors can be induced by uric acid signaling (NFκB or AP-1) following p38 and ERK MAPK activation, leading to enhanced transcription of innate cytokines in addition to the remarkable downregulation of natural IL-1 antagonist, IL-1Ra. In vascular endothelial cells, smooth muscle cells or adipocytes, enhanced transcription of vasoactive substances is known to lead to elevated angiotensin II, MCP-1, endothelin and low availability of nitric oxide which can mediate hypertension. In vascular cells, growth factors such as PDGF are upregulated, leading to smooth muscle cells proliferation and promotion of atherosclerosis. Soluble uric acid has also been shown to induce oxidative stress, alter the redox status of the cell which, on the one hand, activates signaling pathways for gene transcription and, on the other hand, lead to ASC speck formation and inflammasome activation, thus contributing to cleavage of pro-IL-1β. The transcriptional rewiring of uric acid stimulated cells is reminiscent of innate immune memory developed in response to endogenous sterile stimuli, leading to a scenario in which soluble uric acid acts as a silent stimulus that drives epigenetic reprogramming in circulating or tissue resident cells, thereby causing persistent inflammatory effects in response to continuous exposure to uric acid. We propose a model in which cells exposed to high uric acid levels are more prone to develop a strong proinflammatory response when exposed to specific triggers due to innate immune memory, epigenetic changes in exposed cells or persistence of exposure to high uric acid levels.
Oxidative Stress.
Uric acid is an antioxidant that can react with superoxide anion to generate allantoin, peroxynitrite to form triuret, and nitric oxide to form 6-aminouracil25,81. However, the reactions of uric acid with peroxynitrite will also generate the aminocarbonyl radical and the triuretcarbonyl radical.26 Uric acid will also form radicals when reacted with myeloperoxidase.82 Moreover, inside the cell uric acid is prooxidative, and this has been shown in vascular smooth muscle cells, endothelial cells, adipocytes, hepatocytes, pancreatic islet cells and renal tubular cells.83–89 The mechanism involves activation of NADPH oxidase, and in some cell types the NADPH oxidase may translocate to the mitochondria.86,89,90 Interestingly, the effect of soluble urate on mononuclear cells is more complex, as priming of PBMCs with urate results in enhanced IL-1β release in response to LPS, but lesser ROS release91 while another study found no statistically significant effects on IL-1β, SOD2 gene transcription nor total antioxidant capacity of the cell92.
Inflammatory Signaling.
Soluble uric acid has also been found to activate a series of mitogen activated protein kinases (MAP kinases), including p38 and ERK, leading to activation of transcription factors such as NF-κB and AP-1.85,93,94 This is associated with the production of vasoconstrictive substances (including (pro)renin receptor95, endothelin, thromboxane and angiotensin II), a loss of vasodilatory substances (such as nitric oxide), an increase in growth factors (platelet-derived growth factor), and an increase in chemokines (such as monocyte chemoattractant protein-1, MCP-1).84,93,94,96,97 Additionally, reports from experimental studies have provided evidence that elevated uric acid concentrations have proinflammatory effects in various tissues and organs: neutropenic mice have a higher cytokine production upon LPS challenge compared to control animals due to hyperuricemia 98; NFκB was shown to be activated in mice injected intraperitoneally with uric acid in renal or pancreatic cells 47,99; uric acid was revealed to induce NFκB activation in hypothalamic cells and promote inflammation and gliosis with possible consequences of altering the hypothalamic functions in metabolic regulation 100.
Autophagy and inflammasome.
The interplay between inflammasome and autophagy in the post-translational processing and release of IL-1β is also implicated in soluble urate induced effects, in addition to the most widely studied MSU induced inflammation. In vitro data using primary human mononuclear cells revealed a shift in IL-1β and IL-1Ra production at uric acid concentrations ranging from 12.5 to 50 mg/dL101. RNA-sequencing (RNAseq) data in highly pure human monocytes treated for 24h with medium or uric acid confirmed effects such as IL-1β or IL-6 upregulation and IL-1Ra decrease in both RNA and protein levels. Pathway enrichment analysis suggested involvement of the AKT pathway involving mTOR (upregulated genes) and FoxO transcription factors (downregulated genes). AKT phosphorylation was enhanced and PRAS40 phosphorylation was also induced in the presence of uric acid. PRAS40 is a molecule that exerts inhibitory roles on mTOR in non-phosphorylated state: once phosphorylated, PRAS40 dissociates from the Raptor protein, releasing the inhibitory signal on mTOR 102. This coincided with inhibition of autophagy in uric acid primed cells91, in line with mTOR signaling acting as an autophagy inhibition mechanism 103. In line with these data, a recent report described a significant positive correlation between mTOR gene expression circulating serum urate levels in patients with gout68.
Another report in bone marrow derived macrophages in an in vivo model of obstructive nephropathy in mice, showed that soluble urate (180 μM), released in hypoxic conditions, leads to mitochondrial ROS production, ASC speck formation and caspase-1 activation, thereby inducing IL1-β production in an NLRP3 and MyD88 dependent manner104. Soluble uric acid has also been shown to induce ROS in mitochondria from both endothelial cells and hepatocytes86,90. The finding that basal autophagy levels are diminished, is reminiscent of the enhanced cytokine production observed in situations where autophagy was inhibited, therefore showing that autophagy could be a possible new source for intervention in gout. While, based on previously cited reports, autophagy defects are a known cause for IL-1β overproduction in both inflammasome dependent and inflammasome independent manners, it is still not completely understood how IL-1Ra is uncoupled from IL-1β. Pharmacological inhibition of autophagy can diminish IL-1Ra levels but further studies are necessary to show whether these two effects are mechanistically related 105. Moreover, since often the concentrations of uric acid surpass in vivo conditions, new studies are required to validate these cytokine profiles and to assess the relevance in clinical situations.
Intracellular immunometabolic sensors.
