

Abstract
Background
Deep mutational scanning (DMS) studies exploit the mutational landscape of sequence variation by systematically and comprehensively assaying the effect of single amino acid variants (SAVs; also referred to as missense mutations, or non-synonymous Single Nucleotide Variants – missense SNVs or nsSNVs) for particular proteins. We assembled SAV annotations from 22 different DMS experiments and normalized the effect scores to evaluate variant effect prediction methods. Three trained on traditional variant effect data (PolyPhen-2, SIFT, SNAP2), a regression method optimized on DMS data (Envision), and a naïve prediction using conservation information from homologs.
Results
On a set of 32,981 SAVs, all methods captured some aspects of the experimental effect scores, albeit not the same. Traditional methods such as SNAP2 correlated slightly more with measurements and better classified binary states (effect or neutral). Envision appeared to better estimate the precise degree of effect. Most surprising was that the simple naïve conservation approach using PSI-BLAST in many cases outperformed other methods. All methods captured beneficial effects (gain-of-function) significantly worse than deleterious (loss-of-function). For the few proteins with multiple independent experimental measurements, experiments differed substantially, but agreed more with each other than with predictions.
Conclusions
DMS provides a new powerful experimental means of understanding the dynamics of the protein sequence space. As always, promising new beginnings have to overcome challenges. While our results demonstrated that DMS will be crucial to improve variant effect prediction methods, data diversity hindered simplification and generalization.
Keywords: Sequence variation, Variant effect prediction, Deep mutational scanning, Non-synonymous sequence variant, Missense variant, Single nucleotide variant
Background
Recent human sequencing projects conclude that we all carry about 10,000 single amino acid variants (SAVs; also referred to as missense mutations, or non-synonymous Single Nucleotide Variants: nsSNVs) with respect to the “reference genome” and by 20,000 for every pair of unrelated individuals [1, 2]. Many of these SAVs are assumed to be neutral, while others might change protein function, contributing to complex phenotypes and causing diseases. Unfortunately, the gap between SAVs with and without experimental characterization continues to widen [3]: for only one in 10,000 of the known SAVs some experimental information is available [4, 5]. On top, many of those for which something is known may be incorrect disease associations [6]. Without improving the ability to interpret SAV effects, both on the level of the organism and the protein, the promise of precision medicine will remain, importantly unmet [7–10].
Through the increased efficiency of sequencing, a procedure formerly used primarily in silico [11, 12] has become feasible for experiments, namely assessing the effect of all possible SAVs in a protein, i.e. all possible amino acid mutations. In such deep mutational scanning (DMS) studies [13, 14], a sequence library with all possible variants is subjected to selection. In the simplest case, the (logarithmic) difference between sequence frequencies with and without selection pressure yield an effect score for individual or combinations of variants [8, 15–17]. Variants with beneficial and deleterious effect on protein function are discovered together with a quantification of how much effect. Thus, DMS aims at measuring the landscape of functional fitness for select proteins [18].
DMS also screens proteins for improved drug binding, antibody affinity, using non-native chemical stresses, or non-proteinogenic amino acids, and on synthetic proteins [19–26]. Finally, DMS share objectives with directed evolution, benefiting protein engineering [14].
One major challenge for DMS is the development of an assay to measure effect. Evaluating proteins with multiple functions requires multiple assays [8]. For instance, for the Ubiquitin-60S ribosomal protein L40 variant effects have been assessed through their direct impact on yeast growth and through the impaired activation by the E1 enzyme [27, 28]. Similarly, BRCA1 has been assayed through E3 ubiquitin ligase activity and through BARD1 binding and transcript abundance [29, 30]. Even for the same assay, specific experimental conditions might influence measurements [31]. Recently, a protocol for measuring protein abundance has been suggested as a proxy for function and applicable to many proteins [32]. The conclusions from DMS studies are limited by the validity of their functional assays; inferences of more complex effect relationships such as disease risk or clinically actionable pathogenicity often remain too speculative [8, 17]. On top, variants might affect molecular function as assayed by DMS although being clinically benign, i.e. not causing disease.
Long before experimental DMS, prediction methods had addressed the same task in silico [33–41]. These methods were developed on very limited data; many focused on disease-causing SAVs from OMIM [42], others used databases such as PMD [43] cataloguing variants by effect upon protein function or structure. CADD solved the problems of data limitation and bias by considering all mutations that have become fixed in the human population as neutral and a simulated set of all other variants as having an effect [35]. The training dataset determines the type of effect methods can learn. Consequently, methods differ and work only on the type of SAV used for development. Given the limitations in today’s data, all methods have been optimized on relatively small, unrepresentative subsets: fewer than 85,000 of all possible 217 million human SAVs (< 0.04%) have some experimental annotations [44, 45]. Methods agree much more with each other for SAVs with than for those without annotations [46].
DMS datasets constitute a uniquely valuable resource for the evaluation of current SAV effect prediction methods [17, 47, 48], not the least, because most have not used those data. The Fowler lab has, recently, published an excellent analysis of prediction methods on DMS datasets and developed a new regression-based prediction method, Envision, trained only on DMS data [49]. Here, we focus on the analysis of a larger set of DMS studies and present trends in their correlation with SAV effects predicted by four variant effect prediction methods.
Results
DMS studies not complete yet
Our Deep Mutational Scanning (DMS) analyses began with 22 separate experimental datasets from 18 unique proteins, since some experiments were performed on the same protein (Supplementary Online Material (SOM), Fig. S1a, Table S1) [29, 30, 32, 50–65]. In total the set contained 68,447 variants (Fig. S1); 2358 (3%) of these were synonymous, the other 97% constituted SAVs (or missense mutations).
Only ten of the 22 sets (45%) scored some variants for at least 98% of the residues (Table S1). Four DMS studies provided functional scores for over 90% of all possible 19 non-native SAVs. On average, 66% of the residues had SAVs with both deleterious and beneficial effects (Table S2; those two could be seen as “disruptive variants” arching over gain- and loss-of-function). Most SAVs were beneficial for only 3 of 22 studies (14%), for the other 19 studies deleterious outnumbered beneficial SAVs by factors of 1.5–22.5 (Fig. S1b). Due to asymmetries in numbers and experimental fidelity, deleterious and beneficial SAVs were analyzed separately.
