

Abstract
Background
Vaginal atrophy is a frequent complaint of postmenopausal women; symptoms include vaginal dryness, itching, discomfort and painful intercourse. Systemic treatment for these symptoms in the form of oral hormone replacement therapy is not always necessary. An alternative choice is oestrogenic preparations administered vaginally (in the form of creams, pessaries, tablets and the oestradiol‐releasing ring). This is an update of a Chochrane systematic review; the original version was first published in October 2006.
Objectives
The objective of this review was to compare the efficacy and safety of intra‐vaginal oestrogenic preparations in relieving the symptoms of vaginal atrophy in postmenopausal women.
Search methods
We searched the following databases and trials registers to April 2016: Cochrane Gynaecology and Fertility Group Register of trials, The Cochrane Central Register of Controlled Trials (CENTRAL; 2016 issue 4), MEDLINE, Embase, PsycINFO, DARE, the Web of Knowledge, OpenGrey, LILACS, PubMed and reference lists of articles. We also contacted experts and researchers in the field.
Selection criteria
The inclusion criteria were randomised comparisons of oestrogenic preparations administered intravaginally in postmenopausal women for at least 12 weeks for the treatment of symptoms resulting from vaginal atrophy or vaginitis.
Data collection and analysis
Two review authors independently assessed trial eligibility and risk of bias and extracted the data. The primary review outcomes were improvement in symptoms (participant‐assessed), and the adverse event endometrial thickness. Secondary outcomes were improvement in symptoms (clinician‐assessed), other adverse events (breast disorders e.g. breast pain, enlargement or engorgement, total adverse events, excluding breast disorders) and adherence to treatment. We combined data to calculate pooled risk ratios (RRs) (dichotomous outcomes) and mean differences (MDs) (continuous outcomes) and 95% confidence intervals (CIs). Statistical heterogeneity was assessed using the I2 statistic. We assessed the overall quality of the evidence for the main comparisons using GRADE methods.
Main results
We included 30 RCTs (6235 women) comparing different intra‐vaginal oestrogenic preparations with each other and with placebo. The evidence was low to moderate quality; limitations were poor reporting of study methods and serious imprecision (effect estimates with wide confidence intervals)
1. Oestrogen ring versus other regimens
Other regimens included oestrogen cream, oestrogen tablets and placebo. There was no evidence of a difference in improvement in symptoms (participant assessment) either between oestrogen ring and oestrogen cream (odds ratio (OR) 1.33, 95% CI 0.80 to 2.19, two RCTs, n = 341, I2 = 0%, low‐quality evidence) or between oestrogen ring and oestrogen tablets (OR 0.78, 95% CI 0.53 to 1.15, three RCTs, n = 567, I2 = 0%, low‐quality evidence). However, a higher proportion of women reported improvement in symptoms following treatment with oestrogen ring compared with placebo (OR 12.67, 95% CI 3.23 to 49.66, one RCT, n = 67). With respect to endometrial thickness, a higher proportion of women who received oestrogen cream showed evidence of increase in endometrial thickness compared to those who were treated with oestrogen ring (OR 0.36, 95% CI 0.14 to 0.94, two RCTs, n = 273; I2 = 0%, low‐quality evidence). This may have been due to the higher doses of cream used.
2. Oestrogen tablets versus other regimens
Other regimens in this comparison included oestrogen cream, and placebo. There was no evidence of a difference in the proportions of women who reported improvement in symptoms between oestrogen tablets and oestrogen cream (OR 1.06, 95% CI 0.55 to 2.01, two RCTs, n = 208, I2 = 0% low‐quality evidence). A higher proportion of women who were treated with oestrogen tablets reported improvement in symptoms compared to those who received placebo using a fixed‐effect model (OR 12.47, 95% CI 9.81 to 15.84, two RCTs, n = 1638, I2 = 83%, low‐quality evidence); however, using a random‐effect model did not demonstrate any evidence of a difference in the proportions of women who reported improvement between the two treatment groups (OR 5.80, 95% CI 0.88 to 38.29). There was no evidence of a difference in the proportions of women with increase in endometrial thickness between oestrogen tablets and oestrogen cream (OR 0.31, 95% CI 0.06 to 1.60, two RCTs, n = 151, I2 = 0%, low‐quality evidence).
3. Oestrogen cream versus other regimens
Other regimens identified in this comparison included isoflavone gel and placebo. There was no evidence of a difference in the proportions of women with improvement in symptoms between oestrogen cream and isoflavone gel (OR 2.08, 95% CI 0.08 to 53.76, one RCT, n = 50, low‐quality evidence). However, there was evidence of a difference in the proportions of women with improvement in symptoms between oestrogen cream and placebo with more women who received oestrogen cream reporting improvement in symptoms compared to those who were treated with placebo (OR 4.10, 95% CI 1.88 to 8.93, two RCTs, n = 198, I2 = 50%, low‐quality evidence). None of the included studies in this comparison reported data on endometrial thickness.
Authors' conclusions
There was no evidence of a difference in efficacy between the various intravaginal oestrogenic preparations when compared with each other. However, there was low‐quality evidence that intra‐vaginal oestrogenic preparations improve the symptoms of vaginal atrophy in postmenopausal women when compared to placebo. There was low‐quality evidence that oestrogen cream may be associated with an increase in endometrial thickness compared to oestrogen ring; this may have been due to the higher doses of cream used. However there was no evidence of a difference in the overall body of evidence in adverse events between the various oestrogenic preparations compared with each other or with placebo.
Plain language summary
Use by postmenopausal women of creams, pessaries or a vaginal ring to apply oestrogen vaginally for symptoms of vaginal dryness
Review question
Cochrane researchers reviewed the evidence about the efficacy and safety of intravaginal oestrogenic preparations compared with each other or placebo (inactive or sham treatment) in women undergoing treatment for the symptoms of vaginal atrophy.
Background
Vaginal atrophy is a common condition in women after menopause. It causes vaginal dryness and itching and can make intercourse painful. The female hormone oestrogen is a treatment option for vaginal atrophy, but can cause adverse effects such as increased thickness in the lining of the womb (endometrium) which could be due to endometrial hyperplasia or cancer (resulting in vaginal bleeding) and breast tenderness. Oestrogen is available as an oral tablet, skin patch or implant under the skin. Alternatively, women can apply the hormone locally using creams, pessaries (tablets placed in the vagina) or a hormone‐releasing ring placed in the vagina. There is, therefore, the need to evaluate the efficacy and safety of these locally‐administered oestrogenic preparations.
Study characteristics
We found 30 randomised controlled trials comparing intravaginal oestrogenic preparations with one another or with placebo in a total of 6235 postmenopausal women undergoing treatment for the symptoms of vaginal atrophy. The evidence is current to April 2016.
Key results
There was no evidence of a difference in the proportions of women who reported improvement in symptoms of vaginal atrophy between the following treatment comparisons: oestrogen ring and oestrogen cream, oestrogen ring and oestrogen tablets, oestrogen tablets and oestrogen cream, oestrogen cream and isoflavone gel. However, a higher proportion of women reported improvement in symptoms in the following active treatments compared with placebo: oestrogen ring versus placebo, oestrogen tablets versus placebo and oestrogen cream versus placebo. In the case of oestrogen tablets versus placebo and using a random‐effect model for analysis of the data because of substantial heterogeneity, there was no longer evidence of a difference in effect on improvement in symptoms.
With respect to safety, a higher proportion of women who received oestrogen cream showed evidence of increase in endometrial thickness compared to those who were treated with oestrogen ring, which may have been due to the higher doses of cream used. However, there was no evidence of a difference in the proportions of women with increase in thickness of the lining of the womb between oestrogen tablets and oestrogen cream.
Quality of the evidence:
The evidence was of low quality for both improvement in symptoms as reported by women and increase in endometrial thickness. The main limitations of the evidence were poor reporting of study methods, and lack of precision (i.e. effect estimates with wide confidence intervals) in the findings for both outcomes.
Summary of findings
Background
Description of the condition
The ovaries produce a large proportion of circulating oestrogen in women of reproductive age. Before menopause the vagina is made up of thick layers of healthy cells. Oestrogen encourages growth and development of these cells so the vaginal epithelium remains thick and moist and the vagina supple and elastic. However, a dramatic reduction in circulating oestrogen occurs following the loss of ovarian function at the menopause. This oestrogen deficiency induces metabolic and trophic changes. Initially, these may include hot flushes and sweating, mood and sleep disturbances and fatigue. Skin begins to loose its elasticity and bone its strength. Changes occur in the vagina and other genital tissues. These tissues become thinner, drier and less elastic. Low oestrogen levels result in genital areas becoming dry, itchy and more easily irritated. A decrease in blood flow leads to fewer secretions and more dryness and intercourse can become uncomfortable or painful.
Falling oestrogen levels also cause changes in the lower urinary tract: for example the urinary bladder and urethra may display symptoms of urge incontinence, urgency, and frequency. A deterioration in vaginal and urethral tissues is called genitourinary syndrome (Roberts 2016) with a low oestrogen level being the primary cause.
Assessment of vaginal pH is a method for establishing vaginal atrophy (Nilsson 1992). At menopause decreased oestrogen levels cause the vaginal pH to rise to between 6.0 and 7.5 (Caillouette 1997; Crandall 2002). A low premenopausal vaginal pH of 3.5 to 4.5 helps to prevent colonisation with uropathogens. Colonisation can lead to vaginal infection. Therefore, restoration of vaginal pH reflects a clinically important achievement in maintaining the body's natural protection against vaginal infection (Henriksson 1994).
It has been suggested that about 50% of otherwise healthy women over 60 years of age have symptoms related to vaginal atrophy (Iosif 1984). In about 45% of menopausal women vaginal atrophy can be clinically manifest as a syndrome of vaginal dryness, itching, irritation and dyspareunia (painful intercourse) (Bygdeman 1996).
Description of the intervention
Oestrogen‐based hormone therapy (HT) is effective in treating symptoms of vaginal atrophy in postmenopausal women (Campbell 1977). However, only a small percentage (10%) of those who would benefit from oestrogen therapy actually receive it (Berg 1988), since women are often reluctant to volunteer that they have significant vaginal symptoms because of embarrassment (Notelovitz 1997: SOGC 2005). Several forms of HT are available, systemic dosage forms include oral, transdermal preparations, nasal sprays, and injectable (not commonly used), as well as local dosage forms using the vaginal route. Local dosage can be administered in the form of the oestradiol‐releasing vaginal ring, oestrogen‐based vaginal creams, pessaries containing oestriol and a slow‐release 17β‐oestradiol tablet. The harmful effects of HT have resulted in considerable caution in the manner it is currently used (Manson 2016). Current guidelines advise local oestrogen administration over systemic HT if vaginal atrophy is the only indication for treatment ( ACOG 2014; NAMS 2013).
How the intervention might work
Locally administered oestrogenic preparations work directly on the oestrogen‐sensitive tissues of the lower genito‐urinary tract, relieving the symptoms of vaginal atrophy. Local treatment does not induce altered liver metabolism and this makes it possible to use lower doses of oestrogen compared with oral therapy (Heimer 1984). This mechanism of action of local oestrogen results in reduction of systemic adverse effects associated with systemic oestrogen such as bleeding, breast tenderness, and endometrial stimulation (Gerbaldo 1991; Kivovic 1980; Mattson 1989; Mettler 1991). Although local treatment appears to have fewer adverse effects many women consider creams and pessaries to be messy and application times difficult to remember. An alternative treatment is a silicone ring with an oestradiol‐loaded core which can be inserted and left within the vagina to provide a relatively low and constant release of hormone.
Why it is important to do this review
This review evaluated the efficacy and safety of intra‐vaginal oestrogenic preparations for the treatment of vaginal atrophy in postmenopausal women to inform patient‐centred decision making. Different local oestrogenic preparations are currently available for the treatment of vaginal atrophy in postmenopausal women. However, uncertainty still exists as to the efficacy and safety of these various preparations. It has, therefore become imperative to critically appraise the existing empirical evidence related to the efficacy and safety of the various local oestrogenic therapies relative to each other.
This is an update of a Cochrane review first published in 2003 (Suckling 2003), and previously updated in 2006 (Suckling 2006). In the 2006 version,there was evidence that creams, tablets and the oestradiol vaginal ring appeared to be equally effective but more potent than placebo for the symptoms of vaginal atrophy. With respect to safety, there was evidence that the use of conjugated equine oestrogen cream resulted in more incidents of vaginal bleeding than oestrogen ring, as reported by two trials in 274 participants.
Objectives
The objective of this review was to compare the efficacy and safety of intra‐vaginal oestrogenic preparations in relieving the symptoms of vaginal atrophy in postmenopausal women.
Methods
Criteria for considering studies for this review
Types of studies
Published and unpublished randomised controlled trials (RCTs) comparing oestrogenic preparations administered intra‐vaginally for a duration of at least three months in postmenopausal women for the treatment of symptoms resulting from vaginal atrophy or vaginitis were eligible for inclusion. We excluded non‐randomised studies (e.g. studies with evidence of inadequate sequence generation such as alternate days, patient numbers) as they are associated with a high risk of bias.
Types of participants
Postmenopausal women, who had not menstruated for more than 12 months or who had a serum follicle stimulating hormone (FSH) level >= 40 IU/L were eligible for inclusion. Women who had undergone bilateral oophorectomy (removal of both ovaries) were also eligible for inclusion. Women with intercurrent major disease or who had had previous hormone therapy (HT) within three months of commencement of the study were excluded.
Types of interventions
Trials comparing oestrogen supplementation administered intra‐vaginally versus any other active intervention or placebo were eligible for inclusion. These included creams or gels, tablets, vagitories, ovules, pessaries, and an oestradiol‐releasing ring. Duration of treatment must have been at least three months, as this treatment duration should be sufficient to improve vaginal symptoms. For the purpose of the review vagitories, ovules and pessaries were termed as vaginal tablets.
Types of outcome measures
Primary outcomes
1. Improvement in symptoms as assessed by participants
Proportion of women showing cure or improvement (mild symptoms) in most bothersome symptoms relating to atrophy such as dryness, dyspareunia, itching, burning sensation and discomfort at end point (immediately following treatment), assessed using standardised instruments. We grouped symptoms and analysed them as a composite i.e. aggregate symptoms.
2. Endometrial thickness
This is a surrogate for adverse events such as endometrial hyperplasia/dysplasia or cancer. It was assessed as the proportion of women showing evidence of increase in endometrial thickness evaluated through assessment of endometrial stimulation (measured by the progestogen challenge test with withdrawal bleeding, ultrasound measurement of endometrial thickness, assessed at end point.
Secondary outcomes
3. Improvement in symptoms as assessed visually by clinicians
Proportion of women showing cure or improvement (mild symptoms) from clinician evaluation of the appearance of the vagina including vaginal mucosal pallor (pale appearance), petechiae (small red spots on the skin), friability (fragile and delicate tissue) and dryness at end point (immediately after treatment). We grouped symptoms and analysed them as a composite i.e. aggregate symptoms.
4. Improvement in symptoms as assessed by clinicians using laboratory parameters
Measurement of decrease in vaginal pH and assessment of increase in maturation indices, that is cytological assessment for parabasal, intermediate and superficial cells at end points (immediately after treatment). We analysed vaginal pH and maturation indices separately.
5. Other adverse events
Proportion of women with adverse events at end point from treatment, including breast disorders (e.g. breast pain, enlargement or engorgement which may be considered a surrogate marker (indirect indicator) for systemic absorption, and blood oestradiol levels), and total adverse events (excluding breast disorders).
6. Adherence to treatment
Proportion of women adhering to treatment regimen assessed immediately after treatment using participants who completed treatment adherence sheets.
Search methods for identification of studies
We searched for all published and unpublished RCTs of studies comparing intra‐vaginal oestrogen supplementation with any other active intervention or placebo, without language restriction and in consultation with the Gynaecology and Fertility Group Information Specialist.
Electronic searches
-
We searched the following electronic databases, trials registers and websites.
Cochrane Gynaecology and Fertility Specialised Register of Controlled Trials (searched 12 April 2016) (see Appendix 1 for search strategy)
the Cochrane Central Register of Controlled Trials (CENTRAL; The Cochrane Library 2016, Issue 4 (searched 12 April 2016) (see Appendix 2 for search strategy)
MEDLINE via Ovid (from 1966 to 12 April 2016) (see Appendix 3 for search strategy)
Embase via Ovid (1980 to 12 April 2016) (see Appendix 4 for search strategy)
PsycINFO via Ovid (1972 to 12 April 2016) (see Appendix 5 for search strategy
We combined the MEDLINE search with the Cochrane highly sensitive search strategy for identifying randomised trials in MEDLINE (Lefebvre 2011). We combined the Embase and PsycINFO searches with trial filters developed by the Scottish Intercollegiate Guidelines Network (SIGN) sign.ac.uk/methodology/filters.html#random.
-
Other electronic sources of trials included:
-
trials registers for ongoing and registered trials:
clinicaltrials.gov (a service of the US National Institutes of Health) (date of last search 12 April 2016);
who.int/ictrp/search/en/ (The World Health Organisation International Trials Registry Platform search portal) (date of last search 12 April 2016);
DARE (Database of Abstracts of Reviews of Effects) in the Cochrane Library at onlinelibrary.wiley.com/o/cochrane/cochrane_cldare_articles_fs.html (for reference lists from relevant non‐Cochrane reviews) (date of last search April 2016);
the Web of Knowledge wokinfo.com/ (another source of trials and conference abstracts) (date of last search 12 April 2016);
OpenGrey opengrey.eu/ for unpublished literature from Europe (date of last search 12 April 2016);
LILACS database lilacs.bvsalud.org/en/ (for trials from the Portuguese and Spanish speaking world) (date of last search 12 April 2016);
PubMed and Google Scholar (for recent trials not yet indexed in MEDLINE) (date of last search 12 April 2016).
-
There were no restrictions based on language, date of publication or study setting.
Searching other resources
We handsearched reference lists of articles retrieved by the search and contacted experts in the field to obtain additional data. We also handsearched relevant journals and conference abstracts that are not covered in the Cochrane Gynaecology and Fertility Specialised Register, in liaison with the Information Specialist.
Data collection and analysis
Selection of studies
After an initial screen of titles and abstracts retrieved by the search, conducted by ROA and AL, we retrieved the full texts of all potentially eligible studies. Two review authors (ROA and AL) independently examined these full‐text articles for compliance with the inclusion criteria and selected studies eligible for inclusion in the review. We contacted study investigators as required, to clarify study eligibility. We resolved disagreements as to study eligibility by discussion or by consulting a third review author. We documented the selection process with a PRISMA flow diagram (Moher 2009).
Data extraction and management
Two review authors (ROA and AL) independently extracted data from eligible studies using a data extraction form designed and pilot‐tested by the authors. We resolved any disagreements by discussion or by consulting a third review author. Data extracted included study characteristics and outcome data (see data extraction table for details, Appendix 6). We contacted study investigators for further data on methods or results, or both, as required.
Assessment of risk of bias in included studies
Two review authors (ROA and AL) independently assessed the included studies for risk of bias using the Cochrane risk of bias assessment tool to assess: selection (random sequence generation and allocation concealment); performance (blinding of participants and personnel); detection (blinding of outcome assessors); attrition (incomplete outcome data); reporting (selective reporting); and other bias such as imbalance in the numbers of participants randomised at baseline between treatment groups, and differences in demographic characteristics of participants between treatment groups (Higgins 2011). We took care to search for within‐trial selective reporting, such as trials failing to report obvious outcomes, or reporting them in insufficient detail to allow inclusion. We sought published protocols where possible and compared the outcomes between the protocol and the final published study. We resolved disagreements were resolved by discussion or by consulting a third review author. We described all judgements fully and presented the conclusions in the 'Risk of bias' table, which was incorporated into the interpretation of review findings.
Measures of treatment effect
For dichotomous data (e.g. proportion of participants showing improvement in symptoms), we used the numbers of events in the control and intervention groups of each study to calculate Mantel‐Haenszel odds ratios (ORs). For continuous data (e.g. endometrial thickness), we calculated mean differences (MDs) between treatment groups. We reversed the direction of effect of individual studies, if required, to ensure consistency across trials. We presented 95% confidence intervals (CI) for all outcomes. Where data to calculate ORs or MDs were not available, we utilised the most detailed numerical data available that facilitated similar analyses of included studies (e.g. test statistics, P values). We assessed whether the estimates calculated in the review for individual studies were compatible in each case with the estimates reported in the study publications.
Unit of analysis issues
The primary analysis was per woman randomised. If studies reported data that did not allow valid analysis, we contacted study authors for further details; if we could not obtain appropriate data, such data were not included in meta‐analyses.
Dealing with missing data
We analysed the data on an intention‐to‐treat (ITT) basis as far as possible and attempts were made to obtain missing data from the original trialists. Where these were unobtainable, we analysed only the available data.
Assessment of heterogeneity
We considered whether the clinical and methodological characteristics of the included studies were sufficiently similar for meta‐analysis to provide a clinically meaningful summary. We assessed statistical heterogeneity by the measure of the I2 statistic (Higgins 2003). An I2 measurement greater than 50% was taken to indicate substantial heterogeneity (Deeks 2011)
Assessment of reporting biases
In view of the difficulty of detecting and correcting for publication bias and other reporting biases, we minimised their potential impact by ensuring a comprehensive search for eligible studies and by being alert for duplication of data. Where there were 10 or more studies in an analysis, we planned to use a funnel plot to explore the possibility of small study effects (a tendency for estimates of the intervention effect to be more beneficial in smaller studies) (Sterne 2011). However, this was not undertaken because none of the analyses included 10 or more studies.
Data synthesis
Where studies were considered sufficiently similar, we combined the data using a fixed‐effect model in the following comparisons.
Oestrogen ring versus other regimens
Oestrogen tablets versus other regimens
Oestrogen cream or gel versus other regimens
We stratified all comparisons by other regimens.
An increase in the odds of a particular outcome, which may be beneficial (e.g. improvement in symptoms) or detrimental (e.g. adverse events), was displayed graphically in the meta‐analyses to the right of the centre‐line, and a decrease in the odds of an outcome to the left of the centre‐line.
Subgroup analysis and investigation of heterogeneity
We planned to conduct subgroup analyses to determine the separate evidence within the following subgroups.
Duration of symptoms of vaginal atrophy
Severity of symptoms of vaginal atrophy
We could not conduct subgroup analyses, however, due to insufficient data.
Where substantial heterogeneity was detected, we explored possible explanations in sensitivity analyses. We took any statistical heterogeneity into account when interpreting the results, especially where there was any variation in the direction of effect.
Sensitivity analysis
We conducted sensitivity analyses for the primary outcomes (improvement in symptoms as assessed by the women, and endometrial thickness) to determine whether the conclusions were robust to arbitrary decisions made regarding the eligibility and analysis. These analyses included consideration of whether the review conclusions would have differed if:
the summary effect measure was relative risk rather than odds ratio; and
we had adopted a random‐effects model.
Overall quality of the body of evidence: 'Summary of findings' table
We prepared 'Summary of findings' tables using GRADEpro GDT (GRADEpro GDT 2015). These tables evaluated the overall quality of the body of evidence for the main review outcomes (improvement in symptoms as assessed by the women, endometrial thickness, improvement in symptoms as assessed by clinicians and total adverse events), using GRADE criteria (study limitations (i.e. risk of bias), consistency of effect, imprecision, indirectness and publication bias) (Schünemann 2011). Judgements about evidence quality (high, moderate or low) were justified, documented, and incorporated into reporting of results for each outcome.
Results
Description of studies
Results of the search
The searches retrieved a total of 645 articles after removal of duplicates. Fifty‐eight studies (59 references) were potentially eligible and were retrieved in full text. We included 30 studies (31 references) that met our inclusion criteria; we excluded 26 studies, while one study (two references) was classified as 'awaiting classification'. See study tables: Characteristics of included studies, Characteristics of excluded studies, Characteristics of studies awaiting classification.
We have illustrated the process involved in the inclusion and exclusion of studies in a PRISMA flow diagram (Figure 1).
1.
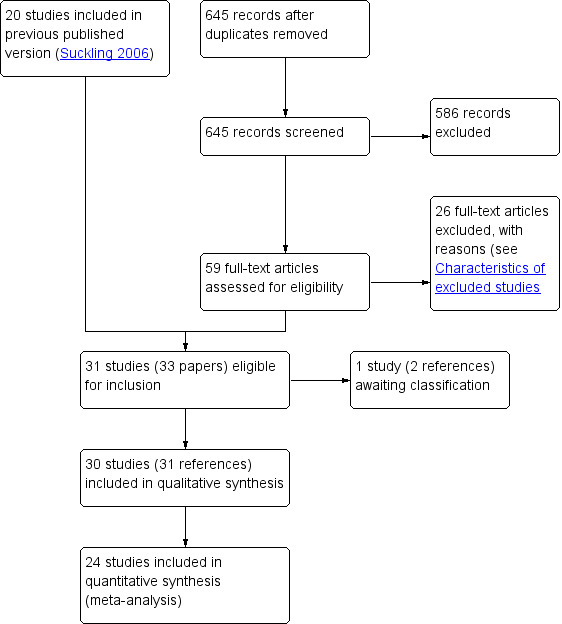
Study flow diagram
Included studies
Study design and setting
We included 30 parallel‐design randomised controlled trials (RCTs). Single centre trials took place in Brazil (Fernandes 2014; Lima 2013), India (Raghunandan 2010), Iran (Hosseinzadeh 2015), Italy (Sardinia) (Dessole 2004), Mexico (Garcia Lara 1993), Sweden (Bygdeman 1996), Thailand (Manonai 2001) and USA (Karp 2012). Multicentre trials took place in Australia (Ayton 1996; Weisberg 2000); Austria, Switzerland and Germany (Casper 1999 study 1); Belgium (Foidart 1991); Canada (Rioux 2000); Canada and USA (Bachmann 2009; Simon 2008); Croatia (Simunic 2003); Denmark (Eriksen 1992; Lose 2000); Germany (Casper 1999 study 2; Griesser 2012); Netherlands (Barentsen 1997); Norway (Dugal 2000); Spain (Cano 2012); Sweden, Finland and Denmark (Henriksson 1994); and USA (Bachmann 2008; Nachtigall 1995; Speroff 2003). Two trials did not state their location (Mac Bride 2014; Nachtigall 1994).