Immunometabolic sensors are increasingly well characterized in disorders associating inflammation, as intracellular metabolites, energy consumption or production are shown to have an interplay with cytokine biology. In this system, AMPK senses variation in AMP to ATP or ADP to ATP ratios and promotes catabolism106. The downstream phosphorylation of substrates induced by AMPK can, however, also impact on inflammatory pathways, such as mTOR signaling or autophagy, therefore AMPK is considered a master regulator of cytokine production in relation to the cellular energy status107. In line with findings above, AMPK signaling was studied in renal and pancreatic cells and was shown to be inhibited by elevated uric acid levels through ROS and mTOR induction108. Furthermore, the effect of uric acid to inhibit AMPK has also been shown in the liver where it is thought to have a role in driving gluconeogenesis, stimulating intrahepatic fat accumulation, and causing insulin resistance24,109.
This effect of AMPK inhibition by urate has also been reported in THP1 human monocytic cell line, which also translated to higher CD68 expression, foam cell formation in response to oxidized LDL cholesterol and activation of NFκB110. In contrast, human primary monocytes were not found to show elevated ROS production, nor was AMPK phosphorylation significantly modified in response to uric acid 91.
In line with AMPK inhibition by uric acid, this effect could be reversed in pancreatic cells by zurampic (inhibitor of urate transporter URAT1)111. Moreover, the oral antidiabetic drug metformin, a known AMPK inducer, rescued the reduced glucose uptake and uric acid induced insulin resistance in muscle cells112 and liver cells109. These results indicate that AMPK is a valid target in gout and hyperuricemia as well. While monocyte data is conflicting, AMPK inhibition is consistent across tissues which are affected by hyperuricemia-associated diseases, such as the kidney, pancreas, muscle and liver.
Innate immune memory in hyperuricemia and functional consequences
The innate branch of the immune system possesses an epigenetically encoded memory of previous insults113. The hypothesis that the immunological memory is not restricted to the adaptive branch of the immune system, was originally introduced in 2011 under the name “trained immunity” 113 and was based on a large body of previous evidence. Trained immunity is now a widely studied process that is based on the epigenetic reprogramming of myeloid or lymphoid cells involved in innate immunity. The general concept behind trained innate immunity refers to the fact that an initial exposure of cells to a stimulus can promote changes in the transcriptional program of the cell, which lead to elevated inflammatory responses in subsequent stimulations. Trained immunity has been mechanistically deciphered for several pathogenic stimuli and has relevance for susceptibility to infection, immunological tolerance or vaccine biology114–116. In addition, sterile stimuli, acting via the same signaling machinery as pathogens, could also induce innate immune memory117. The long-term effects of sterile stressors could be of great importance for a wide range of inflammatory diseases. For example the involvement of epigenetic memory is already well known and extensively studied through the concept of “glycemic memory” in diabetes mellitus 118. Moreover, trained immunity has been shown to be a mechanism that promotes atherosclerosis, in experimental designs using oxidized LDL or Lipoprotein(a) oxidized phospholipids119,120, with epigenetic changes that are persistent in vivo in patients with familial hypercholesterolemia, despite the pharmacological normalization of lipid levels121.
Studies in patients with hyperuricemia have shown that elevated serum urate levels are associated with inflammatory markers such as white blood cells, C reactive protein and circulating innate immune cytokines such as IL-6, IL-1Ra, TNF or IL-18122. CD14+ monocyte counts have been reported to be elevated in hyperuricemic individuals, who displayed higher plasma CCL2 levels and higher expression of adhesion molecule CD11b123. These data indicate that innate immune inflammatory pathways, especially monocyte or macrophage dependent, are upregulated in presence of hyperuricemia. One report on asymptomatic hyperuricemia in male individuals described an inverse correlation between serum urate levels and Natural Killer cell counts124 showing that lymphoid cells could also be associated to changes in urate levels. However, the functional consequences are yet unexplored and further studies on different lymphoid populations are needed.
Having described evidence linking soluble urate inflammatory mechanisms to similar pathways that drive innate immune memory and knowing that hyperuricemia is associated to inflammatory markers, we propose that stimuli such as uric acid or MSU crystals could drive a memory of the innate immune system. Similar to other autoinflammatory conditions, gout patients exhibit a higher cytokine production profile in response to ex vivo stimulations 92,101,125. One study showed that the ex vivo cytokine production was associated to serum urate concentrations in gout patients101, while another study showed a statistically non-significant correlation between serum urate levels and cytokine production capacity92. In vitro pre-exposure to soluble uric acid of peripheral blood mononuclear cells or enriched monocytes potentiates proinflammatory cytokine secretion to a subsequent stimulus in a way that is reversible by broad methylation inhibitors101. In vivo intravenous administration of uric acid in healthy human volunteers was consistent with higher levels of plasma IL-6 after acute lipid ingestion, whereas potently lowering of serum urate with rasburicase did not have a significant effect on IL-6 plasma values after lipid oral tolerance test126. These observations enforce the finding that soluble uric acid works as a silent modulator on monocytes, that, alone, has no remarkable effects on the inflammatory cytokine production in vitro or in vivo but when subsequently challenged, culture supernatants or plasma of healthy volunteers depict elevated proinflammatory cytokines. In line with this hypothesis, a case-control study in gout patients with more than two flares in 12 months versus patients with less than two flares in 12 months found that serum urate values are associated with frequent gout attacks127.
These studies document that both crystalline and soluble uric acid may have a key function in triggering the immune system to a pro-inflammatory pathway that may result in epigenetic imprinting and can facilitate inflammatory reactions in response to subsequent insults. While this is nowadays a concern in the context of chronic inflammatory diseases, there is some data that uric acid may have a protective role in the host response to infections, such as malaria128,129. It is possible that the uricase mutation may have also been a mechanism for protecting against infections in the past 23,130, nevertheless a recent population study failed to show a negative association between hyperuricemia and community acquired pneumonia, urinary tract infection or infection-related mortality 131.
Hyperuricemia and chronic inflammation
Gout.
