

Abstract
Background
Plant genomes are rich in repetitive sequences, and transposable elements (TEs) are the most accumulated of them. This mobile fraction can be distinguished as Class I (retrotransposons) and Class II (transposons). Retrotransposons that are transposed using an intermediate RNA and that accumulate in a “copy-and-paste” manner were screened in three genomes of peppers (Solanaceae). The present study aimed to understand the genome relationships among Capsicum annuum, C. chinense, and C. baccatum, based on a comparative analysis of the function, diversity and chromosome distribution of TE lineages in the Capsicum karyotypes. Due to the great commercial importance of pepper in natura, as a spice or as an ornamental plant, these genomes have been widely sequenced, and all of the assemblies are available in the SolGenomics group. These sequences were used to compare all repetitive fractions from a cytogenomic point of view.
Results
The qualification and quantification of LTR-retrotransposons (LTR-RT) families were contrasted with molecular cytogenetic data, and the results showed a strong genome similarity between C. annuum and C. chinense as compared to C. baccatum. The Gypsy superfamily is more abundant than Copia, especially for Tekay/Del lineage members, including a high representation in C. annuum and C. chinense. On the other hand, C. baccatum accumulates more Athila/Tat sequences. The FISH results showed retrotransposons differentially scattered along chromosomes, except for CRM lineage sequences, which mainly have a proximal accumulation associated with heterochromatin bands.
Conclusions
The results confirm a close genomic relationship between C. annuum and C. chinense in comparison to C. baccatum. Centromeric GC-rich bands may be associated with the accumulation regions of CRM elements, whereas terminal and subterminal AT- and GC-rich bands do not correspond to the accumulation of the retrotransposons in the three Capsicum species tested.
Keywords: Chili peppers Cytogenomics, FISH, Plant genome, Transposable elements
Background
Plant genomes are composed of repetitive and non-repetitive portions, organized in families according to the nature of their sequences, mobility throughout genomes and localization in the chromosomes [1, 2]. Transposable elements are virus-like sequences, and they are grouped into two classes, namely Class I or retrotransposon and Class II or transposon-like. The retrotransposons are the most common elements in plant genomes [1], and they use polygenic chain enzymes, such as reverse transcriptase, for retrotransposition via an intermediate RNA molecule [3–5]. Transposons, on the other hand, use different enzymes, such as transposase (transposons), helicase/replicase (helitrons), polymerase B (polintons) and tyrosine replicase for cryptons [2, 4], to reposition themselves along the genomes.
The LTR-retrotransposons are classified into Copia, Gypsy, Bel-Pao, retrovirus, and endogenous retrovirus superfamilies, and they may be differentiated by the protein domain organization on the polygenic sequence [2, 5, 6]. The superfamilies Copia and Gypsy differ from each other by the location of integrase in the polygenic chain: when the integrase is positioned upstream of the reverse transcriptase the element is recognized as a Copia retrotransposon, whereas a downstream position identifies the Gypsy members [5, 7]. Both superfamilies are subdivided into many lineages [4, 5], with the Gypsy taxon encompassing, for example, clades named of Athila/Tat and chromoviruses with CRM and Del lineages [5, 7]. Similarly, Copia retrotransposons are grouped into other lineages, such as the Ivana/Oryco, Tork and others [5, 7]. Because LTR-RTs present independent activity in chromosomes and different fates in genomes, closely related species may exhibit variability in their occurrence, amount and chromosome distribution, which influence “fluctuations” in DNA C-values (DNA amount in a haploid nucleus, in picograms) [8–10].
The presence of TEs within genomes may influence gene expression and function. Depending on their insertion region, TEs may change the transcript splicing/processing and coding regions (see [11]). In plants, the LTR-RT lineages may be clustered in different chromosome regions, regardless of the superfamily to which they belong. This may be instanced in the accumulation of Copia elements in the proximal chromosomal regions in many plants [12, 13]. Depending on the Gypsy lineage, their position along the chromosomes can be much more diverse. Whereas CRM elements are often localize at centromeric regions [14, 15], Athila/Tat and Del elements were found in heterochromatic and euchromatic regions, often scattered across the chromosomes [14, 16]. However, when the retrotransposons are localized using FISH probes made for superfamilies and not specifically for each Copia or Gypsy lineage, the FISH signals may appear with a scattered profile, as observed in the chromosomes of Copaifera [17]. Studies in samples of Solanaceae have shown that Gypsy retrotransposons may be associated with heterochromatin at pericentromeric regions of Solanum chromosomes [18]. The accumulation of Gypsy-Del elements has been reported in both heterochromatic and euchromatic regions of Capsicum annuum [19].
Capsicum species are an excellent model to investigate the dynamics and distribution of LTR-RTs, because of their relatively large genomes, grouped within 2n = 24 and 26 chromosomes, and DNA C-values range from 3.16 to 5.77 pg, in which the repetitive DNA families may represent more than 70% of the genomes [20]. Capsicum species present a great diversity of repetitive DNA families, especially regarding number and distribution of ribosomal sequences and heterochromatin sites [21–27]. In this context, the present study aims to acknowledge and compare the occurrence and distribution of LTR-RTs on the chromosomes, focusing on Gypsy superfamily that has been predominant in Capsicum genomes, as well as their association with the diversity of other repetitive sequences. Peppers are among the most important vegetables in the world due to their high versatility and wide range of applications in cooking, industry, and decoration [28]. Therefore, large investments have been made to obtain high throughout sequences of C. annuum, C. chinense and C. baccatum [20, 29]. This large data volume has increased the possibility of studying and comparing the genomic organization of these species from the cyto-molecular point of view.
Given the gaps in knowledge regarding the repetitive fractions of Capsicum species, some questions about the dynamics and distribution of LTR-RTs remain unanswered, such as: Are the heterochromatin rich regions collocated with TE rich regions? Do closely related genomes share the same LTR-RT families concerning quantity and chromosome localization? Our discussion focused on the characterization of retrotransposons based on a broad cytogenomic comparison using the repetitive fraction available in the large C. annuum, C. chinense, and C. baccatum datasets.
Results
Comparative analyses based on the conserved domains of transposable elements
The high coverage sequencing scaffolds of Capsicum annuum (3.07Gb), C. chinense (3.22Gb) and C. baccatum (3.01Gb) from Pepper Genome Platform were used for the analysis. The search based on conserved coding domains of polygenic chain (POL) of retrotransposons showed that fractions related with conserved protein domain of reverse transcriptase, integrase and RNase H represents 2.75, 17.14 and 2.47% in the C. annuum, C. chinense and C. baccatum datasets, respectively (Tables 1 and S1). When Class I and II elements were compared, conserved sequences of Class I elements were more abundant in the three datasets. The Class I was more accumulated in C. annuum and C. chinense (> 90%) and less accumulated in C. baccatum (~ 70%). The conserved sequences similar to Class II elements were less represented with < 10% in these datasets (Tables 1 and S1).
Table 1.
