

Summary
The SUSD4 (Sushi domain-containing protein 4) gene encodes a complement inhibitor that is frequently deleted in 1q41q42 microdeletion syndrome, a multisystem congenital disorder that includes neurodevelopmental abnormalities. To understand SUSD4's role in the mammalian nervous system, we analyzed Susd4 knockout (KO) mice. Susd4 KO mice exhibited significant defects in motor performance and significantly higher levels of anxiety-like behaviors. Susd4 KO brain had abnormal “hairy” basket cells surrounding Purkinje neurons within the cerebellum and significantly reduced dendritic spine density in hippocampal pyramidal neurons. Neurons and oligodendrocyte lineage cells of wild-type mice were found to express Susd4 mRNA. Protein expression of the complement component C1q was increased in the brains of Susd4 KO mice. Our data indicate that SUSD4 plays an important role in neuronal functions, possibly via the complement pathway, and that SUSD4 deletion may contribute to the nervous system abnormalities in patients with 1q41q42 deletions.
Subject Areas: Behavioral Neuroscience, Cellular Neuroscience, Components of the Immune System, Neuroscience
Graphical Abstract
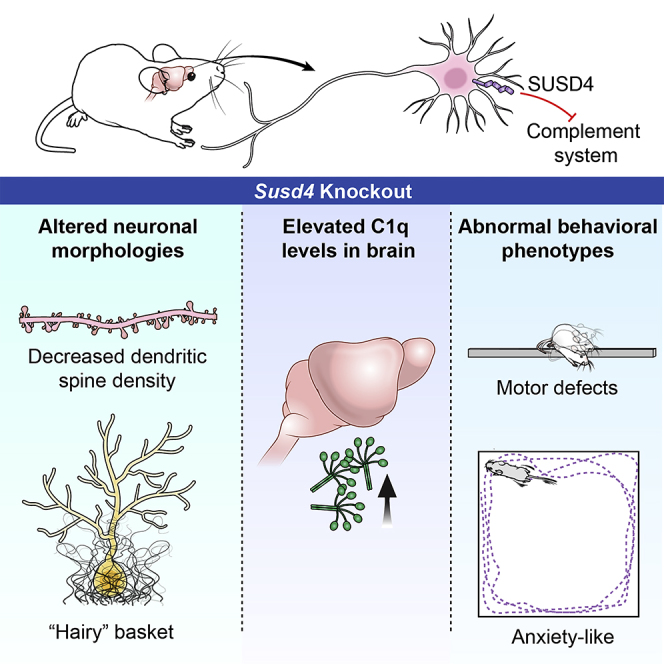
Highlights
-
•
Susd4 is expressed in neurons and oligodendrocyte lineage cells
-
•
Susd4 knockout mice have abnormal hippocampal and cerebellar neuronal morphologies
-
•
Susd4 knockout mice exhibit anxiety-like behaviors and impaired motor function
-
•
Susd4 knockout mice have elevated brain levels of the complement component C1q
Behavioral Neuroscience; Cellular Neuroscience; Components of the Immune System; Neuroscience
Introduction
The complement system, a critical part of the immune system, helps defend against invading pathogens, promote inflammation, and clear damaged cells and debris (Hajishengallis et al., 2017, Holers, 2014). In the central nervous system (CNS), complement components have been found to play important roles in neuron survival and synaptic pruning under both normal and disease conditions (Hong et al., 2016, Lui et al., 2016, Schafer et al., 2012, Stevens et al., 2007). However, the regulatory mechanisms for complement expression and function in the CNS are still largely unknown.
The SUSD4 (Sushi domain-containing protein 4) gene resides on chromosome 1q41 and encodes a 49-kD transmembrane protein containing four extracellular Sushi domain motifs (Tu et al., 2010). The Sushi domain, also known as the complement control protein domain, is commonly involved in protein-protein interactions. Many complement regulators are constructed with complement control protein domains (Gialeli et al., 2018, Kirkitadze and Barlow, 2001). SUSD4 functions as a complement system regulator, as it has been shown to bind to the C1 complex and complement component C1q and block the activation of the complement cascade by interrupting the formation of C3 convertase (Holmquist et al., 2013). However, SUSD4 has also been reported to inhibit the alternative pathway warranting further studies to clarify the SUSD4 mechanisms of action on the complement system.
In humans, the CNS is a major site of SUSD4 expression (Holmquist et al., 2013). Deletion of the SUSD4 gene, together with several other genes, frequently occurs in patients with 1q41q42 microdeletion syndrome (Shaffer et al., 2007), who generally exhibit seizures, significant developmental delay, and intellectual disability, as well as multiple congenital abnormalities, along a variable phenotypic spectrum. It is not known if SUSD4, when included in the deletion interval, contributes to the patients' clinical presentation. Because the physiologic role of SUSD4 in the mammalian nervous system was not yet established, we used Susd4 knockout (KO) mice to identify the neurologic functions of Susd4. We found that Susd4 deletion affects both behavioral phenotypes and neuronal morphology in these mice, demonstrating an important CNS role for SUSD4, and that its deletion may contribute to the phenotypic spectrum of patients with 1q41q42 microdeletion syndrome.
Results
Susd4 KO Mice Exhibit Behavioral Abnormalities
We first determined the relative tissue expression of Susd4 mRNA in wild-type (WT) mice using qPCR (targeting exon 1–2 and exon 5–6 junctions) (Figure 1A). Susd4 mRNA expression was highest in the WT brain and was detectable at lower levels in lung, liver, kidney, and testis, as well as during development from embryonic day 11–17.
Figure 1.

Susd4 KO Mice Exhibit Behavioral Abnormalities
(A) Relative mRNA expression of Susd4 determined by qPCR in a panel of 12 different tissues from WT mice, as well as Susd4 KO mouse brain tissue (far right). qPCR assays for Susd4 were performed using probes spanning exons 1 and 2 (Susd4exon1-2) and exons 5 and 6 (Susd4exon5-6) (n = 3). mRNA expression was normalized to Gapdh mRNA expression. E, embryonic day; N.D., not detectable.
(B) Horizontal balance-beam test. Time for mice to cross 80-cm-long beams of different widths was recorded. Width of the beams: beam 1 is 24 mm; beam 2 is 12 mm; beam 3 is 9.5 mm. Susd4 KO, n = 17; WT, n = 11.