Despite general knowledge that the progression from soluble uric acid to MSU crystals and generation of inflammation is the causal mechanism of gout, some questions remain open in relation to the clinical observations related to gout and hyperuricemia. First, hyperuricemia is a relatively common metabolic condition, yet only a minority of 10-15% of hyperuricemia patients developing symptomatic gout 132. Thus, the factors that trigger the symptomatic inflammatory response still need to be elucidated. Second, when an attack resolves, crystals still remain in the tissues (intercritical gout), that might be contributing to a low grade systemic inflammatory response in the absence of symptoms 133. In addition, as many as 15 or more percent of subjects with hyperuricemia have crystal deposition without symptoms 134,135. Thus, neither hyperuricemia nor MSU deposits are sufficient to trigger full blown inflammation. Moreover, experimental designs usually cannot exclude that in vivo or in vitro effects of high concentrations of uric acid are independent of microcrystal formation. Nevertheless, the parallels between soluble uric acid and crystalline uric acid in inducing the inflammatory response is remarkable and suggests the involvement of a pro-inflammatory state that may involve local as well as systemic effects. Studying these responses in patients has the potential of helping to understand the variability of the phenotype in gout and hyperuricemia and of identifying the missing links that determine the progression from hyperuricemia and crystal deposition to clinically symptomatic gout.
Systemic Conditions Beyond Gout.
The recognition that uric acid has such key proinflammatory roles in innate immunity raises key questions of its importance in other diseases. Indeed, hyperuricemia is a powerful predictor of obesity, metabolic syndrome, nonalcoholic fatty liver and diabetes, and experimental studies tie these inflammatory pathways to the mechanism of disease3,6,24,86. Similar studies have linked serum urate with acute and chronic kidney disease, hypertension, preeclampsia, and cardiovascular disease. 4,5,136 However, a challenging finding is that most Mendelian randomization studies evaluating the relationship of serum uric acid to cardiovascular endpoints have been negative, and clinical trials have also been mixed.4 Nevertheless, there is increasing evidence that lowering uric acid may be beneficial, especially in hyperuricemic individuals with chronic kidney disease who are showing deteriorating renal function.137 Clearly further studies are needed, but the recognition of the involvement of urate in metabolic inflammation offers the potential for novel interventions.
Conclusions
Asymptomatic hyperuricemia is common but has been largely viewed as a metabolic abnormality that increases the risk for gout and kidney stones and is innocent otherwise. Recent studies, however, suggest that the people with asymptomatic hyperuricemia may be at significant risk for cardiometabolic diseases, and that the uric acid may play a contributory role in causing these conditions. The studies summarized here document a major role for both crystalline and soluble urate in driving metabolic inflammation, activating innate immunity and triggering epigenetic pathways that can amplify responses. These studies challenge the concept that hyperuricemia is an “innocent bystander” and raises the possibility that conditions in which hyperuricemia is present may benefit not only from urate lowering therapies, but with ones targeting various aspects of the immune system, including IL-1, autophagy, and epigenetic modifiers. Further studies are needed to better understand how these pathways may influence disease, the genetic factors controlling these responses, and the mechanisms transitioning hyperuricemia to gout and beyond.
Acknowledgements:
LABJ and TOC are supported by a Competitiveness Operational Programme grant of the Romanian Ministry of European Funds (P_37_762, MySMIS 103587). P.B. is supported by NIH/NIDDK, DiaComp, JDRF, Thrasher Research Fund, Center for Women’s Health Research and ISPAD.
Footnotes
Competing interests statement
Dr Johnson is an inventor on several patents and patent applications related to the role of fructose and urate metabolism in hypertension, metabolic syndrome and kidney disease. He also has equity with XORT therapeutics which is developing novel xanthine oxidase inhibitors, and Colorado Research Partners LLC, which is developing inhibitors of fructose metabolism. Finally, he has received honoraria from Eli Lilly, Astra Zeneca and Horizon Pharmaceuticals. Dr. Bjornstad has received consulting fees or speaking honorarium or both from Horizon Pharma, Boehringer Ingelheim, Bayer, and Bristol-Myers Squibb. He also serves on a scientific advisory board for XORTX.
References
- 1.Zhu Y, Pandya BJ & Choi HK Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007-2008. Arthritis and rheumatism 63, 3136–3141, doi: 10.1002/art.30520 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Neogi T & Mikuls TR To Treat or Not to Treat (to Target) in Gout. Ann Intern Med 166, 71–72, doi: 10.7326/M16-2401 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jensen T et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J Hepatol 68, 1063–1075, doi: 10.1016/j.jhep.2018.01.019 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson RJ et al. Hyperuricemia, Acute and Chronic Kidney Disease, Hypertension, and Cardiovascular Disease: Report of a Scientific Workshop Organized by the National Kidney Foundation. Am J Kidney Dis, doi: 10.1053/j.ajkd.2017.12.009 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grayson PC, Kim SY, LaValley M & Choi HK Hyperuricemia and incident hypertension: a systematic review and meta-analysis. Arthritis Care Res (Hoboken) 63, 102–110, doi: 10.1002/acr.20344 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson RJ et al. Sugar, uric acid, and the etiology of diabetes and obesity. Diabetes 62, 