Frequency and relative values of repetitive fraction in the datasets of the three Capsicum genomes
| Species/Lineages | C. annuum | C. chinense | C. baccatum | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| num seq | NR (%) | size bp | size CR% | num seq | NR (%) | size bp | size CR% | num seq | NR (%) | size bp | size CR% | ||
| Copia | Sirevirus | 7421 | 1.69 | 1,339,493 | 1.59 | 9176 | 2.29 | 1,361,598 | 0.26 | 3463 | 0.94 | 546,478 | 0.69 |
| Osser | 45 | 0.01 | 1335 | 0.00 | 65 | 0.02 | 2534 | 0.00 | 6 | 0.00 | 241 | 0.00 | |
| Tork | 5380 | 1.22 | 1,645,586 | 1.95 | 2378 | 0.59 | 357,575 | 0.07 | 8773 | 2.37 | 5,231,667 | 6.58 | |
| Gypsy | Chromovirus | 321,932 | 73.28 | 58,580,214 | 69.41 | 278,519 | 69.43 | 493,702,847 | 95.71 | 78,757 | 21.28 | 15,931,607 | 20.03 |
| Non-chrom. | 15 | 0.00 | 935 | 0.00 | 37 | 0.01 | 1242 | 0.00 | 13 | 0.00 | 970 | 0.00 | |
| OTA | 70,160 | 15.97 | 13,807,983 | 16.36 | 74,530 | 18.58 | 14,025,454 | 2.72 | 226,143 | 61.11 | 34,401,828 | 43.24 | |
| Other LTRs | 12,836 | 2.92 | 5,071,268 | 6.01 | 886 | 0.22 | 44,305 | 0.01 | 3845 | 1.04 | 1,439,107 | 1.81 | |
| Non-LTR-RTs | 0 | 0.00 | 1,866,004 | 2.21 | 3305 | 0.82 | 296,857 | 0.06 | 3648 | 0.99 | 1,201,048 | 1.51 | |
| Endogenous Virus | 1371 | 0.31 | 127,214 | 0.15 | 1392 | 0.35 | 71,067 | 0.01 | 1171 | 0.32 | 185,499 | 0.23 | |
| Transposons | 30,738 | 7.00 | 5,840,435 | 6.92 | 28,627 | 7.14 | 5,135,873 | 1.00 | 29,582 | 7.99 | 7,154,489 | 8.99 | |
| 5S rDNA | 717 | 0.16 | 89,820 | 0.11 | 1370 | 0.34 | 172,715 | 0.03 | 199 | 0.05 | 30,499 | 0.04 | |
| 45S rDNA | 1267 | 0.29 | 1,086,206 | 1.29 | 849 | 0.21 | 637,555 | 0.12 | 18,075 | 4.88 | 14,858,366 | 18.68 | |
Repetitive fraction comparison in Capsicum genomes. Num seq – number of sequences found after the Blast rounds. NR (%) – relative value. Size bp – total length of a repetitive class. Size CR (%) – percentage that the class represents in all the scaffolds
After organizing the Class I sequences as LTR, non-LTR and endogenous retrovirus, the percentages of LTR elements showed more similarity between C. annuum and C. chinense (89.32 and 98.78% respectively) than in C. baccatum with 70.55%. Both non-LTR and ERVs sequences were less accumulated, and they were not equally distributed among species: 2.21 and 0.15% in C. annuum, 0.06 and 0.01% in C. chinense, and 1.51 and 0.23% in C. baccatum (Table 1). Sequences recognized as Copia superfamily members exhibited low representativeness in all three datasets (< 10%), while Gypsy members were the most accumulated. C. annuum and C. chinense exhibited 85.77 and 98.43% of the Gypsy-related sequences respectively, while in C. baccatum, they were 63.27% (Table S1). In Gypsy superfamily, the Tekay/Del elements were predominant in C. annuum and C. chinense, with 67.68 and 95.43% respectively, while in C. baccatum the Tekay/Del elements presented only 15.7%. The Athila/Tat clade was predominant in C. baccatum (43.25%) than in C. annuum and C. chinense at 16.36 and 2.72%, respectively (Tables 1 and S1). Other Gypsy lineages, such as CRM and Galadriel, had lower representation (see Tables 1, S1 and Fig. 1c).
Fig. 1.

Comparison of the relative distribution (%) of repetitive DNA families among three Capsicum datasets. a Note that the Class I elements are more common than Class II and rDNA sequences in the three cases. b LTR-RT elements predominated over non-LTR and ERVs, but note that the C. baccatum genome exhibited 30% fewer sequences than other two datasets. Observe also that Copia superfamily elements were less accumulated (< 10%) than Gypsy ones. c Observe that the accumulated elements were Tekay/Del, Athila/Tat, and CRM, but, except for CRM lineage, there was a big difference in the quantity of the elements in each dataset
The other elements belonging to the non-LTR groups, transposons, for example, were less accumulated in these three datasets, such as in LINE and SINE (< 5%), ERVs (< 1%), CACTA (< 0.1%), hAT (< 2%), MuDR (< 0.1%) and Helitron (< 0.1%). Nevertheless, Sola elements presented an interesting contrast, representing 5.75 and 7.31% in C. annuum and C. baccatum datasets respectively, and only 0.94% in C. chinense dataset. Ribosomal DNA was also estimated, once literature shows a large variation in the number of rDNA sites among these species. Although 5S rDNA showed no great variation among the three species, the 35S rDNA sequences exhibited a contrasting accumulation, with 18.68% in C. baccatum, 1.29% in C. annuum and 0.12% in C. chinense (Tables 1 and S1).
Gypsy autonomous elements dominated the datasets
The search for putative autonomous retrotransposons was focused on Gypsy superfamily members (Tekay/Del, CRM, and Athila/Tat lineages), once their sequences were the most accumulated in the three datasets.
The characterization of retrotransposons was first based on LTR_STRUC [30] output file of Capsicum annuum (considered here as “reference”), which resulted in 254 sequences. From these, only four sequences from CRM, three from Tekay/Del and two from Athila/Tat lineages were characterized. Other 267 sequences that have been identified using the BLAST tool came from C. chinense and C. baccatum datasets, and that were contrasted against C. annuum dataset. The BLAST identified four sequences from CRM, and seven from Tekay/Del lineages as putative autonomous elements.
The sequences characterization was based on LTRs’ presence (Dotter l/b [31]), annotation of GAG and polygenic chain (Artemis [32]) and minimal size of elements (> 3.500 base pairs long). The pseudomolecules that were identified as non-autonomous retrotransposons were those that they did not exhibit one or more LTRs, lost genes from the polygenic chain or ORFs, or had large inverted stretches. After two rounds of alignments (ClustalW [33] followed by Mauve [34]) with putative autonomous sequences, they were organized in groups within each lineage. The Tekay/Del sequences were clustered in five groups, with sequences varying from 8105 to 8902 bp length. The sequences of group 2 were shared between C. annuum and C. chinense, while those of groups 1 and 5 were exclusive for C. annuum, and those of groups 3 and 4 were exclusive for C. baccatum (Figure S1). The CRM sequences were clustered in three groups, varying from 5259 to 7328 bp in length. Except for groups 1 and 2 that appeared in C. annuum and C. baccatum, the others were shared among the three species (Figure S2). The two sequences of the Athila/Tat lineage were distinct from each other, making these two groups exclusive of C. annuum (Figure S3).
The bootstrapped maximum likelihood phylogenetic tree was performed with complete sequences and organized them in three clades: CRM, Athila, and Del (Fig. 2). The CRM sequences were clustered into four groups: A) CRM_2, CRM_4 and CRM_7; B) CRM_5; C) CRM_3 and CRM_6; and D) CRM_1. The Athila sequences presented two groups within C. annuum. The Del elements were more diverse, being organized into three well-supported groups: A) Del_3; B) Del_1 and Del_4; and C) Del_7 and Del_9. The remaining Del sequences did not form well-supported groups (Fig. 2). One sequence was selected from each cluster for a graphical representation (Fig. 3b).
Fig. 2.

Phylogenetic tree based on putative autonomous sequences of Capsicum using the maximum likelihood method with bootstrap 1000. CRM sequences organize four groups (grey, see also the Figure S2) and Athila sequences two groups (blue, see also the Figure S3). The Del sequences were organized in two well-supported groups (orange), corroborating with the MAUVE alignment (Figure S1), and the third group of sequences without well-supported values (green), except for the sequences Del_7 and Del_9 (Figure S1) corroborating with the MAUVE alignment
Fig. 3.

Comparative distribution of the putative autonomous LTR-RTs and the reverse transcriptase sequences along the C. annuum, C. chinense, and C. baccatum datasets. a The Annuum clade, composed by C. annuum and C. chinense and the Baccatum clade (C. baccatum) can be distinguished by the dendrogram on top of the heatmap. In the heatmap, lower and higher accumulation (blue, intermediate and yellow, respectively) represent the amount of conserved sequences found in each dataset. Image shows that Athila/Tat elements accumulated more in C. annuum and C. chinense (marked in yellow and light-yellow), the CRM groups were differentially accumulated, highlighting the absence of CRM III and the predominance of Del I and II in C. baccatum, and bigger accumulation of Del III, IV, and V in C. annuum and C. chinense. The reverse transcriptase of these elements exhibits a similar pattern of distribution than the one observed for the complete elements, Athila/Tat I and II were more accumulate in C. annuum and C. chinense, respectively. CRM I and II were more accumulated in C. chinense, while the group CRM III was more accumulated in C. baccatum. Capsicum annuum and C. chinense had a bigger accumulation of Del I, II and II than C. baccatum, while the groups Del III and IV exhibited more accumulation in C. baccatum. b Graphical representation of LTR-RTs groups. LTR – long terminal repeat, GAG – nucleocapsid, RT – reverse transcriptase, RH – RNAse H, INT – integrase, ASP – aspartase. Asterisks present in Athila illustrations refer to the hallmark ORF for this lineage. Note a difference in extension (bp length) among elements, including GAG and POL positioning, and LTR sizes. Note also that only CRM II exhibits a chromodomain sequence and that all the Del elements present an additional aspartase locus
The clusters of putative autonomous elements were evaluated by their accumulation and distribution in each dataset, in order to compare the probable dynamic of these retrotransposons in the genomes differentiation. In panel 3A, the color scale represents how accumulated these elements are in each dataset (blue represents less accumulation and yellow, more accumulation). In general, C. annuum and C. chinense shared similar sequences in relation to C. baccatum, following the phylogeny proposal for the genus. An exception was observed for Del I (Del_1) group, which on the heatmap (Fig. 3a) exhibited greater accumulation in the C. baccatum genome. The same was observed for the group Del V (Del_8 and Del_10), which despite being composed of sequences from C. baccatum, was more accumulated in C. annuum. When the reverse transcriptase regions of these putative elements were analyzed, this tendency, although not the same as in the complete elements, was retained, with some exceptions such as the accumulation of the group Del II, which was composed only of sequences from C. baccatum (Del_5, Del_6, Del_7, and Del_9), exhibiting more accumulation in C. chinense.
