

Abstract
Type 1 cannabinoid receptors (CB1Rs) are expressed in the dorsal root ganglion (DRG) and contribute to the analgesic effect of cannabinoids. However, the epigenetic mechanism regulating the expression of CB1Rs in neuropathic pain is unknown. G9a (encoded by the Ehmt2 gene), a histone 3 at lysine 9 methyltransferase, is a key chromatin regulator responsible for gene silencing. In this study, we determined G9a's role in regulating CB1R expression in the DRG and in CB1R-mediated analgesic effects in an animal model of neuropathic pain. We show that nerve injury profoundly reduced mRNA levels of CB1Rs but increased the expression of CB2 receptors in the rat DRG. ChIP results indicated increased enrichment of histone 3 at lysine 9 dimethylation, a G9a-catalyzed repressive histone mark, at the promoter regions of the CB1R genes. G9a inhibition in nerve-injured rats not only up-regulated the CB1R expression level in the DRG but also potentiated the analgesic effect of a CB1R agonist on nerve injury-induced pain hypersensitivity. Furthermore, in mice lacking Ehmt2 in DRG neurons, nerve injury failed to reduce CB1R expression in the DRG and to decrease the analgesic effect of the CB1R agonist. Moreover, nerve injury diminished the inhibitory effect of the CB1R agonist on synaptic glutamate release from primary afferent nerves to spinal cord dorsal horn neurons in WT mice but not in mice lacking Ehmt2 in DRG neurons. Our findings reveal that nerve injury diminishes the analgesic effect of CB1R agonists through G9a-mediated CB1R down-regulation in primary sensory neurons.
Keywords: pain, neurobiology, neurophysiology, neuroscience, synapse, dorsal root ganglion
Introduction
Chronic neuropathic pain caused by damage to the nervous system remains a major therapeutic challenge. Cannabinoids produce analgesic effects primarily through activation of type 1 cannabinoid receptors (CB1Rs)3 and type 2 cannabinoid receptors (CB2Rs), both of which are G protein–coupled (1, 2). WIN55,212-2, a mixed CB1R/CB2R agonist, reduces pain hypersensitivity caused by peripheral nerve injury (3–5). Peripheral administration of a specific CB1R agonist, arachidonyl-2′-chloroethylamide (ACEA), inhibits the response of spinal dorsal horn neurons to innocuous and noxious stimuli (6). CB1Rs are widely expressed in the peripheral and central nervous systems, including the dorsal root ganglion (DRG), peripheral and central terminals of DRG neurons, spinal cord dorsal horn, periaqueductal gray, ventral posterolateral thalamus, and cortical regions (7–10). The mRNA and protein levels of CB1Rs in neural tissues are dynamically regulated during development and can be altered under pathological conditions (11, 12). Although the CB1R expression level in the DRG has a large impact on the analgesic effect of cannabinoids (13), it is uncertain how nerve injury alters the expression level of CB1Rs in the DRG and CB1R-mediated analgesic effects.
Gene expression is controlled by various transcription factors and epigenetic machineries, including DNA methylation, histone modifications, and noncoding RNAs (14, 15). Peripheral nerve injury alters the expression levels of thousands of genes, including many G protein–coupled receptors (16–18). Unbiased genome-wide analyses show that nerve injury has only a small effect on DNA methylation levels of the genes in the DRG (19). On the other hand, histone modifications play a key role in nerve injury–induced abnormal gene expression in the DRG and in the transition from acute to chronic pain (17, 20). Histone 3 at lysine 9 dimethylation (H3K9me2), a histone mark usually associated with gene silencing, is the substrate of histone methyltransferase G9a (encoded by the Ehmt2 gene) and G9a-like protein (21, 22). G9a is present in the nucleus of DRG neurons, and traumatic nerve injury increases the expression level and activity of G9a in the DRG (17). G9a in the DRG is responsible for diminished expression of potassium channels and μ-opioid receptors in neuropathic pain (17, 18). However, it is not known whether G9a plays a role in epigenetic regulation of CB1Rs in DRG neurons in neuropathic pain.
In this study, we determined the expression level of CB1Rs in the DRG after nerve injury and the role of G9a in epigenetic silencing of CB1R expression in neuropathic pain. Our study reveals that nerve injury induces a long-lasting reduction in the expression level of CB1Rs in the DRG and that G9a is critically involved in down-regulation of CB1Rs in injured DRGs and in the diminished analgesic effect of CB1R agonists on neuropathic pain. This new information is important for our understanding of epigenetic mechanisms regulating CB1R expression in primary sensory neurons and for the design of better strategies to improve CB1R-mediated cannabinoid analgesic effects on neuropathic pain.
Results
Nerve injury differentially alters the expression level of CB1Rs and CB2Rs in the DRG
Quantitative PCR assay showed that the mRNA level of CB1Rs in the DRG was much lower in spinal nerve ligation (SNL) rats than in sham control rats 5, 10, and 21 days after surgery (p < 0.001, n = 6 rats/group; Fig. 1A). However, SNL had no significant effect on the mRNA level of CB1Rs in the dorsal spinal cord 3 weeks after surgery (Fig. 1B).
Figure 1.

Nerve injury differentially alters the expression levels of CB1Rs and CB2Rs in the DRG and spinal cord. A, real-time PCR data showing the mRNA level of CB1Rs in the rat DRG at days 5, 10, and 21 after sham or SNL surgery. B, the mRNA level of CB1Rs in the rat dorsal spinal cord 21 days after sham or SNL surgery. C, real-time PCR data showing the mRNA level of CB2Rs in the rat DRG on days 5, 10, and 21 after sham or SNL surgery. D, the mRNA level of CB2Rs in the rat dorsal spinal cord 21 days after sham or SNL surgery. Data are shown as means ± S.E. (n = 6 rats/group). Gapdh was used as an endogenous control, and the mean value in the sham group was set to 1. *, p < 0.05; ***, p < 0.001 compared with the sham group at the same time point. Two-way ANOVA followed by Tukey post hoc test was used for A and C. Two-tailed Student's t test was used for B and D.
In contrast, the mRNA level of CB2Rs in the DRG was significantly increased in SNL rats compared with control rats on days 10 and 21, but not on day 5, after surgery (p < 0.001, n = 6 rats/group; Fig. 1C). In addition, SNL significantly increased the mRNA level of CB2Rs in the dorsal spinal cord 3 weeks after surgery (p = 0.0126, n = 6 rats/group; Fig. 1D). These results indicate that peripheral nerve injury induces sustained down-regulation of CB1Rs but increases CB2R expression in the DRG.
Nerve injury increases the enrichment of H3K9me2 in the CB1R promoter
The persistent silencing of CB1R expression in the DRG after nerve injury suggests involvement of an epigenetic mechanism. We have shown previously that SNL increases the expression and activity of G9a in the DRG (17). The G9a-mediated histone modification H3K9me2 is a major repressive histone mark commonly involved in gene silencing in the DRG (17, 20). We next used ChIP-PCR to measure the occupancy of H3K9me2 at the CB1R promoter after SNL. SNL substantially increased the H3K9me2 level at the transcriptional start site (−60/95 bp) of CB1Rs in the DRG 3 weeks after surgery (p = 0.0152, Fig. 2). Furthermore, enrichment of H3K9me2 levels in two other regions, −480/−360 bp (p = 0.0022) and 363/469 bp (p = 0.0043), in the promoter region of CB1Rs in the DRG was also significantly higher in SNL rats than in control rats (Fig. 2). These data suggest that G9a-mediated H3K9me2 is associated with nerve injury–induced CB1R down-regulation in the DRG.
Figure 2.