3307–3315, doi:62/10/3307 [pii] 10.2337/db12-1814 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lv Q et al. High serum uric Acid and increased risk of type 2 diabetes: a systemic review and meta-analysis of prospective cohort studies. PLoS One 8, e56864, doi: 10.1371/journal.pone.0056864 PONE-D-12–32738 [pii] (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuwabara M et al. Asymptomatic Hyperuricemia Without Comorbidities Predicts Cardiometabolic Diseases: Five-Year Japanese Cohort Study. Hypertension, doi: 10.1161/HYPERTENSIONAHA.116.08998 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamanaka H, Japanese Society of G & Nucleic Acid M Japanese guideline for the management of hyperuricemia and gout: second edition. Nucleosides Nucleotides Nucleic Acids 30, 1018–1029, doi: 10.1080/15257770.2011.596496 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Bursill D, Taylor WJ, Terkeltaub R & Dalbeth N The nomenclature of the basic disease elements of gout: A content analysis of contemporary medical journals. Semin Arthritis Rheum 48, 456–461, doi: 10.1016/j.semarthrit.2018.03.017 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bursill D et al. Gout, Hyperuricemia, and Crystal-Associated Disease Network Consensus Statement Regarding Labels and Definitions for Disease Elements in Gout. Arthritis Care Res (Hoboken) 71, 427–434, doi: 10.1002/acr.23607 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kahn K, Serfozo P & Tipton PA Identification of the true product of the urate oxidase reaction. Journal of the American Chemical Society 119, 5435–5442 (1997). [Google Scholar]
- 13.Smith HW From Fish to Philosopher. (Little, Brown and Co, 1953). [Google Scholar]
- 14.Kratzer JT et al. Evolutionary history and metabolic insights of ancient mammalian uricases. Proceedings of the National Academy of Sciences of the United States of America 111, 3763–3768, doi: 10.1073/pnas.1320393111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oda M, Satta Y, Takenaka O & Takahata N Loss of urate oxidase activity in hominoids and its evolutionary implications. Mol Biol Evol 19, 640–653 (2002). [DOI] [PubMed] [Google Scholar]
- 16.Tan PK, Farrar JE, Gaucher EA & Miner JN Coevolution of URAT1 and Uricase during Primate Evolution: Implications for Serum Urate Homeostasis and Gout. Molecular biology and evolution 33, 2193–2200, doi: 10.1093/molbev/msw116 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Orowan E The origin of man. Nature 175, 683–684 (1955). [DOI] [PubMed] [Google Scholar]
- 18.Brooks GW & Mueller E Serum urate concentrations among university professors; relation to drive, achievement, and leadership. Jama 195, 415–418 (1966). [PubMed] [Google Scholar]
- 19.Sutin AR et al. Impulsivity is associated with uric acid: evidence from humans and mice. Biol Psychiatry 75, 31–37, doi: 10.1016/j.biopsych.2013.02.024 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ames BN, Cathcart R, Schwiers E & Hochstein P Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: a hypothesis. Proc Natl Acad Sci U S A 78, 6858–6862 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sevanian A, Davies KJ & Hochstein P Serum urate as an antioxidant for ascorbic acid. Am J Clin Nutr 54, 1129S–1134S (1991). [DOI] [PubMed] [Google Scholar]
- 22.Watanabe S et al. Uric acid, hominoid evolution, and the pathogenesis of salt-sensitivity. Hypertension 40, 355–360 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Johnson RJ & Andrews P Fructose, Uricase, and the Back-to-Africa Hypothesis. Evol Anthropol 19 250–257 (2010). [Google Scholar]
- 24.Cicerchi C et al. Uric acid-dependent inhibition of AMP kinase induces hepatic glucose production in diabetes and starvation: evolutionary implications of the uricase loss in hominids. FASEB J 28, 3339–3350, doi: 10.1096/fj.13-243634 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gersch C et al. Inactivation of nitric oxide by uric acid. Nucleosides Nucleotides Nucleic Acids 27, 967–978 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Imaram W et al. Radicals in the reaction between peroxynitrite and uric acid identified by electron spin resonance spectroscopy and liquid chromatography mass spectrometry. Free Radic Biol Med 49, 275–281, doi:S0891-5849(10)00228-5 [pii] 10.1016/j.freeradbiomed.2010.04.010 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson RJ, Titte S, Cade JR, Rideout BA & Oliver WJ Uric acid, evolution and primitive cultures. Semin Nephrol 25, 3–8 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Kottgen A et al. Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nature genetics 45, 145–154, doi: 10.1038/ng.2500 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakatochi M et al. Genome-wide meta-analysis identifies multiple novel loci associated with serum uric acid levels in Japanese individuals. Commun Biol 2, 115, doi: 10.1038/s42003-019-0339-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Major TJ, Dalbeth N, Stahl EA & Merriman TR An update on the genetics of hyperuricaemia and gout. Nature reviews. Rheumatology 14, 341–353, doi: 10.1038/s41584-018-0004-x (2018). [DOI] [PubMed] [Google Scholar]
- 31.Tin A et al. Large-scale whole-exome sequencing association studies identify rare functional variants influencing serum urate levels. Nature communications 9, 4228, doi: 10.1038/s41467-018-06620-4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson RJ et al. Is there a pathogenetic role for uric acid in hypertension and cardiovascular and renal disease? Hypertension 41, 1183–1190 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Neogi T Gout. Ann Intern Med 165, ITC1–ITC16, doi: 10.7326/AITC201607050 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Choi HK, Atkinson K, Karlson EW, Willett W & Curhan G Purine-rich foods, dairy and protein intake, and the risk of gout in men. The New England journal of medicine 350, 1093–1103, doi: 10.1056/NEJMoa035700 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Quinones GA et al. Effect of insulin on uric acid excretion in humans. Am J Physiol 268, E1–5 (1995). [DOI] [PubMed] [Google Scholar]