Comparative cytogenetics
The sequences of the reverse transcriptase of the most representative LTR-RT lineages were aligned, and four consensus sequences were used for the primer design: three for the Gypsy superfamily (CRM, Tekay/Del, and Athila) and one for the Copia superfamily (Ivana/Oryco). The data is presented in Tables 1 and S1. Fluorescence in situ hybridization assays revealed either scattered or clustered signals, depending on the LTR-RT lineage analyzed. The probe for Ivana/Oryco lineage (Copia) showed a hybridization profile with a few signals scattered along chromosomes, with clear differences between the pairs and, sometimes, with small interstitial and proximal dots (see arrowheads in Fig. 4a, b and Figure S6a-f).
Fig. 4.

FISH using LTR-RTs probes against metaphases and prometaphases of Capsicum species. Chromosomes were counter-stained with DAPI (blue), Copia probes with Cy3–11-dUTP (red) and Gypsy probes labelled with biotin-11-dUTP / avidin–FITC conjugate (green). The Copia Ivana/Oryco probe showed few hybridization signals scattered along chromosomes, with a low accumulated profile in both C. chinense (a) and C. baccatum (b). The Gypsy Tekay/Del probe exhibited hybridization signals dispersed along the chromosomes in the three species, but with a larger accumulation in C. annuum chromosomes (c) than the other two species, such as in C. baccatum (d). The Gypsy Athila/Tat probe showed brighter hybridization signals than Tekay/Del, accumulating in the pericentromeric to interstitial regions of all C. annuum chromosomes (e), differently of C. baccatum because some chromosomes accumulated many signals and others very few (h). The boxes i, ii, iii and iv are highlighting differences in the pericentromeric and interstitial Athila/Tat signals in two C. baccatum chromosomes. The Gypsy CRM probe showed FISH signals accumulated in the centromeric regions, but with two pairs in each species with much less intense signals. Note the arrows in C. baccatum (f) and C. chinense (g). The bar represents 10 μm
The Tekay/Del probe showed scattered signals along the chromosomes in three species (Supplementary figure 4). Although the signals observed in C. annuum (Fig. 4c) and C. chinense (Supplementary figure 4g) were more evident in the interstitial and proximal regions of all the chromosomes, in C. baccatum, the signals accumulated in half of the chromosomes and were very weak in the others (Fig. 4d). These results confirm the observations from bioinformatic analysis, which shows a greater accumulation of Tekay/Del sequences in C. annuum and C. chinense than in C. baccatum.
The Athila/Tat probe also exhibited scattered signals along the chromosomes, being more accumulated in the interstitial regions. In C. annuum, except for a pair with less intense signals, the remainder exhibited brighter signals at the proximal to interstitial (close to proximal) regions, without any signals in terminal ones (Fig. 4e), similar to those found in C. chinense (Supplementary figure 6 g-i). Capsicum baccatum exhibited four chromosomes with less intense signals and brighter FISH signals in the remaining chromosomes. However, four chromosomes showed stronger signals than those observed in C. annuum and C. chinense (Fig. 4h). The CRM probes showed FISH signals accumulated in the proximal regions of all chromosomes in the three species (see Fig. 4 f, g and Supplementary figure 5). Nevertheless, there was a clear difference in signal intensity among chromosome pairs, with a minor signal in a pair of C. baccatum and another in C. chinense (see arrowheads).
The C-CMA/DAPI banding was performed to verify that LTR-RTs’ accumulation areas corresponded to the AT- and GC-rich band regions as well as to check if the diversity in the distribution profiles of repetitive sequences is equivalent in these species. Capsicum annuum showed two pairs without bands, three pairs with terminal C-DAPI+ dots co-located with more intense C-CMA+ bands and terminal C-CMA+ bands in 11 chromosomes, including three pairs with stronger terminal C-CMA+ bands (Fig. 5a, b). Capsicum chinense exhibits a larger number of intense C-CMA+ bands (seven pairs), and of these, two pairs exhibited intense adjacent C-DAPI+ bands. Smaller terminal C-CMA+ bands were observed in 11 chromosomes, and proximal bands were observed as centromeric dots in nine pairs. Some of these bands were evidenced with DAPI and CMA3 staining. Thinner interstitial bands were stained with C-DAPI but not with C-CMA (Fig. 5c, d). Capsicum baccatum showed six pairs with terminal dots and six with centromeric and/or interstitial C-DAPI bands. In three pairs, these bands appeared as CMA+/DAPI+. The strongest terminal C-CMA bands were detected in four pairs in addition to thinner terminal bands in at least one arm of all chromosomes. Proximal centromeric dot bands were observed in 11 pairs, of which only three also appeared as DAPI+ (Fig. 5e, f).
Fig. 5.
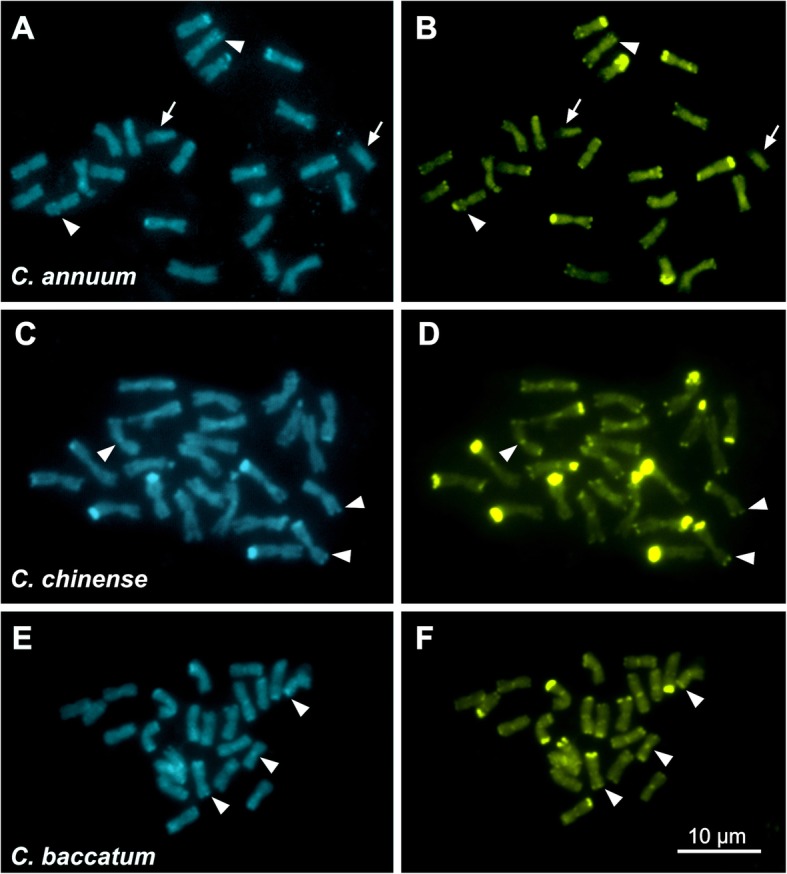
Capsicum species show considerable diversity in the C-CMA/DAPI banding profiles. Observe that C. annuum presents four chromosomes without fluorescent signals. C-DAPI interstitial dots was detected in three chromosome pairs and terminal bands two pairs (a), all of them were co-located with C-CMA bands. Ten pairs exhibit C-CMA signals, being three pairs with stronger terminal and the other as the terminal to subterminal small signals (b). Capsicum chinense showed four pairs with strongest DAPI signals, besides minor centromeric, interstitial and terminal signals on a few chromosomes (c), while C-CMA bands were observed in all the chromosomes, varying as strongest terminal bands in seven pairs, minor centromeric bands in six pairs and as terminal and interstitial dots in ten pairs (d). Some of these bands have been evidenced by DAPI and CMA3 (c, d). Observe that C. baccatum exhibits six pairs with minor terminal and six with centromeric and/or interstitial C-DAPI (e). C-CMA signals were detected in four pairs as strongest terminal bands, but minor terminal bands were observed in all chromosomes, as well as minor centromeric in almost all the chromosomes (f). The bar represents 10 μm
Discussion
Differential accumulation of repetitive DNA families on Capsicum genomes
TEs can move through genomes, representing an evolutionary force that modifies genome structure via mechanisms, such as illegitimate recombination, gene capture, shuffling of regulatory motifs and the generation of new functionality or silencing (see [35]). In the last instance, transposable elements may cause a change in the genomes’ global structure and fluctuations in genome size [10]. They have been useful to compare genomes and karyotypes in evolutionary studies as well as other applicable approaches, such as the study in grapes and blood orange that showed the origin of alterations in the expression of some genes after the insertion of TEs next to them [36].
