

Abstract
Autophagy is an evolutionarily conserved catabolic process that recycles proteins and organelles in a lysosome-dependent manner and is induced as an alternative source of energy and metabolites in response to diverse stresses. Inhibition of autophagy has emerged as an appealing therapeutic strategy in cancer. However, it remains to be explored whether autophagy inhibition is a viable approach for the treatment of hepatocellular carcinoma (HCC). Here, we identify that water-soluble yeast β-D-glucan (WSG) is a novel autophagy inhibitor and exerts significant antitumour efficacy on the inhibition of HCC cells proliferation and metabolism as well as the tumour growth in vivo. We further reveal that WSG inhibits autophagic degradation by increasing lysosomal pH and inhibiting lysosome cathepsins (cathepsin B and cathepsin D) activities, which results in the accumulation of damaged mitochondria and reactive oxygen species (ROS) production. Furthermore, WSG sensitizes HCC cells to apoptosis via the activation of caspase 8 and the transfer of truncated BID (tBID) into mitochondria under nutrient deprivation condition. Of note, administration of WSG as a single agent achieves a significant antitumour effect in xenograft mouse model and DEN/CCl4 (diethylnitrosamine/carbon tetrachloride)-induced primary HCC model without apparent toxicity. Our studies reveal, for the first time, that WSG is a novel autophagy inhibitor with significant antitumour efficacy as a single agent, which has great potential in clinical application for liver cancer therapy.
Keywords: β-D-glucan, Autophagy inhibition, Lysosomal function, Hepatocellular carcinoma, Antitumour
Abbreviations: WSG, water-soluble yeast β-D-glucan; HCC, hepatocellular carcinoma; CQ, chloroquine; ROS, reactive oxygen species; Baf A1, Bafilomycin A1; ATG, autophagy related; GFP, green fluorescent protein; RFP, red fluorescent protein; MAP1LC3B/LC3B, microtubule-associated protein 1 light chain 3; Cat B, cathepsin B; Cat D, cathepsin D; PCC, Pearson correlation coefficient; HBSS, Hank's Balanced Salt Solution; FBS, fetal bovine serum; MMP, mitochondrial membrane potential; tbid, truncated BID
Graphical abstract
Highlights
-
•
Water-soluble yeast β-D-glucan (WSG) exerts direct antitumour activity in HCC
-
•
WSG inhibits autophagic degradation by increasing lysosomal pH
-
•
WSG causes accumulation of damaged mitochondria and ROS production
-
•
WSG sensitizes HCC cells to apoptosis via the activation of caspase 8
1. Introduction
As the most common primary liver cancer, hepatocellular carcinoma (HCC) ranks as the sixth most common cancer and the fourth leading cause of cancer deaths worldwide [1]. There are several drugs used clinically in HCC treatment, such as Sorafenib (in first-line), regorafenib, cabozantinib, and ramucirumab (in second-line) [2], but increasing severe multidrug resistance becomes a major reason for chemotherapy failure. The overall prognosis of HCC remains poor [3] and developing more effective therapeutics for HCC is of critical importance.
Autophagy, an evolutionarily conserved catabolic process and induced in response to nutrient deprivation, hypoxia and diverse cellular stresses [4], enables intracellular contents including proteins, damaged mitochondria and other organelles engulfed by autophagosomes to be degraded and recycled in autolysosomes by lysosomal hydrolases [5,6]. Lysosome-mediated autophagic degradation is important in the maintenance of intracellular metabolic and energetic homeostasis [7]. Dysregulation of autophagy has been closely linked to multiple disease, including cancer [8]. Autophagy has context-dependent roles in HCC [9]. On the one hand, autophagy prevents HCC development by removing damaged organelles and aggregated proteins and preventing genomic instability. On the other hand, once HCC is established, autophagy acts as a pro-survival pathway for HCC cells by providing metabolic building blocks for rapid cell proliferation and promoting viability under stresses, such as hypoxia, nutrient deficiency and chemotherapeutic stress [[10], [11], [12]]. It remains a tremendous challenge to understand the molecular mechanisms of autophagy in different stages of HCC, which may help to provide therapeutic targets for HCC treatment and overcome chemotherapeutic drug resistance [13].
β-D-glucan is a polysaccharide composed of D-glucose monomers and is linked by β-(1 → 3) and β-(1 → 4) or β-(1 → 3) and β-(1 → 6) glycosidic bonds. β-D-glucan is naturally found in bacteria, fungi, algae and cereal [14], with diverse biological effects, such as immune enhancement [15], antitumour [16], antioxidant [17] and anti-hypertension [14]. The cell wall of Saccharomyces cerevisiae is one important source of β-D-glucan. However, yeast β-D-glucan shows extremely low solubility in water, which greatly restricts its application. We developed a method to prepare water-soluble yeast β-D-glucan (WSG) with enhanced solubility [18]. Emerging evidence has shown that β-D-glucan exhibits promising antitumour potential, acting as an immune modulator to enhance the immune system and kill the tumour cells [19,20], or inducing cell cycle arrest and apoptosis, thereby inhibiting tumour invasion, adhesion, and metastasis [21]. In addition, β-D-glucan is used as an adjuvant drug combined with conventional chemotherapeutic drugs in the treatment of cancer [22]. However, it remains to be studied whether β-D-glucan can modulate autophagy against HCC.
In this study, we discover a new antitumour mechanism of WSG by blocking autophagic degradation through increasing lysosomal pH and inhibiting lysosomal cathepsins activities, which results in the accumulation of damaged mitochondria and reactive oxygen species (ROS) production. Furthermore, WSG decreases HCC cells metabolism in glycolysis and TCA cycle and sensitizes HCC cells to apoptosis under nutrient deprivation. More excitingly, WSG significantly inhibits tumour growth in xenograft mouse model and DEN/CCl4 (diethylnitrosamine/carbon tetrachloride)-induced primary HCC model without apparent toxicity. Our findings demonstrate that WSG is a novel autophagy inhibitor and exerts significant antitumour effect with therapeutic potential in the clinical treatment of HCC.
2. Results
2.1. WSG exerts direct inhibition on HCC cell proliferation in vitro and in vivo
The structure of WSG is shown in Fig. 1A. WSG is polymerized by glucose monomers and its main chain is linked by β-(1 → 3)-glycosidic bonds. To examine the direct effect of WSG on HCC cell proliferation, we treated Huh7 cells with WSG at different concentrations and different time points. WSG inhibited Huh7 cells proliferation in a dose-dependent manner and reached an inhibition rate of about 50% at 8 mg/ml (Fig. 1B). We also observed a time-dependent inhibition of cell proliferation by WSG (Fig. 1C). Furthermore, the inhibitory effect of WSG was applicable to several other HCC cell lines in a dose-dependent manner, such as SMMC-7721 cells, HepG2 cells, LM3 cells and human primary HCC cells LIXC 501 cells (Figs. S1A–D). Thereafter, to assess its specificity towards cancer cells, we determined the cytotoxicity of WSG in human normal liver HL-7702 cells. Compared to HCC cells, WSG exhibited a slight inhibition on HL-7702 cells at two different time points (Figs. S1E and F). These results clearly show that WSG specifically inhibits HCC cell proliferation without significant cytotoxicity towards normal liver cells.
Fig. 1.

WSG exerts direct antitumour effects in liver cancer cells and in nude mice. (A) The molecular structure of water-soluble yeast β-D-glucan (WSG). WSG is polymerized by glucose monomers and its main chain is linked by β-(1 → 3)-glycoside bonds. Every nine β-(1 → 3)-glucose units in the main chain linked to one glucose unit in the side chain through β-(1 → 6)-glycoside bonds. (B) Cell viability of Huh7 cells treated with different concentrations of WSG at 48 h. Different concentrations of WSG was compared with control. (C) Cell viability of Huh7 cells at different time points treated with 8 mg/ml WSG. (D) Schematic of workflow for assessment of antitumour effect of WSG in BALB/c male nude mice. Huh7 cells were subcutaneously injected into right and left flanks of nude mice (regarded as day = 0). They were randomly divided into 3 groups on the third day after injection and were administrated with vehicle (n = 9), 150 mg/kg (n = 8) and 300 mg/kg (n = 8) via i. p. Every other day, respectively. Red circles indicate Huh 7 cells. (E) Changes of body weight of nude mice during the period of administration. (F) Images of tumour-bearing nude mice and tumours. (G) The weight of tumours from mice in (F). (H) Changes of tumour volume during the period of administration. (I) Representative images of Ki67 staining in mice tumour (left) and quantification of Ki67 positive cells (right). Scale bar: 50 μm. All data are presented as mean ± SEM. **P < 0.01; ***P < 0.001. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Next, we performed Huh7 cell xenograft model to study the antitumour effect of WSG in vivo. Huh7 cells were subcutaneously injected into right and left flanks of BALB/c male nude mice. Vehicle, 150 mg/kg and 300 mg/kg of WSG were administered through intraperitoneal injection (i.p.) every other day (Fig. 1D). WSG exhibited potent antitumour activity in vivo without significantly affecting body weights during the experiment period (Fig. 1E). The tumour weight and volume were dose-dependently inhibited by WSG (Fig. 1F–H). The tumour growth inhibition rate was higher than 50% in 300 mg/kg WSG group compared to control group (Fig. 1H). In addition, the expression of Ki67, strongly associated with tumour cell proliferation and growth as a proliferation marker in routine pathological examination, was dose-dependently reduced by WSG in tumours (Fig. 1I). Taken together, WSG exerts direct inhibition on HCC cell proliferation in vitro and in vivo without apparent toxicity and side effects.
