

Abstract
Higher expression of the human DNA repair enzyme MUTYH has previously been shown to be strongly associated with reduced survival in a panel of 24 human lymphoblastoid cell lines exposed to the alkylating agent N-methyl-N′-nitro-N-nitrosoguanidine (MNNG). The molecular mechanism of MUTYH-enhanced MNNG cytotoxicity is unclear, because MUTYH has a well-established role in the repair of oxidative DNA lesions. Here, we show in mouse embryonic fibroblasts (MEFs) that this MNNG-dependent phenotype does not involve oxidative DNA damage and occurs independently of both O6-methyl guanine adduct cytotoxicity and MUTYH-dependent glycosylase activity. We found that blocking of abasic (AP) sites abolishes higher survival of Mutyh-deficient (Mutyh−/−) MEFs, but this blockade had no additive cytotoxicity in WT MEFs, suggesting the cytotoxicity is due to MUTYH interactions with MNNG-induced AP sites. We found that recombinant mouse MUTYH tightly binds AP sites opposite all four canonical undamaged bases and stimulated apurinic/apyrimidinic endonuclease 1 (APE1)-mediated DNA incision. Consistent with these observations, we found that stable expression of WT, but not catalytically-inactive MUTYH, enhances MNNG cytotoxicity in Mutyh−/− MEFs and that MUTYH expression enhances MNNG-induced genomic strand breaks. Taken together, these results suggest that MUTYH enhances the rapid accumulation of AP-site intermediates by interacting with APE1, implicating MUTYH as a factor that modulates the delicate process of base-excision repair independently of its glycosylase activity.
Keywords: DNA repair, base excision repair (BER), mutY homolog (MUTYH), DNA methylation, DNA damage, DNA enzyme, DNA damage response, cancer biology, cancer chemoprevention, abasic sites
Introduction
MUTYH,4 the human homolog of mouse Mutyh, is the first base excision repair (BER) enzyme directly implicated in an inherited cancer syndrome, MUTYH-associated polyposis (MAP) (1–3). MUTYH removes adenine mis-incorporated opposite the common DNA oxidation product, 8-oxoguanine (OG) (Fig. 1A), thereby preventing accumulation of G:C to T:A transversion mutations (3, 4). MAP was originally pinpointed as a BER defect because of increased G:C to T:A mutations in the APC gene in tumor tissue (1), demonstrating how a lifelong reduction in DNA repair capacity underlies cancer progression by the accumulation of mutations in other genes. The substrate specificity and cellular role of MUTYH have been extensively studied (3–6). However, accruing evidence over the past decade has implicated a broader role for MUTYH in the cellular response to diverse forms of DNA damage beyond OG:A mispairs (7). MUTYH is implicated in the DNA damage response to alkylating agents (8, 9), DNA cross-linking agents (10), UV radiation (11, 12), hydroxyurea (9, 12), and mitomycin C (13). Furthermore, MUTYH expression appears to inhibit the repair of UV-induced cyclopyrimidine dimers in cells lacking the nucleotide excision repair protein XPA (11).
Figure 1.

Oxidative and alkylative base damage and repair. A, depiction of MUTYH's known canonical role in the repair of OG:A (O:A) lesions in the cell, followed by downstream processing by APE1 and other BER proteins. B, functional groups within dsDNA that are susceptible to methylation by MNNG. Thymine (blue) is paired with adenine (red), and cytosine (teal) is paired with guanine (purple).
High endogenous MUTYH expression in a panel of 24 immortalized human lymphocyte cell lines from healthy human subjects is associated with reduced cell survival to the alkylating agent N-methyl-N′-nitro-N-nitrosoguanidine (MNNG), suggesting MUTYH enhances the cytotoxicity of alkylating agents (8). MNNG induces DNA methyl adducts (Fig. 1B) that are primarily repaired by an alkyl-adenine glycosylase (AAG) or methyl-guanine methyltransferase (MGMT) (14). High basal MUTYH expression was more strongly associated with higher cell death versus genes known to mediate alkylating agent survival, such as AAG and MGMT.
Here, we present evidence suggesting a novel mechanism for MUTYH-dependent cytotoxicity to the alkylating agent MNNG. Our results show that the levels of MNNG associated with MUTYH-dependent toxicity do not generate cellular reactive oxygen species (ROS). In addition, we find in vitro that mouse MUTYH does not have glycosylase activity toward methyl-DNA base adducts. In contrast, our findings show that the levels of genomic strand breaks are increased in WT relative to Mutyh−/− MEFs at 40 min after MNNG treatment, and additionally, there is a decrease in unresolved genomic AP sites in WT versus Mutyh−/− MEFs after 24 h, evidence of a key role of MUTYH in AP site processing. In vitro experiments are consistent with these results showing high affinity of recombinant mouse MUTYH for AP sites and stimulation of APE1 incision. Our observations may be relevant to understanding the more general role of MUTYH in cross-talk with other DNA repair pathways reported by a number of studies (8, 10, 11, 15).
Results
MUTYH expression levels enhance MNNG cytotoxicity
Consistent with a previous report (8), we found that immortalized Mutyh−/− MEFs treated with 67 μm (10 μg/ml) MNNG had significantly higher survival versus wildtype (WT) MEFs, p = 0.0016 (Fig. 2A, 1st and 2nd columns). Several experiments with Mutyh−/− MEFs had unusually high survival; after the experimental results with survival above 50% were discarded as outliers, there still existed a significant difference in survival between these cell lines (p = 0.007), which suggested the effect was robust. To verify the connection between MUTYH expression and MNNG cytotoxicity, we created Mutyh−/− MEF cell lines stably expressing recombinant human MUTYH. Antibiotic-resistant colonies were isolated and screened for MUTYH expression by Western blotting and were categorized as having high or low MUTYH expression as shown in Fig. 2B. Two high MUTYH cell lines had significantly lower survival to MNNG treatment versus three low MUTYH cell lines, p = 0.0048 (Fig. 2A, 3rd and 4th columns), evidence that increasing MUTYH expression levels leads to increasing amounts of MNNG cytotoxicity.
Figure 2.
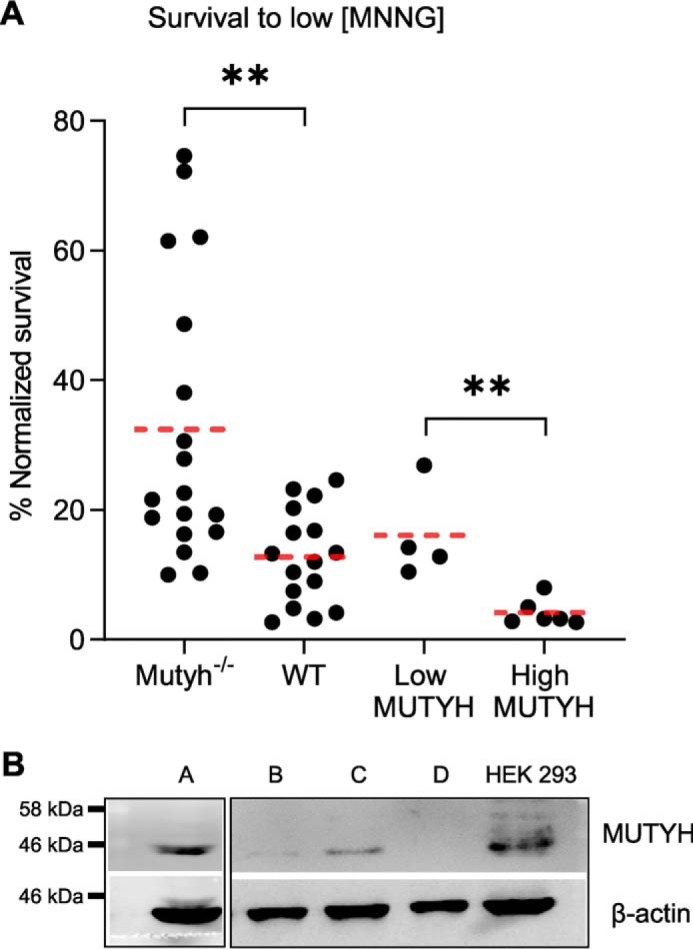
MUTYH expression alters MEF survival to MNNG. A, normalized survival versus control condition 6–7 days after treating with low MNNG (67 μm) in Mutyh−/−, wildtype (WT), and Mutyh−/− MEFs stable cell lines expressing recombinant human MUTYH, classified as low MUTYH (expression) and high MUTYH (expression) based on Western blotting. Data are from at least four biological replicates. **, p < 0.005, t test, significant with Bonferroni correction for multiple comparisons. B, Western blotting of representative Mutyh−/− MEF stable cell lines transfected with the pcDNA3.1 human MUTYH construct after G418 antibiotic selection. HEK-293 cell lysate is shown as a MUTYH-positive control. Clones A and C were categorized as high MUTYH expression. Clones B and D were categorized as low MUTYH expression. The figure is composed of two separate Western blottings using the same antibodies and conditions as detailed under “Experimental procedures.”
MUTYH-mediated cytotoxicity to low MNNG concentrations is independent of ROS
The known activity of MUTYH in oxidative base damage repair suggests that the enhanced MNNG cytotoxicity may be due to induction of ROS. Previous reports indicated that treatment of mammalian cells with 250 μm MNNG induces ROS (16), but it is not known whether lower concentrations also generate ROS. In addition, MUTYH expression has been associated with increased strand breaks and cell death due to oxidative DNA damage (17), which could explain the enhanced toxicity of MUTYH expression with MNNG. However, there is also evidence that MUTYH enhances survival to ROS (18) and that MUTYH expression correlates with increased survival to the alkylating agent temozolomide (TMZ) at a concentration that was shown to induce ROS (19). We thus tested the role of MUTYH and OGG1 status to survival, oxidative DNA damage, and cellular ROS levels, specifically comparing differences at 67 μm MNNG (used in experiments in Fig. 2A) versus higher concentrations previously shown to induce ROS (16).
To directly test the hypothesis that MNNG induces ROS formation in MEF cells under the conditions of our survival assay, we used the redox-sensitive probe 2′,7′-dichloro-dihydrofluorescein diacetate. Oxidative stress of high-concentration MNNG treatment occurs within 1 h of exposure (16). At 1–2 h, 5–50 μm hydrogen peroxide induced ∼5–15% of cells to be categorized as green fluorescence–positive by flow cytometry (Fig. 3B), whereas untreated cells had background fluorescence below 1%, demonstrating sensitive detection of cellular oxidative stress. Equal gating and quantitation of MNNG-treated MEFs showed less than 1% positive fluorescence at all concentrations tested below 333 μm MNNG (Fig. 3A), ∼5-fold higher concentrations than that which mediated increased survival of Mutyh−/−/MEFs shown in Fig. 2A. Differences in the number of fluorescent cells in the 0–167 μm MNNG categories were not significant. Assays at 15 min and 4 h after MNNG treatment, more stringent gating of cells, and concentration dependence analysis failed to detect any evidence of increased ROS below 333 μm MNNG.
Figure 3.

Under conditions of high MNNG, which induce oxidative stress, Mutyh enhances cell survival. A and B, ROS detection using the redox-sensitive fluorescent probe H2-DCFDA in MNNG-treated WT MEFs (A) versus hydrogen peroxide-treated WT MEFs as a positive control (B). Each point represents the percent of fluorescence-positive cells from an independent flow cytometry experiment as detailed under “Experimental procedures.” The trend line represents the mean percent fluorescent cells as a function of concentration. C, 6–7-day survival of WT and Mutyh−/− MEFs at high concentration MNNG from four separate experiments (averaged data from combined 333 and 667 μm treatments). WT MEFs had significantly higher survival under these conditions (**p = 0.01). D, survival of Ogg1−/− MEFs to 67 μm MNNG is not significantly different from their matched parental WT MEFs (p = 0.64). E, detection of MNNG-induced lesions by purified Ogg1 enzyme plasmid nicking assay, where the increased density of the upper open circular (oc) DNA indicates strand nicking versus the closed-circular (cc) plasmid DNA.