The ‘Mobilome’ occupies the largest portion of plants’ genomes and play an important role in the physical and functional aspects of chromosomal structures, such as those of the CR lineage (centromeric retrotransposons) that are associated with chromosomal kinetics [15, 37]. In some monocotyledons, for instance, the Mobilome represents around 75% of the genome, such as 80% in maize [6], and LTR-RTs are the most dynamic elements found within the genome [7, 38]. In Nicotiana attenuata and N. obtusifolia, for example, these elements reach up to 81 and 64% of their respective genomes [39]. The study of LTR-RTs in a chromosomal landscape may assist the understanding of some extent of the regulatory potential of TEs along chromosomes, and may also hold the prospect of its possible application in crop breeding programs, such as peppers. Maize is a good example because several TE families described near the genes have been identified as enhancers or repressors under stressful conditions [40, 41].
Previous studies addressing TEs in the Solanaceae family have shown that the Gypsy elements of Solanum lycopersicum are more abundant than the Copia superfamily members [42], although an approach using only autonomous elements showed a predominance of the Tekay/Del (Gypsy) and Tork (Copia) lineages in this species [43]. This suggests that estimates may vary when considering only autonomous or include sequences of non-autonomous elements. The present results in Capsicum indicate that only 0.002% of the C. annuum dataset corresponds to putative autonomous elements, followed by 0.001% in C. chinense and 0.004% in C. baccatum. The remaining sequences correspond to non-autonomous elements. It is important to highlight that the three genomic datasets were obtained by high-covered sequencing [20, 29], which may support the assembly of the pseudochromosomes as complete elements. Even though the Capsicum sequencing does not cover the entire genome (the sequencing part comprises 87% of the C. annuum, 94% of C. chinense, and 83% of C. baccatum genomes) [20, 44] it can be stated that TEs occupy an important fraction of Capsicum genomes. The major part of coding repetitive fractions relates to fragments of non-autonomous elements, which may be amplified by the activity of the autonomous [36]. This might explain the high percentage of LTR-RT fragments in these three datasets.
According to Qin et al. [29], the Gypsy members were the most abundant LTR retrotransposons in Capsicum, with the highest insertion activity among Solanaceae species. When comparing Gypsy and Copia lineages in this plant group, the percentage of LTR-RTs for C. annuum, C. chinense, and C. baccatum was 89, 98, and 70%, respectively. This result pointing out a predominance of Gypsy (> 70%) over Copia superfamily (< 10%). A contrasting accumulation of Tekay/Del, Athila/Tat and CRM lineages of Gypsy were noted in these three genomes. These data point toward the importance of LTR-RTs’ fate in the process of genome organization and differential accumulation between related species, even after considering that C. annuum and C. chinense (Annuum clade) are closer when compared to C. baccatum of the Baccatum clade (see supplementary figure S7) [45]. Kim et al. [44] reported that Solanum is a closely related genus of Capsicum and shares a common ancestor 19.6 MYA. These authors demonstrated C. annuum and C. chinense share a common ancestor 1.14 MYA, while these species shared 1.74 MYA with C. baccatum. These three species also differ in the accumulation of 35S rDNA, with about 20% more sequences in C. baccatum than in C. annuum and C. chinense, besides the number of rDNA sites in the chromosomes [25]. These genomic differences may be responsible for certain difficulties in performing interspecific crosses between these species of distinct clades because of the pre- and post-zygotic barriers, as reported by Manzur et al. [46] and Cremona et al. [47].
The differential activity of retrotransposons among close genomes was also reported in Helianthus [48] and Solanum [49, 50], and these results corroborate with those obtained in the present study regarding Capsicum. The present data have also shown differences among C. annuum and C. chinense, especially in Tekay/Del, ERVs, and Line-RTE accumulation, suggesting that other elements, besides LTR-RT ones, evolve independently. The differential accumulation of Tnt1 retrotransposons in Nicotiana may be a good example to understand and to support the idea of an independent fate of TEs on genome differentiation [51, 52].
Recovered Del, CRM, and Athila/Tat autonomous elements support the Gypsy LTR-RTs’ predominance
The ability of retrotransposons to activate and invade plant genomes may be associated with some internal and external factors, such as biotic and abiotic stresses, breeding processes, injuries, climatic changes, polyploidization, hybridization, and other events (see [35, 53]). However, the activation and proliferation of TEs may be influenced by the ability to cheat cellular silencing controls [53] and the autonomous elements containing a complete polygenic chain, regulators and both LTRs can do that [54]. The absence of some regions could make these elements non-autonomous. In the present study, more non-autonomous sequences (8-folds) were found in the three Capsicum datasets than potentially autonomous ones. This result suggests that these repetitive element classes may have undergone different events of degeneration along with genomes differentiation.
The putative autonomous elements recovered in the present analysis, i.e., ten sequences of Tekay/Del, two sequences of Athila/Tat of C. annuum and seven of CRM, varied between the datasets. This idea follows the proposal of the independent fate of TEs among genomes [55], and it can be exemplified by the occurrence of some Tekay/Del elements exclusive in C. baccatum compared to three others found in C. annuum and C. chinense. Also, we can mention the thirty-fold difference in CRM amount in C. baccatum in relation to C. annuum and C. chinense. This result is in accordance with Hawkins et al. [56] report, which suggests that in Gossypium species, different lineages of LTR-RTs evolved at different moments along with genome evolutionary history, generating a threefold difference in DNA content among diploid species. In another example, De Castro Nunes et al. [15] observed also a greater accumulation of CRM copies in the diploid Coffea species in comparison to the hybrid tetraploid C. arabica.
Not all LTR-RT rich regions in Capsicum chromosomes are heterochromatin hotspots
It is well established in the literature that TEs, especially LTR-RT superfamilies, occupy “specific” chromosomal regions, with the consensus that Copia elements are distributed preferentially along the chromosomes associated with euchromatin, while Gypsy elements are resident in heterochromatin-rich regions (see [7]). In Coffea, Brachiaria, and Secale, for example, Gypsy probes were located in proximal heterochromatin-rich chromosome regions [17, 57, 58], but in Gossypium species, Gypsy probes were hybridized along chromosomes [59]. However, when the elements are considered according to their phylogenetic positions, i.e., lineages of Copia and Gypsy [5, 7, 60], it becomes evident that there are many differences in the TE distribution profiles, in both plants (see [10, 61]) and animals [62, 63]. Thus, it seems wiser to believe that each element has its characteristics, including chromosomal position, genome impact, epigenetic influence, diversification rate, and other features.
Previous studies using FISH in Capsicum spp. have been restricted to rDNA probes [25], which demonstrated that C. baccatum accumulates more in terminal 35S rDNA sites compared to C. annuum and C. chinense, which exhibited just two to four pairs. Moscone et al. [21, 64], Scaldaferro et al. [24] and Martins et al. [27] reports have shown wide variability in the presence of terminal, interstitial and proximal heterochromatic bands in Capsicum species, such as the large and minor heterochromatic terminal bands in C. annuum, C. chinense, and C. baccatum observed in the present study. FISH results using different LTR-RT probes showed hybridization signals accumulated from proximal (CRM) to interstitial region (Athila/Tat and Tekay/Del), scattered, or minor dots along chromosomes (Tekay/Del, Oryco and Tork), such as in Brachiaria [14]. However, no preferential accumulation or strong signals were found in terminal chromosome regions, suggesting that the LTR-RT families analyzed have no accumulation at regions containing rDNA or terminal heterochromatin in Capsicum chromosomes.
FISH using the Athila/Tat probe strongly hybridized at proximal to interstitial regions in almost all the chromosomes of Capsicum. There was also no evident co-location with heterochromatic regions, although there were small AT- and GC-rich bands in few chromosomes, i.e., without evident correlation with heterochromatin hotspots. In this case, the scattered signals (or dots) observed after FISH with Tekay/Del and Oryco probes are in agreement with the concept of dispersed localization of retroelements within plant genomes, but without dependence on co-localization with heterochromatin blocks. This Athila/Tat dispersion pattern, such as interstitial dots, has been described by Park et al. [65] in C. annuum. Using the Passiflora edulis for comparison, members of Ty3/Gypsy superfamily were the most accumulated, and their sequences appeared scattered along chromosomes, including at the pericentromeric regions [66]. In some Solanaceae species, such as tomato and peppers, elements of the Tekay/Del Gypsy superfamily had a scattered accumulation profile as reported by Park et al. [65], with hybridization in the chromosomes of Solanum lycopersicum (tomato) and Capsicum annuum (pepper), in which pepper had a higher number and more intense signals than those observed in tomato.
