

Abstract
NF-κB is a pro-inflammatory transcription factor that critically regulates immune responses and other distinct cellular pathways. However, many NF-κB–mediated pathways for cell survival and apoptosis signaling in cancer remain to be elucidated. Cell cycle and apoptosis regulatory protein 1 (CARP-1 or CCAR1) is a perinuclear phosphoprotein that regulates signaling induced by anticancer chemotherapy and growth factors. Although previous studies have reported that CARP-1 is a part of the NF-κB proteome, regulation of NF-κB signaling by CARP-1 and the molecular mechanism(s) involved are unclear. Here, we report that CARP-1 directly binds the NF-κB–activating kinase IκB kinase subunit γ (NEMO or NF-κB essential modulator) and regulates the chemotherapy-activated canonical NF-κB pathway. Importantly, blockade of NEMO–CARP-1 binding diminished NF-κB activation, indicated by reduced phosphorylation of its subunit p65/RelA by the chemotherapeutic agent adriamycin (ADR), but not NF-κB activation induced by tumor necrosis factor α (TNFα), interleukin (IL)-1β, or epidermal growth factor. High-throughput screening of a chemical library yielded a small molecule inhibitor of NEMO–CARP-1 binding, termed selective NF-κB inhibitor 1 (SNI)-1). We noted that SNI-1 enhances chemotherapy-dependent growth inhibition of a variety of cancer cells, including human triple-negative breast cancer (TNBC) and patient-derived TNBC cells in vitro, and attenuates chemotherapy-induced secretion of the pro-inflammatory cytokines TNFα, IL-1β, and IL-8. SNI-1 also enhanced ADR or cisplatin inhibition of murine TNBC tumors in vivo and reduced systemic levels of pro-inflammatory cytokines. We conclude that inhibition of NEMO–CARP-1 binding enhances responses of cancer cells to chemotherapy.
Keywords: NF-κB transcription factor, high-throughput screening (HTS), small molecule, breast cancer, cytokine, CARP-1/CCAR1, NEMO/IKKγ, p65/RelA
Introduction
CARP-1/CCAR1 (cell cycle and apoptosis regulatory protein 1) is a ubiquitous ∼130-kDa perinuclear phosphoprotein (1) that has homologs in vertebrates, Apis millifera, and the worm Caenorhabditis elegans. Lst3, the C. elegans ortholog of human CARP-1, is an agonist of Notch signaling that also functions as an inhibitor of the EGFR5–MAPK pathway (2). This EGFR pathway antagonism by Lst3 corroborated our prior findings of CARP-1 requirement for EGFR inhibitor-induced apoptosis (3). Additionally, CARP-1 promoter methylation as well as signaling by protein kinase A regulated CARP-1 expression and function, respectively (3–5). CARP-1 is a phosphoprotein, and although the EGF as well as the ATM kinase signaling target specific serine residues of CARP-1 (6–8), the precise role(s) and kinase(s) of CARP-1 serine phosphorylation remain unclear. CARP-1 binds with the LIM protein Zyxin and regulates apoptosis in response to UV-C irradiation (9), although it also interacts with Necdin to regulate myoblast survival (10). Furthermore, recent studies found CARP-1 as a co-activator of the cell cycle regulatory APC/C E3 ligase (11), the steroid–thyroid family of nuclear receptors (12), the glucocorticoid receptor signaling during adipogenesis, β-catenin in colon cancer metastasis, or neurogenin3-mediated pancreatic endocrine differentiation (13–15). Interestingly, CARP-1 also co-activated tumor suppressor p53 to transduce the DNA damage–induced transcriptional increase of cyclin-dependent kinase inhibitor p21WAF1 in breast cancer cells (12).
Chemotherapeutics such as ADR induce double-strand breaks (DSBs), whereas phosphorylation of H2AX at serine 139 (γ-H2AX) by ATM/ATR functions to repair DSBs (16–18). ADR also promotes apoptosis in part by inducing JNK-dependent γH2AX (19, 20). We found that ADR induced CARP-1 and γH2AX, and depletion of CARP-1 abrogated the γH2AX increase by ADR (21). CARP-1 binds with H2AX, and abrogation of CARP-1/H2AX binding blocked ADR-induced inhibition of TNBC and HeLa cells (21).
NF-κB is a pro-inflammatory transcription factor that is a critical regulator of the immune system, and it is responsive to many stimuli that engage signaling pathways to activate this transcription factor and effect distinct cellular responses (22). Except for C. elegans, the NF-κB signaling components exist in almost all multicellular organisms (23). In mammalian cells, five members of the NF-κB family, including RelA (p65), RelB, c-Rel, p50/p105 (NF-κB1), and p52/p100 (NF-κB2), function by forming homo- and heterodimers. A family of inhibitory proteins, called IκBs, sequester the NF-κB complexes in the cytoplasm. IκBs are phosphorylated by IκB kinase (IKK), which leads to IκB degradation by the ubiquitin–proteasome pathway, followed by release of NF-κB for its translocation to the nucleus where it functions as a transcription factor (23). The IKK complex contains two kinase subunits, IKKα and IKKβ, and an associated regulatory subunit called NEMO (IKKγ). NF-κB regulates cellular homeostasis as well as tumor cell proliferation, survival, metastasis, inflammation, invasion, and angiogenesis, and it often contributes to a resistant phenotype and poor prognosis (24). Although a pro-apoptotic function for NF-κB has also been suggested (25–27), and possibly involves NF-κB regulation of transducers of receptor-mediated apoptosis, a full characterization of the complex molecular details of the apoptotic functions of NF-κB remain to be accomplished. However, therapy-induced DNA damage that causes ATM/ATR activation to promote H2AX-dependent DSB repair also stimulates phosphorylation of NEMO by ATM. The phosphorylated NEMO is mono-ubiquitinated, which triggers its nuclear export and IKK activation in the cytoplasm (28). This therapy-induced activation of canonical NF-κB promotes production of pro-inflammatory cytokines, cell growth, and survival signaling and contributes to therapy resistance.
Because CARP-1 is a regulator of cell growth and survival signaling (1, 3, 12) and a component of the NF-κB proteome (29), and CARP-1 depletion inhibited transcriptional activation of NF-κB by ADR, TNFα, or an experimental CARP-1 functional mimetic (CFM) compound (30), we investigated the molecular mechanism of CARP-1–dependent regulation of NF-κB signaling. We found that CARP-1 directly binds with NEMO, and blockage of this interaction interferes with ADR-induced activation of canonical NF-κB. Pharmacological inhibition of NEMO–CARP-1 binding enhances cisplatin efficacy in part by impacting levels of circulating pro-inflammatory cytokines in immunocompetent mice bearing subcutaneous tumors of murine breast cancer cells.
Results
CARP-1 binds with NEMO
We previously found that TNFα, adriamycin, or CFM-4 compound caused increased transcriptional activation of NF-κB in human TNBC cells, whereas knockdown of CARP-1 attenuated activation of NF-κB by these agents (30). Because adriamycin or epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor Iressa inhibited HBC growth in part by inducing CARP-1 expression (1, 3), and CARP-1 was found to be a part of the NF-κB proteome (29), we investigated whether and how CARP-1 regulates NF-κB signaling. Cellular proteins from the human and murine TNBC or human cervical cancer HeLa cells were immunoprecipitated using anti-CARP-1 (α2) or NEMO antibodies followed by analysis of immunocomplexes by Western blotting (WB) using NEMO or CARP-1 antibodies, respectively. The immunocomplexes derived from using anti-CARP-1 (α2) antibodies contained NEMO protein (Fig. 1A). As also shown in Fig. 1B, CARP-1 protein was present in the immunocomplexes derived from NEMO antibodies. These data in Fig. 1, A and B, demonstrate that CARP-1 interacts with NEMO. We then performed mutagenesis-based analyses to map the interacting epitopes of CARP-1 and NEMO proteins. In the first instance, we utilized constructs expressing myc-His–tagged, nonoverlapping CARP-1 mutants that we have described before (3). Each of the CARP-1 mutant plasmids together with a plasmid expressing GST-tagged NEMO (pEBG–NEMO) were separately transfected in COS-7 cells. Protein lysates were immunoprecipitated using anti-GST antibodies followed by WB with anti-myc tag antibodies. NEMO interacted with the CARP-1(452–654) mutant (Fig. S1A). Next, HBC cells were transfected with various mutants of NEMO (31) together with a plasmid encoding the myc-His–tagged CARP-1(552–654) mutant. Protein lysates were immunoprecipitated using anti-His tag antibodies followed by WB with anti-myc tag antibodies. As shown in Fig. S1B, the CARP-1(552–654) mutant interacted with NEMO(221–405). Additional plasmids expressing myc-His–tagged CARP-1 having in-frame deletions of amino acids 553–599 or 521–566 were generated, and stable neomycin-resistant HBC cells expressing these plasmids were obtained and characterized as detailed under “Experimental procedures.” Generation and characterization of HBC cells stably expressing pcDNA3 vector, myc-His–tagged WT, or CARP-1(600–650) mutant have been described before (3, 21). Myc-tagged proteins were immunoprecipitated from stable sublines expressing vector, WT CARP-1, or CARP-1 mutant proteins followed by WB of immunocomplexes with NEMO antibodies. This experiment revealed CARP-1 amino acids 553–599 harbored NEMO-binding epitope (Fig. S1C). We next generated constructs for expression of GST–NEMO(2–260) and His–TAT–HA-tagged CARP-1 mutant peptides for expression in Escherichia coli as detailed under “Experimental procedures.” These peptides were utilized to determine binding of NEMO(2–260) with various CARP-1 peptides. As shown in Fig. S1D, NEMO(2–260) bound with CARP-1(552–654) and CARP-1(552–580) peptides. Our data in Fig. S1, A–D suggest that CARP-1(552–580) and NEMO(221–260) harbor epitopes for their mutual interaction/binding. On this basis, we generated pcDNA-based recombinant constructs expressing EGFP, EGFP–CARP-1(551–580), GST, GST–NEMO, GST–NEMO(221–261), and GST–NEMO(Δ221–258) proteins, and we utilized each construct to obtain stable, neomycin-resistant HBC or HeLa sublines as detailed under “Experimental procedures” (Fig. S2, A, D–G). Immunoprecipitation and WB experiments further confirmed interaction of CARP-1(551–580) with NEMO (Fig. S2B) and GST–NEMO(221–261) with CARP-1 (Fig. S2H). Stable expression of CARP-1(551–580) results in diminished interaction of endogenous NEMO with CARP-1 (Fig. S2C). Fig. S2I highlights conservation of the NEMO-interacting epitope of CARP-1 proteins deduced from various vertebrates and flies. Interactions of CARP-1 and NEMO and their respective mutants are summarized in Fig. 1, C and D.
Figure 1.

CARP-1 binds with NEMO, and CARP-1 amino acids 553–599 and NEMO amino acids 221–261 harbor respective epitopes for interaction of CARP-1 and NEMO proteins. A, protein complexes from the indicated cells were immunoprecipitated with the noted antibodies followed by the analysis of the immunocomplexes by Western blotting (WB) using anti-NEMO (upper) antibodies. The membrane-containing proteins from whole-cell lysates were then probed with anti-CARP-1 (middle) or anti-NEMO (lower) antibodies for the presence of respective proteins. B, WB analysis of IP protein complexes was derived by using the indicated antibodies from the noted cell lines. The membrane-containing IP proteins were probed with anti-CARP-1 antibodies (upper), and the membrane-containing proteins from whole-cell lysates were probed with anti-NEMO (middle) or anti-CARP-1 (lower) antibodies for the presence of respective proteins. Arrowheads on the left or right, respectively, indicate the presence of the proteins and the molecular weight markers in A and B of each blot. Schematic of CARP-1 WT and its various mutants (C) and NEMO WT and its mutants (D) were utilized in co-IP–WB experiments to elucidate CARP-1 and NEMO interactions and to map the respective minimal epitopes. All CARP-1 proteins have Myc and His6 epitopes at their C termini. All the NEMO proteins, with the exception of 2–260 and 221–261 mutants, harbored 6× Myc epitope at their N termini. NEMO 2–260 and 221–261 mutants had GST epitope at their N termini. Positive interactions are indicated by + and loss/absence of interaction is denoted by −.