Because ROS is generated at high levels of MNNG, we then examined whether there would be a MNNG dose dependence in our cell-survival assays. We found that the differences in survival between WT and Mutyh−/− MEFs were reversed at higher MNNG concentrations (pooled results of 333 and 667 μm; Fig. 3C). At the high MNNG concentrations shown to induce ROS, WT MEFs had significantly higher survival (p = 0.01), suggesting MUTYH has a prosurvival role to oxidative DNA damage induced by high concentrations of MNNG. These results are consistent with previous studies that showed that MUTYH provides protection to hydrogen peroxide and ROS generated by TMZ (18, 19).
We anticipated that induction of ROS by MNNG would also be influenced by the presence of Ogg1 because Ogg1−/− MEFs have been shown to be sensitive to cell death due to oxidative DNA damage (17). Consistent with the lack of ROS generated at low MNNG, we found that Ogg1−/− and WT MEFs had s similar survival to 67 μm MNNG (p = 0.88; Fig. 3D). These results provide further evidence that Mutyh−/− MEF MNNG survival is independent of the OG lesion repair pathway.
Plasmid DNA–nicking assays have been used to assess levels of OG formation induced by chemical treatment in vitro (20). To determine whether the treatment with MNNG concentration that resulted in increased survival of Mutyh−/− MEFs (67 μm) may directly induce formation of OG lesions, plasmid DNA was treated with increasing levels of MNNG and incubated with the human OG glycosylase (hOgg1). As shown in Fig. 3E, at MNNG concentrations of 333 μm and above, a base lesion is formed at a modest level that is removed by hOgg1, but no significant hOgg1-dependent cleavage above a background observed at 67 μm, the concentration used in the cell assays in Fig. 2A.
These data taken together argue against the hypothesis that oxidative DNA damage mediates the higher survival of Mutyh−/− MEFs to MNNG treatment, although due to both the detection limit of these assays and the possibility of rare adducts, we cannot completely rule out the possibility that rare MNNG-induced DNA lesions are substrates for MUTYH. Notably, these results also show that there is a fine balance between the cytotoxic and protective roles of MUTYH that is dependent on the concentrations of MNNG and the generation of ROS.
MUTYH-mediated cytotoxicity to MNNG is independent of O6meG lesions and mismatch repair status
To understand how MUTYH status alters survival to different alkylating agents, we tested the survival of MEFs to TMZ, a cancer chemotherapy agent, and methyl-methanesulfonate (MMS). Although Mutyh−/− MEFs had higher survival to TMZ on average, similar to MNNG, this difference did not reach statistical significance with a TMZ dose of 1.1 mm (p = 0.28, Fig. S1). This concentration likely induces oxidative DNA damage (19), but lower concentrations did not induce significant cell death regardless of MEF genotype. Thus, the survival of MEFs to these TMZ concentrations may be a result of competing prosurvival and pro-death activities of MUTYH. Mutyh−/− MEFs have increased survival to MNNG and decreased survival to 2 mm MMS in parallel wells of the same set of experiments (p < 0.001, Fig. S1).
MUTYH interacts with mismatch repair (MMR) proteins in both bacteria and mammalian cells (21, 22), and alkylating agent cytotoxicity depends on O6-methylguanine (O6mG) lesions and the MMR pathway (23, 24). To test for MUTYH involvement in the repair of O6mG lesions, we used the MGMT inhibitor O6-benzylguanine (BG), which leads to increased levels of O6mG in genomic DNA and MMR-dependent cell death (25). Although BG treatment altered MNNG cytotoxicity, the effect of BG and MUTYH status was independent and did not interact, evidence that MUTYH does not alter MNNG survival via interactions with O6mG lesion repair (Fig. S2).
MUTYH and its homologs are adenine glycosylases that have strong preferences for OG in the opposite strand, but they will also remove A in G:A and C:A mispairs (26–29). To test the hypothesis that mammalian Mutyh can remove adenine mispaired opposite O6mG lesions formed after MNNG treatment, we conducted in vitro glycosylase cleavage assays as described previously (30) using purified mouse Mutyh enzyme. A 30-bp dsDNA duplex containing a centrally-located O6mG:A lesion was treated with Mutyh under single-turnover (STO) conditions ([DNA] < [enzyme]) for 30 min at 37 °C (Fig. S3); however, no excision activity was detected under these conditions.
Mammalian Mutyh does not possess methyl-base glycosylase activity in vitro
To our knowledge, the ability of MUTYH to excise methylated bases has not been tested. Because MUTYH-mediated strand scission can enhance cell death (17, 32), one possible mechanism for MUTYH-dependent, MNNG-induced toxicity could be the recognition and excision of a methylated base. To test this, we adapted a LC-tandem MS (LC-MS/MS) approach (33) to detect and quantify glycosylase-dependent methyl-base excision from MNNG-treated calf thymus genomic DNA. This approach gave sufficient sensitivity to detect five methylpurine adducts predominantly formed by MNNG in the low picomole range: O6mG, 7-methylguanine (7mG); 7-methyladenine (7mA); 3-methyladenine (3mA); and 1-methyladenine (1mA).
To gauge the extent of methyl-base damage after in vitro MNNG treatment of genomic DNA, samples were treated with 5 n HCl to hydrolyze acid–labile lesions (Fig. 4, 1st panel). LC-MS/MS analysis predominantly detected 1mA, 3mA, and 7mG lesions, with 7mG and 3mA comprising ∼75% of the total methylated purines, similar to previously reported values (34). AAG, the only known glycosylase in humans to excise alkylated purine bases (14), was used as a positive control for methylpurine excision activity.
Figure 4.

Recombinant Mutyh does not possess methyl-base glycosylase activity. The 1st five panels are representative LC traces for each condition (HCl, no enzyme (No Enz, negative control), AAG (positive control), WT Mutyh, and D207N Mutyh), with signal intensity monitored by MS counts. The last panel is a bar graph summarizing the detection of methylated bases quantified by LC-MS/MS for each experimental condition. All reactions were performed in experimental triplicates, and error bars represent the standard deviation from the average.
To test for Mutyh-specific glycosylase activity, WT Mutyh and a catalytically-inactive variant, D207N Mutyh, were used. Mutyh Asp-207, homologous to Asp-222 in humans and Asp-144 in Geobacillus stearothermophilus MutY, is required for catalysis of adenine excision (35, 36). The corresponding variants in G. stearothermophilus (D144N) and Escherichia coli (D138N) have high affinity for OG:A mispairs similar to WT enzyme (35, 37), and D222N MUTYH has no detectable glycosylase activity (38). After 1 h at 37 °C, AAG excised predominantly 3mA and 7mG as expected, with no detectable activity for the other lesions tested (Fig. 4 and supplementary Table S1). Minimal amounts of released methylpurine bases were detected in reactions with purified Mutyh enzymes, similar to levels observed in control reactions with no enzyme.
To support these results, we tested for unbiased cleavage of methylated lesions by implementing the in vitro glycosylase cleavage assay described above. MNNG-damaged DNA was treated with either purified AAG or Mutyh under STO conditions for 30–60 min at 37 °C (Fig. S4). AAG was found to excise multiple methylpurine lesions, with no excision activity detected for Mutyh, corroborating results from the LC-MS/MS assay.
Taken together, these data do not support methylpurine glycosylase activity in MUTYH-dependent MNNG cytotoxicity. One possibility that cannot be completely ruled out is that a rare DNA methyl adduct is a substrate for MUTYH or that MUTYH is involved in the repair of methyl-phosphodiester lesions, which are reported to form 12–17% of the DNA lesions by MNNG (34).
AP site blocker OTX abolishes the enhanced MNNG survival of Mutyh−/−MEFs
BER glycosylases bind their AP site products tightly, and MUTYH binds the AP site analog THF opposite OG with sub-nanomolar affinity (39). Methoxyamine (MX) potentiates the cytotoxicity of alkylating agents by forming a reversible covalent bond with AP sites (40–42), and it is currently being evaluated in clinical chemotherapy trials (43). The small molecule OTX forms covalent bonds by a similar mechanism to MX (44) and is neutral at physiological pH; thus, it does not introduce confounding neutralization salts into experimental conditions. To determine whether MUTYH-mediated cytotoxicity to MNNG is mediated by AP site interactions, we pretreated WT and Mutyh−/− MEFs with OTX before MNNG treatment and measured differences in cell survival. We found that OTX treatment had virtually no effect on the survival of WT MEFs to MNNG (7.8% versus 8.5% survival, not significant), but that OTX abolished the enhanced survival of Mutyh−/− MEFs to MNNG (21.1 to 5.8% survival), p = 0.005 (Fig. 5A). The complete restoration of WT levels of MNNG toxicity in Mutyh−/− MEFs by the addition of OTX, combined with the complete lack of additive OTX toxicity in WT MEFs, is evidence that both MUTYH and OTX enhance the toxicity of MNNG by a common molecular mechanism.
Figure 5.

Evidence of AP site interactions in MUTYH-mediated MNNG toxicity. A, cell survival data of WT and Mutyh−/− MEFs treated with 33 μm MNNG with and without 3 mm OTX or with OTX alone. The enhanced survival of Mutyh−/− MEFs to MNNG is abolished by OTX (1st column versus 2nd column, p = 0.005) but has no effect on WT MEFs (4th column versus 5th column). OTX alone is significantly more toxic to Mutyh−/− versus WT MEFs (3rd column versus 6th column, p = 0.011). B, ARP assay quantification of AP sites in genomic DNA extracted from MEFs treated with MNNG versus untreated control. There was a significant increase in reactive AP sites in Mutyh−/− MEFs (1st column versus 2nd column) but not in WT MEFs upon MNNG treatment (p < 0.001, t test). C, diagram summarizing experimental results, which highlights the lack of an additive effect between the small molecule OTX and MUTYH, suggesting they potentiate MNNG cell death by a similar mechanism.
Cellular DNA accumulates a significant number of AP sites without exogenous damaging agents (45). If MUTYH interacts with AP sites in genomic DNA, we hypothesized that OTX alone may be more toxic in Mutyh−/− MEFs versus WT MEFs. The data in Fig. 5A confirm this, with 21.1% Mutyh−/− MEF survival versus 51.0% WT MEF survival to OTX alone (p = 0.011). Indeed, Mutyh−/− MEFs survival to OTX alone is similar to treatment with 33 μm MNNG alone (23.4% versus 21.1%), evidence that AP site interactions are a significant contributor to OTX/MNNG toxicity in Mutyh−/− MEFs, and as expected, the combined treatment is synergistic (5.8% survival). Importantly, MUTYH-dependent differences in survival to the AP site blocker OTX without an alkylating agent present is evidence that alkylation per se does not underlie MUTYH toxicity. These data suggest that MUTYH interactions with AP sites enhance MNNG cytotoxicity.