2.2. WSG inhibits autophagic flux at its late stage
Next, we investigated whether WSG exert its antitumour effects through inducing cell cycle arrest. Flow cytometry assays of propidium iodide (PI) DNA staining revealed that WSG treatment for 48 h did not have significant effect on the distribution of cell cycle in Huh7 cells (Fig. S1G). Consistently, the protein expressions of STAT 3, cyclin-dependent kinases inhibitors p21 and p27 and cyclins, such as Cyclin D1, Cyclin E1, Cyclin A2 and Cyclin B1, did not change significantly (Fig. S1H). Together, WSG inhibits tumour cell proliferation without causing cell cycle arrest.
To investigate the underlying mechanisms responsible for the antitumour effects of WSG, we performed transcriptomic RNA sequencing (RNA-Seq) analysis on Huh7 cells treated with WSG. We found 535 differentially expressed genes (DEGs), among which 144 genes were up-regulated and 391 genes were down-regulated compared to control group (Fig. S1I). Interestingly, we found that the autophagy-related genes, such as Map1lc3a, Map1lc3b2, Sqstm1, Ctsb and Ctsd, were significantly up-regulated (Fig. 2A). To further investigate the effect of WSG on autophagy, we first tested WSG treatment on GFP-LC3 puncta in Huh7 cells, a well-known marker of autophagosomes. As shown in Fig. 2B, WSG enhanced the GFP-LC3 puncta formation significantly. Then we examined the changes of the LC3 conversion by western blotting, and the protein level of LC3-II was markedly increased after WSG treatment (Fig. 2C). The increase of GFP-LC3 puncta and LC3-II levels demonstrated that autophagosomes were accumulated upon WSG treatment, which may be a result of increased formation of autophagosomes (increased autophagic flux) or blockage of autophagosomes degradation (inhibition of autophagy at its late stage). We next tested the expression of p62 protein, a selective autophagy receptor for degradation of ubiquitinated substrates as they interact with LC3 and polyubiquitin [23]. We found that p62 was also increased, consistent with RNA-Seq analysis. Similarly, the protein levels of LC3-II and p62 were increased in SMMC-7721 cells (Fig. S2A). Interestingly, the proteins upstream of autophagic process, such as BECN1, ATG5 (autophagy-related 5), mTOR and AMPK, did not change significantly (Fig. 2C). These data indicated that WSG may inhibit autophagy at its late stage.
Fig. 2.

WSG inhibits autophagic flux at its late stage. (A) The expression of autophagy-related genes from transcriptomic RNA-Seq data in control and WSG (8 mg/ml) treated Huh7 cells. (B) Fluorescence imaging and quantification of GFP-LC3 puncta in Huh7 cells treated with 8 mg/ml WSG for 48 h. The number of GFP-LC3B puncta per cell was counted in 15 cells each condition and data are representative of 3 independent experiments. Scale bar: 5 μm. (C) Expression of LC3, p62 and the proteins upstream autophagic process in Huh7 cells upon WSG treatment for 48 h. Quantification of LC3-II/GAPDH, p62/GAPDH, p-AMPK/AMPK and p-mTOR/mTOR were based on 3 independent experiments. (D) Blockage of autophagic flux by WSG. Huh7 cells were treated with WSG (8 mg/ml) for 48 h. Baf A1 (bafilomycin A1, 50 nM) treated cells for 6 h before cell lysis. Quantification of LC3-II/GAPDH and p62/GAPDH were based on 3 independent experiments. (E) Representative fluorescence images of WSG and CQ (chloroquine) on autophagosome maturation and autolysosome formation. Huh7 cells were treated with 8 mg/ml WSG for 48 h or CQ (25 mM) for 6 h. More than 15 cells were counted in each condition and data are representative of 3 independent experiments. Scale bar: 5 μm. All data are presented as mean ± SEM. *P < 0.05; **P < 0.01.
Next, we used autophagy inhibitor Baf A1 (Bafilomycin A1), a vacuolar H+ ATPase inhibitor that inhibits the fusion between autophagosomes and lysosomes, to evaluate the effect of WSG on autophagic flux. There was no further increase in LC3-II levels with combined treatment of WSG with 50 nM Baf A1 (Fig. 2D), demonstrating that WSG inhibits the late stage of autophagy rather than promoting autophagosome formation. To further verify that WSG inhibits autophagic late stage, we used a tandem fluorescence RFP-GFP-LC3 reporter system to monitor autophagosome maturation and autolysosome formation. In a normal autophagic process, it gives rise to more red-only puncta when the fluorescence of GFP is quenched due to low lysosomal pH, whereas RFP is acid-insensitive. Otherwise, it appears to be more yellow puncta when the late stage of autophagy is blocked, which may result from the inhibition of the fusion between autophagosomes and lysosomes or the impairment of lysosomal function [24]. As shown in Fig. 2E, there was a significant increase in autophagosome numbers (yellow puncta) without changes in autolysosome numbers (red puncta) after WSG exposure. This effect was similar to that of the autophagy inhibitor chloroquine (CQ), which inhibits lysosomal hydrolases and prevents autophagic degradation by increasing lysosomal pH. Taken together, these results suggest that WSG blocks autophagic flux at its late stage by inhibiting the fusion between autophagosomes and lysosomes or impairing the lysosomal function.
2.3. WSG impairs lysosome function by increasing lysosomal pH and inhibiting cathepsin activities
To confirm our hypothesis, we examined the autophagosome-lysosome fusion through the colocalization of the autophagosomal marker GFP-LC3 with lysosome marker LAMP1 [25]. As shown in Fig. 3A, nutrient deprivation with HBSS (Hank's Balanced Salt Solution) treatment induced a remarkable colocalization of GFP-LC3 puncta with LAMP1 (yellow puncta) with a high PCC (Pearson correlation coefficient) of 0.60, suggesting that the fusion between autophagosomes and lysosomes was normal during starvation upon autophagy activation. In contrast, Baf A1, which prevents autophagosome-lysosome fusion, exhibited a significant separation of GFP-LC3 puncta and LAMP1 (PCC = 0.34). Of note, we found that WSG treated cells exhibited increased yellow puncta similar to the effect of nutrient deprivation, indicating that the fusion between autophagosomes and lysosomes was not inhibited by WSG treatment (Fig. 3A).
Fig. 3.