Different from the other LTR-RT probes, the CRM probe exhibited intense signals in the proximal regions, associated with centromeres, and in Capsicum chromosomes, these regions were rich in CMA+ and DAPI+ signals. The centromeric retrotransposon lineage of chromovirus (also called centromeric retrotransposon of maize or CRM) occurs preferentially in proximal chromosome regions. CRMs carry particular domains called chromodomain (CHRomatin Organization MOdifer DOMAIN) and CR motifs that have potential to interact with the CENH3 centromeric protein and to participate in the centromere function [37, 67]. FISH using CRM lineage probes have been described in several plant species, for example in some monocotyledon groups [58, 68, 69], suggesting that besides the association with specific centromeric proteins, this accumulation may also be associated with recombination-poor regions.
Conclusions
This comparative cytogenomic analysis using the three most economically important Capsicum species showed great diversity in genome composition, although there is a closer approximation between C. annuum and C. chinense when compared to C. baccatum, as suggested by the phylogeny. The dataset screening of these three species showed that there was a differential accumulation of transposable elements, especially those from the lineages Tekay/Del, CRM and Athila/Tat from Gypsy, while those of the Copia superfamily were underrepresented. From a chromosomal point of view, these transposable elements were dispersed along the chromosomes (Copia) as well as in blocks (Gypsy), highlighting those of the CRM lineage that predominated the centromeric region. Another aspect is that LTR-RTs are not always associated with heterochromatin-rich regions. These data support the idea of the independent fate of LTR-RTs. Such genomic and chromosomal differences between closely related species should be taken into account in breeding programs, as they may interfere with the success of interspecific crosses and the introgression of agronomic traits of interest. Capsicum spp. proved to be a good model for most studies on the repetitive fraction, from both genomic and chromosomal points of view, considering the diversity in the accumulation and genomic distribution of LTR-RTs.
Methods
Plant materials
Seeds of Capsicum annuum cv. Criollo de Morelos (accession GBUEL145), C. chinense (accession GBUEL27) and C. baccatum (accession GBUEL118), identified by Dr. Leandro S. A. Gonçalves, were obtained from the gene bank of Londrina State University. The samples were sowed in 128-cell polystyrene trays containing the substrate Vivatto®. Ten seedlings of each species were grown in the Cytogenetics and Plant Diversity Laboratory greenhouse.
Genomic analysis
The following three genomes used for bioinformatics analysis were obtained from Pepper Genome Platform (http://peppergenome.snu.ac.kr/): scaffolds from Capsicum annuum v.1.6, C. chinense v.1.2, and C. baccatum v.1.2. Files were used to search for autonomous and non-autonomous LTR-RTs. For genomic comparisons, a database was built to run a local BLAST, which contained all the TEs conserved protein sequences from REXdb [5], GypsyDB [61], RepBase [70] and NCBI (http://www.ncbi.nlm.nih.gov/), containing 283,676 protein sequences. To identify the rDNA sequences, a second database was compiled comprising nucleotides 35S and 5S rDNA sequences from different organisms, obtained from NCBI, containing 1652 sequences.
The repetitive fraction (transposable elements, 35 and 5S rDNA) evaluation was conducted by the Blast version 2.2.28+, comparing the genomic dataset against the local databases. The parameters used were E-value 10e-4, max target seqs 1 and the remaining were set by program default. The results obtained are plotted in Table S1, which includes quantitative and qualitative estimates according to the phylogeny proposal by Neumann et al. [5] as well as 35S and 5S rDNA fractions. The aim was to recognize and to differentiate retrotransposons in different lineages, and produce a more refined in situ hybridization, and to support the idea of the independent fate of these elements. The sequences were grouped into the superfamilies Copia (Ale, Alesia, Angela, Bianca, Bryco, Lyco, Gymco, Ikeros, Ivana, Osser, SIRE, TAR and Tork lineages) and Gypsy (CRM, Chlamyvir, Galadriel, Tcn1, Reina, Tekay/Del, Athila, Tat, Ogre, Retand, Phygy, and Selgy lineages). Due to their similarity, Athila, Tat, Ogre, and Retand members have been referred as Athila/Tat clade.
Autonomous and non-autonomous estimates and primer design
LTR_STRUC [30] has been used to compare and search for LTR-retrotransposons in the reference genome of Capsicum annuum. Putative retrotransposons sequences were then classified into Gypsy and Copia superfamilies according to their similarity measured against the Gypsy Database protein domains (http://www.gydb.org/index.php/Main_Page) by Genewise alignment [71] and annotated with Artemis [32]. The putative elements were then used as a database for BLAST rounds against the other datasets.
The BLAST output files were organized with sequences greater than 3500 bp and more than 70% identity, and they were used to search for sequences with both LTRs with the Dotter l/b using program default parameters [31]. Subsequently, sequences that carried both LTRs were submitted to the online BLAST at NCBI to search for the presence of conserved domains. The sequences were also screened by the presence of stop codon by the Pfam online tool [72]. The complete nucleotide sequences with the correct protein order were aligned with the MAUVE [34] program and grouped as per the graphic similarity. These data were then validated by aligning them with CLUSTALW [33]. From each group, a representative sequence was chosen for the graphic annotation using the IBS [73] program. To understand the relationships among these sequences, the alignment was used to construct a bootstrapped maximum likelihood (ML) phylogenetic tree (1000 bootstraps) in MegaX [74]. The ML tree was generated with GTR (general time-reversible) mutation model, gamma-distributed and invariant sites (G + I) rates among sites, and the heuristic method was nearest-neighbor-interchange (NNI). To understand how these putative autonomous elements are dispersed and accumulated along with the datasets, HeatMapper tool [75] was used in the three datasets through two approaches, one being with a representative sequence from each group of putative autonomous elements and the other with the reverse transcriptase region of these groups.
The most conserved stretch of the reverse transcriptase of each LTR-RT lineage was used for primer designing with the custom primers, OligoPerfect TM Designer tool of Thermo Fisher Scientific (http://tools.thermofisher.com, see Table 2). The primers’ viability was assessed with the PCR primer stats tool (http://www.bioinformatics.org).
Table 2.
List of primers of reverse transcriptase sequences, that have been designed for probes obtaining and FISH
| Elements | Primers |
|---|---|
| Athila/Tat - RT |
F 5′ GGGTGGTATTGCTTCTTGGA 3′ R 5′ GAATCACCTACCACAGAG 3′ |
| CRM - RT |
F 5′ CCACCAACAAGATAACGG 3′ R 5′ CCATCCATTCATAGAGACC 3′ |
| Tekay/Del - RT |
F 5′ GTTCAGGGTGCCAAGTGT 3′ R 5’GGGCGTTAGTCAACCTGAAG 3′ |
| Ivana/Oryco - RT |
F 5′ GGTTCAAGGATCGGTTGATAG 3′ R 5′ GTTGAGCTTGCACACCATGT3’ |
RT Reverse transcriptase
DNA extraction, PCR and LTR-RT probes
DNA was isolated from young leaves of each species using the cetyltrimethylammonium bromide (CTAB) method [76], purified with phenol:chloroform (1:1, v/v), chloroform:isoamyl alcohol (24:1, v/v) and RNase (1 mg mL− 1) and precipitated in 100% absolute ethanol. The samples were eluted in 10 mM Tris–HCl pH 8 and the concentrations were estimated using a NanoDrop 2000 Spectrophotometer (Thermo Scientific).
The LTR-RT probes were obtained by PCR using specific primers for each lineage, with C. annuum as a DNA template. A standard PCR [5 U μL− 1 Taq polymerase (0.5 μL), 10× buffer (2.5 μL), 50 mM MgCl2 (1.5 μL), 10 mM dNTP (1 μL), 5 mM primers (2 μL each) and H2O up to a final volume of 25 μL] was used under the following conditions: 94 °C for 2 min, 30 cycles of 94 °C for 40 s, 59 °C for 40 s and 72 °C for 1 min, and a final extension of 72 °C for 10 min. The reactions were tested via electrophoresis in an agarose gel at 3 V cm− 1 and stained with ethidium bromide. The probes for each LTR-RT lineage were obtained through the re-amplification of PCR products that involved labeling with biotin-11-dUTP (Gypsy families) or Cy3-dUTP (Copia families).
Cytogenetic analysis
The root tips were pretreated with 0.5% colchicine (1 h 30 min) and fixed in ethanol-acetic acid (3:1, v:v). The fixed material was treated in a solution of 2% cellulase and 20% pectinase and squashed in a drop of 60% acetic acid. After liquid nitrogen freezing, the coverslips were removed, and the slides were air-dried.