Some correlation achieved by all methods
SetCommon constituted a subset of all 22 datasets with 32,981 effect SAVs (17,781 deleterious) for which we had predictions from each method (Table 1). Although all predictions differed from the experiments, all correlated slightly positively for deleterious SAVs (Spearman ρ ≥ 0.1, Fig. 1a-c, Tables 2, S3). The 95% confidence intervals (CIs) of methods did not overlap, and their differences were statistically significant (Table S4).
Table 1.
Number of SAVs in aggregated datasetsa
| Number of SAVs | ||||
|---|---|---|---|---|
| Total | Neutral | Deleterious | Beneficial | |
| SetAll | 66,089 | 818 b | 45,382 | 19,889 |
| SetCommon | 32,981 | 0 | 17,781 | 15,200 |
| SetCommonSyn90 | 15,621 | 8926 | 4545 | 2150 |
| SetCommonSyn95 | 15,621 | 10,587 | 3209 | 1825 |
| SetCommonSyn99 | 15,621 | 13,506 | 1548 | 567 |
aSetAll depicts the total number of SAVs collected, while SetCommon contains only SAVs with predictions from every analyzed method. SetCommonSyn contains all SAVs with predictions where a thresholding scheme could be applied to yield classification of SAVs into neutral and effect (see Methods). The number of SAVs in every single DMS experiment are depicted in Fig. S1 and Table S1
bThe ccdB set classifies variant effect in categories and contains 818 non-synonymous variants which fall in the same category as the wild-type. Hence these SAVs could be considered neutral
Fig. 1.
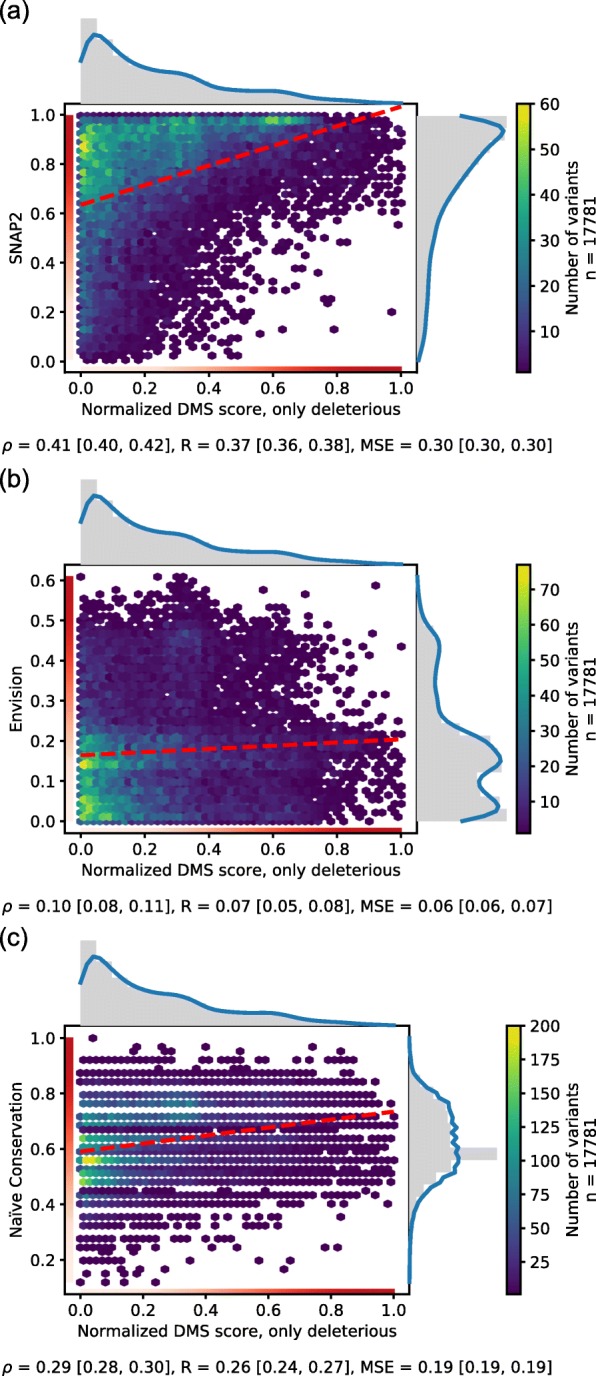
DMS experiments vs. variant effect predictions. In a hexbin plot, 17,781 deleterious effect SAVs in SetCommon were compared to normalized scores for three prediction methods (SNAP2 [38], Envision [49], and Naïve Conservation). Values on both axes range from 0 (neutral) to 1 (maximal effect) as denoted by the gradient from white (neutral) to red (effect). Dashed red lines give linear least-squared regressions. Marginals denote distributions of experimental and predicted scores with a kernel density estimation overlaid in blue. The footer denotes Spearman ρ, Pearson R and the mean squared error together with the respective 95% confidence intervals. The method scores are given on the y-axes and reveal the method: a SNAP2, b Envision – the only method trained on DMS data, c Naïve Conservation read off PSI-BLAST profiles
Table 2.
Pearson ρ and mean squared error (MSE) for methods on SetCommona
| deleterious SAVs (n = 17,781) | beneficial SAVs (n = 15,200) | |||
|---|---|---|---|---|
| ρ | MSE | ρ | MSE | |
| SNAP2 | 0.41 [0.40, 0.42] | 0.3 [0.30, 0.30] | 0.02 [0.01, 0.04] | 0.23 [0.23, 0.24] |
| Envision | 0.1 [0.08, 0.11] | 0.06 [0.06, 0.07] | −0.14 [−0.16, − 0.13] | 0.05 [0.04, 0.05] |
| Naïve Conservation | 0.29 [0.27, 0.30] | 0.19 [0.19, 0.19] | −0.08 [− 0.09, − 0.06] | 0.19 [0.19, 0.20] |
aSetCommon denotes the set of SAVs with predictions from every method (see Methods). ρ denotes Spearman ρ (higher is better), MSE the mean squared error (lower is better, Methods, SOM_Note3). Values in brackets are 95% confidence intervals
Both SIFT [39] and PolyPhen-2 [37] are optimized for capturing binary effects, not correlations, as confirmed by recent studies [47, 49]. Consequently, analysis for these was confined to binary predictions. SNAP2 [38] and Envision [49] scores appeared, overall, less binary (Figs. 1a-b). SNAP2 distributions were skewed toward high effect, while Envision also succeeded in detecting SAVs with less pronounced effects (Fig. 1a-b). Predictions by Naïve Conservation, based on PSI-BLAST profiles, correlated more with the DMS experiments than Envision (Fig. 1c).