Nerve injury increases the enrichment of H3K9me2 at the promoter region of CB1Rs. ChIP-PCR quantification data show the levels of H3K9me2 at three genomic regions around the transcriptional start site of CB1Rs in rat DRG tissues 21 days after sham or SNL surgery. n = 6 DRG samples from 12 rats in each group. Data were normalized to the total H3 value and are presented as means ± S.E. The mean value in the sham group was set to 1. *, p < 0.05; **, p < 0.01 compared with the sham group (Mann–Whitney rank-sum test).
Inhibition of G9a activity restores CB1R expression in the DRG and potentiates the analgesic effect of the CB1R agonist on neuropathic pain
We next determined whether increased G9a activity contributes to nerve injury–induced down-regulation of CB1Rs in the DRG. We treated SNL rats with daily intrathecal injections of 10 μg of UNC0638 or DMSO (as the vehicle control) for 7 days. The efficacy of intrathecal UNC0638, a highly specific G9a inhibitor, has been confirmed in our previous study (17). Treatment with UNC0638 largely restored the mRNA level of CB1Rs in the injured L5 and L6 DRGs (n = 8 rats/group, Fig. 3A). However, treatment with UNC0638 had no effect on the mRNA level of CB1Rs in the spinal cord (n = 6 rats/group, Fig. 3B). Also, the mRNA level of CB1Rs in the DRG did not differ significantly between sham rats treated with vehicle and sham rats treated with UNC0638 (n = 8 rats/group, Fig. 3A).
Figure 3.

G9a inhibition reverses the reduction in CB1R expression in the DRG after nerve injury. A, the mRNA level of CB1Rs in the DRG from sham and SNL rats treated with UNC0638 or DMSO (control (Cont)) (n = 8 rats/group). Drug treatment began 21 days after surgery, and the ipsilateral L5 and L6 DRG tissues were obtained 24 h after the last UNC0638 or DMSO injection. The mRNA level of CB1Rs was normalized to that of GAPDH in the same samples, and the mean value of CB1R levels in sham control rats was considered to be 1. B, the mRNA level of CB1Rs in the dorsal spinal cord from sham and SNL rats treated with UNC0638 or DMSO (n = 6 rats/group). Data are shown as means ± S.E. ***, p < 0.001 compared with the sham-treated group or DMSO-treated group (one-way ANOVA followed by Tukey post hoc test).
We used immunoblotting and DRG tissues from CB1R conditional knockout mice (generated by crossing female Cnr1flox/flox mice with male AdvillinCre/+ mice) as stringent controls to validate three CB1R antibodies commonly used in the literature, including those from Alomone Labs, Cayman Chemical, and Frontier Institute Co. (see supporting information). However, we were unable to validate the specificity of these three antibodies because they detected similar protein bands in the DRG tissues from WT and CB1R conditional knockout mice (supporting information). For this reason, we did not include our immunoblot data for CB1R protein quantification.
We then determined whether the analgesic effect of the CB1R agonist was altered in SNL rats and whether such a change was due to augmented G9a activity. We measured the effect of ACEA, a specific CB1R agonist (23, 24), administered intrathecally on pain hypersensitivity in SNL rats 7 days after intrathecal treatment with UNC0638 or DMSO. In DMSO-treated SNL rats, intrathecal injection of 10, 50, or 100 μg of ACEA produced little effect on tactile allodynia, measured with von Frey filaments, or mechanical hyperalgesia, tested with a noxious pressure stimulus (n = 10 rats/group, Fig. 4). In contrast, the inhibitory effects of ACEA injected intrathecally on allodynia and hyperalgesia were significantly potentiated in UNC0638-treated SNL rats (n = 10 rats/group, Fig. 4). Treatment with UNC0638 also significantly increased the baseline tactile and pressure withdrawal thresholds of SNL rats, as we reported previously (17, 18). These results suggest that augmented G9a activity mediates nerve injury–induced CB1R down-regulation in the DRG and the diminished analgesic effect of CB1R agonists on neuropathic pain.
Figure 4.
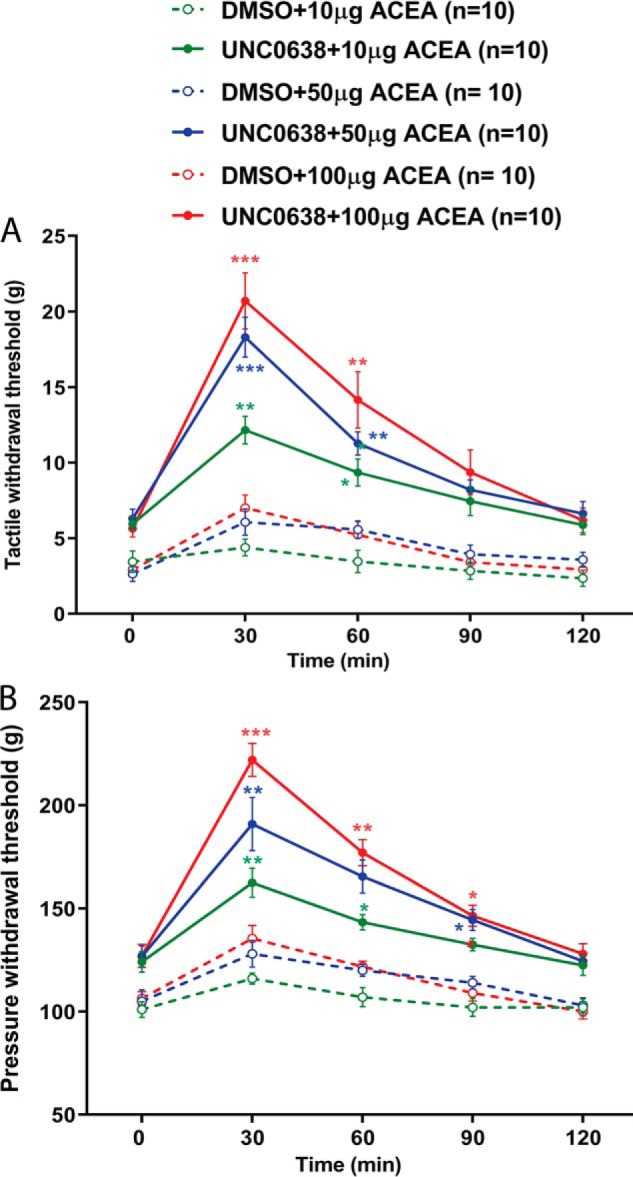
G9a inhibition potentiates the analgesic effect of the CB1R agonist diminished by nerve injury. A and B, time course of the effect of intrathecal ACEA on the tactile (A) and pressure (B) withdrawal thresholds in SNL rats treated with UNC0638 or DMSO (n = 10 rats/group). Data are shown as means ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001 compared with the respective baseline at time 0 (two-way ANOVA followed by Tukey post hoc test).
G9a in DRG neurons is required for nerve injury–induced silencing of CB1R expression and the diminished analgesic effect of the CB1R agonist
To directly determine the role of G9a in DRG neurons in reduced CB1R expression and the diminished analgesic effect of the CB1R agonist caused by nerve injury, we generated Ehmt2 conditional knockout mice (Ehmt2-cKO) by crossing Ehmt2-floxed mice with a primary sensory neuron–specific Cre mouse line, Advillin-Cre (17, 25), so that Ehmt2 was selectively ablated from DRG neurons. Selective ablation of the G9a protein in the DRG in Ehmt2-cKO mice has been demonstrated previously (17). In WT mice, SNI significantly reduced the mRNA level of CB1Rs in the DRG (p < 0.001, n = 6 mice/group, Fig. 5). In contrast, in Ehmt2-cKO mice, SNI had no significant effect on the mRNA level of CB1Rs in the DRG (Fig. 5). Furthermore, the mRNA level of CB1Rs in the DRG did not differ significantly between sham-treated WT mice and sham-treated Ehmt2-cKO mice (Fig. 5).