WSG impairs lysosome function by increasing lysosomal pH and inhibiting cathepsins activities without affecting the fusion between autophagosomes and lysosomes. (A) The colocalization of GFP-LC3 and LAMP1. Huh7 cells transfected with GFP-LC3 were treated with WSG (8 mg/ml) for 48 h or Baf A1 (50 nM) for 6 h before fixed in total medium. And HBSS medium was changed 3 h before cells were fixed. PCC (Pearson correlation coefficient), a statistic to quantify the extent of colocalization of the two signals, is calculated using Image J software from 3 independent experiments. Scale bar: 5 μm. (B) The effect of WSG on lysosomal pH. Huh7 cells were treated with WSG (8 mg/ml) for 48 h or CQ (25 mM) for 6 h. At the end of treatment, cells were treated with 2 μM LysoSensor Green DND-189 to evaluate lysosomal pH change. The fluorescence intensity is calculated using Image J software from 3 independent experiments. Scale bar: 5 μm. (C) The protein levels of Cat D (cathepsin D) and Cat B (cathepsin B) in Huh7 cells upon WSG treatment for 48 h. (D) Representative transmission electron microscopy (TEM) images in Huh7 cells treated with WSG (8 mg/ml) for 48 h. The arrow in left panel indicates autophagic vacuole contained electron dense cellular contents or organelles at various stages of degradation. The arrows in right panel indicate autophagic vacuoles contained electron translucent and morphologically intact cellular contents or organelles. (E) Expression of ubiquitinated proteins in Huh7 cells upon WSG treatment for 48 h. All data are presented as mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
The low lysosomal pH is required for maintaining lysosomal function. Next we evaluated the lysosomal pH by using lysosensor DND-189, which exhibited a pH-dependent increase in fluorescence intensity upon acidification [26]. WSG significantly decreased the fluorescence, indicating an increase of lysosomal pH. This effect was similar to that of CQ, albeit to a much lesser extent (Fig. 3B). Cathepsins, the major group of proteases inside lysosome, plays an important role in maintaining cellular homeostasis by degrading and recycling cellular contents, which is especially important for the execution of autophagy [27]. They are synthesized as inactive pro-cathepsins and can be activated by other proteolytic enzymes or autohydrolyzed to form active cathepsins under acidic conditions [27]. The roles of cathepsins has been extensively investigated in cancer development, and overexpression and hypersecretion of pro-cathepsins/cathepsins have been shown to induce proliferation and migration in numerous cancers [[28], [29]]. We next examined the maturation process of cathepsin D (Cat D) and cathepsin B (Cat B) by western blotting. Consistent with the increased lysosomal pH, the mature form of Cat D and Cat B reduced dramatically in a dose-dependent manner upon WSG treatment both in Huh7 cells (Fig. 3C) and SMMC-7721 cells (Fig. S2B), suggesting an impaired function in autolysosomal degradation of intracellular components. Next, we conducted transmission electron microscopy (TEM) analysis and found there was a significant increase in autophagic vacuoles which contained cellular contents or organelles appearing morphologically intact in WSG treated Huh7 cells, illustrating that the degradation function of autolysosomes was impaired (Fig. 3D). Previous studies reported that lysosomal dysfunction can result in an increased level of ubiquitinated proteins [30,31]. As expected, WSG significantly increased the accumulation of ubiquitinated proteins in Fig. 3E. Taken together, all these results demonstrate that WSG impairs lysosomal function by increasing its pH and inhibiting cathepsins activities, which blocks the degradation of autophagic cargos.
2.4. Autophagy inhibition by WSG decreases the metabolism in glycolysis and the TCA cycle
Autophagy plays a critical role in cancer to support tumour metabolism, given that the degradation of autophagic cargos provides a variety of metabolic materials for recycling into various metabolic pathways of cancer cells [32,33]. Metabolic reprogramming is a core hallmark of cancer. Increased aerobic glycolysis, known as Warburg effect, is often observed along with up-regulation of glucose transporters and glucose uptake in various cancers [34]. As the centre of oxidative phosphorylation, TCA cycle is also upregulated to meet the requirements of cellular energy production, biosynthesis and redox balance in cancer cells. Emerging studies demonstrate that the TCA cycle is a potential target for cancer therapy despite early dogma that cancer cells appear to bypass the TCA cycle and mainly utilize aerobic glycolysis [35,36]. Our transcriptomic RNA-Seq data showed that the expression levels of most metabolism-related genes were significantly down-regulated upon WSG treatment (Fig. 4A). And the glucose consumption was reduced dramatically in Huh7 cells in a dose-dependent manner (Fig. 4B), consistent with the decreased expression of glucose transporter genes Slc2a1 and Slc2a4 in Fig. 4A.
Fig. 4.

Autophagy inhibition by WSG decreases the metabolism in glycolysis and the TCA cycle pathways. (A) The expression of metabolism-related genes from transcriptomic RNA-Seq data in control and WSG (8 mg/ml) treated Huh7 cells. (B) The glucose consumption in collected medium after treatment with WSG at 48 h in Huh7 cells. (C)13C distribution of glycolysis and the first turn of the TCA cycle with 50% U–13C-glucose (labelled at all six carbons) and 50% glucose (unlabelled). All colored circles indicate 13C-labelled carbon atoms from the labelled glucose. Striped circles indicate 13C-labelled carbons are converted from the labelled glucose in glycolysis, red circles indicate 13C-labelled carbons are obtained from pyruvate dehydrogenase (PDH), and blue circles indicate 13C-labelled carbons are converted from pyruvate carboxylase (PC) or malate enzyme (ME) to catalyze metabolic pathways. G-6-P, glucose-6-phosphate; F-6-P, fructose-6-phosphate; F-1,6–2P, fructose-1,6-diphosphate; GAP, glyceraldehyde; ME, malate enzyme. (D) Fraction of the labelled metabolites of M+3 from 13C-glucose in glycolysis by WSG treatment for 48 h in Huh7 cells. (E) Fraction of the labelled metabolites of M+2 from 13C-glucose in TCA cycle by WSG treatment for 48 h in Huh7 cells. (F) Fraction of the labelled metabolites of M+3 from 13C-glucose in TCA cycle by WSG treatment for 48 h in Huh7 cells. All data are presented as mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
To further investigate the effect of WSG on cellular metabolism, we traced the cellular metabolic flux changes in Huh7 cells using gas chromatography and mass spectrometry (GC-MS) techniques after incubated the cells with the medium containing 50% stable isotope labelled U–13C-glucose (uniform labelled glucose, labelled at all six carbons). Interestingly, the enriched labelled carbon in both glycolysis and the TCA cycle from the labelled glucose were clearly decreased dose-dependently (Figs. S3A and B). And the M+3 (13C labelled at all three positions) fraction of enriched labelled metabolites in glycolysis, such as pyruvate, alanine and lactate, were significantly reduced compared with control (Fig. 4C and D), suggesting that glycolysis flux was apparently inhibited upon WSG treatment. In addition, pyruvate enters the TCA cycle through pyruvate dehydrogenase (PDH), pyruvate carboxylase (PC) or malate enzyme (ME). PDH produces metabolites of M+2 (13C labelled at two positions), whereas PC or ME produces metabolites of M+3, including malate, asparate and fumarate, in the first turn [37]. As shown in Fig. 4E and F, WSG treatment significantly reduced M+2 and M+3 fraction of enriched labelled metabolites in TCA cycle. These results demonstrated that tumour cell metabolism was significantly decreased in glycolysis and the TCA cycle upon WSG treatment, consistent with our previous results that WSG inhibited autophagic degradation and reduced metabolic materials for recycling.
2.5. Blockage of autophagic degradation by WSG causes damaged mitochondria accumulation and ROS production, which sensitizes HCC cells to apoptosis under nutrient deprivation condition
Autophagy is a critical adaptive factor for tumour cells to survive diverse stresses by removing damaged mitochondria, controlling ROS production, and reducing apoptosis [38]. Mitochondria, where the TCA cycle takes place in eukaryotic organisms, is essential for ATP production, redox balance and biosynthesis of key intermediates in various tumours [36]. From our TEM analysis, we noted that the morphology of mitochondria changed significantly in WSG treated Huh7 cells. Compared to control, the mitochondria appeared to be swollen and round, of which the matrix became bright or electron-translucent and the cristae was shorter, broken and arranged loosely (Fig. 5A). Besides, the mitochondrial membrane potential (MMP), an important indicator of mitochondrial health, was significantly decreased and the ROS production was increased in a dose-dependent manner upon WSG treatment (Fig. 5B and C). And the ratio of ATP/AMP and ATP/ADP was also reduced (Fig. 5D) analysed by liquid chromatography with tandem mass spectrometry (LC-MS/MS). Consistent with decreased TCA cycle metabolism, these results suggested that WSG caused mitochondria dysfunction. Autophagy is an important way to remove damaged mitochondria, whereas WSG inhibited autophagic degradation according to our previous results. As shown in Fig. 5A, the number of damaged mitochondria in WSG treated cells was more than control, indicating an impaired lysosomal function to remove damaged mitochondria.
Fig. 5.