For fluorescence in situ hybridization, slides received a mix containing a solution (30 μL) composed of 100% formamide (15 μL), 50% polyethylene glycol (6 μL), 20× SSC (3 μL), 100 ng of calf thymus DNA (1 μL), 10% SDS (1 μL) and 100 ng of probes (4 μL). The mix was denatured at 90 °C for 10 min, and hybridization was performed at 37 °C for 24 h in a humid chamber. Post-hybridization washes were carried out with 70% stringency, using an SSC buffer, with pH 7.0. After the probe detection with an avidin-fluorescein isothiocyanate (FITC) conjugate, washes were performed in 4× SSC/0.2% Tween-20 at room temperature. The slides were mounted with 25 μL of DABCO, a solution composed of glycerol (90%), 1,4-diaza-bicyclo (2.2.2)-octane (2.3%), 20 mM Tris–HCl, pH 8.0 (2%), 2.5 mM MgCl2 (4%) and distilled water (1.7%) in addition to 1 μL of 2 μg mL− 1 4,6′-diamidino-2-phenylindole (DAPI).
For C-CMA/DAPI banding, the samples were incubated in a sequence of 45% acetic acid (8 min), saturated solution of Ba(OH)2 at room temperature (8 min) and in 2× saline sodium citrate at 60 °C (1 h and 30 min). After this, the slides were stained with a CMA3 for 90 min and DAPI for 30 min [77] and mounted in a solution of McIlvaine buffer plus glycerol (1:1, v:v) with 2.5 mM MgCl2.
The chromosome images were acquired in greyscale with a Leica DM4500 B microscope coupled with a DFC300FX camera, pseudo-colored (blue for DAPI, greenish-yellow for FITC and red for Cy3) and contrasted using GIMP 2.8 Linux.
Supplementary information
Additional file 1: Figure S1. Mauve alignment of Tekay/Del sequences from Capsicum genomes. Figure S2. Mauve alignment of CRM sequences from Capsicum genomes. Figure S3. Mauve alignment of Athila sequences from Capsicum genomes. Figure S4. FISH using LTR-RTs probes against metaphases and prometaphases of Capsicum species. Figure S5. FISH using LTR-RTs probes against metaphases and prometaphases of Capsicum species. Figure S6. FISH using LTR-RTs probes against metaphases and prometaphases of Capsicum species. Table S1. Frequency and relative values of repetitive fraction in the three Capsicum genomes. Figure S7. Phylogeny for Capsicum adapted from Carizzo-Garcia et al. [45]
Acknowledgments
ALLV, LSAG, and RR thank CNPq for the productivity scholarship (numbers 309902/2018-5, 307911/2018-7 and 307569/2017-9, respectively) and RR for the scholarship FAPERJ E-26/202.985/2017.
Abbreviations
- CRM
Centromeric Retrotransposon of Maize
- ERV
Endogenous Retrovirus
- FISH
Fluorescence in situ hybridization
- LINEs
Long Interspersed Elements
- LTR-RT
LTR-retrotransposons
- LTRs
Long Terminal Repeats
- pg
Picograms
- rDNA
Ribosomal DNA
- SDS
Sodium Dodecyl Sulfate
- SINEs
Short interspersed nuclear elements
- SSC
Saline-Sodium Citrate
- TEs
Transposable elements
Authors’ contributions
RA performed all the experiments, wrote and corrected the manuscript. VYB and LAC performed bioinformatic analysis. LSA G and RR were responsible for varieties of supply, plant maintenance, analyses, and manuscript corrections. ALLV designed the study, checked the data analyses, and organized, wrote and corrected the manuscript, as the head of the group. All authors have read and approved the manuscript.
Funding
This study was partially financed by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES) - Finance Code 001. The authors thank the Brazilian agencies FINEP, Fundação Araucária, CNPq, CAPES, and ProPPG-UEL for their financial support.
Availability of data and materials
Original sequences of these three genomes can be accessed at Pepper Genome Platform (http://peppergenome.snu.ac.kr/).
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12864-020-6618-9.
References
- 1.Heslop-Harrison JS, Schwarzacher T. Organisation of the plant genome in chromosomes. Plant J. 2011;66(1):18–33. doi: 10.1111/j.1365-313X.2011.04544.x. [DOI] [PubMed] [Google Scholar]
- 2.Bennetzen JL, Wang H. The contributions of transposable elements to the structure, function, and evolution of plant genomes. Annu Rev Plant Biol. 2014;65:505–530. doi: 10.1146/annurev-arplant-050213-035811. [DOI] [PubMed] [Google Scholar]
- 3.Kazazian HH. Mobile elements: drivers of genome evolution. Science. 2004;303(5664):1626–1632. doi: 10.1126/science.1089670. [DOI] [PubMed] [Google Scholar]
- 4.Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, et al. A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 2007;8(12):973. doi: 10.1038/nrg2165. [DOI] [PubMed] [Google Scholar]
- 5.Neumann P, Novák P, Hoštáková N, Macas J. Systematic survey of plant LTR-retrotransposons elucidates phylogenetic relationships of their polyprotein domains and provides a reference for element classification. Mob DNA. 2019;10(1):1. doi: 10.1186/s13100-018-0144-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, et al. The B73 maize genome: complexity, diversity, and dynamics. Science. 2009;326(5956):1112–1115. doi: 10.1126/science.1178534. [DOI] [PubMed] [Google Scholar]
- 7.Orozco-Arias S, Isaza G, Guyot R. Retrotransposons in plant genomes: structure, identification, and classification through bioinformatics and machine learning. Int J Mol Sci. 2019;20(15):3837. doi: 10.3390/ijms20153837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bennetzen JL. Mechanisms and rates of genome expansion and contraction in flowering plants. Genetica. 2002;115(1):29–36. doi: 10.1023/a:1016015913350. [DOI] [PubMed] [Google Scholar]
- 9.Dodsworth S, Jang TS, Struebig M, Chase MW, Weiss-Schneeweiss H, Leitch AR. Genome-wide repeat dynamics reflect phylogenetic distance in closely related allotetraploid Nicotiana (Solanaceae) Plant Syst Evol. 2017;303(8):1013–1020. doi: 10.1007/s00606-016-1356-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Souza TB, Chaluvadi SR, Johnen L, Marques A, González-Elizondo MS, Bennetzen JL, Vanzela ALL. Analysis of retrotransposon abundance, diversity and distribution in holocentric Eleocharis (Cyperaceae) genomes. Ann Bot. 2018;122(2):279–290. doi: 10.1093/aob/mcy066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hirsch CD, Springer NM. Transposable element influences on gene expression in plants. Biochim Biophys Acta Gene Regul Mech. 2017;1860(1):157–165. doi: 10.1016/j.bbagrm.2016.05.010. [DOI] [PubMed] [Google Scholar]
- 12.Heslop-Harrison JS, Brandes A, Schwarzacher T. Tandemly repeated DNA sequences and centromeric chromosomal regions of Arabidopsis species. Chromosom Res. 2003;11:241–253. doi: 10.1023/a:1022998709969. [DOI] [PubMed] [Google Scholar]
- 13.Underwood CJ, Henderson IR, Martienssen RA. Genetic and epigenetic variation of transposable elements in Arabidopsis. Curr Opin Plant Biol. 2017;36:135–141. doi: 10.1016/j.pbi.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Santos FC, Guyot R, Do Valle CB, Chiari L, Techio VH, Heslop-Harrison P, Vanzela ALL. Chromosomal distribution and evolution of abundant retrotransposons in plants: gypsy elements in diploid and polyploid Brachiaria forage grasses. Chromosom Res. 2015;23(3):571–582. doi: 10.1007/s10577-015-9492-6. [DOI] [PubMed] [Google Scholar]
- 15.De Castro NR, Orozco-Arias S, Crouzillat D, Mueller LA, Strickler SR, Descombes P, Vanzela AL. Structure and distribution of centromeric retrotransposons at diploid and allotetraploid Coffea centromeric and pericentromeric regions. Front Plant Sci. 2018;9:175. doi: 10.3389/fpls.2018.00175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mlinarec J, Franjević D, Harapin J, Besendorfer V. The impact of the Tekay chromoviral elements on genome organisation and evolution of Anemone sl (Ranunculaceae) Plant Biol. 2016;18(2):332–347. doi: 10.1111/plb.12393. [DOI] [PubMed] [Google Scholar]