Envision might approximate experimental values best
When evaluating methods by the numerical difference between experimental and predicted variant effect scores (mean squared error, MSE), Envision appeared best, followed at considerable distance by Naïve Conservation and SNAP2 (Fig. 1, Table 2). However, its low MSE partially originated from predicting no SAV with strong effect (the highest Envision score was 61% of the possible maximum – 0.61). This resembled the experimental distribution skewed towards low effect (Fig. 1b, gray distributions next to x- and y-axes). Indeed, shuffling the prediction scores yielded the same MSE (Fig. S2a). Predicting a normal distribution around the experimental mean, performed slightly worse but still better than all other prediction methods (Fig. S2b). When considering each DMS measurement separately, Envision also appeared to perform best except for the transcriptional coactivator YAP1 (YAP1) with the most uniform distribution of effect scores (similar number of lowest, medium, and strongest effects observed; Fig. S3b, Table S5).
All classification methods detect increasing effect strength
Do methods work better for SAVs with stronger observed effect? Toward this end, the experimental scores were sorted into 20 bins of increasing effect strength, and the effect predictions in each bin (here referred to as recall) were monitored for all prediction methods. All classification methods tended to reach higher recall levels for SAVs with stronger effects (Fig. 2a, higher values toward the right). Furthermore, all methods also show an increase without a clear saturation point showing that the range of increasing effect strength is detected. For some methods the difference between the least- and most-effect bins was higher than for others, i.e. their predictions distinguished more between high and low experimental scores (Fig. 2b).
Fig. 2.
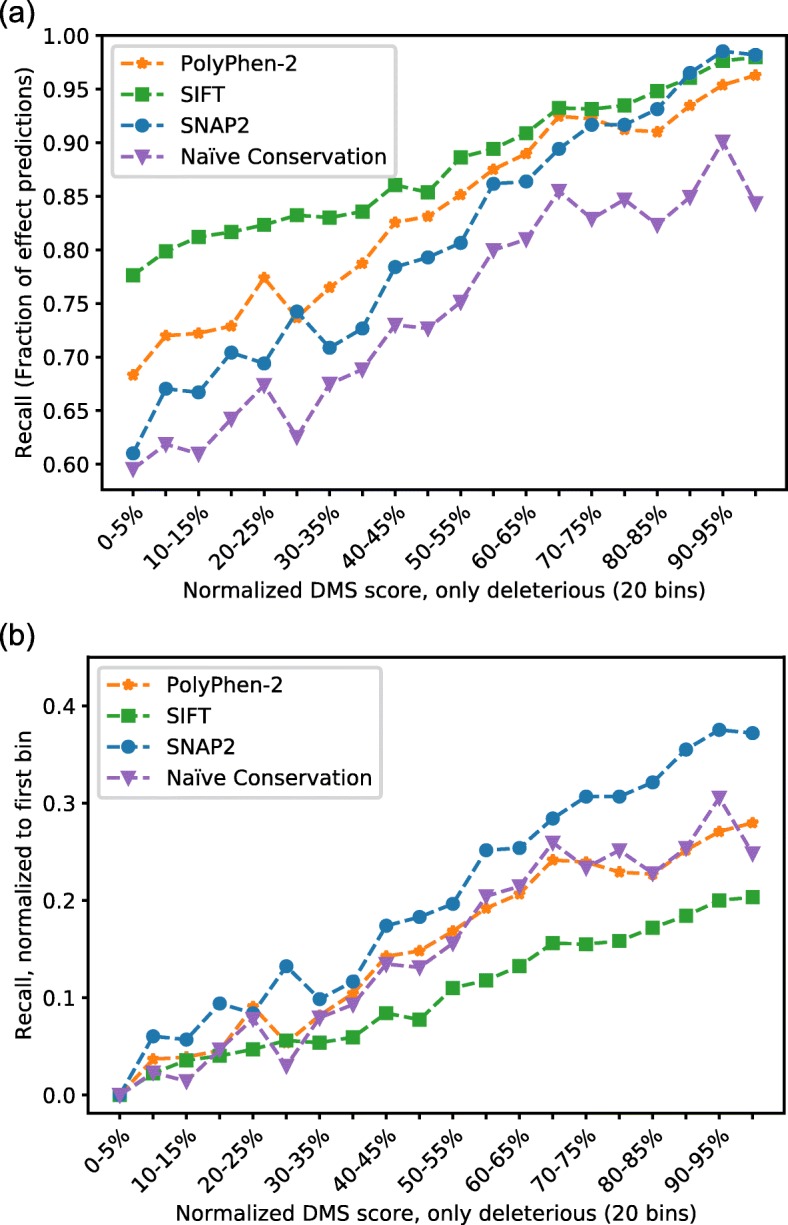
Recall proportional to deleterious DMS effect scores. The continuous normalized DMS scores with deleterious effect in SetCommon were split into 20 bins of equal size. a In each bin the fraction of SAVs predicted as having an effect by the binary classification methods (PolyPhen-2 [37], SIFT [39] and SNAP2 [38]) was shown. Naïve Conservation read off PSI-BLAST profiles was treated as an effect prediction when scores were above 0. For all other methods the default score thresholds were applied. b shows the values adjusted for the amount of effect predicted in the first bin
Beneficial effects difficult to predict
Unlike for deleterious SAVs, no method correlated, on average, with beneficial effect SAVs (− 0.14 ≤ ρ ≤ 0.02, Tables 2, S6, Fig. S4). Furthermore, most methods essentially predicted similar numbers or lower numbers of effect variants irrespective of the observed effect strength with the exception of SNAP2 that detected some high effect SAVs (Fig. S5). The conservation-based prediction also decreased substantially from a Spearman ρ of 0.29 for deleterious to − 0.08 for beneficial SAVs (Table 2, Fig. S4c). SNAP2 scores were shifted more toward lower effect than for deleterious SAVs (Fig. 1a and Fig. S4a, gray distributions). In contrast to Spearman ρ, the MSE for beneficial effect SAVs was similar to that for deleterious SAVs. Envision again was by far best (MSE = 0.05, Tables 2, S7, Fig. S6). However, although Envision used 25% beneficial effect SAVs for development (SOM_Note1), the correlation was much lower for beneficial than for deleterious SAVs (ρ = − 0.14 versus 0.1).