(C) Accelerating-rotarod test. Latency time for mice to fall from the rotarod was recorded. Susd4 KO, n = 17; WT, n = 15.
(D–F) Elevated zero-maze test. (D) Time mice spent in closed and open arms of the maze was recorded for 5 min. (E) Number of open-arm entries by mice was recorded for 5 min. (F) Total distance mice traveled in the open arms was recorded for 5 min Susd4 KO, n = 17; WT, n = 15.
(G–I) Open-field test. (G) Representative tracing of the path traveled by a WT and a Susd4 KO mouse over 30 min in the test. The blue dot represents the starting point and the red dot represents the endpoint of the path. (H) Total distance mice traveled was recorded for 30 min. (I) Time mice spent in the center of the field was recorded for 30 min Susd4 KO, n = 17; WT, n = 15.
(J) Cylinder test. Number of rears by mice was recorded for 90 s Susd4 KO, n = 17; WT, n = 15.
(K and L) Hole-board test. (K) Total distance mice traveled was recorded for 5 min. (L) Number of head pokes by mice into holes was recorded for 5 min Susd4 KO, n = 17; WT, n = 15.
Data represent the mean ± SEM. ∗: p < 0.05; ∗∗: p < 0.005; ∗∗∗: p < 0.0005; ∗∗∗∗: p < 0.0001. For a complete behavioral phenotyping battery, see also Table S1 and Figure S1.
To identify the physiologic functions of the Susd4 gene, we utilized a Susd4 KO mouse model (Tang et al., 2010). Expression of Susd4 mRNA was not detectable in Susd4 KO mouse brain, confirming the presence of a null Susd4 allele (Figure 1A, far right). Susd4 KO mice were viable, fertile, and grew normally (Tang et al., 2010) (see also Table S1). We subsequently performed behavioral phenotyping to determine if the Susd4 deletion would affect nervous-system functions. Compared with WT mice, Susd4 KO mice exhibited defects in motor and coordination function, as revealed by horizontal balance-beam and accelerating-rotarod tests. In the balance-beam test, as the width of the balance beam decreased, which increased the demand for fine locomotor control, the mean time that the Susd4 KO mice spent to cross the beam was significantly longer than that of WT mice (Figure 1B). In the accelerating-rotarod test, the mean latency time that Susd4 KO mice stayed on the rotarod as it accelerated was significantly shorter, indicating poorer motor coordination, than that of WT mice (Figure 1C). Gross sensorimotor and locomotor functions were not affected as assessed by Shirpa screening and DigiGait tests (Table S1, Figures S1A and S1B).
Further behavioral testing indicated that Susd4 KO mice exhibited anxiety-like behavioral phenotypes (Crawley, 1985, Holter et al., 2015). In the elevated zero-maze test Susd4 KO mice spent significantly more mean time in the closed arms (and significantly less mean time in the open arms) (Figure 1D), and the mean frequency of open-arm entries (Figure 1E) and the mean distance traveled in the open arms were significantly less than those observed in WT mice (Figure 1F). In the open-field test, both the mean total distance traveled (Figures 1G and 1H) and the mean time spent in the center of the field were significantly less for Susd4 KO mice than for WT mice (Figures 1G and 1I). In addition, the Susd4 KO mice undertook significantly reduced exploratory activity in the cylinder test (mean number of rears; Figure 1J) and the hole-board test (mean total distance traveled; Figure 1K) and mean number of head-poking into holes (Figure 1L) compared with WT mice. Learning and memory functions were generally not impaired in the Susd4 KO mice as revealed by Morris water maze testing (Figure S1B).
Susd4 KO Mice Exhibit Abnormal Neuronal Morphology and Hypertrophic Microglia
Bielschowsky silver staining is a histochemical method to visualize nerve fibers and has been used in studies of Alzheimer's disease and other neurodegenerative disorders to identify axonal or neurofibrillary pathology (Spittaels et al., 1999, Switzer, 2000, Yamamoto and Hirano, 1986). Using this stain, we found an abnormal morphology of basket cells, termed “hairy” (Erickson-Davis et al., 2010), indicating a dense and tangled axonal plexus surrounding Purkinje cell soma, in the cerebellum of Susd4 KO mice that was not found in the cerebellum of WT mice (Figure 2A). Changes in neurofilament heavy chain phosphorylation status is an indicator for axonal degeneration in neurological diseases and can be detected with the SMI-31 antibody (Dale and Garcia, 2012, Petzold, 2005, Stone et al., 2019, Wang et al., 2001). Immunostaining with SMI-31 revealed that the hairy basket cell dendrites enclosing the cell bodies of calbindin-positive Purkinje neurons in the Susd4 KO mice prominently expressed phosphorylated neurofilament heavy chain (Figures 2B and 2C). Abnormal hairy basket cells are believed to be associated with Purkinje cell degeneration (Erickson-Davis et al., 2010), although no significant difference in the numbers of cerebellar Purkinje cells in Susd4 KO and WT mice was found (Figure S2). The abnormal basket cell morphology may be related to the motor function defects observed in Susd4 KO mice.
Figure 2.

Susd4 KO Mice Exhibit Abnormal Neuronal Morphology and Hypertrophic Microglia
(A) Bielschowsky silver staining of cerebellum. Top panels, representative sagittal view of the whole cerebellum for a WT and a Susd4 KO mouse. Arrows indicate the dense-appearing basket cell layer in the Susd4 KO mouse cerebellum relative to the WT tissue. Scale bar, 200 μm. Lower panels, representative higher magnification view shows Purkinje cell layer and basket cells (arrows). Note “hairy” appearance of basket cells in the Susd4 KO specimen. Scale bar, 20 μm
(B) Representative immunostaining with SMI-31antibody, labeling phosphorylated neurofilament heavy chain (green), and calbindin antibody, labeling Purkinje cells (red), in cerebellum sections from Susd4 KO and WT mice; scale bar, 20 μm.
(C) Representative higher-magnification view showing “hairy” basket cell dendrites (SMI-31 immunostaining in green) surrounding Purkinje cell soma (calbindin immunostaining in red); scale bar, 10 μm.
(D) Golgi-Cox staining of brains. Representative images of dendritic spines for WT and Susd4 KO mice.