WSG leads to mitochondria dysfunction and sensitizes liver cancer cells to apoptosis via the intrinsic and extrinsic pathways under nutrient deprivation. (A) Representative TEM images of mitochondria in Huh7 cells treated with WSG (8 mg/ml) for 48 h. The arrows indicate normal mitochondria (left panel) and abnormal mitochondria (right panel). (B–C) Fluorescent images of mitochondrial membrane potential (MMP) (B) and reactive oxygen species (ROS) (C) in Huh7 cells treated with WSG for 48 h. Cells were stained with 50 nM TMRM (Tetramethylrhodamine, a cell-permeant dye that accumulates in active mitochondria with intact membrane potentials) or 5 μM DHE (dihydroethidium, a fluorescent probe for the detection of ROS generation), respectively. The fluorescence intensity was calculated in 20 individual fields per well from six individual wells. (D) The ratio of ATP/ADP and ATP/AMP (normalized to control) in Huh7 cells treated with WSG for 48 h. (E) Effect of WSG on apoptosis by annexin-V and propidium iodide (PI) double staining. Huh7 cells were treated with 8 mg/ml WSG in the presence or absence of fetal bovine serum (FBS) for 48 h. (F) Expression of LC3, p62, cathepins and caspases 8 in Huh7 cells treated with 8 mg/ml WSG in the presence or absence of FBS for 48 h (left panel). Increased lysosomal pH leads to decreased lysosomal cathepsins maturation, followed by inhibition of autophagic degradation, which then leads to the accumulation of autophagic cargos and triggers caspase 8-mediated apoptosis upon WSG treatment under nutrient deprivation (right panel). (G) Caspase 8 activity in Huh7 cells treated with WSG under FBS starvation for 48 h. (H) WSG induced the truncated BID (tBID) to transfer into mitochondria. C: cytosol; M: mitochondria. All data are presented as mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.
Since nutrient deprivation is one of the most pervasive microenvironment stresses in solid tumours, autophagy enables tumour cells to be more tolerant to such extreme conditions to escape from cancer therapy [39]. To investigate whether WSG-mediated autophagy inhibition could affect the survival of HCC cells under nutrition deficiency, we treated Huh7 cells with WSG in the presence or absence of the fetal bovine serum (FBS). Interestingly, we found that there was no obvious apoptosis with WSG treatment or serum starvation alone, whereas a combination of WSG treatment with serum starvation led to significant apoptosis (Fig. 5E). Furthermore, the increased protein levels of LC3-II and p62 and reduced mature form of Cat D and Cat B indicated that autophagy was blocked and lysosomal function was impaired upon WSG treatment under serum starvation (Fig. 5F), consistent with our previous results under complete medium. Moreover, we found that caspase 8 was activated a dose-dependent manner upon WSG treatment under serum starvation (Fig. 5F and G).
The apoptosis can be induced via the extrinsic pathway or the intrinsic pathway. It has been reported that the two pathways converge on the cleavage of the protein BID into the truncated BID (tBID) by activated caspase 8, which results in mitochondrial outer membrane permeabilization (MOMP) and the release of cytochrome c into the cytosol by various intracellular stimuli, including oxidative stress, hypoxia, DNA damage, and nutrient deprivation [40,41]. Next, we isolated mitochondria from cytosol and found caspase 8 activation significantly increased the formation of tBID in the mitochondria upon WSG treatment under serum starvation (Fig. 5H). These results indicated that HCC cells were more sensitive to apoptosis upon WSG treatment under nutrient deprivation condition via the intrinsic and extrinsic pathways, suggesting that WSG had a more effective antitumour effect in extreme tumour living conditions, such as nutrient deprivation and hypoxia in the tumour microenvironment.
2.6. WSG attenuates DEN/CCl4-induced primary liver cancer tumorigenesis
To investigate whether WSG-inhibited autophagy in vitro translates into anti-tumour effects in vivo, we performed DEN/CCl4 (diethylnitrosamine/carbon tetrachloride)-induced primary hepatocellular carcinoma model in C57BL/6 mice. After injection of DEN and CCl4 at the 3rd and 8th week respectively, mice were divided into four groups and then administrated with 50 mg/kg WSG, 25 mg/kg CQ, 50 mg/kg WSG and 25 mg/kg CQ or vehicle through intraperitoneal injection at the 12th to 39th week (Fig. 6A). Mice were sacrificed at the 39th week. As expected, the body weights of four groups had no significant difference and we did not observed apparent behaviour alterations among these four groups (Fig. S4A), suggesting that the administrated concentration of WSG was well tolerated without apparent toxicity. WSG or CQ treated mice had significantly smaller liver weight and the ratio of liver/body weight (Fig. S4B and Fig. 6C), moreover, total tumour numbers, the incidence of tumours larger than 5 mm and the max tumour volume were all decreased significantly compared to control group (Fig. 6D–F). In addition, the expression of Ki67 was significantly reduced in the treatment groups compared to the control (Fig. 6G and H). Although the combination of WSG and CQ did not show synergistic antitumour effects, WSG alone achieved similar antitumour effects to CQ. Next, we performed western blot analysis in tumour tissuess and observed that exposure of WSG, CQ, or the combination of both compounds increased the protein level of LC3-II and p62, and reduced the mature form of Cat B than control groups, indicating effective autophagy inhibition and lysosomal dysfunction by WSG treatment (Fig. 6I). Furthermore, the cleaved caspase 8 was also increased in WSG treated groups, indicating that WSG triggered cell apoptosis to inhibit tumour growth (Fig. 6I). All together, these data demonstrated that WSG was a potential antitumour agent in treatment of HCC by inhibiting autophagic degradation and inducing tumour cell apoptosis in vivo.
Fig. 6.

WSG attenuates DEN-CCl4-induced primary liver cancer tumorigenesis. (A) Schematic of workflow for assessment of antitumour effect of WSG in C57BL/6 mice. Mice were treated with CCl4 twice a week (i.p.) for three months. WSG or CQ was administrated (i.p.) at the dose of 50 mg/kg or 25 mg/kg three times a week, respectively. (B) Representative images of livers and hematoxylin-eosin (HE) staining images of livers. Red-dotted circles indicate tumours. Black-dotted lines indicate the boundary of normal tissues and tumour tissues. N, normal tissue; T, tumour tissue. Scale bar: 50 μm. (C–F) Shows the ration of liver weight/body weight (C), total tumour number (D), the number of tumours bigger than 5 mm (E) and max tumour volume (F). (G) Representative images of Ki67 staining in liver. Scale bar: 50 μm. (H) Quantification of Ki67 positive cells in (G). More than 10 fields were counted in each mice and data are representative of 5 mice in each group. (I) Expression of LC3, p62, Cat B and caspase 8 in tumours from at least 3 different mice in each groups. All data are presented as mean ± SEM. *P < 0.05; **P < 0.01. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
2.7. Discussion
HCC is one of the leading causes of cancer death worldwide. Despite considerable improvements in HCC treatment, clinical drug treatment is limited due to drug resistance and the five-year survival rate still remains unsatisfactory. A growing number of studies showed that autophagy is one of the risk factors of HCC and targeting autophagy has been explored as a potential therapeutic strategy for HCC [13]. In this study, we found WSG significantly inhibited HCC cell growth, glycolysis and TCA metabolism, and induced HCC cell apoptosis under nutrient deprivation by inhibiting autophagy and impairing lysosomal function (Fig. 7). In addition, WSG exerted remarkable tumour inhibition activity in our xenograft mouse model and DEN/CCl4-induced primary HCC mouse model. Our study has demonstrated, for the first time, that WSG is a novel autophagy inhibitor with great potential for HCC treatment.
Fig. 7.

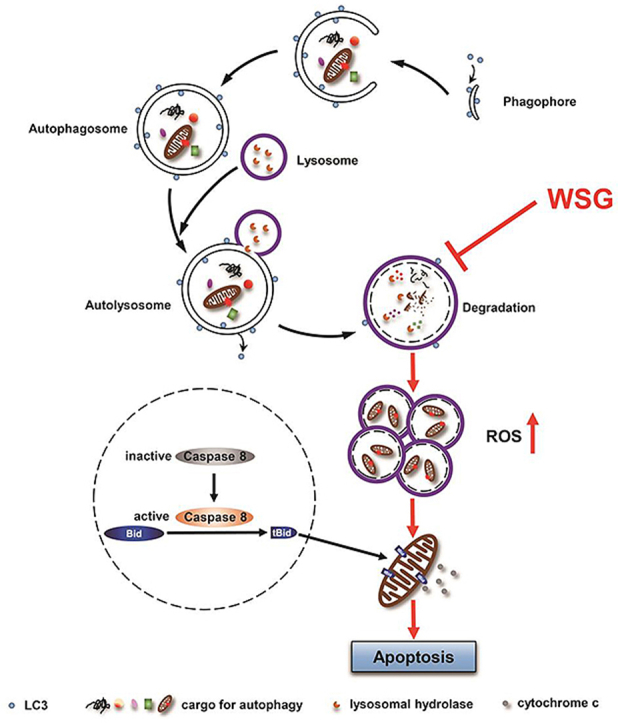
A working model of antitumour effect of WSG by inhibiting autophagic degradation. WSG inhibits autophagy flux by inhibiting lysosome acidification and lysosome cathepsins activities without affecting the fusion between autophagosomes and lysosomes. The blockage of autophagic degradation leads to the accumulation of damaged mitochondria and reactive oxygen species (ROS) production. Furthermore, WSG sensitizes liver cancer cells to apoptosis via the cleavage of the protein BID into the truncated BID (tBID) by activated caspase 8, which results in activation of the mitochondrial apoptosis pathway under nutrient deprivation.