Participants
The studies included 6235 postmenopausal women with symptoms of vaginal atrophy. The mean age across studies ranged from 45 to 66 years. Most of the included trials required that women had any, or all symptoms of urogenital atrophy: vaginal dryness with or without dyspareunia, pruritus, dysuria and or urgency; and signs of atrophic vaginitis, including: pallor (pale appearance to skin), petechiae, friability (fragile and delicate skin) and dryness. Other inclusion criteria included being naturally menopausal for at least one year, or surgically menopausal (bilateral oophorectomy) for at least one year. Exclusion criteria for most studies included: known to have hormone‐dependent neoplasia and women who had taken systemic or vaginal oestrogens within three to six months of commencement of the study. Full details of the inclusion and exclusion criteria are found in the Characteristics of included studies.
Interventions
The trials included a wide variety of interventions under three broad comparisons:
-
Oestrogen ring versus other regimens:
oestrogen ring versus oestrogen cream (Ayton 1996; Barentsen 1997; Nachtigall 1995);
oestrogen ring versus oestrogen tablets (Casper 1999 study 1; Henriksson 1994; Lose 2000; Weisberg 2000);
oestrogen ring versus placebo ring (Casper 1999 study 2; Karp 2012; Speroff 2003).
-
Oestrogen tablets versus other regimens:
oestrogen tablets versus oestrogen cream (Ayton 1996; Hosseinzadeh 2015; Manonai 2001; Rioux 2000);
oestrogen tablets versus placebo tablets (Bachmann 2008; Dessole 2004; Eriksen 1992; Foidart 1991; Garcia Lara 1993; Griesser 2012); Simon 2008; Simunic 2003);
oestradiol tablets versus oestriol tablets: (Dugal 2000).
-
Oestrogen cream versus other regimens:
oestrogen cream versus non‐hormonal local bio adhesive vaginal moisturising gel (Bygdeman 1996; Nachtigall 1994);
oestrogen cream versus isoflavone gel (Lima 2013);
oestrogen cream versus non‐hormonal lubricant gel (Raghunandan 2010);
oestrogen cream versus placebo cream (Bachmann 2009; Cano 2012).
The duration of intervention was 12 weeks in most of the included studies. In other studies, the duration varied between 13 weeks (Speroff 2003), 15 weeks (Nachtigall 1995), four months (Garcia Lara 1993), six months (Dessole 2004; Dugal 2000; Foidart 1991; Lose 2000; Rioux 2000) and 12 months (Simunic 2003).
Outcomes
In the previous update, the review authors excluded one study on the basis of not reporting outcomes relevant to the review, and we did not include this study in the current update either (Tolino 1990). In the current update, we did not exclude any studies for not reporting the review's relevant outcomes. Symptoms reported by the included studies were many and disparate, therefore we grouped symptoms found to be consistent across studies as composites, where appropriate. For participant‐assessed improvement, the symptoms found to be consistent across studies were dryness, itching, dysuria and dyspareunia. For clinician‐assessed improvement, the components of composite were vaginal mucosal pallor (pale appearance), petechiae (small red spots on the skin), friability (fragile and delicate tissue) and dryness. The components of breast disorders were: breast pain, enlargement and engorgement. We considered consistency in directions of effect estimates of individual components before we reported them as composites. Thus all symptoms grouped as composites have similar directions in effect estimates.
Primary outcomes
The primary outcomes for this review are improvement in symptoms as assessed by the women, and endometrial thickness (adverse event).
Twelve of the included studies measured improvement in symptoms, defined as the proportion of women showing cure or improvement (mild symptoms) in most bothersome symptoms relating to atrophy (participant‐assessed at end point i.e. immediately after treatment) (Ayton 1996; Barentsen 1997; Cano 2012; Casper 1999 study 2; Garcia Lara 1993; Henriksson 1994; Hosseinzadeh 2015; Lima 2013; Lose 2000; Manonai 2001; Simunic 2003; Weisberg 2000). We assessed the severity of symptoms relating to atrophy at end point using the visual analogue scale (VAS) (or its modified form) in all the studies that reported this outcome. The VAS is a psychometric response scale for subjective assessment of certain attributes such as pain, which cannot be measured directly; it has been found to be sufficiently reliable and valid as a measuring instrument.
Four of the included studies assessed endometrial thickness (Ayton 1996; Manonai 2001; Nachtigall 1995; Rioux 2000).
Secondary outcomes
The review's secondary outcome measures are improvement in symptoms as assessed by clinicians through physical examination of the vagina, improvement in symptoms as assessed by clinicians using laboratory parameters (decrease in vaginal pH and increase in maturation indices), other adverse events (breast disorders and total adverse events) and adherence to treatment.
Eleven of the included studies measured improvement in symptoms (clinician‐assessed) (Ayton 1996; Barentsen 1997; Cano 2012; Casper 1999 study 2; Garcia Lara 1993; Griesser 2012; Henriksson 1994; Hosseinzadeh 2015; Lose 2000; Nachtigall 1995; Simunic 2003).
Eleven of the included studies assessed improvement in symptoms (decrease in vaginal pH) (Bachmann 2009; Barentsen 1997; Bygdeman 1996; Henriksson 1994; Cano 2012; Casper 1999 study 2; Dessole 2004; Griesser 2012; Karp 2012; Manonai 2001; Nachtigall 1994).
Eight of the included studies measured improvement in symptoms (increase in maturation indices) (Ayton 1996; Barentsen 1997; Cano 2012; Casper 1999 study 2; Griesser 2012; Karp 2012; Manonai 2001; Speroff 2003).
Five of the included studies measured adverse events (breast disorders) (Casper 1999 study 1; Dugal 2000; Henriksson 1994; Lose 2000; Nachtigall 1995) while four included studies measured total adverse events (Karp 2012; Nachtigall 1995; Raghunandan 2010; Simon 2008).
Five of the included studies assessed adherence to treatment (Ayton 1996; Dugal 2000; Henriksson 1994; Manonai 2001; Nachtigall 1995).
Six of the included studies either did not report any of the review's outcomes (Fernandes 2014) or measured them in non‐usable forms such as median (range), mean (SE) or mean without SD, dichotomous as against continuous data, reporting of only the level of statistical significance (P values), etc (Bachmann 2008; Mac Bride 2014; Casper 1999 study 2; Eriksen 1992; Foidart 1991;). We did not, therefore, include these studies in quantitative synthesis (meta‐analyses). We included Fernandes 2014 in the review, although it did not report any of the review's outcomes; the study focused mainly on sexual function‐related outcomes such as arousal, lubrication, satisfaction and orgasm.
Excluded studies
We excluded 26 studies from the review for the following reasons:
seven studies were not RCTs;
10 studies did not administer treatment for up to 12 weeks;
five studies did not make use of relevant interventions;
three studies did not include relevant participants;
one study did not report relevant outcomes (this was used as an exclusion criterion in the last update; studies were, however, not excluded on the basis of not reporting relevant outcomes in the current update).
Risk of bias in included studies
2.
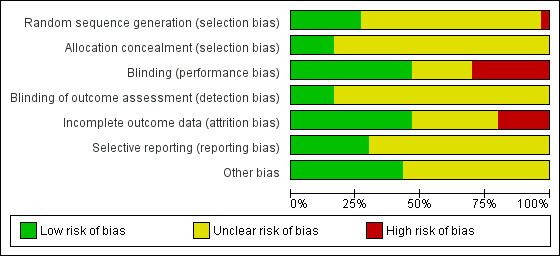
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies
3.
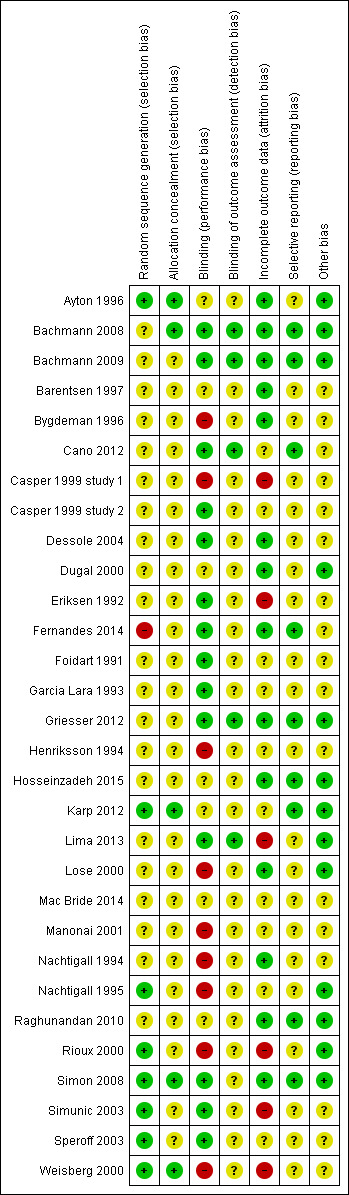
Risk of bias summary: review authors' judgements about each risk of bias item for each included study
Allocation
Sequence generation
Eight studies were at low risk of selection bias related to sequence generation, as they used computer randomisation or a random numbers table (Ayton 1996; Karp 2012; Nachtigall 1995; Rioux 2000; Simon 2008; Simunic 2003; Speroff 2003; Weisberg 2000). Twenty‐one studies did not describe the method used and were at unclear risk of this bias (Bachmann 2008; Bachmann 2009; Barentsen 1997; Mac Bride 2014; Bygdeman 1996; Cano 2012; Casper 1999 study 1; Casper 1999 study 2; Dessole 2004; Dugal 2000; Eriksen 1992; Foidart 1991; Garcia Lara 1993; Griesser 2012; Henriksson 1994; Hosseinzadeh 2015; Lima 2013; Lose 2000; Manonai 2001; Nachtigall 1994; Raghunandan 2010). We rated the remaining study as being at high risk of bias because the study authors stated that participants were given a number according to their order of inclusion in the study (Fernandes 2014).
Allocation concealment
Five studies were at low risk of selection bias related to allocation concealment, as they used sequentially‐numbered sealed opaque envelopes (Ayton 1996; Bachmann 2008; Karp 2012; Simon 2008; Weisberg 2000). The remaining 25 studies failed to describe methods of allocation concealment and we rated these as at unclear risk of bias for this domain (Bachmann 2009; Barentsen 1997; Mac Bride 2014; Bygdeman 1996; Cano 2012; Casper 1999 study 1; Casper 1999 study 2; Dessole 2004; Dugal 2000; Eriksen 1992; Fernandes 2014; Foidart 1991; Garcia Lara 1993; Griesser 2012; Henriksson 1994; Hosseinzadeh 2015; Lima 2013; Lose 2000; Manonai 2001; Nachtigall 1994; Nachtigall 1995; Raghunandan 2010; Rioux 2000; Simunic 2003; Speroff 2003).
Blinding
We considered that blinding might influence both the primary and secondary outcomes as some of them were subjectively assessed either by participants or personnel. Most of the included studies reported at least one of both primary and secondary outcomes. Fourteen studies were at low risk of performance bias because both participants and personnel were blinded (double‐blinded trials) (Bachmann 2008; Bachmann 2009; Cano 2012; Casper 1999 study 2; Dessole 2004; Eriksen 1992; Fernandes 2014; Foidart 1991; Garcia Lara 1993; Griesser 2012; Lima 2013; Simon 2008; Simunic 2003; Speroff 2003); seven studies did not report whether or not participants or personnel, or both, were blinded and we, therefore, rated them as unclear (Ayton 1996; Barentsen 1997; Mac Bride 2014; Dugal 2000; Hosseinzadeh 2015; Karp 2012; Raghunandan 2010). The remaining nine studies were open‐label trials and we, therefore, assessed them as being at high risk of performance bias (Bygdeman 1996; Casper 1999 study 1; Henriksson 1994; Lose 2000; Manonai 2001; Nachtigall 1994; Nachtigall 1995; Rioux 2000; Weisberg 2000).
In the domain of detection bias, we assessed five studies as being at low risk of bias because the outcome assessors were blinded as well as participants and personnel (some of the outcomes were assessed by participants and personnel) (Bachmann 2008; Bachmann 2009; Cano 2012; Griesser 2012; Lima 2013). In the remaining 25 studies, outcome assessment by participants, clinicians and assessors was not completely blinded and we, therefore, assessed them as unclear with respect to blinding of outcome assessment (detection bias) (Ayton 1996; Barentsen 1997; Mac Bride 2014; Bygdeman 1996; Casper 1999 study 1; Casper 1999 study 2; Dessole 2004; Dugal 2000; Eriksen 1992; Fernandes 2014; Foidart 1991; Garcia Lara 1993; Henriksson 1994; Hosseinzadeh 2015; Karp 2012; Lose 2000; Manonai 2001; Nachtigall 1994; Nachtigall 1995; Raghunandan 2010; Rioux 2000; Simon 2008; Simunic 2003; Speroff 2003; Weisberg 2000).
Incomplete outcome data
We rated 14 trials at low risk of bias with respect to attrition bias either because there were no withdrawals or losses to follow‐up or because they analysed all or most (> 95%) of the women randomised (Ayton 1996; Bachmann 2008; Bachmann 2009; Barentsen 1997; Bygdeman 1996; Dessole 2004; Dugal 2000; Fernandes 2014; Griesser 2012; Hosseinzadeh 2015; Lose 2000; Nachtigall 1994; Raghunandan 2010; Simon 2008). In 10 studies, there was insufficient evidence to make a conclusive judgement in relation to attrition bias and we, therefore, assessed them as unclear (Mac Bride 2014; Cano 2012; Casper 1999 study 2; Foidart 1991; Garcia Lara 1993; Henriksson 1994; Karp 2012; Manonai 2001; Nachtigall 1995; Speroff 2003). In the remaining six studies, the proportions of withdrawals, or reasons for withdrawals or losses to follow‐up differed substantially between the treatment groups and we did not analyse the data on the basis of ITT. We therefore rated these studies as being at high risk with respect to attrition bias (Casper 1999 study 1; Eriksen 1992; Lima 2013; Rioux 2000; Simunic 2003; Weisberg 2000).
Selective reporting
No protocol of any included study was available for assessment in relation to pre‐specified outcome measures. We therefore assessed this domain of bias using the information available in the methods sections of the published papers. We rated nine studies as being at low risk of selective reporting bias, as they reported all outcomes pre‐specified in the methods section (Bachmann 2008; Bachmann 2009; Cano 2012; Fernandes 2014; Griesser 2012; Hosseinzadeh 2015; Karp 2012; Raghunandan 2010; Simon 2008). The remaining 21 studies we rated at unclear risk of selective reporting bias, as there was insufficient information to make a conclusive judgement (Ayton 1996; Barentsen 1997; Mac Bride 2014; Bygdeman 1996; Casper 1999 study 1; Casper 1999 study 2; Dessole 2004; Dugal 2000; Eriksen 1992; Foidart 1991; Garcia Lara 1993; Henriksson 1994; Lima 2013; Lose 2000; Manonai 2001; Nachtigall 1994; Nachtigall 1995; Rioux 2000; Simunic 2003; Speroff 2003; Weisberg 2000).
Other potential sources of bias
We assessed 13 studies as being at low risk of within‐study bias as baseline demographic characteristics such as age and BMI were similar or the numbers of participants randomised to treatment groups were balanced at baseline (Ayton 1996; Bachmann 2008; Bachmann 2009; Dugal 2000; Griesser 2012; Hosseinzadeh 2015; Karp 2012; Lima 2013; Lose 2000; Nachtigall 1995; Raghunandan 2010; Rioux 2000; Simon 2008) The remaining 17 studies did not report sufficient information to make a conclusive judgement in relation to within‐study bias (Barentsen 1997; Mac Bride 2014; Bygdeman 1996; Cano 2012; Casper 1999 study 1; Casper 1999 study 2; Dessole 2004; Eriksen 1992; Fernandes 2014; Foidart 1991; Garcia Lara 1993; Henriksson 1994; Manonai 2001; Nachtigall 1994; Simunic 2003; Speroff 2003; Weisberg 2000).
Effects of interventions
See: Table 1; Table 2; Table 3
Summary of findings for the main comparison. Oestrogen ring compared to other regimens for vaginal atrophy in postmenopausal women.
| Oestrogen ring compared to other regimens for vaginal atrophy in postmenopausal women | ||||||
| Patient or population: postmenopausal women with vaginal atrophy Settings: outpatient clinic Intervention: oestrogen ring Comparison: other regimen | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Other regimen | Oestrogen ring | |||||
| Improvement in symptoms (participant‐assessed) (oestrogen ring vs oestrogen cream) | 717 per 1000 | 771 per 1000 (670 to 847) | OR 1.33 (0.80 to 2.19) | 341 ( 2studies) | ⊕⊕⊝⊝ low1,2 | |
| Improvement in symptoms (participant‐assessed) (oestrogen ring vs oestrogen tablets) | 582 per 1000 | 521 per 1000 (425 to 616) | OR 0.78 (0.53 to 1.15) | 567 (3 studies) | ⊕⊕⊝⊝ low1,2 | |
| Endometrial thickness (oestrogen ring vs oestrogen cream) | 117 per 1000 | 46 per 1000 (18 to 111) | OR 0.36 (0.14 to 0.94) | 273 (2 studies) | ⊕⊕⊝⊝ low1,3 | |
| Improvement in symptoms (clinician‐assessed) (oestrogen ring vs oestrogen cream) | 706 per 1000 | 714 per 1000 (627 to 786) | OR 1.04 (0.70 to 1.53) | 533 (3 studies) | ⊕⊕⊝⊝ low1,2 | |
| Improvement in symptoms (clinician‐assessed) (oestrogen ring vs oestrogen tablets) | 636 per 1000 | 717 per 1000 (611 to 802) | OR 1.45 (0.90 to 2.32) | 397 (2 studies) | ⊕⊕⊝⊝ low1,2 | |
| Adverse events (total adverse events) (oestrogen ring vs oestrogen cream) | 364 per 1000 | 335 per 1000 (212 to 483) | OR 0.88 (0.47 to 1.63) | 192 (1 study) | ⊕⊕⊝⊝ low2,3 | |
| Adverse events (total adverse events) (oestrogen ring vs placebo) | 444 per 1000 | 264 per 1000 (81 to 587) | OR 0.45 (0.11 to 1.78) | 37 (1 study) | ⊕⊕⊕⊝ moderate3 | |
| *The basis for the assumed risk is the median control group risk across studies. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Downgraded by 1 level as most risk of bias domains were rated either as unclear or high 2 Downgraded by 1 level due to effect estimate with wide confidence interval 3 Downgraded by 1 level due to small sample size
Summary of findings 2. Oestrogen tablets compared to other regimens for vaginal atrophy in postmenopausal women.
| Oestrogen tablets compared to other regimens for vaginal atrophy in postmenopausal women | ||||||
| Patient or population: postmenopausal women with vaginal atrophy Settings: outpatient clinic Intervention: oestrogen tablets Comparison: other regimen | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Other regimen | Oestrogen tablets | |||||
| Improvement in symptoms (participant‐assessed) (oestrogen tablets vs oestrogen cream) | 683 per 1000 | 695 per 1000 (542 to 812) | OR 1.06 (0.55 to 2.01) | 208 (2 studies) | ⊕⊕⊝⊝ low2,3 | |
| Improvement in symptoms (participant‐assessed) (oestrogen tablets vs placebo) | 294 per 1000 | 839 per 1000 (803 to 868) | OR 12.47 (9.81 to 15.84) | 1638 (2 studies) | ⊕⊕⊝⊝ low1,4 | Using a random effects model, there was no evidence of a difference in effect: OR 5.80, 95% CI 0.88 to 38.29 |
| Endometrial thickness (oestrogen tablets vs oestrogen cream) | 80 per 1000 | 26 per 1000 (5 to 122) | OR 0.31 (0.06 to 1.60) | 151 (2 studies) | ⊕⊕⊝⊝ low2,3 | |
| Improvement in symptoms (clinician‐assessed) (oestrogen tablets vs oestrogen cream) | 697 per 1000 | 699 per 1000 (612 to 774) | OR 1.03 (0.70 to 1.52) | 528 (3 studies) | ⊕⊕⊝⊝ low1,3 | |
| Improvement in symptoms (clinician‐assessed) (oestrogen tablets vs placebo) | 262 per 1000 | 820 per 1000 (787 to 849) | OR 12.85 (10.39 to 15.89) | 2078 (4 studies) | ⊕⊕⊝⊝ low1,5 | |
| Adverse events (total adverse events) (oestrogen tablets vs placebo) | 19 per 1000 | 24 per 1000 (5 to 115) | OR 1.27 (0.24 to 6.69) | 309 (1 study) | ⊕⊕⊕⊝ moderate3 | |
| *The basis for the assumed risk is the median control group risk across studies. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Downgraded by 1 level as most risk of bias domains were assessed either as unclear or high 2 Downgraded by 1 level due to small sample size 3 Downgraded by 1 level due to effect estimate with wide confidence interval 4 Downgraded by 1 level due to substantial heterogeneity among studies (I2 = 83%) 5 Downgraded by 1 level due to substantial heterogeneity among studies (I2 = 90%)
Summary of findings 3. Oestrogen cream compared to other regimens for vaginal atrophy in postmenopausal women.
| Oestrogen cream compared to other regimens for vaginal atrophy in postmenopausal women | ||||||
| Patient or population: postmenopausal women with vaginal atrophy Settings: outpatient clinic Intervention: oestrogen cream Comparison: other regimen | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Other regimen | Oestrogen cream | |||||
| Improvement in symptoms (participant‐assessed) (oestrogen cream vs isoflavone gel) | 967 per 1000 | 984 per 1000 (701 to 999) | OR 2.08 (0.08 to 53.76) | 50 (1 study) | ⊕⊕⊝⊝ low2,3 | |
| Improvement in symptoms (participant‐assessed) oestrogen cream vs placebo) | 685 per 1000 | 899 per 1000 (803 to 951) | OR 4.10 (1.88 to 8.93) | 198 (2 studies) | ⊕⊕⊝⊝ low1,2 | |
|
Endometrial thickness not reported |
‐ | ‐ | Not estimable | ‐ | ‐ | |
| Improvement in symptoms (clinician‐assessed) (oestrogen cream vs placebo) | 646 per 1000 | 857 per 1000 (728 to 931) | OR 3.29 (1.47 to 7.36) | 153 (1 study) | ⊕⊕⊝⊝ low1,2 | |
| Adverse events (total adverse events) (oestrogen cream vs non‐hormonal lubricant gel) | ‐ | ‐ | OR 10.67 (0.54 to 209.64) | 50 (1 study) | ⊕⊕⊝⊝ low2,3 | |
| *The basis for the assumed risk is the median control group risk across studies. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Downgraded by 1 level as most risk of bias domains were assessed either as unclear or high 2 Downgraded by 1 level due to small sample size 3 Downgraded by 1 level due to effect estimate with wide confidence interval
1. Oestrogen ring versus other regimens
Primary outcomes
1.1 Improvement in symptoms (participant‐assessed at end point)
1.1. Analysis.
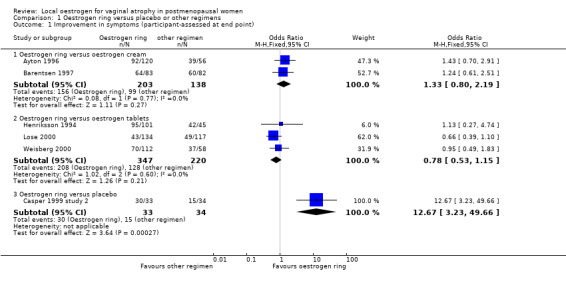
Comparison 1 Oestrogen ring versus placebo or other regimens, Outcome 1 Improvement in symptoms (participant‐assessed at end point).
4.
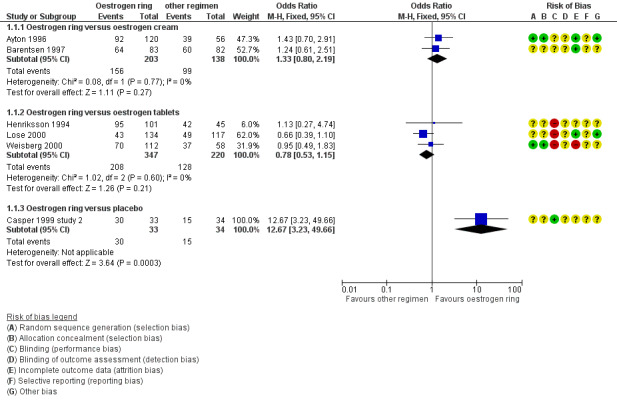
Forest plot of comparison: 1 Oestrogen ring versus placebo or other regimens, outcome: 1.1 Improvement in symptoms (participant‐assessed at end point).
1.1.1 Oestrogen ring versus oestrogen cream
There was no evidence of a difference in the proportions of women who reported improvement in symptoms between women who were treated with oestrogen ring and those who received oestrogen cream (OR 1.33, 95% CI 0.80 to 2.19, two RCTs, n = 341, I2 = 0%, low‐quality evidence). The evidence suggests that if the chance of improvement in symptoms following treatment with oestrogen cream is assumed to be 72%, the chance following treatment with oestrogen ring would be between 67% and 85%. On sensitivity analysis using risk ratio (RR) (RR 1.08, 95% CI 0.94 to 1.23) or a random‐effects model (OR 1.33, 95% CI 0.81 to 2.19), there was no change in the direction of the effect estimate or the evidence.