Figure 5.

Genetic deletion of Ehmt2 in DRG neurons restores the expression level of CB1Rs in the mouse DRG reduced by nerve injury. Quantitative PCR analysis shows the mRNA levels of CB1Rs in the DRG from Ehmt2-cKO and WT mice subjected to SNI or sham surgery (n = 6 mice/group). Data are shown as means ± S.E. ***, p < 0.001 compared with the WT sham group (one-way ANOVA followed by Dunnett post hoc test).
SNI substantially decreased the withdrawal thresholds of the ipsilateral hind paw in response to application of von Frey filaments and noxious pressure and heat stimuli, indicating pain hypersensitivity, in seven WT control mice 3 weeks after surgery (Fig. 6). The tactile (0.84 ± 0.06 versus 0.82 ± 0.04 g, t = 0.21, p = 0.85) and pressure (78.08 ± 4.25 versus 77.02 ± 5.04 g, t = 1.72, p = 0.13) withdrawal thresholds did not differ significantly between the SNI and sham control mice 3 weeks after surgery (n = 8 mice/group), similar to what we reported previously for Ehmt2-cKO mice (17, 18). In WT mice subjected to SNI, intraperitoneal injection of 1 mg/kg and 5 mg/kg of the CB1R agonist ACEA had no significant effect on the mechanical and thermal withdrawal thresholds (n = 7 mice, Fig. 6). In contrast, intraperitoneal injection of ACEA at both 1 mg/kg and 5 mg/kg significantly increased the mechanical and thermal withdrawal thresholds in Ehmt2-cKO mice 3 weeks after SNI (n = 8 mice, Fig. 6). These data provide unambiguous evidence that G9a in DRG neurons is indispensable for nerve injury–induced down-regulation of CB1Rs and the diminished analgesic effect of the CB1R agonist on neuropathic pain.
Figure 6.
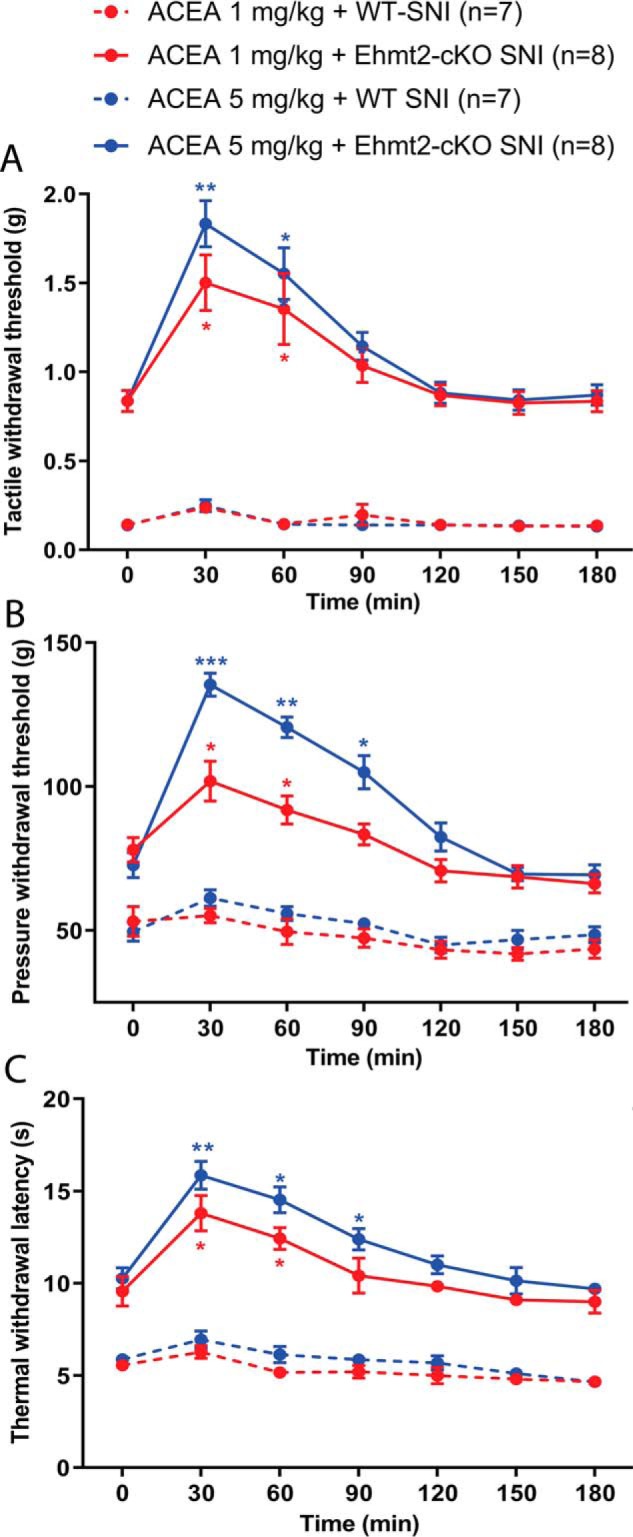
Ablation of Ehmt2 in DRG neurons augments the analgesic effect of the CB1R agonist diminished by nerve injury. A–C, time course of the effect of intraperitoneal injection of ACEA (1 and 5 mg/kg) on the tactile (A) and pressure (B) withdrawal thresholds and thermal sensitivity (C) of 8 Ehmt2-cKO and 7 WT control mice subjected to SNI. Data are shown as means ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001 compared with the respective baseline at time 0 (two-way ANOVA followed by Tukey post hoc test).
Ablation of G9a in DRG neurons potentiates the inhibitory effect of the CB1R agonist on synaptic glutamate release from primary afferent nerves after nerve injury
CB1Rs are expressed presynaptically at primary afferent nerve terminals, and CB1R activation can reduce nociceptive input to the spinal cord (9, 10, 24). In the spinal cord, the mixed CB1R/CB2R agonist WIN55,212-2 reduces synaptic transmission, and this effect is blocked by the CB1R antagonist SR141716A (26). We thus used Ehmt2-cKO mice to determine whether G9a in DRG neurons influences the inhibitory effect of the CB1R agonist on glutamatergic input from primary afferent nerves to spinal dorsal horn neurons in neuropathic pain. In WT mice receiving sham surgery, bath application of 50–200 nm but not 10–20 nm ACEA significantly reduced the amplitude of monosynaptically evoked excitatory postsynaptic currents (EPSCs) of spinal lamina II neurons (n = 12 neurons, Fig. 7, A and B). SNI markedly increased the baseline amplitude of EPSCs of lamina II neurons monosynaptically evoked from dorsal root stimulation, in agreement with our previous reports (27, 28). Strikingly, in WT mice subjected to SNI, bath application of up to 200 nm ACEA had no significant inhibitory effect on the amplitude of evoked EPSCs of spinal dorsal horn neurons (n = 11 neurons, Fig. 7, A and B).
Figure 7.

Genetic ablation of Ehmt2 in DRG neurons restores the inhibitory effect of the CB1R agonist on glutamate release from primary afferent terminals diminished by nerve injury. A–C, representative recording traces and mean data showing the inhibitory effect of 10–200 nm ACEA on the amplitude of EPSCs of dorsal horn neurons evoked monosynaptically from dorsal root stimulation in WT (A and B) and Ehmt2-cKO (A and C) mice subjected to sham or SNI surgery. In the top panel of B, the baseline amplitude of EPSCs was different between the sham and SNI groups, and the effect of ACEA was normalized to the respective baseline amplitude (percent inhibition) for comparison in the bottom panel. The number of neurons tested in each group is indicated in B and C. Data are shown as means ± S.E. *, p < 0.05 compared with the respective baseline value. #, p < 0.05 compared with the WT sham group at the same ACEA concentration (two-way ANOVA followed by Tukey post hoc test).