- 36.Lee SM et al. Low serum uric acid level is a risk factor for death in incident hemodialysis patients. Am J Nephrol 29, 79–85, doi:000151292 [pii] 10.1159/000151292 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suliman ME et al. J-shaped mortality relationship for uric acid in CKD. Am J Kidney Dis 48, 761–771 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Kuo CF, Grainge MJ, Zhang W & Doherty M Global epidemiology of gout: prevalence, incidence and risk factors. Nature reviews. Rheumatology 11, 649–662, doi: 10.1038/nrrheum.2015.91 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Faires JS & McCarty DJ Acute arthritis in man and dog after intrasynovial injection of sodium urate crystals. Lancet 280, 682–685 (1962). [DOI] [PubMed] [Google Scholar]
- 40.Shimizu T, Hori H, Umeyama M & Shimizu K Characteristics of gout patients according to the laterality of nephrolithiasis: A cross-sectional study using helical computed tomography. Int J Rheum Dis 22, 567–573, doi: 10.1111/1756-185X.13443 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Landgren AJ et al. Incidence of and risk factors for nephrolithiasis in patients with gout and the general population, a cohort study. Arthritis research & therapy 19, 173, doi: 10.1186/s13075-017-1376-z (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marchini GS, Sarkissian C, Tian D, Gebreselassie S & Monga M Gout, stone composition and urinary stone risk: a matched case comparative study. The Journal of urology 189, 1334–1339, doi: 10.1016/j.juro.2012.09.102 (2013). [DOI] [PubMed] [Google Scholar]
- 43.Hahn K, Kanbay M, Lanaspa MA, Johnson RJ & Ejaz AA Serum uric acid and acute kidney injury: A mini review. J Adv Res 8, 529–536, doi: 10.1016/j.jare.2016.09.006 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shimada M et al. A novel role for uric acid in acute kidney injury associated with tumour lysis syndrome. Nephrol Dial Transplant 24, 2960–2964, doi:gfp330 [pii] 10.1093/ndt/gfp330 (2009). [DOI] [PubMed] [Google Scholar]
- 45.Roncal-Jimenez C et al. Heat Stress Nephropathy From Exercise-Induced Uric Acid Crystalluria: A Perspective on Mesoamerican Nephropathy. Am J Kidney Dis 67, 20–30, doi: 10.1053/j.ajkd.2015.08.021 (2016). [DOI] [PubMed] [Google Scholar]
- 46.Ryu ES et al. Uric acid-induced phenotypic transition of renal tubular cells as a novel mechanism of chronic kidney disease. Am J Physiol Renal Physiol 304, F471–480, doi:ajprenal.00560.2012 [pii] 10.1152/ajprenal.00560.2012 (2013). [DOI] [PubMed] [Google Scholar]
- 47.Zhou Y et al. Uric acid induces renal inflammation via activating tubular NF-kappaB signaling pathway. PLoS One 7, e39738, doi: 10.1371/journal.pone.0039738 PONE-D-12–06772 [pii] (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Verzola D et al. Uric acid promotes apoptosis in human proximal tubule cells by oxidative stress and the activation of NADPH oxidase NOX 4. PLoS One 9, e115210, doi: 10.1371/journal.pone.0115210 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Keenan T et al. Causal Assessment of Serum Urate Levels in Cardiometabolic Diseases Through a Mendelian Randomization Study. J Am Coll Cardiol 67, 407–416, doi: 10.1016/j.jacc.2015.10.086 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.White J et al. Plasma urate concentration and risk of coronary heart disease: a Mendelian randomisation analysis. Lancet Diabetes Endocrinol 4, 327–336, doi: 10.1016/S2213-8587(15)00386-1 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jordan DM et al. No causal effects of serum urate levels on the risk of chronic kidney disease: A Mendelian randomization study. PLoS medicine 16, e1002725, doi: 10.1371/journal.pmed.1002725 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Johnson RJ, Merriman T & Lanaspa MA Causal or Noncausal Relationship of Uric Acid With Diabetes. Diabetes 64, 2720–2722, doi: 10.2337/db15-0532 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ter Horst R et al. Host and Environmental Factors Influencing Individual Human Cytokine Responses. Cell 167, 1111–1124 e1113, doi: 10.1016/j.cell.2016.10.018 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li Y et al. A Functional Genomics Approach to Understand Variation in Cytokine Production in Humans. Cell 167, 1099–1110 e1014, doi: 10.1016/j.cell.2016.10.017 (2016). [DOI] [PubMed] [Google Scholar]
- 55.Su X, Xu B, Yan B, Qiao X & Wang L Effects of uric acid-lowering therapy in patients with chronic kidney disease: A meta-analysis. PLoS One 12, e0187550, doi: 10.1371/journal.pone.0187550 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Qu LH, Jiang H & Chen JH Effect of uric acid-lowering therapy on blood pressure: systematic review and meta-analysis. Ann Med 49, 142–156, doi: 10.1080/07853890.2016.1243803 (2017). [DOI] [PubMed] [Google Scholar]
- 57.Martinon F, Petrilli V, Mayor A, Tardivel A & Tschopp J Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241, doi: 10.1038/nature04516 (2006). [DOI] [PubMed] [Google Scholar]
- 58.Dinarello CA A clinical perspective of IL-1beta as the gatekeeper of inflammation. European journal of immunology 41, 1203–1217, doi: 10.1002/eji.201141550 (2011). [DOI] [PubMed] [Google Scholar]
- 59.Dinarello CA The history of fever, leukocytic pyrogen and interleukin-1. Temperature (Austin) 2, 8–16, doi: 10.1080/23328940.2015.1017086 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Netea MG et al. Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood 113, 2324–2335, doi: 10.1182/blood-2008-03-146720 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gross O, Thomas CJ, Guarda G & Tschopp J The inflammasome: an integrated view. Immunological reviews 243, 136–151, doi: 10.1111/j.1600-065X.2011.01046.x (2011). [DOI] [PubMed] [Google Scholar]
- 62.Martinon F Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241 (2006). [DOI] [PubMed] [Google Scholar]