1.1.2 Oestrogen ring versus oestrogen tablets
Similarly, there was no evidence of a difference in the proportions of women who reported improvement in symptoms between women who received oestrogen ring and those who were treated with oestrogen tablets (OR 0.78, 95% CI 0.53 to 1.15, three RCTs, n = 567, I2 = 0%, low‐quality evidence). The evidence suggests that if the chance of improvement in symptoms following treatment with oestrogen tablets is assumed to be 58%, the chance following treatment with oestrogen ring would be between 43% and 62%. A similar pattern was observed in the direction of the effect estimate with no change in the evidence on sensitivity analysis using RR (RR 0.92, 95% CI 0.81 to 1.05) or a random‐effects model (OR 0.78, 95% CI 0.53 to 1.15).
1.1.3 Oestrogen ring versus placebo
A higher proportion of women reported improvement in symptoms following treatment with oestrogen ring compared with placebo (OR 12.67, 95% CI 3.23 to 49.66, one RCT, n = 67). On sensitivity analysis, there was no difference in the direction of the effect estimate or the evidence using RR (RR 2.06, 95% CI 1.39 to 3.05) or a random‐effects model (OR 12.67, 95% CI 3.23 to 49.66).
1.2. Endometrial thickness
1.2. Analysis.
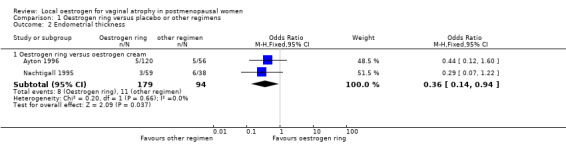
Comparison 1 Oestrogen ring versus placebo or other regimens, Outcome 2 Endometrial thickness.
1.2.1. Oestrogen ring versus oestrogen cream
A higher proportion of women who received oestrogen cream showed evidence of increase in endometrial thickness compared to those who were treated with oestrogen ring (OR 0.36, 95% CI 0.14 to 0.94, two RCTs, n = 273; I2 = 0%, low‐quality evidence). The evidence suggests that if the risk of increase in endometrial thickness following treatment with oestrogen cream is assumed to be 12%, the risk following treatment with oestrogen ring would be between 2% and 11%. Both studies used higher doses of cream than is currently recommended in clinical practice, which may have caused systemic absorption and increase in endometrial thickness. In one trial, 1 g of oestrogen cream was administered as a preparation containing 0.625 mg of conjugated equine oestrogen every night for 12 weeks (Ayton 1996). In the other trial, 2 g of the preparation was administered three times weekly for 12 weeks (Nachtigall 1995).
There was no change to the direction of the effect estimate or the evidence on sensitivity analysis using RR (RR 0.39, 95% CI 0.16 to 0.95) or a random‐effects model (OR 0.37, 95% CI 0.14 to 0.96).
Secondary outcomes
1.3. Improvement in symptoms (clinician‐assessed at end point)
1.3. Analysis.
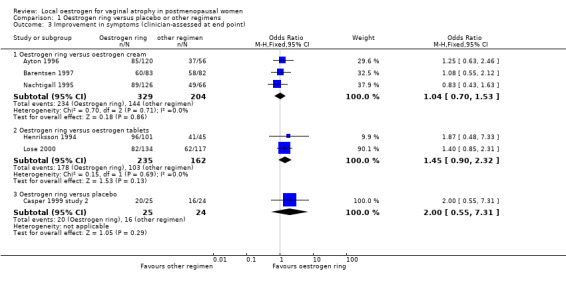
Comparison 1 Oestrogen ring versus placebo or other regimens, Outcome 3 Improvement in symptoms (clinician‐assessed at end point).
1.3.1. Oestrogen ring versus oestrogen cream
There was no evidence of a difference in improvement in symptoms as assessed by clinicians at 12 weeks between women who received oestrogen ring and those who were treated with oestrogen cream (OR 1.04, 95% CI 0.70 to 1.53, three RCTs, n = 533, I2 = 0%, low‐quality evidence). The evidence suggests that if the chance of improvement in symptoms following treatment with oestrogen cream is assumed to be 71%, the chance following treatment with oestrogen ring would be between 63% and 79%.
1.3.2. Oestrogen ring versus oestrogen tablets
Similarly, there was no evidence of a difference in improvement in symptoms between women who received oestrogen ring and those who were treated with oestrogen tablets, (OR 1.45, 95% CI 0.90 to 2.32, two RCTs, n = 397, I2 = 0%, low‐quality evidence). The evidence suggests that if the chance of improvement in symptoms following treatment with oestrogen tablets is assumed to be 64%, the chance following treatment with oestrogen ring would be between 61% and 80%.
1.3.3. Oestrogen ring versus placebo
There was no difference in improvement in symptoms between women who received oestrogen ring and those who received placebo ring (OR 2.00, 95% CI 0.55 to 7.31, one RCT, n = 49).
1.4. Improvement in symptoms (decrease in vaginal pH at end point)
1.4. Analysis.
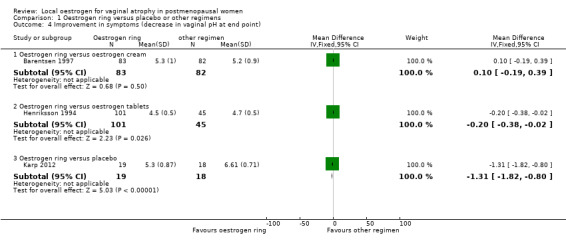
Comparison 1 Oestrogen ring versus placebo or other regimens, Outcome 4 Improvement in symptoms (decrease in vaginal pH at end point).
1.4.1. Oestrogen ring versus oestrogen cream
There was no evidence of a difference in improvement in symptoms as assessed using decrease in values of vaginal pH (difference in values at baseline and at 12 weeks) between women who were treated with oestrogen ring and those who received oestrogen cream (MD 0.10, 95% CI ‐0.19 to 0.39, one RCT, n = 165).
1.4.2. Oestrogen ring versus oestrogen tablets
Women who were treated with oestrogen ring demonstrated evidence of improvement in symptoms with a lower mean difference (MD) in vaginal pH (better outcome) compared with those who received oestrogen tablets (MD ‐0.20, 95% CI ‐0.38 to ‐0.02, one RCT, n = 146).
1.4.3. Oestrogen ring versus placebo
There was evidence of a difference in improvement in symptoms between women who received oestrogen ring and those who were treated with placebo, with women on oestrogen ring recording a lower MD in vaginal pH (better outcome) compared with women on placebo (MD ‐1.31, 95% CI ‐1.82 to ‐0.80, one RCT, n = 37). A second study reported this outcome but not in a form that allowed inclusion in meta‐analysis.
1.5. Improvement in symptoms (increase in maturation indices at end point)
1.5. Analysis.
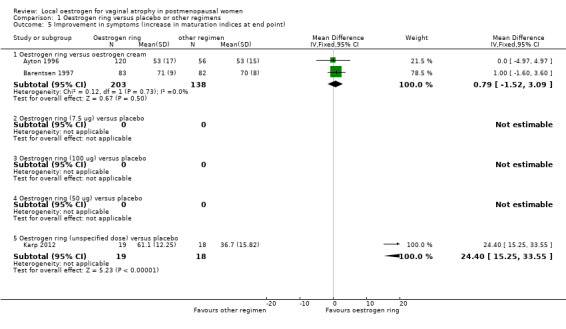
Comparison 1 Oestrogen ring versus placebo or other regimens, Outcome 5 Improvement in symptoms (increase in maturation indices at end point).
1.5.1. Oestrogen ring versus oestrogen cream
There was no evidence of a difference in improvement in symptoms as assessed using increase in values of vaginal maturation indices at end point between women who received oestrogen ring and those who were treated with oestrogen cream (MD 0.79, 95% CI ‐1.52 to 3.09, two RCTs, n = 341, I2 = 0%).
1.5.2. Oestrogen ring (7.5 µg) versus oestrogen tablets
Result of analysis not estimable: no usable data were available.
1.5.3. Oestrogen ring (100 µg) versus placebo
Result of analysis not estimable: no usable data were available.
1.5.4. Oestrogen ring (50 µg) versus placebo
Result of analysis not estimable: no usable data were available.
1.5.5. Oestrogen ring (unspecified dose) versus placebo
Women who received oestrogen ring demonstrated evidence of a higher MD in maturation indices (better outcome) compared with those who were treated with placebo (MD 24.40, 95% CI 15.25 to 33.55, one RCT, n = 37).
1.6. Adverse events (breast disorders)
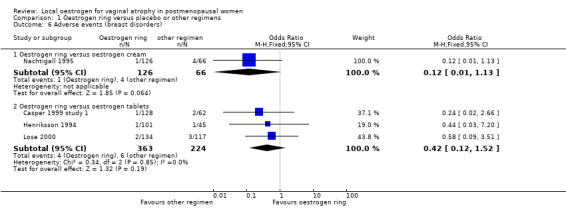
1.6. Analysis.
Comparison 1 Oestrogen ring versus placebo or other regimens, Outcome 6 Adverse events (breast disorders).
1.6.1. Oestrogen ring versus oestrogen cream
There was no evidence of a difference in the proportions of women with breast disorders between the two treatment groups (OR 0.12, 95% CI 0.01 to 1.13, one RCT, n = 192).
1.6.2. Oestrogen ring versus tablets
There was no evidence of a difference in the proportions of women with breast disorders between women who received oestrogen ring and those who were treated with oestrogen tablets (OR 0.42, 95% CI 0.12 to 1.52, three RCTs, n = 587, I2 = 0%).
1.7. Adverse events (total adverse events)
1.7. Analysis.
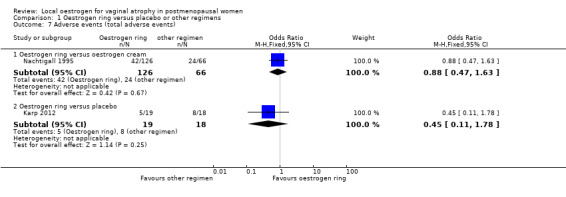
Comparison 1 Oestrogen ring versus placebo or other regimens, Outcome 7 Adverse events (total adverse events).
1.7.1. Oestrogen ring versus oestrogen cream
There was no evidence of a difference in the proportions of women with total adverse events between the two treatment groups (OR 0.88, 95% CI 0.47 to 1.63, one RCT, n = 192, low‐quality evidence). The evidence suggests that if the risk of total adverse events following treatment with oestrogen cream is assumed to be 36%, the risk following treatment with oestrogen ring would be between 21% and 48%.
1.7.2. Oestrogen ring versus placebo
There was no evidence of a difference in the proportions of women with total adverse events between the two treatment groups (OR 0.45, 95% CI 0.11 to 1.78, one RCT, n = 37, moderate‐quality evidence). The evidence suggests that if the risk of total adverse events following treatment with placebo is assumed to be 44%, the risk following treatment with oestrogen ring would be between 8% and 59%.
1.8. Adherence to treatment
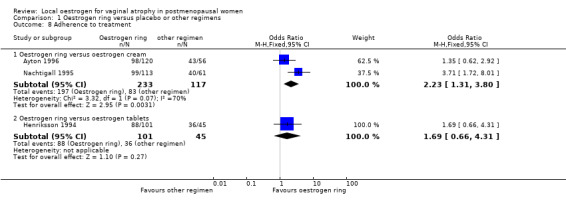
1.8. Analysis.
Comparison 1 Oestrogen ring versus placebo or other regimens, Outcome 8 Adherence to treatment.
1.8.1. Oestrogen ring versus oestrogen cream
There was evidence of a higher proportion of adherence to treatment among women who were treated with oestrogen ring compared with those who received oestrogen cream (OR 2.23, 95% CI 1.31 to 3.80, two RCTs, n = 350, I2 = 70%) The presence of substantial heterogeneity was explored in a sensitivity analysis: there was no change in the evidence on sensitivity analysis using RR (RR 1.19, 95% CI 1.05 to 1.35) or a random‐effects model (OR 2.24, 95% CI 0.83 to 6.05). There were no variations in the directions of effect estimates of individual trials included in the meta analysis.
1.8.2. Oestrogen ring versus oestrogen tablets
There was no evidence of a difference in the proportions of women who adhered to the treatment regimen between the two treatment groups (OR 1.69, 95% CI 0.66 to 4.31, one RCT, n = 146).
2. Oestrogen tablets versus other regimens
Primary outcomes
2.1. Improvement in symptoms (participant‐assessed at end point)
2.1. Analysis.
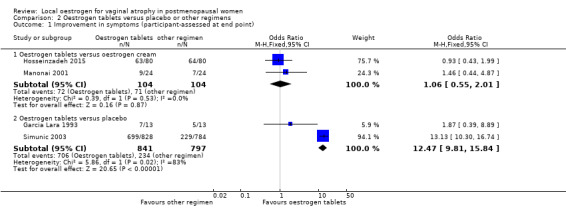
Comparison 2 Oestrogen tablets versus placebo or other regimens, Outcome 1 Improvement in symptoms (participant‐assessed at end point).
5.
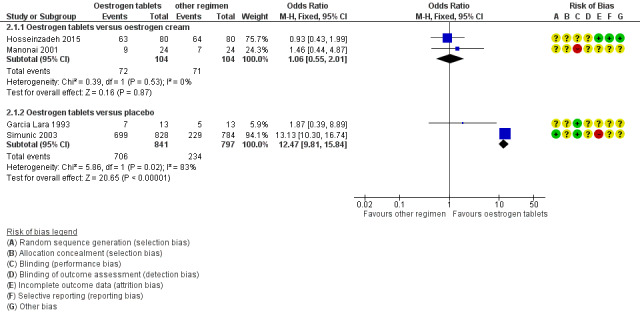
Forest plot of comparison: 2 Oestrogen tablets versus placebo or other regimens, outcome: 2.1 Improvement in symptoms (participant‐assessed at end point).
2.1.1. Oestrogen tablets versus oestrogen cream
There was no evidence of a difference in the proportions of women who reported improvement in symptoms between the two treatment groups (OR 1.06, 95% CI 0.55 to 2.01, two RCTs, n = 208, I2 = 0% low‐quality evidence). The evidence suggests that if the chance of improvement in symptoms following oestrogen tablets is assumed to be 68%, the chance following oestrogen cream would be between 54% and 81%. There was no difference in the above evidence on sensitivity analysis using RR (RR 1.01, 95% CI 0.86 to 1.20) or a random‐effects model (OR 1.06, 95% CI 0.55 to 2.01).
2.1.2. Oestrogen tablets versus placebo
There was evidence of a difference in the proportions of women who reported improvement in symptoms between the two treatment groups with a higher proportion reporting improvement in the oestrogen tablet group (OR 12.47, 95% CI 9.81 to 15.84, two RCTs, n = 1638, I2 = 83%, low‐quality evidence). The evidence suggests that if the chance of improvement following placebo is assumed to be 29%, the chance following oestrogen tablets would be between 80% and 87%. We explored the presence of substantial heterogeneity between the two studies that contributed data to the meta analysis using sensitivity analysis. The results on using RR were similar to the results above, showing evidence of a difference in improvement in symptoms between the two treatment groups; however, a random‐effects model showed no evidence of a difference in the proportions of women who reported improvement in symptoms between the two treatment groups (OR 5.80, 95% CI 0.88 to 38.29). In addition, although there were no variations in the directions of the effect estimates between the two studies, one of the studies demonstrated a substantial difference in symptom improvement between the two treatment groups while the other showed no evidence of a difference. A close look at the two studies showed some differences with respect to participants and doses of interventions. One of the studies (Garcia Lara 1993 included women aged 43 to 45 years and treated them with vaginal ovules of oestradiol 3.5 mg, two per week in the first three weeks and one per week for the remaining weeks over a period of four months. The second study (Simunic 2003) included women between 51 and 66 years who were treated with 25 µg of micronised 17β‐oestradiol vaginal tablet once a day over a period of two weeks and then twice a week for the remaining 12 months. This study was assessed as at high risk of attrition bias as the proportions of withdrawals differed between the two treatment groups and data were not analysed on the basis of ITT.
2.2. Endometrial thickness
2.2. Analysis.
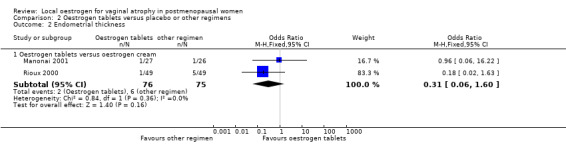
Comparison 2 Oestrogen tablets versus placebo or other regimens, Outcome 2 Endometrial thickness.
2.2.1. Oestrogen tablets versus oestrogen cream
There was no evidence of a difference in the proportions of women with increase in endometrial thickness between the two treatment groups (OR 0.31, 95% CI 0.06 to 1.60, two RCTs, n = 151, I2 = 0%, low‐quality evidence). This suggests that if the risk of increase in endometrial thickness following oestrogen cream is assumed to be 8%, the risk following oestrogen tablets would be between 5% and 12%. On sensitivity analysis, the evidence was the same using either RR (RR 0.33, 95% CI 0.07 to 1.59) or a random‐effects model (OR 0.34, 95% CI 0.06 to 1.92).
Secondary outcomes
2.3. Improvement in symptoms (clinician‐assessed at end point)
2.3. Analysis.
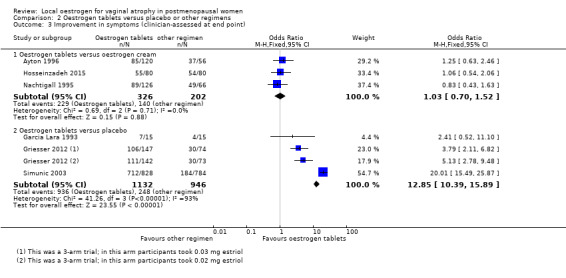
Comparison 2 Oestrogen tablets versus placebo or other regimens, Outcome 3 Improvement in symptoms (clinician‐assessed at end point).
2.3.1. Oestrogen tablets versus oestrogen cream
There was no evidence of a difference in the proportions of women with improvement in symptoms between the two treatment groups as assessed by the clinicians (OR 1.03, 95% CI 0.70 to 1.52, three RCTs, n = 528, I2 = 0%, low‐quality evidence). The evidence suggests that if the chance of improvement following oestrogen cream is assumed to be 70%, the chance following oestrogen tablets would be between 61% and 77%.
2.3.2. Oestrogen tablets versus placebo
A higher proportion of women who were treated with oestrogen tablets showed evidence of improvement in symptoms when compared to those who received placebo (OR 12.85, 95% CI 10.39 to 15.89, four RCTs, n = 2078, I2 = 93%, low‐quality evidence). The evidence suggests that if the chance of improvement following placebo is assumed to be 26%, the chance following oestrogen tablets would be between 79% and 85%. There was no change in the evidence on sensitivity analysis using RR (RR 3.10, 95% CI 2.78 to 3.46) or a random‐effects model (OR 6.07, 95% CI 2.07 to 17.85). A further sensitivity analysis removing Simunic 2003 which was at high risk of attrition bias eliminated the heterogeneity and the benefit of the intervention remained. We suggest that the magnitude of the effect seen should be interpreted with caution.
2.4. Improvement in symptoms (decrease in vaginal pH at end point)
2.4. Analysis.
Comparison 2 Oestrogen tablets versus placebo or other regimens, Outcome 4 Improvement in symptoms (decrease in vaginal pH at end point).
2.4.1. Oestrogen tablets versus oestrogen cream
There was no evidence of a difference in improvement in symptoms between the two treatment groups as assessed using the values of vaginal pH (MD 0.20, 95% CI ‐0.12 to 0.52, one RCT, n = 48).
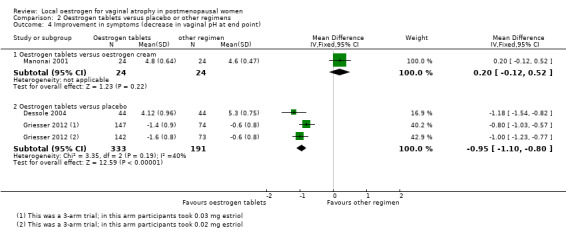
2.4.2. Oestrogen tablets versus placebo
Women who were treated with oestrogen tablets demonstrated evidence of improvement in symptoms with a lower mean difference (MD) in vaginal pH (better outcome) compared with those who received placebo (MD ‐0.95, 95% CI ‐1.10 to ‐0.80, three RCTs, n = 524, I2 = 40%).
2.5. Improvement in symptoms (increase in maturation indices at end point)
2.5. Analysis.
Comparison 2 Oestrogen tablets versus placebo or other regimens, Outcome 5 Improvement in symptoms (increase in maturation indices at end point).
2.5.1. Oestrogen tablets versus oestrogen cream
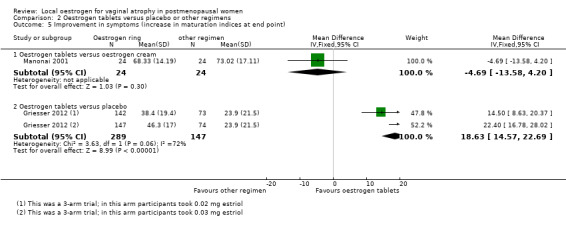
There was no evidence of a difference in improvement in symptoms between the two treatment groups as assessed using the values of vaginal maturation indices (MD ‐4.69, 95% CI ‐13.58 to 4.20, one RCT, n = 48).
2.5.2. Oestrogen tablets versus placebo
Women who were treated with oestrogen tablets demonstrated evidence of improvement in symptoms with a higher mean difference in maturation indices (better outcome) compared with those who received placebo (MD 18.63, 95% CI 14.57 to 22.69, two RCTs, n = 436, I2 = 72%). The evidence did not differ on sensitivity analysis using a random‐effects model (MD 18.50, 95% CI 10.76 to 26.24). In addition, there were no variations in the directions of the effect estimates of individual studies.
2.6. Adverse events (breast disorders)
2.6. Analysis.
Comparison 2 Oestrogen tablets versus placebo or other regimens, Outcome 6 Adverse events (breast disorders).
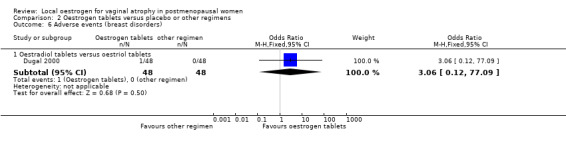
2.6.1. Oestradiol tablets versus oestriol tablets
There was no evidence of a difference in the proportions of women with breast disorders between the two treatment groups (OR 3.06, 95% CI 0.12 to 77.09, one RCT, n = 96).
2.7. Adverse events (total adverse events)
2.7. Analysis.
Comparison 2 Oestrogen tablets versus placebo or other regimens, Outcome 7 Adverse events (total adverse events).
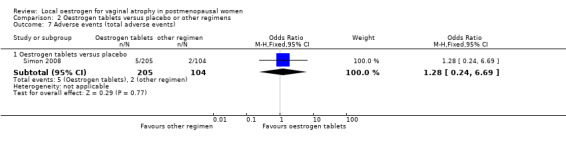
2.7.1. Oestrogen tablets versus placebo
There was no evidence of a difference in the proportions of women with total adverse events between the two treatment groups (OR 1.27, 95% CI 0.24 to 6.69, one RCT, n = 309, moderate‐quality evidence). The evidence suggests that if the risk of adverse events following placebo is assumed to be 2%, the risk following oestrogen tablets would be between 1% and 12%.
2.8. Adherence to treatment
2.8. Analysis.

Comparison 2 Oestrogen tablets versus placebo or other regimens, Outcome 8 Adherence to treatment.
2.8.1. Oestrogen tablets versus oestrogen cream
There was no evidence of a difference in the proportions of women who adhered to the treatment protocol between the two treatment groups (OR 1.90, 95% CI 0.41 to 8.94, one RCT, n = 53).
2.8.2. Oestradiol tablets versus oestriol tablets
A higher proportion of women who were treated with oestradiol tablets adhered to the treatment protocol when compared with those who received oestriol tablets (OR 2.69, 95% CI 1.15 to 6.31, one RCT, n = 96).
3. Oestrogen cream versus other regimens
Primary outcomes
3.1. Improvement in symptoms (participant‐assessed at end point)
3.1. Analysis.
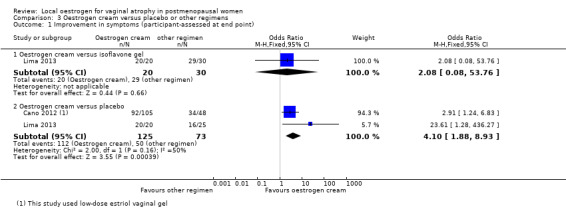
Comparison 3 Oestrogen cream versus placebo or other regimens, Outcome 1 Improvement in symptoms (participant‐assessed at end point).
6.
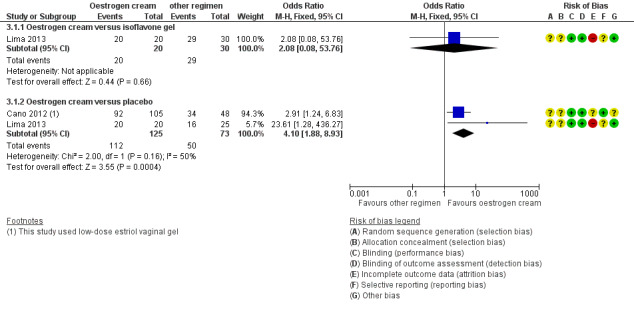
Forest plot of comparison: 3 Oestrogen cream versus placebo or other regimens, outcome: 3.1 Improvement in symptoms (participant‐assessed at end point).
3.1.1. Oestrogen cream versus isoflavone gel
There was no evidence of a difference in the proportions of women with improvement in symptoms between the two treatment groups (OR 2.08, 95% CI 0.08 to 53.76, one RCT, n = 50, low‐quality evidence). The evidence suggests that if the chance of improvement following isoflavone gel is assumed to be 97%, the chance following oestrogen cream would be between 70% and 100%. This evidence did not change on sensitivity analysis using RR (RR 1.03, 95% CI 0.92 to 1.14) or a random‐effects model (OR 2.08, 95% CI 0.08 to 53.76).