In the light of understanding the pro-survival role of autophagy in tumour cells under various stresses, autophagy inhibition has been explored as an appealing therapeutic strategy in cancer. Multiple steps in the autophagy pathway provide potential drug targets for cancer therapy. Lysosome inhibitors CQ and HCQ (hydroxychloroquine) are currently the only clinically available drugs targeting autophagy in cancer therapy, however, the inconsistent inhibition of autophagy among patients and dose-limiting toxicities limited further clinical application of the HCQ therapy [4]. The next generation lysosome inhibitors are also in development, such as Lys05, which is a more potent autophagy inhibitor than CQ and HCQ due to its single-agent antitumour activity and greater deacidification capacity of the lysosome [42]. Inhibitors against autophagy regulators, such as VPS34 (vacuolar sorting protein 34 that forms a complex with BECN1) [[43], [44], [45]], ULK1 (human homolog of ATG1) [46] and ATG4B (in conjunction with ATG7) [47], have been reported to inhibit tumour growth in preclinical mouse models. Although there are many ongoing clinical trials targeting autophagy for cancer treatment, none of them focuses on HCC. Thus, it is of great significance for clinical trials targeting autophagy using novel autophagy inhibitors with lower toxicity and better therapeutic effect for HCC therapy [9].
β-D-glucans possess strong immune-modulatory activities, which have been proven by in vitro and in vivo clinical trials based on animals and humans [14]. We found WSG exerted prominent antitumour effects in immunodeficient BALB/c nude mice (Fig. 1F–H), which lack a thymus and are unable to produce T-cells, suggesting that the direct antitumour effects of WSG was independent of the immune system. Next, we determined whether autophagy plays an important role in the direct antitumour mechanism of β-D-glucan against HCC. Our results suggested that WSG was a novel autophagy inhibitor and inhibited autophagic flux at its late stage in HCC cells (Fig. 2).
To fully understand the possible mechanism of WSG on autophagy inhibition, we examined the autolysosome formation and lysosomal function upon WSG treatment. Lysosome is an intracellular acidic organelle and its low luminal pH (4–5) is indispensable for lysosomal enzyme activation and cargo degradation. It has been reported that an increase in lysosomal pH can impair the fusion of lysosomes with autophagosomes [48]. However, another study has also confirmed that autophagosome-lysosome fusion and autolysosome acidification are two separable processes and observed that the vesicle fusion remains active after genetically depleting individual subunits of the V-ATPase, which is lysosomal proton pump to maintain the low lysosomal luminal pH [49]. Cathepsins are among the most important lysosomal hydrolases that degrade lysosomal proteins at an acidic pH [29]. These proteins are first synthesized as inactive pre-pro-cathepsins in rough endoplasmic reticulum, where they are removed of the signal peptide and glycosylated to yield pro-cathepsins. Pro-cathepsins are transported to the Golgi and undergo further glycosylation and phosphorylation to form mannose-6-phosphate proteins, which are targeted to lysosomes by mannose-6-phosphate receptors and then are hydrolyzed at the acidic lysosomal environment to form active and mature cathepsins [[28], [50]]. Our data strongly suggest that WSG blocks autophagic flux by increasing lysosomal pH and inhibiting cathepsins activities without affecting the fusion between autophagosomes and lysosomes (Fig. 3A–C).
Autophagy can degrade diverse substrates to feed into various metabolic pathways that implicated its potential role in supporting tumour metabolism. Previous study has provided evidence that autophagy is important for Ras-mediated oncogenic transformation due to its maintenance of glycolysis [51]. Knockdown of ATG7 results in decreased glycolysis in chronic myeloid leukemia cells, demonstrating the importance of autophagy in sustaining glycolysis [52]. Our metabolic flux assays using labelled glucose showed that glycolysis and TCA metabolism were significantly decreased upon WSG-induced autophagy inhibition (Fig. 4). In addition, autophagy is also essential for maintaining mitochondrial quality control and removing damaged proteins and organelles, particularly mitochondria [33]. Autophagy deficiency increased the accumulation of defective mitochondria and impaired tumour development in Kras-driven model [53]. Of note, there was significant accumulation of damaged mitochondria and ROS production in cancer cells treated with WSG (Fig. 5A–C), along with the accumulation of autophagic cargos including ubiquitinated proteins (Fig. 3D and E).
It is well established that autophagy promotes tumour cell survival during periods of stresses, such as hypoxia, nutrient deficiency and therapeutic resistance. Based on the fact that caspase 8 was activated both in FBS-free culture condition (Fig. 5F) and tumour tissues from C57BL/6 mice (Fig. 6I), we demonstrated that WSG sensitized HCC cells to apoptosis under nutrient deprivation. Previous findings have demonstrated a link between autophagy and the extrinsic apoptotic pathway mediated by p62, which binds caspase 8 to enable its aggregation and activation [54]. However, we found that caspase 8 activation was independent of p62 upon WSG treatment under serum starvation when we knocked down p62 (data not shown). It has also reported that the autophagy inhibitor CQ can sensitize breast cancer cells to chemotherapy independent of autophagy [55]. It is conceivable that the action of WSG to inhibit autophagy may partially contribute to the apoptosis induced by WSG under nutrient deprivation.
In summary, we have uncovered a novel mechanism underlying antitumour effects of β-D-glucan that WSG suppresses autophagy by increasing lysosomal pH and inhibiting lysosome cathepsins, resulting in the blockage of autophagic degradation. The accumulation of damaged mitochondria finally leads to ROS production. Furthermore, WSG sensitizes HCC cells to apoptosis via the intrinsic and extrinsic pathways under nutrient deprivation (Fig. 7). Our study has demonstrated for the first time that WSG is a natural autophagy inhibitor with a great potential in clinical application for liver cancer therapy.
3. Materials and methods
3.1. Cell culture
Huh7 cells, SMMC-7721 cells, LM3 cells and HL-7702 cells were purchased from the Cell Bank of Chinese Academy of Sciences, Shanghai, China. LIXC501 cells were the primary tumour cells that separated from human primary liver tumour tissues of the early stage HCC patients. These cells were cultured in Dulbecco's Modified Eagle Medium (Hyclone) supplemented with 10% FBS (Hyclone) and 1% penicillin-streptomycin (Gibco) at 37 °C in a 5% CO2 humidified atmosphere. WSG was dissolved in medium and then filtered the medium through 0.22 μm Steriflip filter (Millipore) for cell culture. All experiments were performed at least 3 independent experiments or 3 repeats.
3.2. Cell viability assay
2 × 103 cells/well was seeded in 96-well plates for 6 repeats. After adherent, cells were treated with WSG-contained medium for different time points. Then Cell Counting Kit with a catalogue number of #OCPA(C) 20132001(2) (Obio Technology, Shanghai, China) was added to each well at the indicated time according to the manufacturer's instruction.
3.3. Mouse models
All animal experimental protocols were approved by Institutional Animal Care and Use Committee at Shanghai Institute of Nutrition and Health, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences.
3.4. Xenograft tumour growth mouse model
Huh7 cells (2 × 106), resuspended in 75 μl DMEM and ECM gel matrix, were injected subcutaneously into both sides of 5-week-old BALB/c male nude mice (Shanghai Super B&K laboratory animal Corp). Mice were randomly divided into 3 groups on the third day after injection and were administrated with vehicle (0.9% NaCl solution), 150 mg/kg and 300 mg/kg WSG via intraperitoneal injection every other day for 4 weeks. The tumour volume and body weight were recorded at the same time of administration. Tumours were collected and weighted after the mice were sacrificed. Tumour volumes were calculated using formula V = 0.52 × a × b2 where “a” is tumour length and “b” is tumour width. The animals were maintained in individually ventilated cages at 23–25 °C, a relative humidity of 50–60% under specific pathogen-free environment.