- 17.Gaeta ML, Yuyama PM, Sartori D, Fungaro MHP, Vanzela ALL. Occurrence and chromosome distribution of retroelementsand NUPT sequences in Copaifera langsdorffii Desf. (Caesalpinioideae) Chromosom Res. 2010;18:515–524. doi: 10.1007/s10577-010-9131-1. [DOI] [PubMed] [Google Scholar]
- 18.Yang TJ, Lee S, Chang SB, Yu Y, de Jong JH, Wing RA. Indepth sequence analysis of the centromeric region of tomatochromosome 12: identification of a large CAA block and char-acterization of centromeric retrotranposons. Chromosoma. 2005;114:103–117. doi: 10.1007/s00412-005-0342-8. [DOI] [PubMed] [Google Scholar]
- 19.Park M, Park J, Kim S, Kwon J-K, Park HM, Bae IH, Yang T-J, Lee Y-H, Kang B-C, Choi D. Evolution of the large genome in Capsicum annuum occurred through accumulation of single-type long terminal repeat retrotransposons and their derivatives. Plant J. 2012;69:1018–1029. doi: 10.1111/j.1365-313X.2011.04851.x. [DOI] [PubMed] [Google Scholar]
- 20.Kim S, Park M, Yeom SI, Kim YM, Lee JM, Lee HA, et al. Genome sequence of the hot pepper provides insights into the evolution of pungency in Capsicum species. Nat Genet. 2014;46(3):270. doi: 10.1038/ng.2877. [DOI] [PubMed] [Google Scholar]
- 21.Moscone EA, Lambrou M, Hunziker AT, Ehrendorfer F. Giemsa C-banded karyotypes in Capsicum (Solanaceae) Plant Syst Evol. 1993;186(3–4):213–229. [Google Scholar]
- 22.Park YK, Kim BD, Kim BS, Armstrong KC, Kim NS. Karyotyping of the chromosomes and physical mapping of the 5S rRNA and 18S-26S rRNA gene families in five different species in Capsicum. Genes Genet Syst. 1999;74(4):149–157. [Google Scholar]
- 23.Park YK, Park KC, Park CH, Kim NS. Chromosomal localization and sequence variation of 5S rRNA gene in five Capsicum species. Molecules Cells. 2000;10(1):18–24. doi: 10.1007/s10059-000-0018-4. [DOI] [PubMed] [Google Scholar]
- 24.Scaldaferro MA, Grabiele M, Moscone EA. Heterochromatin type, amount and distribution in wild species of chili peppers (Capsicum, Solanaceae) Genet Resour Crop Evol. 2013;60(2):693–709. [Google Scholar]
- 25.Scaldaferro MA, da Cruz MVR, Cecchini NM, Moscone EA. FISH and AgNor mapping of the 45S and 5S rRNA genes in wild and cultivated species of Capsicum (Solananceae) Genome. 2015;59(2):95–113. doi: 10.1139/gen-2015-0099. [DOI] [PubMed] [Google Scholar]
- 26.Aguilera PM, Debat HJ, Grabiele M. An integrated physical map of the cultivated hot chili pepper, Capsicum baccatum var. pendulum. Int J Agric Biol. 2017. 10.17957/IJAB/15.0303.
- 27.Martins LDV, Peron AP, Lopes ÂCDA, Gomes RLF, Carvalho RD, Feitoza LDL. Heterochromatin distribution and histone modification patterns of H4K5 acetylation and H3S10 phosphorylation in Capsicum L. Crop Breed Appl Biotechnol. 2018;18(2):161–168. [Google Scholar]
- 28.Moreira AFP, Ruas PM, De Fátima Ruas C, Baba VY, Giordani W, Arruda IM, et al. Genetic diversity, population structure and genetic parameters of fruit traits in Capsicum chinense. Sci Hortic. 2018;236:1–9. [Google Scholar]
- 29.Qin C, Yu C, Shen Y, Fang X, Chen L, Min J, et al. Whole-genome sequencing of cultivated and wild peppers provides insights into Capsicum domestication and specialization. Proc Natl Acad Sci. 2014;111(14):5135–5140. doi: 10.1073/pnas.1400975111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCarthy EM, McDonald JF. LTR_STRUC: a novel search and identification program for LTR retrotransposons. Bioinformatics. 2003;19(3):362–367. doi: 10.1093/bioinformatics/btf878. [DOI] [PubMed] [Google Scholar]
- 31.Sonnhammer EL, Durbin R. A dot-matrix program with dynamic threshold control suited for genomic DNA and protein sequence analysis. Gene. 1995;167(1–2):GC1–GC10. doi: 10.1016/0378-1119(95)00714-8. [DOI] [PubMed] [Google Scholar]
- 32.Carver T, Harris SR, Berriman M, Parkhill J, McQuillan JA. Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics. 2011;28(4):464–469. doi: 10.1093/bioinformatics/btr703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Darling AC, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14(7):1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Galindo-González L, Mhiri C, Deyholos MK, Grandbastien MA. LTR-retrotransposons in plants: engines of evolution. Gene. 2017;626:14–25. doi: 10.1016/j.gene.2017.04.051. [DOI] [PubMed] [Google Scholar]
- 36.Lisch D. How important are transposons for plant evolution? Nat Rev Genet. 2013;14(1):49. doi: 10.1038/nrg3374. [DOI] [PubMed] [Google Scholar]
- 37.Neumann P, Navrátilová A, Koblížková A, Kejnovský E, Hřibová E, Hobza R, Macas J. Plant centromeric retrotransposons: a structural and cytogenetic perspective. Mob DNA. 2011;2(1):4. doi: 10.1186/1759-8753-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Negi P, Rai AN, Suprasanna P. Moving through the stressed genome: emerging regulatory roles for transposons in plant stress response. Front Plant Sci. 2016;7:1448. doi: 10.3389/fpls.2016.01448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu S, Brockmöller T, Navarro-Quezada A, Kuhl H, Gase K, Ling Z, et al. Wild tobacco genomes reveal the evolution of nicotine biosynthesis. Proc Natl Acad Sci. 2017;114(23):6133–6138. doi: 10.1073/pnas.1700073114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Makarevitch I, Waters AJ, West PT, Stitzer M, Hirsch CN, Ross-Ibarra J, Springer NM. Transposable elements contribute to activation of maize genes in response to abiotic stress. PLoS Genet. 2015;11(1):e1004915. doi: 10.1371/journal.pgen.1004915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mao H, Wang H, Liu S, Li Z, Yang X, Yan J, et al. A transposable element in a NAC gene is associated with drought tolerance in maize seedlings. Nat Commun. 2015;6:8326. doi: 10.1038/ncomms9326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jouffroy O, Saha S, Mueller L, Quesneville H, Maumus F. Comprehensive repeatome annotation reveals strong potential impact of repetitive elements on tomato ripening. BMC Genomics. 2016;17(1):624. doi: 10.1186/s12864-016-2980-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paz RC, Kozaczek ME, Rosli HG, Andino NP, Sanchez-Puerta MV. Diversity, distribution and dynamics of full-length Copia and gypsy LTR retroelements in Solanum lycopersicum. Genetica. 2017;145(4–5):417–430. doi: 10.1007/s10709-017-9977-7. [DOI] [PubMed] [Google Scholar]
- 44.Kim S, Park J, Yeom SI, Kim YM, Seo E, Kim KT, Kim MS, Lee JM, Cheong K, Shin HS, Kim SB. New reference genome sequences of hot pepper reveal the massive evolution of plant disease-resistance genes by retroduplication. Genome Biol. 2017;18(1):210. doi: 10.1186/s13059-017-1341-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carrizo García C, Barfuss MH, Sehr EM, Barboza GE, Samuel R, Moscone EA, Ehrendorfer F. Phylogenetic relationships, diversification and expansion of chili peppers (Capsicum, Solanaceae) Ann Bot. 2016;118(1):35–51. doi: 10.1093/aob/mcw079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Manzur JP, Fita A, Prohens J, Rodríguez-Burruezo A. Successful wide hybridization and introgression breeding in a diverse set of common peppers (Capsicum annuum) using different cultivated Ají (C. baccatum) accessions as donor parents. PLoS One. 2015;10(12):e0144142. doi: 10.1371/journal.pone.0144142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cremona G, Iovene M, Festa G, Conicella C, Parisi M. Production of embryo rescued hybrids between the landrace “Friariello” (Capsicum annuum var. annuum) and C. baccatum var. pendulum: phenotypic and cytological characterization. Euphytica. 2018;214(8):129. [Google Scholar]
- 48.Staton SE, Bakken BH, Blackman BK, Chapman MA, Kane NC, Tang S, Burke JM. The sunflower (Helianthus annuus L.) genome reflects a recent history of biased accumulation of transposable elements. Plant J. 2012;72(1):142–153. doi: 10.1111/j.1365-313X.2012.05072.x. [DOI] [PubMed] [Google Scholar]