3.1.2. Oestrogen cream versus placebo
There was evidence of a difference in the proportions of women with improvement in symptoms between the two treatment groups with more women in the oestrogen cream group reporting improvement in symptoms compared to those in the placebo group (OR 4.10, 95% CI 1.88 to 8.93, two RCTs, n = 198, I2 = 50%, low‐quality evidence). The evidence suggests that if the chance of improvement following placebo is assumed to be 69%, the chance following oestrogen cream would be between 80% and 95%. There was no change in the evidence on sensitivity analysis using RR (RR 1.31, 95% CI 1.11 to 1.54). However, a random‐effects model did not show any evidence of a difference in symptom improvement between the two treatment groups (OR 5.34, 95% CI 0.76 to 37.49) although there were no variations in the direction of the effect estimates of individual studies.
3.2. Endometrial thickness
This outcome was not reported by any of the included studies.
Secondary outcomes
3.3. Improvement in symptoms (clinician‐assessed at end point)
3.3. Analysis.
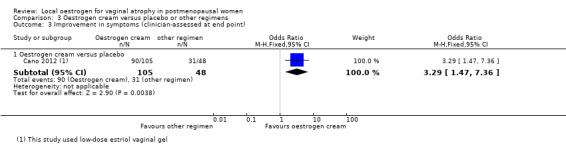
Comparison 3 Oestrogen cream versus placebo or other regimens, Outcome 3 Improvement in symptoms (clinician‐assessed at end point).
3.3.1. Oestrogen cream versus placebo
A higher proportion of women who were treated with oestrogen cream demonstrated improvement in symptoms as assessed by the clinicians, compared to those who received placebo (OR 3.29, 95% CI 1.47 to 7.36, one RCT, n = 153, low‐quality evidence). The evidence suggests that if the chance of improvement following placebo is assumed to be 65%, the chance following oestrogen cream would be between 73% and 93%.
3.4. Improvement in symptoms (decrease in vaginal pH at end point)
3.4. Analysis.
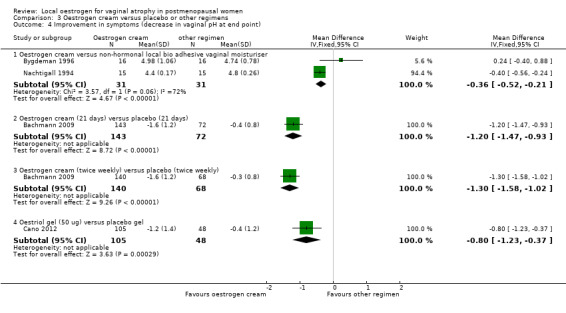
Comparison 3 Oestrogen cream versus placebo or other regimens, Outcome 4 Improvement in symptoms (decrease in vaginal pH at end point).
3.4.1. Oestrogen cream versus non‐hormonal local bio adhesive vaginal moisturising gel
There was evidence of a lower mean difference value (better outcome) in women who received oestrogen cream compared with those who were treated with non‐hormonal local bio adhesive vaginal moisturising gel (MD ‐0.36, 95% CI ‐0.52 to ‐0.21, two RCTS, n = 62, I2 = 72%). The presence of substantial heterogeneity was explored in a sensitivity analysis using a random effects model and there was no evidence of a difference in improvement in symptoms (decrease in vaginal pH) between the two treatment groups (MD ‐0.16, 95% CI ‐0.77 to 0.45). However, there were no variations in the direction of the effect estimate of individual studies. The standard deviations reported in Nachtigall 1994 appear unusually small. When this study was removed from the analysis there was no evidence of a difference between the groups using either analysis model.
3.4.2. Oestrogen cream (21 days) versus placebo (21 days)
Women who were treated with oestrogen cream daily for 21 days demonstrated evidence of improvement in symptoms with a lower mean difference in vaginal pH (better outcome) compared with those who received placebo for the same number of days (MD ‐1.20, 95% CI ‐1.47 to ‐0.93, one RCT, n = 215).
3.4.3. Oestrogen cream (twice weekly) versus placebo (twice weekly)
There was evidence of a lower mean difference value (better outcome) in women who received oestrogen cream twice weekly compared with those who were treated with placebo twice weekly (MD ‐1.30, 95% CI ‐1.58 to ‐1.02, one RCT, n = 208).
3.4.4. Oestriol gel (50 ug) versus placebo
Women who were treated with oestriol gel (50 ug) demonstrated evidence of improvement in symptoms with a lower mean difference in vaginal pH (better outcome) compared with those who received an equivalent dose of placebo (MD ‐0.80, 95% CI ‐1.23 to ‐0.37, one RCT, n = 153).
3.5. Improvement in symptoms (increase in maturation indices at end point)
3.5. Analysis.
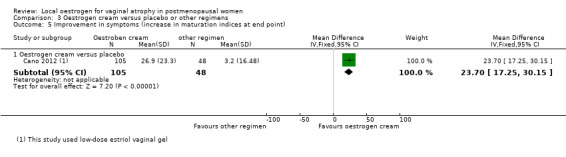
Comparison 3 Oestrogen cream versus placebo or other regimens, Outcome 5 Improvement in symptoms (increase in maturation indices at end point).
3.5.1. Oestrogen cream versus placebo
Women who were treated with oestrogen cream demonstrated evidence of improvement in symptoms with a higher mean difference in maturation indices (better outcome) compared to those who received placebo (MD 23.70, 95% CI 17.25 to 30.15, one RCT, n = 153).
3.6. Adverse events (breast disorders
None of the included studies reported this outcome.
3.7. Adverse events (total adverse events)
3.7. Analysis.
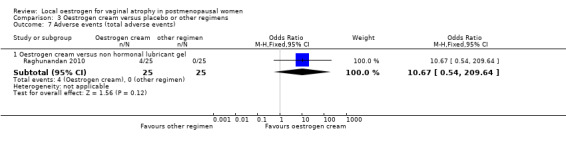
Comparison 3 Oestrogen cream versus placebo or other regimens, Outcome 7 Adverse events (total adverse events).
3.7.1. Oestrogen cream versus non‐hormonal lubricant gel
There was no evidence of a difference in the proportions of women with total adverse events between the two treatment groups (OR 10.67, 95% CI 0.54 to 209.64, one RCT, n = 50).
3.8. Adherence to treatment
This outcome was not reported by any of the included studies.
No subgroup analysis was undertaken for any outcomes as there were insufficient data in the included studies.
Discussion
Summary of main results
This review assessed the efficacy and safety of available intra‐vaginal oestrogenic preparations, in particular the oestradiol‐releasing ring versus oestrogen creams, tablets or placebo, oestrogen tablets versus oestrogen cream, placebo or oestriol tablets and oestrogen cream versus isoflavone gel, non‐hormonal local bio adhesive vaginal moisturising gel, non‐hormonal lubricant gel or placebo.
We assessed efficacy using participant‐reported improvement in symptoms, clinician‐assessed improvement in symptoms, and laboratory as well as cytological evidence of improvement in symptoms; decrease in vaginal pH; and increase in vaginal maturation indices. We assessed safety using evidence of increase in endometrial thickness; incidents of breast disorders, such as breast pain and engorgement; and total adverse events associated with the treatment.
From the overall body of the findings, there was no conclusive evidence of a difference in efficacy between the various oestrogenic preparations compared with each other. However, findings from the review showed that oestrogenic preparations were associated with better efficacy in terms of improvement in symptoms compared with sham treatment or placebo. With respect to safety, there was no conclusive evidence of a difference in the main adverse events (endometrial thickness, breast disorders and total adverse events) between oestrogenic preparations versus each other or placebo. Although two small trials (Ayton 1996; Nachtigall 1995; n = 273) reported an increase in endometrial thickness in women who were treated with oestrogen cream compared to those who received oestrogen ring, the evidence was of low quality due to a high level of uncertainty associated with the effect estimate. In addition the studies used higher doses of cream than now recommended in clinical practice. In one trial, 1 g of oestrogen cream was administered as a preparation containing 0.625 mg of conjugated equine oestrogen every night for 12 weeks (Ayton 1996). In the other trial, 2 g of the preparation was administered three times weekly for 12 weeks (Nachtigall 1995). The recommended dose of conjugated equine oestrogen administered vaginally is 0.625 mg daily for one to two weeks as an induction therapy, followed by low doses for maintenance therapy (ACOG 2014). Typically this would be twice weekly in a clinical setting. Both studies thus used higher doses of cream than currently recommended in clinical practice which may have caused systemic absorption and increase in endometrial thickness. Further research using recommended doses of vaginal cream would be useful to confirm or refute this finding.
Effect estimates for both efficacy and safety measures were small with wide confidence intervals. The quality of the evidence for the GRADE‐specific outcomes (participant‐assessed improvement in symptoms, endometrial thickness, clinician‐assessed improvement in symptoms and total adverse events) was either low (for most of the outcomes) or moderate, indicating a high level of uncertainty in the effect estimates which may change with further research.
Overall completeness and applicability of evidence
This review set out to examine the evidence in respect of the efficacy and safety associated with intra‐vaginal oestrogenic preparations in relieving the symptoms of vaginal atrophy in postmenopausal women. All the included studies investigated postmenopausal women with symptoms of vaginal atrophy in a broad range of comparisons, including oestrogenic preparations compared with each other, placebo or non oestrogenic preparations. Although diverse outcome measures were investigated by the included studies, a number of them were relevant to the review's main objectives. However, most of the comparisons were investigated in small trials which were not adequately powered to detect effect estimates with low levels of uncertainty. In addition, the disparate nature of the treatment regimens made it difficult to combine data in meta analyses. Similarly, six of the included studies could not be included in quantitative synthesis because either they did not report outcome measures relevant to the review or reported them in non usable form. Thus the evidence was largely based on a small number of underpowered trials resulting in effect estimates with wide confidence intervals. Most of the included studies did not investigate endometrial thickness, which was one of the review's primary outcomes. Similarly, few included studies investigated other adverse events (breast disorders and total adverse events). In all the included studies, data were either not reported on long‐term outcomes or reported in non usable forms. We could not investigate the separate evidence of the effect of symptom duration and severity on the efficacy and safety of the various oestrogenic preparations due to insufficient data. This review excluded women with a history of severe atrophy and breast or endometrial cancer. Thus the efficacy and safety of local vaginal oestrogen were not assessed in this group of women. From a clinical point of view, it would be useful to consider these women in a future update of the review.
Quality of the evidence
All the identified interventions were subsumed under three main comparisons: oestrogen ring versus other regimens, oestrogen tablets versus other regimens and oestrogen cream versus other regimens. Four outcome measures were included in the 'Summary of findings' tables: improvement in symptoms (participant‐assessed), endometrial thickness, improvement in symptoms (clinician‐assessment) and adverse events (total adverse events). In oestrogen ring versus other regimens, data were available on all the GRADE‐specific outcomes. The quality of the evidence was low for all the outcomes except total adverse events, with moderate‐quality evidence. In the second comparison, oestrogen tablets versus other regimens, all the GRADE‐specific outcomes were assessed. A similar pattern of findings were observed as in the first comparison. In the third and final comparison, oestrogen cream versus other regimens, findings on one of the GRADE‐specific outcomes (endometrial thickness) were not incorporated into the table. The remaining outcomes were all of low quality.
Low‐quality evidence implies that further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate while moderate quality evidence while moderate‐quality evidence means that further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate.
The main limitations of the evidence were poor reporting (a majority of the included studies had most risk of bias domains assessed as unclear due to insufficient information) and imprecision due to small sample sizes or effect estimates with wide confidence intervals, or both.
Potential biases in the review process
We searched all the important databases and imposed no language restriction in the course of the search. However, we were mindful of the fact that these database searches might not have identified all the potentially eligible trials.
Agreements and disagreements with other studies or reviews
We found one systematic review which examined the efficacy and safety of vaginal oestrogen for genitourinary syndrome of menopause (Rahn 2014). In the review, 14 studies (n = 4232) compared vaginal oestrogen with placebo while 18 studies (n = 2236) compared one type of vaginal oestrogen preparation with another. In the first comparison (vaginal oestrogen versus placebo), women who were treated with vaginal oestrogen at the recommended doses consistently demonstrated evidence of more benefits with improvement in both genital and urinary symptoms compared to those who received placebo. The review, however, found no conclusive evidence of a difference in endometrial safety between the two treatment groups. In the second comparison (one vaginal oestrogen versus another), the review did not find evidence of differences in efficacy and safety between the different vaginal oestrogen preparations administered at the recommended doses and frequencies. These findings are consistent with those of this review.
In the 2013 position statement of the North American Menopause Society (NAMS), which is based on cumulative evidence from many studies, intra‐vaginal oestrogenic preparations (oestrogen vaginal ring, oestrogen tablets and oestrogen cream) are considered to be equally effective and well tolerated in the management of postmenopausal women with symptoms of vulvovaginal atrophy (Lindah 2014; NAMS 2013). The UK National Institute for Health and Care Excellence (NICE), in its 2015 guidance for the management of menopause, recommends the use of vaginal oestrogen for the treatment of postmenopausal women with symptoms of urogenital atrophy without any preference for any oestrogenic preparations with respect to efficacy and safety (NICE 2015). Similarly, the Society of Obstetricians and Gynaecologists of Canada states in its clinical practice guideline: "Conjugated estrogen cream, an intravaginal sustained‐release oestradiol ring, and low‐dose oestradiol vaginal tablets are recommended as effective treatment for vaginal atrophy." (SOGC 2014). These findings are in agreement with those of this review which did not find evidence of a difference in efficacy and safety between the various oestrogenic preparations when compared with each other.
Authors' conclusions
Implications for practice.
There is no conclusive evidence of a difference in efficacy between the various intravaginal oestrogenic preparations when compared with each other. However, there is low‐quality evidence that intra‐vaginal oestrogenic preparations improve the symptoms of vaginal atrophy in postmenopausal women when compared to placebo. There is low‐quality evidence that oestrogen cream may be associated with increase in endometrial thickness compared to oestrogen ring, but this may have been due to the higher doses used. However there is no conclusive evidence of a difference in the overall body of evidence in adverse events between the various oestrogenic preparations compared with each other or with placebo. As previously noted, the low quality of the evidence resulted from poor reporting, with the review authors assessing a majority of the included studies as unclear in most risk of bias domains, due to insufficient information; and imprecision, due to small sample sizes or effect estimates with wide confidence intervals, or both.
These results should be applied in the context of improvement of the symptoms of vaginal atrophy. Intra‐vaginal oestrogenic preparations in the forms of creams, pessaries, tablets and the oestradiol‐releasing ring are shown to be effective for the symptoms of vaginal atrophy. There are few trials comparing an intervention with placebo. The oestradiol vaginal ring can be considered as an effective and practical alternative to creams, pessaries and tablets. Dienoestrol cream has been withdrawn worldwide, and conjugated equine oestrogen cream has been withdrawn in New Zealand and Australia; 17ß oestradiol tablets in the dose of 25 µg have also been withdrawn worldwide and replaced with the lower dose 10 µg tablet. This is not available in New Zealand.
Women using intra‐vaginal oestrogenic preparations who have postmenopausal bleeding should have endometrial investigation.
Implications for research.
More well powered RCTs conducted in accordance with the CONSORT statement are required to adequately address the balance of efficacy and safety of the individual oestrogenic preparations for the management of postmenopausal women with symptoms of vaginal atrophy (Schultz 2010). Although not a subject of this review, there is the need for RCTs on the efficacy and safety of intra‐vaginal oestrogenic preparations in women with severe vaginal atrophy following treatment for breast or endometrial cancer. In addition, findings from this review can be further enriched by future trials which examine the long‐term efficacy and safety of treatment. Intra‐vaginal oestrogenic preparations versus placebo should be investigated further. Future trials of intra‐vaginal oestrogenic administration should consider investigating serum oestradiol levels, as some of the adverse events associated with treatment might be surrogate markers, indicating systemic absorption.
What's new
| Date | Event | Description |
|---|---|---|
| 4 November 2016 | Review declared as stable | Further evidence is unlikely to change the conclusions of this review. |
History
Protocol first published: Issue 2, 1999 Review first published: Issue 4, 2003
| Date | Event | Description |
|---|---|---|
| 30 August 2016 | New search has been performed | The review has been updated, and 10 new studies added (Bachmann 2008; Bachmann 2009; Mac Bride 2014; Cano 2012; Fernandes 2014; Griesser 2012; Karp 2012; Lima 2013; Raghunandan 2010; Simon 2008) |
| 30 August 2016 | New citation required but conclusions have not changed | The addition of new evidence did not change the conclusions of this review. |
| 7 November 2008 | Amended | Converted to new review format. |
| 13 June 2006 | New citation required and conclusions have changed | Substantive amendment |
Acknowledgements
Thanks to members of the Editorial office of Cochrane Gynaecology and Fertility who assisted at all stages of the review. The authors thank Dr Ray Kennedy and Dr Jane Suckling for their contributions to previous versions of this review.
Appendices
Appendix 1. Cochrane Gynaecology and Fertility specialised register
PROCITE platform
From inception until 12.04.16
Keywords CONTAINS "*Vaginitis" or "vaginosis symptoms" or "vaginosis" or "vaginal atrophy" or "vaginal dryness" or "vaginal lubrication" or "vaginal symptoms" or "atrophic vaginitis" or "atrophy" or "dyspareunia" or "uro‐genital symptoms" or "urogenital atrophy" or "urogenital symptoms" or "Vulvar Atrophy" or "vulvo‐vaginal symptoms" or "vulvodynia" or "Vulvovaginal atrophy" or Title CONTAINS "*Vaginitis" or "vaginosis symptoms" or "vaginosis" or "vaginal atrophy" or "vaginal dryness" or "vaginal lubrication" or "vaginal symptoms" or "atrophic vaginitis" or "atrophy" or "dyspareunia" or "uro‐genital symptoms" or "urogenital atrophy" or "urogenital symptoms" or "vulvo‐vaginal symptoms" or "vulvodynia" or "Vulvovaginal atrophy "
AND
Keywords CONTAINS "vaginal capsules" or "vaginal estradiol" or "vaginal gel" or "vaginal pessary" or "vaginal tablet" or "vaginal tablets" or "vaginal ring" or "low dose estradiol" or "oestrodiol" or "oestrogen" or "estradiol" or "estradiol cream" or "Estriol‐" or "estrogen" or "*Estrogens" or "estrogen therapy" or "17‐beta estradiol" or "intravaginal estradiol tablets" or "intravaginal" or Title CONTAINS "vaginal capsules" or "vaginal estradiol" or "vaginal gel" or "vaginal pessary" or "vaginal tablet" or "vaginal tablets" or "vaginal ring" or "low dose estradiol" or "oestrodiol" or "oestrogen" or "estradiol" or "estradiol cream" or "Estriol‐" or "estrogen" or "*Estrogens" or "estrogen therapy" or "17‐beta estradiol" or "intravaginal estradiol tablets" or "intravaginal"
Appendix 2. CENTRAL search strategy
CRSO web platform
From inception until 12.04.16
#1MESH DESCRIPTOR Atrophic Vaginitis EXPLODE ALL TREES (2) #2MESH DESCRIPTOR Dyspareunia EXPLODE TREES (294) #3MESH DESCRIPTOR Vaginitis EXPLODE ALL TREES (676) #4pruritis:TI,AB,KY (150) #5((vagin* adj2 dry*)):TI,AB,KY (232) #6(urogenital atroph* or urogenital symptom*):TI,AB,KY (79) #7(menopaus* adj2 symptom*):TI,AB,KY (745) #8(postmenopaus* adj2 symptom*):TI,AB,KY (139) #9Dyspareuni*:TI,AB,KY (442) #10(urogenital ageing):TI,AB,KY (2) #11(urogenital disorder*):TI,AB,KY (8) #12#1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR #9 OR #10 OR #11 (2220) #13MESH DESCRIPTOR Estrogens EXPLODE ALL TREES (6147) #14((*vagina* adj3 administ*)):TI,AB,KY (1464) #15MESH DESCRIPTOR Vaginal Creams, Foams, and Jellies EXPLODE ALL TREES (302) #16((*vagin* adj2 cream*)):TI,AB,KY (442) #17(vaginal ring*):TI,AB,KY (158) #18(vagina* pessar*):TI,AB,KY (161) #19(vagina* tablet*):TI,AB,KY (220) #20vagitories:TI,AB,KY (6) #21(vagina* gel*):TI,AB,KY (197) #22(vagina* capsule*):TI,AB,KY (25) #23ovule*:TI,AB,KY (66) #24(oestradiol or oestrogen):TI,AB,KY (1845) #25oestrogenic:TI,AB,KY (80) #26estradiol:TI,AB,KY (6910) #2717B‐estradiol:TI,AB,KY (18) #28((oestriol or estriol)):TI,AB,KY (350) #29((dienoestrol or replens)):TI,AB,KY (14) #30#13 OR #14 OR #15 OR #16 OR #17 OR #18 OR #19 OR #20 OR #21 OR #22 OR #23 OR #24 OR #25 OR #26 OR #27 OR #28 OR #29 (11557) #31#12 AND #30 (860)
Appendix 3. MEDLINE search strategy
Database: Ovid MEDLINE(R) In‐Process & Other Non‐Indexed Citations, Ovid MEDLINE(R) Daily and Ovid MEDLINE(R)
OVID platform
From inception until 12.04.16
1 Vaginitis/ (3265) 2 ($vagin$ adj2 atroph$).tw. (619) 3 Sexual Dysfunction, Physiological/ or Dyspareunia/ or vaginism.tw. (9388) 4 pruritis.tw. or Pruritus/ (9794) 5 ($vagin$ adj2 dry$).tw. (789) 6 (urogenital atrophy or urogenital symptom$).tw. (397) 7 (menopaus$ adj2 symptom$).tw. (3660) 8 (postmenopaus$ adj2 symptom$).tw. (697) 9 Dyspareuni$.tw. (2895) 10 urogenital ageing.tw. (11) 11 urogenital disorder$.tw. (96) 12 or/1‐11 (29104) 13 estrogen.tw. or exp Estrogens/ (202231) 14 ($vagina$ adj3 administ$).tw. (1361) 15 "Vaginal Creams, Foams and Jellies"/ (1104) 16 (vagin$ adj2 cream$).tw. (330) 17 vaginal ring$.tw. (730) 18 vaginal pessar$.tw. (309) 19 vagina$ tablet$.tw. (362) 20 vagitories.tw. (11) 21 vagina$ gel$.tw. (372) 22 vagina$ capsule$.tw. (30) 23 ovule$.tw. (2038) 24 (oestradiol or oestrogen).tw. (24894) 25 oestrogenic.tw. (1620) 26 estradiol.tw. (71063) 27 17B‐estradiol.tw. (54) 28 (oestriol or estriol).tw. (4948) 29 (dienoestrol or replens).tw. (77) 30 or/13‐29 (233413) 31 randomized controlled trial.pt. (413263) 32 controlled clinical trial.pt. (90520) 33 randomized.ab. (343103) 34 placebo.tw. (173470) 35 clinical trials as topic.sh. (176075) 36 randomly.ab. (246887) 37 trial.ti. (149165) 38 (crossover or cross‐over or cross over).tw. (66938) 39 or/31‐38 (1033187) 40 exp animals/ not humans.sh. (4224815) 41 39 not 40 (950151) 42 12 and 30 and 41 (1095)
Appendix 4. Embase search strategy
OVID platform
From inception until 12.04.16
1 exp Vaginitis/ (12609) 2 ($vagin$ adj2 atroph$).tw. (983) 3 exp female sexual dysfunction/ (10020) 4 vaginism.tw. (42) 5 pruritis.tw. (1600) 6 exp female genital pruritus/ (37) 7 ($vagin$ adj2 dry$).tw. (1296) 8 (urogenital atrophy or urogenital symptom$).tw. (587) 9 (menopaus$ adj1 symptom$).tw. (4400) 10 (postmenopaus$ adj2 symptom$).tw. (932) 11 Dyspareuni$.tw. (5064) 12 urogenital ageing.tw. (20) 13 urogenital disorder$.tw. (143) 14 or/1‐13 (31094) 15 ($vagina$ adj3 administ$).tw. (1582) 16 vaginal cream$.tw. (327) 17 vaginal ring$.tw. (983) 18 vagina$ pessar$.tw. (425) 19 vagina$ tablet$.tw. (501) 20 vagina$ capsule$.tw. (49) 21 (oestradiol or oestrogen).tw. (25871) 22 estradiol.tw. (77487) 23 oestrogenic.tw. (1656) 24 17B‐estradiol.tw. (392) 25 (oestriol or estriol).tw. (4663) 26 (dienoestrol or replens).tw. (183) 27 exp estrogen/ (230358) 28 estrogen$.tw. (137342) 29 vagitories.tw. (10) 30 ovule$.tw. (1865) 31 vagina$ gel$.tw. (519) 32 or/15‐31 (305624) 33 Clinical Trial/ (855936) 34 Randomized Controlled Trial/ (397801) 35 exp randomization/ (70046) 36 Single Blind Procedure/ (21855) 37 Double Blind Procedure/ (127422) 38 Crossover Procedure/ (46656) 39 Placebo/ (272777) 40 Randomi?ed controlled trial$.tw. (132820) 41 Rct.tw. (19837) 42 random allocation.tw. (1502) 43 randomly allocated.tw. (24326) 44 allocated randomly.tw. (2097) 45 (allocated adj2 random).tw. (750) 46 Single blind$.tw. (17141) 47 Double blind$.tw. (160313) 48 ((treble or triple) adj blind$).tw. (535) 49 placebo$.tw. (229991) 50 prospective study/ (328346) 51 or/33‐50 (1556516) 52 case study/ (37152) 53 case report.tw. (302831) 54 abstract report/ or letter/ (955011) 55 or/52‐54 (1288092) 56 51 not 55 (1515756) 57 14 and 32 and 56 (2651)
Appendix 5. PsycINFO search strategy
OVID platform
From inception until 12.04.16
1 vaginitis.tw. (41) 2 (vagina$ adj2 atroph$).tw. (38) 3 pruritis.tw. (40) 4 (vagina$ adj3 dry$).tw. (125) 5 (urogenital atrophy or urogenital symptom$).tw. (16) 6 (menopaus$ adj1 symptom$).tw. (531) 7 (postmenopaus$ adj1 symptom$).tw. (31) 8 exp Sexual Function Disturbances/ or exp Dyspareunia/ (7800) 9 exp Pruritus/ (196) 10 Sexual Dysfunction.tw. (4376) 11 Dyspareunia.tw. (471) 12 Pruritus.tw. (254) 13 or/1‐12 (10331) 14 exp Estrogens/ (5390) 15 (intravaginal$ adj3 administ$).tw. (7) 16 vaginal cream$.tw. (6) 17 vaginal ring$.tw. (37) 18 (vagina$ adj3 pessar$).tw. (4) 19 (vagina$ adj2 tablet$).tw. (5) 20 vagina$ capsule$.tw. (1) 21 (oestradiol or oestrogen).tw. (796) 22 oestrogenic.tw. (33) 23 17B‐estradiol.tw. (19) 24 oestriol.tw. (3) 25 estrogen$.tw. (6421) 26 (dienoestrol or replens).tw. (2) 27 or/14‐26 (8562) 28 13 and 27 (284) 29 random.tw. (42844) 30 control.tw. (332835) 31 double‐blind.tw. (18599) 32 clinical trials/ (8391) 33 placebo/ (3991) 34 exp Treatment/ (606250) 35 or/29‐34 (929103) 36 28 and 35 (188)
Appendix 6. Data extraction and eligibility form
| Data extraction and eligibility form for JS157 Reviewers: see notes in italics before relevant sections | |||||||||
| Review ID | Date form completed | ||||||||