MUTYH status alters cellular AP site processing in response to MNNG
To investigate the effect of MUTYH on AP site processing in cellular genomic DNA after MNNG treatment, we measured genomic AP sites using an aldehyde-reactive probe (ARP) assay (46), where a biotin-labeled small molecule reacts with the free aldehyde form of AP site intermediates. We tested WT versus Mutyh−/− MEF genomic DNA extracted 24 h after treatment with 67 μm MNNG or control buffer, as detailed under “Experimental procedures.” There was a significant increase in detection of reactive AP sites in Mutyh−/− MEFs but not in WT MEFs upon MNNG treatment shown in Fig. 5B (p < 0.001, t test). The higher survival to MNNG in Mutyh−/− MEFs is associated with higher levels of free AP sites after 24 h. These repair intermediates have not been processed by downstream AP endonucleases such as APE1, which catalyze the formation of DNA single-strand breaks that can be cytotoxic (41). These data are consistent with previous work that demonstrates that MUTYH enhances APE1 activity (47, 48), as the presence of MUTYH leads to both fewer unprocessed AP sites and enhanced MNNG cytotoxicity.
Mammalian Mutyh does not form Schiff base cross-links with AP site-containing DNA
The high-binding affinity of MUTYH to AP sites (39), the differential effect of OTX with MUTYH present, and the MUTYH-associated cytotoxicity of MEFs to MNNG led us to speculate that MUTYH may be forming cytotoxic covalent cross-links to AP sites in mammalian cells, similar to the mechanism of OTX adduct formation. Previous studies have shown that the slow dissociation of E. coli MutY from AP sites gives sufficient time for lysine residues near the enzyme-active site to form transient Schiff base cross-links with the AP site ribose sugar (49–51). This is typical of bifunctional glycosylases, but with MutY, further β or β-δ elimination leading to strand scission is extremely weak and possibly an artifact of heat treatment (51, 52), consistent with classification of MutY as a monofunctional glycosylase (51).
To test for the ability of mammalian MutY homologs to form Schiff base intermediates with DNA, we implemented in vitro trapping assays to reduce and separate covalently cross-linked complexes by denaturing gel electrophoresis using Mutyh. Importantly, the primary lysine involved in MutY cross-linking at position 142 (49) is not conserved in mammalian enzymes. However, Lys-20 in MutY has also been implicated in cross-linking (53), and the homologous MUTYH position is highly conserved in mammalian species, including humans (Lys-94) and mice (Lys-79). Cross-linking experiments between MutY enzymes and 30-bp duplexes containing an OG:A lesion were performed as described previously (49, 51, 53) using either NaBH4 or NaCNBH3 as reducing agents (Fig. 6). When OG:A-containing DNA was treated with excess E. coli MutY in the presence of 90 mm reducing agent, ∼80% of the DNA was trapped in a covalent complex with enzyme in a concentration-dependent manner, similar to previously reported findings. Under the same conditions, WT MUTYH was not found to form trappable complexes with AP site-containing DNA, post-adenine excision (Fig. 6). Similar results were found with MNNG-treated 30-bp duplexes with T:A in place of OG:A. Taken together, these data establish that mammalian MutY enzymes do not form transient Schiff base intermediates with AP sites in vitro.
Figure 6.

Trapping of transient Mutyh–DNA complexes with reducing agents. In vitro cross-linking assays with bacterial and mammalian MutY enzymes with OG:A-containing 30-bp DNA (A) or MNNG-treated T:A-containing 30-bp DNA (B) using 90 mm of either NaBH4 or NaCNBH3 as reducing agents. A, 1st and 2nd lanes, DNA with E. coli MutY and NaBH4; 3rd lane, DNA E. coli MutY and NaCNBH3; 4th to 6th lanes are the same conditions as the 1st to 3rd lanes, but with WT mouse Mutyh. B, 1st lane, no enzyme control; 2nd lane, DNA with mouse Mutyh; 3rd lane, DNA with mouse MUTYH and NaBH4. C, proposed mechanism for formation of a transient Schiff base intermediate between MutY and substrate DNA, followed by reductive “trapping” of the enzyme–DNA complex.
Mouse Mutyh binds to AP sites analogs opposite all four canonical bases
We hypothesized that MUTYH recognition of AP sites and subsequent downstream processing by APE1 could explain MUTYH-mediated cytotoxicity to MNNG. Jansson et al. (10) have previously reported that Schizosaccharomyces pombe MUTYH homolog, Myh1, accumulates and tightly binds to chromatin in cells damaged with the chemotherapeutic phleomycin. Notably, phleomycin exerts its cytotoxic effects through a free radical-based mechanism that generates oxidized AP sites opposite purines, subsequently leading to DNA strand breaks (54). To measure Mutyh binding affinity to AP sites, we employed a fluorescence polarization-based assay (55, 56) to measure apparent dissociation constants (Kd) between Mutyh and a fluorescently-tagged 30-bp DNA containing a centrally-located base opposite the AP site analog tetrahydrofuran (THF), under conditions where [DNA] < [enzyme]. With increasing enzyme concentrations, the rate at which free fluorescently-tagged DNA tumbles in solution is reduced by bound enzyme, thus increasing the polarization. The experimentally measured polarization depends on the polarization of both the free and bound DNA, as well as the fraction of the two species present in solution for a given enzyme concentration. Fitting these data to a one-site binding isotherm enabled us to determine the enzyme concentration at which the substrate is 50% bound or the apparent Kd value. A representative plot illustrating the binding of Mutyh to 30-bp A:THF- and T:THF-containing duplex DNA is shown in Fig. 7.
Figure 7.

Mutyh exhibits high affinity for AP sites. Representative fluorescence polarization data used for apparent Kd determination with Mutyh and X:THF-containing DNA, where X = T or A. Table 1 lists the experimentally determined apparent Kd values for a given base pair context.
The apparent dissociation constants determined via this assay are summarized in Table 1. Mutyh's affinity for G:THF-containing duplexes was found to be similar to that previously published using electrophoretic mobility shift assays (39). Mouse MUTYH showed a slight preference for binding to THF opposite purines over pyrimidines, which is expected given the enzyme's native activity toward A:OG lesions in the cell. However, we did not anticipate a narrow distribution of apparent Kd values for all four bases opposite THF (∼50–140 nm), especially when compared with values we report for nonspecific, undamaged DNA in the same sequence (>600 nm). Initial binding titrations with D207N mouse Mutyh with the same THF duplex using fluorescence polarization indicated that affinity that was too high to be measured accurately based on experimental limitations of the required DNA concentration. To more accurately assess the binding affinity between D207N Mutyh and THF-containing DNA, an electrophoretic mobility shift assay (EMSA) was employed that allowed use of a lower DNA duplex concentration ([DNA] < Kd) (39, 57). Notably, both methods provide similar values for WT Mutyh with a G:THF duplex. The apparent Kd values determined via EMSA for D207N Mutyh are reported in Table 1. Interestingly, D207N was found to bind THF-containing DNA ∼5-, 50-, and 300-fold tighter than WT MUTYH for THF opposite T, C, and G, respectively; presumably due to a gain of electrostatically favorable interactions between the asparagine amide side chain and the reduced abasic site analog.
Table 1.
Apparent dissociation constants (Kd) for WT and D207N Mutyh with DNA containing an abasic site analog (THF)
All values represent the average of three separate experiments, and the error is the standard deviation from the average.
| Central bp | Mutyh | D207N |
|---|---|---|
| nm | nm | |
| G:THF | 50 ± 16a,b | 0.16 ± 0.04c |
| C:THF | 110 ± 30a | 2.3 ± 1.8c |
| T:THF | 137 ± 7a | 23 ± 11c |
| A:THF | 80 ± 10a | NDd |
| OG:THF | <0.05c,e | NDd |
| G:C | > 600a | > 600c |
a Experimental values measured using fluorescent polarization assays as detailed under “Experimental procedures.” The DNA duplex concentration was 5 nm for all experiments.
b Value determined using fluorescence polarization is similar to that for electrophoretic mobility shift assays determined previously (60 ± 10 nm) by Pope et al. (39).
c Experimental values measured using electrophoretic mobility shift assays as detailed under “Experimental procedures.” DNA duplex concentration was 5 pm for all experiments.
d ND means not determined.
e Data were previously determined by Pope et al. (39).
APE1 activity is stimulated by WT Mutyh, but not D207N Mutyh
A “passing of the baton” model has often been used to describe the cascade of events involved between BER enzymes, where one enzyme processes its DNA substrate to produce an intermediate complex and that intermediate is then handed off to the next enzyme in the pathway (58). MUTYH amino acids 295–318 make specific physical interactions with APE1 (59), which stimulates both MUTYH turnover (60, 61) and APE1 excision activity (48). Furthermore, interactions between MUTYH and APE1 are dramatically weakened upon mutation of APE1 Asn-212, an active-site residue critical to abasic site recognition (48, 62).
Previous work showed that truncated MUTYH enhances the activity of APE1 (48). We adapted the above-described glycosylase assay to measure the initial rate of APE1 incision of THF-containing DNA, either alone or in the presence of full-length Mutyh bound to substrate DNA. The initial rate (νo) of APE1-dependent incision was determined with THF-containing substrates in EDTA-free reaction buffer supplemented with 10 mm MgCl2 under multiple-turnover conditions ([DNA] > [APE1]) (Fig. 8 and Table 2). Next, THF-containing DNA was preincubated with an equimolar amount of DNA glycosylase for 10 min at 37 °C, in the absence of APE1. APE1 was then added to the glycosylase/DNA mixture to initiate AP site incision that was monitored as a function of time, and the impact on νo was assessed. These experiments sought to compare how APE1 activity is either stimulated or deterred in the presence of a given glycosylase. The results from these assays are summarized in Fig. 8 and Table 2.
Figure 8.
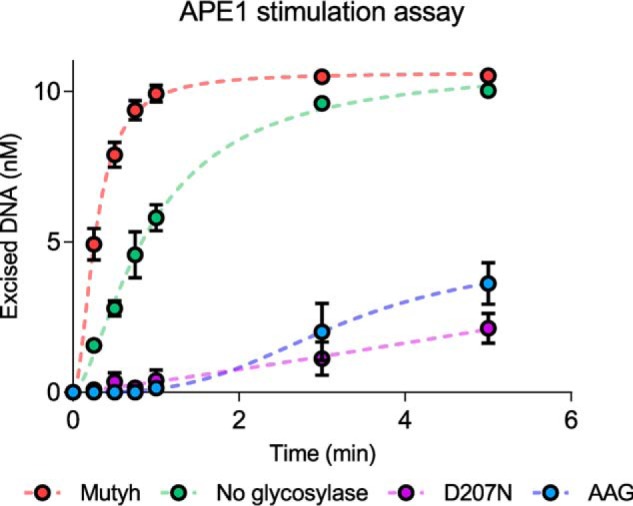
Mutyh stimulates APE1 activity. Representative data for in vitro APE1 stimulation assays performed with either glycosylase-free or with glycosylase-bound duplex DNA containing a centrally-located T:THF lesion. Reactions were conducted at 37 °C for 40 min under multiple turnover conditions and quenched by the addition of 0.2 m NaOH at preselected time points, as described under “Experimental procedures.” Dashed lines are included for illustration purposes, and do not represent fits to experimental data.
Table 2.