In contrast, in Ehmt2-cKO mice subjected to SNI and sham surgery, bath application of 50–200 nm ACEA similarly attenuated the amplitude of monosynaptically evoked EPSCs of lamina II neurons (Fig. 7, A and C). The inhibitory effect of ACEA on the amplitude of evoked EPSCs in lamina II neurons did not differ significantly between SNI Ehmt2-cKO mice (n = 12 neurons) and sham-treated Ehmt2-cKO mice (n = 10 neurons). These findings clearly indicate that G9a in DRG neurons is responsible for nerve injury–induced reduction in CB1R-mediated inhibition of primary afferent input to spinal dorsal horn neurons.
Discussion
Our study demonstrates that nerve injury induced long-lasting CB1R down-regulation in the DRG and diminished the analgesic effect of the CB1R agonist on neuropathic pain. We found that, in both rats and mice, nerve injury caused by SNL and SNI, respectively, consistently reduced the expression of CB1Rs in the DRG. Our quantitative PCR results are consistent with unbiased RNA-Seq data showing that CB1R expression is significantly reduced in the DRG after nerve injury (17). A previous study, using only three rats per group, reported that L5 SNL did not alter the mRNA level of CB1Rs in the injured DRG but did reduce the percentage of CB1R/IB4 and CB1R/CGRP double-labeled DRG neurons (29). We also showed that the analgesic effect of the CB1R agonist on nerve injury–induced pain hypersensitivity was diminished, which can be explained by down-regulation of CB1Rs in the DRG. Increased glutamatergic synaptic input from DRG neurons is important for the development of neuropathic pain, and inhibition of this nociceptive input is an effective approach for treating neuropathic pain (27, 30). Similar to other Gi/o-coupled receptors (31), CB1Rs, upon activation, inhibit calcium channels in various types of neurons (32–34). The CB1R agonist ACEA has been shown to inhibit the response of spinal dorsal horn neurons to noxious stimuli (24). Similarly, we showed that ACEA reduced synaptic glutamate release from primary afferent terminals in sham-treated WT mice, but this effect was diminished after nerve injury. These findings suggest that nerve injury-induced CB1R down-regulation in the DRG reduces CB1R-mediated inhibition of excitatory synaptic transmission from primary sensory neurons to spinal dorsal horn neurons.
Furthermore, our study provides unequivocal evidence that G9a-mediated H3K9m2 is essential for nerve injury–induced reduction in CB1R expression in the DRG. G9a-mediated histone lysine methylation plays an important role in gene expression changes in the DRG caused by nerve injury (17). G9a is generally known for mediating H3K9m2-related gene silencing and transcriptional repression (35–37). We found that nerve injury increased the enrichment of H3K9me2 not only at the transcriptional start site (−60 to 95 bp) but also to its upstream (−480 to −360 bp) and downstream (363 to 469 bp) in the promoter region of CB1Rs. This finding suggests that increased histone methylation by G9a is dispersed in the promoter region of CB1Rs in the injured DRG. We showed that inhibiting G9a activity using UNC0638 reversed the nerve injury-reduced expression levels of CB1Rs in the rat DRG. Importantly, in mice in which G9a was genetically ablated in DRG neurons, nerve injury failed to reduce CB1R expression in the DRG. Because G9a inhibition or knockout had no effect on the expression of CB1Rs in the DRG in sham-treated animals, it is possible that G9a activity in normal DRG neurons is too low to regulate basal CB1R expression.
Our study also demonstrates a crucial role of G9a in DRG neurons in the diminished inhibitory effects of the CB1R agonist on nociception and on peripheral glutamatergic input to spinal dorsal horn neurons in neuropathic pain. We found that G9a inhibition or genetic ablation of Ehmt2 in DRG neurons potentiated the antinociceptive effect of ACEA, a specific CB1R agonist. In addition, the inhibitory effect of ACEA on glutamatergic input from primary afferent terminals in the spinal dorsal horn was normalized in Ehmt2-cKO mice, indicating an essential role of G9a in nerve injury–induced silencing of CB1R expression at central terminals of DRG neurons.
We have shown that 638 genes are regulated by G9a in the injured DRG (17). Although our study demonstrates an important role of G9a in nerve injury–induced CB1R down-regulation, other genes altered by G9a manipulation could indirectly affect the CB1R signaling and its analgesic action. Also, it is unclear whether certain transcriptional repressors, such as REST/NRSF, are involved in nerve injury-induced CB1R down-regulation. Although G9a and REST/NRSF often act coordinately on gene silencing (28, 38, 39), further studies are needed to define whether REST/NRSF is part of the G9a-containing repressor complex involved in CB1R silencing in the injured DRG. We reported previously that G9a is responsible for the diminished μ-opioid receptor expression in the DRG and the opioid analgesic effect after nerve injury (18). This study provides another example of the critical role of G9a in sustained down-regulation of CB1Rs, another important analgesic target and G protein–coupled receptor. Thus, peripheral nerve injury is generally associated with G9a-mediated epigenetic silencing of endogenous antinociceptive genes in primary sensory neurons, which may contribute to the development of chronic neuropathic pain.
Although some clinical studies report that cannabinoids reduce chronic neuropathic pain (40, 41), others show that cannabinoid compounds have very limited analgesic efficacy for treating neuropathic pain in patients (42–45). Notably, cannabis contains more than 400 chemically distinct entities, and not all cannabinoid compounds produce analgesia via CB1Rs. For example, the analgesic effect of cannabidiol on chronic pain is mediated primarily via glycine receptors (46). Also, the analgesic effect of Δ(9)-tetrahydrocannabinol on acute pain is still intact in CB1R KO mice (47). Interestingly, we found that the mRNA level of CB2Rs in the DRG was increased 10 and 21 days but not 5 days after nerve injury, which may explain the efficacy of certain cannabinoid compounds in neuropathic pain conditions. Our finding is consistent with previous reports showing that traumatic nerve injury increases CB2R expression levels in DRG neurons and the spinal cord (48–50) and provides further support for development of CB2R agonists for treating chronic neuropathic pain (51, 52). Because G9a is generally involved in gene silencing, it is unlikely that G9a plays a role in CB2R up-regulation in the injured DRG. Indeed, we found that G9a inhibition with UNC0638 did not attenuate the nerve injury–induced increase in the mRNA level of CB2Rs in the rat DRG (17). The epigenetic mechanism responsible for CB2R up-regulation in the injured DRG needs to be investigated in future studies.
In summary, our findings reveal G9a in primary sensory neurons as an important epigenetic regulator for control of CB1R expression in neuropathic pain. G9a is the key chromatin modulator responsible for down-regulation of CB1Rs in the injured DRG and the diminished analgesic effect of CB1R agonists on neuropathic pain. Systemically administering specific CB1R agonists may have little clinical use because of their undesirable adverse effects in the central nervous system, such as dependence and cognitive impairment. Nevertheless, G9a inhibitors could conceivably be used to restore CB1R expression in injured DRG neurons, thus potentiating the analgesic effect of cannabinoids and peripherally restricted CB1R agonists on neuropathic pain.