Experimental agreement sets the benchmark for prediction methods
The above comparisons of experimental and predicted SAV effects raise the question of what agreement can realistically be obtained. One proxy for an answer is the comparison of different DMS studies conducted on the same protein. Such data were available for 11 measurements on 4 proteins (Table S8, Fig. S7); unfortunately, Envision predictions were available for only one of those proteins (BRCA1). For deleterious SAVs, the lowest correlation was that between two measurements on breast cancer type 1 susceptibility protein, BRCA1 and BRCA1_2015_E3 (ρ = 0.21, Fig. S7b). Rather than experimental noise, the low correlation might also originate from different experimental setups employed for multi-functional proteins such as BRCA1. The strong correlation (ρ = 0.93) between two experiments that measured the same condition for bla (beta-lactamase TEM precursor; bla and bla_2014, Fig. S7h) provided a single case in strong support of such an explanation. To compare prediction methods and experiments, we assessed the difference in ρ and MSE for each combination of the 11 measurements (Fig. 3). Experiments clearly agreed more with each other than with SNAP2 and Naïve Conservation on the same datasets (Fig. 3: all values negative).
Fig. 3.
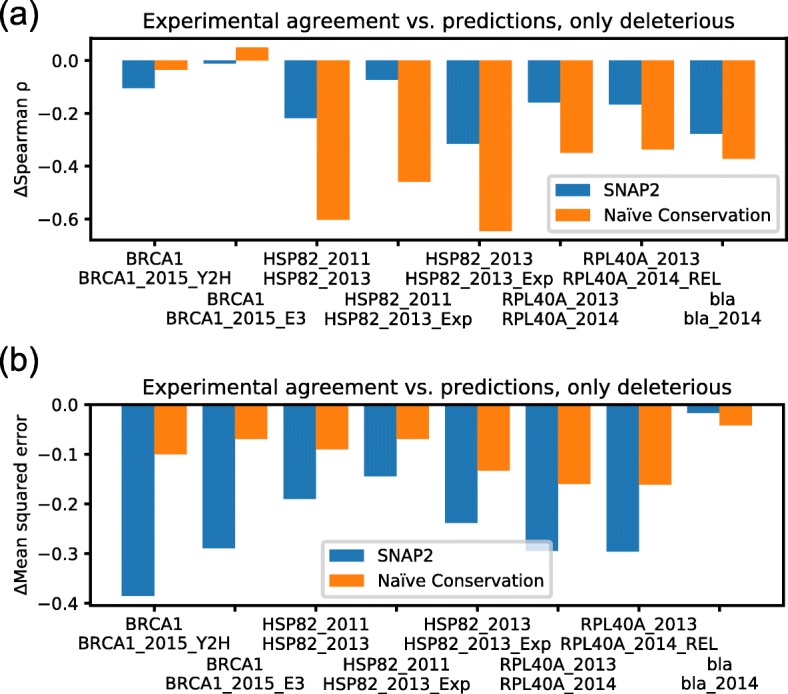
Experimental agreement vs. predictions. For every pair of experimental measurements on the same protein (Table S1), the agreement between two experiments and that between each experiment and the predictions of SNAP2 and Naïve Conservation are compared. a ∆ρ = 0.5*(ρ(× 1,p1) + ρ(× 2,p2)) - ρ(× 1,× 2), (b) ∆MSE = MSE(× 1,× 2) - 0.5*(MSE(× 1,p1) + MSE(× 2,p2)). Where × 1/× 2 are the experiments and p1/p2 the predictions on the two experiments, all of which are calculated based on the largest possible set of SAVs. Negative values on the y-axes thus imply that the agreement between experiments is higher than that between experiment and prediction, positive values that predictions agree more
Experiments did not correlate at all with each other for beneficial effect (mean ρ = 0.03) although the MSE remained low (mean MSE = 0.05, Table S8, Fig. S8). The major issue for this comparison was the small number of only 572 SAVs.
Assessment of binary classification (neutral/effect) similar to regression
Scores from binary classification methods (neutral or effect) are often assessed through receiver operating characteristic (ROC) curves avoiding to choose particular thresholds to distinguish neutral and effect. Toward this end, we assigned classes to SAVs through normalization by experimental measurements of synonymous variants [60] (Methods). Other solutions are feasible, each with their own ad hoc parameter choices and flaws implying that the following results provide one snapshot instead of a sustained method ranking.
On the 3209 deleterious effect SAVs of SetCommonSyn95 (10,587 neutral, Table 1, Fig. S9), SNAP2 achieved the highest area under the curve (AUC, 0.76, 95% CI [0.75, 0.77]). It was the only method statistically significantly better than Naïve Conservation (0.73 [0.72, 0.74], Figs. 4, S10 Table S9). Precision-recall curves also highlighted the smooth transition of SNAP2 scores opposed to those for Naïve Conservation although the peak performance was similar for both (Fig. S11). Envision - not developed for this task - performed better than random, but clearly worse than the classification methods (AUC = 0.55 [0.54, 0.56]). However, the four proteins considered here (BRCA1, PPARG, PTEN and TPMT), also correlated above average for SNAP2, PolyPhen-2 and SIFT (Table S3). Using different thresholds in severity to classify SAVs did not qualitatively change these major findings (SetCommonSyn90, SetCommonSyn99, Fig. S12a-b).