| Review title | |||||||||
| Review author name / ID | |||||||||
| Co‐reviewer name / ID | |||||||||
| If any other references are found to the same trial, code first paper as A; code any further papers found as B, C, etc.; link any found for listing in RevMan | |||||||||
| Study identifier: Davar 2012 | Author: | Year of publication: | |||||||
| Title | |||||||||
| Contact author details: | |||||||||
| Eligibility | |||||||||
| RCT | [ ] yes [ ] no [ ] unclear |
Describe: | |||||||
| Relevant participants | [ ] yes x [ ] no [ ] unclear |
Describe: | |||||||
| Relevant interventions | [ ] yes x [ ] no [ ] unclear |
Describe: | |||||||
| Relevant outcome measures | [ ] yes [ ] no X [ ] unclear |
Describe: | |||||||
| INCLUDE IN REVIEW? | [ ] yes [ ] no X | If no, give reason: | |||||||
| Notes: | |||||||||
| Characteristics of included studies | |||||||||
| Participants | |||||||||
| Diagnostic criteria (definition of eligibility) | Inclusion criteria: Exclusion criteria: |
||||||||
| Group A | Group B | ||||||||
| Number of participants at randomisation | |||||||||
| Number analysed at outcome | |||||||||
| Withdrawals/Exclusions | |||||||||
| Age (mean, SD) | |||||||||
| Setting e.g. fertility clinic | |||||||||
| Country | |||||||||
| Interventions | |||||||||
| Describe these including mode of delivery, route, doses, timing. Quote from paper if possible | |||||||||
| Group A | Group B | ||||||||
| Intervention | |||||||||
| Standard treatment | |||||||||
| Treatment length | |||||||||
| Follow up length | |||||||||
| Loss to follow up | |||||||||
| Other info regarding treatment | |||||||||
| Quality assessment | |||||||||
| Refer to Cochrane Handbook for Systematic Reviews of Interventions, table 8.5a | |||||||||
| Selection bias | |||||||||
| Random sequence generation | [ ] low risk [ ] high risk [ ] unclear risk → more information required |
Describe: | |||||||
| Allocation sequence concealment | [ ] low risk [ ] high risk [ ] unclear risk → more information required |
Describe: | |||||||
| Performance bias | |||||||||
| Blinding of participants and personnel | [ ] low risk [ ] high risk [ ] unclear risk → more information required |
Describe: | |||||||
| Detection bias | |||||||||
| Blinding of outcome assessment (patient‐reported outcomes) |
[ ] low risk [ ] high risk [ ] unclear risk → more information required |
Describe: | |||||||
| Attrition bias | |||||||||
| Due to amount, nature or handling of incomplete outcome data | [ ] low risk [ ] high risk [ ] unclear risk → more information required |
Describe: | |||||||
| Reporting bias | |||||||||
| Selective reporting: check methods against reported outcomes | [ ] low risk [ ] high risk [ ] unclear risk → more information required |
Describe: | |||||||
| Other sources of bias | |||||||||
| Sources of bias such as differences in demographic characteristics | [ ] low risk [ ] high risk [ ] unclear risk → more information required |
Describe: | |||||||
| Is there anything not reported? If information is missing, record this, so that it is apparent that the information is missing, not just not extracted | |||||||||
| Incomplete outcome data | |||||||||
| Description of outcomes (per woman randomised) | |||||||||
| Indicate the reported outcomes and describe as appropriate | |||||||||
| Describe: | |||||||||
| Describe: | |||||||||
| Describe: | |||||||||
| Describe: | |||||||||
| Describe: | |||||||||
| Results | |||||||||
| Record summary data for each intervention group (e.g. 2×2 table for dichotomous data; means and SDs for continuous data). | |||||||||
| Participants (number allocated and completed) |
No. in treatment group: | No. in control group: | |||||||
| Dichotomous outcomes | No of events | No of participants | No of events | No of participants | |||||
| Continuous outcomes | Mean, SD | No of participants | Mean; SD | No of participants | |||||
| Note any other results reported but not listed as outcome measures: | |||||||||
| Miscellaneous | |||||||||
| Funding source stated | |||||||||
| Ethical approval obtained | |||||||||
| Written consents obtained from participants | |||||||||
| Key conclusions of the study authors. | |||||||||
| Miscellaneous comments from the study authors. | |||||||||
| References to other relevant studies. | |||||||||
| Correspondence required. | |||||||||
| Miscellaneous comments by the review authors. | |||||||||
Data and analyses
Comparison 1. Oestrogen ring versus placebo or other regimens.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Improvement in symptoms (participant‐assessed at end point) | 6 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 Oestrogen ring versus oestrogen cream | 2 | 341 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.33 [0.80, 2.19] |
| 1.2 Oestrogen ring versus oestrogen tablets | 3 | 567 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.78 [0.53, 1.15] |
| 1.3 Oestrogen ring versus placebo | 1 | 67 | Odds Ratio (M‐H, Fixed, 95% CI) | 12.67 [3.23, 49.66] |
| 2 Endometrial thickness | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 Oestrogen ring versus oestrogen cream | 2 | 273 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.36 [0.14, 0.94] |
| 3 Improvement in symptoms (clinician‐assessed at end point) | 6 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 Oestrogen ring versus oestrogen cream | 3 | 533 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.70, 1.53] |
| 3.2 Oestrogen ring versus oestrogen tablets | 2 | 397 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.45 [0.90, 2.32] |
| 3.3 Oestrogen ring versus placebo | 1 | 49 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.0 [0.55, 7.31] |
| 4 Improvement in symptoms (decrease in vaginal pH at end point) | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4.1 Oestrogen ring versus oestrogen cream | 1 | 165 | Mean Difference (IV, Fixed, 95% CI) | 0.10 [‐0.19, 0.39] |
| 4.2 Oestrogen ring versus oestrogen tablets | 1 | 146 | Mean Difference (IV, Fixed, 95% CI) | ‐0.20 [‐0.38, ‐0.02] |
| 4.3 Oestrogen ring versus placebo | 1 | 37 | Mean Difference (IV, Fixed, 95% CI) | ‐1.31 [‐1.82, ‐0.80] |
| 5 Improvement in symptoms (increase in maturation indices at end point) | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.1 Oestrogen ring versus oestrogen cream | 2 | 341 | Mean Difference (IV, Fixed, 95% CI) | 0.79 [‐1.52, 3.09] |
| 5.2 Oestrogen ring (7.5 ug) versus placebo | 0 | 0 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 5.3 Oestrogen ring (100 ug) versus placebo | 0 | 0 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 5.4 Oestrogen ring (50 ug) versus placebo | 0 | 0 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 5.5 Oestrogen ring (unspecified dose) versus placebo | 1 | 37 | Mean Difference (IV, Fixed, 95% CI) | 24.4 [15.25, 33.55] |
| 6 Adverse events (breast disorders) | 4 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 6.1 Oestrogen ring versus oestrogen cream | 1 | 192 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.12 [0.01, 1.13] |
| 6.2 Oestrogen ring versus oestrogen tablets | 3 | 587 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.42 [0.12, 1.52] |
| 7 Adverse events (total adverse events) | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 7.1 Oestrogen ring versus oestrogen cream | 1 | 192 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.88 [0.47, 1.63] |
| 7.2 Oestrogen ring versus placebo | 1 | 37 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.45 [0.11, 1.78] |
| 8 Adherence to treatment | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 8.1 Oestrogen ring versus oestrogen cream | 2 | 350 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.23 [1.31, 3.80] |
| 8.2 Oestrogen ring versus oestrogen tablets | 1 | 146 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.69 [0.66, 4.31] |
Comparison 2. Oestrogen tablets versus placebo or other regimens.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Improvement in symptoms (participant‐assessed at end point) | 4 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 Oestrogen tablets versus oestrogen cream | 2 | 208 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.55, 2.01] |
| 1.2 Oestrogen tablets versus placebo | 2 | 1638 | Odds Ratio (M‐H, Fixed, 95% CI) | 12.47 [9.81, 15.84] |
| 2 Endometrial thickness | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 Oestrogen tablets versus oestrogen cream | 2 | 151 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.31 [0.06, 1.60] |
| 3 Improvement in symptoms (clinician‐assessed at end point) | 6 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 Oestrogen tablets versus oestrogen cream | 3 | 528 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.70, 1.52] |
| 3.2 Oestrogen tablets versus placebo | 3 | 2078 | Odds Ratio (M‐H, Fixed, 95% CI) | 12.85 [10.39, 15.89] |
| 4 Improvement in symptoms (decrease in vaginal pH at end point) | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4.1 Oestrogen tablets versus oestrogen cream | 1 | 48 | Mean Difference (IV, Fixed, 95% CI) | 0.20 [‐0.12, 0.52] |
| 4.2 Oestrogen tablets versus placebo | 2 | 524 | Mean Difference (IV, Fixed, 95% CI) | ‐0.95 [‐1.10, ‐0.80] |
| 5 Improvement in symptoms (increase in maturation indices at end point) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.1 Oestrogen tablets versus oestrogen cream | 1 | 48 | Mean Difference (IV, Fixed, 95% CI) | ‐4.69 [‐13.58, 4.20] |
| 5.2 Oestrogen tablets versus placebo | 1 | 436 | Mean Difference (IV, Fixed, 95% CI) | 18.63 [14.57, 22.69] |
| 6 Adverse events (breast disorders) | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 6.1 Oestradiol tablets versus oestriol tablets | 1 | 96 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.06 [0.12, 77.09] |
| 7 Adverse events (total adverse events) | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 7.1 Oestrogen tablets versus placebo | 1 | 309 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.28 [0.24, 6.69] |
| 8 Adherence to treatment | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 8.1 Oestrogen tablets versus oestrogen cream | 1 | 53 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.90 [0.41, 8.94] |
| 8.2 Oestradiol tablets versus oestriol tablets | 1 | 96 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.69 [1.15, 6.31] |
Comparison 3. Oestrogen cream versus placebo or other regimens.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Improvement in symptoms (participant‐assessed at end point) | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 Oestrogen cream versus isoflavone gel | 1 | 50 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.08 [0.08, 53.76] |
| 1.2 Oestrogen cream versus placebo | 2 | 198 | Odds Ratio (M‐H, Fixed, 95% CI) | 4.10 [1.88, 8.93] |
| 2 Endometrial thickness | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3 Improvement in symptoms (clinician‐assessed at end point) | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 Oestrogen cream versus placebo | 1 | 153 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.29 [1.47, 7.36] |
| 4 Improvement in symptoms (decrease in vaginal pH at end point) | 4 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4.1 Oestrogen cream versus non‐hormonal local bio adhesive vaginal moisturiser | 2 | 62 | Mean Difference (IV, Fixed, 95% CI) | ‐0.36 [‐0.52, ‐0.21] |
| 4.2 Oestrogen cream (21 days) versus placebo (21 days) | 1 | 215 | Mean Difference (IV, Fixed, 95% CI) | ‐1.20 [‐1.47, ‐0.93] |
| 4.3 Oestrogen cream (twice weekly) versus placebo (twice weekly) | 1 | 208 | Mean Difference (IV, Fixed, 95% CI) | ‐1.30 [‐1.58, ‐1.02] |
| 4.4 Oestriol gel (50 ug) versus placebo gel | 1 | 153 | Mean Difference (IV, Fixed, 95% CI) | ‐0.8 [‐1.23, ‐0.37] |
| 5 Improvement in symptoms (increase in maturation indices at end point) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.1 Oestrogen cream versus placebo | 1 | 153 | Mean Difference (IV, Fixed, 95% CI) | 23.7 [17.25, 30.15] |
| 6 Adverse events (breast disorders) | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 7 Adverse events (total adverse events) | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 7.1 Oestrogen cream versus non hormonal lubricant gel | 1 | 50 | Odds Ratio (M‐H, Fixed, 95% CI) | 10.67 [0.54, 209.64] |
| 8 Adherence to treatment | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Ayton 1996.
| Methods | Randomisation on a 2:1 basis, via a random number‐generating computer programme using sequential envelopes (sealed, opaque) at each trial centre Participants not blinded. Pathologist blinded Multi‐centre (3), parallel design Number of women randomised: n = 194 Number of women analysed: n = 176 (ITT), n = 166 (PP) Number of withdrawals: n = 18; Estring group: 9 due to adverse experiences; nausea, headache, abdominal pain, backache, urinary infection, vulval discomfort, candida, and vaginal bleeding, 2 women: ring fell out Premarin group: 7 (5 women due to abdominal and pelvic discomfort, backache, premenstrual syndrome symptoms, breast discomfort and candida and 2 lost to follow‐up Power calculation reported and analysis by intention‐to‐treat and per‐protocol analysis Source of funding: Pharmacia | |
| Participants | Inclusion criteria: postmenopausal, symptoms of urogenital atrophy (vaginal dryness with or without dyspareunia, pruritus, dysuria and/or urgency and signs of atrophic vaginitis, including pallor, petechiae, friability and/or dryness) Age: 36‐86 years (mean age 59) Source of participants: response to advertisement Exclusion criteria: history of hysterectomy, bilateral oophorectomy, hormone‐dependant neoplasia, vaginal bleeding after the initial progestogen challenge test or of unknown origin, sex hormone treatment in the preceding 3 months, grade II‐III vaginal prolapse, thrombo‐embolic disease and liver disease Location: Sydney and Melbourne, Australia | |
| Interventions | Treatment: oestradiol vaginal ring (Estring), inserted high in the vagina by the investigator at the inclusion visit with instructions to remain in situ continuously for 12 weeks. Women were allowed to remove the ring for a short time if coitus desired. Ring: silicone core contains 2 mg of micronised 17β oestradiol, uniform sustained release of 5‐10 mcg per 24 hours of oestradiol for 3 months Control: 0.625 mg conjugated equine oestrogen (Premarin) cream vaginally graduated applicator used to insert 1 g every night for 3 weeks followed by 1 week free of treatment, repeating this 4‐week cycle twice to produce a total of 12 weeks' treatment Duration: 12 weeks | |
| Outcomes | Cure and response rate, maturation value and vaginal pH, vaginal and vulval irritation/ulceration, intercurrent vaginal bleeding (indication of endometrial thickness), adherence to treatment, discomfort, acceptance of treatment delivery | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random sequence generation |
| Allocation concealment (selection bias) | Low risk | Allocation concealed by sealed opaque envelope |
| Blinding (performance bias) | Unclear risk | Not reported |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Some outcome assessors (pathologist) were blinded; unclear whether participants were blinded as some outcomes were self assessed |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Data analysed using ITT |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to make a conclusive judgement |
| Other bias | Low risk | Treatment groups were balanced at baseline |
Bachmann 2008.
| Methods | A 3‐arm parallel RCT | |
| Participants | 230 postmenopausal women with atrophic vaginitis Age (mean): Group A: 58.3 (7.4); Group B: 57.7 (6.5); Group C: 57.6 (4.8) Inclusion criteria: women aged 45 years or older with moderate to severe vaginal dryness and soreness were enrolled. All participants had serum E2 concentrations of 20 pg/mL or less, with 5% or less superficial vaginal cells. Participants were also required to be at least 12 months postmenopausal, with an endometrial thickness of 5 mm or less as determined by transvaginal ultrasonography Exclusion criteria: known or suspected history of breast carcinoma, hormone‐dependent tumour, genital bleeding of unknown cause, acute thrombophlebitis or thromboembolic disorder associated with oestrogen use, vaginal infection requiring treatment, allergy to the test drug or its constituents, or any serious disease or chronic condition that could interfere with study compliance were among the criteria for exclusion. The use of any investigational drug within the 30 days preceding screening, any homeopathic preparation within the 7 days preceding study drug initiation, and any exogenous corticosteroid or sex hormones within the 8 weeks preceding study drug initiation were prohibited |
|
| Interventions |
Group A: 25 mcg oestradiol tablet (n = 91). 1 tablet inserted into the vagina daily for 14 days, then twice per week Group B: 10 mcg tablet (n = 92). 1 tablet inserted into the vagina daily for 14 days, then twice per week Group C: placebo (n = 47). 1 tablet inserted into the vaginal daily for 14 days, then twice per week Duration: 12 weeks; follow up: 52 weeks |
|
| Outcomes | Vaginal pH, maturation index, vaginal health, vaginal symptoms, adverse events | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | It was not reported what was used in generating the randomisation code, however it was stated that, "A randomization code was generated and assigned in blocks of five" |
| Allocation concealment (selection bias) | Low risk | It was reported that, "A sealed envelope with the randomization number and identity of the treatment for each participant was given to each investigator. Allocations were concealed in sealed envelope" |
| Blinding (performance bias) | Low risk | Trial was a "double‐blind, placebo‐controlled, parallel‐group 12‐week study" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Some outcomes assessed by participants who were blinded. Adverse events, symptoms and other outcomes assessed by investigators who also were blinded. Also endometrial biopsy results assessed by “two independent pathologists who were masked to treatment group and each others' interpretation |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Analysis of data was based on ITT |
| Selective reporting (reporting bias) | Low risk | All the outcomes specified in the methods section ware reported |
| Other bias | Low risk | It was reported that, "Demographic and baseline characteristics were similar across treatment groups, with the exception of a slightly higher percentage of white participants in the 25 mcg E2" group |
Bachmann 2009.
| Methods | A 4‐arm parallel RCT | |
| Participants | 423 postmenopausal women with moderate to severe symptoms of vaginal atrophy Age (mean, SD): Group A: 57.7 (5.8); Group B: 58.0 (5.8); Group C: 57.5 (5.5); Group D: 58.7 (5.8) Inclusion criteria: the trial enrolled generally healthy postmenopausal women (aged 45‐80) with an intact uterus and symptoms of moderate to severe vaginal atrophy. These were defined as the following: a baseline composite score (at the initial screening visit) of at least 5 (1 = mild, 2 = moderate, 3 = severe) for the 4 symptoms of vaginal dryness, itching, burning and dyspareunia (at least one of these symptoms had to be moderate or severe); a total score of 15 or less on the Genital Health Clinical Evaluation (GHCE), a tool used to evaluate six parameters (vaginal pH, fluid secretion, moisture, vaginal rugosity, mucosal colour, and epithelial mucosa) scored on a scale of 1‐4; vaginal pH of at least 5; and a clinical diagnosis of atrophic vaginitis, defined as 0% to 5% superficial cells on vaginal cytologic smear. Additional inclusion criteria included a serum oestradiol concentration of 30 pg/mL or less and a serum follicle stimulating hormone level greater than the lower limit of normal for postmenopausal women at the given laboratory Exclusion criteria: the use of an intrauterine device within 3 months of screening or the use of any oral, vaginal, or transdermal medication containing oestrogens, androgens, or progestins within 8 weeks of screening. Women who had used vaginal moisturisers, lubricants, jellies, ointments, douches, herbal medications, over‐the‐counter preparations, home remedies, or natural oestrogen products (ie, soy products) for the treatment of menopausal symptoms agreed to refrain from their use for a minimum of 7 days before screening. Participants who currently used more than two antihypertensive medications, had used any investigational drug or device within 30 days of screening, or had urogynecologic surgery within 3 months of screening were also excluded. |
|
| Interventions |
Group A: conjugated oestrogen (21/7) (n = 143). 0.3 mg cream applied once daily (21 days on/7 days off) Group B: placebo (21/7) (n = 72). 0.3 mg cream applied once daily (21 days on/7 days off) Group C: conjugated oestrogen (2 x /wk) (n = 140). 0.3 mg cream applied twice weekly Group D: placebo (2 x /wk) (n = 68). 0.3 mg cream applied twice weekly Duration: 12 weeks |
|
| Outcomes | Participant‐reported symptoms, vaginal health, vaginal pH, maturation index, adverse events | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported; it was only stated that, "participants were randomly assigned to one of four treatment regimens" |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information on method used in allocation concealment |
| Blinding (performance bias) | Low risk | It was reported that, "The first phase was double‐blind, randomized, and placebo‐controlled" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Some outcomes were assessed by participants (adverse events, symptom scores) and participants were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Rates and reasons for withdrawals were similar across treatment groups; data was analysed using the modified ITTA: "The primary efficacy analyses at week 12 were done using the modified intent‐to‐treat (MITT)" |
| Selective reporting (reporting bias) | Low risk | All the outcomes specified in the methods section were reported |
| Other bias | Low risk | "There were no differences between treatment groups in demographic and clinical characteristics at baseline" |
Barentsen 1997.
| Methods | Women were allocated to one of two treatment schedules via a central randomisation list Open label, cross‐over design with single blinding for cytological evaluation Number of women randomised: n = 168 Number of women analysed n = 165 Number of withdrawals: n = 27; 11 in ring group, 16 in cream group, reasons included; protocol violations such as wrong inclusion, interruption of treatment, later visits than allowed, premature withdrawal from treatment and miscellaneous reasons such as itching, eczema, allergic reaction (2 ring women, 3 cream women) and one woman lost ring Power calculation for sample size reported and ITT analysis performed for some outcomes Source of funding: Pharmacia | |
| Participants | Inclusion criteria: 2 years after spontaneous or surgical menopause (bilateral oophorectomy) and symptoms of atrophic vaginitis including vaginal dryness Age: not stated Source of participants: clinics in 12 centres Exclusion criteria: known contra‐indications or precautions for oestrogen therapy and women with sex hormone treatment during last 3 months Location: Netherlands | |
| Interventions | Treatment: oestradiol ring (Estring) containing 2 mg micronised 17β oestradiol with a constant release of 7.5 mcg oestradiol/24 hours for 90 days Control: estriol cream (Synapause) containing 1 mg estriol/G of cream 0.5 mg daily for 2 weeks followed by maintenance dose 0.5 mg 3 times a week Duration: 3 months + 3 months (first period data only used) | |
| Outcomes | Maturation value, vaginal pH, cured or improved symptoms, responders (parabasal cells decreased > 25%), adverse events, vaginal irritation administration form, preference | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Central randomisation but no further details reported |
| Allocation concealment (selection bias) | Unclear risk | No details reported |
| Blinding (performance bias) | Unclear risk | No details reported |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Single blinding but some outcomes were self assessed |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Data analysis based on ITT |
| Selective reporting (reporting bias) | Unclear risk | Insufficient details to make a conclusive judgement |
| Other bias | Unclear risk | Insufficient details to make a conclusive judgement |
Bygdeman 1996.
| Methods | Open label randomisation method Single‐centre, parallel‐group design, no blinding Number of women randomised: n = 40 Number of women analysed: n = 39 No power calculation given and analysis not by ITT Source of funding: not stated | |
| Participants | Inclusion criteria: postmenopausal women complaining of vaginal dryness, natural menopause oophorectomy Age: 43‐76 (mean 58.3 years) Source of participants: not stated Exclusion criteria: hormonal‐dependent tumours, known or suspected other serious diseases, abnormal genital bleeding, past history of active thromboembolic disorder, vaginal infection, HRT in last 3 months, vaginal use of douche or lubricant Location: Sweden | |
| Interventions | Treatment: a non‐hormonal local bio adhesive vaginal gel composed of purified water enmeshed in a carbomer‐polycarbophil system (Replens) one vaginal application 3 times a week for 3 months Control: an oestrogenic cream (0.01%); Dienoestrol (Cilag), 0.5 mg daily for 2 weeks then 3 times a week for 3 months Duration: 3 months | |
| Outcomes | Vaginal pH, vaginal dryness index, pruritis, dyspareunia, overall feeling, adverse events | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information to make a conclusive judgement |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to make a conclusive judgement |
| Blinding (performance bias) | High risk | Open label trial |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Open label trial and some of the outcomes were self‐assessed; but unclear whether outcome assessors were blinded or not |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Only one participant withdrew from the trial |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to make a conclusive judgement |
| Other bias | Unclear risk | Insufficient information to make a conclusive judgement |
Cano 2012.