Experimental procedures
Rat model of neuropathic pain and intrathecal cannulation
All experimental protocols were approved by the Institutional Animal Care and Use Committee of the University of Texas MD Anderson Cancer Center (approval no. 882-RN02). Male Sprague-Dawley rats (8–10 weeks old, Harlan Laboratories) were used in this study. L5 and L6 SNL was used as an experimental model of neuropathic pain, as described previously (53). In brief, we induced anesthesia with 2%–3% isoflurane inhalation, isolated the left L5 and L6 spinal nerves, and ligated them with sutures under a surgical microscope. Sham control rats underwent the same procedure except for nerve ligation. Pain hypersensitivity developed 10–14 days after SNL and lasted at least 8 weeks. All animals were housed (2–3 rats/cage) in a controlled environment (24 °C ± 2 °C, 12:12 h light-dark cycles) and received standard laboratory food and clean water ad libitum. At the end of the experiments, animals were deeply anesthetized with isoflurane inhalation and then rapidly decapitated.
Two weeks after SNL surgery, we inserted intrathecal catheters in some SNL rats anesthetized with 2%–3% isoflurane via a nose cone. Briefly, a puncture was made in the atlanto-occipital membrane, and a polyethylene (PE-10) catheter was introduced, with the caudal tip placed at the lumbar spinal cord level (54). The rats were allowed to recover for at least 5 days before the catheters were used for intrathecal injections. Drugs were injected intrathecally at a volume of 10 μl, followed by a 5-μl saline flush. UNC0638 (Sigma-Aldrich), a selective G9a inhibitor (17, 18), and ACEA (Sigma-Aldrich), a highly specific CB1R agonist (23, 24), were dissolved in DMSO. Drug treatment started 3 weeks after SNL, when the chronic pain was well-established. The doses of these agents were selected from previous studies (17, 18, 55, 56).
Generation of Ehmt2 conditional knockout mice and mouse model of neuropathic pain
Ehmt2 cKO mice were generated as described previously (17, 18). In brief, we deleted the Ehmt2 gene in DRG neurons by crossing female Ehmt2flox/flox mice with male AdvillinCre/+ mice, a primary sensory neuron–specific Cre line (25, 57). Tail biopsies were used for genotyping 3 weeks after birth. Cre-negative floxed littermates were used as WT control mice, and all mice were of the C57BL/6J genetic background. Age-matched adult (8- to 10-week-old) males and females were used for final experiments.
Spared nerve injury (SNI) was performed in mice as a model of neuropathic pain as described previously (18, 58). The mice were anesthetized with 2%–3% isoflurane, and an incision was made on the left lateral thigh to locate the sciatic nerve. The common peroneal and tibial nerves were ligated and sectioned (leaving the sural nerve intact) under a surgical microscope. The sham mice were subjected to the same procedure without the nerve injury. Final behavioral studies and spinal cord slice recordings were performed 2–3 weeks after surgery.
Behavioral assessment of nociception in rodents
To quantify tactile allodynia, we applied von Frey filaments (Stoelting, Wood Dale, IL) to the animals' left hind paw (ipsilateral to SNL or SNI). Rodents were placed individually in suspended chambers on a mesh floor. Calibrated von Frey filaments were applied vertically to the plantar surface of the left hind paw with sufficient force to bend the filaments for 6 s. The tactile stimulus producing a 50% likelihood of withdrawal was calculated using the “up–down” method, as described previously (59).
To measure mechanical nociception, we used the paw pressure test on the left hind paw. The Analgesy-Meter device (Ugo Basile) was pressed to trigger a motor that applied a constantly increasing force to the left hind paw. When the animal displayed a withdrawal response, the pedal was released immediately to record the nociceptive withdrawal threshold (60).
Hind paw thermal sensitivity in rodents was assessed using a radiant heat source (IITC Life Science, Woodland Hills, CA). The light that generated noxious heat was focused onto the plantar surface of the left hind paw (18, 54). The withdrawal latency was registered on a timer when the hind paw moved away abruptly from the noxious heat. The investigators performing behavioral measurements were blinded to the drug treatments and genotypes.
RNA isolation and real-time PCR assay
Total RNA was isolated from the left lumbar DRG and dorsal spinal cord tissues using TRIsure (BIO-38032, Bioline, Taunton, MA). Reverse transcription of 1 μg of RNA treated with RNase-free DNase (79254, Qiagen, Hilden, Germany) was performed using the RevertAid RT Reverse Transcription Kit (#K1619, Thermo Fisher Scientific, Waltham, MA). 2 μl of complementary DNA diluted five times was added to a 20-μl reaction volume with SYBR Green PCR Mix (A25780, Thermo Fisher Scientific). Real-time PCR was carried out using a Quant Studio 7 Flex Real-Time PCR System (Applied Biosystems, Waltham, MA). The thermal cycling conditions were as follows: 1 cycle at 95 °C for 10 min, 40 cycles at 95 °C for 15 s, and 1 cycle at 60 °C for 45 s. The following primers were used: rat CB1R forward, AGA CAC CAC CTT CCG TAC CAT CAC; rat CB1R reverse, GGA GTT GTC TCC TGC GGT CAT CTT T; rat CB2R forward, TGA GGT GAA AAC CAC CAC AGG GC; rat CB2R reverse, GTC CAG GAC CCC TCT CTT CTG; rat Gapdh forward, CAT CCC AGA GCT GAA CGG GAA G; rat Gapdh reverse, GTC CTC AGT GTA GCC CAG GAT GC; mouse CB1R forward, CAG CAG GAG ACA CAA CCA AC; mouse CB1R reverse, TTC TCC AGA ACC GTG AAG GTG; mouse Tuba1a forward, CCA CTA CAC CAT TGG CAA GGA GA; and mouse Tuba1a reverse, GGA GGT GAA GCC AGA GCC AGT.
ChIP assay
ChIP assays were performed as described previously (28). In brief, L5 and L6 DRG tissues were isolated and cross-linked with 2 mm disuccinimidyl glutarate for 35 min, followed by 2% formaldehyde for 20 min. The DRG tissues were homogenized with lysis buffer and sonicated into fragments of 200–1000 bp using a water bath sonicator (Qsonica, Newtown, CT) at 4 °C (40 cycles of 30 s on and 30 s off). Chromatin was pulled down using Dynabeads Protein G magnetic beads (10003D, Thermo Fisher Scientific) conjugated with an anti-H3K9me2 antibody (ab1220, Abcam, Cambridge, MA) or anti-H3 antibody (ab1791, Abcam). DNA was recovered using the QIAquick PCR Purification Kit (58106, Qiagen) after decross-linking and was used for real-time PCR. The primers used were as follows: rat ChIP CB1R (−480/−360 bp) forward, GAT GCA CAT GCT CAG GGG AGA CT; rat ChIP CB1R (−480/−360 bp) reverse, CTG TGG GGA CCT CGG AGG TC; rat ChIP CB1R (−60/95 bp) forward, GGG AAA GAG GCT TCA TGT TGA CAT G; rat ChIP CB1R (−60/95 bp) reverse, CAT ACC TCA GCC ATG GGT GCT CC; rat ChIP CB1R (363/469 bp) forward, GCT GAA AAT AAG ACC TCA TGG TGG; and rat ChIP CB1R (363/469 bp) reverse, GTC CGC TGG GAG ATG GGT AAC AG.
Spinal cord slice preparation and electrophysiological recordings
Electrophysiological recordings in spinal cord slices were performed as described previously (54, 57). The lumbar spinal cord tissues were removed rapidly via laminectomy from mice anesthetized with 2%–3% isoflurane. The spinal cords at L5 and L6 were sliced transversely to a thickness of 400 μm using a vibratome in ice-cold sucrose artificial cerebrospinal fluid presaturated with 95% O2 and 5% CO2. The spinal cord slices were incubated in Krebs solution oxygenated with 95% O2 and 5% CO2 at 34 °C for at least 1 h before recordings.