Interference of CARP-1 interaction with NEMO enhances adriamycin efficacy in part through attenuation of RelA activation
To determine if and the extent of CARP-1 interaction with NEMO regulated cell growth signaling, we utilized the above-described stable HBC and HeLa cells that express EGFP, EGFP–CARP-1(551–580), GST, GST–NEMO(221–261), and CARP-1(Δ553–599) proteins. Competition of endogenous CARP-1 binding with NEMO by overexpressing CARP-1(551–580) or NEMO(221–261) resulted in a generally greater loss of cell viabilities following treatments with the chemotherapeutics adriamycin, cisplatin, 5-fluorouracil (5-FU), or an experimental compound CFM-4.16 when compared with the respective vector-expressing cells (Fig. 2, A–C). We next clarified whether perturbation of CARP-1 binding with NEMO impacted NF-κB signaling. We utilized HBC cells that stably express myc-His–tagged WT CARP-1 or CARP-1(Δ553–599) mutant proteins. These cells were either untreated or separately treated with adriamycin, CFM-4.16, TNFα, EGF, or IL-1β followed by analysis of cell lysates by WB for expression of serine 536 phosphorylated or total p65/RelA as indicated under “Experimental procedures.” All the agents provoked a robust increase in RelA activation in cells expressing WT CARP-1 (Fig. 2D). Serine 536 phosphorylation of p65 however was diminished in cells expressing CARP-1(Δ553–599) that were treated with adriamycin or CFM-4.16 but not EGF, TNFα, or IL-1β (Fig. 2D). These data suggest that NF-κB signaling involving p65 activation in the presence of adriamycin or CFM-4.16 involves CARP-1 interaction with NEMO. Because adriamycin and CFM-4.16 function in part by promoting DNA damage (21), and NEMO regulates activation of canonical NF-κB following DNA damage (28, 31), our findings would suggest the involvement of CARP-1 binding with NEMO for DNA damage–induced activation of the canonical NF-κB pathway. DNA damage–induced signaling promotes NEMO sumoylation and its translocation to the nucleus, followed by phosphorylation by the ATM/ATR kinase that results in NEMO mono-ubiquitination and nuclear export along with ATM to activate IKK kinase in cytosol (28, 31, 32). The fact that CARP-1 is a perinuclear protein (1), it remains to be clarified whether CARP-1 interaction with NEMO regulates nuclear and/or cytoplasmic translocation of NEMO following DNA damage.
Figure 2.

Interference of CARP-1 binding with NEMO enhances chemotherapy efficacy in part by inhibiting activation of p65/RelA. A–C, indicated cell lines were treated with DMSO (Control) or with the noted dose and time of indicated agents. Determination of viable/live cells was carried out by MTT assays as detailed under “Experimental procedures.” The bar chart columns represent the means of two independent experiments; bars indicate S.E. A and C, * and @, p ≤ 0.001 relative to respective vector sublines. D, cells stably expressing myc-His–tagged WT CARP-1 or CARP-1 (Δ553–599) mutant were either treated with DMSO (Control) or with various agents for indicated doses and times. Cell lysates were then analyzed by WB for levels of phosphorylated and total p65/RelA as described under “Experimental procedures.” Arrowheads on the left or right indicate the presence of proteins or molecular weight markers, respectively.
We next investigated whether expression of the CARP-1(Δ551–599) mutant also interfered with activities of other key transducers of the canonical NF-κB pathway. HBC cells stably expressing WT CARP-1 or CARP-1(Δ551–599) mutant were separately treated with DMSO (control), adriamycin, CFM-4.16, or TNFα for a shorter (1 h) or longer (6 h) duration. WB analyses revealed a robust activation of p65/RelA, α/β, and γ subunits of IKK occurred in cells expressing WT CARP-1 that were treated with adriamycin, CFM-4.16, or TNFα over short (1 h) or long (6 h) durations (Fig. 3). Consistent with our data in Fig. 2D, activation of p65 was diminished in HBC cells expressing CARP-1(Δ551–599) that were treated with adriamycin or CFM-4.16 (Fig. 3B). Of note is that although a robust loss of p65 activation occurred in CFM-4.16 or adriamycin-treated HBC cells expressing CARP-1(Δ551–599) that were treated over a longer (6 h) period, a moderate reduction in p65 activities also occurred in these cells that were treated over a shorter (1 h) period. Interestingly, expression of CARP-1(Δ551–599) resulted in diminished serine 85 phosphorylation of IKKγ/NEMO regardless of the agent or duration of treatment, whereas activities of IKKα/β were diminished in cells that were treated with the respective agent for a short (1 h) duration. However, a robust IKKα/β activation occurred in HBC cells expressing CARP-1(Δ551–599) over a longer (6 h) treatment with CFM-4.16 or adriamycin, but not TNFα. p65 activation was also noted in HBC cells expressing GST–NEMO following treatments with IL-1β, EGF, and adriamycin (Fig. S3A). Interference of CARP-1 binding with NEMO in the HBC cells with stable expression of NEMO(221–261) fragment resulted in attenuated p65 activation when treated with adriamycin but not EGF or IL-1β (Fig. S3A). In addition, our confocal microscopy-based in situ analysis revealed a reduction in serine 85 phosphorylation of IKKγ/NEMO in adriamycin, CFM-4.16, or TNFα-treated HBC cells that express CARP-1(Δ551–599) when compared with IKKγ/NEMO activation in adriamycin, CFM-4.16, or TNFα-treated HBC cells that express WT CARP-1 (Fig. S3, B and C). Moreover, a 6-h but not a 1-h treatment with either of the agents provoked a robust activation of stress-activated protein kinase/MAPK JNK1/2 in HBC cells expressing WT or the Δ551–599 mutant of CARP-1 (Fig. 3B). These data collectively suggest that expression of CARP-1(Δ551–599) interferes with serine 85 phosphorylation of IKKγ/NEMO in the presence of adriamycin, CFM-4.16, or TNFα. Because serine 85 phosphorylation of NEMO by ATM kinase is required for NF-κB activation following DNA damage (28), and CARP-1 is a perinuclear protein (1), attenuation of NEMO phosphorylation at serine 85 in CFM-4.16 or adriamycin-treated HBC cells that express CARP-1(Δ551–599) would suggest that CARP-1 binding with NEMO is likely required for ATM-dependent phosphorylation of IKKγ/NEMO and subsequent activation of IKK and p65 in cells treated with DNA damage–inducing agents.
Figure 3.

Interference of CARP-1 binding with NEMO inhibits activation of canonical NF-κB signaling. Indicated cells stably expressing WT or mutant CARP-1 protein were treated essentially as in Fig. 2D for 1-h (A) or 6-h (B) durations. Cell lysates were then analyzed by WB for levels of CARP-1, phosphorylated and total p65/RelA, NEMO, IKKβ, and JNK1/2 proteins as described under “Experimental procedures.” The WB membranes in A and B were probed with anti-actin antibodies to assess protein loading. Arrowheads on the left or right of each blot in A and B indicate the presence of proteins or molecular weight markers, respectively.
Kinetics of CARP-1 binding with NEMO and identification of pharmacological inhibitors of NEMO–CARP-1 interaction
We conducted computational modeling and SPR studies to investigate the binding kinetics of CARP-1(551–580) and NEMO(221–261) peptides following our previously described methods (21). Because the crystal structure of CARP-1 remains to be resolved, we utilized SWISS-MODEL (33) that indicated a 51.6% identity of CARP-1(551–600) to TET2 resulting in a random coil domain (Fig. 4A). The crystal structure of NEMO is characterized and permitted us to obtain the NEMO(221–261) structure from PDB code 3CL3 (34) as shown in Fig. 4B. Docking of these two models using ZDOCK 3.0.2 with IRaPPA re-ranking (35) and the top three predictions (Fig. 4, C–E) were retained for further analysis via molecular dynamics (MD). Because small peptides have significantly more conformational freedom afforded compared with an entire protein, a larger fluctuation in the backbone root mean square deviation (RMSD) calculations is observed. Smaller values reflect greater stability of each complex throughout the simulations (Fig. S4). After solvation, equilibration, and heating, the structures undergo significant conformational change as expected to relieve clashes from docking. Complex 1 (Fig. S4A) shows a smooth increase throughout the 24-ns time course of the production run until an RMSD of roughly 10 Å after 7 ns. Beyond this point, the RMSD did not deviate significantly indicating a stable complex was reached. This was reflected in the histogram analysis by the Gaussian curve observed with a peak at an RMSD of 10 Å. No other dominant pose was observed. In complex 2 (Fig. S4B), there was significantly smaller shift in structure from the initial pose. The RMSD initially rose to ∼8 Å, but the structure relaxed to an area where the backbone RMSD leveled off at around 6 Å. Once again, the histogram analysis shows a Gaussian distribution with a peak at an RMSD of 6 Å for the highest occurrence. For complex 3 (Fig. S4C), an equilibrium was not reached as indicated by the continually rising backbone RMSD. Histogram analysis did not show any significantly dominant conformer, confirming no equilibrium was reached. Furthermore, we conducted binding energy calculations using MM-GBSA/PBSA to determine the potential of these two peptides to interact in a biological setting (Table 1). Calculations were taken from 200 snapshots sampled from the last 5 ns of simulation. The calculated binding energies for all three complexes was very similar because the difference between MM-GBSA and MM-PBSA values was not large. These data support the idea that CARP-1(551–600) and NEMO(221–261) peptides are likely to form a relatively strong interaction in a biological setting.
Figure 4.

Computational analyses of CARP-1(551–600) binding with NEMO(221–261). A, Swiss Model image of CARP-1(551–600). B, PDB code 3CL3 image of NEMO(221–261). C–E, three top-scoring docked complexes of CARP-1(551–600) (gray)/NEMO(221–261) (green) in descending order: C, D, and then E.
Table 1.
Calculation of binding energies (BE) (kcal/mol) of the CARP-1(551–600)/NEMO(221–261) using MM-GBSA and MM-PBSA
| Complex 1 |
Complex 2 |
Complex 3 |
||||
|---|---|---|---|---|---|---|
| BE | S.D. | BE | S.D. | BE | S.D. | |
| kcal/mol | kcal/mol | kcal/mol | ||||
| MM-GBSA | −53.7 | 5.9 | −66.1 | 5.9 | −44.3 | 8.2 |
| MM-PBSA | −59.0 | 6.5 | −75.5 | 8.1 | −55.1 | 9.6 |
The predicted kinetics of interaction of CARP-1(551–600) and NEMO(221–261) epitopes was further validated by utilizing the respective chemically-synthesized peptides to determine their in-solution binding by SPR technology as described under “Experimental procedures” (21). As shown in Fig. 5A, this experiment revealed an equilibrium dissociation constant (KD value) of 1.02 × 10−7 m (Ka = 2.07 × 103 m−1·s−1 and Kd = 2.12 × 10−4 s−1). On the collective basis of our data in Fig. 1, and the biophysical and SPR data above, we developed in vitro binding assays utilizing chemically-synthesized CARP-1 and NEMO peptides. In the first instance, we utilized CARP-1(551–580) and NEMO(221–261) peptides to carry out buffer optimization and DMSO tolerance of the assay as detailed under “Experimental procedures”. The optimal binding was noted with PBS or PBS plus 0.01% bovine skin gelatin (BSG), and the presence of 2.5% DMSO did not affect this binding (Fig. S5, A and B). The presence of 0.01% Tween minimizes nonspecific binding and generates higher reproducibility as noted by smaller error bars in Fig. S5A. For the purpose of HTS, we further adapted our binding assay for use in ELISA-based Alpha screen strategy (AlphaLISA; PerkinElmer Life Sciences) by utilizing Flag–CARP-1(546–580) and biotin–NEMO(221–261) peptides as noted under “Experimental procedures.” As shown in Fig. 5B, the assay demonstrated a robust interaction. The assay also demonstrated a Z′ factor of >0.5 indicating a suitable robustness threshold. HTS yielded two small-molecule inhibitors (SMI) of CARP-1(546–580) binding with NEMO(221–261). Because interference of CARP-1 binding with NEMO resulted in attenuation of RelA activation and NF-κB signaling (Fig. 3), the compounds 1-(3,4-dihydroxyphenyl)-2-{(1-(4-methylphenyl)-1H-tetrazol-5-yl)thio}ethanone and 2-{[(4-methoxyphenyl)sulfonyl]amino}-N-(2-phenylethyl)benzamide (Fig. 5, C and D) were labeled as Selective NF-κB Inhibitors (SNI)-1 and -2, respectively. Interestingly, SNI-1 elicited a biphasic IC50 of ∼300 nm, whereas the IC50 for SNI-2 was ∼25 μm in our AlphaLISA assay (Fig. 5, C and D). Although the precise reason for the biphasic IC50 for the SNI-1 compound is not known, of note is that the SNI-1 compound inhibited binding of CARP-1(546–580) with NEMO(221–261) with an IC50 that appears closer to the dissociation constant (KD) of CARP-1(551–580) and NEMO(221–260) peptides noted in the SPR assay (Fig. 5A). For this reason, we chose to investigate properties of SNI-1 compound further in biochemical and biological assays in vitro.
Figure 5.