| Methods | 2‐arm parallel RCT | |
| Participants | 167 postmenopausal women with vaginal atrophy Age (mean): Group A: 56.5; Group B: 57.2 Inclusion criteria: postmenopausal with at least 2 years of amenorrhoea caused by either natural or surgical menopause (bilateral oophorectomy). They also presented symptoms and signs of atrophy of the vaginal mucosa including as a minimum, vaginal dryness and at least one sign of the condition verified by the investigator Exclusion criteria: history of malignant or pre‐malignant lesions of the breasts or endometrium, malignant colon or hepatic tumours, malignant melanoma, venous thromboembolic disorders (deep vein thrombosis, pulmonary embolism) or arterial thromboembolic disorders (ischaemic heart disease, myocardial infarction, cerebrovascular accident), peripheral arterial disease, mesenteric artery thrombosis, renal artery thrombosis, or coagulopathies |
|
| Interventions |
Group A: low‐dose estriol vaginal gel (n = 114). 1 g of vaginal gel containing 50 µg of estriol Group B: placebo (n = 53). 1 g of placebo vaginal gel Duration: 12 weeks |
|
| Outcomes | Changes in vaginal atrophy symptomatology, vaginal pH, maturation index, adverse event, adherence to treatment | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported, it was only stated that, "Eligible women were randomized in a ratio of 2:1....." |
| Blinding (performance bias) | Low risk | Study was described as, "randomized, doubleblind, placebo‐controlled study." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Participant‐reported outcomes ‐ a double‐blind trial |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Proportions of and reasons for withdrawals differ between the two treatment groups and data was not analysed on the basis of ITT |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes reported |
| Other bias | Unclear risk | Participants similar in baseline demographic characteristics but numbers not balanced at randomisation |
Casper 1999 study 1.
| Methods | Method of randomisation not stated Open label, parallel design, multicentre (14) participants randomised 2:1 ratio Pathologists blinded Number of women randomised: n = 219 (Estring 147, pessaries 72) Number of women analysed: n = 190/171 (ring 116, pessary 55) Number of withdrawals: n = 29 (ring 19, suppository 10). 19 more women excluded (ring 12, suppository 7) Reasons for withdrawal: not stated No power calculation No ITT Source of funding: Pharmacia and Upjohn | |
| Participants | Inclusion criteria: at least 2 years after spontaneous or surgical menopause presenting with one or more signs and symptoms of atrophic vaginitis due to oestrogen deficiency, pruritus vulvae, dyspareunia, dysuria, urinary urgency; on examination; petechiae, friability or vaginal dryness Age: not stated Source of participants: not stated Exclusion criteria: if received sex hormone therapy within previous 3 months, or had severe hepatic or renal diseases, oestrogen dependant neoplasms and urinary tract infections despite antibiotics, or had endometrial thickness > 5 mm or vaginal ulceration, irritation or bleeding from causes other than epithelial atrophy Location: Austria, Switzerland, Germany | |
| Interventions | Treatment: oestradiol‐releasing silicone ring (Estring) core containing 2 mg 17β‐oestradiol releasing 7.5 mcg/24 hours for 90 days Control: oestradiol pessary 0.5 mg (Ovestin) Duration: 3 months | |
| Outcomes | Vaginal pH, response, adverse events, adherence to treatment | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding (performance bias) | High risk | Open label trial |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Single blinding but some outcomes were self‐assessed |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Proportions of withdrawals differed between the two treatment groups and data analysis was not based on ITT |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to make a conclusive judgement |
| Other bias | Unclear risk | Insufficient information to make a conclusive judgement |
Casper 1999 study 2.
| Methods | Method of randomisation: not stated Double‐blind, parallel design, placebo controlled Number of participants: randomised: n = 84 Number of participants analysed: n = 67 (33 in ring group and 34 in placebo group) Number of withdrawals: 4 participants excluded due to protocol violation (3 in ring group and 1 in placebo group). 13 withdrawals due to participant request (6 in ring group and 4 placebo group), adverse events (1 case of profuse eczema in placebo group), lack of efficacy (1 case in placebo group) and abandonment (1 case in oestradiol group) No power calculation and no ITT analysis Source of funding: Pharmacia and Upjohn | |
| Participants | Inclusion and exclusion criteria as per study 1 Location: Germany | |
| Interventions | Treatment: oestradiol‐releasing silicone ring (Estring) per study 1 Control: placebo ring | |
| Outcomes | Maturation value, vaginal pH, endometrial thickness, freedom of symptoms (dyspareunia, pallor, petechiae, friability, vaginal dryness) | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding (performance bias) | Low risk | Double‐blinded trial |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Some outcomes were self‐assessed but unclear whether other outcome assessors were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Data analysed on the basis of ITT |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to make a conclusive judgement |
| Other bias | Unclear risk | Insufficient information to make a conclusive judgement |
Dessole 2004.
| Methods | Method of randomisation: not stated Double‐blind, placebo‐controlled. Number of participants randomised: n = 88 Number of participants analysed: n = 88 Number of withdrawals: 4 in treatment group and 7 in placebo group (discomfort experienced: 2 in treatment and 2 in placebo group, localised adverse reactions (burning and itching) 2 in treatment group and 1 in placebo group; and 4 in the control group did not benefit from therapy Power calculation and ITT analysis performed Source of funding: not stated | |
| Participants | Inclusion criteria: participants presented with symptoms and signs of urinary stress incontinence, vaginal atrophy, and histories of recurrent urinary tract infections. None had received oestrogen before the study Age: 54‐62 years Source of participants: not stated Exclusion criteria: women with anatomical lesions of the urogenital tract, such as uterovaginal prolapse, cystocele, and rectocele of grade II or III, presence of severe systemic disorders, thromboembolic diseases, bilary lithiasis, previous breast or uterine cancer, abnormal uterine bleeding, and body mass index of 25 kg/m2 or higher Location: Sardinia | |
| Interventions | Treatment: intravaginal oestradiol ovules (1 mg) once daily for 2 weeks and then 2 ovules once weekly for 6 months Control: placebo vaginal ovules same regimen Duration: 6 months | |
| Outcomes | Vaginal dryness, dyspareunia, vaginal pH, KPI of vaginal epithelium | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding (performance bias) | Low risk | Double‐blinded, placebo‐controlled trial |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Some outcomes were self‐assessed but unclear whether other outcome assessors were blinded e.g. the pathologists |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Data analysed on the basis of ITT |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to make a conclusive judgement |
| Other bias | Unclear risk | Insufficient information to make a conclusive judgement |
Dugal 2000.
| Methods | Randomisation method not stated Parallel, multi centre, single blinded study Number of women randomised: n = 96 Number of women analysed: n = 85 Number of withdrawals: n = 11 (6 women in the tablet group; 3 adverse events (paraesthesia, leukorrhoea, endometrial disorder), 2 non‐compliance, 1 medical problems (hypothyroidism); 5 women in the vagitories group (2 treatment ineffective, 2 did not attend clinic visits, 1 personal problem) Power calculation for sample size performed and analysis by ITT Source of funding: Nova Nordisk Pharmaceutical | |
| Participants | Inclusion criteria: women with signs and symptoms of vaginal atrophy and did not require systemic oestrogen therapy for treatment of vasomotor symptoms or prophylaxis of osteoporosis and had not experienced vaginal bleeding for 1 year Age: 50‐70 years Source of participants: not stated Exclusion criteria: women who had taken systemic or vaginal oestrogens within 6 months of the study, history of breast or endometrial cancer, abnormal genital bleeding, acute thrombophlebitis or thromboembolic disorders associated with previous oestrogen use or current urinary or vaginal infection Location: Norway | |
| Interventions | Treatment: oestradiol vaginal tablet 25 mcg 17β oestradiol Control: estriol vagitories (0.5 mg estriol) once daily for 2 weeks and twice weekly thereafter. Duration: 6 months | |
| Outcomes | Vaginal dryness, maturation, adverse effects, leakage of medicine, requirement of sanitary wear, ease of use, hygenic | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding (performance bias) | Unclear risk | Not reported |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Single‐blinded study but some outcomes were self‐assessed |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Data analysed on the basis of ITT |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to make a conclusive judgement |
| Other bias | Low risk | Treatment groups were balanced at baseline |
Eriksen 1992.
| Methods | Randomisation method not stated Placebo‐controlled, double‐blinding Number of centres: not stated Number of women randomised: n = 164 (81 treatment 83 placebo) Number of women analysed: n = 154 Number of withdrawals: n = 10 (6 in tablet group and 4 in placebo) Reasons for withdrawal: aggravation of lichen sclerosis, dysuria, attack of anxiety, disliked the administration form, vaginal bleeding, slight depression, aggravation of articular pain, no effect of treatment, pain in the outer genital region No power calculation and no ITT Source of funding: not stated | |
| Participants | Inclusion criteria: women suffering from vaginal symptoms related to postmenopausal atrophy Age: 45‐70 years Source of participants: outpatients Exclusion criteria: history of cancer or thromboembolic episodes, vaginal bleeding of unknown origin, pregnancy, oestrogen treatment for the duration of at least 1 month before participation Location: Denmark | |
| Interventions | Treatment: 25 mcg 17β‐oestradiol tablet (Vagifem) once daily for 2 weeks, then twice a week for 10 weeks Control: placebo, once daily for 2 weeks then twice a week for 2 weeks Duration: 3 months | |
| Outcomes | Moderate‐severe symptoms of vaginal atrophy; dryness, itching and burning, dyspareunia, adverse events | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding (performance bias) | Low risk | Double‐blinded trial |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double‐blinded trial but unclear whether other outcome assessors were blinded e.g. the pathologists |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Proportion of withdrawals differs between the two treatment groups and analysis was not on the basis of ITT |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to make a conclusive judgement |
| Other bias | Unclear risk | Insufficient information to make a conclusive judgement |
Fernandes 2014.
| Methods | A 4‐arm parallel RCT | |
| Participants | 80 postmenopausal women with symptoms of vaginal atrophy Age (mean, SD): Group A: 57.0 (5.4); Group B: 56.2 (5.3); Group C: 56.4 (4.8); Group D: 57.7 (4.7) Inclusion criteria: women aged 40–70 years with physiological menopause and a history of amenorrhoea for > 3 years with a follicle‐stimulating hormone level of > 30 mIU/mL. They had not taken hormonal treatment for menopausal symptoms in the past 6 months, had shown normal Pap smears and mammograms for the past 12 months, and had complaints compatible with the symptoms of vaginal atrophy (vaginal dryness, vulvovaginal irritation/itching, and pain at sexual activity 6 months ago) Exclusion criteria: women who were expected to undergo an oophorectomy or hysterectomy and those with a body mass index < 18.5 kg/m2 or > 30 kg/m2. "We excluded those women with a contraindication for the use of estrogen or testosterone, namely those with a history of myocardial infarction, severe hypertension, diabetes mellitus, thromboembolic disease, liver failure, ulcerative colitis, Crohn’s disease, breast or endometrial cancer, fibrocystic breast disease with atypical hyperplasia, genital bleeding of unknown origin, a family history of breast cancer, endometrial hyperplasia, or positive serology for human immunodeficiency virus, hepatitis B, or C". Finally, women were excluded if they had a vaginal infection at the time of their gynecological examination. |
|
| Interventions |
Group A: acid polyacrylic (n = 20) Vaginal cream with polyacrylic acid (Vagidrat®, Myralis Pharma Ltd, Aguai, Sao Paulo, Brazil) one vaginal applicator with 3 g cream per application. Group B: testosterone (n = 20) Vaginal cream with testosterone propionate: 1 vaginal applicator with 1 g of cream per application containing 300 μg testosterone propionate prepared using testosterone micronised powder in an emollient cream with silicone to keep the cream iso‐osmolar Group C: estrogen vaginal cream (n = 20) Vaginal cream with conjugated estrogens (Premarin®, Wyeth Pharmaceuticals, Itapevi, São Paulo, Brazil): one vaginal applicator with 1 g of cream per application containing 0.625 mg conjugated estrogens Group D: lubricant (placebo; n = 20) Lubricant with glycerin gel (K‐Y jelly Johnson & Johnson, São José dos Campos, São José dos Campos, Brazil) 3 g in one applicator per application adjusted to maintain similarity with the polyacrylic acid application. This group was used as a control for the 3 other treatment groups Duration: 12 weeks |
|
| Outcomes | Adverse event (allergic vaginitis) | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | It was stated that, "All participants were given a number (1–80) according to their order of inclusion in the study." |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information on method used in allocation concealment |
| Blinding (performance bias) | Low risk | It was reported that, "Dispensation of the topical agent was done by a gynecologist who was not part of the selection/interview team" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information was reported on the blinding of outcome assessor |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | It was reported that, "Data were analyzed according to intention to treat, including all participants in each group" |
| Selective reporting (reporting bias) | Low risk | All the outcomes specified in the methods section were reported |
| Other bias | Unclear risk | It was reported that, "There were no significant differences between groups in terms of age, time after menopause, skin color, smoking habits, numbers of pregnancies, or socioeconomic status" |
Foidart 1991.
| Methods | Randomised to control or placebo group according to randomisation code Parallel group design with double blinding at 2 centres Number of women randomised: n = 109 (56 to treatment 53 to placebo) Number of withdrawals: not stated No power calculation and no ITT analysis Source of funding: not stated | |
| Participants | Inclusion criteria: moderate to severe urogenital and systemic postmenopausal complaints and follicle stimulating and oestradiol (E2) serum levels within postmenopausal range, last menses one year prior to treatment, score at least 6 points on UGI (urogenital scale) and 20 on KI (Kupperman Index) Age: 32‐66 (mean 54.9) Exclusion criteria: not stated Location: Belgium | |
| Interventions | Treatment: vaginal suppository containing 3.5 mg oestriol (E3) or placebo, twice weekly for 3 weeks followed by 1 suppository weekly for 6 months Duration: 6 months | |
| Outcomes | Efficacy: UGI score, vaginal pH Safety: change in endometrium | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Sequence was said to have been generated through randomisation code, no further details were reported |
| Allocation concealment (selection bias) | Unclear risk | No details reported |
| Blinding (performance bias) | Low risk | Double‐blinded trial |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Unclear whether outcome assessors other than the participants were blinded e.g. the pathologists |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Withdrawals were not reported and unclear whether data were analysed on the basis of ITT |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to make a conclusive judgement |
| Other bias | Unclear risk | Insufficient information to make a conclusive judgement |
Garcia Lara 1993.
| Methods | Randomisation not specified, list used consecutive numbers. Double‐blinding, single‐centre, parallel group design Number of women randomised: n = 30 Number of women analysed: n = 26 No power calculation and no ITT analysis Source of funding: not stated | |
| Participants | Inclusion criteria: > 1 year menopausal symptoms, vasomotor instability Age: 43‐47 years (mean 45) Source of participants: hospital clinic Exclusion criteria: no vasomotor instability, psychiatric illness, oral contraceptives or hormonal treatment within 4 months of trial commencement Location: Mexico | |
| Interventions | Treatment: vaginal ovules of oestradiol 3.5 mg. First 3 weeks 2 per week and remainder 1 per week Control: placebo Duration: 4 months | |
| Outcomes | Improvement in symptoms (none or light) | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Sequence was said to have been generated using consecutive numbers, no further details were reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding (performance bias) | Low risk | Double‐blinded trial |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Unclear whether other outcome assessors other than the participants were blinded e.g. the pathologists |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Proportion of withdrawals per treatment group was not reported and data were not analysed on the basis of ITT |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to make a conclusive judgement |
| Other bias | Unclear risk | Insufficient information to make a conclusive judgement |
Griesser 2012.
| Methods | 3‐arm parallel design RCT | |
| Participants | 436 postmenopausal women with vaginal atrophy Age (mean, SD): Group A: 64.9 (8.1); Group B:65.4 (7.3); Group C: 64.8 (7.8) Inclusion criteria: postmenopausal women (last menstrual period more than 12 months ago or having undergone bilateral ovariectomy) aged 18 years or older with a clinical diagnosis of vaginal atrophy, a vaginal maturation index (VMI) < 40% and a vaginal pH value > 5 were eligible for inclusion. At least one subjective symptom of vaginal atrophy (dryness, pain/burning sensation, pruritus, discharge, dyspareunia) had to be rated at a score of ≥ 65 on the visual analogue scale (VAS) Exclusion criteria: hormone replacement therapy, therapy with phytoestrogens or local vaginal hormonal therapy during 12 weeks preceding baseline, as well as current or suspected estrogen‐dependent malignant tumour, a Pap smear ≥ III, endometrial thickness > 5 mm, current or suspected vaginal infection, current symptomatic urinary tract infection, existing or previous breast cancer or suspicion thereof, undiagnosed bleeding in the genital area, current venous thromboembolic disease, known severe renal insufficiency or hypersensitivity to estriol or any of the excipients (hard fat and emulsifiers) of the study medication |
|
| Interventions |
Group A: estriol pessary 0.2 mg (n = 142). Estriol pessary 0.2 mg once‐daily application or 20 days, followed by twice‐weekly administration for a further 9 weeks as a maintenance therapy Group B: estriol pessary 0.03 mg (n = 147) (same administration as in Group A) Group C: placebo (n = 147). Treatment protocol as described for Group A Duration: 12 weeks |
|
| Outcomes | Improvement in atrophy symptoms, vaginal pH, adverse events, tolerability | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding (performance bias) | Low risk | Stated as “double blind” |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Stated as “double blind” (for participant‐ and clinician‐assessed outcomes) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Proportions of and reasons for withdrawals fairly balanced between the treatment groups |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes reported |
| Other bias | Low risk | Baseline demographic characteristic (age) was similar between the groups |
Henriksson 1994.
| Methods | Open, parallel‐group, single‐blinded comparative trial with active control, randomised in the proportions 2:1 Number of centres: 9 Number of women randomised: n = 165 Number of women analysed: n = 146 Number of withdrawals: n = 19; 8 women withdrew before 12 weeks treatment (6 in the ring group and 2 in the pessary group). In 2 cases the ring fell out, and in 4 the ring was taken out due to adverse effects (fever, pain, pruritus, urticaria, impaired asthma, and too short vagina). In the pessary group 1 woman was lost and 1 refused to take pessaries due to burning mucosa and disturbed sleep. 1 woman from the ring group was excluded from per protocol analysis because of wrong randomisation; 4 excluded due to loss of ring before visit 3. In pessary group; 6 excluded from per protocol analysis at visit 3; one had taken hormonal treatment after visit 2, 5 had forgotten to take pessaries before visit 3, 3 women were excluded from per protocol analysis at visit 2, 2 had not taken all pessaries prescribed, and one taken pessaries without removing plastic wrapping. Power calculation for sample size performed and analysis by ITT | |
| Participants | Inclusion criteria: postmenopausal women at least 2 years after spontaneous or surgical (bilateral oophorectomy) menopause, complaining of oestrogen deficiency symptoms of atrophic vaginitis, signs of atrophic vaginal mucosa Age: 45‐80 years (mean 59) Source of participants: centres Exclusion criteria: oestrogen‐dependant neoplasia, abnormal vaginal bleeding of unknown origin, acute or chronic liver disease, acute intermittent porphyria, thromboembolic disease, sex hormone treatment during preceding 3 months, uterovaginal prolapse (grade II to III) and significant bacteruria Location: outpatients; 4 in Sweden, 3 in Finland and 2 in Denmark | |
| Interventions | Treatment: silicone rubber vaginal ring (silastic) containing 2 mg of micronised 17β‐oestradiol releasing 6.5 to 9.5 mcg per 24 hours over a 3‐month period Control: vaginal pessaries (Ovesterin) 0.5 mg estriol daily for 3 weeks. weeks 4‐12; 1 pessary twice weekly Duration: 3 months | |
| Outcomes | Cured/improved subjects' symptoms, physician assessment of mucosa, maturation value, vaginal pH, adverse events, withdrawals, adherence administration form, sexual discomfort, no other discomfort | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding (performance bias) | High risk | Open label trial |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Single‐blinded trial but some of the outcomes were participant‐assessed |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Data were analysed on the basis of ITT |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to make a conclusive judgement |
| Other bias | Unclear risk | Treatment groups were balanced at baseline |
Hosseinzadeh 2015.
| Methods | 2‐arm parallel RCT Number of centres: 1 Number of women randomised: 160 Number of women analysed: ?160 Number of withdrawals: ?0 |
|
| Participants |
Inclusion criteria: 160 postmenopausal women with clinical diagnosis of vaginal atrophy Age (mean, SD): 50‐70 years: Group A: 56.55 ± 8.63; Group B: 55.28 ± 6.12 BMI (kg/m2): Group A: 23.21 ± 3; Group B: 25 22.48 ± 2.56 Exclusion criteria: history of carcinoma of the breast or endometrium, abnormal genital bleeding, acute thrombophlebitis, or thromboembolic disorders associated with previous oestrogen therapy, treated with systemic or vaginal oestrogen within 6 months of the study, or had any contraindication for oestrogen therapy |
|
| Interventions |
Group A: vaginal oestrogen cream (1 tube per night for 14 nights, then 1 tube 2 nights in 1 week for 10 weeks) Group B: Vagifem (oestrogen tablet) 25 mcg tablets (with similar treatment plan) |
|
| Outcomes | Severity of vaginal atrophy (assessed by gynaecologist), 4 main symptoms of atrophic vaginitis: dysuria, dyspareunia, vaginal itching and dryness (participant self report), Satisfaction with treatment, acceptability of treatment (pain, vaginal leakage, need for sanitary towels, user friendliness), adverse events | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "women were randomly divided into Vagifem (from Novo Nordisk) or vaginal estrogen cream (Equin from Actoverco) treatment groups (80 women in each group)" |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding (performance bias) | Unclear risk | Not reported but participants were likely to be unblinded since the 2 treatments required different administration |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Gynaecologists were “unaware of the treatment group” and so there is a low risk of bias for the gynaecologist assessment of vaginal atrophy but other outcomes were answered by participant self‐report |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | It appears that there were no dropouts after randomisation |
| Selective reporting (reporting bias) | Low risk | All outcomes pre‐specified in the methods section were reported |
| Other bias | Low risk | Baseline demographic characteristics similar in both treatment groups |
Karp 2012.
| Methods | 3‐arm parallel design RCT | |
| Participants | 65 women undergoing vaginal reconstructive surgery and also with evidence of symptomatic vaginal atrophy and pelvic organ prolapse Age (mean, SD): Group A: 65 (7.4); Group B: 66 (7.9); Group C: 65 (7.8) Inclusion criteria: postmenopausal women at least 2 years after spontaneous or surgical menopause with symptomatic urogenital atrophy and pelvic organ prolapse who opted to undergo reconstructive vaginal surgery. Eligible candidates had to have at least one symptom (vaginal dryness, vulvar pruritus, dyspareunia, dysuria, or urinary urgency) and/or sign (vaginal pallor, petechiae, friability, or vaginal dryness) of atrophic vaginitis Exclusion criteria: contraindications to oestrogen use (vaginal bleeding, oestrogen‐dependent cancers, hepatic or thrombotic disease), allergies to silicone and/or oestradiol, absence of vulvovaginal atrophy on examination and/or vaginal pH of less than or equal to 4.0, or use of vaginal or systemic oestrogen in the previous 6 months |
|
| Interventions |
Group A: oestradiol‐releasing vaginal ring (n = 22). This was applied immediately after surgery Group B: placebo ring (n = 21). As in Group A Group C: no ring (n = 22) Duration: 12 weeks |
|
| Outcomes | Vaginal pH, maturation index, objective signs of atrophy, subjective bother of atrophy, adverse events | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random assignment via computer generation in "blocks of 20 to one of 3 groups" |
| Allocation concealment (selection bias) | Low risk | Allocation of randomisation group was made by the primary author via sealed envelopes on the day of surgery once the participant was under anaesthesia in the operating room |
| Blinding (performance bias) | Unclear risk | Participants were blinded but unclear whether the clinicians were blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Some of the outcome assessors were blinded to the treatment protocols but unclear whether clinician‐assessed outcomes were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Attrition came from the 2 groups with vaginal rings in place. No attrition from the control group and analysis was not based on ITT |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes reported |
| Other bias | Low risk | Baseline demographic characteristic (age) was similar between the treatment groups |
Lima 2013.
| Methods | 3‐arm parallel design | |
| Participants | 90 postmenopausal women without a hysterectomy and with vaginal atrophy Age (mean): Group A: 56; Group B: 57; Group C: 57 Inclusion criteria: non‐hysterectomised, postmenopausal (2 or more years since final menstrual cycle) women who were 45 years of age or older with symptoms of vaginal dryness and/or pruritus, pain/soreness, vulvar or vaginal burning, and dyspareunia. All participants were required to have serum E2 levels less than 20 pg/mL, follicle‐stimulating hormone levels greater than 40 mIU/mL, no superficial cells on vaginal cytology, an endometrial thickness of less than 4.0 mm as assessed by transvaginal ultrasonography, and a normal mammography during the 6 months leading up to study entry Exclusion criteria: use of any investigational drug or exogenous sex hormones within the 6 months leading up to study drug initiation, or current use of corticosteroids, known or suspected history of hormone‐dependent tumour, breast carcinoma, genital bleeding of unknown cause, acute thromboembolic disorder associated with oestrogen use, vaginal infection requiring treatment, allergy to the test drug or its constituents, hot flushes, and any serious disease or chronic condition that could interfere with study compliance |
|
| Interventions |
Group A: conjugated equine estrogen cream (n = 30) 0.5 g corresponding to 0.3 mg administered vaginally at bedtime Group B: isoflavone vaginal gel (n = 30) 1 g of isoflavone gel or 1 g administered vaginally at bedtime Group C: placebo (n = 30) 1 g of placebo administered vaginally at bedtime Duration: 12 weeks |
|
| Outcomes | Maturation index, vaginal atrophy symptomatology | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding (performance bias) | Low risk | Reported as .."double‐blind, randomized, placebo‐controlled, clinical trial.." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Reported as "double" blind and outcomes were either assessed by participants or clinicians |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 1/3 of participants discontinued from CEE cream group, 17% from placebo group and none from isoflavone group. Likely to cause bias and analysis was not based on ITT although it was reported that "All data reported at week 0 and week 12 are from the intent‐to‐treat analyses, with missing values for each individual computed using the last observation carried forward approach" |
| Selective reporting (reporting bias) | Unclear risk | Other than endometrial safety, no adverse events are reported |
| Other bias | Low risk | "No significant differences were observed among the three groups regarding age, age at and time since menopause, height, weight or BMI" |
Lose 2000.