Whole-cell patch-clamp recordings were performed using a glass pipette (5–10 megohms) filled with an internal solution containing 135.0 mm potassium gluconate, 5.0 mm tetraethylammonium, 2.0 mm MgCl2, 0.5 mm CaCl2, 5.0 mm HEPES, 5.0 mm EGTA, 5.0 mm Mg-ATP, 0.5 mm Na-GTP, and 10.0 mm lidocaine N-ethyl bromide. The spinal lamina II outer neurons on the ipsilateral (left) side were chosen for recording because they receive nociceptive input predominantly from primary sensory nerves (54, 61). We used electrical stimulation (0.2 ms, 0.6 mA, and 0.1 Hz) of the dorsal root to evoke EPSCs at a holding potential of −60 mV. EPSCs were considered monosynaptic when the latency of evoked EPSCs was constant and a 20-Hz electrical stimulation did not cause conduction failure (62, 63). During the recording, the slices were continuously perfused with Krebs solution at 3.0 ml/min at 34 °C. The input resistance was monitored, and the recording was abandoned when the resistance changed more than 15%.
Statistical analysis
All data are expressed as means ± S.E. Analyses of the drug's effect on the amplitude of evoked EPSCs were performed using Clampfit 9.2 software (Axon Instruments, Union City, CA). We recorded only one neuron from each spinal cord slice and used three to four mice for each group. ACEA (10–200 nm) was bath-applied in an ascending order, each for 6 min. The amplitude of EPSCs was quantified after it had stabilized at a reduced level. A two-tailed Student's t test or Mann–Whitney rank-sum test was used to compare two groups, and one-way or two-way analysis of variance (ANOVA) followed by Dunnett or Tukey post hoc test was used to determine the differences between more than two groups. p < 0.05 was considered statistically significant.
Author contributions
Y. L., J. Z., L. C., S.-R. C., H. C., and G. Z. data curation; Y. L., J. Z., L. C., and S.-R. C. formal analysis; Y. L., J. Z., L. C., S.-R. C., and H. C. investigation; Y. L. and S.-R. C. writing-original draft; G. Z. validation; H.-L. P. conceptualization; H.-L. P. supervision; H.-L. P. funding acquisition; H.-L. P. writing-review and editing.
Supplementary Material
This study was supported by National Institutes on Drug Abuse Grant DA041711 (to H.-L. P.), National Institutes of Health Grant NS101880, and by the N. G. and Helen T. Hawkins Endowment. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supporting Methods, Results, and References.
- CB1R
- type 1 cannabinoid receptor
- CB2R
- type 2 cannabinoid receptor
- ACEA
- arachidonyl-2′-chloroethylamide
- DRG
- dorsal root ganglion
- H3K9me2
- histone 3 at lysine 9 dimethylation
- NRSF
- neuron-restrictive silencing factor
- REST
- RE1-silencing transcription factor
- SNL
- spinal nerve ligation
- cKO
- conditional knockout
- SNI
- spared nerve injury
- EPSC
- excitatory postsynaptic current
- ANOVA
- analysis of variance.
References
- 1. Matsuda L. A., Lolait S. J., Brownstein M. J., Young A. C., and Bonner T. I. (1990) Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 346, 561–564 10.1038/346561a0 [DOI] [PubMed] [Google Scholar]
- 2. Munro S., Thomas K. L., and Abu-Shaar M. (1993) Molecular characterization of a peripheral receptor for cannabinoids. Nature 365, 61–65 10.1038/365061a0 [DOI] [PubMed] [Google Scholar]
- 3. Bridges D., Ahmad K., and Rice A. S. (2001) The synthetic cannabinoid WIN55,212-2 attenuates hyperalgesia and allodynia in a rat model of neuropathic pain. Br. J. Pharmacol. 133, 586–594 10.1038/sj.bjp.0704110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fox A., Kesingland A., Gentry C., McNair K., Patel S., Urban L., and James I. (2001) The role of central and peripheral Cannabinoid1 receptors in the antihyperalgesic activity of cannabinoids in a model of neuropathic pain. Pain 92, 91–100 10.1016/S0304-3959(00)00474-7 [DOI] [PubMed] [Google Scholar]
- 5. Herzberg U., Eliav E., Bennett G. J., and Kopin I. J. (1997) The analgesic effects of R(+)-WIN 55,212-2 mesylate, a high affinity cannabinoid agonist, in a rat model of neuropathic pain. Neurosci. Lett. 221, 157–160 10.1016/S0304-3940(96)13308-5 [DOI] [PubMed] [Google Scholar]
- 6. Kelly S., Jhaveri M. D., Sagar D. R., Kendall D. A., and Chapman V. (2003) Activation of peripheral cannabinoid CB1 receptors inhibits mechanically evoked responses of spinal neurons in noninflamed rats and rats with hindpaw inflammation. Eur. J. Neurosci. 18, 2239–2243 10.1046/j.1460-9568.2003.02957.x [DOI] [PubMed] [Google Scholar]
- 7. Tsou K., Brown S., Sañudo-Peña M. C., Mackie K., and Walker J. M. (1998) Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience 83, 393–411 10.1016/S0306-4522(97)00436-3 [DOI] [PubMed] [Google Scholar]
- 8. Hohmann A. G., and Herkenham M. (1999) Localization of central cannabinoid CB1 receptor messenger RNA in neuronal subpopulations of rat dorsal root ganglia: a double-label in situ hybridization study. Neuroscience 90, 923–931 10.1016/S0306-4522(98)00524-7 [DOI] [PubMed] [Google Scholar]
- 9. Salio C., Fischer J., Franzoni M. F., and Conrath M. (2002) Pre- and postsynaptic localizations of the CB1 cannabinoid receptor in the dorsal horn of the rat spinal cord. Neuroscience 110, 755–764 10.1016/S0306-4522(01)00584-X [DOI] [PubMed] [Google Scholar]
- 10. Veress G., Meszar Z., Muszil D., Avelino A., Matesz K., Mackie K., and Nagy I. (2013) Characterisation of cannabinoid 1 receptor expression in the perikarya, and peripheral and spinal processes of primary sensory neurons. Brain Struct. Funct. 218, 733–750 10.1007/s00429-012-0425-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Laprairie R. B., Kelly M. E., and Denovan-Wright E. M. (2012) The dynamic nature of type 1 cannabinoid receptor (CB(1)) gene transcription. Br. J. Pharmacol. 167, 1583–1595 10.1111/j.1476-5381.2012.02175.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. D'Addario C., Di Francesco A., Pucci M., Finazzi Agrò A., and Maccarrone M. (2013) Epigenetic mechanisms and endocannabinoid signalling. FEBS J. 280, 1905–1917 10.1111/febs.12125 [DOI] [PubMed] [Google Scholar]
- 13. Agarwal N., Pacher P., Tegeder I., Amaya F., Constantin C. E., Brenner G. J., Rubino T., Michalski C. W., Marsicano G., Monory K., Mackie K., Marian C., Batkai S., Parolaro D., Fischer M. J., et al. (2007) Cannabinoids mediate analgesia largely via peripheral type 1 cannabinoid receptors in nociceptors. Nat. Neurosci. 10, 870–879 10.1038/nn1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Herre M., and Korb E. (2019) The chromatin landscape of neuronal plasticity. Curr. Opin. Neurobiol. 59, 79–86 10.1016/j.conb.2019.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jiang Y., Langley B., Lubin F. D., Renthal W., Wood M. A., Yasui D. H., Kumar A., Nestler E. J., Akbarian S., and Beckel-Mitchener A. C. (2008) Epigenetics in the nervous system. J. Neurosci. 28, 11753–11759 10.1523/JNEUROSCI.3797-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang H., Sun H., Della Penna K., Benz R. J., Xu J., Gerhold D. L., Holder D. J., and Koblan K. S. (2002) Chronic neuropathic pain is accompanied by global changes in gene expression and shares pathobiology with neurodegenerative diseases. Neuroscience 114, 529–546 10.1016/S0306-4522(02)00341-X [DOI] [PubMed] [Google Scholar]