Observed rate constants (ν0) determined for APE1 with X:THF-containing duplex DNA substrate (where X = G, C, or T)
Duplex DNA was preincubated with no additional enzyme (APE1 alone) or with the DNA glycosylase specified below. Reported rate constants have been averaged over three separate experiments, and the error is the standard deviation from the average. All reactions were performed at 37 °C under multiple-turnover conditions ([DNA] > [enzyme]) with EDTA-free buffer supplemented with 10 mm MgCl2.
| Enzyme | T:THF |
C:THF |
G:THF |
|||
|---|---|---|---|---|---|---|
| ν0(min−1) | Fold-change | ν0(min−1) | Fold-change | ν0(min−1) | Fold-change | |
| APE1 alone | 2.7 ± 0.2 | 0.2 ± 0.02 | 0.4 ± 0.03 | |||
| APE1 with WT Mutyh | 8.9 ± 0.5 | 3.3 | 0.4 ± 0.03 | 2 | 0.7 ± 0.02 | 1.8 |
| APE1 with D207N Mutyh | 0.4 ± 0.05 | 0.1 | 0.1 ± 0.005 | 0.5 | ||
| APE1 with AAG | 0.8 ± 0.007 | 0.3 | 0.2 ± 0.02 | 1 | ||
APE1 activity was found to be enhanced by ∼3-fold in the presence of Mutyh-bound T:THF-containing DNA when compared with the measured activity for APE1 acting on T:THF-containing DNA alone, consistent with previous work (48). Surprisingly, APE1 activity was found to be inhibited in the presence of the same concentration of D207N Mutyh bound to the same DNA substrate, reducing the observed rate of excision 16-fold, consistent with the observed tighter binding of D207N shown in Table 1. Furthermore, a similar inhibition was observed when measuring APE1 activity on AAG-bound DNA, resulting in an approximate 11-fold reduction in measured activity. These results are intriguing given that Asp-207 of Mutyh (Asp-222 of human MUTYH) is not known to participate in the hand-off process between Mutyh-bound AP sites and APE1, although D207N binding to THF-containing DNA is significantly tighter than WT Mutyh binding. Furthermore, the Mutyh-dependent stimulation of APE1 activity appears to be robust, as similar rate enhancements in the presence of Mutyh were observed for a 30-bp DNA duplex containing THF paired opposite C or G, albeit with weaker overall APE1 activity observed when compared with THF incision opposite T. These results highlight a novel relationship between WT Mutyh-specific enhancement of APE1 activity that is relatively indiscriminate of base pair context.
WT MUTYH enhanced MNNG cytotoxicity, but D222N MUTYH does not
The above biochemical experiments with purified Mutyh and APE1 indicate that there are specific MUTYH–APE1 interactions that enhance APE1 strand scission activity at AP sites but that this effect is abolished with the D207N variant. To test whether there are differences in the ability of WT versus the catalytically-inactive human MUTYH to stimulate MNNG cell death, we compared MNNG survival of Mutyh−/− MEFs expressing the equivalent D222N human MUTYH isoform (Fig. 9A). Western blotting verified that protein expression of D222N cell lines assayed was similar to MUTYH-High cell lines (Fig. 9A, inset). Survival of D222N MEFs was not significantly different from the parental Mutyh−/− MEFs, 34% versus 32.5%, respectively, but it was significantly higher versus matched transgenic expression of WT MUTYH (p < 0.005; Fig. 9A), 5th column versus 4th column). Thus, MUTYH enhancement of MNNG cytotoxicity is not present when the D222N MUTYH isoform is expressed.
Figure 9.

MNNG-dependent cytotoxicity depends on MUTYH catalytic residue Asp-222 and leads to genomic DNA strand breaks. A, survival to low MNNG (67 μm) in Mutyh−/− MEF stable cell lines expressing recombinant D222N (5th column) versus WT MUTYH (4th column). Data from Fig. 2A are represented in the 1st to 4th columns. Inset: Western blotting of WT MUTYH expression from Fig. 2B versus D222N stable cell line MUTYH expression. There is significantly higher survival in cells expressing D222N versus WT MUTYH (4th column versus 5th column, **, p < 0.005, t test, significant with Bonferroni correction for multiple comparisons). B, MUTYH increases genomic strand breaks in MNNG-treated MEFs as quantified by alkaline gel electrophoresis. Genomic DNA was extracted 40 min after MNNG treatment, quantified, incubated in an alkaline loading buffer, and run on a 0.8% agarose gel overnight at pH 12.4 as detailed under “Experimental procedures.” M = molecular weight marker. C, quantification of total sample band density in the lower region (tail) versus the upper band from two biological replicates (five gels). WT cells had significantly more DNA in the lower region at 67 μm versus Mutyh−/− cells (**, t test, p < 0.01), indicative of increased strand breaks in WT versus Mutyh−/− MEFs. Trial 5 is shown, and all other gels and data are shown in Fig. S5.
MUTYH expression increases MNNG-induced genomic strand breaks
Mutyh enhances APE1 strand scission activity in vitro (Fig. 8). To test whether cellular MUTYH increases MNNG-induced DNA strand breaks, we employed alkaline gel electrophoresis of DNA extracted from MEFs 40 min after MNNG treatment (44). There is a visible increase in the migration of genomic DNA with increasing MNNG concentrations, evidence of enhanced DNA strand breaks (Fig. 9B). DNA from WT MEFs have a visibly greater amount of this migration versus Mutyh−/− MEFs, and quantification of DNA in the lower “tail” region versus the primary upper band is significantly increased in WT versus Mutyh−/− MEFs at 67 μm MNNG (Fig. 9C and Fig. S5).
Discussion
In an unbiased whole-genome analysis of gene expression, Fry et al. (8) found an association between high MUTYH expression and reduced MNNG survival in immortalized human lymphocytes. Studies with small-molecule AP site blockers (41, 63, 64) and the genetic alteration of alkyl-DNA glycosylases (65, 66) indicate dysregulation of AP site processing can mediate significant cytotoxicity, particularly upon alkylating agent treatment (67). A common theme in studies of the role of AP sites in alkylating agent cytotoxicity is the delicate balance of AP site repair factors, because the enhancement of the prosurvival activity of AAG and APE1 can lead to the accumulation of cytotoxic AP site repair intermediates, PARP activation, and cell death (42, 66, 68–70). There is also evidence that MNNG induces cell death independent of MMR via dsDNA breaks formed by closely-spaced single-strand breaks induced by BER intermediates (23).
Mutyh has high affinity for the AP site mimic THF across all base-pairing contexts (Fig. 7 and Table 1), and Mutyh enhances APE1-mediated strand scission at these sites by ∼3-fold (Fig. 8). Previous work found APE1 strand scission activity was stimulated by ∼3-fold with an N-terminal MUTYH fragment (48), independent confirmation of a highly significant effect. The higher survival of MEFs to OTX alone with MUTYH present (Fig. 5A) suggests a positive role for MUTYH in AP site processing. Lack of MUTYH significantly reduces genomic DNA strand breaks induced by MNNG after 40 min (Fig. 9, A and B), but it increases unresolved AP site intermediates after 24 h (Fig. 5B), both data consistent with the model that MUTYH has a significant role in enhancing AP site processing. We propose that MUTYH potentiates alkylating agent cytotoxicity via an enhancement of AP site processing that would subsequently overwhelm downstream repair factors (Fig. 10).
Figure 10.

Proposed model of MUTYH-mediated enhancement of MNNG cytotoxicity, with and without OTX.
There is increasing evidence that AP sites are oxidized, modified, and form DNA–DNA and DNA–protein cross-links that are biologically relevant (71–75). Both the NEIL3 glycosylase and AAG help resolve inter-strand cross-links (76, 77), and the newly-discovered HMCES protein shields AP sites from cytotoxic processing (78). Our data do not rule out the possibility that specific AP site modifications may be preferentially bound by MUTYH or even modified within the MUTYH active site. There is evidence that polymerase β can only process unmodified 5′-dRP intermediates via short-patch BER, and other modified intermediates must be processed by long-patch BER (79). MUTYH has been shown to channel bound AP sites into the long-patch base excision repair pathway (80), and interacts with both proliferating cell nuclear antigen (PCNA) and replication protein A (RPA) (59), so one speculative possibility is that these interactions play a positive role in channeling specifically-modified forms of AP sites away from nonproductive short-patch BER.
An interesting alternative hypothesis is that MUTYH possesses glycosylase activity on methylated bases. This is supported by evidence that MUTYH cytotoxicity is abolished in the catalytically-inactive variant (Fig. 9A), but MS analysis of MNNG-treated DNA incubated with Mutyh did not detect methyl-base excision activity (Fig. 4). Our cellular and biochemical data also appear to contradict this model. First, if MUTYH-mediated MNNG cytotoxicity was due to methyl-base glycosylase activity, this would not explain why OTX alone is more toxic in cells without MUTYH. Second, MUTYH should create more AP sites and thus would be predicted to enhance OTX toxicity, but this does not occur (Fig. 5A). Also, MUTYH expression is associated with fewer AP sites 24 h after MNNG treatment (2nd column versus 4th column in Fig. 5B). These data support a model where AP site resolution is enhanced by MUTYH, which contradicts a model where MUTYH creates new AP sites.
The lack of MNNG-induced cytotoxicity due to D222N MUTYH expression, together with the lack of APE1 enhancement by the purified mouse D207N variant, is consistent with molecular evidence that APE1 and MUTYH have a tightly-regulated interaction. Importantly, the purified D207N protein has higher affinity to AP site analogs versus the WT enzyme (Table 1), but expression of the analogous human D222N variant does not induce a cytotoxic effect (Fig. 9A), suggesting that passive blockade of AP sites by MUTYH does not underlie its cytotoxic effect. Luncsford et al. (48) demonstrate that MUTYH–APE1-binding interactions depend on APE1 residues Asn-212 and Gln-137, which are paradoxically on the opposite sides of the APE1 structure. Asn-212 sits directly in the APE1-active site (62), suggesting dynamic interactions between MUTYH and APE1 are occurring. Furthermore, MUTYH interactions with Hus1 enhance APE1–MUTYH interactions (48), and Hus1 interactions with MUTYH are abolished by two Hus1 amino acid changes (13). These results indicate that protein–protein interactions between MUTYH, APE1, and Hus1 appear to be easily disrupted by subtle molecular changes, suggesting that the MUTYH D222N active-site mutant could also alter APE1 interactions. Taken together with cellular data supporting the interaction of MUTYH with AP sites, these data suggest MUTYH and APE1 are intimately linked in the processing of AP site damage.
Inherited variants of MUTYH lead to colorectal cancer via the slow accumulation of single base mutations due to oxidative DNA damage (1, 4, 7). Surprisingly, there is also clinical and cellular evidence that MUTYH deficiency leads to chromosomal aberrations (81–84). Our data suggest MUTYH may have a positive role in resolving DNA strand break intermediates not limited to oxidative DNA damage. This leaves open a speculative possibility that germ line variants in MUTYH that alter APE1–MUTYH interactions may promote tumorigenesis via a pathway distinct from defects in oxidative DNA damage repair. Furthermore, MUTYH rapidly initiates PARP1 activation upon oxidative DNA damage treatment (17, 85, 86), although the exact molecular mechanism is unknown. This rapid activation of PARP1 is not consistent with MUTYH activity on OG:A substrates, because OG:A is not the direct result of guanine oxidation and is only created after a subsequent replication cycle (7). Thus, a speculative but simple explanation of the role of MUTYH in rapid PARP1 activation is that it is mediated by a separate function involving AP site processing, which is supported by evidence that MUTYH increases MNNG-induced DNA strand breaks (Fig. 9, B and C). PARP1 recognizes and binds to AP sites, but is not strongly activated until the 5′-dRP product of APE1 strand scission is created (87, 88). Given that alkylating agents primarily induce cell death via PARP1 activation (89), it logically follows that MUTYH stimulation of APE1 would enhance alkylating agent cytotoxicity.
Previous work has implicated MUTYH in the response to diverse forms of DNA damage beyond OG:A mispairs (9–13). Here, we have thoroughly investigated the interaction of MUTYH with MNNG damage, and we have found a common DNA repair intermediate to be central to MUTYH involvement. It is possible that MUTYH's role in AP site processing underlies a broader relationship between MUTYH and other forms of DNA damage and repair. For example, the recent report of MUTYH involvement in the repair of UV-induced damage (11) could possibly be explained by the formation of AP sites by UV irradiation (78). Given that AP site blockers are under investigation in cancer chemotherapy clinical trials (43), this suggests broader implications of MUTYH status in predicting clinical responses to DNA-damaging chemotherapeutics.