| Methods | Randomisation by central office using numbers sequentially, no blinding, parallel design, multicentre (27) Number of women randomised: n = 254 Number of women analysed: n = 251 (134 ring, 117 pessary) Number of withdrawals: n = 8 (5 in ring group and 3 in pessary group due to adverse events) Power calculation for sample size performed and analysis by ITT Source of funding: Pharmacia and Upjohn | |
| Participants | Inclusion criteria: women who report with one bothersome lower tract symptom appearing at least 2 years after spontaneous or surgical postmenopause Age: 66 (mean) Source of participants: 26 clinics of practicing gynaecologists and one outpatient department Exclusion criteria: known or suspected oestrogen dependant neoplasia ovarian or mammary, ovarian or corpus uteri malignancies, vaginal bleeding of unknown origin, clinically significant disease, acute or intermittent porphyria, uterovaginal prolapse of grade II or III, sex or hormone treatment within last 6 months, previous participation in clinical trials within 3 months prior to inclusion, signs of vaginal irritation other than atrophy‐derived or signs of vaginal ulceration Location: Denmark | |
| Interventions | Treatment: oestradiol releasing vaginal ring (Estring) with constant release of 7.5 mg oestradiol per 24 hours for 3‐month period Control: oestriol vaginal pessaries (Ovestin) 0.5 mg daily for 3 weeks, followed by 1 pessary every second day for the rest of the 24‐week period | |
| Outcomes | Urgency, frequency, urge incontinence, stress incontinence; nocturia, dysuria, vaginal dryness, dyspareunia, adverse events, assessment of administration | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation by central office using numbers sequentially no further details were reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding (performance bias) | High risk | Open label (no blinding) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Unclear whether other outcome assessors e.g. the pathologists were blinded or not |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Data were analysed on the basis of ITT |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to make a conclusive judgement |
| Other bias | Low risk | Treatment groups were balanced at baseline |
Mac Bride 2014.
| Methods | 2‐arm parallel RCT | |
| Participants | 38 postmenopausal women with bothersome symptoms of vulvo vaginal atrophy Inclusion criteria: healthy postmenopausal women with bothersome symptoms of vulvo vaginal atrophy Exclusion criteria: not reported |
|
| Interventions |
Group A: very low dose oestradiol vaginal cream (n = 19) applied to the vagina daily for 2 weeks followed by twice weekly for 10 weeks Group B: placebo (n = 19) applied as described for the active treatment group Duration: 12 weeks |
|
| Outcomes | Vaginal pH, maturation index, symptoms relating to atrophy | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information was reported on randomisation process |
| Allocation concealment (selection bias) | Unclear risk | No information was provided on allocation concealment |
| Blinding (performance bias) | Unclear risk | No information was given with respect to the blinding of participants and/or personnel |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information was provided on the blinding of outcome assessment |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient information was given on withdrawals/losses to follow‐up per treatment groups |
| Selective reporting (reporting bias) | Unclear risk | Although all the outcomes specified in the methods section ware reported no data were, however, available on any of the outcomes |
| Other bias | Unclear risk | Insufficient information was reported to make a conclusive judgement |
Manonai 2001.
| Methods | Open label, randomised, no blinding Number of women randomised: n = 53 (27 tablet, 26 cream) Number of women analysed: n = 48 Number of withdrawals: n = 5 (tablet group; 2 due to concerns about hormonal contents and 1 due to vaginal bleeding) (cream group; 2 due to pelvic discomfort) No power calculation and no ITT analysis Source of funding: not stated | |
| Participants | Inclusion criteria: healthy, natural postmenopausal, urogenital symptoms, periods ceased for 1 year Age: 45‐70 Source of participants: menopause clinic Exclusion criteria: pathology of urogenital tract, marked cystoceles or urethrocystoceles (bladder herniation into vaginal canal), symptoms of detrusor instability, presence or history of liver or renal disorders and prior use of sex hormone within 6 months of study Location: Bangkok, Thailand | |
| Interventions | Treatment: oestradiol vaginal tablet (25 mcg) daily for 2 weeks and then once a week for 10 weeks Control: conjugated oestrogen cream 0.625 mg daily for 2 weeks and then 1 g twice per week for 10 weeks Duration: 3 months | |
| Outcomes | KPI index (karyopyknotic), maturation value, endometrial thickness, vaginal health index, vaginal pH, symptoms of vaginal atrophy (dryness, burning/pain, dyspareunia, frequency, nocturia, stress incontinence), endometrial proliferation, plasma oestradiol levels, satisfaction, adherence to treatment, pelvic discomfort | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding (performance bias) | High risk | Open label (no blinding) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Unclear whether other outcome assessors such as the pathologists were blinded or not |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Proportions and reasons for withdrawals differ between the two treatment groups and data were not analysed on the basis of ITT |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to make a conclusive judgement |
| Other bias | Unclear risk | Insufficient information to make a conclusive judgement |
Nachtigall 1994.
| Methods | Randomisation method not stated Open‐label, single‐centre, parallel design, participants not blinded Number of women randomised: n = 30 Number of women analysed: n = 30 Number of withdrawals: not stated No power calculation and no ITT analysis Source of funding: Columbia Labs | |
| Participants | Inclusion criteria: > 1 year past last menstrual period, not on hormone therapy, cancer free and experiencing vaginal discomfort or dyspareunia Age: not stated Source of participants: not stated Exclusion criteria: not stated Location: not stated | |
| Interventions | Treatment: non‐hormonal local bio adhesive vaginal moisturiser (Replens) 3 times per week Control: Premarin cream (conjugated oestrogen vaginal cream containing 0.625 mg/g) 2 g every day Duration: 12 weeks | |
| Outcomes | Vaginal moisture, fluid volume, elasticity, pH | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding (performance bias) | High risk | Open label (no blinding) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Unclear whether outcome assessors such as the pathologists were blinded or not |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No withdrawals were reported in the treatment groups |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to make a conclusive judgement |
| Other bias | Unclear risk | Insufficient information to make a conclusive judgement |
Nachtigall 1995.
| Methods | Women were randomly assigned in a 2:1 ratio using separate computer‐generated randomisation lists Open‐label, multi‐centre (10), parallel‐group design with single blinding (assessor, cytology) Number of women randomised: n = 196 Number of women analysed: n = 192 (126 ring, 66 cream) Number of participants completed 12‐week follow‐up : n = 173 (113 in vaginal ring group and 60 in cream group) Number of withdrawals: n = 23, 16 from the vaginal ring group and 7 from the vaginal cream group Reasons for withdrawal: in vaginal ring group were adverse events (5), protocol violation (4), unwilling to continue (3), other reasons (4); and in the vaginal cream group, protocol violation (5), unwilling to continue (1), and other reasons (1) Power calculation for sample size reported and ITT analysis performed for some outcomes Source of funding: Pharmacia | |
| Participants | Inclusion criteria: naturally menopausal for at least a year, oophorectomised 1 year or more or post‐hysterectomy (with 1 or both ovaries remaining; 55 years or older; FSH level of at least 40 mIU/mL; have symptoms of vaginal dryness, one or more signs (pallor, petechiae, friability, or lack of vaginal moisture) Source of participants: not given Exclusion criteria: allergy or contra‐indication to oestrogen use; gonadal hormone treatment within the previous 3 months; investigational drug use within 30 days, bleeding after the progestogen challenge; evidence of endometrial stimulation by pelvic sonography; known or suspected oestrogen‐dependant neoplasms; abnormal vaginal bleeding of unknown origin; acute or chronic liver disease; acute intermittent porphyria; history of thrombophlebitis; thrombosis; thrombo‐embolic disorder; or cerebral stroke; history of genital warts; or grade 2 or 3 uterine prolapses extending to or beyond the introitus Location: USA | |
| Interventions | Treatment: oestradiol vaginal ring (Estring) 7.5 mcg of oestradiol per 24 hours inserted for 12 weeks followed by a 3‐week period of no medication Control: conjugated oestrogen vaginal cream (Premarin) contains 0.625 mg/g conjugated equine oestrogen, 2 g 3 times weekly for 12 weeks followed by a 3‐week period of no medication Duration: 15 weeks | |
| Outcomes | Improvement in vaginal atrophy > 25% reduction in cells: basal/parabasal and intermediate, physician evaluation, vaginal pH, participant assessment, end tissue response, challenge test, sonography, end biopsies (for evidence of endometrial hyperplasia), adverse events, product comfort, ease of use and overall rating, adherence to treatment | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random sequence |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding (performance bias) | High risk | Open label trial |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Single‐blinded trial (cytologist blinded) but some outcomes were participant‐assessed |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Proportions of withdrawals differ between treatment groups and data were not analysed on the basis of ITT for all outcomes |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to make a conclusive judgement |
| Other bias | Low risk | Treatment groups were balanced at baseline |
Raghunandan 2010.
| Methods | A 3‐arm parallel RCT but only 2 arms of the trial were relevant | |
| Participants | 50 postmenopausal women with urogenital and sexual dysfunction Age (mean, SD): Group A: 52.16 (7.53); Group B: 51.60 (5.66) Inclusion criteria: postmenopausal women in the age group of 40–65 years with symptoms of urogenital and sexual dysfunction, who have undergone spontaneous amenorrhoea at least 12 months prior to screening or have undergone surgical menopause at least 6 weeks prior, were included in the study. Urogenital disorders included vaginal atropy, vulvitis, urethritis, dyspareunia, recurrent urinary tract infections, and urinary incontinence symptoms. Sexual dysfunction disorders included sexual pain disorder or a problem with sexual desire, arousal, or orgasm that causes distress Exclusion criteria: women with any known contraindication to HRT as well as those using any hormonal product within 6 weeks of screening visit were excluded from the study |
|
| Interventions |
Group A: local oestrogen cream (n = 25). 25 women were given Premarin cream preparation locally once daily application of 1 gm of cream containing 0.625 mg of conjugated equine oestrogen for 2 weeks followed by twice weekly application for further 10 weeks Group B: non‐hormonal lubricant (n = 25). 25 women were given non‐hormonal lubricant gel locally, once‐daily application of 1 g of gel for 2 weeks followed by twice weekly application for further 10 weeks |
|
| Outcomes | Adverse event (total events) | |
| Notes | Most outcome data were reported in non‐usable form e.g. mean percentage, etc. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information on randomisation process |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information on method used in allocation concealment |
| Blinding (performance bias) | Unclear risk | No information was reported on performance bias |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information was reported on the blinding of outcome assessor |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | It was stated that "All women analyzed at completion of 12 weeks of therapy" |
| Selective reporting (reporting bias) | Low risk | All the outcomes specified in the methods section ware reported |
| Other bias | Low risk | "All the study parameters in study groups 1 and 2 and in the control group were comparable with each other at the initiation of therapy" |
Rioux 2000.
| Methods | Open‐label, parallel, multi‐centre (6 centres) study. Women randomised using a predetermined, computer‐generated scheme. No blinding Number of women randomised: n = 159 (80 tablets 79 cream) Number of women analysed: n = 126 (72 tablets and 54 cream) Number of withdrawals: n = 33 (8 in tablet group: 4 due to adverse effects, 2 due to non‐compliance, 1 withdrawal of consent, 1 E2 level did not meet criteria. 25 in cream group: 14 due to adverse effects, 8 non‐compliance, 2 messy application of cream and 1 E2 level did not meet eligibility criteria) No power calculation and no intention to treat analysis. Source of funding: Novo Nordisk Pharmaceutical | |
| Participants | Inclusion criteria: intact uteri and two or more vaginal symptoms (dryness, soreness, or irritation) rated as moderate to severe. 1 year past menopause and have serum E2 concentrations of 30 pg/ml (110 pmol/L) and FSH 40 IU/L or more. Age: 42‐85 years (mean 57) Source of participants: not stated Exclusion criteria: known or suspected breast cancer, oestrogen‐dependent neoplasia, positive or suspicious mammogram results or any systemic malignant disease, abnormal vaginal bleeding, uterine bleeding of unknown cause or history of thrombolytic disorder. During 3 months prior to study women were not to have received hormone therapy Location: Canada | |
| Interventions | Treatment: 17β oestradiol vaginal tablets (25 mcg) (Vagifem) once daily for 2 weeks, then once a week Control: conjugated equine oestrogen cream (Premarin) 2 g daily (equivalent to 1.25 mg conjugated equine oestrogens) for 21 days, withheld treatment for 7 days, and then repeated regimen Duration: 6 months | |
| Outcomes | Dryness, irritation and soreness; adverse events (e.g. evidence of endometrial thickness), plasma oestradiol levels, ease of administration | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Women randomised using a predetermined, computer‐generated scheme |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding (performance bias) | High risk | Open label (no blinding) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Unclear whether other outcome assessors such as the pathologists were blinded or not |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Proportions of withdrawals differed between the treatment groups and data were not analysed on the basis of ITT |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to make a conclusive judgement |
| Other bias | Low risk | Treatments groups balanced at baseline |
Simon 2008.
| Methods | A 2‐arm parallel RCT | |
| Participants | 309 postmenopausal women with vaginal atrophy Age (mean, SD): Group A: 57.5 (5.64); Group B: 57.7 (5.27) Inclusion criteria: all participants were required to have serum E2 levels less than 20 pg/mL, follicle stimulating hormone levels more than 40 mIU/mL, 5% or more superficial cells in vaginal cytology, vaginal pH more than 5.0, an endometrial thickness of less than 4.0 mm as assessed by transvaginal ultrasonography, and a normal mammogram within the 6 months before study entry Exclusion criteria: known or suspected history of breast carcinoma, hormone‐dependent tumour, genital bleeding of unknown cause, acute thrombophlebitis or thromboembolic disorder associated with oestrogen use, vaginal infection requiring treatment, allergy to the test drug or its constituents, or any serious disease or chronic condition that could interfere with study compliance. The use of any investigational drug within the 30 days preceding screening, exogenous sex hormones within 3 months before study drug initiation, or current use of corticosteroids were prohibited |
|
| Interventions |
Group A: oestrogen vaginal tablet (n = 205). one vaginal tablet (10 ug oestradiol) inserted daily for 14 days and subsequently one tablet twice per week Group B: placebo (n = 104). Placebo identical in appearance to the active drug was inserted as described for the treatment group Duration: 12 weeks |
|
| Outcomes | Vaginal pH, adverse events | |
| Notes | Some outcome data were reported in non‐usable form e.g. mean without SD | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | It was stated that "A central telephone system was used for randomization" |
| Allocation concealment (selection bias) | Low risk | It was reported that "Copies of the randomization codes were kept in sealed envelopes at the site as well as by the clinical trial sponsor" |
| Blinding (performance bias) | Low risk | Study was reported as "'double‐blind, randomized, parallel‐group, placebo controlled, multicenter trial" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information was reported on the blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All data reported at week 12 and week 52 are from the ITT analyses, with missing values for each individual imputed using the last observation carried forward approach |
| Selective reporting (reporting bias) | Low risk | All the outcomes specified in the methods section ware reported |
| Other bias | Low risk | It was reported that "Demographics and baseline characteristics were comparable across treatment groups" |
Simunic 2003.
| Methods | Multicentre, double‐blind, placebo‐controlled. Women randomised by the method of random number generator
Number of women randomised: n = 1612 (treatment group n = 828 placebo group n = 784) Number of women analysed: n = 1567. Number of withdrawals: n = 45 (11 failure of favourable effects, 10 due to side effects, 19 fear of cancer, 5 in placebo group ? reason) No power calculation and no ITT analysis Source of funding: not stated |
|
| Participants |
Inclusion criteria: urogenital complaints with at least 1 year's history of postmenopause. The women should not have been subjected to any oestrogen replacement treatment for at least 6 months
Age: 51‐66 years
Source of participants: not stated
Exclusion criteria: any systemic disease or infection, suspected or proven malignant disease, unexpected uterine bleeding, previous hysterectomy or surgical correction for genuine stress urinary incontinence, or acute urogynecological infection. Location: Croatia |
|
| Interventions | Treatment: 25 ug of micronised 17β‐oestradiol (Vagifem) vaginal tablet Control: placebo vaginal tablet Duration: women were treated once a day over a period of 2 weeks, and then twice a week for the remaining 12 months | |
| Outcomes | Burning, recurrent vaginitis, petechiae, dyspareunia, vaginal atrophy, serum oestradiol, adverse events, success (participant and physician) | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised by the method of random number generator |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding (performance bias) | Low risk | Double‐blinded, placebo‐controlled trial |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Unclear whether other outcome assessors such as the pathologists were blinded |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Proportions of withdrawals differed between the 2 treatment groups and data were not analysed on the basis of ITT |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to make a conclusive judgement |
| Other bias | Unclear risk | Insufficient information to make a conclusive judgement |
Speroff 2003.
| Methods | Double‐blinded, multicentre (35 centres). Randomisation schedule generated with the SAS ProcPlan. Randomised in blocks of 6‐13 weeks of treatment Number of women randomised: n = 333 Number of women analysed: n = 325 Number of withdrawals: n = 8 women who failed to provide post baseline data. Power calculation and ITT analysis performed Source of funding: Warner Chilcott, a division of Galen Holdings PLC. Dr Speroff owns stock in the company | |
| Participants | Inclusion criteria: a postmenopausal woman, with or without a uterus who had had at least 7 moderate to severe hot flushes a day or an average of at least 56 moderate to severe vasomotor symptoms per week for the 2 weeks before randomisation. In addition, a woman with a uterus was required to have amenorrhoea for more than 12 months before randomisation; if she had amenorrhoea for less than 12 months but at least 6 months, she was also required to have a follicle stimulating hormone of at least 40 IU and an E2 level of no more than 20 pg/mL. A woman was also eligible if she had had a hysterectomy and bilateral oophorectomy performed more than 6 weeks before randomisation Age: Mean age 51.7 years Source of participants: not stated Exclusion criteria: past or current thromboembolic disorder, or cerebrovascular accident; endometriosis; allergy or intolerance to previous HRT, including past or current oestrogen dependant neoplasia; abnormal uninvestigated vaginal bleeding within 6 months of randomisation; and known or suspected pregnancy. Previous treatment with any of the following was also reason for exclusion: oestrogen, progestogen, androgen, or systemic corticosteroids by the oral route within 8 weeks of screening, by transdermal or buccal delivery within 4 weeks of screening, or by injection within 6 months of screening; hormone pellets or implants inserted within the previous 5 years or an implant removed within the last 3 months; unopposed ERT for 6 months or more in the woman with an intact uterus; or selective oestrogen receptor modulators within 8 weeks of screening Location: USA | |
| Interventions | Treatment: vaginal ring delivering 50 ug/day of E2 (n = 113) or 100 ug/day of E2 (n = 112) Control: placebo ring (n = 108) Duration: 13 weeks | |
| Outcomes | Maturation index, vaginal dryness; overall satisfaction | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated SAS programme |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding (performance bias) | Low risk | Double‐blinded trial |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Unclear whether other outcome assessors such as the pathologists were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Proportion of withdrawals per treatment group not reported and number of women analysed differed from the number randomised |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to make a conclusive judgement |
| Other bias | Unclear risk | Insufficient information to make a conclusive judgement |
Weisberg 2000.
| Methods | Open label, parallel, 2:1 distribution. Computer‐generated programme allocation. Opening sequential envelopes. Single blinding by independent cytopathologist Number of women randomised: n = 185 Number of women analysed: n = 146 Number of withdrawals: n = 39 (32 ring (15 due to adverse effects); 7 tablet (2 due to adverse effects)). No power calculation and no ITT analysis. Source of funding: Pharmacia and Upjohn | |
| Participants | Inclusion criteria: 2 years postmenopausal with significant symptoms or objective signs of urogenital atrophy (vaginal dryness, genital pruritus, dyspareunia, dysuria, urinary frequency, urgency or nocturia, have an endometrium equal or less than 5 mm thickness on USS and a negative progestogen challenge test Exclusion criteria: hysterectomised or significant uterine prolapse, received hormonal treatment within previous 3 months, experienced bleeding after the progestogen challenge test or had vaginal bleeding of unknown origin. Women with clinically significant hepatic or kidney disease, acute or intermittent porphyria or a confirmed history of thrombo‐embolic disease Age: 46‐81 years Source of participants: clinics and advertisements in the local press Location: Australia | |
| Interventions | Treatment: oestradiol vaginal ring containing 2 mg micronised 17β oestradiol releasing 8 mcg per 24 hours; equally 0.7 mg over 3 months Control: vaginal tablet (Vagifem) a mucoadhesive tablet with an applicator for insertion containing 25 mcg of 17β oestradiol, one tablet daily for 2 weeks and twice weekly as maintenance from week 3‐48 | |
| Outcomes | Oestradiol levels | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated sequence |
| Allocation concealment (selection bias) | Low risk | Allocation concealed in envelopes |
| Blinding (performance bias) | High risk | Open label trial |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Single‐blinded trial but some outcomes were participant‐assessed |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Proportions of withdrawals differed between the treatment groups and data were not analysed on the basis of ITT |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to make a conclusive judgement |
| Other bias | Unclear risk | Insufficient information to make a conclusive judgement |
ITT: intention‐to‐treat KPI index: karyopyknotic index PP: per protocol RCT: randomised controlled trial
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Akrivis 2003 | Design not relevant: not a RCT |
| Bazin 2011 | Duration of intervention was less than 12 weeks |
| Chakravorty 1998 | Design not relevant: no control or placebo group |
| Chompootaweep 1998 | Duration of intervention less than 12 weeks: study only 8 weeks in length |
| Cicinelli 2003 | Intervention not relevant |
| Delgado 2016 | Duration of treatment was less than 12 weeks |
| Dyer 1982 | Intervention not relevant: dose response study |
| Gass 2013 | Design not relevant: survey |
| Gupta 2008 | Participants not relevant: included healthy postmenopausal women with no symptoms of vaginal atrophy |
| Heimer 1992 | Duration of intervention less than 12 weeks |
| Henriksson 1996 | Design not relevant: no comparison group |
| Holmgren 1989 | Intervention not relevant: dose response study |
| Jaisamrarn 2013 | Duration of intervention was less than 12 weeks |
| Jokar 2016 | Duration of treatment was less than 12 weeks |
| Long 2006 | Participants not relevant: hysterectomised postmenopausal women |
| Marx 2004 | Intervention not relevant: oral oestrogen |
| Mattsson 1983 | Design not relevant: not a true RCT |
| Mattsson 1989 | Duration of intervention less than 12 weeks: study only 9 weeks in length |
| Mazur 2005 | Duration of intervention less than 12 weeks: study only 4 weeks in length |
| Mettler 1991 | Intervention not relevant: dose response |
| Nilsson 1992 | Duration of intervention less than 12 weeks: study only 4 weeks in length |
| Plotti 1994 | Design not relevant: not a true RCT |
| Raymundo 2004 | Design not relevant: no randomisation |
| Tolino 1990 | Outcomes not relevant |
| Vaccaro 2013 | Duration of interventions was less than 12 weeks |
| Vartiainen 1993 | Design not relevant: not a true RCT |
Characteristics of studies awaiting assessment [ordered by study ID]
Kroll 2014.
| Methods | 2‐arm parallel RCT |
| Participants | 472 sexually active postmenopausal women experiencing dyspareunia |
| Interventions |
Group A: 15 mcg oestradiol vaginal cream 0.003% once daily for 2 weeks followed by 3 times per week for an additional 10 weeks Group B: “vehicle vaginal cream” (placebo) with same administration as group A |
| Outcomes | Participant‐assessed severity of dyspareunia Vaginal pH Vaginal maturation indices Clinician assessment of vaginal health Self‐assessed severity of vaginal dryness and vaginal bleeding from sexual activity |
| Notes | This is a conference abstract, need to ask for the data for the reported outcomes (P values only given) |
Differences between protocol and review
In the 2016 update the authors re‐evaluated the importance of the outcomes and specified that they were either primary or secondary. Improvement of vaginal symptoms according to participants' own assessment was made a primary outcome, as this reflects the participants’ own experiences. The primary safety outcome became endometrial thickness, as this is an important concern relating to local oestrogen administration. Other outcomes were specified as secondary: improvement in symptoms according to clinician assessment or laboratory parameters, other adverse events and adherence to treatment. The 2016 update also grouped symptoms so the assessed outcome was an aggregate measure. Previous versions of the review assessed individual symptoms and did not distinguish between primary and secondary outcomes.
Contributions of authors
In the 2016 update, Anne Lethaby and Reuben Ayeleke selected trials for inclusion, performed independent data extraction, quality assessment of the included trials, data entry and updating of the review's text. Helen Roberts provided clinical advice and commented on the review.
In the earlier versions of the review:
Jane Suckling performed initial searches of databases for trials, was involved in selecting trials for inclusion, performed independent data extraction and quality assessment of the included trials, and was responsible for statistical analysis and interpretation of the data.
Anne Lethaby was involved in writing the protocol, performed initial searches of databases for trials, was involved in selecting trials for inclusion, performed independent data extraction and quality assessment of the included trials, and commented on drafts of the review.
Ray Kennedy was involved in writing the protocol and selecting trials for inclusion.
Helen Roberts provided clinical expertise and assisted in writing implications for practice and research.
Sources of support
Internal sources
Department of Obstetrics and Gynaecology, University of Auckland, New Zealand.
External sources
Health Research Council, Auckland, New Zealand.