- 63.Misawa T et al. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nature immunology 14, 454–460, doi: 10.1038/ni.2550 (2013). [DOI] [PubMed] [Google Scholar]
- 64.Joosten LA et al. Engagement of fatty acids with Toll-like receptor 2 drives interleukin-1beta production via the ASC/caspase 1 pathway in monosodium urate monohydrate crystal-induced gouty arthritis. Arthritis and rheumatism 62, 3237–3248, doi: 10.1002/art.27667 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jhang JJ, Cheng YT, Ho CY & Yen GC Monosodium urate crystals trigger Nrf2- and heme oxygenase-1-dependent inflammation in THP-1 cells. Cell Mol Immunol 12, 424–434, doi: 10.1038/cmi.2014.65 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim SK, Choe JY & Park KY Enhanced p62 Is Responsible for Mitochondrial Pathway-Dependent Apoptosis and Interleukin-1beta Production at the Early Phase by Monosodium Urate Crystals in Murine Macrophage. Inflammation 39, 1603–1616, doi: 10.1007/s10753-016-0387-2 (2016). [DOI] [PubMed] [Google Scholar]
- 67.Wang Y, Viollet B, Terkeltaub R & Liu-Bryan R AMP-activated protein kinase suppresses urate crystal-induced inflammation and transduces colchicine effects in macrophages. Annals of the rheumatic diseases, doi: 10.1136/annrheumdis-2014-206074 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vazirpanah N et al. mTOR inhibition by metformin impacts monosodium urate crystal-induced inflammation and cell death in gout: a prelude to a new add-on therapy? Annals of the rheumatic diseases 78, 663–671, doi: 10.1136/annrheumdis-2018-214656 (2019). [DOI] [PubMed] [Google Scholar]
- 69.Netea MG, van de Veerdonk FL, van der Meer JW, Dinarello CA & Joosten LA Inflammasome-independent regulation of IL-1-family cytokines. Annual review of immunology 33, 49–77, doi: 10.1146/annurev-immunol-032414-112306 (2015). [DOI] [PubMed] [Google Scholar]
- 70.Cho JS et al. Neutrophil-derived IL-1beta is sufficient for abscess formation in immunity against Staphylococcus aureus in mice. PLoS pathogens 8, e1003047, doi: 10.1371/journal.ppat.1003047 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schreiber A et al. Neutrophil serine proteases promote IL-1beta generation and injury in necrotizing crescentic glomerulonephritis. Journal of the American Society of Nephrology : JASN 23, 470–482, doi: 10.1681/ASN.2010080892 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Joosten LA et al. Alpha-1-anti-trypsin-Fc fusion protein ameliorates gouty arthritis by reducing release and extracellular processing of IL-1beta and by the induction of endogenous IL-1Ra. Annals of the rheumatic diseases, doi: 10.1136/annrheumdis-2014-206966 (2015). [DOI] [PubMed] [Google Scholar]
- 73.Cumpelik A, Ankli B, Zecher D & Schifferli JA Neutrophil microvesicles resolve gout by inhibiting C5a-mediated priming of the inflammasome. Annals of the rheumatic diseases 75, 1236–1245, doi: 10.1136/annrheumdis-2015-207338 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schauer C et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nature medicine 20, 511–517, doi: 10.1038/nm.3547 (2014). [DOI] [PubMed] [Google Scholar]
- 75.Schorn C et al. Bonding the foe - NETting neutrophils immobilize the pro-inflammatory monosodium urate crystals. Frontiers in immunology 3, 376, doi: 10.3389/fimmu.2012.00376 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hahn J et al. Aggregated neutrophil extracellular traps resolve inflammation by proteolysis of cytokines and chemokines and protection from antiproteases. FASEB journal : official publication of the Federation of American Societies for Experimental Biology, fj201800752R, doi: 10.1096/fj.201800752R (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Reber LL, Gaudenzio N, Starkl P & Galli SJ Neutrophils are not required for resolution of acute gouty arthritis in mice. Nature medicine 22, 1382–1384, doi: 10.1038/nm.4216 (2016). [DOI] [PubMed] [Google Scholar]
- 78.Reinwald C et al. Reply to “Neutrophils are not required for resolution of acute gouty arthritis in mice”. Nature medicine 22, 1384–1386, doi: 10.1038/nm.4217 (2016). [DOI] [PubMed] [Google Scholar]
- 79.Desai J, Steiger S & Anders HJ Molecular Pathophysiology of Gout. Trends Mol Med 23, 756–768, doi: 10.1016/j.molmed.2017.06.005 (2017). [DOI] [PubMed] [Google Scholar]
- 80.Manfredi AA, Ramirez GA, Rovere-Querini P & Maugeri N The Neutrophil’s Choice: Phagocytose vs Make Neutrophil Extracellular Traps. Frontiers in immunology 9, 288, doi: 10.3389/fimmu.2018.00288 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gersch C et al. Reactions of peroxynitrite with uric acid: formation of reactive intermediates, alkylated products and triuret, and in vivo production of triuret under conditions of oxidative stress. Nucleosides Nucleotides Nucleic Acids 28, 118–149, doi:908701251 [pii] 10.1080/15257770902736400 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Meotti FC et al. Urate as a physiological substrate for myeloperoxidase: implications for hyperuricemia and inflammation. J Biol Chem 286, 12901–12911, doi:M110.172460 [pii] 10.1074/jbc.M110.172460 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Corry DB et al. Uric acid stimulates vascular smooth muscle cell proliferation and oxidative stress via the vascular renin-angiotensin system. J Hypertens 26, 269–275 (2008). [DOI] [PubMed] [Google Scholar]
- 84.Yu MA, Sanchez-Lozada LG, Johnson RJ & Kang DH Oxidative stress with an activation of the renin-angiotensin system in human vascular endothelial cells as a novel mechanism of uric acid-induced endothelial dysfunction. J Hypertens 28, 1234–1242 (2010). [PubMed] [Google Scholar]
- 85.Sautin YY, Nakagawa T, Zharikov S & Johnson RJ Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress. Am J Physiol Cell Physiol 293, C584–596, doi:00600.2006 [pii] 10.1152/ajpcell.00600.2006 (2007). [DOI] [PubMed] [Google Scholar]