- 17. Laumet G., Garriga J., Chen S. R., Zhang Y., Li D. P., Smith T. M., Dong Y., Jelinek J., Cesaroni M., Issa J. P., and Pan H. L. (2015) G9a is essential for epigenetic silencing of K+ channel genes in acute-to-chronic pain transition. Nat. Neurosci. 18, 1746–1755 10.1038/nn.4165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang Y., Chen S. R., Laumet G., Chen H., and Pan H. L. (2016) Nerve injury diminishes opioid analgesia through lysine methyltransferase-mediated transcriptional repression of μ-opioid receptors in primary sensory neurons. J. Biol. Chem. 291, 8475–8485 10.1074/jbc.M115.711812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Garriga J., Laumet G., Chen S. R., Zhang Y., Madzo J., Issa J. J., Pan H. L., and Jelinek J. (2018) Nerve injury-induced chronic pain is associated with persistent DNA methylation reprogramming in dorsal root ganglion. J. Neurosci. 38, 6090–6101 10.1523/JNEUROSCI.2616-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang J., and Pan H.-L. (2018) in Epigenetics of Chronic Pain (Bai G., and Ren K., eds.) pp. 85–98, Academic Press, London, United Kingdom [Google Scholar]
- 21. Tachibana M., Sugimoto K., Fukushima T., and Shinkai Y. (2001) Set domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. J. Biol. Chem. 276, 25309–25317 10.1074/jbc.M101914200 [DOI] [PubMed] [Google Scholar]
- 22. Tachibana M., Ueda J., Fukuda M., Takeda N., Ohta T., Iwanari H., Sakihama T., Kodama T., Hamakubo T., and Shinkai Y. (2005) Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev. 19, 815–826 10.1101/gad.1284005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hillard C. J., Manna S., Greenberg M. J., DiCamelli R., Ross R. A., Stevenson L. A., Murphy V., Pertwee R. G., and Campbell W. B. (1999) Synthesis and characterization of potent and selective agonists of the neuronal cannabinoid receptor (CB1). J. Pharmacol. Exp. Ther. 289, 1427–1433 [PubMed] [Google Scholar]
- 24. Kelly S., and Chapman V. (2001) Selective cannabinoid CB1 receptor activation inhibits spinal nociceptive transmission in vivo. J. Neurophysiol. 86, 3061–3064 10.1152/jn.2001.86.6.3061 [DOI] [PubMed] [Google Scholar]
- 25. Zhou X., Wang L., Hasegawa H., Amin P., Han B. X., Kaneko S., He Y., and Wang F. (2010) Deletion of PIK3C3/Vps34 in sensory neurons causes rapid neurodegeneration by disrupting the endosomal but not the autophagic pathway. Proc. Natl. Acad. Sci. U.S.A. 107, 9424–9429 10.1073/pnas.0914725107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Morisset V., and Urban L. (2001) Cannabinoid-induced presynaptic inhibition of glutamatergic EPSCs in substantia gelatinosa neurons of the rat spinal cord. J. Neurophysiol. 86, 40–48 10.1152/jn.2001.86.1.40 [DOI] [PubMed] [Google Scholar]
- 27. Chen J., Li L., Chen S. R., Chen H., Xie J. D., Sirrieh R. E., MacLean D. M., Zhang Y., Zhou M. H., Jayaraman V., and Pan H. L. (2018) The α2δ-1-NMDA receptor complex is critically involved in neuropathic pain development and gabapentin therapeutic actions. Cell Rep. 22, 2307–2321 10.1016/j.celrep.2018.02.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang J., Chen S. R., Chen H., and Pan H. L. (2018) RE1-silencing transcription factor controls the acute-to-chronic neuropathic pain transition and Chrm2 receptor gene expression in primary sensory neurons. J. Biol. Chem. 293, 19078–19091 10.1074/jbc.RA118.005846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mitrirattanakul S., Ramakul N., Guerrero A. V., Matsuka Y., Ono T., Iwase H., Mackie K., Faull K. F., and Spigelman I. (2006) Site-specific increases in peripheral cannabinoid receptors and their endogenous ligands in a model of neuropathic pain. Pain 126, 102–114 10.1016/j.pain.2006.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Deng M., Chen S. R., and Pan H. L. (2019) Presynaptic NMDA receptors control nociceptive transmission at the spinal cord level in neuropathic pain. Cell. Mol. Life Sci. 76, 1889–1899 10.1007/s00018-019-03047-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pan H. L., Wu Z. Z., Zhou H. Y., Chen S. R., Zhang H. M., and Li D. P. (2008) Modulation of pain transmission by G-protein-coupled receptors. Pharmacol. Ther. 117, 141–161 10.1016/j.pharmthera.2007.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brown S. P., Safo P. K., and Regehr W. G. (2004) Endocannabinoids inhibit transmission at granule cell to Purkinje cell synapses by modulating three types of presynaptic calcium channels. J. Neurosci. 24, 5623–5631 10.1523/JNEUROSCI.0918-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Twitchell W., Brown S., and Mackie K. (1997) Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J. Neurophysiol. 78, 43–50 10.1152/jn.1997.78.1.43 [DOI] [PubMed] [Google Scholar]
- 34. Khasabova I. A., Harding-Rose C., Simone D. A., and Seybold V. S. (2004) Differential effects of CB1 and opioid agonists on two populations of adult rat dorsal root ganglion neurons. J. Neurosci. 24, 1744–1753 10.1523/JNEUROSCI.4298-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Barski A., Cuddapah S., Cui K., Roh T. Y., Schones D. E., Wang Z., Wei G., Chepelev I., and Zhao K. (2007) High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837 10.1016/j.cell.2007.05.009 [DOI] [PubMed] [Google Scholar]
- 36. Peters A. H., Kubicek S., Mechtler K., O'Sullivan R. J., Derijck A. A., Perez-Burgos L., Kohlmaier A., Opravil S., Tachibana M., Shinkai Y., Martens J. H., and Jenuwein T. (2003) Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol. Cell 12, 1577–1589 10.1016/S1097-2765(03)00477-5 [DOI] [PubMed] [Google Scholar]
- 37. Tachibana M., Sugimoto K., Nozaki M., Ueda J., Ohta T., Ohki M., Fukuda M., Takeda N., Niida H., Kato H., and Shinkai Y. (2002) G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 16, 1779–1791 10.1101/gad.989402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Roopra A., Qazi R., Schoenike B., Daley T. J., and Morrison J. F. (2004) Localized domains of G9a-mediated histone methylation are required for silencing of neuronal genes. Mol. Cell 14, 727–738 10.1016/j.molcel.2004.05.026 [DOI] [PubMed] [Google Scholar]
- 39. Tahiliani M., Mei P., Fang R., Leonor T., Rutenberg M., Shimizu F., Li J., Rao A., and Shi Y. (2007) The histone H3K4 demethylase SMCX links REST target genes to X-linked mental retardation. Nature 447, 601–605 10.1038/nature05823 [DOI] [PubMed] [Google Scholar]