3.5. DEN/CCl4-induced primary liver cancer mouse model
Three-week-old male C57BL/6 mice were purchased from Shanghai Model Organisms Center, China. C57BL/6 mice were injected with DEN (Sigma, 50 μg/g, i. p.) when they were 3 weeks old. The mice were randomly divided into 4 groups and treated with CCl4 (5 μl/g of 9:1 mixture of olive oil and CCl4, i. p.) at 8th week twice a week for 3 months and their body weight was recorded at the same time. WSG and CQ (Chloroquine diphosphate salt, Sigma) were administrated (i.p.) at the dose of 50 mg/kg and 25 mg/kg dissolved in vehicle (0.9% NaCl solution), respectively, at 12 weeks of age three times a week. Mice were killed under isoflurane anaesthesia at 39th week. Livers and tumours were rapidly excised, weighted, freshly frozen in liquid nitrogen and stored at −80 °C until immunoblot analysis. A small part of the livers were fixed in formalin for further immunohistochemistry and hematoxylin and eosin staining.
3.6. Cell cycle assay
Huh7 cells were treated WSG (8 mg/ml) for 48 h. Digestion of adherent cells using 0.5% trypsin (Gibco). Cells were washed with Dulbecco's phosphate buffered saline (PBS) twice and resuspended into single cell suspension and fixed with chilled 70% ethanol overnight at −4 °C. Before analysis, the cells were stained with Propidium Iodide (PI) by using Cell Cycle and Apoptosis Analysis Kit (Beyotime Biotechnology, C1052) according to the manufacturer's protocol. The cells were analysed using the FACS flow cytometry (FACSCalibur; Becton Dickinson, USA)
3.7. Western blotting
Western blotting was performed as described previously [56]. The following antibodies were used for western blotting: STAT 3 (Abcam, ab109085), p-STAT 3 (phospho S727, Abcam, ab30647), p21 (Proteintech, 10355-1-AP), p27 (Proteintech, 25614-1-AP), Cyclin D1 (Abcam, ab134175), Cyclin E1 (Abcam, ab133266), Cyclin B1 (Abcam, ab181593), Cyclin A2 (Abcam, ab181591), LC3 (Cell Signalling Technology, 2775S), p62 (Abcam, ab91526), p-AMPK (Thr172, Cell Signalling Technology, 2535S), AMPK (Cell Signalling Technology, 5832T), p-mTOR (Ser2448, Cell Signalling Technology, 5536P), mTOR (Cell Signalling Technology, 2983P), BECN1 (Santa Cruz, sc-48341), ATG5 (Proteintech, 10181-2-AP), GAPDH (Proteintech, 60004-1-Ig), Cathepsin D (Cell Signalling Technology, 2284), Cathepsin B (Cell Signalling Technology, 31718), Ub (Santa Cruz, sc-8017), Caspase 8 (Proteintech, 13423-1-AP), Bid (Cell Signalling Technology, 2002T), VDAC1/2 (Proteintech, 10866-1-AP), ki67 (Abcam, ab15580), LAMP1 (Cell Signalling Technology, 9091S).
3.8. RNA-sequence analysis
Huh7 cells were treated with 8 mg/ml WSG and were lysed with TRIzol reagent. The library preparation and sequencing were carried out by Shenzhen Genomics Institute BGI (Wuhan, China). We measured the expression of the transcript isoforms using RSEM, and used the NOISeq method to screen differentially expressed genes (DEGs) between 2 groups. Microarray data is available here: https://www.biosino.org/node/project/detail/OEP000452.
3.9. Transmission electron electron microscopy
Huh7 cells were treated with WSG (8 mg/ml) in complete medium for 48 h and fixed by 2.5% glutaraldehyde at 4 °C overnight. Samples for TEM imaging were prepared and stained as described previously [57] and observed under transmission electron microscope transmission electron microscopy (FEI Tecnau G2 spirit).
3.10. Immunofluorescence and fluorescence microscopy
For GFP-LC3 and lamp1 colocalization, Huh7 cells were transfected with GFP-LC3 plasmid for 48 h and cells were then seeded to a cover glass slide chamber and treated with WSG, HBSS (Gibco) or Baf A1 (Selleck). Immunofluorescence staining was performed as previously described [57].
For autophagic flux analysis, Huh7 cells were seeded and grown overnight on a 20-mm-glass-bottom cell-culture dish (NEST, 801001). The formation of autolysosomes in cells treated with WSG and CQ was detected using the Premo Autophagy Tandem Sensor RFP-GFP-LC3B Kit (Thermo Fisher Scientific, P36239) according to the manufacturer's protocol. Nuclei were stained in FBS-free DMEM medium contained 5 ng/ml hoechst (Thermo Fisher scientific) at 37 °C for 10 min.
For GFP-LC3 puncta assay, cells were transfected with GFP-LC3 plasmid for 48 h, and then plated on a 20-mm-glass-bottom cell-culture dish and treated with WSG. All fluorescent images were taken using confocal microscopy (Carl Zeiss Meditec, Inc.) and quantified using Image J software.
3.11. Lysosomal pH assay
Huh7 cells (6 × 104) were plated on a 20-mm-glass-bottom cell-culture dish and treated with WSG or CQ. Then cells were incubated with 2 μM LysoSensor Green DND-189 (Shanghai Yeasen) reagent at 37 °C for 45 min. Nuclei were stained in FBS-free DMEM medium containing 5 ng/ml hoechst at 37 °C for 10 min. Fluorescent images were taken using confocal microscopy.
3.12. Labelled 13C-glucose assay and analysis by GC-MS
1.5 × 106 Huh 7 cells were seeded in 10 cm dish. After cells were adherent, the medium was changed with U–13C-glucose medium which contains low glucose DMEM (1 g/L glucose, no glutamine, Gibco, 11054-020), 1 g/L U–13C-glucose (Cambridge Isotope Laboratory), 1 mM pyruvate, 2 mM l-glutamine, 10% FBS and 1% penicillin-streptomycin with or without WSG. It was regarded as 0 h at this time point. U–13C-glucose medium after cell culture was collected at 24 h and 48 h. The cell plates were immediately put on dry ice and added with 2 ml 50% methanol (precooled at −80 °C) for 20 min. Then scraped off the cells with cell scraper and transferred the cell lysate into tubes on dry ice. 1.5 ml chloroform was added into the tubes and samples were then vortexed and centrifuged at 14000 g for 15 min at 4 °C. The supernatant were lyophilized in a vacuum concentrator and resolved with 70 μl O-isobutylhydroxylamine (20 mg/ml) at 85 °C by metal bath for 20 min 30 μl derivative reagent N-tert-Butyldimethylsilyl-N-Methytrifluoro-acetamide (Sigma) was added to each tube at 85 °C by metal bath for 1 h. Samples were centrifuged at 14000 g for 15 min at 4 °C. The supernatant was for GC-MS analysis.
GC-MS analysis was performed as described previously [58]. A Shimadzu QP-2010 Ultra GC-MS was programmed with an injection temperature of 250 °C injection split ratio 1/10 and injected with 1 μl of sample. The GC column used was a 30 m × 0.25 mm × 0.25 mm HP-5ms. GC-MS data were analysed to determine isotope labelling and quantities of metabolites and implemented in MATLAB [59].
3.13. Glucose consumption assay
Collected medium from labelled 13C-glucose was used to measure the glucose consumption by d-Glucose Assay Kit (R-Biopharm, 10716251035) according to the manufacturer's protocol.
3.14. Measurement of mitochondrial membrane potential (MMP) and reactive oxygen species (ROS)
1.5 × 104 Huh7 cells were seeded in 48-well plates and treated with different concentration of WSG for 48 h. Cells were stained as previously described [56]. The MMP or ROS was determined by High-Content Screening (HCS, Thermo scientific).
3.15. ATP, ADP, and AMP detection by LC–MS/MS
We used TSQ Vantage LC-MS interfaced with Ultimate 3000 Liquid Chromatography system (Thermo Scientific) and Triple quadrupole mass spectrometry to detect ATP, ADP and AMP of cells. Polar metabolites were extracted according to the previous protocol [60]. Samples and standards were measured using a TSQ Vantage equipped with a HILIC column (Amide 4.6 × 100 mm ID 3.5 μm; Part No: 186004868, Waters). Compounds were eluted from the column with a linear gradient as flows: 0–3 min, 15% A; 3–16 min, to 50%A; 16–20 min, to 85% A; 20–23 min, to 15% A; 23–25 min, 15% A. All samples were measured in negative mode.
3.16. Caspase 8 activity
Huh7 cells were treated with WSG (8 mg/ml) in FBS-free medium for 48 h. Caspase 8 activity was measured with Caspase 8 Activity Assay Kit (Beyotime Biotechnology, C1151) according to the manufacturer's protocol.