(E) Quantification of the number of spines per micrometer dendrite on secondary or tertiary branches from both apical and basal parts of the pyramidal cells in hippocampal CA regions of WT and Susd4 KO Golgi-stained mouse brains. Data represent the mean ± SEM. n = 3 mice per group; ∗∗∗∗: p < 0.0001.
(F) Iba1immunostaining (red) for microglia. Top panels, representative view of hippocampal regions from WT and Susd4 KO mice. Scale bar, 40 μm. Lower panels, representative higher magnification view shows ramified resting microglia (arrows in top panel) in WT in contrast with hypertrophic microglia (arrow heads in top panel) in Susd4 KO. CA, cornu ammonis. Scale bar, 10 μm.
(G) Quantification of Iba1+ cells per square millimeter in hippocampal regions from WT and Susd4 KO mice. Data represent the mean ± SEM. n = 3 mice per group. ∗: p < 0.05.
(H) Quantification of Iba1 fluorescence intensity/microglia from the stained hippocampal sections. Data represent the mean ± SEM. WT values were set to 1.0. n = 3 mice per group. ∗∗∗∗: p < 0.0001. Nuclei were visualized with DAPI staining (blue) for (B), (C), and (F).
The Golgi-Cox impregnation method is a classical technique used to reveal the morphology of whole individual neurons, including dendritic spines (Mancuso et al., 2013, Rosoklija et al., 2014). Dendritic spines are small protrusions that receive inputs from axons at the synapse, which are important for regulation of neuron activities and connections. Abnormalities in dendritic spines, such as changes in spine number or density, may lead to neuron degeneration and cognitive disorders (Fiala et al., 2002, Penzes et al., 2011, Skaper et al., 2017). Using the Golgi-Cox staining technique, we found a significant reduction in dendritic spine density on the pyramidal cells in hippocampal CA region in Susd4 KO mice compared with WT mice (Figures 2D and 2E).
Microglia, resident immune cells in the brain, are involved in neuronal synaptic pruning mediated by complement factors (Tenner et al., 2018). Immunostaining of microglia with Iba1 antibodies revealed dense hypertrophic morphology of microglia in the Susd4 KO hippocampal sections compared with the classic ramified morphology of resting microglia in the WT hippocampus (Figure 2F). The Susd4 KO microglia were increased in number and stained more intensely with Iba1 antibody (Figures 2G and 2H). These results suggest activation of microglia in the Susd4 KO hippocampi, a process that has been correlated with elevated synaptic pruning (Hong et al., 2016).
Susd4 Is Highly Expressed in Hippocampal Neurons and Cerebellar Purkinje Cells
Although Susd4 is highly expressed in brain, its normal cellular expression in hippocampus and cerebellum—areas of abnormal neuronal morphology in Susd4 KO mice—was not known. We initially tested three different commercial SUSD4 antibodies on mouse brain tissue sections, and all showed the same immunoreactivity in tissues from Susd4 KO mice compared with WT mice, indicating a lack of sufficient antibody specificity. Therefore, we used the RNAscope Multiplex Fluorescent Assay to probe the cellular expression of Susd4 mRNA. This in situ RNA hybridization assay allows simultaneous detection of three different mRNA targets per sample, with each target probe binding to a specific mRNA, each of which is then labeled with different fluorescence detectors. Initially, we used probes for Susd4 mRNA, Gfap mRNA (specific for astrocytes), and Rbfox (also known as NeuN) mRNA (specific for most neuronal types). As a specificity control for the Susd4 probe we also hybridized sections from the Susd4 KO mice, which showed no Susd4 mRNA (Figure 3A). The WT mouse brain hippocampal CA layer and dentate gyrus were highly positive for NeuN mRNA (red) and Susd4 mRNA (green) (Figure 3B). Gfap mRNA (cyan) was diffusely distributed (Figure 3B). At a higher-resolution view, Susd4 mRNA was co-localized in cells expressing NeuN mRNA (Figure 3C) but not in cells expressing Gfap mRNA (Figure 3D). In the cerebellum of WT mice, the granular layer, which is adjacent to the Purkinje cell layer, was highly positive for NeuN mRNA (red). Purkinje cells do not express NeuN (Mullen et al., 1992). In WT mice, both the granular layer and Purkinje cell layer regions were positive for Susd4 mRNA (green), with the large Purkinje cells showing an intense reaction. Gfap mRNA (cyan) expression was not enriched in either of these layers (Figures 3E and 3F). These results indicate that Susd4 mRNA has relatively high expression in both hippocampal neurons and cerebellar Purkinje cells, which are areas that exhibited abnormal neuronal morphology in Susd4 KO mice.
Figure 3.

Susd4 mRNA is Expressed in Neuronal Cells in WT Brain
The RNAscope Multiplex Fluorescent Assay was performed with sagittal brain sections from WT and Susd4 KO mice. Sets of probes were used to detect mRNA expression of Susd4 (green), NeuN (red), and Gfap (cyan) in situ. Nuclei were visualized with DAPI staining (blue).
(A) Representative images from cortex sections show the assay specifically detects Susd4 mRNA in WT but not in Susd4 KO, whereas NeuN mRNA is detected in both. Scale bar, 20 μm.
(B) Susd4 mRNA is highly expressed in the hippocampus. Scale bar, 100 μm. CA, cornu ammonis; DG, dentate gyrus.
(C and D) Representative higher-magnification views show that Susd4 mRNA colocalizes with NeuN (C) but not Gfap (D). Scale bar, 10 μm.
(E) Susd4mRNA is highly expressed in Purkinje cells not the granular cells in cerebellum. Scale bar, 40 μm. PCL, Purkinje cell layer; GL, granular layer.
(F) Representative higher-magnification view shows the high expression of Susd4 mRNA in Purkinje neurons. Scale bar, 10 μm.