Kinetics of CARP-1 binding with NEMO and identification of pharmacological inhibitors of CARP-1 interaction with NEMO. A, SPR sensogram showing binding of CARP-1(551–580) and NEMO(221–260) peptides as detailed under “Experimental procedures.” B, solution phase binding of Flag–CARP-1(546–580) and biotin–NEMO(221–261) peptides. The histogram shows fluorescence signal following binding of the two peptides over three noted times using AlphaLisa assay format as described under “Experimental procedures.” C and D, structure, percent inhibition of binding of the Flag–CARP-1(546–580), and *biotin-NEMO(221–261) peptides by respective compound, chemical name, formula, molecular weight, and our laboratory abbreviated name of each compound that was identified following HTS as detailed under “Experimental procedures.” E, SNI-1 binds CARP-1. Left panel, His–TAT–HA–CARP-1(551–580) was affinity-purified and immobilized on Ni-NTA beads, with or without SNI-1, washed three times with RIPA buffer to remove free compound, and then allowed to bind with affinity-purified GST–NEMO(221–261) as under “Experimental procedures.” Right panel, GST–NEMO(221–261) peptide was affinity-purified and immobilized on GSH-Sepharose, incubated with or without SNI-1, washed with RIPA buffer as above, and then allowed to bind with affinity-purified His–TAT–HA–CARP-1(551–580). The complexes were analyzed by SDS-PAGE followed by WB with noted antibodies in respective top and middle blots. The lower blots in each panel indicate respective input peptides. F, SNI-1 does not affect NEMO interaction with RIPK1. HBC cells were untreated (Control) or treated with the indicated agents for the noted dose and time. Protein complexes were immunoprecipitated (IP) with the noted antibodies followed by the analysis of the immunocomplexes by Western blotting (WB) using anti-CARP-1 (upper blot), anti-NEMO (middle blot), and anti-RIPK1 (lower blot) antibodies. Arrowheads on the left or right side of each blot in the left panel indicate the presence of proteins or molecular weight markers, respectively.
We next clarified the biochemical mechanism of inhibition of CARP-1(551–580) binding with NEMO(221–261) by SNI-1. Here, we utilized E. coli-expressed GST–NEMO(221–261) and His–TAT–HA–CARP-1(551–580) peptides in IP–WB assays as described under “Experimental procedures.” As shown in Fig. 5E, incubation of SNI-1 with His–TAT–HA–CARP-1(551–580) peptide that was immobilized with Ni-NTA beads abrogated binding of GST–NEMO(221–261) with His–TAT–HA–CARP-1(551–580) peptide. Incubation of SNI-1 with GST–NEMO(221–261) peptide that was immobilized with GST-Sepharose beads in contrast failed to abrogate binding of His–TAT–HA–CARP-1(551–580) with the GST–NEMO(221–261) peptide. These data suggest that SNI-1 binds with CARP-1(551–580) epitope and prevents binding of NEMO(221–261) with CARP-1(551–580). In light of our findings in Fig. 3 demonstrating CARP-1 involvement in ATM-dependent NEMO phosphorylation in the presence of adriamycin, and because cytosolic ATM/NEMO/RIPK1 also regulates NF-κB response to DNA damage (36), we determined whether CARP-1 was also involved in adriamycin-induced NEMO/RIPK1 signaling. IP–WB analyses utilizing GST–NEMO-expressing HBC cells revealed that NEMO interacted with CARP-1 or RIPK1 in untreated, control, and adriamycin-treated cells (Fig. 5F). Presence of SNI-1 alone or in combination with adriamycin abrogated NEMO interaction with CARP-1 but not with RIPK1 (Fig. 5F). Thus, targeting of CARP-1 interaction with NEMO does not impact the NEMO–RIPK-1 interaction. Because adriamycin activates ATM to regulate canonical NF-κB and DSB repair pathways, it remains to be clarified whether CARP-1 regulates RIPK1 signaling dependent and/or independent of ATM. The data in Fig. 5 collectively demonstrate that SNI-1 binds with CARP-1 and is a novel SMI of CARP-1 binding with NEMO.
SNI-1 enhances efficacy of DNA damage-inducing chemotherapeutics and inhibits secretion of pro-inflammatory and oncogenic cytokines by cancer cells in vitro and in vivo
We next investigated the potential of SNI-1 as a novel inhibitor of cancer cell growth. Although SNI-1 doses of 1.0, 2.5, 5.0, or 10.0 μm over a 24-h period caused a modest ∼10–20% loss of viability of the human MDA–MB-231 or the murine 4T1 TNBC cells, treatments of these TNBC cells with a 5.0 μm dose of SNI-1 over a 72-h period revealed an IC50 of ∼4.0–4.5 μm (Fig. S6A). SNI-1 treatments also resulted in reduced viabilities of diffuse large B-cell and follicular cell lymphoma cells with IC50 values of ∼10.0 and 7.5 μm (Fig. S6, B and C). Given that adriamycin inhibited growth of MDA–MB-231 TNBC cells with an IC50 of ∼3 μm (37), we treated human and murine TNBC cells as well as human PDX-derived TNBC cells with a 5 μm dose of adriamycin as a single agent or in combination with various doses of SNI-1 over a period of 24 h. Treatments of human and murine TNBC cells with a combination of adriamycin and SNI-1 caused a significantly greater loss of cell viabilities when compared with cells treated with either agent alone (Fig. 6A). A statistically significant and greater loss of viability of human PDX-derived TNBC cells, however, also occurred in the presence of 5 μm each of SNI-1 and adriamycin when compared with either compound alone (Fig. 6A). Although a 5 μm dose of adriamycin for 48 h elicited ∼30% inhibition of mammospheres derived from human TNBC PDX tumors, a 2.5 or 5.0 μm dose of SNI-1 failed to inhibit growth of mammospheres derived from human PDX tumors (Fig. 6B). A statistically significant and greater loss of viability of mammospheres derived from human TNBC PDX tumors, however, occurred in the combined presence of SNI-1 and adriamycin when compared with either compound alone (Fig. 6B). Adriamycin induces DSBs and activates NF-κB signaling that likely functions to promote DSB repair, survival, and eventual resistance of surviving cancer cells (21, 23, 24, 30). Our data would suggest that abrogation of NF-κB activation by SNI-1 likely interferes with cell survival with a consequent increase in adriamycin-induced viability loss of the TNBC cells. Chemotherapeutics such as 5-FU, cisplatin, as well as ionizing radiation also activate NF-κB signaling in various cancer cells (38). Because cisplatin forms covalent bonds with DNA resulting in intra-strand DNA adducts and cross-links that in turn block transcription and replication (39), we next clarified whether SNI-1 also interferes with cisplatin-dependent NF-κB signaling to enhance anti-cancer efficacy of cisplatin. To test this possibility, we treated parental and adriamycin-resistant MDA–MB-231 and 4T1 TNBC cells (40) with cisplatin, SNI-1, or a combination followed by measurement of cell viabilities as above. Treatments with a 10 μm dose of cisplatin for 24 h caused a moderate 20–30% loss of viability of the TNBC cells, whereas a cisplatin and SNI-1 combination elicited a marked, statistically significant loss of viabilities of these cells when compared with cells treated with either agent alone (Fig. 6C). Interestingly, treatments of adriamycin-resistant TNBC cells with a combination of cisplatin and SNI-1 also resulted in a greater loss of their viabilities when compared with cells that were treated with either agent alone (Fig. 6D). Similarly, a combination of adriamycin and SNI-1 also provoked a greater loss of viability of the parental and cisplatin-resistant TNBC cells when compared with cells that were treated with either agent (Fig. S6D). Because cisplatin is a front line clinical agent for treatment of nonsmall cell lung, renal, and pancreatic cancers, as well as a subset of BRCA-mutant TNBCs, our proof–of–concept studies demonstrate a greater loss of viabilities of these cells when exposed to cisplatin together with SNI-1 (Fig. 6E and Fig. S6E). Moreover, because oxaliplatin is utilized for treatment of colon cancers, our studies also revealed a greater growth inhibition of different colon cancer cells that were treated with a combination of oxaliplatin and SNI-1 when compared with cells that were treated with either agent alone (Fig. S6F). Consistent with our data with TNBC cells, treatments of human cervical cancer HeLa cells with a combination of adriamycin and SNI-1 also provoked a greater loss of their viabilities when compared with SNI-1 or adriamycin-treated cells (Fig. 6F). We generated Crisper-based knockout of CARP-1 in HeLa cells (HeLa CARP-1 KO cells), as described under “Experimental procedures.” In addition, we obtained and utilized HeLa NEMO KO cells (41) in experiments below. Because the interference of NEMO–CARP-1 interaction inhibited NF-κB signaling (Fig. 3), we first clarified whether SNI-1 inhibited adriamycin-induced transcriptional activation of NF-κB. For this purpose, we utilized WT and NEMO (KO) HeLa cells in conjunction with NF-κB–TATA–Luc reporter plasmid as detailed under “Experimental procedures.” Fig. S6G shows reduced NF-κB transcriptional activity in adriamycin-treated NEMO (KO) cells when compared with their adriamycin-treated WT counterparts. Consistent with activation of NF-κB–mediated survival signaling by adriamycin, treatments of the HeLa CARP-1 KO cells (HeLa CARP-1−/−) or HeLa NEMO KO cells (HeLa NEMO−/−) with adriamycin resulted in a significant increase or decrease, respectively, in their viabilities when compared with the viabilities of the adriamycin-treated WT HeLa cells (Fig. 6F). Interestingly, although SNI-1 treatments resulted in a moderate ∼30% reduction in the viabilities of WT HeLa cells, a further significant loss of viabilities of NEMO KO cells was noted following treatment with SNI-1 when compared with the similarly-treated WT HeLa cells (Fig. 6F). Importantly, as also shown in Fig. 6F, SNI-1 failed to provoke any loss of viabilities of HeLa CARP-1 KO cells suggesting a requirement of CARP-1 for transduction of signaling by SNI-1. In light of the fact that adriamycin also activates ATM-dependent H2AX (γH2AX) to promote DSB repair signaling (18), our WB analyses revealed a robust γH2AX level in adriamycin-treated HeLa cells regardless of the absence of CARP-1 or NEMO proteins (Fig. S6H). Thus, our data in Fig. 6 suggest that although CARP-1 regulates adriamycin-induced canonical NF-κB signaling, CARP-1 is required for signaling by SNI-1. A combination of SNI-1 and the DNA damage–inducing chemotherapeutics is therefore a superior strategy for inhibiting growth of a variety of cancer cells, including the drug-resistant TNBC cells.
Figure 6.

SNI-1 enhances anti-cancer efficacy of chemotherapy in vitro, and CARP-1 is required for cell growth suppression by SNI-1. A–F, cell viability was determined by MTT assay following treatments of cells with vehicle/DMSO (Control) or the indicated times and doses of various agents. The columns in each histogram indicate percent of live/viable cells relative to their DMSO-treated controls and represent the means of two to three independent experiments; bars, S.E. *, **, ***, p < 0.001 relative to respective cells treated with chemotherapy only (A–D) or SNI-1 only (E); *, **, and ***, p < 0.05. 0.01, and 0.001, respectively, relative to corresponding WT cells (F).
Because SNI-1 in combination with genotoxic chemotherapeutics provoked a greater loss of viability of a number of cancer cells and because SNI-1 alone also caused a moderate inhibition of cell growth, our WB analysis revealed stimulation of apoptosis in cells exposed to SNI-1 as noted by elevated CARP-1 levels and cleavage of poly(ADP-ribose) polymerase or caspase-3 (Fig. 7, A–D). Although treatments with adriamycin or cisplatin, but not SNI-1, also provoked a robust increase in p65/RelA activation and phosphorylation of NEMO, the presence of SNI-1 generally resulted in diminished p65 activation and NEMO phosphorylation by adriamycin or cisplatin (Fig. 7, A–E). Adriamycin, but not SNI-1, treatment caused p65 activation in the nuclear compartment, whereas NEMO phosphorylation occurred in both the nuclear and cytosolic compartments (Fig. 7F). As expected, the presence of SNI-1 interfered with adriamycin-induced activation of p65 and NEMO phosphorylation and resulted in diminished cytosolic p65 levels (Fig. 7F). Because disruption of NEMO–CARP-1 interaction impacted adriamycin-induced p65 activation (Fig. 3), and caspase-3 activation occurred in cells treated with adriamycin, SNI-1, or a combination (Fig. 7, A–D), we next clarified whether NEMO was required for p65 and caspase activation by adriamycin. As expected, adriamycin provoked a robust p65 activation in HeLa WT, but not NEMO (KO), cells, whereas caspase-3 cleavage occurred in cells that were treated with adriamycin, SNI-1, or a combination regardless of NEMO (Fig. S7). Moreover, because SNI-1 binds with CARP-1 (Fig. 5E) and CARP-1 interacts with RIPK1 (42), and because SNI-1 did not interfere with RIPK1 interaction with NEMO (Fig. 5F), we next clarified whether SNI-1 also regulated RIPK1 signaling. Our WB analysis revealed that ADR or SNI-1 induced expression of cleaved RIPK1 (Fig. 7G). Interestingly, a combination of ADR and SNI-1 provoked a robust increase in cleaved RIPK1 (Fig. 7G). Cleavage of RIPK1 and caspase 3 will be consistent with prior studies demonstrating apoptosis signaling by DSB-inducing genotoxic chemotherapeutics that promote RIPK1 cleavage and activation of pro-apoptotic caspases-3, -6, and -8 (43). Although the mechanisms of apoptosis by SNI-1 and adriamycin combination remain to be clarified, our data support a conclusion that blockage of CARP-1 binding with NEMO interferes with chemotherapy-activated NEMO phosphorylation and p65/RelA activation to attenuate canonical NF-κB signaling.