Fig. 4.
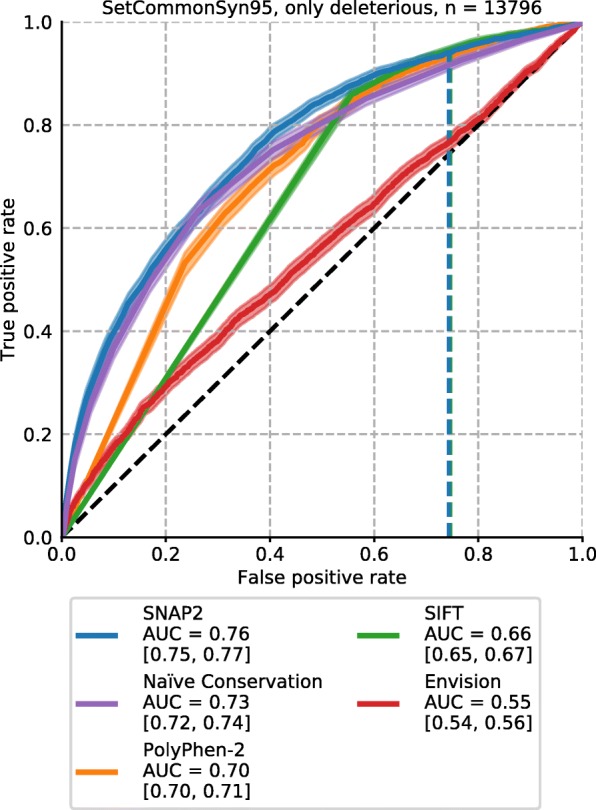
Classification performance of all prediction methods. Shown are ROC curves for 13,796 deleterious effect SAVs which were classified into either neutral, defined by the middle 95% of the scores from synonymous variants, or effect (SetCommonSyn95). Shaded areas around lines denote 95% confidence intervals. The legend denotes the AUC for each of the five prediction methods, along with the 95% confidence intervals. Horizontal dashed lines denote the default score threshold used by SNAP2 (blue) and SIFT (green)
At their default thresholds SIFT, PolyPhen-2, and SNAP2 consider over two thirds of the neutral variants to have an effect. Interestingly, the behavior of Envision trained on DMS data was the reverse as previously illustrated by the maximal scores reaching only up to 61% of the possible maximal values (and thereby contributing to a seemingly low MSE).
Beneficial SAVs were also difficult to classify: PolyPhen-2 and SNAP2 performed best with AUC = 0.62, followed by SIFT, while Envision predictions were not better than random (Fig. S13, Fig. S12c-d, Table S9). Naïve Conservation also performed significantly worse at a level of random predictions.
Discussion
No clear winner in predicting effect variants
We compared the predictions of five methods with SAV effects determined by DMS experiments. SNAP2 was trained on binary classification data (effect or neutral). Nevertheless, predictions have been shown to correlate with effect strength [5, 66, 67]. To a degree, the Deep Mutational Scanning (DMS) data replicated this finding, highlighting that even methods trained for classification capture aspects of effect strength.
Sorting DMS scores into 20 bins and including classification methods SIFT and PolyPhen-2 in the analysis, all methods indicated better recognition of high effect SAVs. This finding might be attributed to the bias of classifications methods towards high effect variants, a common criticism in the field [68–71]. We observed the same trend for Naïve Conservation exclusively using PSI-BLAST profiles to predict SAV effects. This emphasized the importance of this signal but to some extend also explained the traditional classification methods’ bias since they all rely on this input.
The significantly better performance of Envision in estimating the precise degree of effect especially suggested value in this approach. However, the low MSE was largely explained by that Envision correctly predicted the overall distribution of experimental scores. Thus, the definite distinction between ‘good prediction’ and ‘advantageous bias’ remained elusive.
When treating DMS effect scores as binary assignments (neutral or effect), ROC curves highlighted the high false positive rates of the evaluated classification methods. A similar perspective on over-prediction has recently been observed for ClinVar data [69]. Over-prediction might be encouraged by the way many users of prediction methods mistakenly chose their tools, namely by testing a small set of SAVs they know have an effect and valuing methods highest when they predict effects for more of those.
Family conservation carries most important signal
Most surprising was the overall good performance of Naïve Conservation. Disease causing SAVs from OMIM typically affect the most conserved residues [46], and machine-learning based predictions have been criticized to largely capture conservation [17, 70, 72–74]. Furthermore, simple conservation patterns can capture aspects of variant effects [75]. Our findings partially validated this for DMS experiments, although the effect distributions observed by DMS and predicted by Naïve Conservation differed substantially (Fig. 1c, gray distributions). Another recent analysis also found a method heavily relying on evolutionary information as one of the best performers on DMS data, although more sophisticated than our naïve approach [48, 76].
Beneficial effects neither correctly predicted, nor consistent between experiments
The bad correlation and classification performance of beneficial effect SAVs by all methods suggested those to have distinctly different signatures than deleterious SAVs, missed by current approaches. Generally, SAVs with neutral or beneficial effects are often not recognized well [69, 77]. In part, this is attributable to the lack of respective experimentally verified data useable for training sets. For beneficial effect variants, the rise of DMS studies could help to alleviate this problem and lead to the development of less biased methods.
Agreement between experimental studies was particularly low for beneficial effect SAVs. Maybe DMS assays are still biased towards measuring deleterious effects. These results put the seemingly poor predictions of beneficial SAVs into perspective. Generally, the wide variation of correlation between experiments for different datasets/proteins has also been observed in another recent DMS analysis [48].