- 40. Berman J. S., Symonds C., and Birch R. (2004) Efficacy of two cannabis based medicinal extracts for relief of central neuropathic pain from brachial plexus avulsion: results of a randomised controlled trial. Pain 112, 299–306 10.1016/j.pain.2004.09.013 [DOI] [PubMed] [Google Scholar]
- 41. Karst M., Salim K., Burstein S., Conrad I., Hoy L., and Schneider U. (2003) Analgesic effect of the synthetic cannabinoid CT-3 on chronic neuropathic pain: a randomized controlled trial. JAMA 290, 1757–1762 10.1001/jama.290.13.1757 [DOI] [PubMed] [Google Scholar]
- 42. Stockings E., Campbell G., Hall W. D., Nielsen S., Zagic D., Rahman R., Murnion B., Farrell M., Weier M., and Degenhardt L. (2018) Cannabis and cannabinoids for the treatment of people with chronic noncancer pain conditions: a systematic review and meta-analysis of controlled and observational studies. Pain 159, 1932–1954 10.1097/j.pain.0000000000001293 [DOI] [PubMed] [Google Scholar]
- 43. Häuser W., Petzke F., and Fitzcharles M. A. (2018) Efficacy, tolerability and safety of cannabis-based medicines for chronic pain management: an overview of systematic reviews. Eur. J. Pain 22, 455–470 10.1002/ejp.1118 [DOI] [PubMed] [Google Scholar]
- 44. Lötsch J., Weyer-Menkhoff I., and Tegeder I. (2018) Current evidence of cannabinoid-based analgesia obtained in preclinical and human experimental settings. Eur. J. Pain 22, 471–484 10.1002/ejp.1148 [DOI] [PubMed] [Google Scholar]
- 45. Nugent S. M., Morasco B. J., O'Neil M. E., Freeman M., Low A., Kondo K., Elven C., Zakher B., Motu'apuaka M., Paynter R., and Kansagara D. (2017) The effects of cannabis among adults with chronic pain and an overview of general harms: a systematic review. Ann. Intern. Med. 167, 319–331 10.7326/M17-0155 [DOI] [PubMed] [Google Scholar]
- 46. Xiong W., Cui T., Cheng K., Yang F., Chen S. R., Willenbring D., Guan Y., Pan H. L., Ren K., Xu Y., and Zhang L. (2012) Cannabinoids suppress inflammatory and neuropathic pain by targeting α3 glycine receptors. J. Exp. Med. 209, 1121–1134 10.1084/jem.20120242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zimmer A., Zimmer A. M., Hohmann A. G., Herkenham M., and Bonner T. I. (1999) Increased mortality, hypoactivity, and hypoalgesia in cannabinoid CB1 receptor knockout mice. Proc. Natl. Acad. Sci. U.S.A. 96, 5780–5785 10.1073/pnas.96.10.5780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Beltramo M., Bernardini N., Bertorelli R., Campanella M., Nicolussi E., Fredduzzi S., and Reggiani A. (2006) CB2 receptor-mediated antihyperalgesia: possible direct involvement of neural mechanisms. Eur. J. Neurosci. 23, 1530–1538 10.1111/j.1460-9568.2006.04684.x [DOI] [PubMed] [Google Scholar]
- 49. Wotherspoon G., Fox A., McIntyre P., Colley S., Bevan S., and Winter J. (2005) Peripheral nerve injury induces cannabinoid receptor 2 protein expression in rat sensory neurons. Neuroscience 135, 235–245 10.1016/j.neuroscience.2005.06.009 [DOI] [PubMed] [Google Scholar]
- 50. Zhang J., Hoffert C., Vu H. K., Groblewski T., Ahmad S., and O'Donnell D. (2003) Induction of CB2 receptor expression in the rat spinal cord of neuropathic but not inflammatory chronic pain models. Eur. J. Neurosci. 17, 2750–2754 10.1046/j.1460-9568.2003.02704.x [DOI] [PubMed] [Google Scholar]
- 51. Gutierrez T., Crystal J. D., Zvonok A. M., Makriyannis A., and Hohmann A. G. (2011) Self-medication of a cannabinoid CB2 agonist in an animal model of neuropathic pain. Pain 152, 1976–1987 10.1016/j.pain.2011.03.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Petrov R. R., Knight L., Chen S. R., Wager-Miller J., McDaniel S. W., Diaz F., Barth F., Pan H. L., Mackie K., Cavasotto C. N., and Diaz P. (2013) Mastering tricyclic ring systems for desirable functional cannabinoid activity. Eur. J. Med. Chem. 69, 881–907 10.1016/j.ejmech.2013.09.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kim S. H., and Chung J. M. (1992) An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 50, 355–363 10.1016/0304-3959(92)90041-9 [DOI] [PubMed] [Google Scholar]
- 54. Chen S. R., Hu Y. M., Chen H., and Pan H. L. (2014) Calcineurin inhibitor induces pain hypersensitivity by potentiating pre- and postsynaptic NMDA receptor activity in spinal cords. J. Physiol. 592, 215–227 10.1113/jphysiol.2013.263814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kelly S., and Chapman V. (2003) Cannabinoid CB1 receptor inhibition of mechanically evoked responses of spinal neurones in control rats, but not in rats with hindpaw inflammation. Eur. J. Pharmacol. 474, 209–216 10.1016/S0014-2999(03)02085-5 [DOI] [PubMed] [Google Scholar]
- 56. Jones M. R., Wang Z. Y., and Bjorling D. E. (2015) Intrathecal cannabinoid-1 receptor agonist prevents referred hyperalgesia in acute acrolein-induced cystitis in rats. Am. J. Clin. Exp. Urol. 3, 28–35 [PMC free article] [PubMed] [Google Scholar]
- 57. Sun J., Chen S. R., Chen H., and Pan H. L. (2019) μ-Opioid receptors in primary sensory neurons are essential for opioid analgesic effect on acute and inflammatory pain and opioid-induced hyperalgesia. J. Physiol. 597, 1661–1675 10.1113/JP277428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Laedermann C. J., Pertin M., Suter M. R., and Decosterd I. (2014) Voltage-gated sodium channel expression in mouse DRG after SNI leads to re-evaluation of projections of injured fibers. Mol. Pain 10, 19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chaplan S. R., Bach F. W., Pogrel J. W., Chung J. M., and Yaksh T. L. (1994) Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 53, 55–63 10.1016/0165-0270(94)90144-9 [DOI] [PubMed] [Google Scholar]
- 60. Chen S. R., Cai Y. Q., and Pan H. L. (2009) Plasticity and emerging role of BKCa channels in nociceptive control in neuropathic pain. J. Neurochem. 110, 352–362 10.1111/j.1471-4159.2009.06138.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pan Y. Z., and Pan H. L. (2004) Primary afferent stimulation differentially potentiates excitatory and inhibitory inputs to spinal lamina II outer and inner neurons. J. Neurophysiol. 91, 2413–2421 10.1152/jn.01242.2003 [DOI] [PubMed] [Google Scholar]
- 62. Zhou H. Y., Chen S. R., Chen H., and Pan H. L. (2008) Sustained inhibition of neurotransmitter release from nontransient receptor potential vanilloid type 1-expressing primary afferents by μ-opioid receptor activation-enkephalin in the spinal cord. J. Pharmacol. Exp. Ther. 327, 375–382 10.1124/jpet.108.141226 [DOI] [PubMed] [Google Scholar]
- 63. Zhou H. Y., Chen S. R., Chen H., and Pan H. L. (2010) Opioid-induced long-term potentiation in the spinal cord is a presynaptic event. J. Neurosci. 30, 4460–4466 10.1523/JNEUROSCI.5857-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.