We next determined if Susd4 mRNA is also expressed in the oligodendrocyte lineage using an Olig2 probe (Zhou et al., 2000). Our results indicated that a subset of Olig2-expressing cells also express Susd4 mRNA in the cortex and corpus callosum (Figures S3A and S3B). Approximately 35% of the total Olig2+ cells in the cortex and 7.5% of the total Olig2+ in the corpus callosum were also positive for Susd4 (Figures S3C and S3D), similar to the levels of oligodendrocyte progenitors in these regions (Young et al., 2013). Although Olig2+ cells were reduced in the Susd4 KO cortex compared with WT, myelin basic protein (MBP) expression was found to be marginally increased in the Susd4 KO cerebrum, suggesting the level of myelination was not adversely affected (Figures S3C and S3E). Numbers of Olig2+ cells in the corpus callosum were similar in Susd4 KO and WT mice (Figure S3D).
Brain C1q Protein Expression Is Increased in Susd4 KO Mice
In the nervous system, C1q is known to play critical roles in neuron dendritic pruning and synaptogenesis (Bialas and Stevens, 2013, Chu et al., 2010, Michailidou et al., 2015, Perry and O'Connor, 2008). To determine if SUSD4 expression affects C1q levels in the brain, we investigated C1q protein expression in Susd4 KO mice by both immunostaining and western blotting. Immunostaining of hippocampal sections showed that C1q fluorescence intensity was increased 1.5-fold in the Susd4 KO mice, when compared with WT mice (Figures 4A–4C). In the cerebellum, C1q protein was found to be relatively highly expressed in the Purkinje cells of the anterobasal lobes of the Susd4 KO mice compared with WT (Figure 4D).
Figure 4.

Brain C1q Protein Expression Is Increased in Susd4 KO Mice
(A and B) Representative immunostaining for C1q (green) in the hippocampus sections of Susd4 KO (A) and WT (B) mice; scale bar, 100 μm.
(C) Quantification of C1q fluorescence intensity from the stained hippocampus sections. Data represent the mean ± SEM. WT values were set to 1.0. n = 3 mice per group. ∗∗∗∗: p < 0.0001.
(D) Representative immunostaining for C1q (green) and calbindin (red) in the cerebellum sections of Susd4 KO and WT mice. Scale bar, 40 μm. Nuclei were visualized with DAPI staining (blue) for (A), (B), and (D).
(E) Western blot analysis of C1q polypeptide levels in cerebellum lysates obtained from three WT and three Susd4 KO mice. β-Actin was simultaneously detected on the same blot as a loading control. L, molecular weight ladder.
(F) Quantification of C1q polypeptide expression from western blots. Expression was normalized to β-actin expression. Data represent the mean ± SEM. WT values were set to 1.0. ∗: p < 0.05.
(G) Representative immunostaining for C1q (green) and VGAT (red) in the WT and Susd4 KO hippocampal sections. Circles indicate the C1q and VGAT colocalized puncta in the sections. Arrow heads point to dendrite colocalized C1q and VGAT in the Susd4 KO section. Scale bar, 5 μm.
(H) Quantification using ImageJ of percentage VGAT colocalized with C1q. Data represent the mean ± SEM. n = 3 mice per group. ∗∗∗∗: p < 0.0001.
(I) Western blot analysis of VGAT in hippocampal lysates obtained from three WT and four Susd4 KO mice. β-Actin was detected on the same blot as a loading control.
(J) Quantification of VGAT protein levels from western blots. Expression was normalized to β-actin expression. WT values were set to 1.0. ∗∗: p < 0.005.
Western blotting with a monoclonal antibody to C1q indicated a 1.7-fold increase of constituent C1q expression in the cerebellum from the Susd4 KO mice when compared with the cerebellum from WT mice (Figures 4E and 4F). C1qa mRNA levels in hippocampus and cerebellum of the Susd4 KO mice compared with that of WT mice were not significantly different (Figures S4A and S4B), suggesting that the difference in protein levels was post-transcriptional. We investigated downstream of the complement pathway by determining complement C3 levels by western blotting of hippocampal extracts from the Susd4 KO and WT mice. The level of C3 was not significantly different nor was there evidence of altered C3 proteolysis (Figures S4C and S4D).
We determined, through co-staining of C1q and synaptic markers on Susd4 KO and WT hippocampal sections, if there was increased deposition of C1q on neuronal synapses, which might lead to increased synaptic pruning in the Susd4 KO brain. Co-localization analysis of inhibitory synaptic marker vesicular GABA transporter (VGAT) (Stephan et al., 2013) and C1q revealed a 2.5-fold increase of double-positive synapses in the Susd4 KO (Figures 4G and 4H). Western blotting demonstrated that VGAT levels were decreased by about 20% in the hippocampus from the Susd4 KO mice when compared with the hippocampus from WT mice (Figures 4I and 4J).
Co-localization analysis of excitatory synaptic marker, vesicular glutamate transporter 1 (VGlut1) and C1q revealed a 1.6-fold increase of double-positive synapses in the Susd4 KO hippocampal sections (Figures S4G and S4H) (Stephan et al., 2013). The total VGlut1 protein level measured by western blotting was not statistically significant between Susd4 KO and the WT hippocampus (Figures S4I and S4J). These results show that, in the absence of SUSD4, C1q levels are elevated in the brain with increased deposition at synapses, which may lead to synaptic loss through pruning.
Discussion
SUSD4 has been identified as a novel complement regulator that binds to the complement component C1q (Holmquist et al., 2013, Tu et al., 2010); however, its in vivo roles in the CNS are still largely unknown. Here, in the Susd4 KO mouse model we found increased C1q levels in the brain, activation status of microglia, neuronal morphology in the hippocampus and cerebellum, and motor and anxiety-like behavioral phenotypes.
The abnormal behavioral changes indicative of increased anxiety and decreased motor function observed in the Susd4 KO mice may be a reflection of morphologic changes in brain regions, such as the hippocampus, an area known to be involved in anxiety (Bannerman et al., 2014), and the cerebellum, the control center for motor function and coordination (Manto et al., 2012). Indeed, we found that brains from Susd4 KO mice were characterized by abnormal “hairy” basket cells surrounding Purkinje cell soma in the cerebellum, as well as decreased dendritic spine density on the pyramidal cells in the CA region of hippocampus.