Figure 7.
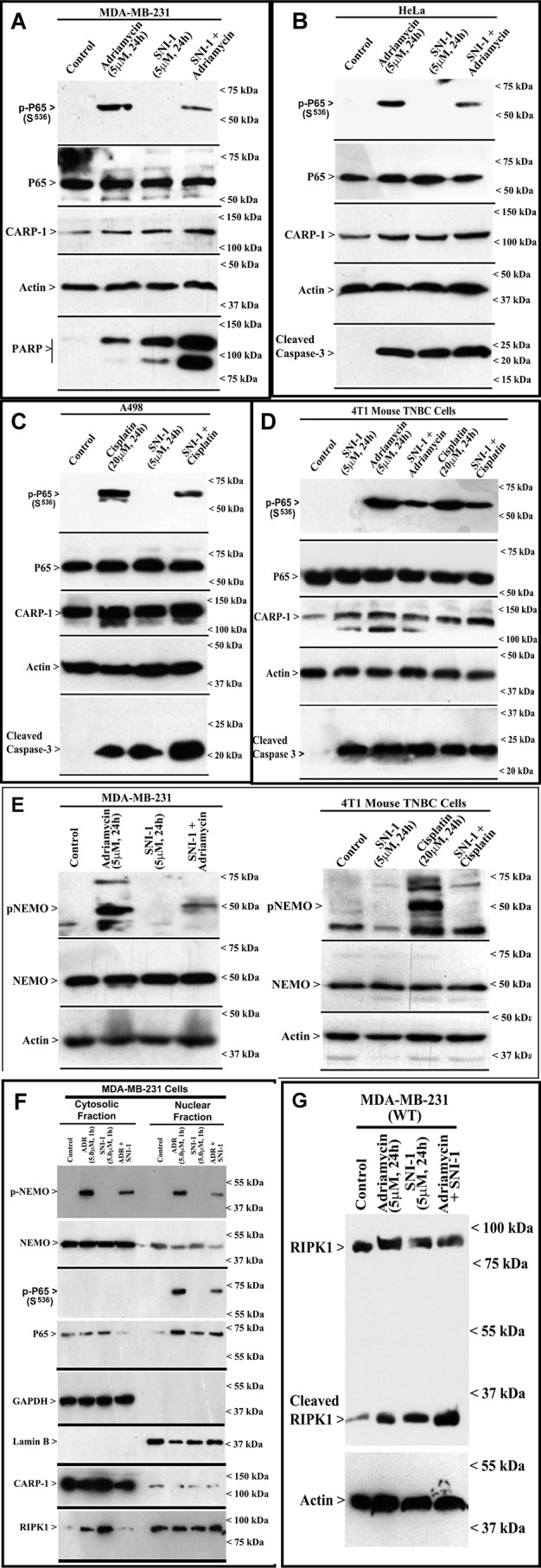
SNI-1 attenuates chemotherapy-induced phosphorylation/activation of RelA and NEMO, promotes RIPK1 cleavage, and enhances CARP-1 levels and apoptosis. A–G, indicated cells were either treated with DMSO (Control) or treated with noted times and doses of respective agents. F, cellular proteins were first separated into cytosolic and nuclear fractions. The cell lysates were then analyzed by WB for levels of phospho-p65, p65, CARP-1, PARP, cleaved caspase-3, RIPK1, actin, lamin B, GAPDH, phospho-NEMO, and NEMO proteins as described under “Experimental procedures.”
RelA regulates transcriptional activation of NF-κB target genes, including several pro-inflammatory cytokines through the canonical and atypical pathways (32). Moreover, the DNA damage-inducing chemotherapeutics such as adriamycin, cisplatin, or 5-FU induce inflammatory cytokines that function in part to promote survival and resistance of cancer cells (44). We next determined whether and the extent to which the presence of SNI-1 would attenuate chemotherapy-induced secretion of pro-inflammatory cytokines by cancer cells. Treatments with SNI-1 provoked a modest increase in levels of pro-inflammatory cytokines TNFα and IL-8 in culture media of human TNBC cells when compared with the levels of these cytokines in the media from the respective untreated cells (Fig. 8, A–D and H). SNI-1 treatments, however, failed to cause an increase in secretion of TNFα and IL-1β in murine TNBC cells (Fig. 8, E–G). As expected, treatments of parental and chemo-resistant human and murine TNBC cells and the parental renal cancer cells with adriamycin, 5-FU, or cisplatin provoked a robust increase in levels of TNFα, IL-8, and IL-1β in the media of the respective cell line. Consistent with attenuation of p65/RelA activation in cells that were exposed to a combination of SNI-1 and adriamycin or cisplatin, the presence of SNI-1 also caused a robust decline in secretion of genotoxic chemotherapy-induced pro-inflammatory cytokines TNFα, IL-8, and IL-1β (Fig. 8, A–H). Our data in Figs. 7 and 8 collectively suggest that pharmacological blockage of CARP-1–NEMO binding functions to enhance chemotherapy efficacy in part by promoting superior growth inhibition of cancer cells and reducing activation of canonical NF-κB. Inhibition of canonical NF-κB, in turn, diminishes production of chemotherapy-induced inflammation and survival-promoting cytokines.
Figure 8.

SNI-1 attenuates chemotherapy-induced secretion of pro-inflammatory cytokines in vitro. A–H, indicated cells were either treated with DMSO (Control) or treated with noted times and doses of respective agents. The media from the cells were analyzed by ELISA for levels of different pro-inflammatory cytokines as under “Experimental procedures.”
To investigate the therapeutic potential of SNI-1, we conducted in vivo studies to determine the efficacy and potency of SNI-1 alone or in combination with adriamycin or cisplatin as described under “Experimental procedures” and our previously published methods (40, 45). As shown in Fig. 9 and Fig. S8, all the treatment groups except the SNI-1 treatment group showed tumor growth inhibition as indicated by the reduced median tumor volume compared with the control group. With the exception of cisplatin and cisplatin with SNI-1 arms, the groups treated with either SNI-1 or ADR as single agents or as a combination failed to reach effective T/C throughout the treatment period (Fig. 9 and Fig. S8). There was a sustained and reduced median tumor volume noted in adriamycin plus SNI-1 and cisplatin plus SNI-1 groups compared with respective single agent–treated groups (Fig. S8B), Of particular note was the response generated by cisplatin plus the SNI-1–treated group. This group reached a therapeutic T/C of <42% from day 7 until the end of treatment on day 18. In addition, consistent with our findings in Fig. 8, E and F, ELISA-based analyses revealed that treatments with adriamycin or cisplatin, but not SNI-1, robustly stimulated serum levels of pro-inflammatory cytokines TNFα and IL-1β (Fig. 9C and Fig. S8, C and D). Adriamycin and SNI-1 combination provoked a decline in serum levels of these cytokines when compared with their levels in sera derived from adriamycin-treated animals (Fig. S8, C and D). A combination of SNI-1 and cisplatin treatments, however, elicited a rather robust decline in serum levels of both TNFα and IL-1β when compared with their levels in sera derived from animals treated with cisplatin only (Fig. 9C and Fig. S8, C and D). Furthermore, immunohistochemical analyses of the tumors derived from animals treated with cisplatin, but not SNI-1, revealed the presence of phosphorylated p65/RelA, whereas a decline in the levels of phosphorylated p65 was noted in tumors derived from animals treated with cisplatin plus SNI-1 (Fig. 9D and Fig. S9A). Consistent with our in vitro data in Fig. 7, tumors derived from animals treated with cisplatin, SNI-1, or a combination revealed the presence of cleaved caspase-3 when compared with tumors derived from untreated controls (Fig. 9D and Fig. S9A). Interestingly, and in contrast to our in vitro data with the cancer cell models, our in vivo studies revealed that although SNI-1 administration caused absent to minimal inhibition of tumor growth, it did not provoke toxicities in the animals. Although hematoxylin and eosin staining of various tissues, including lungs, spleen, heart, liver, kidneys, and bone marrow of the animals treated with SNI-1, did not indicate microscopic alterations, immunohistochemical staining of these tissues also failed to show the presence of activated caspase-3 (Fig. S9B). The data in Figs. S8 and S9 and in Fig. 9 collectively suggest that SNI-1 is likely safe and bioavailable with minimal to absent systemic toxicities. SNI-1 functions to enhance anti-tumor efficacy of cisplatin in vivo, in part by robustly inhibiting p65/RelA activation, lowering systemic levels of pro-inflammatory cytokines, and inducing tumor apoptosis (Fig. 10).
Figure 9.

SNI-1 enhances tumor suppression by cisplatin in part by attenuating systemic levels of pro-inflammatory cytokines and promoting tumor apoptosis. Histogram columns showing median tumor volume (A) or percent T/C (B) of the TNBC (4T1) xenograft-bearing mice treated with the indicated agents. The xenograft establishment, treatment, and analysis procedures were carried out essentially as detailed under “Experimental procedures.” C, serum levels of noted pro-inflammatory cytokines. The columns in histograms indicate noted systemic cytokine levels in two representative animals from each of the control and treatment groups; bars, S.E. D, immunohistochemical staining of the noted proteins in the tumors derived from mice with median tumor volumes from each of the control and treatment groups in A was carried out as detailed under “Experimental procedures.” Bar, 200 mm.
Figure 10.
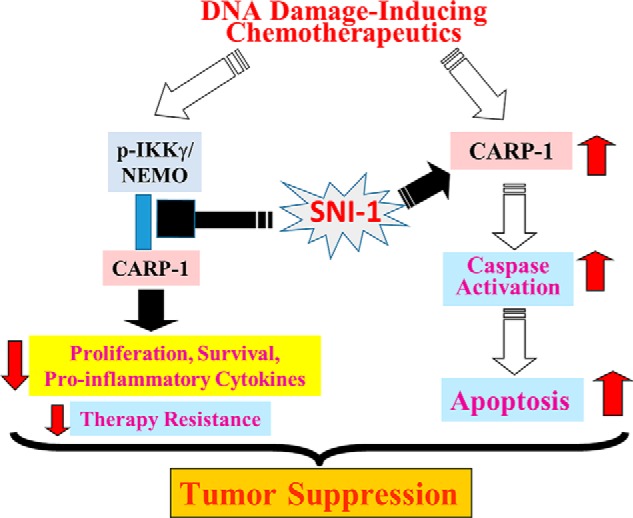
Schematic of mechanism of action of SNI-1.
Discussion
In this report we describe for the first time that CARP-1/CCAR1 is a regulator of canonical NF-κB signaling. CARP-1 regulates chemotherapy-induced canonical NF-κB signaling in part by binding with the NEMO/IKKγ. Although CARP-1 binding with NEMO was reported in a previous proteomics-based study (29), neither the molecular mechanism(s) nor the functional consequences of this interaction were elucidated. Here, we employed mutagenesis-based studies to define the molecular basis of this interaction. We found that CARP-1 amino acids 551–580 harbor the minimal epitope for its interaction with NEMO, whereas amino acids 221–261 of the NEMO protein contained the CARP-1–interacting epitope. CARP-1 interaction with NEMO was functionally significant because expression of CARP-1(Δ553–599) interfered with activation of RelA by adriamycin or the CFM-4.16 compound but not TNFα, IL2-β, or EGF. Moreover, stable expression of CARP-1(551–580) or NEMO(221–261) peptides that would in principle compete/interfere with binding of endogenous CARP-1 and NEMO proteins resulted in significantly higher loss of viabilities of cells treated with adriamycin, cisplatin, 5-FU, or CFM-4.16.
Adriamycin or our CFM-4.16 compound promotes apoptosis in part by inducing DNA damage (21). The cellular DNA damage response (DDR) involves activation of ATM kinase and its downstream target H2AX and nucleus to cytoplasm activation of canonical NF-κB. For a robust DDR, NEMO is SUMOylated and translocates to the nucleus. NEMO is then phosphorylated by ATM kinase and then is monoubiquitylated, followed by nuclear export of the NEMO/ATM complex and activation of cytoplasmic IKK (32). Interestingly, DNA damage per se is not necessary for NEMO SUMOylation. Other stress conditions, such as oxidative stress, ethanol exposure, heat shock, and electric shock, also induce NEMO SUMOylation (46).