- 49.Di Filippo M, Traini A, D'Agostino N, Frusciante L, Chiusano ML. Euchromatic and heterochromatic compositional properties emerging from the analysis of Solanum lycopersicum BAC sequences. Gene. 2012;499(1):176–181. doi: 10.1016/j.gene.2012.02.044. [DOI] [PubMed] [Google Scholar]
- 50.Bolger A, Scossa F, Bolger ME, Lanz C, Maumus F, Tohge T, Fich EA. The genome of the stress-tolerant wild tomato species Solanum pennellii. Nat Genet. 2014;46(9):1034. doi: 10.1038/ng.3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vernhettes S, Grandbastien MA, Casacuberta JM. The evolutionary analysis of the Tnt1 retrotransposon in Nicotiana species reveals the high variability of its regulatory sequences. Mol Biol Evol. 1998;15(7):827–836. doi: 10.1093/oxfordjournals.molbev.a025988. [DOI] [PubMed] [Google Scholar]
- 52.Melayah D, Lim KY, Bonnivard E, Chalhoub B, Borne FDD, Mhiri C, Grandbastien MA. Distribution of the Tnt1 retrotransposon family in the amphidiploid tobacco (Nicotiana tabacum) and its wild Nicotiana relatives. Biol J Linn Soc. 2004;82(4):639–649. [Google Scholar]
- 53.Casacuberta E, González J. The impact of transposable elements in environmental adaptation. Mol Ecol. 2013;22(6):1503–1517. doi: 10.1111/mec.12170. [DOI] [PubMed] [Google Scholar]
- 54.Kumar A, Bennetzen JL. Plant retrotransposons. Annu Rev Genet. 1999;33(1):479–532. doi: 10.1146/annurev.genet.33.1.479. [DOI] [PubMed] [Google Scholar]
- 55.Jurka J, Bao W, Kojima KK. Families of transposable elements, population structure and the origin of species. Biol Direct. 2011;6(1):44. doi: 10.1186/1745-6150-6-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hawkins JS, Proulx SR, Rapp RA, Wendel JF. Rapid DNA loss as a counterbalance to genome expansion through retrotransposon proliferation in plants. Proc Natl Acad Sci. 2009;106(42):17811–17816. doi: 10.1073/pnas.0904339106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yuyama PM, Pereira LFP, Dos Santos TB, Sera T, Vilas-Boas LA, Lopes FR, et al. FISH using a gag-like fragment probe reveals a common Ty 3-gypsy-like retrotransposon in genome of Coffea species. Genome. 2012;55(12):825–833. doi: 10.1139/gen-2012-0081. [DOI] [PubMed] [Google Scholar]
- 58.Zhang H, Koblížková A, Wang K, Gong Z, Oliveira L, Torres GA, et al. Boom-bust turnovers of megabase-sized centromeric DNA in Solanum species: rapid evolution of DNA sequences associated with centromeres. Plant Cell. 2014;26(4):1436–1447. doi: 10.1105/tpc.114.123877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lu H, Cui X, Liu Z, Liu Y, Wang X, Zhou Z. Discovery and annotation of a novel transposable element family in Gossypium. BMC Plant Biol. 2018;18(1):307. doi: 10.1186/s12870-018-1519-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Llorens C, Futami R, Covelli L, Domínguez-Escribá L, Viu JM, Tamarit D, Aguilar-Rodríguez J, Vicente-Ripolles M, Fuster G, Bernet GP, Maumus F. The Gypsy Database (GyDB) of mobile genetic elements: release 2.0. Nucleic Acids Res. 2010;39(suppl_1):D70–D74. doi: 10.1093/nar/gkq1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ma B, Xin Y, Kuang L, He N. Distribution and characteristics of transposable elements in the mulberry genome. Plant Genome. 2019. 10.3835/plantgenome2018.12.0094. [DOI] [PMC free article] [PubMed]
- 62.Schemberger MO, Nogaroto V, Almeida MC, Artoni RF, Valente GT, Martins C, et al. Sequence analyses and chromosomal distribution of the Tc1/mariner element in Parodontidae fish (Teleostei: Characiformes) Gene. 2016;593(2):308–314. doi: 10.1016/j.gene.2016.08.034. [DOI] [PubMed] [Google Scholar]
- 63.Schemberger MO, Nascimento VD, Coan R, Ramos É, Nogaroto V, Ziemniczak K, et al. DNA transposon invasion and microsatellite accumulation guide W chromosome differentiation in a Neotropical fish genome. Chromosoma. 2019;128:1–14. doi: 10.1007/s00412-019-00721-9. [DOI] [PubMed] [Google Scholar]
- 64.Moscone EA, Lambrou M, Ehrendorfer F. Fluorescent chromosome banding in the cultivated species of Capsicum (Solanaceae) Plant Syst Evol. 1996;202(1–2):37–63. [Google Scholar]
- 65.Park M, Jo S, Kwon JK, Park J, Ahn JH, Kim S, Kim BD. Comparative analysis of pepper and tomato reveals euchromatin expansion of pepper genome caused by differential accumulation of Ty3/gypsy-like elements. BMC Genomics. 2011;12(1):85. doi: 10.1186/1471-2164-12-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pamponét VCC, Souza MM, Silva GS, Micheli F, de Melo CAF, de Oliveira SG, Corrêa RX. Low coverage sequencing for repetitive DNA analysis in Passiflora edulis Sims: cytogenomic characterization of transposable elements and satellite DNA. BMC Genomics. 2019;20(1):262. doi: 10.1186/s12864-019-5576-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Houben A, Schroeder-Reiter E, Nagaki K, Nasuda S, Wanner G, Murata M, et al. CENH3 interacts with the centromeric retrotransposon cereba and GC-rich satellites and locates to centromeric substructures in barley. Chromosoma. 2007;116(3):275–283. doi: 10.1007/s00412-007-0102-z. [DOI] [PubMed] [Google Scholar]
- 68.Nagaki K, Neumann P, Zhang D, Ouyang S, Buell CR, Cheng Z, Jiang J. Structure, divergence, and distribution of the CRR centromeric retrotransposon family in rice. Mol Biol Evol. 2005;22:845–855. doi: 10.1093/molbev/msi069. [DOI] [PubMed] [Google Scholar]
- 69.Vogel JP, Garvin DF, Mockler TC, Schmutz J, Rokhsar D, Bevan MW, Barry K, et al. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature. 2010;463:763–768. doi: 10.1038/nature08747. [DOI] [PubMed] [Google Scholar]
- 70.Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase update, a database of eukaryotic repetitive elements. Cytogenetic Genome Res. 2005;110(1–4):462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- 71.Birney E, Clamp M, Durbin R. GeneWise and genomewise. Genome Res. 2004;14(5):988–995. doi: 10.1101/gr.1865504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.El-Gebali S, Mistry J, Bateman A, Eddy SR, Luciani A, Potter SC, Qureshi M, Richardson LJ, Salazar GA, Smart A, Sonnhammer EL. The Pfam protein families database in 2019. Nucleic Acids Res. 2018;47(D1):D427–D432. doi: 10.1093/nar/gky995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu W, Xie Y, Ma J, Luo X, Nie P, Zuo Z, Lahrmann U, Zhao Q, Zheng Y, Zhao Y, Xue Y. IBS: an illustrator for the presentation and visualization of biological sequences. Bioinformatics. 2015;31(20):3359–3361. doi: 10.1093/bioinformatics/btv362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35(6):1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Babicki S, Arndt D, Marcu A, Liang Y, Grant JR, Maciejewski A, Wishart DS. Heatmapper: web-enabled heat mapping for all. Nucleic Acids Res. 2016. (epub ahead of print). 10.1093/nar/gkw419. [DOI] [PMC free article] [PubMed]
- 76.Doyle JJ, Doyle JL. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemical Bull. 1987;19:11–15. [Google Scholar]
- 77.Schwarzacher T, Ambros P, Schweizer D. Application of Giemsa banding to orchid karyotype analysis. Plant Syst Evol. 1980;134(3–4):293–297. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. Mauve alignment of Tekay/Del sequences from Capsicum genomes. Figure S2. Mauve alignment of CRM sequences from Capsicum genomes. Figure S3. Mauve alignment of Athila sequences from Capsicum genomes. Figure S4. FISH using LTR-RTs probes against metaphases and prometaphases of Capsicum species. Figure S5. FISH using LTR-RTs probes against metaphases and prometaphases of Capsicum species. Figure S6. FISH using LTR-RTs probes against metaphases and prometaphases of Capsicum species. Table S1. Frequency and relative values of repetitive fraction in the three Capsicum genomes. Figure S7. Phylogeny for Capsicum adapted from Carizzo-Garcia et al. [45]
Data Availability Statement
Original sequences of these three genomes can be accessed at Pepper Genome Platform (http://peppergenome.snu.ac.kr/).