3.17. Apoptosis assay
Huh7 cells were treated with WSG (8 mg/ml) in complete medium or FBS-free medium for 48 h. Digestion of adherent cells using 0.5% trypsin (Gibco) and suspended cells were also collected. Cells were stained with FITC Annexin V and PI by using FITC Annexin V Apoptosis Detection Kit (BD Pharmingen, 556547) according to the manufacturer's protocol. Analyse by flow cytometry within 1 h.
3.18. Mitochondria isolation
Huh7 cells were treated with WSG (8 mg/ml) in FBS-free medium for 48 h. Mitochondria was isolated using Cell Mitochondria Isolation Kit (Beyotime Biotechnology, C3601) according to the manufacturer's protocol.
3.19. Statistical analysis
Data are presented as mean ± SEM. Statistical significance was analysed using the unpaired two-tailed Student's t-test at least 3 independent experiments using GraphPad Prism (GraphPad Software, USA). P-value < 0.05 was considered statistically significant. *P < 0.05; **P < 0.01; ***P < 0.001.
Declaration of competing interest
The authors declare no competing financial interests.
Acknowledgments
We would like to acknowledge the stimulating discussion with Dr. Wen-Xing Ding at University of Kansas Medical Center, Kansas, USA. This work was financially supported by National Key R&D Program of China administered by Chinese Ministry of Science and Technology (MOST) (2016YFD0400205, 2018YFA0800300), the National Natural Science Foundation of China (31671231 and 91857112). We acknowledge the help from molecular biology core laboratory, animal facilities, and mass spectrometry facilities at Shanghai Institutes of Nutrition and Health (SINH), CAS, Shanghai.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2020.101495.
Contributor Information
Qiang Wang, Email: wangqiang06@caas.cn.
Yongzhen Tao, Email: yztao01@sibs.ac.c.
Huiyong Yin, Email: hyyin@sibs.ac.cn.
Author contributions
H·Y.Y. and Y.Z.T. designed this study. N.N.W., G.J.L., M.L., X.X.H., C.Z.Y. and Q.C.T. performed experiments. N.N.W., Y.Z.T. and H·Y.Y. wrote the paper. H.Z.L, W.Q.B. and Q.W. provided and prepared β-glucan samples. X.S. helped RNA-seq data analysis.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Graf D., Vallbohmer D., Knoefel W.T., Kropil P., Antoch G., Sagir A. Multimodal treatment of hepatocellular carcinoma. Eur. J. Intern. Med. 2014;25:430–437. doi: 10.1016/j.ejim.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 2.Llovet J.M., Montal R., Villanueva A. Randomized trials and endpoints in advanced HCC: role of PFS as a surrogate of survival. J. Hepatol. 2019;70:1262–1277. doi: 10.1016/j.jhep.2019.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kanwal F., Singal A.G. Surveillance for hepatocellular carcinoma: current best practice and future direction. Gastroenterology. 2019;157:54–64. doi: 10.1053/j.gastro.2019.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levy J.M.M., Towers C.G., Thorburn A. Targeting autophagy in cancer. Nat. Rev. Canc. 2017;17:528–542. doi: 10.1038/nrc.2017.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mizushima N., Yoshimori T., Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011;27:107–132. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 6.Rubinsztein D.C., Marino G., Kroemer G. Autophagy and aging. Cell. 2011;146:682–695. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 7.Singh R., Cuervo A.M. Autophagy in the cellular energetic balance. Cell Metabol. 2011;13:495–504. doi: 10.1016/j.cmet.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mizushima N., Levine B., Cuervo A.M., Klionsky D.J. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee Y.J., Jang B.K. The role of autophagy in hepatocellular carcinoma. Int. J. Mol. Sci. 2015;16:26629–26643. doi: 10.3390/ijms161125984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang Z.J., Chee C.E., Huang S., Sinicrope F.A. The role of autophagy in cancer: therapeutic implications. Mol. Canc. Therapeut. 2011;10:1533–1541. doi: 10.1158/1535-7163.MCT-11-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guntuku L., Gangasani J.K., Thummuri D., Borkar R.M., Manavathi B., Ragampeta S. IITZ-01, a novel potent lysosomotropic autophagy inhibitor, has single-agent antitumour efficacy in triple-negative breast cancer in vitro and in vivo. Oncogene. 2019;38:581–595. doi: 10.1038/s41388-018-0446-2. [DOI] [PubMed] [Google Scholar]
- 12.Sui X., Chen R., Wang Z., Huang Z., Kong N., Zhang M. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 2013;4:e838. doi: 10.1038/cddis.2013.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang S., Yang L., Li X., Li B., Li Y., Zhang X. New insights into autophagy in hepatocellular carcinoma: mechanisms and therapeutic strategies. Am. J. Cancer Res. 2019;9:1329–1353. [PMC free article] [PubMed] [Google Scholar]
- 14.Bashir K.M.I., Choi J.S. Clinical and physiological perspectives of beta-glucans: the past, present, and future. Int. J. Mol. Sci. 2017;18 doi: 10.3390/ijms18091906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee J.G., Kim Y.S., Lee Y.J., Ahn H.Y., Kim M., Kim M. Effect of immune-enhancing enteral nutrition enriched with or without beta-glucan on immunomodulation in critically ill patients. Nutrients. 2016;8 doi: 10.3390/nu8060336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoon T.J., Kim T.J., Lee H., Shin K.S., Yun Y.P., Moon W.K. Anti-tumour metastatic activity of beta-glucan purified from mutated Saccharomyces cerevisiae. Int. Immunopharm. 2008;8:36–42. doi: 10.1016/j.intimp.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 17.Saluk-Juszczak J., Krolewska K., Wachowicz B. beta-glucan from Saccharomyces cerevisiae as a blood platelet antioxidant. Platelets. 2010;21:451–459. doi: 10.3109/09537101003780032. [DOI] [PubMed] [Google Scholar]
- 18.Liu H., Li Y., Gao J., Shi A., Liu L., Hu H. Effects of microfluidization with ionic liquids on the solubilization and structure of beta-D-glucan. Int. J. Biol. Macromol. 2016;84:394–401. doi: 10.1016/j.ijbiomac.2015.12.014. [DOI] [PubMed] [Google Scholar]
- 19.Chan G.C., Chan W.K., Sze D.M. The effects of beta-glucan on human immune and cancer cells. J. Hematol. Oncol. 2009;2:25. doi: 10.1186/1756-8722-2-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tian J., Ma J., Ma K., Guo H., Baidoo S.E., Zhang Y. beta-Glucan enhances antitumour immune responses by regulating differentiation and function of monocytic myeloid-derived suppressor cells. Eur. J. Immunol. 2013;43:1220–1230. doi: 10.1002/eji.201242841. [DOI] [PubMed] [Google Scholar]
- 21.Xu H., Zou S., Xu X. The beta-glucan from Lentinus edodes suppresses cell proliferation and promotes apoptosis in estrogen receptor positive breast cancers. Oncotarget. 2017;8:86693–86709. doi: 10.18632/oncotarget.21411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y., Li Q., Wang J., Cheng F., Huang X., Cheng Y. Polysaccharide from Lentinus edodes combined with oxaliplatin possesses the synergy and attenuation effect in hepatocellular carcinoma. Canc. Lett. 2016;377:117–125. doi: 10.1016/j.canlet.2016.04.037. [DOI] [PubMed] [Google Scholar]
- 23.Katsuragi Y., Ichimura Y., Komatsu M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015;282:4672–4678. doi: 10.1111/febs.13540. [DOI] [PubMed] [Google Scholar]
- 24.Zhou J., Hu S.E., Tan S.H., Cao R., Chen Y., Xia D. Andrographolide sensitizes cisplatin-induced apoptosis via suppression of autophagosome-lysosome fusion in human cancer cells. Autophagy. 2012;8:338–349. doi: 10.4161/auto.18721. [DOI] [PubMed] [Google Scholar]
- 25.Terasawa K., Tomabechi Y., Ikeda M., Ehara H., Kukimoto-Niino M., Wakiyama M. Lysosome-associated membrane proteins-1 and -2 (LAMP-1 and LAMP-2) assemble via distinct modes. Biochem. Biophys. Res. Commun. 2016;479:489–495. doi: 10.1016/j.bbrc.2016.09.093. [DOI] [PubMed] [Google Scholar]