Although DNA damage–induced NEMO translocation to and from the nucleus is a hallmark of IKK activation in the canonical NF-κB pathway, the molecular mechanism(s) regulating nuclear–cytoplasmic shuttling of NEMO have yet to be fully clarified. In this regard, a recent report has revealed that IPO3 (also known as importin 3, transportin 2, TRN2, or TNPO2) functions as a critical NEMO nuclear import receptor during DDR (47). IPO3 facilitates NEMO nuclear translocation in a manner dependent on two distinct nuclear localization signal (NLS) sequences in the human NEMO protein. Although human NEMO NLS1 and NLS2 sequences have been mapped to positions 254–257 and 357–360, respectively, the murine NEMO protein harbors only the NLS2 sequence and lacks NLS1 (47). The murine CARP-1 and NEMO proteins interact (Fig. 1A), and NEMO-binding epitopes of human and murine CARP-1 are identical (Fig. S2H). Furthermore, because disruption of CARP-1 binding with NEMO by SNI-1 resulted in loss of chemotherapy-induced activation of p65/RelA (Fig. 7) in both human and murine cells, it would collectively suggest that CARP-1 binding with NEMO is independent of NEMO NLS1. Moreover, ATM kinase also rapidly translocates to the nucleus following induction of DSBs that involves binding with importin α1/β1 heterodimer that depends on a distinct NLS in the ATM protein (48). A recent study further highlighted genotoxic stress-induced mono-ubiquitination of NEMO by an E3-ligase TRIM37 (49). Genotoxic stress-induced ATM activation resulted in phosphorylation of TRIM31 in cytosol, which induced a complex with TRAF6, and consequent nuclear import. Disruption of TRAF6 binding with TRIM31 resulted in increased sensitivity to chemotherapeutics in part due to diminished NEMO mono-ubiquitination in the nucleus. Whether CARP-1 also binds with ATM, TRAF6, TRIM31, or another E3 ligase is currently unclear. CARP-1, however, directly binds with NEMO. Because CARP-1 is a perinuclear protein, it also not clear whether CARP-1 binding with NEMO functions to regulate nuclear import of NEMO following DNA damage. However, abrogation of CARP-1 binding with NEMO resulted in diminished NEMO phosphorylation (Figs. 3 and 7). Because ATM kinase promotes serine 85 phosphorylation of NEMO in the nucleus following activation of DNA damage signaling, it is likely that CARP-1 binding with NEMO serves to facilitate ATM-dependent phosphorylation of NEMO. This possibility is also supported by our WB data in Fig. 3 and confocal imaging (Fig. S3) where CFM-4.16 or adriamycin-treated cells that express the CARP-1(Δ553–599) mutant had diminished NEMO serine 85 phosphorylation and cytoplasmic accumulation when compared with their similarly treated counterparts expressing WT CARP-1. Because NEMO phosphorylation often precedes its mono-ubiquitination, it is also unclear whether and the extent to which CARP-1 binding with NEMO regulates NEMO mono-ubiquitination. Nevertheless, our current findings collectively would support the hypothesis that CARP-1 binding with NEMO facilitates ATM-mediated NEMO phosphorylation. This signaling likely promotes the nuclear export of the NEMO/ATM complex for activation of canonical NF-κB by genotoxic stress to modulate apoptotic response (28). Activation of this pathway, results in elevated levels of pro-inflammatory cytokines that contribute to therapy resistance (47).
Although more than 700 different inhibitors (aspirin to IκBα super repressor) of NF-κB have been reported (50), thus far no NF-κB blocker has been approved for human use. Given NF-κB's physiological roles in immunity, inflammation, and cellular homeostasis, a selective inhibition of therapy-induced NF-κB activation without affecting the immunity, inflammation, and homeostasis signaling would be desirable. Functional studies would then be necessary for determining the optimal regulator/transducer in this complex pathway, as well as identification of opportunities for synergistic agents to augment their efficacy and minimize resistance mechanisms. In this context, our proof–of–concept preliminary studies provide evidence of selective activation of chemotherapy-dependent canonical NF-κB signaling by CARP-1–NEMO interactions. Moreover, a number of prior reports have highlighted targeting of NEMO for inhibition of inflammation regulated by canonical NF-κB signaling. For example, targeting of NEMO by endogenous proteins such as Hsp70 (51) and cell-permeable NEMO-binding domain peptides of IKKα and IKKβ (52, 53), the peptides corresponding to the leucine zipper and the coiled-coil 2 (CC2) regions of NEMO (54) have been documented. Furthermore, a novel small molecule that targets the NEMO ubiquitin–binding domain was recently reported (55). In addition, the medicinal compound Withaferin disrupted ubiquitin-based NEMO reorganization by regulating its covalent modification and binding with ubiquitin, and it also targeted IKKβ to inhibit NF-κB signaling and associated inflammatory responses (56–58).
Our high-throughput chemical biological studies resulted in identification of novel, small molecular compounds. The compound SNI-1 binds with CARP-1 and interferes with the NEMO–CARP-1 interaction (Fig. 5). Although SNI-1 does not bind with NEMO, by disrupting CARP-1 binding with NEMO, it causes loss of chemotherapy-induced phosphorylation of NEMO. Because NEMO phosphorylation is often a prerequisite for NEMO ubiquitination and nuclear export to promote chemotherapy-induced activation of NF-κB, SNI-1 would not be expected to interfere with functions of cytoplasmic NEMO often necessary for activation of canonical NF-κB following growth factor or cytokine-dependent cellular homeostasis. In this regard, our data demonstrate that the presence of SNI-1 affects only the chemotherapy-induced p65/RelA activation. Because chemotherapy often activates canonical NF-κB to promote survival and production of pro-inflammatory cytokines, the presence of SNI-1 also attenuates secretion of chemotherapy-induced inflammatory cytokines in vitro as well as systemically in TNBC tumor-bearing animals in vivo. Of note here is that similar to chemotherapy, SNI-1 is able to moderately inhibit cancer cell growth in vitro in part by inducing apoptosis as indicated by increasing levels of activated/cleaved caspase-3 (Fig. 7). Interestingly, and in contrast to the in vitro cell models, our in vivo studies revealed that although SNI-1 administration caused absent to minimal inhibition of tumor growth, it did not provoke toxicities in the animals either. Although hematoxylin and eosin staining of various tissues, including lungs, spleen, heart, liver, kidneys, and bone marrow of the animals treated with SNI-1, did not indicate microscopic alterations, immunohistochemical staining of these tissues also failed to show the presence of activated caspase-3 (Fig. S9), albeit caspase-3 activation was noted in tissues derived from chemotherapy-treated animals (data not shown). These findings collectively underscore a suitable safety profile of SNI-1 for further development and testing.
In summary, we demonstrate that CARP-1 is a novel, endogenous regulator of chemotherapy-induced canonical NF-κB activation. Pharmacological inhibition of CARP-1 binding with NEMO enhances genotoxic chemotherapy efficacy in vitro and in vivo, in part by attenuating activation of canonical NF-κB, and secretion of NF-κB activated pro-inflammatory cytokines. Our lead compound, SNI-1, represents a novel tool to investigate canonical NF-κB signaling with the potential for translational development to target chemotherapy-induced cancer survival and resistance mechanisms.
Experimental procedures
Materials
DMEM, Eagle's minimal essential medium, and antibiotics (penicillin and streptomycin) were purchased from Invitrogen. Fetal bovine serum (FBS) was purchased from Denville Scientific Inc. (Metuchen, NJ), and DMSO was purchased from Thermo Fisher Scientific (Fair Lawn, NJ). Chemiluminescence reagent was purchased from Amersham Biosciences, and the protein assay kit was purchased from Bio-Rad. Structure and synthesis of the CFM-4 analog CFM-4.16 has been described previously (40). Clinical grade ADR, cisplatin, oxaliplatin, and 5-fluouracil were obtained from the Harper Hospital Pharmacy, Wayne State University, Detroit, MI. The SNI-1 and -2 compounds 1-(3,4-dihydroxyphenyl)-2-{(1-(4-methylphenyl)-1H-tetrazol-5-yl)thio}ethanone and 2-{[(4-methoxyphenyl)sulfonyl]amino}-N-(2-phenylethyl)benzamide, respectively, that inhibited CARP-1/NEMO binding in the HTS (see below) were purchased from ChemBridge, San Diego, CA. SNI-1 compound of >98% purity was also synthesized by Otava Chemicals, Toronto, Canada. 5-Dimethylthiazol-2-yl-2,5-diphenyltetrazolium bromide (MTT), anti-FLAG tag, and anti-actin antibodies were purchased from Sigma. The affinity-purified anti-CARP-1 (α1 and α2) polyclonal antibodies have been described (1). Anti-EGFP and phospho(Ser-85) NEMO antibodies were purchased from Abcam, Cambridge, MA, and anti-HA–tag antibodies were purchased from Biolegend, San Diego, CA. Antibodies for GST-tag, Myc-tag, His6-tag, total NEMO, phospho(Ser-536), total p65RelA, phospho(Ser-176/Ser-180), IKKα/β and total IKKβ, phospho and total JNK1/2, phospho(Tyr-705), total STAT3, and RIPK1 were purchased from Cell Signaling, Beverley, MA.
Recombinant plasmid constructs
The plasmids for expression of myc-His–tagged WT CARP-1 (clone 6.1.2), CARP-1(Δ600–650), CARP-1(1–198), CARP-1(97–454), CARP-1(452–654), CARP-1(603–898), and CARP-1(896–1150) have been described before (1, 3, 21). Additional pcDNA-based plasmids for expression of myc-His–tagged CARP-1(Δ553–599), CARP-1(Δ521–566), CARP-1(452–625), CARP-1(452–610), CARP-1(452–552), CARP-1(552–654), CARP-1(552–640), CARP-1(552–625), CARP-1(552–610), CARP-1(552–580), CARP-1(571–600), CARP-1(591–620), pcDNA3–EGFP, pcDNA3–EGFP, CARP-1(551–580), pcDNA3–GST, pcDNA3–GST–NEMO, pcDNA3–GST–NEMO(221–261), and pcDNA3–GST–NEMO (Δ221–258) were generated by standard molecular biological and cloning manipulations. Plasmids encoding WT and mutant NEMO proteins with 6x myc epitopes at the N terminus have been described (31) and were kindly provided by Dr. Shigeki Miyamoto, Department of Pharmacology, University of Wisconsin-Madison, Madison, WI. These plasmids encoded NEMO ΔN120 (lacking the N-terminal 220 amino acids), NEMO ΔC125 (lacking the C-terminal 25 amino acids), NEMO C417R, and NEMO D406V mutant proteins. Recombinant plasmids encoding GST-tagged NEMO(WT), NEMO(2–260), NEMO(221–261), NEMO(221–317), and NEMO(296–419) were generated by PCR amplification of NEMO cDNA fragments and their subsequent subcloning in the pEBG vector plasmid. The NEMO(2–260) cDNA was cloned in pGEX-4T-1 vector to generate bacterially (E. coli)-expressed GST–NEMO(2–260) protein. Additional CARP-1 cDNA fragments were cloned in pTAT-HA vector (5) to generate bacterially (E. coli)-expressed His–TAT–HA-tagged CARP-1(552–654), CARP-1(552–580), CARP-1(571–600), CARP-1(591–620), CARP-1(611–640), and CARP-1(631–660) proteins. The NF-κB TATA-Luc plasmid harboring 5× NF-κB consensus enhancer sequences positioned upstream of TATA sequences that collectively drive firefly luciferase reporter as well as the plasmid for expression of Renilla luciferase (pTK/Renilla Luc) were purchased from Stratagene, Inc. (La Jolla, CA) and Promega, Inc. (Madison, WI), respectively. All the recombinant plasmids were sequenced to confirm the accuracy and validity of various inserts/epitopes.