The phenomenon of “hairy” basket cells in the cerebellum has been specifically described in degenerative human brains of individuals with cerebellar essential tremor that are characterized by progressive loss of Purkinje cells and degenerative axonal abnormalities (Axelrad et al., 2008, Babij et al., 2013, Kuo et al., 2017). Basket cells are GABAergic inhibitory interneurons distributed across different regions of the brain, including the cortex, hippocampus, and cerebellum. Basket cells send inhibitory synapses to the soma of target cells to control their overall potentials. Purkinje cells are the sole output of motor coordination from the cerebellar cortex (Erickson-Davis et al., 2010). Although the mechanism of increased “hairiness” of basket cells is not clear, one possibility is that it represents an accumulation of converging and reorganizing of basket-cell processes recruited from damaged Purkinje cells (Erickson-Davis et al., 2010). Similar hairy basket formation was detected in epileptic hippocampus specimens owing to loss of affected pyramidal cells innervated by the basket cells (Arellano et al., 2004, Buckmaster and Jongen-Relo, 1999). However, in the Susd4 KO model, no apparent loss of Purkinje cells in the cerebellum occurred, indicating that SUSD4 may influence innervative synaptic formation between Purkinje and basket cells through a different mechanism.
Dendritic spines are membranous protrusions that neurons use to form the postsynaptic component of the excitatory synapse (Nimchinsky et al., 2002, Rochefort and Konnerth, 2012, von Bohlen Und Halbach, 2009). Given that spines are the site for neurons to receive synaptic transmission, changes in spine morphology and numbers can be a fundamental indicator of physiological or pathological changes in the brain (Berry and Nedivi, 2017, Herms and Dorostkar, 2016, Moser et al., 1994, Perez-Cruz et al., 2011). The molecular mechanisms that regulate spine genesis (synaptogenesis) and removal (synaptic pruning) have been intensively studied (Kilinc, 2018, Lu and Van Vactor, 2013, Sudhof, 2018). Accumulating evidence has revealed that components of the complement system (C1q, C3, and C4) and microglia play important roles in regulation of synaptic refinement and elimination during developmental synaptogenesis in the CNS (Mastellos, 2014, Presumey et al., 2017, Schafer et al., 2012, Tenner et al., 2018). Activation of complement under pathological conditions can lead to excessive synaptic pruning by microglia and neuron degeneration, as has been reported for Alzheimer's disease and other cognitive or behavioral disorders (Depboylu et al., 2011, Lian et al., 2015, Pavlovski et al., 2012). In Susd4 KO mouse brain, we found decreased dendritic spine density in hippocampal pyramidal neurons. SUSD4 has been shown to bind to C1q, a subunit of the first component (C1 complex) for the classic complement pathway, and inhibit formation of downstream C3 convertase (Holmquist et al., 2013). The increased C1q protein expression and neuronal synaptic deposition, hypertrophic microglia, and reduced synaptic density of neurons in Susd4 KO mice suggest that Susd4 may regulate complement-dependent synaptic pruning by microglia.
The exact mechanism leading to the increase of C1q in the Susd4 KO brain has not been established. The Susd4 deletion does not affect C1qa mRNA levels indicating that the mechanism is likely post-transcriptional. One possibility is that SUSD4 can alter the C1q levels directly by accelerating C1q degradation, as described for other complement control protein-containing regulators, such as DAF/CR1 on the C3/C5 convertase and CSMD1/factor I on C4b/C3b (Escudero-Esparza et al., 2013, Noris and Remuzzi, 2013, Seya et al., 1985).
The human SUSD4 locus resides on chromosome 1q41 and is frequently deleted in patients with 1q41q42 microdeletion syndrome (Shaffer et al., 2007). Clinical features, which occur along a variable phenotypic spectrum, generally include severe developmental delay, seizures, as well as multiple congenital abnormalities. DISP1 (Jun et al., 2013, Kantarci et al., 2010), TP53PB3 (Zak et al., 2016), WDR26 (Skraban et al., 2017, Yanagishita et al., 2019), and FOXO28 (Cassina et al., 2015, Yanagishita et al., 2019) have each been implicated as genes responsible for some features of the deletion syndrome. Although SUSD4 was included in the original description of the smallest region of deletion overlap among patients with 1q41q42 microdeletion syndrome (Shaffer et al., 2007), further refinement has indicated that SUSD4 is unlikely to be responsible for major features of the disease (Jun et al., 2013). However, the demonstration here that Susd4 has important functions in the mammalian CNS raises the possibility that it may be a contributory factor in neurologic symptom severity when it is deleted in conjunction with the major causative genes in the patients with 1q41q42 microdeletion syndrome.
In summary, we have demonstrated substantial neurologic abnormalities in mice in which the Susd4 gene has been deleted. These findings suggest an important role for SUSD4 in the CNS and that it may act through regulation of C1q functions in the CNS. Finally, the linkage of Susd4 expression to mammalian CNS function implicates the deletion of the SUSD4 gene as a potential phenotypic modifier in 1q41q42 microdeletion syndrome.
Limitation of the Study
We have shown that deletion of SUSD4 increases the levels of C1q in the brain. Although previously shown as an in vitro complement system inhibitor, the mechanism by which SUSD4 regulates the complement system and C1q levels in brain is not known and is a subject for future research.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Kate Frost and Camille Wang for assistance in the early stages of the project. This work was supported by the Intramural Research Programs of the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases (R.L.P.), and the National Human Genome Research Institute (C.J.T.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author Contributions
Conceptualization, H.Z., C.J.T., and R.L.P; Investigation, H.Z., L.E.M., C.B., and G.T.; Writing – Original Draft, H.Z.; Writing – Review & Editing, C.J.T. and R.L.P.; Supervision, C.J.T. and R.L.P.
Declaration of Interests
The authors declare no competing interests.
Published: March 27, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.100957.
Contributor Information
Cynthia J. Tifft, Email: cynthiat@mail.nih.gov.
Richard L. Proia, Email: proia@nih.gov.