Experimental procedures
MEF cell lines
Mutyh−/− and Ogg−/− MEF cell lines were a kind gift from Yali Xie, and their creation was described previously (90). The presence of the targeted Mutyh deletion insert in exon 6 was verified via PCR analysis of genomic DNA, as shown in Fig. S6. Cell lines were incubated at 37 °C and 5% CO2, with high-glucose Dulbecco's modified Eagle's medium (Invitrogen), with 10% fetal bovine serum (FBS), 1% nonessential amino acids, and 1% GlutMax (Invitrogen). The 535-amino acid human MUTYH protein isoform (RefSeq NP_001041636.1 encoded by the type 1 (α3) mRNA isoform NM_001048171.1) was cloned into a pcDNA3.1 vector (Invitrogen) and was transiently transfected with Attractene transfection reagent (Qiagen) into Mutyh−/− MEFs. Cells were treated with 400–800 μg/ml G418 antibiotic, and after 7–14 days, single resistant colonies were isolated. MUTYH gene expression was then confirmed and evaluated through Western blotting using the Abcam 4D10 mouse mAb at 1:240 dilution and Santa Cruz Biotechnology β-actin mouse mAb sc-47778-HRP at 1:1000 dilution.
Cellular survival assays
4–16 h after plating 5–10 × 104 cells per well in six-well plates, cells were exposed to the indicated concentrations of MNNG. Pretreatment with OTX or BG was done 2 h before alkylating agent treatment as indicated. Cells were counted 6–7 days after addition of the alkylating agent, using trypan blue exclusion. Counts were normalized to untreated cells that were plated and grown in parallel.
Plasmid nicking assay
Plasmid DNA was prepared using standard midi-prep kits (Promega). Prior to the hOgg1 nicking assay, DNA was tested for pre-existing lesions by treatment with Fpg and Ogg1 enzyme in excess and purified as described previously (91, 92). Lack of endogenous DNA damage was verified by agarose gel electrophoresis to detect the presence of open-circular (nicked) plasmid DNA. These aliquots were then treated with MNNG concentrations as indicated in 400 mm Tris, pH 7.6, at 37 °C for 45 min. Typical enzyme treatment was in 20 mm Tris, 10 mm EDTA, 100 μg/ml BSA using 10 μm active fraction hOgg1 enzyme in a 30-μl reaction volume with 300 ng of 4.7-kb plasmid DNA and incubating at 37 °C for 10 min. 100 ng of DNA with 1× loading dye was run on a 1% agarose gel at 70 V for 1 h to differentiate closed-circular and open-circular plasmid DNA.
H2-DCFDA redox-sensitive probe assay
The DCF probe (Thermo Fisher Scientific, catalog no. D399) was dissolved in DMSO to 20 mm. MEF cells were incubated with 10 μm DCF probe for 30 min before DNA damage treatment (either MNNG or a hydrogen peroxide positive control) and harvested at time points after damage by incubation in 0.25% trypsin for 4 min, quenching with FBS, rinsed with ice-cold PBS twice, and then analyzed by flow cytometry versus untreated controls. The percent of cells with fluorescence above background was quantified by applying equal gating across the different conditions within a single experiment, and the values were normalized to the hydrogen peroxide–positive control.
Preparation of DNA substrates
6-Carboxyfluorescein (6-FAM) 5′-end–labeled DNA oligonucleotides were purchased from Eurofins Genomics, and the unlabeled DNA oligonucleotides were purchased from Integrated DNA Technologies. Oligonucleotides were purified via HPLC using a Dionex DNAPac PA-100 ion-exchange column on a Beckman Nouveau Gold HPLC system. One 30-nucleotide) DNA sequence was used in this study: 5′-CTGTAACGGGAGCTYGTGGCTCCATGATCG-3′, where Y = C, A, or an abasic site analog, THF. This strand was annealed to a 30-nucleotide complement strand: 5′-CGATCATGGAGCCACXAGCTCCCGTTACAG-3′, where X = G, C, T, A, or OG. For preparation of radiolabeled substrates, 5 pmol of the Y-containing DNA strand was 5′-end–labeled with 32P by adding 30 μCi of [γ-32P]ATP and T4 polynucleotide kinase (New England Biolabs). The radiolabeled oligonucleotides were purified using Illustra MicroSpin G-50 columns (GE Healthcare). Appropriate amounts of unlabeled Y-containing oligonucleotides were added to the radiolabeled strand and mixed with a 10% excess of the complementary strand in annealing buffer (20 mm Tris-HCl, pH 7.6, 10 mm EDTA, and 150 mm NaCl) to produce a final duplex concentration of 100 nm. 6-FAM–labeled duplexes were also mixed with 10% excess complementary strand in annealing buffer. DNA mixtures were incubated at 90 °C for 5 min, followed by slow cooling to 4 °C overnight. Additional details and reagents used provided in the Supplemental Materials and Methods.
Methylation of DNA substrates in vitro
DNA substrates for in vitro assays were damaged with MNNG as described previously (93). Briefly, 400 mm MNNG was prepared in DMSO and diluted with an equal volume of 30 mm sodium acetate, pH 5.0, to a final concentration of 200 mm. Typically, 8 μmol of MNNG was added to 1 mg/ml DNA in buffer (20 mm Tris-HCl, 20 mm NaCl, 20 mm DTT, 0.2 m EDTA) and incubated in the dark for 2 h at room temperature.
Expression and purification of recombinant WT and D207N murine Mutyh
Overexpression and purification of Mutyh proteins were performed as reported previously (39). A pET28a vector containing the Mutyh gene that expresses Mutyh with an N-terminal hexa-His tag was used. D207N Mutyh was constructed by introduction of a G–to–A mutation at position 644 in the Mutyh gene using the antisense primer 5′-GTAAAACGTTCCCATTCACCACACCGGTTAC-3′ followed by Kunkel mutagenesis (Transcriptic) and confirmed by DNA sequencing. Mutyh was overexpressed in BL21(DE3) cells. Purified protein fractions were run on an SDS-PAGE to ascertain purity and found to be greater than 95% pure. The concentration of total protein was determined using an estimated ϵ280 = 85,370 m−1 cm−1 (ExPASy), and the concentration of the iron–sulfur cluster was estimated using ϵ410 = 17,000 m−1 cm−1. WT Mutyh concentrations were corrected for percent activity (57). D207N Mutyh concentration was corrected to reflect the active-binding fraction using a binding titration assay with OG:A-containing 30-bp substrate (57).
LC-MS/MS assay
To assay for glycosidic cleavage of methylated bases from MNNG-damaged DNA in vitro, a PLC-MS/MS analysis was adapted from the Eichman lab (33). 1 mg of purified calf thymus DNA was damaged with MNNG as described above. The methylated DNA was then ethanol-precipitated and resuspended in 1 ml of Tris-EDTA, pH 8.0 (TE), buffer before being dialyzed against additional TE buffer three times at 4 °C to remove any spontaneously hydrolyzed DNA bases. 10 μg of the methylated calf thymus DNA was incubated with either 5 n HCl or 5 μm enzyme for 1 h at 37 °C. Enzymatic reactions contained 50 mm HEPES, pH 7.5, 100 mm KCl, 10 mm DTT, 2 mm EDTA, and 0.1 mg/ml BSA in a final 50-μl reaction volume. Reactions were terminated by addition of 50 μl of stop buffer (0.5 mg/ml salmon DNA, 1 mg/ml BSA, 1 m NaCl), followed by ethanol precipitation of the DNA. The supernatant was evaporated to dryness, and the residue was reconstituted in 50 μl of LC-MS grade water.
Separation and detection of the nucleobases were performed using an Agilent 6490 triple quadrupole mass spectrometer equipped with an Agilent 1290 infinity UHPLC system and an Agilent ZORBAX Eclipse Plus C18 column (2.1 × 50-mm inner diameter, 1.8-μm particle size). The column temperature was set at 40 °C. The aqueous mobile phase A used for UHPLC separation was LC-MS grade water with 0.1% formic acid (v/v). The organic mobile phase B was 90% acetonitrile with 0.1% formic acid in water (v/v). An optimized 6-min binary gradient was used: 0.0–2.0 min, 0% B; 2.0–3.0 min, 0–20% B; 3.0–4.0 min, 20–90% B; 4.0–4.9 min, 90% B; 4.9–5.0 min, 90–0% B; 5.0–6.0 min, 0% B. The flow rate was set at 0.3 ml/min. An electrospray ionization source was used and operated in positive ion mode. The MS source parameters were as follows: the drying gas temperature and sheath gas temperature were both set at 200 °C; drying gas flow rate and sheath gas flow rate were set at 11 and 7 liters/min, respectively; nebulizer pressure was set at 25 p.s.i.; the capillary voltage and fragmentor voltage were set at 1800 and 280 V, respectively. The nucleobases were quantified using multiple reaction monitoring (MRM) mode of the triple quadrupole mass spectrometer. Protonated species were monitored as the precursor ions of the analytes. Product ions for MRM transitions were determined by conducting a product ion scan for each analyte. The MRM transitions monitored were as follows: 166 → 134 for O6mG; 166 → 124 for 7mG; 166 → 149 for both O6mG and 7mG; 150 → 109 for 1mA; 150 → 123 for 3mA; 150 → 79 for 7mA; and 150 → 133 for both 1mA and 3mA. Data processing was performed using Agilent MassHunter Quantitative Analysis software.
DNA-protein cross-linking assay
To assay for transiently-formed Schiff base intermediates between enzymes and DNA substrates, an approach was used as described previously (49, 51, 53). Assays were performed using the DNA substrate duplexes described above, where Y = A, X = T or OG, and the Y-containing strand was 32P-5′-end–labeled. MNNG-damaged DNA duplexes were prepared as described above. 90 mm NaBH4 and NaCNBH3 was used to reduce and trap enzyme–DNA complexes.
ARP assay
To determine the number of AP sites present in genomic DNA, MEFs were treated with 67 μm MNNG or 300 mm Tris buffer (control) for 24 h, and DNA was isolated with DNAzol (Thermo Fisher Scientific) per the manufacturer's protocol. DNA was quantified by agarose gel electrophoresis and normalized for equal loading. AP sites were quantified versus supplied standards using kit DK02-12 from Dojindo Molecular Technologies, according to the manufacturer's protocol.
Fluorescence polarization assay
Quantitative fluorescence polarization assays (31, 56) were performed using the 6-FAM 5′-end–labeled DNA substrate duplexes described above, where Y = THF, and X = G, C, T, or A. Reactions contained 5 nm duplex, 20 mm Tris-HCl, pH 7.6, 100 mm NaCl, 1 mm EDTA, 1 mm DTT, 5% glycerol, 0.1 mg/ml BSA, and varying amounts of WT Mutyh in a reaction volume of 50 μl. Enzyme aliquots of varying concentrations were prepared at 4 °C with dilution buffer containing 20 mm Tris-HCl, pH 7.6, 10 mm EDTA, and 20% glycerol. Samples containing DNA and enzyme were incubated at 25 °C for 20 min. Samples were then distributed equally across a black, 384-well, low-flange microplate (Greiner Bio-One) before being subjected to fluorescence polarization analysis on a CLARIOstar microplate reader (BMG LABTECH, Germany) using the following scan settings: dichroic, F:LP 504; excitation, 482-16; emission, 530–540; target temperature, 25 °C. Focus and gain adjustments for each channel were made using a reference well containing 6-FAM-end–labeled DNA in buffer with no enzyme. Apparent Kd values were determined by fitting the data (percent bound substrate versus log[enzyme]) to the equation for one-site ligand binding using the program Grafit version 5.0.2 (Erithacus Software). Apparent Kd values were determined from three separate experiments.