Conclusions
Deep mutational scanning (DMS) studies set out to explore the relation between protein sequence and molecular function. We collected 22 DMS experiments and focused on single amino acid variants (SAVs, also referred to as missense mutations or non-synonymous SNVs). Most studies probe only a small subset of all possible variants (for a protein with N residues, there are 19*N non-native SAVs). Two experiments probing the same protein tended to agree more with each other than with predictions for deleterious effect (Fig. 3). Nonetheless, experiments also disagreed significantly (Table S8). No single measure captured all aspects of the comparison between experiments and predictions, e.g. the ranking of methods changed crucially depending on the measure used to compare (Table 2, SOM_Note2).
We analyzed five variant effect prediction methods: Envision was trained on DMS data, PolyPhen-2, SIFT and SNAP2 were methods developed to classify into effect/neutral, and Naïve Conservation (essentially using PSI-Blast conservation to predict effect/neutral) was added to gauge the importance of evolutionary conservation for the prediction. For deleterious SAVs, all methods reached slightly positive Spearman ρ correlations with the DMS experiments (Fig. 1). The classification method SNAP2 correlated most with effect strength, although most of the correlation was explained by simple conservation. The lowest mean squared error (MSE) was achieved by Envision. Its MSE was as low as that between experiments, although most of the low MSE could be explained by correctly predicting the distribution of scores (Fig. 1, Fig. S2a). All methods performed better on SAVs with deleterious (akin to loss-of-function) than with beneficial (gain-of-function) effect. However, experimental agreement was also almost non-existing for beneficial effects.
Although binary classification methods, surprisingly, captured aspects of non-binary measurements, they performed much better for the binary classification task (projecting DMS results onto neutral vs. effect; Fig. 4). Notably, Naïve Conservation captured effect better than some more advanced tools. Methods performed better for SAVs with stronger experimental effect scores (Fig. 2: higher toward right), although most classifiers tended to substantially over-predict at their default scores (Fig. 4). Overall, our analyses confirm some of the trends from other reviews of DMS data [48, 49].
The challenge for the next generation of prediction methods will be to learn from the diversity of DMS. To give just one example: OMIM, a popular source of training data, contained ~ 11,000 SAVs referenced in dbSNP (02/2019, [78]). This is a magnitude matched by a single large DMS experiment. The generality of a single SAV might not be comparable between the sets, yet DMS opens up variant effect prediction to new methodologies, possibly even to deep learning approaches [79, 80]. The enriched data might also allow methods to distinguish between toggle and rheostat positions [73]. Furthermore, DMS studies contain many beneficial effect SAVs that have, so far, been underrepresented. Finally, DMS focuses on molecular function, i.e. some of the disruptive SAVs (deleterious or beneficial) might correspond to clinically benign SAVs. Nevertheless, DMS will likely give rise to new methods better predicting SAV effects upon molecular protein function and upon organisms. In fact, growth-based DMS assays have been shown to be predictive of human disease SAVs in a recent study [48]. Therefore, a combination of experimental data with new prediction methods might be what is needed to attain the goals of precision medicine.
Methods
Dataset collection
Figure 5 sketches the basic workflow of this analysis. We retrieved all DMS datasets available by June 2019 that report over 100 SAVs available from the literature. Functional effect scores were taken directly from the supplemental material published or requested from the authors (Table S10). The data were formatted in a variety of formats including Excel, and tab- or comma-separated files. Scores were manually mapped either to the UniProtKB identifier given in the publication or to its closest BLAST match (Table S11) [44, 81]. Six of the 22 experiments contained up to five substitutions (pairwise sequence identity ≥98%); those were maintained for prediction. We refer to the combined data as SetAll (66,089 SAVs) supplemented by SetCommon with 32,981 SAVs for which we had a prediction from every method tested (Table 1). SetCommon contained SAVs from ten of the 22 experiments: YAP1, MAPK1, BRCA1, CCR5, CXCR4, GAL4, PPARG, PTEN, TPMT, and Ube4b (Table S1). During completion of this manuscript, MaveDB, a centralized resource of multiplexed assays of variant effect has been published [8, 82]. MaveDB identifiers exist for ten of our 22 datasets (November 2019, Table S10).
Fig. 5.

Concept of analysis. Experimental scores of variant effects (missense mutations, or single amino acid variants, labelled SAVs) from Deep Mutational Scanning (DMS) experiments were compared to in silico prediction methods. Envision was the only method developed on DMS data; it provides continuous scores mirroring the DMS data. SIFT, PolyPhen-2 can be evaluated as binary classification methods. SNAP2 is a classification method but provides continuous scores that can also be used. Naïve Conservation is provided as a baseline for both cases
SetAll contained several proteins with multiple independent experimental measurements. Inclusion of additional sets analyzed previously [49], yielded a total of three measurements for Hsp82 and BRCA1 and two for both beta-lactamase and ubiquitin (Table S1) [27, 28, 83]. Performance measures were calculated only on SAVs and not between DMS measurements from the same publication. For analysis of beneficial effect SAVs, all studies on Hsp82 had to be excluded since the sets contain only three of those SAVs each.
Processing functional effect scores
Several DMS studies provide multiple effect scores for the same protein of which we decided on only one per set (Table S12). In the following processing, effect scores were left as provided by the authors as much as possible but adjusted such that the wild-type score for each measurement (Table S13) became 0, and larger values denoted more effect. Next, scores were interpolated, separately for each of the 22 DMS measurements, to lie between 0 and 1 (highest effect). This interpolation did not affect Spearman ρ or the mean squared error within each dataset. Beneficial and deleterious effects had to be analyzed separately because experimental assays were not symmetrical and further normalization might over- or underrepresent effects. The resulting score distributions differed significantly between experiments (e.g. in contrast to the more homogeneous subset used previously [49]).
We also created sets with binary classifications (effect vs. neutral) from all DMS studies with synonymous variants: The middle 95% of effect score values from synonymous variants was used to define which SAVs were considered neutral. All SAVs outside this range were considered as effect. We applied the same procedure using 90% or 99% of synonymous variants’ values and refer to the thresholding schemes as syn90, syn95, and syn99. Applying these schemes to the four experiments in SetCommon which have synonymous variants (BRCA1, PTEN, TPMT, PPARG) yields SetCommonSyn90|95|99. Again, deleterious and beneficial effect SAVs were analyzed separately.