Supplemental Information
References
- Arellano J.I., Munoz A., Ballesteros-Yanez I., Sola R.G., DeFelipe J. Histopathology and reorganization of chandelier cells in the human epileptic sclerotic hippocampus. Brain. 2004;127:45–64. doi: 10.1093/brain/awh004. [DOI] [PubMed] [Google Scholar]
- Axelrad J.E., Louis E.D., Honig L.S., Flores I., Ross G.W., Pahwa R., Lyons K.E., Faust P.L., Vonsattel J.P. Reduced Purkinje cell number in essential tremor: a postmortem study. Arch. Neurol. 2008;65:101–107. doi: 10.1001/archneurol.2007.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babij R., Lee M., Cortes E., Vonsattel J.P., Faust P.L., Louis E.D. Purkinje cell axonal anatomy: quantifying morphometric changes in essential tremor versus control brains. Brain. 2013;136:3051–3061. doi: 10.1093/brain/awt238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannerman D.M., Sprengel R., Sanderson D.J., McHugh S.B., Rawlins J.N., Monyer H., Seeburg P.H. Hippocampal synaptic plasticity, spatial memory and anxiety. Nat. Rev. Neurosci. 2014;15:181–192. doi: 10.1038/nrn3677. [DOI] [PubMed] [Google Scholar]
- Berry K.P., Nedivi E. Spine dynamics: are they all the same? Neuron. 2017;96:43–55. doi: 10.1016/j.neuron.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bialas A.R., Stevens B. TGF-beta signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat. Neurosci. 2013;16:1773–1782. doi: 10.1038/nn.3560. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Buckmaster P.S., Jongen-Relo A.L. Highly specific neuron loss preserves lateral inhibitory circuits in the dentate gyrus of kainate-induced epileptic rats. J. Neurosci. 1999;19:9519–9529. doi: 10.1523/JNEUROSCI.19-21-09519.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassina M., Rigon C., Casarin A., Vicenzi V., Salviati L., Clementi M. FBXO28 is a critical gene of the 1q41q42 microdeletion syndrome. Am. J. Med. Genet. A. 2015;167:1418–1420. doi: 10.1002/ajmg.a.37033. [DOI] [PubMed] [Google Scholar]
- Chu Y., Jin X., Parada I., Pesic A., Stevens B., Barres B., Prince D.A. Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc. Natl. Acad. Sci. U S A. 2010;107:7975–7980. doi: 10.1073/pnas.0913449107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawley J.N. Exploratory behavior models of anxiety in mice. Neurosci. Biobehav. Rev. 1985;9:37–44. doi: 10.1016/0149-7634(85)90030-2. [DOI] [PubMed] [Google Scholar]
- Dale J.M., Garcia M.L. Neurofilament phosphorylation during development and disease: which came first, the phosphorylation or the accumulation? J. Amino Acids. 2012;2012:382107. doi: 10.1155/2012/382107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depboylu C., Schafer M.K., Arias-Carrion O., Oertel W.H., Weihe E., Hoglinger G.U. Possible involvement of complement factor C1q in the clearance of extracellular neuromelanin from the substantia nigra in Parkinson disease. J. Neuropathol. Exp. Neurol. 2011;70:125–132. doi: 10.1097/NEN.0b013e31820805b9. [DOI] [PubMed] [Google Scholar]
- Erickson-Davis C.R., Faust P.L., Vonsattel J.P., Gupta S., Honig L.S., Louis E.D. "Hairy baskets" associated with degenerative Purkinje cell changes in essential tremor. J. Neuropathol. Exp. Neurol. 2010;69:262–271. doi: 10.1097/NEN.0b013e3181d1ad04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escudero-Esparza A., Kalchishkova N., Kurbasic E., Jiang W.G., Blom A.M. The novel complement inhibitor human CUB and Sushi multiple domains 1 (CSMD1) protein promotes factor I-mediated degradation of C4b and C3b and inhibits the membrane attack complex assembly. FASEB J. 2013;27:5083–5093. doi: 10.1096/fj.13-230706. [DOI] [PubMed] [Google Scholar]
- Fiala J.C., Spacek J., Harris K.M. Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res. Brain Res. Rev. 2002;39:29–54. doi: 10.1016/s0165-0173(02)00158-3. [DOI] [PubMed] [Google Scholar]
- Gialeli C., Gungor B., Blom A.M. Novel potential inhibitors of complement system and their roles in complement regulation and beyond. Mol. Immunol. 2018;102:73–83. doi: 10.1016/j.molimm.2018.05.023. [DOI] [PubMed] [Google Scholar]
- Hajishengallis G., Reis E.S., Mastellos D.C., Ricklin D., Lambris J.D. Novel mechanisms and functions of complement. Nat. Immunol. 2017;18:1288–1298. doi: 10.1038/ni.3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herms J., Dorostkar M.M. Dendritic spine pathology in neurodegenerative diseases. Annu. Rev. Pathol. 2016;11:221–250. doi: 10.1146/annurev-pathol-012615-044216. [DOI] [PubMed] [Google Scholar]
- Holers V.M. Complement and its receptors: new insights into human disease. Annu. Rev. Immunol. 2014;32:433–459. doi: 10.1146/annurev-immunol-032713-120154. [DOI] [PubMed] [Google Scholar]
- Holmquist E., Okroj M., Nodin B., Jirstrom K., Blom A.M. Sushi domain-containing protein 4 (SUSD4) inhibits complement by disrupting the formation of the classical C3 convertase. FASEB J. 2013;27:2355–2366. doi: 10.1096/fj.12-222042. [DOI] [PubMed] [Google Scholar]
- Holter S.M., Einicke J., Sperling B., Zimprich A., Garrett L., Fuchs H., Gailus-Durner V., Hrabe de Angelis M., Wurst W. Tests for anxiety-related behavior in mice. Curr. Protoc. Mouse Biol. 2015;5:291–309. doi: 10.1002/9780470942390.mo150010. [DOI] [PubMed] [Google Scholar]
- Hong S., Beja-Glasser V.F., Nfonoyim B.M., Frouin A., Li S., Ramakrishnan S., Merry K.M., Shi Q., Rosenthal A., Barres B.A. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352:712–716. doi: 10.1126/science.aad8373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun K.R., Hur Y.J., Lee J.N., Kim H.R., Shin J.H., Oh S.H., Lee J.Y., Seo E.J. Clinical characterization of DISP1 haploinsufficiency: a case report. Eur. J. Med. Genet. 2013;56:309–313. doi: 10.1016/j.ejmg.2013.03.007. [DOI] [PubMed] [Google Scholar]