Electrophoretic mobility shift assay
Quantitative electrophoretic mobility shift assays were performed as described previously (57). Reactions were performed using the same conditions as the above described fluorescence polarization except with 5 pm duplex DNA. Reported values for WT Mutyh binding to G:THF were similar to those previously reported (39) and were used as validation for direct comparison to values determined by fluorescence polarization.
APE1 stimulation assay
Assays for glycosylase-dependent APE1 stimulation were performed under multiple-turnover conditions ([DNA] ≫ [APE1]) in a manner similar to that described previously (48). 32P-5′-end–labeled 30-bp DNA duplexes were prepared as described above, where Y = THF and X = G, C, or T. Mutyh enzymes were purified as described above, whereas APE1 and AAG were commercially available (New England Biolabs). The reaction buffer contained 20 mm Tris-HCl, pH 7.6, 10 mm MgCl2, 100 μg/ml BSA, 30 mm NaCl, and 20 nm duplex DNA. Reaction mixtures were equilibrated at 37 °C for 5 min before the addition of 20 nm (Mutyh and D207N Mutyh) or 192 pm (AAG) glycosylase prepared in dilution buffer (20 mm Tris-HCl, 20% glycerol, pH 7.6) and equilibrated for an additional 10 min at 37 °C. APE1 was added to reaction tubes to initiate the time course at a final concentration of 33 pm. Aliquots were removed at designated times (15 s to 40 min) and quenched by the addition of 0.2 m NaOH with subsequent heating for 5 min at 95 °C before being analyzed by denaturing PAGE as described above. Product concentration was plotted as a function of time, and initial rates (νo) were determined by fitting the data using linear regression.
Alkaline gel electrophoresis of genomic DNA
Based on the work of Luke et al. (44), MEFs were treated with increasing concentrations of MNNG as indicated, and genomic DNA was extracted with DNAzol (Invitrogen) 40 min after treatment per the manufacturer's protocol. DNA was quantified by both UV absorbance at 260 nm and standard agarose gel electrophoresis and adjusted to a concentration of 200 ng/μl. Samples were denatured with a pH 12.8 alkaline loading buffer (100 mm NaOH, 50 mm HEPES) for 20 min at 37 °C and then mixed with standard 6× loading buffer. A 0.8% agarose gel was prepared with 50 mm NaCl and 1 mm EDTA and soaked in a mild alkaline running buffer (30 mm NaOH, 1 mm EDTA, pH 12.4) for at least 1 h. 10 μl (2 μg) of DNA was loaded in wells and run at 25–30 V overnight at 4 °C. The gel was neutralized in 400 mm Tris, pH 7.5, at room temperature for 30 min, then stained with EtBr or SYBR Safe for 1 h, briefly destained, and imaged. Quantification of strand breaks was completed using ImageQuant software (GE Healthcare).
Quantification and statistical analysis
Data are expressed as ±S.D. unless otherwise stated. Statistical tests were performed using the Student's t test (GraphPad) and analysis of variance (Vassarstats.net).
Author contributions
A. G. R., D. M. B., C. B. L., and S. S. D. conceptualization; A. G. R., D. M. B., X. M., and G. X. validation; A. G. R., D. M. B., X. M., G. X., A. N. R., and P. L. M. investigation; A. G. R., D. M. B., X. M., G. X., P. L. M., C. B. L., and S. S. D. methodology; A. G. R. and D. M. B. writing-original draft; A. G. R., D. M. B., C. B. L., and S. S. D. writing-review and editing; C. B. L. and S. S. D. resources; C. B. L. and S. S. D. software; C. B. L. and S. S. D. supervision; C. B. L. and S. S. D. funding acquisition; C. B. L. and S. S. D. project administration.
Supplementary Material
Acknowledgments
We thank Tracy Yang for assisting with MEF cell culture experiments; Jazmin Stenson for assisting with the ARP assay; Angela Monterrubio for Mutyh protein preparation for the O6meG:A assay; and Ryan Giordano for assistance with statistical analysis.
This work was supported by National Institutes of Health Grants 2RO1CA067985 (to S. S. D.) and RO1GM049077 (to C. B. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S6, Table S1, and supporting Materials and methods.
- MUTYH
- MutY human homolog
- BER
- base excision repair
- MNNG
- N-methyl-N′-nitro-N-nitrosoguanidine
- Mutyh
- MutY mouse homolog
- AP
- apurinic/apyrimidinic (abasic) site
- OG
- 8-oxoguanine
- MEF
- mouse embryonic fibroblast
- OTX
- O-(tetrahydro-2H-pyran-2-yl)-hydroxylamine
- OGG1
- OG glycosylase 1
- bp
- base pair
- ROS
- reactive oxygen species
- MMR
- mismatch repair
- ARP
- aldehyde reactive probe
- ANOVA
- analysis of variance
- H2DCFDA
- 2,7-dichlorodihydrofluorescein diacetate
- LC
- liquid chromatography
- THF
- tetrahydrofuran
- MAP
- MUTYH-associated polyposis
- MX
- methoxyamine
- PARP
- poly(ADP-ribose) polymerase
- 6-FAM
- 6-carboxyfluorescein
- TMZ
- temozolomide
- MGMT
- methylguanine methyltransferase
- BG
- O6-benzylguanine
- STO
- single-turnover
- AAG
- alkyl-adenine glycosylase
- hOgg1
- human OG glycosylase 1
- MMS
- methyl-methanesulfonate
- 7mG
- 7-methylguanine
- 7mA
- 7-methyladenine
- 3mA
- 3-methyladenine
- 1mA
- 1-methyladenine
- FBS
- fetal bovine serum
- UHPLC
- ultra-HPLC
- MRM
- multiple reaction monitoring.
References
- 1. Al-Tassan N., Chmiel N. H., Maynard J., Fleming N., Livingston A. L., Williams G. T., Hodges A. K., Davies D. R., David S. S., Sampson J. R., and Cheadle J. P. (2002) Inherited variants of MYH associated with somatic G:C→T:A mutations in colorectal tumors. Nat. Genet. 30, 227–232 10.1038/ng828 [DOI] [PubMed] [Google Scholar]
- 2. Cheadle J. P., and Sampson J. R. (2007) MUTYH-associated polyposis–from defect in base excision repair to clinical genetic testing. DNA Repair 6, 274–279 10.1016/j.dnarep.2006.11.001 [DOI] [PubMed] [Google Scholar]
- 3. David S. S., O'Shea V. L., and Kundu S. (2007) Base-excision repair of oxidative DNA damage. Nature 447, 941–950 10.1038/nature05978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Banda D. M., Nuñez N. N., Burnside M. A., Bradshaw K. M., and David S. S. (2017) Repair of 8-oxoG:A mismatches by the MUTYH glycosylase: mechanism, metals and medicine. Free Radic. Biol. Med. 107, 202–215 10.1016/j.freeradbiomed.2017.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Markkanen E., Dorn J., and Hübscher U. (2013) MUTYH DNA glycosylase: the rationale for removing undamaged bases from the DNA. Front. Genet. 4, 18 10.3389/fgene.2013.00018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Oka S., and Nakabeppu Y. (2011) DNA glycosylase encoded by MUTYH functions as a molecular switch for programmed cell death under oxidative stress to suppress tumorigenesis. Cancer Sci. 102, 677–682 10.1111/j.1349-7006.2011.01869.x [DOI] [PubMed] [Google Scholar]
- 7. Raetz A. G., and David S. S. (2019) When you're strange: unusual features of the MUTYH glycosylase and implications in cancer. DNA Repair 80, 16–25 10.1016/j.dnarep.2019.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fry R. C., Svensson J. P., Valiathan C., Wang E., Hogan B. J., Bhattacharya S., Bugni J. M., Whittaker C. A., and Samson L. D. (2008) Genomic predictors of interindividual differences in response to DNA damaging agents. Genes Dev. 22, 2621–2626 10.1101/gad.1688508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jansson K., Warringer J., Farewell A., Park H. O., Hoe K. L., Kim D. U., Hayles J., and Sunnerhagen P. (2008) The tumor suppressor homolog in fission yeast, myh1(+), displays a strong interaction with the checkpoint gene rad1(+). Mutat. Res. 644, 48–55 10.1016/j.mrfmmm.2008.07.001 [DOI] [PubMed] [Google Scholar]
- 10. Jansson K., Alao J. P., Viktorsson K., Warringer J., Lewensohn R., and Sunnerhagen P. (2013) A role for Myh1 in DNA repair after treatment with strand-breaking and crosslinking chemotherapeutic agents. Environ. Mol. Mutagen 54, 327–337 10.1002/em.21784 [DOI] [PubMed] [Google Scholar]
- 11. Mazouzi A., Battistini F., Moser S. C., Ferreira da Silva J., Wiedner M., Owusu M., Lardeau C. H., Ringler A., Weil B., Neesen J., Orozco M., Kubicek S., and Loizou J. I. (2017) Repair of UV-induced DNA damage independent of nucleotide excision repair is masked by MUTYH. Mol. Cell 68, 797–807.e7 10.1016/j.molcel.2017.10.021 [DOI] [PubMed] [Google Scholar]
- 12. Hahm S. H., Park J. H., Ko S. I., Lee Y. R., Chung I. S., Chung J. H., Kang L. W., and Han Y. S. (2011) Knock-down of human MutY homolog (hMYH) decreases phosphorylation of checkpoint kinase 1 (Chk1) induced by hydroxyurea and UV treatment. BMB Rep. 44, 352–357 10.5483/BMBRep.2011.44.5.352 [DOI] [PubMed] [Google Scholar]
- 13. Lim P. X., Patel D. R., Poisson K. E., Basuita M., Tsai C., Lyndaker A. M., Hwang B. J., Lu A. L., and Weiss R. S. (2015) Genome protection by the 9–1-1 complex subunit HUS1 requires clamp formation, DNA contacts, and ATR signaling-independent effector functions. J. Biol. Chem. 290, 14826–14840 10.1074/jbc.M114.630640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fu D., Calvo J. A., and Samson L. D. (2012) Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat. Rev. Cancer 12, 104–120 10.1038/nrc3185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Russo M. T., De Luca G., Casorelli I., Degan P., Molatore S., Barone F., Mazzei F., Pannellini T., Musiani P., and Bignami M. (2009) Role of MUTYH and MSH2 in the control of oxidative DNA damage, genetic instability, and tumorigenesis. Cancer Res. 69, 4372–4379 10.1158/0008-5472.CAN-08-3292 [DOI] [PubMed] [Google Scholar]
- 16. Chiu L. Y., Ho F. M., Shiah S. G., Chang Y., and Lin W. W. (2011) Oxidative stress initiates DNA damager MNNG-induced poly(ADP-ribose)polymerase-1-dependent parthanatos cell death. Biochem. Pharmacol. 81, 459–470 10.1016/j.bcp.2010.10.016 [DOI] [PubMed] [Google Scholar]
- 17. Oka S., Ohno M., Tsuchimoto D., Sakumi K., Furuichi M., and Nakabeppu Y. (2008) Two distinct pathways of cell death triggered by oxidative damage to nuclear and mitochondrial DNAs. EMBO J. 27, 421–432 10.1038/sj.emboj.7601975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hwang B. J., Shi G., and Lu A. L. (2014) Mammalian MutY homolog (MYH or MUTYH) protects cells from oxidative DNA damage. DNA Repair 13, 10–21 10.1016/j.dnarep.2013.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Svilar D., Dyavaiah M., Brown A. R., Tang J. B., Li J., McDonald P. R., Shun T. Y., Braganza A., Wang X. H., Maniar S., St Croix C. M., Lazo J. S., Pollack I. F., Begley T. J., and Sobol R. W. (2012) Alkylation sensitivity screens reveal a conserved cross-species functionome. Mol. Cancer Res. 10, 1580–1596 10.1158/1541-7786.MCR-12-0168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pastoriza-Gallego M., Armier J., and Sarasin A. (2007) Transcription through 8-oxoguanine in DNA repair-proficient and Csb(−)/Ogg1(−) DNA repair-deficient mouse embryonic fibroblasts is dependent upon promoter strength and sequence context. Mutagenesis 22, 343–351 10.1093/mutage/gem024 [DOI] [PubMed] [Google Scholar]