- 86.Lanaspa MA et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: potential role in fructose-dependent and -independent fatty liver. J Biol Chem 287, 40732–40744, doi:M112.399899 [pii] 10.1074/jbc.M112.399899 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Roncal-Jimenez CA et al. Sucrose induces fatty liver and pancreatic inflammation in male breeder rats independent of excess energy intake. Metabolism 60, 1259–1270, doi:S0026-0495(11)00020-5 [pii] 10.1016/j.metabol.2011.01.008 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cirillo P et al. Ketohexokinase-dependent metabolism of fructose induces proinflammatory mediators in proximal tubular cells. J Am Soc Nephrol 20, 545–553, doi:ASN.2008060576 [pii] 10.1681/ASN.2008060576 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Choi YJ et al. Uric acid induces endothelial dysfunction by vascular insulin resistance associated with the impairment of nitric oxide synthesis. FASEB J 28, 3197–3204, doi: 10.1096/fj.13-247148 (2014). [DOI] [PubMed] [Google Scholar]
- 90.Sanchez-Lozada LG et al. Uric Acid-Induced Endothelial Dysfunction Is Associated with Mitochondrial Alterations and Decreased Intracellular ATP Concentrations. Nephron Exp Nephrol 121, e71–e78, doi:000345509 [pii] 10.1159/000345509 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Crisan TO et al. Uric acid priming in human monocytes is driven by the AKT-PRAS40 autophagy pathway. Proc Natl Acad Sci U S A 114, 5485–5490, doi: 10.1073/pnas.1620910114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Alberts BM et al. Secretion of IL-1beta From Monocytes in Gout Is Redox Independent. Frontiers in immunology 10, 70, doi: 10.3389/fimmu.2019.00070 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kang DH, Park SK, Lee IK & Johnson RJ Uric acid-induced C-reactive protein expression: implication on cell proliferation and nitric oxide production of human vascular cells. J Am Soc Nephrol 16, 3553–3562 (2005). [DOI] [PubMed] [Google Scholar]
- 94.Kanellis J et al. Uric acid stimulates monocyte chemoattractant protein-1 production in vascular smooth muscle cells via mitogen-activated protein kinase and cyclooxygenase-2. Hypertension 41, 1287–1293 (2003). [DOI] [PubMed] [Google Scholar]
- 95.Xu C et al. Activation of Renal (Pro)Renin Receptor Contributes to High Fructose-Induced Salt Sensitivity. Hypertension 69, 339–348, doi: 10.1161/HYPERTENSIONAHA.116.08240 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kang DH et al. A role for uric acid in the progression of renal disease. J Am Soc Nephrol 13, 2888–2897 (2002). [DOI] [PubMed] [Google Scholar]
- 97.Rao GN, Corson MA & Berk BC Uric acid stimulates vascular smooth muscle cell proliferation by increasing platelet-derived growth factor A-chain expression. J Biol Chem 266, 8604–8608 (1991). [PubMed] [Google Scholar]
- 98.Netea MG, Kullberg BJ, Blok WL, Netea RT & van der Meer JW The role of hyperuricemia in the increased cytokine production after lipopolysaccharide challenge in neutropenic mice. Blood 89, 577–582 (1997). [PubMed] [Google Scholar]
- 99.Jia L et al. Hyperuricemia causes pancreatic beta-cell death and dysfunction through NF-kappaB signaling pathway. PloS one 8, e78284, doi: 10.1371/journal.pone.0078284 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lu W et al. Uric Acid Produces an Inflammatory Response through Activation of NF-kappaB in the Hypothalamus: Implications for the Pathogenesis of Metabolic Disorders. Scientific reports 5, 12144, doi: 10.1038/srep12144 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Crisan TO et al. Soluble uric acid primes TLR-induced proinflammatory cytokine production by human primary cells via inhibition of IL-1Ra. Annals of the rheumatic diseases, doi: 10.1136/annrheumdis-2014-206564 (2015). [DOI] [PubMed] [Google Scholar]
- 102.Wiza C, Nascimento EB & Ouwens DM Role of PRAS40 in Akt and mTOR signaling in health and disease. American journal of physiology. Endocrinology and metabolism 302, E1453–1460, doi: 10.1152/ajpendo.00660.2011 (2012). [DOI] [PubMed] [Google Scholar]
- 103.Dunlop EA & Tee AR mTOR and autophagy: a dynamic relationship governed by nutrients and energy. Semin Cell Dev Biol 36, 121–129, doi: 10.1016/j.semcdb.2014.08.006 (2014). [DOI] [PubMed] [Google Scholar]
- 104.Braga TT et al. Soluble Uric Acid Activates the NLRP3 Inflammasome. Scientific reports 7, 39884, doi: 10.1038/srep39884 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Buffen K et al. Autophagy suppresses host adaptive immune responses toward Borrelia burgdorferi. Journal of leukocyte biology 100, 589–598, doi: 10.1189/jlb.4A0715-331R (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Garcia D & Shaw RJ AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Molecular cell 66, 789–800, doi: 10.1016/j.molcel.2017.05.032 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.O’Neill LA & Hardie DG Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature 493, 346–355, doi: 10.1038/nature11862 (2013). [DOI] [PubMed] [Google Scholar]
- 108.Zhang Y et al. Uric acid induces oxidative stress and growth inhibition by activating adenosine monophosphate-activated protein kinase and extracellular signal-regulated kinase signal pathways in pancreatic beta cells. Molecular and cellular endocrinology 375, 89–96, doi: 10.1016/j.mce.2013.04.027 (2013). [DOI] [PubMed] [Google Scholar]
- 109.Lanaspa MA et al. Counteracting roles of AMP deaminase and AMP kinase in the development of fatty liver. PLoS One 7, e48801, doi: 10.1371/journal.pone.0048801 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Luo C et al. High Uric Acid Activates the ROS-AMPK Pathway, Impairs CD68 Expression and Inhibits OxLDL-Induced Foam-Cell Formation in a Human Monocytic Cell Line, THP-1. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology 40, 538–548, doi: 10.1159/000452567 (2016). [DOI] [PubMed] [Google Scholar]
- 111.Xin Y et al. Zurampic Protects Pancreatic beta-Cells from High Uric Acid Induced-Damage by Inhibiting URAT1 and Inactivating the ROS/AMPK/ERK Pathways. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology 47, 1074–1083, doi: 10.1159/000490184 (2018). [DOI] [PubMed] [Google Scholar]