- Kantarci S., Ackerman K.G., Russell M.K., Longoni M., Sougnez C., Noonan K.M., Hatchwell E., Zhang X., Pieretti Vanmarcke R., Anyane-Yeboa K. Characterization of the chromosome 1q41q42.12 region, and the candidate gene DISP1, in patients with CDH. Am. J. Med. Genet. A. 2010;152A:2493–2504. doi: 10.1002/ajmg.a.33618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilinc D. The emerging role of mechanics in synapse formation and plasticity. Front. Cell Neurosci. 2018;12:483. doi: 10.3389/fncel.2018.00483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkitadze M.D., Barlow P.N. Structure and flexibility of the multiple domain proteins that regulate complement activation. Immunol. Rev. 2001;180:146–161. doi: 10.1034/j.1600-065x.2001.1800113.x. [DOI] [PubMed] [Google Scholar]
- Kuo S.H., Lin C.Y., Wang J., Sims P.A., Pan M.K., Liou J.Y., Lee D., Tate W.J., Kelly G.C., Louis E.D. Climbing fiber-Purkinje cell synaptic pathology in tremor and cerebellar degenerative diseases. Acta Neuropathol. 2017;133:121–138. doi: 10.1007/s00401-016-1626-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian H., Yang L., Cole A., Sun L., Chiang A.C., Fowler S.W., Shim D.J., Rodriguez-Rivera J., Taglialatela G., Jankowsky J.L. NFkappaB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer's disease. Neuron. 2015;85:101–115. doi: 10.1016/j.neuron.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C.S., Van Vactor D. Genetic analysis of synaptogenesis. In: Rubenstein John, Rakic Pasko., editors. Comprehensive Developmental Neuroscience: Cellular Migration and Formation of Neuronal Connections. Academic Press; 2013. pp. 537–577. [Google Scholar]
- Lui H., Zhang J., Makinson S.R., Cahill M.K., Kelley K.W., Huang H.Y., Shang Y., Oldham M.C., Martens L.H., Gao F. Progranulin deficiency promotes circuit-specific synaptic pruning by microglia via complement activation. Cell. 2016;165:921–935. doi: 10.1016/j.cell.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso J.J., Chen Y., Li X., Xue Z., Wong S.T. Methods of dendritic spine detection: from Golgi to high-resolution optical imaging. Neuroscience. 2013;251:129–140. doi: 10.1016/j.neuroscience.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manto M., Bower J.M., Conforto A.B., Delgado-Garcia J.M., da Guarda S.N., Gerwig M., Habas C., Hagura N., Ivry R.B., Marien P. Consensus paper: roles of the cerebellum in motor control--the diversity of ideas on cerebellar involvement in movement. Cerebellum. 2012;11:457–487. doi: 10.1007/s12311-011-0331-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastellos D.C. Complement emerges as a masterful regulator of CNS homeostasis, neural synaptic plasticity and cognitive function. Exp. Neurol. 2014;261:469–474. doi: 10.1016/j.expneurol.2014.06.019. [DOI] [PubMed] [Google Scholar]
- Michailidou I., Willems J.G., Kooi E.J., van Eden C., Gold S.M., Geurts J.J., Baas F., Huitinga I., Ramaglia V. Complement C1q-C3 associated synaptic changes in multiple sclerosis hippocampus. Ann. Neurol. 2015;77:1007–1026. doi: 10.1002/ana.24398. [DOI] [PubMed] [Google Scholar]
- Moser M.B., Trommald M., Andersen P. An increase in dendritic spine density on hippocampal CA1 pyramidal cells following spatial learning in adult rats suggests the formation of new synapses. Proc. Natl. Acad. Sci. U S A. 1994;91:12673–12675. doi: 10.1073/pnas.91.26.12673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen R.J., Buck C.R., Smith A.M. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 1992;116:201–211. doi: 10.1242/dev.116.1.201. [DOI] [PubMed] [Google Scholar]
- Nimchinsky E.A., Sabatini B.L., Svoboda K. Structure and function of dendritic spines. Annu. Rev. Physiol. 2002;64:313–353. doi: 10.1146/annurev.physiol.64.081501.160008. [DOI] [PubMed] [Google Scholar]
- Noris M., Remuzzi G. Overview of complement activation and regulation. Semin. Nephrol. 2013;33:479–492. doi: 10.1016/j.semnephrol.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlovski D., Thundyil J., Monk P.N., Wetsel R.A., Taylor S.M., Woodruff T.M. Generation of complement component C5a by ischemic neurons promotes neuronal apoptosis. FASEB J. 2012;26:3680–3690. doi: 10.1096/fj.11-202382. [DOI] [PubMed] [Google Scholar]
- Penzes P., Cahill M.E., Jones K.A., VanLeeuwen J.E., Woolfrey K.M. Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 2011;14:285–293. doi: 10.1038/nn.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Cruz C., Nolte M.W., van Gaalen M.M., Rustay N.R., Termont A., Tanghe A., Kirchhoff F., Ebert U. Reduced spine density in specific regions of CA1 pyramidal neurons in two transgenic mouse models of Alzheimer's disease. J. Neurosci. 2011;31:3926–3934. doi: 10.1523/JNEUROSCI.6142-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry V.H., O'Connor V. C1q: the perfect complement for a synaptic feast? Nat. Rev. Neurosci. 2008;9:807–811. doi: 10.1038/nrn2394. [DOI] [PubMed] [Google Scholar]
- Petzold A. Neurofilament phosphoforms: surrogate markers for axonal injury, degeneration and loss. J. Neurol. Sci. 2005;233:183–198. doi: 10.1016/j.jns.2005.03.015. [DOI] [PubMed] [Google Scholar]
- Presumey J., Bialas A.R., Carroll M.C. Complement system in neural synapse elimination in development and disease. Adv. Immunol. 2017;135:53–79. doi: 10.1016/bs.ai.2017.06.004. [DOI] [PubMed] [Google Scholar]