- 21. Bai H., and Lu A. L. (2007) Physical and functional interactions between Escherichia coli MutY glycosylase and mismatch repair protein MutS. J. Bacteriol. 189, 902–910 10.1128/JB.01513-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gu Y., Parker A., Wilson T. M., Bai H., Chang D. Y., and Lu A. L. (2002) Human MutY homolog, a DNA glycosylase involved in base excision repair, physically and functionally interacts with mismatch repair proteins human MutS homolog 2/human MutS homolog 6. J. Biol. Chem. 277, 11135–11142 10.1074/jbc.M108618200 [DOI] [PubMed] [Google Scholar]
- 23. Stojic L., Cejka P., and Jiricny J. (2005) High doses of SN1 type methylating agents activate DNA damage signaling cascades that are largely independent of mismatch repair. Cell Cycle 4, 473–477 10.4161/cc.4.3.1528 [DOI] [PubMed] [Google Scholar]
- 24. Stojic L., Mojas N., Cejka P., Di Pietro M., Ferrari S., Marra G., and Jiricny J. (2004) Mismatch repair-dependent G2 checkpoint induced by low doses of SN1 type methylating agents requires the ATR kinase. Genes Dev. 18, 1331–1344 10.1101/gad.294404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dolan M. E., Moschel R. C., and Pegg A. E. (1990) Depletion of mammalian O6-alkylguanine-DNA alkyltransferase activity by O6-benzylguanine provides a means to evaluate the role of this protein in protection against carcinogenic and therapeutic alkylating agents. Proc. Natl. Acad. Sci. U.S.A. 87, 5368–5372 10.1073/pnas.87.14.5368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lu A. L., Yuen D. S., and Cillo J. (1996) Catalytic mechanism and DNA substrate recognition of Escherichia coli MutY protein. J. Biol. Chem. 271, 24138–24143 10.1074/jbc.271.39.24138 [DOI] [PubMed] [Google Scholar]
- 27. McGoldrick J. P., Yeh Y. C., Solomon M., Essigmann J. M., and Lu A. L. (1995) Characterization of a mammalian homolog of the Escherichia coli MutY mismatch repair protein. Mol. Cell. Biol. 15, 989–996 10.1128/MCB.15.2.989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tsai-Wu J. J., Liu H. F., and Lu A. L. (1992) Escherichia coli MutY protein has both N-glycosylase and apurinic/apyrimidinic endonuclease activities on A.C and A.G mispairs. Proc. Natl. Acad. Sci. U.S.A. 89, 8779–8783 10.1073/pnas.89.18.8779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nghiem Y., Cabrera M., Cupples C. G., and Miller J. H. (1988) The mutY gene: a mutator locus in Escherichia coli that generates G.C—-T.A transversions. Proc. Natl. Acad. Sci. U.S.A. 85, 2709–2713 10.1073/pnas.85.8.2709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Porello S. L., Leyes A. E., and David S. S. (1998) Single-turnover and pre-steady-state kinetics of the reaction of the adenine glycosylase MutY with mismatch-containing DNA substrates. Biochemistry 37, 14756–14764 10.1021/bi981594+ [DOI] [PubMed] [Google Scholar]
- 31. Lundblad J. R., Laurance M., and Goodman R. H. (1996) Fluorescence polarization analysis of protein–DNA and protein–protein interactions. Mol. Endocrinol. 10, 607–612 10.1210/mend.10.6.8776720 [DOI] [PubMed] [Google Scholar]
- 32. Martin S. A., McCabe N., Mullarkey M., Cummins R., Burgess D. J., Nakabeppu Y., Oka S., Kay E., Lord C. J., and Ashworth A. (2010) DNA polymerases as potential therapeutic targets for cancers deficient in the DNA mismatch repair proteins MSH2 or MLH1. Cancer Cell 17, 235–248 10.1016/j.ccr.2009.12.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mullins E. A., Rubinson E. H., Pereira K. N., Calcutt M. W., Christov P. P., and Eichman B. F. (2013) An HPLC-tandem mass spectrometry method for simultaneous detection of alkylated base excision repair products. Methods 64, 59–66 10.1016/j.ymeth.2013.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wyatt M. D., and Pittman D. L. (2006) Methylating agents and DNA repair responses: methylated bases and sources of strand breaks. Chem. Res. Toxicol. 19, 1580–1594 10.1021/tx060164e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fromme J. C., Banerjee A., Huang S. J., and Verdine G. L. (2004) Structural basis for removal of adenine mispaired with 8-oxoguanine by MutY adenine DNA glycosylase. Nature 427, 652–656 10.1038/nature02306 [DOI] [PubMed] [Google Scholar]
- 36. Woods R. D., O'Shea V. L., Chu A., Cao S., Richards J. L., Horvath M. P., and David S. S. (2016) Structure and stereochemistry of the base excision repair glycosylase MutY reveal a mechanism similar to retaining glycosidases. Nucleic Acids Res. 44, 801–810 10.1093/nar/gkv1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wright P. M., Yu J., Cillo J., and Lu A. L. (1999) The active site of the Escherichia coli MutY DNA adenine glycosylase. J. Biol. Chem. 274, 29011–29018 10.1074/jbc.274.41.29011 [DOI] [PubMed] [Google Scholar]
- 38. Kundu S., Brinkmeyer M. K., Livingston A. L., and David S. S. (2009) Adenine removal activity and bacterial complementation with the human MutY homologue (MUTYH) and Y165C, G382D, P391L and Q324R variants associated with colorectal cancer. DNA Repair 8, 1400–1410 10.1016/j.dnarep.2009.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pope M. A., and David S. S. (2005) DNA damage recognition and repair by the murine MutY homologue. DNA Repair 4, 91–102 10.1016/j.dnarep.2004.08.004 [DOI] [PubMed] [Google Scholar]
- 40. Liu L., Markowitz S., and Gerson S. L. (1996) Mismatch repair mutations override alkyltransferase in conferring resistance to temozolomide but not to 1,3-bis(2-chloroethyl)nitrosourea. Cancer Res. 56, 5375–5379 [PubMed] [Google Scholar]
- 41. Liu L., Taverna P., Whitacre C. M., Chatterjee S., and Gerson S. L. (1999) Pharmacologic disruption of base excision repair sensitizes mismatch repair-deficient and -proficient colon cancer cells to methylating agents. Clin. Cancer Res. 5, 2908–2917 [PubMed] [Google Scholar]
- 42. Tang J. B., Svilar D., Trivedi R. N., Wang X. H., Goellner E. M., Moore B., Hamilton R. L., Banze L. A., Brown A. R., and Sobol R. W. (2011) N-Methylpurine DNA glycosylase and DNA polymerase β modulate BER inhibitor potentiation of glioma cells to temozolomide. Neuro-Oncol. 13, 471–486 10.1093/neuonc/nor011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Caimi P. F., Cooper B. W., William B. M., Dowlati A., Barr P. M., Fu P., Pink J., Xu Y., Lazarus H. M., de Lima M., and Gerson S. L. (2017) Phase I clinical trial of the base excision repair inhibitor methoxyamine in combination with fludarabine for patients with advanced hematologic malignancies. Oncotarget 8, 79864–79875 10.18632/oncotarget.20094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Luke A. M., Chastain P. D., Pachkowski B. F., Afonin V., Takeda S., Kaufman D. G., Swenberg J. A., and Nakamura J. (2010) Accumulation of true single-strand breaks and AP sites in base excision repair deficient cells. Mutat. Res. 694, 65–71 10.1016/j.mrfmmm.2010.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Atamna H., Cheung I., and Ames B. N. (2000) A method for detecting abasic sites in living cells: age-dependent changes in base excision repair. Proc. Natl. Acad. Sci. U.S.A. 97, 686–691 10.1073/pnas.97.2.686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kubo K., Ide H., Wallace S. S., and Kow Y. W. (1992) A novel sensitive and specific assay for abasic sites, the most commonly produced DNA lesion. Biochemistry 31, 3703–3708 10.1021/bi00129a020 [DOI] [PubMed] [Google Scholar]
- 47. Tominaga Y., Ushijima Y., Tsuchimoto D., Mishima M., Shirakawa M., Hirano S., Sakumi K., and Nakabeppu Y. (2004) MUTYH prevents OGG1 or APEX1 from inappropriately processing its substrate or reaction product with its C-terminal domain. Nucleic Acids Res. 32, 3198–3211 10.1093/nar/gkh642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Luncsford P. J., Manvilla B. A., Patterson D. N., Malik S. S., Jin J., Hwang B. J., Gunther R., Kalvakolanu S., Lipinski L. J., Yuan W., Lu W., Drohat A. C., Lu A. L., and Toth E. A. (2013) Coordination of MYH DNA glycosylase and APE1 endonuclease activities via physical interactions. DNA Repair 12, 1043–1052 10.1016/j.dnarep.2013.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Williams S. D., and David S. S. (1999) Formation of a Schiff base intermediate is not required for the adenine glycosylase activity of Escherichia coli MutY. Biochemistry 38, 15417–15424 10.1021/bi992013z [DOI] [PubMed] [Google Scholar]
- 50. Williams S. D., and David S. S. (2000) A single engineered point mutation in the adenine glycosylase MutY confers bifunctional glycosylase/AP lyase activity. Biochemistry 39, 10098–10109 10.1021/bi0004652 [DOI] [PubMed] [Google Scholar]
- 51. Williams S. D., and David S. S. (1998) Evidence that MutY is a monofunctional glycosylase capable of forming a covalent Schiff base intermediate with substrate DNA. Nucleic Acids Res. 26, 5123–5133 10.1093/nar/26.22.5123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zharkov D. O., and Grollman A. P. (1998) MutY DNA glycosylase: base release and intermediate complex formation. Biochemistry 37, 12384–12394 10.1021/bi981066y [DOI] [PubMed] [Google Scholar]
- 53. Manuel R. C., Hitomi K., Arvai A. S., House P. G., Kurtz A. J., Dodson M. L., McCullough A. K., Tainer J. A., and Lloyd R. S. (2004) Reaction intermediates in the catalytic mechanism of Escherichia coli MutY DNA glycosylase. J. Biol. Chem. 279, 46930–46939 10.1074/jbc.M403944200 [DOI] [PubMed] [Google Scholar]
- 54. Povirk L. F. (1996) DNA damage and mutagenesis by radiomimetic DNA-cleaving agents: bleomycin, neocarzinostatin and other enediynes. Mutat. Res. 355, 71–89 10.1016/0027-5107(96)00023-1 [DOI] [PubMed] [Google Scholar]
- 55. Prystay L., Gosselin M., and Banks P. (2001) Determination of equilibrium dissociation constants in fluorescence polarization. J. Biomol. Screen. 6, 141–150 10.1177/108705710100600304 [DOI] [PubMed] [Google Scholar]
- 56. Rossi A. M., and Taylor C. W. (2011) Analysis of protein–ligand interactions by fluorescence polarization. Nat. Protoc. 6, 365–387 10.1038/nprot.2011.305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Porello S. L., Williams S. D., Kuhn H., Michaels M. L., and David S. S. (1996) Specific recognition of substrate analogs by the DNA mismatch repair enzyme MutY. J. Am. Chem. Soc. 118, 10684–10692 10.1021/ja9602206 [DOI] [Google Scholar]