- 26.Li D.L., Wang Z.V., Ding G., Tan W., Luo X., Criollo A. Doxorubicin blocks cardiomyocyte autophagic flux by inhibiting lysosome acidification. Circulation. 2016;133:1668–1687. doi: 10.1161/CIRCULATIONAHA.115.017443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jung M., Lee J., Seo H.Y., Lim J.S., Kim E.K. Cathepsin inhibition-induced lysosomal dysfunction enhances pancreatic beta-cell apoptosis in high glucose. PloS One. 2015;10 doi: 10.1371/journal.pone.0116972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Achour O., Bridiau N., Kacem M., Delatouche R., Bordenave-Juchereau S., Sannier F. Cathepsin D activity and selectivity in the acidic conditions of a tumor microenvironment: utilization in the development of a novel Cathepsin D substrate for simultaneous cancer diagnosis and therapy. Biochimie. 2013;95:2010–2017. doi: 10.1016/j.biochi.2013.07.010. [DOI] [PubMed] [Google Scholar]
- 29.Liaudet-Coopman E., Beaujouin M., Derocq D., Garcia M., Glondu-Lassis M., Laurent-Matha V. Cathepsin D: newly discovered functions of a long-standing aspartic protease in cancer and apoptosis. Canc. Lett. 2006;237:167–179. doi: 10.1016/j.canlet.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 30.Carew J.S., Medina E.C., Esquivel J.A., 2nd, Mahalingam D., Swords R., Kelly K. Autophagy inhibition enhances vorinostat-induced apoptosis via ubiquitinated protein accumulation. J. Cell Mol. Med. 2010;14:2448–2459. doi: 10.1111/j.1582-4934.2009.00832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lao Y., Wan G., Liu Z., Wang X., Ruan P., Xu W. The natural compound oblongifolin C inhibits autophagic flux and enhances antitumour efficacy of nutrient deprivation. Autophagy. 2014;10:736–749. doi: 10.4161/auto.28034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rabinowitz J.D., White E. Autophagy and metabolism. Science (New York, NY) 2010;330:1344–1348. doi: 10.1126/science.1193497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.White E., Mehnert J.M., Chan C.S. Autophagy, metabolism, and cancer. Clinical cancer research : an. official journal of the American Association for Cancer Research. 2015;21:5037–5046. doi: 10.1158/1078-0432.CCR-15-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 35.Anderson N.M., Mucka P., Kern J.G., Feng H. The emerging role and targetability of the TCA cycle in cancer metabolism. Protein & cell. 2018;9:216–237. doi: 10.1007/s13238-017-0451-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weinberg S.E., Chandel N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015;11:9–15. doi: 10.1038/nchembio.1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bruntz R.C., Lane A.N., Higashi R.M., Fan T.W. Exploring cancer metabolism using stable isotope-resolved metabolomics (SIRM) J. Biol. Chem. 2017;292:11601–11609. doi: 10.1074/jbc.R117.776054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chang Y., Yan W., He X., Zhang L., Li C., Huang H. miR-375 inhibits autophagy and reduces viability of hepatocellular carcinoma cells under hypoxic conditions. Gastroenterology. 2012;143 doi: 10.1053/j.gastro.2012.04.009. 177-87.e8. [DOI] [PubMed] [Google Scholar]
- 39.Amaravadi R.K., Thompson C.B. The roles of therapy-induced autophagy and necrosis in cancer treatment. Clin. Canc. Res.: Off. J. Am. Assoc. Cancer Res. 2007;13:7271–7279. doi: 10.1158/1078-0432.CCR-07-1595. [DOI] [PubMed] [Google Scholar]
- 40.Favaloro B., Allocati N., Graziano V., Di Ilio C., De Laurenzi V. Role of apoptosis in disease. Aging. 2012;4:330–349. doi: 10.18632/aging.100459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mukhopadhyay S., Panda P.K., Sinha N., Das D.N., Bhutia S.K. Autophagy and apoptosis: where do they meet? Apoptosis : an Int. J. Program. Cell Death. 2014;19:555–566. doi: 10.1007/s10495-014-0967-2. [DOI] [PubMed] [Google Scholar]
- 42.McAfee Q., Zhang Z., Samanta A., Levi S.M., Ma X.H., Piao S. Autophagy inhibitor Lys05 has single-agent antitumour activity and reproduces the phenotype of a genetic autophagy deficiency. Proc. Natl. Acad. Sci. U. S. A. 2012;109:8253–8258. doi: 10.1073/pnas.1118193109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bago R., Malik N., Munson M.J., Prescott A.R., Davies P., Sommer E. Characterization of VPS34-IN1, a selective inhibitor of Vps34, reveals that the phosphatidylinositol 3-phosphate-binding SGK3 protein kinase is a downstream target of class III phosphoinositide 3-kinase. Biochem. J. 2014;463:413–427. doi: 10.1042/BJ20140889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dowdle W.E., Nyfeler B., Nagel J., Elling R.A., Liu S., Triantafellow E. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat. Cell Biol. 2014;16:1069–1079. doi: 10.1038/ncb3053. [DOI] [PubMed] [Google Scholar]
- 45.Ronan B., Flamand O., Vescovi L., Dureuil C., Durand L., Fassy F. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat. Chem. Biol. 2014;10:1013–1019. doi: 10.1038/nchembio.1681. [DOI] [PubMed] [Google Scholar]
- 46.Egan D.F., Chun M.G., Vamos M., Zou H., Rong J., Miller C.J. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol. Cell. 2015;59:285–297. doi: 10.1016/j.molcel.2015.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Akin D., Wang S.K., Habibzadegah-Tari P., Law B., Ostrov D., Li M. A novel ATG4B antagonist inhibits autophagy and has a negative impact on osteosarcoma tumours. Autophagy. 2014;10:2021–2035. doi: 10.4161/auto.32229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kawai A., Uchiyama H., Takano S., Nakamura N., Ohkuma S. Autophagosome-lysosome fusion depends on the pH in acidic compartments in CHO cells. Autophagy. 2007;3:154–157. doi: 10.4161/auto.3634. [DOI] [PubMed] [Google Scholar]
- 49.Mauvezin C., Neufeld T.P. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy. 2015;11:1437–1438. doi: 10.1080/15548627.2015.1066957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen S., Dong H., Yang S., Guo H. Cathepsins in digestive cancers. Oncotarget. 2017;8:41690–41700. doi: 10.18632/oncotarget.16677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lock R., Roy S., Kenific C.M., Su J.S., Salas E., Ronen S.M. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Biol. Cell. 2011;22:165–178. doi: 10.1091/mbc.E10-06-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Karvela M., Baquero P., Kuntz E.M., Mukhopadhyay A., Mitchell R., Allan E.K. ATG7 regulates energy metabolism, differentiation and survival of Philadelphia-chromosome-positive cells. Autophagy. 2016;12:936–948. doi: 10.1080/15548627.2016.1162359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guo J.Y., Chen H.Y., Mathew R., Fan J., Strohecker A.M., Karsli-Uzunbas G. Activated Ras requires autophagy to maintain oxidative metabolism and tumourigenesis. Gene Dev. 2011;25:460–470. doi: 10.1101/gad.2016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jin Z., Li Y., Pitti R., Lawrence D., Pham V.C., Lill J.R. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell. 2009;137:721–735. doi: 10.1016/j.cell.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 55.Maycotte P., Aryal S., Cummings C.T., Thorburn J., Morgan M.J., Thorburn A. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy. 2012;8:200–212. doi: 10.4161/auto.8.2.18554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li X., Cui X.X., Chen Y.J., Wu T.T., Xu H., Yin H. Therapeutic potential of a prolyl hydroxylase inhibitor FG-4592 for Parkinson's diseases in vitro and in vivo: regulation of redox biology and mitochondrial function. Front. Aging Neurosci. 2018;10:121. doi: 10.3389/fnagi.2018.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhao X., Fang Y., Yang Y., Qin Y., Wu P., Wang T. Elaiophylin, a novel autophagy inhibitor, exerts antitumour activity as a single agent in ovarian cancer cells. Autophagy. 2015;11:1849–1863. doi: 10.1080/15548627.2015.1017185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ma L., Tao Y., Duran A., Llado V., Galvez A., Barger J.F. Control of nutrient stress-induced metabolic reprogramming by PKCzeta in tumourigenesis. Cell. 2013;152:599–611. doi: 10.1016/j.cell.2012.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Portnoy V.A., Scott D.A., Lewis N.E., Tarasova Y., Osterman A.L., Palsson B.O. Deletion of genes encoding cytochrome oxidases and quinol monooxygenase blocks the aerobic-anaerobic shift in Escherichia coli K-12 MG1655. Appl. Environ. Microbiol. 2010;76:6529–6540. doi: 10.1128/AEM.01178-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yuan M., Breitkopf S.B., Yang X., Asara J.M. A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat. Protoc. 2012;7:872–881. doi: 10.1038/nprot.2012.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.