Performance measures
Experiments and predictions were compared through three measures (SOM_Note3, SOM_Note2): (1) mean squared error (MSE) calculated with the scikit-learn metrics module [84]; (2) Pearson R (pearsonr) and (3) Spearman ρ (spearmanr) both calculated with the SciPy stats module [85]. For convenience linear least-squares regression lines (linregress) were added to the correlation plots. Pearson R was added for ease of comparison to others but not discussed as it is not robust and most datasets violated both its validity assumptions (normal distribution & absence of significant outliers [86]). We further found no evidence to supplement MSE by a measure more robust to outliers (SOM_Note2). 95% confidence intervals (CIs) for R, ρ and MSE were estimated using a percentile bootstrap with 1000 random samples with replacement.
The performance of binary predictions (effect vs. neutral) was measured through receiver operating characteristic (ROC) curves and the area under those curves (AUC) calculated through the pROC package in R, which was also used to calculate 95% confidence intervals of ROC (ci.se) and AUC (ci.auc) [87, 88]. Additionally, precision-recall curves were created using scikit-learn (precision-recall-curve). These are defined with TP as true positives (predicted and observed as effect), FP as false positives (predicted as effect, observed as neutral), and FN as false negatives (predicted neutral, observed effect): Precision = TP/(TP + FP), Recall = True Positive Rate = TP/(TP + FN) and False Positive Rate = FP/(FP + TN).
Prediction methods
The sequences determined during dataset collection were used as input to a set of commonly used variant effect prediction methods. Each method was run to predict the effect of all 19 non-native amino acids at every position in the protein. SNAP2 [38] was run locally using default parameters on UniProtKB (Release 2018_09). SIFT version 6.2.1 [39] was run locally (UniProtKB/TrEMBL Release 2018_10). PolyPhen-2 [37] predictions were retrieved from the webserver in batch mode with classification model humdiv on genome assembly GRCh37/hg19 and default parameters [89]. Predictions failed for all relevant residues of the three DMS studies on Hsp82. Envision [49] predictions were retrieved online which requires UniProtKB identifiers as input [90]. Therefore, Envision predictions could be analyzed only for ten proteins (Table S14). While SNAP2 and SIFT predicted all SAVs, PolyPhen-2 and Envision failed for some residues, shrinking the size of the datasets. We always report performance on the largest common subset of SAVs per dataset.
As a baseline, predictions were also created by running PSI-BLAST with three iterations on UniProtKB (Release 2018_09). Scores from the resulting profile (position-specific scoring matrix) had their signs flipped and were then directly used as a measure of effect, i.e. less frequent substitutions have a higher effect than conserved ones. We refer to this method as Naïve Conservation. The prediction was not intended to be the most accurate conservation score possible but rather to represent a suitable baseline since (PSI-)BLAST results are used in some way as input feature by all methods analyzed here.
For SIFT, scores were reversed such that higher values implied higher effect. The same was done for Envision predictions of deleterious effect. Envision predictions of beneficial effect were treated separately and mapped to the range of [0,0.2]. This yielded the same performance than scaling between [0,1] or no scaling (SOM_Note4). Finally, prediction scores of all methods were adjusted to lie between 0 (no effect) and 1 (highest effect) using the theoretical maximum and minimum prediction value of every method.
Supplementary information
Additional file 1. Supporting Online Material (SOM) containing additional figures, tables and notes.
Acknowledgements
The authors wish to thank all groups that work on DMS and readily provided their data, either as part of their manuscript or swiftly upon personal contact. Thanks also to Michael Bernhofer and Maria Littmann (both TUM) for helpful discussions, to Inga Weise (TUM) for administrative support and to Tim Karl (TUM) for help with hard- and software. Particular thanks to the anonymous reviewers who helped importantly to clarify the path through the data plethora.
Abbreviations
- AUC
Area under the ROC curve
- CI
Confidence interval
- DMS
Deep mutational scanning
- MSE
Mean squared error
- ROC
Receiver operating characteristic
- SAV
Single amino acid variant
Authors’ contributions
TW performed initial analyses and data curation on a smaller set of datasets. JR collected additional datasets, improved on the methodology and expanded the analyses. JR and BR wrote the manuscript. BR conceptualized and supervised the work. All authors read and approved the final manuscript.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) – project number 640508. This work was also supported by the DFG and the Technical University of Munich (TUM) in the framework of the Open Access Publishing Program. Further funding was provided by the Bavarian Ministry for Education through funding to the TUM paying for the positions of the authors. The funding bodies did not play any role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials
The datasets generated and code for their analysis are available on Mendeley Data (10.17632/2rwrkp7mfk.1).
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12859-020-3439-4.