- 58. Wilson S. H., and Kunkel T. A. (2000) Passing the baton in base excision repair. Nat. Struct. Biol. 7, 176–178 10.1038/73260 [DOI] [PubMed] [Google Scholar]
- 59. Parker A., Gu Y., Mahoney W., Lee S. H., Singh K. K., and Lu A. L. (2001) Human homolog of the MutY repair protein (hMYH) physically interacts with proteins involved in long patch DNA base excision repair. J. Biol. Chem. 276, 5547–5555 10.1074/jbc.M008463200 [DOI] [PubMed] [Google Scholar]
- 60. Pope M. A., Chmiel N. H., and David S. S. (2005) Insight into the functional consequences of hMYH variants associated with colorectal cancer: distinct differences in the adenine glycosylase activity and the response to AP endonucleases of Y150C and G365D murine MYH. DNA Repair 4, 315–325 10.1016/j.dnarep.2004.10.003 [DOI] [PubMed] [Google Scholar]
- 61. Yang H., Clendenin W. M., Wong D., Demple B., Slupska M. M., Chiang J. H., and Miller J. H. (2001) Enhanced activity of adenine-DNA glycosylase (Myh) by apurinic/apyrimidinic endonuclease (Ape1) in mammalian base excision repair of an A/GO mismatch. Nucleic Acids Res. 29, 743–752 10.1093/nar/29.3.743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rothwell D. G., and Hickson I. D. (1996) Asparagine 212 is essential for abasic site recognition by the human DNA repair endonuclease HAP1. Nucleic Acids Res. 24, 4217–4221 10.1093/nar/24.21.4217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Liu L., Yan L., Donze J. R., and Gerson S. L. (2003) Blockage of abasic site repair enhances antitumor efficacy of 1,3-bis-(2-chloroethyl)-1-nitrosourea in colon tumor xenografts. Mol. Cancer Ther. 2, 1061–1066 [PubMed] [Google Scholar]
- 64. Taverna P., Liu L., Hwang H. S., Hanson A. J., Kinsella T. J., and Gerson S. L. (2001) Methoxyamine potentiates DNA single-strand breaks and double strand breaks induced by temozolomide in colon cancer cells. Mutat. Res. 485, 269–281 10.1016/S0921-8777(01)00076-3 [DOI] [PubMed] [Google Scholar]
- 65. Calvo J. A., Moroski-Erkul C. A., Lake A., Eichinger L. W., Shah D., Jhun I., Limsirichai P., Bronson R. T., Christiani D. C., Meira L. B., and Samson L. D. (2013) Aag DNA glycosylase promotes alkylation-induced tissue damage mediated by Parp1. PLoS Genet. 9, e1003413 10.1371/journal.pgen.1003413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Posnick L. M., and Samson L. D. (1999) Imbalanced base excision repair increases spontaneous mutation and alkylation sensitivity in Escherichia coli. J. Bacteriol. 181, 6763–6771 10.1128/JB.181.21.6763-6771.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bobola M. S., Kolstoe D. D., Blank A., Chamberlain M. C., and Silber J. R. (2012) Repair of 3-methyladenine and abasic sites by base excision repair mediates glioblastoma resistance to temozolomide. Front. Oncol. 2, 176 10.3389/fonc.2012.00176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Troll C. J., Adhikary S., Cueff M., Mitra I., Eichman B. F., and Camps M. (2014) Interplay between base excision repair activity and toxicity of 3-methyladenine DNA glycosylases in an E. coli complementation system. Mutat. Res. 763, 64–73 10.1016/j.mrfmmm.2014.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mutamba J. T., Svilar D., Prasongtanakij S., Wang X. H., Lin Y. C., Dedon P. C., Sobol R. W., and Engelward B. P. (2011) XRCC1 and base excision repair balance in response to nitric oxide. DNA Repair 10, 1282–1293 10.1016/j.dnarep.2011.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Rinne M. L., He Y., Pachkowski B. F., Nakamura J., and Kelley M. R. (2005) N-Methylpurine DNA glycosylase overexpression increases alkylation sensitivity by rapidly removing nontoxic 7-methylguanine adducts. Nucleic Acids Res. 33, 2859–2867 10.1093/nar/gki601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Laverty D. J., Averill A. M., Doublié S., and Greenberg M. M. (2017) The a-rule and deletion formation during abasic and oxidized abasic site bypass by DNA polymerase θ. ACS Chem. Biol. 12, 1584–1592 10.1021/acschembio.7b00211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Quiñones J. L., and Demple B. (2016) When DNA repair goes wrong: BER-generated DNA–protein crosslinks to oxidative lesions. DNA Repair 44, 103–109 10.1016/j.dnarep.2016.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yang Z., Price N. E., Johnson K. M., Wang Y., and Gates K. S. (2017) Interstrand cross-links arising from strand breaks at true abasic sites in duplex DNA. Nucleic Acids Res. 45, 6275–6283 10.1093/nar/gkx394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ensminger M., Iloff L., Ebel C., Nikolova T., Kaina B., and Lbrich M. (2014) DNA breaks and chromosomal aberrations arise when replication meets base excision repair. J. Cell Biol. 206, 29–43 10.1083/jcb.201312078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Yang K., Park D., Tretyakova N. Y., and Greenberg M. M. (2018) Histone tails decrease N7-methyl-2′-deoxyguanosine depurination and yield DNA–protein cross-links in nucleosome core particles and cells. Proc. Natl. Acad. Sci. U.S.A. 115, E11212–E11220 10.1073/pnas.1813338115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Semlow D. R., Zhang J., Budzowska M., Drohat A. C., and Walter J. C. (2016) Replication-dependent unhooking of DNA interstrand cross-links by the NEIL3 glycosylase. Cell 167, 498–511.e14 10.1016/j.cell.2016.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Admiraal S. J., and O'Brien P. J. (2015) Base excision repair enzymes protect abasic sites in duplex DNA from interstrand cross-links. Biochemistry 54, 1849–1857 10.1021/bi501491z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mohni K. N., Wessel S. R., Zhao R., Wojciechowski A. C., Luzwick J. W., Layden H., Eichman B. F., Thompson P. S., Mehta K. P. M., and Cortez D. (2019) HMCES maintains genome integrity by shielding abasic sites in single-strand DNA. Cell 176, 144–153.e13 10.1016/j.cell.2018.10.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Nakamura J., La D. K., and Swenberg J. A. (2000) 5′-Nicked apurinic/apyrimidinic sites are resistant to β-elimination by β-polymerase and are persistent in human cultured cells after oxidative stress. J. Biol. Chem. 275, 5323–5328 10.1074/jbc.275.8.5323 [DOI] [PubMed] [Google Scholar]
- 80. van Loon B., and Hübscher U. (2009) An 8-oxo-guanine repair pathway coordinated by MUTYH glycosylase and DNA polymerase λ. Proc. Natl. Acad. Sci. U.S.A. 106, 18201–18206 10.1073/pnas.0907280106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Grasso F., Ruggieri V., De Luca G., Leopardi P., Mancuso M. T., Casorelli I., Pichierri P., Karran P., and Bignami M. (2015) MUTYH mediates the toxicity of combined DNA 6-thioguanine and UVA radiation. Oncotarget 6, 7481–7492 10.18632/oncotarget.3037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Xie Y., Yang H., Miller J. H., Shih D. M., Hicks G. G., Xie J., and Shiu R. P. (2008) Cells deficient in oxidative DNA damage repair genes Myh and Ogg1 are sensitive to oxidants with increased G(2)/M arrest and multinucleation. Carcinogenesis 29, 722–728 10.1093/carcin/bgn033 [DOI] [PubMed] [Google Scholar]
- 83. Middeldorp A., van Puijenbroek M., Nielsen M., Corver W. E., Jordanova E. S., ter Haar N., Tops C. M., Vasen H. F., Lips E. H., van Eijk R., Hes F. J., Oosting J., Wijnen J., van Wezel T., and Morreau H. (2008) High frequency of copy-neutral LOH in MUTYH-associated polyposis carcinomas. J. Pathol. 216, 25–31 10.1002/path.2375 [DOI] [PubMed] [Google Scholar]
- 84. Lipton L., Halford S. E., Johnson V., Novelli M. R., Jones A., Cummings C., Barclay E., Sieber O., Sadat A., Bisgaard M. L., Hodgson S. V., Aaltonen L. A., Thomas H. J., and Tomlinson I. P. (2003) Carcinogenesis in MYH-associated polyposis follows a distinct genetic pathway. Cancer Res. 63, 7595–7599 [PubMed] [Google Scholar]
- 85. Shen Y., McMackin M. Z., Shan Y., Raetz A., David S., and Cortopassi G. (2016) Frataxin deficiency promotes excess microglial DNA damage and inflammation that is rescued by PJ34. PLoS ONE 11, e0151026 10.1371/journal.pone.0151026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Giovannini S., Weller M. C., Repmann S., Moch H., and Jiricny J. (2019) Synthetic lethality between BRCA1 deficiency and poly(ADP-ribose) polymerase inhibition is modulated by processing of endogenous oxidative DNA damage. Nucleic Acids Res. 47, 9132–9143 10.1093/nar/gkz624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Khodyreva S. N., Prasad R., Ilina E. S., Sukhanova M. V., Kutuzov M. M., Liu Y., Hou E. W., Wilson S. H., and Lavrik O. I. (2010) Apurinic/apyrimidinic (AP) site recognition by the 5′-dRP/AP lyase in poly(ADP-ribose) polymerase-1 (PARP-1). Proc. Natl. Acad. Sci. U.S.A. 107, 22090–22095 10.1073/pnas.1009182107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Prasad R., Horton J. K., Chastain P. D. 2nd, Gassman N. R., Freudenthal B. D., Hou E. W., and Wilson S. H. (2014) Suicidal cross-linking of PARP-1 to AP site intermediates in cells undergoing base excision repair. Nucleic Acids Res. 42, 6337–6351 10.1093/nar/gku288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Fouquerel E., and Sobol R. W. (2014) ARTD1 (PARP1) activation and NAD(+) in DNA repair and cell death. DNA Repair 23, 27–32 10.1016/j.dnarep.2014.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Xie Y., Yang H., Cunanan C., Okamoto K., Shibata D., Pan J., Barnes D. E., Lindahl T., McIlhatton M., Fishel R., and Miller J. H. (2004) Deficiencies in mouse Myh and Ogg1 result in tumor predisposition and G to T mutations in codon 12 of the K-ras oncogene in lung tumors. Cancer Res. 64, 3096–3102 10.1158/0008-5472.CAN-03-3834 [DOI] [PubMed] [Google Scholar]
- 91. Leipold M. D., Muller J. G., Burrows C. J., and David S. S. (2000) Removal of hydantoin products of 8-oxoguanine oxidation by the Escherichia coli DNA repair enzyme, FPG. Biochemistry 39, 14984–14992 10.1021/bi0017982 [DOI] [PubMed] [Google Scholar]
- 92. Leipold M. D., Workman H., Muller J. G., Burrows C. J., and David S. S. (2003) Recognition and removal of oxidized guanines in duplex DNA by the base excision repair enzymes hOGG1, yOGG1, and yOGG2. Biochemistry 42, 11373–11381 10.1021/bi034951b [DOI] [PubMed] [Google Scholar]
- 93. Van Houten B., and Sancar A. (1987) Repair of N-methyl-N′-nitro-N-nitrosoguanidine-induced DNA damage by ABC excinuclease. J. Bacteriol. 169, 540–545 10.1128/JB.169.2.540-545.1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.