Declarations of interest
Anne Lethaby: no conflicts of interest to declare Reuben Ayeleke: no conflicts of interest to declare Helen Roberts: no conflicts of interest to declare
Stable (no update expected for reasons given in 'What's new')
References
References to studies included in this review
Ayton 1996 {published data only}
- Ayton R, Darling G, Murkies A, Farrell E, Weisberg E, Selinus I, Fraser S. A comparative study of safety and efficacy of continuous low dose oestradiol released from a vaginal ring compared with conjugated equine oestrogen vaginal cream in the treatment of postmenopausal urogenital atrophy. British Journal of Obstetrics and Gynaecology 1996;103:351‐8. [DOI] [PubMed] [Google Scholar]
Bachmann 2008 {published data only}
- Bachmann G, Lobo RA, Gut R, Nachtigall L, Notelovitz M. Efficacy of low‐dose estradiol vaginal tablets in the treatment of atrophic vaginitis. A randomized controlled trial. Obstetrics & Gynecology 2008;111(1):67‐76. [DOI] [PubMed] [Google Scholar]
Bachmann 2009 {published data only}
- Bahmann G, Bouchard C, Hoppe D, Ranganath R, Altomare C, Vieweg A, et al. Efficacy and safety of low‐dose regimens of conjugated estrogens cream administered vaginally. Menopause: The Journal of The North American Menopause Society 2009;16(4):719‐727. [DOI: 10.1097/gme.0b013e3181a48c4e] [DOI] [PubMed] [Google Scholar]
Barentsen 1997 {published data only}
- Barentsen R, Weijer PHM, Schram JHN. Continuous low dose estradiol released from a vaginal ring versus estriol vaginal cream for urogenital atrophy. European Journal of Obstetrics and Gynecology and Reproductive Biology 1997;71:73‐80. [DOI] [PubMed] [Google Scholar]
Bygdeman 1996 {published data only}
- Bygdeman M, Swahn ML. Replens versus dienoestrol cream in the symptomatic treatment of vaginal atrophy in postmenopausal women. Maturitas 1996;23(3):259‐63. [DOI] [PubMed] [Google Scholar]
Cano 2012 {published data only}
- Cano A, Estévez J, Usandizaga, R, Gallo JL, Guinot M, Delgado JL, et al. The therapeutic effect of a new ultra low concentration estriol gel formulation (0.005% estriol vaginal gel) on symptoms and signs of postmenopausal vaginal atrophy: results from a pivotal phase III study. Menopause 2012;19(10):1130‐9. [DOI] [PubMed] [Google Scholar]
Casper 1999 study 1 {published data only}
- Casper F, Petri E. Local treatment of urogenital atrophy with an estradiol‐releasing vaginal ring: a comparative and placebo‐controlled multicenter study. International Urogynecology Journal 1999;10:171‐6. [DOI] [PubMed] [Google Scholar]
Casper 1999 study 2 {published data only}
- Casper F, Petri E. Local treatment of urogenital atrophy with an estradiol‐releasing vaginal ring: a comparative and placebo‐controlled multicenter study. International Urogynecology Journal 1999;10:171‐6. [DOI] [PubMed] [Google Scholar]
Dessole 2004 {published data only}
- Dessole S, Rubattu G, Ambrosini G, Gallo O, Capobianco G, Cherchi P, et al. Efficacy of low‐dose intravaginal estradiol on urogenital aging in postmenopausal women. Menopause 2004;11:49‐56. [DOI] [PubMed] [Google Scholar]
Dugal 2000 {published data only}
- Dugal R, Hesla K, Sordal T, Aase KH, Lilleeidet O, Wickstrom E. Comparisons of usefulness of estradiol vaginal tablets and estriol vagitories for treatment of vaginal atrophy. Acta Obstetricia et Gynecologica Scandinavica 2000;79(4):293‐7. [PubMed] [Google Scholar]
Eriksen 1992 {published data only}
- Eriksen PS, Rasmussen H. Low‐dose 17B‐estradiol vaginal tablets in the treatment of atrophic vaginitis: a double‐blind placebo controlled study. European Journal of Obstetrics and Gynecology and Reproductive Biology 1991;44:137‐44. [DOI] [PubMed] [Google Scholar]
Fernandes 2014 {published data only}
- Fernandes T, Costa‐Paiva LH, Pinto‐Neto AM. Efficacy of vaginally applied estrogen, testosterone, or polyacrylic acid on sexual function in postmenopausal women: a randomised controlled trial. Journal of Sexual Medicine 2014;11:1262‐70. [DOI] [PubMed] [Google Scholar]
Foidart 1991 {published data only}
- Foidart JM, Vervliet J, Buytaert PH. Efficacy of sustained‐release vaginal oestriol in alleviating urogenital and systemic climacteric complaints. Maturitas 1991;13:99‐107. [DOI] [PubMed] [Google Scholar]
Garcia Lara 1993 {published data only}
- Garcia Lara E. Efficiency of vaginal ovules of estriol for treatment of symptoms of menopause [Eficacia y seguridad del uso de estriol, ovulos vaginales, en al tratamiento de sintomas menopausicos]. Investigacion Medica Internacional 1993;19:159‐65. [Google Scholar]
Griesser 2012 {published data only}
- Griesser H, Skonietzki S, Fischer T, Fielder K, Suesskind M. Low dose estriol pessaries for the treatment of vaginal atrophy: a double‐blind placebo‐controlled trial investigating the efficacy of pessaries containing 0.2 mg and 0.03 mg estriol. Maturitas 2012;71(4):360‐8. [DOI] [PubMed] [Google Scholar]
Henriksson 1994 {published data only}
- Henrikson L, Stjerquist M, Boquist L, Alander U, Selinus I. A comparative multicenter study of the effects of continuous low dose estradiol released from a new vaginal ring versus estriol vaginal pessaries in postmenopausal women with symptoms and signs of urogenital atrophy. American Journal of Obstetrics and Gynecology 1994;171(3):625‐32. [DOI] [PubMed] [Google Scholar]
Hosseinzadeh 2015 {published data only}
- Daneshmand F, Hosseinzadeh P, Ghahiri A, Ghasemi M. A comparative study of vaginal estrogen cream and sustained‐released estradiol vaginal tablet (Vagifem) in the treatment of atrophic vaginitis among postmenopausal women. Iranian Journal of Reproductive Medicine 2014;12(6 SUPPL. 1):12‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosseinzadeh P, Ghahiri A, Daneshmand F, Ghasemi M. A comparative study of vaginal estrogen cream and sustained‐release estradiol vaginal tablet (Vagifem) in the treatment of atrophic vaginitis in Isfahan, Iran in 2010‐2012. Journal of Research in Medical Sciences 2015;20(12):1160‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Karp 2012 {published data only}
- Karp DR, Jean‐Michel M, Johnston Y, Suciu G, Aguilar VC, Davila GW. A randomized clinical trial of the impact of local estrogen on postoperative tissue quality after vaginal reconstructive surgery. Female Pelvic Medicine & Reconstructive Surgery 2012;18(4):211‐5. [DOI] [PubMed] [Google Scholar]
Lima 2013 {published data only}
- Lima SM, Yamada SS, Reis BF, Postigo S, Galvão da Silva MA, Aoki T. Effective treatment of vaginal atrophy with isoflavone vaginal gel. Maturitas 2013;74(3):252‐8. [DOI] [PubMed] [Google Scholar]
Lose 2000 {published data only}
- Lose G, Englev E. Oestradiol‐releasing vaginal ring versus oestriol vaginal pessaries in the treatment of bothersome lower urinary tract symptoms. British Journal of Obstetrics and Gynaecology 2000;107(8):1029‐34. [DOI] [PubMed] [Google Scholar]
Mac Bride 2014 {published data only}
- Mac Bride MB, Shuster L, Rhodes D, Grossardt B, Warndahl R, Debra B. Low dose vaginal estrogens for the treatment of vulvovaginal atrophy: a randomized, placebo‐controlled trial evaluating effect of estriol, estradiol or placebo on vulvovaginal atrophy symptoms. Journal of Sexual Medicine 2014;11:220‐1. [Google Scholar]
Manonai 2001 {published data only}
- Manonai J, Theppisai U, Suthutvoravut S, Udomsubpayakul U, Chittacharoen A. The effect of estradiol vaginal tablet and conjugated estrogen cream on urogenital symptoms in postmenopausal women: a comparative study. Journal of Obstetrics and Gynaecology 2001;27(5):255‐60. [DOI] [PubMed] [Google Scholar]
Nachtigall 1994 {published data only}
- Nachtigall L. Comparative study: Replens versus local estrogen in menopausal women. Fertility and Sterility 1994;61:178‐80. [DOI] [PubMed] [Google Scholar]
Nachtigall 1995 {published data only}
- Bachmann G, Notelovitz M, Nachtigall L, Birgerson L. A comparative study of a low‐dose estradiol vaginal ring and conjugated estrogen cream for postmenopausal urogenital atrophy. Primary Care Update for Ob/Gyns 1997;4(3):109‐15. [Google Scholar]
- Nachtigall L. Clinical trial of the estradiol vaginal ring in the US. Maturitas 1995;22 Suppl:43‐7. [DOI] [PubMed] [Google Scholar]
Raghunandan 2010 {published data only}
- Raghunandan C, Agrawal S, Dubey P, Choudhury M, Jain A. A comparative study of the effects of local estrogen with or without local testosterone on vulvovaginal and sexual dysfunction in postmenopausal women. Journal of Sexual Medicine 2010;7:1284‐90. [10. 1111/1. 1743‐6109.2009.01667.x] [DOI] [PubMed] [Google Scholar]
Rioux 2000 {published data only}
- Rioux J, Devlin C, Gelfand M, Steinberg W, Hepburn D. 17B estradiol vaginal tablet versus conjugated equine estrogen vaginal cream to relieve menopausal atrophic vaginitis. Menopause: The Journal of the North American Society 2000;7(3):156‐61. [DOI] [PubMed] [Google Scholar]
Simon 2008 {published data only}
- Simon J, Nachtigall L, Gut R, Lang E, Archer DF, Utian W. Effective treatment of vaginal atrophy with an ultra‐low‐dose estradiol vaginal tablet. Obstetrics & Gynecology 2008;112(5):1053‐1060. [DOI] [PubMed] [Google Scholar]
Simunic 2003 {published data only}
- Simunic V, Banovic I, Ciglar S, Jeren L, Pavicic Baldani D, Sprem M. Local estrogen treatment in patients with urogenital symptoms. International Journal of Gynecology and Obstetrics 2003;82:187‐97. [DOI] [PubMed] [Google Scholar]
Speroff 2003 {published data only}
- Speroff L. Efficacy and tolerability of a novel estradiol vaginal ring for relief of menopausal symptoms. Obstetrics & Gynecology 2003;102(4):823‐34. [DOI] [PubMed] [Google Scholar]
Weisberg 2000 {unpublished data only}
References to studies excluded from this review
Akrivis 2003 {published data only}
- Akrivis C, Varras M, Thodos A, Hadjopoulos G, Bellou A, Antoniou N. Action of 25 ug 17B‐oestradiol vaginal tablets in the treatment of vaginal atrophy in Greek postmenopausal women; clinical study. Clinical Experimental Obstetrics and Gynecology 2003;4:229‐3. [PubMed] [Google Scholar]
Bazin 2011 {published data only}
- Bazin S, Lefebvre J, Fortier M, Brisson J, Brouillette F, Bujold E, et al. [Évaluation d’une crème oestrogénique vaginale dans le traitement de la vestibulodynie provoquée : essai randomisé à double insu]. Journal of Obstetrics and Gynaecology Canada 2011;33(8):838‐43. [DOI] [PubMed] [Google Scholar]
Chakravorty 1998 {published data only}
- Chakravorty R, Chakravorty PS. Urogenital ageing in Indian postmenopausal women and choice of HRT. Abstracts of the 14th Annual meeting of the ESHRE, Goteborg 1998;13(Abstract Book 1):333‐4. [Google Scholar]
Chompootaweep 1998 {published data only}
- Chompootaweep S, Nunthapisud P, Trivijitsilp P, Sentrakul P, Dusitsin N. The use of two estrogen preparations (a combined contraceptive pill versus conjugated estrogen cream) intravaginally to treat urogenital symptoms in postmenopausal Thai women: a comparative study. Clinical Pharmacology Therapy 1998;64:204. [DOI] [PubMed] [Google Scholar]
Cicinelli 2003 {published data only}
- Cicinelli E, Naro E, Ziegler D, Matteo M, Morgese S, Galantino P, et al. Placement of the vaginal 17 B ‐estradiol tablets in the inner or outer one third of the vagina affects the preferential delivery of 17B‐estradiol towards the uterus or periurethral areas, thereby modifying efficacy and endometrial safety. American Journal of Obstetrics and Gynecology 2003;July:55‐8. [DOI] [PubMed] [Google Scholar]
Delgado 2016 {published data only}
- Delgado JL, Estevez, J, Radicioni, M, et al. Pharmacokinetics and preliminary efficacy of two vaginal gel formulations of ultra‐low‐dose estriol in postmenopausal women. Climacteric 2016;19(2):172‐180. [DOI] [PubMed] [Google Scholar]
Dyer 1982 {published data only}
- Dyer G, Young O, Townend P, Collins W, Whitehead M, Jelowitz J. Dose related changes in vaginal cytology after topical conjugated equine oestrogens. British Medical Journal 1982;284(6318):789. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gass 2013 {published data only}
- Gass M, Cochrane BB, Larson JC, et al. Impact of discontinuing hormone therapy on sexual activity and vaginal symptoms in postmenopausal women. Menopause 2013;20(12):1337. [Google Scholar]
Gupta 2008 {published data only}
- Gupta P, Ozel B, Stanczyk FZ, et al. The effect of transdermal and vaginal estrogen therapy on markers of postmenopausal estrogen status. Menopause, 2008;15(1):94‐97. [DOI: 10.1097/gme.0b013e318148b98b] [DOI] [PubMed] [Google Scholar]
Heimer 1992 {published data only}
- Heimer G, Englund D. Effects of vaginally administered oestriol on post‐menopausal urogenital disorders: a cytohormonal study. Maturitas 1992;14(3):171‐9. [DOI] [PubMed] [Google Scholar]
Henriksson 1996 {published data only}
- Henriksson L, Stjernquist M, Boquist L, Cedergren I, Selinus I. A one year multicenter study of efficacy and safety of a continuous, low dose, estradiol releasing vaginal ring in postmenopausal women with symptoms and signs of urogenital aging. American Journal of Obstetrics and Gynecology 1996;174(1):85‐92. [DOI] [PubMed] [Google Scholar]
Holmgren 1989 {published data only}
- Holmgren P, Lindskog M, von‐Schoultz B. Vaginal rings for continuous low dose release of oestradiol in the treatment of urogenital atrophy. Maturitas 1989;11(1):55‐63. [DOI] [PubMed] [Google Scholar]
Jaisamrarn 2013 {published data only}
- Jaisamrarn U, Triratanachat S, Chaikittisilpa S, Grob P, Prasauskas V, Taechakraichana N. Ultra‐low‐dose estriol and lactobacilli in the local treatment of postmenopausal vaginal atrophy. Climacteric. 2013;16(3):347‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
Jokar 2016 {published data only}
- Jokar A, Davari T, Asadi N, et al. Comparison of the hyaluronic acid vaginal cream and conjugated estrogen used in treatment of vaginal atrophy of menopause women: a randomized controlled clinical trial. International Journal of Community Based Nursing and Midwifery 2016;4(1):69‐78. [PMC free article] [PubMed] [Google Scholar]
Long 2006 {published data only}
- Long C, Liu C, Hsu S, et al. A randomized comparative study of the effects of oral and topical estrogen therapy on the vaginal vascularization and sexual function in hysterectomized postmenopausal women. Menopause 2006;13(5):737‐43. [DOI: ] [DOI] [PubMed] [Google Scholar]
Marx 2004 {published data only}
- Marx P, Schade G, Wilbourn S, Blank S, Moyer D, Nett R. Low dose (0.3 mg) synthetic conjugated estrogens A is effective for magnaging atrophic vaginitis. Maturitas 2004;47:47‐54. [DOI] [PubMed] [Google Scholar]
Mattsson 1983 {published data only}
- Mattsson L, Cullberg G. A clinical evaluation of treatment with estriol vaginal cream versus suppository in postmenopausal women. Acta Obstetricia et Gynecologica Scandinavica 1983;62(5):397‐401. [DOI] [PubMed] [Google Scholar]
Mattsson 1989 {published data only}
- Mattsson LA, Cullberg G, Eriksson O, Knutsson F. Vaginal administration of low‐dose oestradiol‐effects on the endometrium and vaginal cytology. Maturitas 1989;11:217. [DOI] [PubMed] [Google Scholar]
Mazur 2005 {published data only}
- Mazur D, Vens‐Cappell B, Lohmann K, Breckwolt M. Fractionated use of a 17B estradiol cream for the treatment of vaginal atrophy in postmenopausal women. Geburtsh Frauenheilk 2005;65:584‐9. [Google Scholar]
Mettler 1991 {published data only}
- Mettler L, Olsen P. Long term treatment of atrophic vaginitis with low dose oestradiol vaginal tablets. Maturitas 1991;14(1):22‐31. [DOI] [PubMed] [Google Scholar]
Nilsson 1992 {published data only}
- Nilsson K, Heimer G. Low‐dose oestradiol in the treatment of urogenital oestrogen deficiency: A pharmacokinetic and pharmacodynamic study. Maturitas 1992;15:121‐7. [DOI] [PubMed] [Google Scholar]
Plotti 1994 {published data only}
- Plotti G, Gozzi G, Bobbio C, Giulianella G, Buttini G, Italiana P. Cetyltrimethylammonium naproxenate as vaginal douches in postmenopausal vulvovaginal distrophies. Minerva Ginecologica 1994;46(10):583‐6. [PubMed] [Google Scholar]
Raymundo 2004 {published data only}
- Raymundo N, Yu‐Cheng B, Zi‐yan H, Huey Lai C, Leung K, Subramaniam R, et al. Treatment of atrophic vaginitis with topical conjugated equine estrogens in postmenopausal Asian women. Climacteric 2004;7:312‐8. [DOI] [PubMed] [Google Scholar]
Tolino 1990 {published data only}
- Tolino A, Ronsini S, Granata P, Gallo F, Riccio S, Montemagno U. Topical treatment with estriol in postmenopausal atrophic vaginitis. Revue Francaise de Gynecologie et d'Obstetrique 1990;85(12):692‐7. [PubMed] [Google Scholar]
Vaccaro 2013 {published data only}
- Vaccaro CM, Mutema GK, Fellner AN, Crisp CC, Estanol MV, Kleeman SD, et al. Histologic and cytologic effects of vaginal estrogen in women with pelvic organ prolapse: a randomised controlled trial. Female Pelvic Medicine & Reconstructive Surgery 2013;19(1):34‐9. [DOI] [PubMed] [Google Scholar]
Vartiainen 1993 {published data only}
- Vartiainen J, Wahlstrom T, Nilsson C‐G. Effects and acceptability of a new 17B‐oestradiol releasing vaginal ring in the treatment of postmenopausal complaints. Maturitas 1993;17:129‐37. [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
Kroll 2014 {published data only}
- Kroll R, Carlyon T, Thomas HM, Reape KZ. Efficacy and safety of a new low‐dose estradiol vaginal cream administered 3 times a week for the treatment of dyspareunia associated with menopause. Menopause 2014;21(12):1353. [Google Scholar]
- Portman D, Carlyon T, Thomas HM, Reape KZ. Efficacy and safety of a new low‐dose estradiol vaginal cream administered 2 times a week for the treatment of vaginal dryness associated with menopause. Menopause 2014;21(12):1360. [Google Scholar]
Additional references
ACOG 2014
- The American College of Obstetricians and Gynecologists. Management of menopausal symptoms. Obstettrics & Gynecology 2014;123(1):202‐16. [DOI] [PubMed] [Google Scholar]
Berg 1988
- Berg G, Gottqall T, Hammar M, Lindgren R. Climacteric symptoms among women aged 60‐62 in Linkoping, Sweden in 1986. Maturitas 1988;10:193‐9. [DOI] [PubMed] [Google Scholar]
Caillouette 1997
- Caillouette JC, Sharp CF, Zimmerman GJ, Subir R. Vaginal pH as a marker for bacterial pathogens and menopausal status. American Journal of Obstetrics and Gynecology 1997;176:1270‐5. [DOI] [PubMed] [Google Scholar]
Campbell 1977
- Campbell S, Whitehead M. Oestrogen therapy and the menopausal syndrome. Clinics in Obstetrics & Gynaecology 1977;4(1):31‐47. [PubMed] [Google Scholar]
Crandall 2002
- Crandall C. Vaginal estrogen preparations: A review of safety and efficacy for vaginal atrophy. Jounal of Women's Health 2002;11:857‐77. [DOI] [PubMed] [Google Scholar]
Deeks 2011
- Deeks JJ, Higgins JPT, Altman DG (editors). Chapter 9: Analysing data and undertaking meta‐analyses. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Gerbaldo 1991
- Gerbaldo D, Ferraiolo A, Croce S, Truini M, Capitanio GL. Endometrial morphology after 12 months of vaginal oestriol therapy in postmenopausal women. Maturitas 1991;13:269‐74. [DOI] [PubMed] [Google Scholar]
GRADEpro GDT 2015 [Computer program]
- GRADE Working Group, McMaster University. GRADEpro GDT. Hamilton (ON): GRADE Working Group, McMaster University, 2015.
Heimer 1984
- Heimer G, Englund D. Estriol: absorption after long‐term vaginal treatment and gastrointestinal absorption as influenced by a meal. Acta Obstetricia et Gynecologia Scandinavica 1984;63:563‐7. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Altman DG, Sterne JAC (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Iosif 1984
- Iosif CS, Bekassy Z. Prevalence of genito‐urinary symptoms in the late menopause. Acta Obstetricia et Gynaecologica Scandinavica 1984;63:257‐60. [DOI] [PubMed] [Google Scholar]
Kivovic 1980
- Kivovic PM, Cotes‐Prieto J, Milojevic S, Haspels AA, Aljinovic A. The treatment of postmenopausal vaginal atrophy with Ovestin vaginal cream or suppositories: clinical, endocrinological and safety aspects. Maturitas 1980;2:275‐82. [DOI] [PubMed] [Google Scholar]
Lefebvre 2011
- Lefebvre C, Manheimer E, Glanville J. Chapter 6: Searching for studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Lindah 2014
- Lindah SH. Reviewing the options for local estrogen treatment of vaginal atrophy. International Journal of Women's Health 2014;6:307‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Manson 2016
- Manson JAE, Kaunitz AM. Menopause management — getting clinical care back on track. New England Journal of Medicine 2016;374:803‐6. [DOI] [PubMed] [Google Scholar]
Mattson 1989
- Mattson L, Culberge G, Eriksson O, Knutsson F. Vaginal administration of low‐dose oestradiol ‐ effects on the endometrium and vaginal cytology. Maturitas 1989;11:217‐22. [DOI] [PubMed] [Google Scholar]
Moher 2009
- Moher D, Liberati A, Tetzlaff J, Altman DG, The PRISMA Group. Preferred reporting items for systematic reviews and meta‐analyses: The PRISMA Statement. PLoS Medicine 2009;6(7):e1000097. [DOI: 10.1371/journal.pmed1000097] [DOI] [PMC free article] [PubMed] [Google Scholar]
NAMS 2013
- The North American Menopause Society. Management of symptomatic vulvovaginal atrophy: 2013 position statement of the North American Menopause Society. Menopause 2013;20(9):888‐902. [DOI] [PubMed] [Google Scholar]
NICE 2015
- The National Institute for Health and Care Excellence. Menopause: diagnosis and management. NICE Guideline 2015; Vol. NG23. [https://www.nice.org.uk/guidance/ng23/resources/menopause‐diagnosis‐and‐management‐1837330217413. Acessed: 23/06/2016] [PubMed]
Notelovitz 1997
- Notelovitz M. Urogenital aging: solutions in clinical practice. International Journal of Gynecology and Obstetrics 1997;59:Suppl:35‐9. [DOI] [PubMed] [Google Scholar]
Rahn 2014
- Rahn DD, Carberry C, Sanses TV, et al. Vaginal Estrogen for Genitourinary Syndromeof Menopause. Obstetrics & Gynecology 2014;124(6):1147‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
Roberts 2016
- Roberts H, Hickey M. Managing the menopause: An update. Maturitas 2016;86:53–58. [DOI] [PubMed] [Google Scholar]
Schultz 2010
- Schulz KF, Altman DG, Moher D, for the CONSORT Group. CONSORT 2010 Statement: updated guidelines for reporting parallel group randomised trials. Annals of Internal Medicine 2010;152:Epub 24 March. [DOI] [PubMed] [Google Scholar]
Schünemann 2011
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In: Higgins JPT, Green S (editors), Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
SOGC 2005
- The Society of Obstetricians and Gynaecologists of Canada. The detection and management of vaginal atrophy. International Journal of Gynecology and Obstetrics 2005;88:222‐8. [DOI] [PubMed] [Google Scholar]
SOGC 2014
- The Society of Obstetricians and Gynaecologists of Canada. Managing Menopause. Journal of Obstetrics and Gynaecology Canada 2014;36(9 Suppl 2):S3. [Google Scholar]
Sterne 2011
- Sterne JAC, Egger M, Moher D (editors). Chapter 10: Addressing reporting biases. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Intervention. Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
References to other published versions of this review
Suckling 2003
- Suckling J, Lethaby A, Kennedy R. Local oestrogen for vaginal atrophy in postmenopausal women. Cochrane Database of Systematic Reviews 2003, Issue 4. [DOI: 10.1002/14651858.CD001500] [DOI] [PubMed] [Google Scholar]
Suckling 2006
- Suckling JA, Kennedy R, Lethaby A, Roberts H. Local oestrogen for vaginal atrophy in postmenopausal women. Cochrane Database of Systematic Reviews 2006, Issue 4. [DOI: 10.1002/14651858.CD001500.pub2] [DOI] [PubMed] [Google Scholar]