Cell lines and cell culture
Routine maintenance and culture of MDA–MB-468 and MDA–MB-231 (both lack estrogen receptors and have mutant p53), SUM-149, SUM-1315, and HCC1937 (all three have mutant BRCA1), human TNBC, human cervical cancer HeLa, human pancreatic cancer PANC-1, human diffuse large B-cell lymphoma WSU-DLCL2, human follicular lymphoma WSU-FSCCL, human clear cell renal carcinoma A498, human colon cancer HT-29, SW620, HCT-116, HCT-116 (p53−/−), colon epithelial IEC-6 cells, and monkey kidney COS-7 cells were carried out essentially as described previously (1, 3, 11, 21, 45). HeLa and MDA–MB-468 cells having CRISPR-based CARP-1 knockouts were generated and characterized on a fee–for–service basis by Biocytogen Corp., Wakefield, MA. HeLa cells having CRISPR-based NEMO knockouts have been described before (41) and were kindly provided by Dr. Zhengfan Jiang, School of Life Sciences, Peking University, Beijing, China. The murine TNBC cell line 4T1 that was derived from a spontaneously arising BALB/c mammary tumor was obtained from the Karmanos Cancer Institute and maintained in culture as described before (40). Generation, characterization, and culture of drug (ADR or cisplatin)-resistant human TNBC MDA–MB-468 and MDA–MB-231 cells as well as ADR-resistant murine 4T1 cells has been detailed before (40). All the cell culture media were also supplemented with 10% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin, and the cells were maintained at 37 °C and 5% CO2. For cell growth and MTT studies, the cells were cultured in fresh media with 5–10% FBS prior to their treatments with various agents. Generation and characterization of MDA–MB-468 cells expressing reduced CARP-1 have been described before (1). The stable sublines were generated by transfecting the MDA–MB-468 and HeLa cells with the pcDNA3 vectors pcDNA3-CARP-1(WT), pcDNA3-EGFP, and pcDNA3-GST and the various Myc-His, EGFP, or GST-tagged mutants of CARP-1 as well as NEMO followed by selection in the presence of 800 μg/ml neomycin using methods described previously (1, 3, 11, 21, 45). The cell lysates from WT, untransfected cells, neomycin-resistant pools, or individual sublines were then subjected to IP and WB analyses as below. Three well-characterized TNBC PDX tumors (TM00089, TM00098, and TM00091) were purchased from The Jackson Laboratory and were routinely maintained/passaged at the Karmanos Cancer Institute Animal Model and Therapeutic Evaluation Core (AMTEC).
Three-dimensional mammosphere assays
The PDX tumor (TM00098) cells were dissociated from the tumor fragments and cultured for two-dimensional and mammosphere studies in vitro as we have done previously (40). Briefly, the cells were washed twice in 1× PBS, trypsinized, and pelleted at 200 × g at room temperature. Cells were then resuspended in 5 ml of mammosphere media (DMEM/F-12 containing 2 mm l-glutamine, 100 units/ml penicillin, 100 units/ml streptomycin, 1× B27 supplement, 20 ng/ml recombinant human EGF (Sigma) and 10 ng/ml recombinant human basic fibroblast growth factor (R&D Systems)). Approximately 5000 viable cells per ml were then seeded in an ultra-low adherent 60-mm plate and incubated at 37 °C and 5% CO2 for 2 weeks without disturbing the plates. After the mammospheres were formed, fresh media with or without adriamycin (5 μm), SNI-1 (2.5 or 5.0 μm), or a combination of these agents was added, and the cells were incubated for additional 48 h at 37 °C and 5% CO2. The mammospheres in the untreated and treated plates were photographed, and the cells were then dissociated to determine their viabilities by the MTT assay as described (40).
Cell viability, immunoprecipitation, luciferase, and Western blotting assays
500–1000 cells were seeded in each well of 96-well plates and then either untreated (control) or treated with various agents for the noted times. After treatment, MTT reagent was added at 0.5 mg/ml concentration for 2–4 h at 37 °C. DMSO was added to solubilize formazan, and the plate was read at 570 nm in a plate reader. The histograms indicating levels of cell viability were generated by plotting the net absorbances as described (1, 3). Next, logarithmically growing cells were either untreated or treated with different agents for various time periods. The cells were lysed to prepare protein extracts. Immunoprecipitation was carried out by incubating ∼1 mg of the protein lysate with appropriate antibodies. For GST-pulldowns, GST–NEMO or various His–TAT–HA-tagged CARP-1 peptides were generated in E. coli BL21 cells as we described previously (5, 21). Briefly, bacterial pellet was lysed in 100–200 μl of BPER buffer (Thermo Fisher Scientific) with DNase I at room temperature, and the supernatant was checked for expression of respective fusion peptides by WB. Following confirmation of expression, 5–20 μl of lysate expressing GST fusion protein was first incubated with 20 μl of precleared GSH-Sepharose in a final volume of 100 μl at 4 °C for 2 h with constant rotation. The Sepharose beads were spun at 800 × g for 2 min, and the pellet was washed two to four times with 100–200 μl of RIPA buffer with 0.5 m NaCl. The beads were spun again as above and mixed with 5–20 μl of E. coli lysate expressing His–TAT–HA CARP-1 peptides. The reactions were incubated further at 4 °C for 2 h with constant rotation. The peptide-bound Sepharose beads were pelleted and washed with 100–200 μl of RIPA buffer with 0.1 m NaCl for 2–4 washes. If necessary, additional washes with 0.05 m NaCl buffer were carried out. In some instances, the complexes were incubated with a small molecule compound (see below) followed by additional NaCl washes as above. After the final wash, the Sepharose/protein complexes were spun and then resuspended in SDS loading buffer for electrophoresis on 12–15% SDS-PAGE, followed by WB with appropriate antibodies. Alternatively, a similar pulldown strategy was carried out by immobilizing the His–TAT–HA peptides on the Ni-NTA matrix, followed by washing, incubating with a small molecule compound, and/or E. coli lysates with GST-tagged proteins, SDS-PAGE analysis, and WB with anti-GST tag antibodies. Luciferase assays were performed essentially as described before (30). Briefly, 3 × 105 cells in culture media minus FBS were plated in 12- or 24-well plates and transfected with a combination of pTK/Renilla Luc and NF-κB–TATA–Luc plasmids. Five hours post-transfection, FBS was added to media, and cells were allowed to grow for 18 h. Cells were then treated with DMSO (control), adriamycin, SNI-1, or a combination for 1 h, harvested, and lysed, and the Renilla and firefly luciferase activities were measured using the Dual-Luciferase assay kit (Promega) following the vendor's guidelines.
Immunofluorescence staining and confocal microscopy
Cells were plated onto chamber slides 24 h prior to treatment. Following treatment of cells with respective compounds, the adherent cells were fixed with 5% formaldehyde for 10 min and then washed with PBS. Samples were blocked (0.5% Nonidet P-40, 5% milk powder, 1% fetal bovine serum) for 30 min. After a single wash with PBS, cells were incubated with primary antibodies for 45–60 min. Cells were washed with PBS and then incubated with secondary antibodies for another 45–60 min, followed by washing with PBS and mounting with 0.1 μg/ml 4′,6′-diamidino-2-phenylindole (DAPI) containing mounting solution. For confocal imaging, cells were first fixed with paraformaldehyde, stained for CARP-1 by myc-tag antibodies (green), NEMO by phospho-NEMO antibodies (red), and DAPI (blue) for nuclear staining. Immunofluorescent or confocal images were taken using Zeiss LSM 510 Meta NLO (×63), essentially as described before (21).
Cytokine ELISAs
Secretion of pro-inflammatory cytokines TNFα, IL8, and IL-1β in untreated and treated human TNBC, and renal cancer cells and mouse 4T1 TNBC cells as well as in sera of 4T1 tumor-bearing BALB/c mice was quantitatively measured by 96-well Quantikine colorimetric ELISA-based assays following the manufacturer's (R&D Systems, Minneapolis, MN) suggested methods and guidelines.
Kinetics of CARP-1–NEMO interaction
In the absence of available X-ray crystal structures for CARP1, we first performed homology modeling on its known sequence to build a suitable protein model utilizing the methodology recently detailed by us (21). Briefly, SWISS-MODEL (33) was used to build homology models for CARP1(551–600) that harbors the epitope for interaction with NEMO. A crystal structure for NEMO has been elucidated, and thus the structure of NEMO(221–261) that interacts with CARP1 was obtained from PDB code 3CL3 (34). Protein–protein docking was performed using ZDOCK 3.0.2f with IRaPPA re-ranking (35). The top three predictions for each complex were further subjected to molecular dynamics (MD) using the AMBER14 package to relieve clashes resulting from docking (59). MD calculations were conducted as reported previously with a 24-ns production run for each complex (21).
Next, the kinetics of CARP-1 binding with NEMO were determined by SPR technology (Profacgen, Shirley, NY). Briefly, CARP-1(551–580) peptide (NH2-HRPEETHKGRTVPAHVETVVLFFPDVWHCL-COOH) was dissolved in water, and various concentrations of CARP-1 peptide were manually printed onto the bare gold-coated (thickness 47 nm) PlexArray Nanocapture Sensor Chip (Plexera Bioscience, Seattle, WA) at 40% humidity. Each concentration was printed in replicate, and each spot contained 0.2 μl of sample solution. The chip was incubated in 80% humidity at 4 °C overnight and rinsed with 10× PBST for 10 min, 1× PBST for 10 min, and deionized water twice for 10 min. The chip was then blocked with 5% (w/v) nonfat milk in water overnight and washed with 10× PBST for 10 min, 1× PBST for 10 min, and deionized water twice for 10 min before being dried under a stream of nitrogen prior to use. The binding reactions with NEMO(221–260) peptide (NH2-EEKRKLAQLQVAYHQLFQEYDNHIKSSVVGSERKRGMQLE-COOH) were performed in PBST buffer (0.01 m PBS (0.138 m NaCl; 0.0027 m KCl), 0.05% Tween 20, pH 7.4). SPRi measurements were performed with PlexArray HT (Plexera Bioscience, Seattle). Collimated light (660 nm) passes through the coupling prism reflects off the SPR-active gold surface and is received by the CCD camera. Buffers and samples were injected by a nonpulsatile piston pump into the 30-μl flow cell that was mounted on the coupling prism. Each measurement cycle contained four steps: washing with PBST running buffer at a constant rate of 2 μl/s to obtain a stable baseline; sample injection at 5 μl/s for binding; surface washing with PBST at 2 μl/s for 300 s; and regeneration with 0.5% (v/v) H3PO4 at 2 μl/s for 300 s. All the measurements were performed at 25 °C. The signal changes after binding and washing (in amplitude unit) were recorded as the assay value. Selected protein-grafted regions in the SPR images were analyzed, and the average reflectivity variations of the chosen areas were plotted as a function of time. Real-time binding signals were recorded and analyzed by Data Analysis Module (Plexera Bioscience, Seattle, WA). Kinetic analysis was performed using BIAevaluation 4.1 software (Biacore, Inc.).
Association and dissociation rate constants were calculated by numerical integration and global fitting to a 1:1 interaction model and the equation: dRU(t)/dt = kaC(Rmax − RU(t)) − kdRU(t), where RU(t) is the response at time t; Rmax is the maximum response; C is the concentration of analyte in solution; ka is the association rate constant; kd is the dissociation rate constant; and RU (0) = 0.
AlphaLISA assay for high-throughput screening
For screening a library of chemical compounds, we developed and optimized a fee-for-service ELISA-based assay for use in a 384-well format (SAMDI Tech, Chicago, IL). The assay development involved buffer optimization by testing peptide binding in PBS, PBS + 0.01% Tween, PBS + 0.01% BSG, PBS + 0.01% Tween, 0.01% BSG, and a proprietary buffer 79389 (BPS Bioscience, San Diego, CA). The assay utilized streptavidin donor and anti-FLAG acceptor beads (PerkinElmer Life Sciences) in conjunction with Flag-tagged CARP-1(546–580) and biotin-tagged NEMO(221–261) peptides that were chemically synthesized to >95% purity (Peptides America, Fairfax, VA). The peptides were dissolved in water, and the binding reactions consisted of 100 nm Flag-tagged CARP-1(546–580) with 1000, 500, 250, 125, 62.5, 31.25, 15.125, or 0 nm biotin-tagged NEMO(221–261) peptide in BPS buffer. The reaction was carried out at room temperature for a 60-min incubation of the peptide pair, followed by 0, 30, and 90 min incubation with AlphaLisa beads. In addition, binding reaction containing different concentrations of biotin-tagged NEMO(221–261) peptide was incubated with 100 nm Flag-tagged CARP-1(546–580) peptide in the absence or presence of 2.5% DMSO. The assay signal (fluorescence) was measured at 680 nm excitation and 615 nm emission wavelengths to determine assay robustness and DMSO tolerance. Next, 10,240 total compounds from a Chembridge diversity set were screened in pools of 8 (5 μm final concentration) with a final concentration of 1% DMSO, 100 nm each peptide utilizing 384-well OptiPlates. The plates were read on a Pherastar FS plate reader. Positive control wells that lacked compound and negative controls were run absent the biotinylated peptide. Hits were identified as those wells showing a % inhibition of >3 standard deviations from the average inhibition across the plate. Those wells were further analyzed to investigate the eight compounds individually in duplicate. The hits revealed during this confirmation step were then analyzed in a dose-response experiment with 50 μm top concentrations of compound with a 3-fold 10-point dilution series in duplicate.