- Rochefort N.L., Konnerth A. Dendritic spines: from structure to in vivo function. EMBO Rep. 2012;13:699–708. doi: 10.1038/embor.2012.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosoklija G.B., Petrushevski V.M., Stankov A., Dika A., Jakovski Z., Pavlovski G., Davcheva N., Lipkin R., Schnieder T., Scobie K. Reliable and durable Golgi staining of brain tissue from human autopsies and experimental animals. J. Neurosci. Methods. 2014;230:20–29. doi: 10.1016/j.jneumeth.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer D.P., Lehrman E.K., Kautzman A.G., Koyama R., Mardinly A.R., Yamasaki R., Ransohoff R.M., Greenberg M.E., Barres B.A., Stevens B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seya T., Holers V.M., Atkinson J.P. Purification and functional analysis of the polymorphic variants of the C3b/C4b receptor (CR1) and comparison with H, C4b-binding protein (C4bp), and decay accelerating factor (DAF) J. Immunol. 1985;135:2661–2667. [PubMed] [Google Scholar]
- Shaffer L.G., Theisen A., Bejjani B.A., Ballif B.C., Aylsworth A.S., Lim C., McDonald M., Ellison J.W., Kostiner D., Saitta S. The discovery of microdeletion syndromes in the post-genomic era: review of the methodology and characterization of a new 1q41q42 microdeletion syndrome. Genet. Med. 2007;9:607–616. doi: 10.1097/gim.0b013e3181484b49. [DOI] [PubMed] [Google Scholar]
- Skaper S.D., Facci L., Zusso M., Giusti P. Synaptic plasticity, dementia and Alzheimer disease. CNS Neurol. Disord. Drug Targets. 2017;16:220–233. doi: 10.2174/1871527316666170113120853. [DOI] [PubMed] [Google Scholar]
- Skraban C.M., Wells C.F., Markose P., Cho M.T., Nesbitt A.I., Au P.Y.B., Begtrup A., Bernat J.A., Bird L.M., Cao K. WDR26 haploinsufficiency causes a recognizable syndrome of intellectual disability, seizures, abnormal gait, and distinctive facial features. Am. J. Hum. Genet. 2017;101:139–148. doi: 10.1016/j.ajhg.2017.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spittaels K., Van den Haute C., Van Dorpe J., Bruynseels K., Vandezande K., Laenen I., Geerts H., Mercken M., Sciot R., Van Lommel A. Prominent axonopathy in the brain and spinal cord of transgenic mice overexpressing four-repeat human tau protein. Am. J. Pathol. 1999;155:2153–2165. doi: 10.1016/S0002-9440(10)65533-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan A.H., Madison D.V., Mateos J.M., Fraser D.A., Lovelett E.A., Coutellier L., Kim L., Tsai H.H., Huang E.J., Rowitch D.H. A dramatic increase of C1q protein in the CNS during normal aging. J. Neurosci. 2013;33:13460–13474. doi: 10.1523/JNEUROSCI.1333-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens B., Allen N.J., Vazquez L.E., Howell G.R., Christopherson K.S., Nouri N., Micheva K.D., Mehalow A.K., Huberman A.D., Stafford B. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- Stone S., Yue Y., Stanojlovic M., Wu S., Karsenty G., Lin W. Neuron-specific PERK inactivation exacerbates neurodegeneration during experimental autoimmune encephalomyelitis. JCI Insight. 2019;4:1–17. doi: 10.1172/jci.insight.124232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof T.C. Towards an understanding of synapse formation. Neuron. 2018;100:276–293. doi: 10.1016/j.neuron.2018.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Switzer R.C., 3rd Application of silver degeneration stains for neurotoxicity testing. Toxicol. Pathol. 2000;28:70–83. doi: 10.1177/019262330002800109. [DOI] [PubMed] [Google Scholar]
- Tang T., Li L., Tang J., Li Y., Lin W.Y., Martin F., Grant D., Solloway M., Parker L., Ye W. A mouse knockout library for secreted and transmembrane proteins. Nat. Biotechnol. 2010;28:749–755. doi: 10.1038/nbt.1644. [DOI] [PubMed] [Google Scholar]
- Tenner A.J., Stevens B., Woodruff T.M. New tricks for an ancient system: physiological and pathological roles of complement in the CNS. Mol. Immunol. 2018;102:3–13. doi: 10.1016/j.molimm.2018.06.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Z., Cohen M., Bu H., Lin F. Tissue distribution and functional analysis of Sushi domain-containing protein 4. Am. J. Pathol. 2010;176:2378–2384. doi: 10.2353/ajpath.2010.091036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bohlen Und Halbach O. Structure and function of dendritic spines within the hippocampus. Ann. Anat. 2009;191:518–531. doi: 10.1016/j.aanat.2009.08.006. [DOI] [PubMed] [Google Scholar]
- Wang J., Tung Y.C., Wang Y., Li X.T., Iqbal K., Grundke-Iqbal I. Hyperphosphorylation and accumulation of neurofilament proteins in Alzheimer disease brain and in okadaic acid-treated SY5Y cells. FEBS Lett. 2001;507:81–87. doi: 10.1016/s0014-5793(01)02944-1. [DOI] [PubMed] [Google Scholar]
- Yamamoto T., Hirano A. A comparative study of modified Bielschowsky, Bodian and thioflavin S stains on Alzheimer's neurofibrillary tangles. Neuropathol. Appl. Neurobiol. 1986;12:3–9. doi: 10.1111/j.1365-2990.1986.tb00677.x. [DOI] [PubMed] [Google Scholar]
- Yanagishita T., Yamamoto-Shimojima K., Nakano S., Sasaki T., Shigematsu H., Imai K., Yamamoto T. Phenotypic features of 1q41q42 microdeletion including WDR26 and FBXO28 are clinically recognizable: the first case from Japan. Brain Dev. 2019;41:452–455. doi: 10.1016/j.braindev.2018.12.006. [DOI] [PubMed] [Google Scholar]
- Young K.M., Psachoulia K., Tripathi R.B., Dunn S.J., Cossell L., Attwell D., Tohyama K., Richardson W.D. Oligodendrocyte dynamics in the healthy adult CNS: evidence for myelin remodeling. Neuron. 2013;77:873–885. doi: 10.1016/j.neuron.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zak J., Vives V., Szumska D., Vernet A., Schneider J.E., Miller P., Slee E.A., Joss S., Lacassie Y., Chen E. ASPP2 deficiency causes features of 1q41q42 microdeletion syndrome. Cell Death Differ. 2016;23:1973–1984. doi: 10.1038/cdd.2016.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q., Wang S., Anderson D.J. Identification of a novel family of oligodendrocyte lineage-specific basic helix-loop-helix transcription factors. Neuron. 2000;25:331–343. doi: 10.1016/s0896-6273(00)80898-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.