- 112.Yuan H et al. Metformin ameliorates high uric acid-induced insulin resistance in skeletal muscle cells. Molecular and cellular endocrinology 443, 138–145, doi: 10.1016/j.mce.2016.12.025 (2017). [DOI] [PubMed] [Google Scholar]
- 113.Netea MG, Quintin J & van der Meer JW Trained immunity: a memory for innate host defense. Cell host & microbe 9, 355–361, doi: 10.1016/j.chom.2011.04.006 (2011). [DOI] [PubMed] [Google Scholar]
- 114.Saeed S et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 345, 1251086, doi: 10.1126/science.1251086 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Novakovic B et al. beta-Glucan Reverses the Epigenetic State of LPS-Induced Immunological Tolerance. Cell 167, 1354–1368 e1314, doi: 10.1016/j.cell.2016.09.034 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Blok BA, Arts RJ, van Crevel R, Benn CS & Netea MG Trained innate immunity as underlying mechanism for the long-term, nonspecific effects of vaccines. Journal of leukocyte biology, doi: 10.1189/jlb.5RI0315-096R (2015). [DOI] [PubMed] [Google Scholar]
- 117.Crisan TO, Netea MG & Joosten LA Innate immune memory: Implications for host responses to damage-associated molecular patterns. European journal of immunology 46, 817–828, doi: 10.1002/eji.201545497 (2016). [DOI] [PubMed] [Google Scholar]
- 118.Rajasekar P, O’Neill CL, Eeles L, Stitt AW & Medina RJ Epigenetic Changes in Endothelial Progenitors as a Possible Cellular Basis for Glycemic Memory in Diabetic Vascular Complications. Journal of diabetes research 2015, 436879, doi: 10.1155/2015/436879 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bekkering S, Joosten LA, van der Meer JW, Netea MG & Riksen NP Trained innate immunity and atherosclerosis. Current opinion in lipidology 24, 487–492, doi: 10.1097/MOL.0000000000000023 (2013). [DOI] [PubMed] [Google Scholar]
- 120.van der Valk FM et al. Oxidized Phospholipids on Lipoprotein(a) Elicit Arterial Wall Inflammation and an Inflammatory Monocyte Response in Humans. Circulation 134, 611–624, doi: 10.1161/CIRCULATIONAHA.116.020838 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bekkering S et al. Treatment with Statins Does Not Revert Trained Immunity in Patients with Familial Hypercholesterolemia. Cell metabolism 30, 1–2, doi: 10.1016/j.cmet.2019.05.014 (2019). [DOI] [PubMed] [Google Scholar]
- 122.Ruggiero C et al. Uric acid and inflammatory markers. Eur Heart J 27, 1174–1181, doi: 10.1093/eurheartj/ehi879 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Grainger R, McLaughlin RJ, Harrison AA & Harper JL Hyperuricaemia elevates circulating CCL2 levels and primes monocyte trafficking in subjects with inter-critical gout. Rheumatology 52, 1018–1021, doi: 10.1093/rheumatology/kes326 (2013). [DOI] [PubMed] [Google Scholar]
- 124.Gao L et al. Male asymptomatic hyperuricemia patients display a lower number of NKG2D+ NK cells before and after a low-purine diet. Medicine (Baltimore) 97, e13668, doi: 10.1097/MD.0000000000013668 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mylona EE et al. Enhanced interleukin-1beta production of PBMCs from patients with gout after stimulation with Toll-like receptor-2 ligands and urate crystals. Arthritis research & therapy 14, R158, doi: 10.1186/ar3898 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tanaka T et al. A double blind placebo controlled randomized trial of the effect of acute uric acid changes on inflammatory markers in humans: A pilot study. PloS one 12, e0181100, doi: 10.1371/journal.pone.0181100 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Abhishek A, Valdes AM, Zhang W & Doherty M Association of Serum Uric Acid and Disease Duration With Frequent Gout Attacks: A Case-Control Study. Arthritis Care Res (Hoboken) 68, 1573–1577, doi: 10.1002/acr.22855 (2016). [DOI] [PubMed] [Google Scholar]
- 128.Griffith JW, Sun T, McIntosh MT & Bucala R Pure Hemozoin is inflammatory in vivo and activates the NALP3 inflammasome via release of uric acid. J Immunol 183, 5208–5220, doi: 10.4049/jimmunol.0713552 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Orengo JM et al. Uric acid is a mediator of the Plasmodium falciparum-induced inflammatory response. PLoS ONE 4, e5194, doi: 10.1371/journal.pone.0005194 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Shriner D et al. Evolutionary context for the association of gamma-globin, serum uric acid, and hypertension in African Americans. BMC Med Genet 16, 103, doi: 10.1186/s12881-015-0249-z (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Spaetgens B et al. Risk of infections in patients with gout: a population-based cohort study. Scientific reports 7, 1429, doi: 10.1038/s41598-017-01588-5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bardin T & Richette P Definition of hyperuricemia and gouty conditions. Current opinion in rheumatology 26, 186–191, doi: 10.1097/BOR.0000000000000028 (2014). [DOI] [PubMed] [Google Scholar]
- 133.Inaba S, Sautin Y, Garcia GE & Johnson RJ What can asymptomatic hyperuricaemia and systemic inflammation in the absence of gout tell us? Rheumatology (Oxford) 52, 963–965, doi:ket001 [pii] 10.1093/rheumatology/ket001 (2013). [DOI] [PubMed] [Google Scholar]
- 134.Dalbeth N et al. Urate crystal deposition in asymptomatic hyperuricaemia and symptomatic gout: a dual energy CT study. Annals of the rheumatic diseases 74, 908–911, doi: 10.1136/annrheumdis-2014-206397 (2015). [DOI] [PubMed] [Google Scholar]
- 135.Perez-Ruiz F, Marimon E & Chinchilla SP Hyperuricaemia with deposition: latest evidence and therapeutic approach. Ther Adv Musculoskelet Dis 7, 225–233, doi: 10.1177/1759720X15599734 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Asghar ZA et al. Maternal fructose drives placental uric acid production leading to adverse fetal outcomes. Sci Rep 6, 25091, doi: 10.1038/srep25091 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sato Y et al. The case for uric acid- lowering treatment in patients with hyperuricaemia and CKD. Nature Rev Nephrol in press (2019). [DOI] [PubMed] [Google Scholar]