Establishment of TNBC cell-derived xenografts in syngeneic mice
Generation of 4T1 TNBC cell-derived subcutaneous xenografts in BALB/c mice were performed according to our previously published methods and protocols approved by the Institutional Animal Care & Use Committee at the Wayne State University (40, 45). Then 6–8-week-old BALB/cAnNCr female mice were purchased from Charles River Laboratories (Horsham, PA). Following suitable acclimation of animals, 1 × 106 4T1 TNBC cells were resuspended in 200 μl of sterile saline and implanted in the flanks using a 27-gauge needle. Tumors were allowed to grow to 500–1000 mg (∼10 days) and then aseptically harvested, minced into 3–4-mm3 (30 mg) fragments, and transplanted subcutaneously into naive recipient mice using a standard 12-gauge trocar to serially maintain the tumor in vivo. For efficacy studies, tumors serially maintained in vivo were aseptically harvested and minced into 3–4-mm3 (30 mg) fragments and then bilaterally transplanted subcutaneously along the flanks using a standard 12-gauge trocar. Mice were then randomly assigned to either the control or one of five treatment arms (n = 6 mice/group) as follows: no treatment; SNI-1 (70 mg/kg/dose; quaque die 1–13 via intraperitoneal injection; total dose 910 mg/kg); adriamycin (4 mg/kg/dose; Day 1, 5, 10, and 14 via intravenous injection; total dose 16 mg/kg); cisplatin (CIS) (3 mg/kg/dose; Day 1, 5, 10, and 14 via intravenous injection; total dose 12 mg/kg); and SNI-1 + ADR or SNI-1 + CIS on matching respective single arm schedules. For the combination arm, SNI-1 was administered first, followed within 1 h by either ADR or CIS. A pilot dose-route determination conducted with SNI-1 in nontumor-bearing mice using a solubilized formulation (5% DMSO, 5% ethanol, 2% Tween 80 (all v/v) with double-distilled water) found that the compound was not suitable for chronic intravenous administration. Therefore, for the efficacy studies reported here, SNI-1 was formulated in 8% DMSO (v/v) and 8% Cremophor (v/v) in cell grade water, pH 4, as diluent. Clinical grade ADR stock (2 mg/ml) was diluted to the appropriate concentration with cell grade water, pH 4, and clinical grade CIS (1 mg/ml) diluted with USP 0.9% saline, pH 6. All mice were monitored daily for changes in condition and body weight. Tumors were measured three times weekly by caliper, and tumor volume (in milligrams) was calculated using the following formula: (A × B2)/2, where A and B are the tumor length and width (in millimeters), respectively. End points for assessing antitumor activity consisted of qualitative determinations via tumor growth inhibition (%T/C) where T is the median tumor volume of treated mice and C is the median tumor volume of control mice on any given day of measurement. According to NCI-accepted criteria from the National Institutes of Health, a treatment is considered effective if the T/C is <42%. Highly-active agents produce T/C values <20%. Efficacy was also assessed quantitatively using tumor growth delay (T/C) defined as the difference between the median time (in days) required for the treatment group tumors to reach 1000 mg and the median time (days) for the control group tumors to reach the same volume. After the last treatment, tumor tissue and samples from various organs (spleen, liver, kidney, heart, bone, and lungs) were collected. Additionally, the whole-blood samples were obtained from a representative tumor-bearing mouse from each group via terminal cardiac puncture. We performed immunohistochemical analyses of the tumor and tissue samples for expression of activated RelA (serine 536 phosphorylated p65), total RelA, cleaved caspase-3, and CARP-1 proteins. The levels of pro-inflammatory cytokines in the sera were analyzed by sandwich ELISAs following methods detailed above and previously published by us (40, 45).
Statistical analyses
The statistical analyses were performed using Prism 6.0 software. The data were expressed as means± S.E. and analyzed using two-tailed Student's test or one-way analysis of variance followed by a post hoc test. A p value of <0.05 was considered statistically significant.
Author contributions
J. V., S. C. S., V. T. C., M. M., P. M., E. L., S. D., J. W. G., and L. A. P. data curation; J. V., S. C. S., V. T. C., M. M., P. M., E. L., S. D., J. W. G., and L. A. P. investigation; P. M. formal analysis; J. W. G. methodology; A. K. R. conceptualization; A. K. R. supervision; A. K. R. funding acquisition; J. V., A. K. R. writing-original draft.
Supplementary Material
Acknowledgments
We gratefully acknowledge Amro Aboukameel and Dr. Vishnu T. Undyala for technical assistance with certain experiments. We also gratefully acknowledge ongoing discussions with Dr. Karin Przyklenk, Wayne State University Cardiovascular Research Institute, regarding involvement of CARP-1/NEMO signaling in cardiomyocyte models. The Karmanos Cancer Institute was recipient of NCI support grant from the National Institutes of Health Cancer Center.
This work was supported by a Department of Veterans Affairs merit review grant (to A. K. R.) and Wayne State University Molecular Therapeutics Seed Money Grant (to A. K. R.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S9.
- EGFR
- epidermal growth factor receptor
- NF-κB
- nuclear factor-κB
- TNFα
- tumor necrosis factor α
- IL-8
- interleukin-8
- IL-1β
- interleukin 1β
- 5-FU
- 5-fluouracil
- IKK
- inhibitory κB kinase
- NEMO
- NF-κB essential modulator (also known as IKKγ)
- EGFP
- enhanced green fluorescent protein
- GST
- glutathione S-transferase
- FBS
- fetal bovine serum
- TAT
- trans-activation of transcription tag
- HBC
- human breast cancer
- TNBC
- triple-negative breast cancer
- SNI-1
- selective NF-κB inhibitor-1
- ADR
- adriamycin/doxorubicin
- CIS
- cisplatin
- MTT
- 5-dimethylthiazol-2-yl-2,5-diphenyltetrazolium bromide
- SPR
- surface plasmon resonance
- RMSD
- root mean square deviation
- DMEM
- Dulbecco's modified Eagle's medium
- WB
- Western blotting
- DSB
- double-strand break
- CFM
- CARP-1 functional mimetic
- PDX
- patient-derived
- DDR
- DNA damage response
- ATM
- ataxia telangiectasia mutated
- ATR
- ATM and Rad3-related
- HTS
- high-throughput screening
- SMI
- small-molecule inhibitor
- Ni-NTA
- nickel-nitrilotriacetic acid
- NLS
- nuclear localization signal
- DAPI
- 4′,6′-diamidino-2-phenylindole
- PDB
- Protein Data Bank
- MD
- molecular dynamics
- BSG
- bovine skin gelatin
- CIS
- cisplatin
- MM-GBSA
- molecular mechanics- generalized born and surface area
- MM-PBSA
- molecular mechanics- Poisson-Boltzmann surface area.
References
- 1. Rishi A. K., Zhang L., Boyanapalli M., Wali A., Mohammad R. M., Yu Y., Fontana J. A., Hatfield J. S., Dawson M. I., Majumdar A. P., and Reichert U. (2003) Identification and characterization of a cell cycle and apoptosis regulatory protein-1 as a novel mediator of apoptosis signaling by retinoid CD437. J. Biol. Chem. 278, 33422–33435 10.1074/jbc.M303173200 [DOI] [PubMed] [Google Scholar]
- 2. Yoo A. S., Bais C., and Greenwald I. (2004) Crosstalk between the EGFR and LIN-12/Notch pathways in C. elegans vulval development. Science 303, 663–666 10.1126/science.1091639 [DOI] [PubMed] [Google Scholar]
- 3. Rishi A. K., Zhang L., Yu Y., Jiang Y., Nautiyal J., Wali A., Fontana J. A., Levi E., and Majumdar A. P. (2006) Cell cycle- and apoptosis-regulatory protein-1 is involved in apoptosis signaling by epidermal growth factor receptor. J. Biol. Chem. 281, 13188–13198 10.1074/jbc.M512279200 [DOI] [PubMed] [Google Scholar]
- 4. Jiang Y., Puliyappadamba V. T., Zhang L., Wu W., Wali A., Yaffe M. B., Fontana J. A., and Rishi A. K. (2010) A novel mechanism of cell growth regulation by cell cycle and apoptosis regulatory protein (CARP)-1. J. Mol. Signal. 5, 7 10.1186/1750-2187-5-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang L., Levi E., Majumder P., Yu Y., Aboukameel A., Du J., Xu H., Mohammad R., Hatfield J. S., Wali A., Adsay V., Majumdar A. P., and Rishi A. K. (2007) Transactivator of transcription-tagged cell cycle and apoptosis regulatory protein-1 peptides suppress the growth of human breast cancer cells in vitro and in vivo. Mol. Cancer Ther. 6, 1661–1672 10.1158/1535-7163.MCT-06-0653 [DOI] [PubMed] [Google Scholar]
- 6. Beausoleil S. A., Jedrychowski M., Schwartz D., Elias J. E., Villén J., Li J., Cohn M. A., Cantley L. C., and Gygi S. P. (2004) Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc. Natl. Acad. Sci. U.S.A. 101, 12130–12135 10.1073/pnas.0404720101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blagoev B., Kratchmarova I., Ong S. E., Nielsen M., Foster L. J., and Mann M. (2003) A proteomics strategy to elucidate functional protein–protein interactions applied to EGF signaling. Nat. Biotechnol. 21, 315–318 10.1038/nbt790 [DOI] [PubMed] [Google Scholar]
- 8. Matsuoka S., Ballif B. A., Smogorzewska A., McDonald E. R. 3rd, Hurov K. E., Luo J., Bakalarski C. E., Zhao Z., Solimini N., Lerenthal Y., Shiloh Y., Gygi S. P., and Elledge S. J. (2007) ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316, 1160–1166 10.1126/science.1140321 [DOI] [PubMed] [Google Scholar]
- 9. Hervy M., Hoffman L. M., Jensen C. C., Smith M., and Beckerle M. C. (2010) The LIM protein zyxin binds CARP-1 and promotes apoptosis. Genes Cancer 1, 506–515 10.1177/1947601910376192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. François S., D'Orlando C., Fatone T., Touvier T., Pessina P., Meneveri R., and Brunelli S. (2012) Necdin enhances myoblasts survival by facilitating the degradation of the mediator of apoptosis CCAR1/CARP1. PLoS ONE 7, e43335 10.1371/journal.pone.0043335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Puliyappadamba V. T., Wu W., Bevis D., Zhang L., Polin L., Kilkuskie R., Finley R. L. Jr, Larsen S. D., Levi E., Miller F. R., Wali A., and Rishi A. K. (2011) Antagonists of anaphase-promoting complex (APC)–2-cell cycle and apoptosis regulatory protein (CARP)-1 interaction are novel regulators of cell growth and apoptosis. J. Biol. Chem. 286, 38000–38017 10.1074/jbc.M111.222398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim J. H., Yang C. K., Heo K., Roeder R. G., An W., and Stallcup M. R. (2008) CCAR1, a key regulator of mediator complex recruitment to nuclear receptor transcription complexes. Mol. Cell 31, 510–519 10.1016/j.molcel.2008.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ou C. Y., Kim J. H., Yang C. K., and Stallcup M. R. (2009) Requirement of cell cycle and apoptosis regulator 1 for target gene activation by Wnt and β-catenin and for anchorage-independent growth of human colon carcinoma cells. J. Biol. Chem. 284, 20629–20637 10.1074/jbc.M109.014332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ou C. Y., Chen T. C., Lee J. V., Wang J. C., and Stallcup M. R. (2014) Coregulator cell cycle and apoptosis regulator 1 (CCAR1) positively regulates adipocyte differentiation through the glucocorticoid signaling pathway. J. Biol. Chem. 289, 17078–17086 10.1074/jbc.M114.548081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lu C. K., Lai Y. C., Lin Y. F., Chen H. R., and Chiang M. K. (2012) CCAR1 is required for Ngn3-mediated endocrine differentiation. Biochem. Biophys. Res. Commun. 418, 307–312 10.1016/j.bbrc.2012.01.016 [DOI] [PubMed] [Google Scholar]
- 16. Pommier Y., Leo E., Zhang H., and Marchand C. (2010) DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 17, 421–433 10.1016/j.chembiol.2010.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fornari F. A., Randolph J. K., Yalowich J. C., Ritke M. K., and Gewirtz D. A. (1994) Interference by doxorubicin with DNA unwinding in MCF-7 breast tumor cells. Mol. Pharmacol. 45, 649–656 [PubMed] [Google Scholar]
- 18. Podhorecka M., Skladanowski A., and Bozko P. (2010) H2AX phosphorylation: its role in DNA damage response and cancer therapy. J. Nucleic Acids 2010, 920161 10.4061/2010/920161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Picco V., and Pagès G. (2013) Linking JNK activity to the DNA damage response. Genes Cancer 4, 360–368 10.1177/1947601913486347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lu C., Zhu F., Cho Y. Y., Tang F., Zykova T., Ma W. Y., Bode A. M., and Dong Z. (2006) Cell apoptosis: requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3. Mol. Cell 23, 121–132 10.1016/j.molcel.2006.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sekhar S. C., Venkatesh J., Cheriyan V. T., Muthu M., Levi E., Assad H., Meister P., Undyala V. V., Gauld J. W., and Rishi A. K. (2019) A H2AX(−)CARP-1 interaction regulates apoptosis signaling following DNA damage. Cancers 11, E221 10.3390/cancers11020221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Graef I. A., Gastier J. M., Francke U., and Crabtree G. R. (2001) Evolutionary relationships among Rel domains indicate functional diversification by recombination. Proc. Natl. Acad. Sci. U.S.A. 98, 5740–5745 10.1073/pnas.101602398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang Q., Lenardo M. J., and Baltimore D. (2017) 30 years of NF-κB: a blossoming of relevance to human pathobiology. Cell 168, 37–57 10.1016/j.cell.2016.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu J., Yang D., Minemoto Y., Leitges M., Rosner M. R., and Lin A. (2006) NF-κB is required for UV-induced JNK activation via induction of PKCδ. Mol. Cell 21, 467–480 10.1016/j.molcel.2005.12.020 [DOI] [PubMed] [Google Scholar]
- 25. Shou Y., Li N., Li L., Borowitz J. L., and Isom G. E. (2002) NF-κB–mediated up-regulation of Bcl-X(S) and Bax contributes to cytochrome c release in cyanide-induced apoptosis. J. Neurochem. 81, 842–852 10.1046/j.1471-4159.2002.00880.x [DOI] [PubMed] [Google Scholar]
- 26. Martin A. G., Trama J., Crighton D., Ryan K. M., and Fearnhead H. O. (2009) Activation of p73 and induction of Noxa by DNA damage requires NF-κB. Aging 1, 335–349 10.18632/aging.100026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ryan K. M., Ernst M. K., Rice N. R., and Vousden K. H. (2000) Role of NF-κB in p53-mediated programmed cell death. Nature 404, 892–897 10.1038/35009130 [DOI] [PubMed] [Google Scholar]
- 28. Wu Z. H., Shi Y., Tibbetts R. S., and Miyamoto S. (2006) Molecular linkage between the kinase ATM and NF-κB signaling in response to genotoxic stimuli. Science 311, 1141–1146 10.1126/science.1121513 [DOI] [PubMed] [Google Scholar]
- 29. Bouwmeester T., Bauch A., Ruffner H., Angrand P. O., Bergamini G., Croughton K., Cruciat C., Eberhard D., Gagneur J., Ghidelli S., Hopf C., Huhse B., Mangano R., Michon A. M., Schirle M., et al. (2004) A physical and functional map of the human TNF-α/NF-κB signal transduction pathway. Nat. Cell Biol. 6, 97–105 10.1038/ncb1086 [DOI] [PubMed] [Google Scholar]
- 30. Muthu M., Cheriyan V. T., Munie S., Levi E., Frank J., Ashour A. E., Singh M., and Rishi A. K. (2014) Mechanisms of neuroblastoma cell growth inhibition by CARP-1 functional mimetics. PLoS ONE 9, e102567 10.1371/journal.pone.0102567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huang T. T., Wuerzberger-Davis S. M., Wu Z. H., and Miyamoto S. (2003) Sequential modification of NEMO/IKKγ by SUMO-1 and ubiquitin mediates NF-κB activation by genotoxic stress. Cell 115, 565–576 10.1016/S0092-8674(03)00895-X [DOI] [PubMed] [Google Scholar]
- 32. Perkins N. D. (2007) Integrating cell-signalling pathways with NF-κB and IKK function. Nat. Rev. Mol. Cell Biol. 8, 49–62 10.1038/nrm2083 [DOI] [PubMed] [Google Scholar]
- 33. Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F. T., de Beer T. A. P., Rempfer C., Bordoli L., Lepore R., and Schwede T. (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 10.1093/nar/gky427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bagnéris C., Ageichik A. V., Cronin N., Wallace B., Collins M., Boshoff C., Waksman G., and Barrett T. (2008) Crystal structure of a vFlip-IKKγ complex: insights into viral activation of the IKK signalosome. Mol. Cell 30, 620–631 10.1016/j.molcel.2008.04.029 [DOI] [PubMed] [Google Scholar]
- 35. Pierce B. G., Wiehe K., Hwang H., Kim B. H., Vreven T., and Weng Z. (2014) ZDOCK server: interactive docking prediction of protein–protein complexes and symmetric multimers. Bioinformatics 30, 1771–1773 10.1093/bioinformatics/btu097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang Y., Xia F., Hermance N., Mabb A., Simonson S., Morrissey S., Gandhi P., Munson M., Miyamoto S., and Kelliher M. A. (2011) A cytosolic ATM/NEMO/RIP1 complex recruits TAK1 to mediate the NF-κB and p38 mitogen-activated protein kinase (MAPK)/MAPK-activated protein 2 responses to DNA damage. Mol. Cell. Biol. 31, 2774–2786 10.1128/MCB.01139-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wen S. H., Su S. C., Liou B. H., Lin C. H., and Lee K. R. (2018) Sulbactam-enhanced cytotoxicity of doxorubicin in breast cancer cells. Cancer Cell Int. 18, 128 10.1186/s12935-018-0625-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang W., Mani A. M., and Wu Z. H. (2017) DNA damage–induced nuclear factor-κB activation and its roles in cancer progression. J. Cancer Metastasis Treat. 3, 45–59 10.20517/2394-4722.2017.03 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rocha C. R. R., Silva M. M., Quinet A., Cabral-Neto J. B., and Menck C. F. M. (2018) DNA repair pathways and cisplatin resistance: an intimate relationship. Clinics 73, e478s 10.6061/clinics/2018/e478s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cheriyan V. T., Muthu M., Patel K., Sekhar S., Rajeswaran W., Larsen S. D., Polin L., Levi E., Singh M., and Rishi A. K. (2016) CARP-1 functional mimetics are novel inhibitors of drug-resistant triple negative breast cancers. Oncotarget 7, 73370–73388 10.18632/oncotarget.12333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fang R., Wang C., Jiang Q., Lv M., Gao P., Yu X., Mu P., Zhang R., Bi S., Feng J. M., and Jiang Z. (2017) NEMO-IKKβ are essential for IRF3 and NF-κB activation in the cGAS-STING pathway. J. Immunol. 199, 3222–3233 10.4049/jimmunol.1700699 [DOI] [PubMed] [Google Scholar]
- 42. Muthu M., Somagoni J., Cheriyan V. T., Munie S., Levi E., Ashour A. E., Yassin A. E., Alafeefy A. M., Sochacki P., Polin L. A., Reddy K. B., Larsen S. D., Singh M., and Rishi A. K. (2015) Identification and testing of novel CARP-1 functional mimetic compounds as inhibitors of non-small cell lung and triple-negative breast cancers. J. Biomed. Nanotechnol. 11, 1608–1627 10.1166/jbn.2015.2099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. van Raam B. J., Ehrnhoefer D. E., Hayden M. R., and Salvesen G. S. (2013) Intrinsic cleavage of receptor-interacting protein kinase-1 by caspase-6. Cell Death Differ. 20, 86–96 10.1038/cdd.2012.98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vyas D., Laput G., and Vyas A. K. (2014) Chemotherapy-enhanced inflammation may lead to the failure of therapy and metastasis. Onco Targets Ther. 7, 1015–1023 10.2147/OTT.S60114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cheriyan V. T., Alsaab H. O., Sekhar S., Stieber C., Kesharwani P., Sau S., Muthu M., Polin L. A., Levi E., Iyer A. K., and Rishi A. K. (2017) A CARP-1 functional mimetic loaded vitamin E-TPGS micellar nano-formulation for inhibition of renal cell carcinoma. Oncotarget 8, 104928–104945 10.18632/oncotarget.20650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wuerzberger-Davis S. M., Nakamura Y., Seufzer B. J., and Miyamoto S. (2007) NF-κB activation by combinations of NEMO SUMOylation and ATM activation stresses in the absence of DNA damage. Oncogene 26, 641–651 10.1038/sj.onc.1209815 [DOI] [PubMed] [Google Scholar]
- 47. Hwang B., McCool K., Wan J., Wuerzberger-Davis S. M., Young E. W., Choi E. Y., Cingolani G., Weaver B. A., and Miyamoto S. (2015) IPO3-mediated nonclassical nuclear import of NF-κB essential modulator (NEMO) drives DNA damage-dependent NF-κB activation. J. Biol. Chem. 290, 17967–17984 10.1074/jbc.M115.645960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Young D. B., Jonnalagadda J., Gatei M., Jans D. A., Meyn S., and Khanna K. K. (2005) Identification of domains of ataxia-telangiectasia mutated required for nuclear localization and chromatin association. J. Biol. Chem. 280, 27587–27594 10.1074/jbc.M411689200 [DOI] [PubMed] [Google Scholar]
- 49. Wu G., Song L., Zhu J., Hu Y., Cao L., Tan Z., Zhang S., Li Z., and Li J. (2018) An ATM/TRIM37/NEMO axis counteracts genotoxicity by activating nuclear-to-cytoplasmic NF-κB signaling. Cancer Res. 78, 6399–6412 10.1158/0008-5472.CAN-18-2063 [DOI] [PubMed] [Google Scholar]
- 50. Gilmore T. D., and Herscovitch M. (2006) Inhibitors of NF-κB signaling: 785 and counting. Oncogene 25, 6887–6899 10.1038/sj.onc.1209982 [DOI] [PubMed] [Google Scholar]
- 51. Ran R., Lu A., Zhang L., Tang Y., Zhu H., Xu H., Feng Y., Han C., Zhou G., Rigby A. C., and Sharp F. R. (2004) Hsp70 promotes TNF-mediated apoptosis by binding IKKγ and impairing NF-κB survival signaling. Genes Dev. 18, 1466–1481 10.1101/gad.1188204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. May M. J., D'Acquisto F., Madge L. A., Glöckner J., Pober J. S., and Ghosh S. (2000) Selective inhibition of NF-κB activation by a peptide that blocks the interaction of NEMO with the IκB kinase complex. Science 289, 1550–1554 10.1126/science.289.5484.1550 [DOI] [PubMed] [Google Scholar]
- 53. Dai S., Hirayama T., Abbas S., and Abu-Amer Y. (2004) The IκB kinase (IKK) inhibitor, NEMO-binding domain peptide, blocks osteoclastogenesis and bone erosion in inflammatory arthritis. J. Biol. Chem. 279, 37219–37222 10.1074/jbc.C400258200 [DOI] [PubMed] [Google Scholar]
- 54. Agou F., Courtois G., Chiaravalli J., Baleux F., Coïc Y. M., Traincard F., Israël A., and Véron M. (2004) Inhibition of NF-κB activation by peptides targeting NF-κB essential modulator (nemo) oligomerization. J. Biol. Chem. 279, 54248–54257 10.1074/jbc.M406423200 [DOI] [PubMed] [Google Scholar]
- 55. Vincendeau M., Hadian K., Messias A. C., Brenke J. K., Halander J., Griesbach R., Greczmiel U., Bertossi A., Stehle R., Nagel D., Demski K., Velvarska H., Niessing D., Geerlof A., Sattler M., and Krappmann D. (2016) Inhibition of canonical NF-κB signaling by a small molecule targeting NEMO–ubiquitin interaction. Sci. Rep. 6, 18934 10.1038/srep18934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hooper C., Jackson S. S., Coughlin E. E., Coon J. J., and Miyamoto S. (2014) Covalent modification of the NF-κB essential modulator (NEMO) by a chemical compound can regulate its ubiquitin binding properties in vitro. J. Biol. Chem. 289, 33161–33174 10.1074/jbc.M114.582478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jackson S. S., Oberley C., Hooper C. P., Grindle K., Wuerzberger-Davis S., Wolff J., McCool K., Rui L., and Miyamoto S. (2015) Withaferin A disrupts ubiquitin-based NEMO reorganization induced by canonical NF-κB signaling. Exp. Cell Res. 331, 58–72 10.1016/j.yexcr.2014.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Heyninck K., Lahtela-Kakkonen M., Van der Veken P., Haegeman G., and Vanden Berghe W. (2014) Withaferin A inhibits NF-κB activation by targeting cysteine 179 in IKβ. Biochem Pharmacol. 91, 501–509 10.1016/j.bcp.2014.08.004 [DOI] [PubMed] [Google Scholar]
- 59. Case D. A., Babin V., Berryman J. T., Betz R. M., Cai Q., Cerutti D. S., Cheatham T. E. 3rd., Darden T. A., Duke R. E., Gohlke H., Goetz A. W., Gusarov S., Homeyer N., Janowski P., Kaus J., et al. (2014) AMBER 14, University of California, San Francisco, CA [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.