References
- 1.Tennessen Jacob A., Bigham Abigail W., O’Connor Timothy D., Fu Wenqing, Kenny Eimear E., Gravel Simon, McGee Sean, Do Ron, Liu Xiaoming, Jun Goo, Kang Hyun Min, Jordan Daniel, Leal Suzanne M., Gabriel Stacey, Rieder Mark J., Abecasis Goncalo, Altshuler David, Nickerson Deborah A., Boerwinkle Eric, Sunyaev Shamil, Bustamante Carlos D., Bamshad Michael J., Akey Joshua M. Evolution and Functional Impact of Rare Coding Variation from Deep Sequencing of Human Exomes. Science. 2012;337(6090):64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.The 1000 Genomes Project Consortium A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manolio TA, Fowler DM, Starita LM, Haendel MA, MacArthur DG, Biesecker LG, Worthey E, Chisholm RL, Green ED, Jacob HJ, et al. Bedside Back to bench: building bridges between basic and clinical genomic research. Cell. 2017;169:6–12. doi: 10.1016/j.cell.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Beer TAP, Laskowski RA, Parks SL, Sipos B, Goldman N, Thornton JM. Amino acid changes in disease-associated variants differ radically from variants observed in the 1000 genomes project dataset. PLoS Comput Biol. 2013;9. [DOI] [PMC free article] [PubMed]
- 5.Mahlich Y, Reeb J, Hecht M, Schelling M, De Beer TAP, Bromberg Y, Rost B. Common sequence variants affect molecular function more than rare variants? Sci Rep. 2017;7:1608. doi: 10.1038/s41598-017-01054-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rost B, Radivojac P, Bromberg Y. Protein function in precision medicine: deep understanding with machine learning. FEBS Lett. 2016;590:2327–2341. doi: 10.1002/1873-3468.12307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Starita LM, Ahituv N, Dunham MJ, Kitzman JO, Roth FP, Seelig G, Shendure J, Fowler DM. Variant interpretation: functional assays to the rescue. Am J Hum Genet. 2017;101:315–325. doi: 10.1016/j.ajhg.2017.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Capriotti E, Ozturk K, Carter H. Integrating molecular networks with genetic variant interpretation for precision medicine. Wiley Interdiscip Rev Syst Biol Med. 2019;11(3):e1443. doi: 10.1002/wsbm.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Daneshjou R, Wang Y, Bromberg Y, Bovo S, Martelli PL, Babbi G, Lena PD, Casadio R, Edwards M, Gifford D, et al. Working toward precision medicine: predicting phenotypes from exomes in the critical assessment of genome interpretation (CAGI) challenges. Hum Mutat. 2017;38:1182–1192. doi: 10.1002/humu.23280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bromberg Y, Yachdav G, Rost B. SNAP predicts effect of mutations on protein function. Bioinformatics (Oxford, England) 2008;24:2397–2398. doi: 10.1093/bioinformatics/btn435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hecht M, Bromberg Y, Rost B. News from the protein mutability landscape. J Mol Biol. 2013;425:3937–3948. doi: 10.1016/j.jmb.2013.07.028. [DOI] [PubMed] [Google Scholar]
- 13.Hietpas R, Roscoe B, Jiang L, Bolon DNA. Fitness analyses of all possible point mutations for regions of genes in yeast. Nat Protoc. 2012;7:1382–1396. doi: 10.1038/nprot.2012.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wrenbeck EE, Faber MS, Whitehead TA. Deep sequencing methods for protein engineering and design. Curr Opin Struct Biol. 2017;45:36–44. doi: 10.1016/j.sbi.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Araya Carlos L., Fowler Douglas M. Deep mutational scanning: assessing protein function on a massive scale. Trends in Biotechnology. 2011;29(9):435–442. doi: 10.1016/j.tibtech.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fowler DM, Stephany JJ, Fields S. Measuring the activity of protein variants on a large scale using deep mutational scanning. Nat Protoc. 2014;9:2267–2284. doi: 10.1038/nprot.2014.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fowler DM, Fields S. Deep mutational scanning: a new style of protein science. Nat Methods. 2014;11:801–807. doi: 10.1038/nmeth.3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eyre-Walker A, Keightley PD. The distribution of fitness effects of new mutations. Nat Rev Genet. 2007;8:610–618. doi: 10.1038/nrg2146. [DOI] [PubMed] [Google Scholar]
- 19.Forsyth CM, Juan V, Akamatsu Y, DuBridge RB, Doan M, Ivanov AV, Zhiyuan M, Polakoff D, Razo J, Wilson K, et al. Deep mutational scanning of an antibody against epidermal growth factor receptor using mammalian cell display and massively parallel pyrosequencing. MAbs. 2013;5:523–532. doi: 10.4161/mabs.24979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mavor D, Barlow K, Thompson S, Barad BA, Bonny AR, Cario CL, Gaskins G, Liu Z, Deming L, Axen SD, et al. Determination of ubiquitin fitness landscapes under different chemical stresses in a classroom setting. eLife. 2016;5:1–23. doi: 10.7554/eLife.15802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mavor D, Barlow KA, Asarnow D, Birman Y, Britain D, Chen W, Green EM, Kenner LR, Mensa B, Morinishi LS, et al. Extending chemical perturbations of the ubiquitin fitness landscape in a classroom setting reveals new constraints on sequence tolerance. Biology Open. 2018;7:bio036103. doi: 10.1242/bio.036103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tinberg CE, Khare SD, Dou J, Doyle L, Nelson JW, Schena A, Jankowski W, Kalodimos CG, Johnsson K, Stoddard BL, et al. Computational design of ligand-binding proteins with high affinity and selectivity. Nature. 2013;501:212–216. doi: 10.1038/nature12443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Procko E, Hedman R, Hamilton K, Seetharaman J, Fleishman SJ, Su M, Aramini J, Kornhaber G, Hunt JF, Tong L, et al. Computational design of a protein-based enzyme inhibitor. J Mol Biol. 2013;425:3563–3575. doi: 10.1016/j.jmb.2013.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Whitehead TA, Chevalier A, Song Y, Dreyfus C, Fleishman SJ, De Mattos C, Myers CA, Kamisetty H, Blair P, Wilson IA, et al. Optimization of affinity, specificity and function of designed influenza inhibitors using deep sequencing. Nat Biotechnol. 2012;30:543–548. doi: 10.1038/nbt.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fujino Y, Fujita R, Wada K, Fujishige K, Kanamori T, Hunt L, Shimizu Y, Ueda T. Robust in vitro affinity maturation strategy based on interface-focused high-throughput mutational scanning. Biochem Biophys Res Commun. 2012;428:395–400. doi: 10.1016/j.bbrc.2012.10.066. [DOI] [PubMed] [Google Scholar]
- 26.Rogers JM, Passioura T, Suga H. Nonproteinogenic deep mutational scanning of linear and cyclic peptides. Proc Natl Acad Sci. 2018;115:201809901. doi: 10.1073/pnas.1809901115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roscoe BP, Thayer KM, Zeldovich KB, Fushman D, Bolon DNA. Analyses of the effects of all ubiquitin point mutants on yeast growth rate. J Mol Biol. 2013;425:1363–1377. doi: 10.1016/j.jmb.2013.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roscoe BP, Bolon DNA. Systematic exploration of ubiquitin sequence, E1 activation efficiency, and experimental fitness in yeast. J Mol Biol. 2014;18:1199–1216. doi: 10.1016/j.jmb.2014.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Starita LM, Young DL, Islam M, Kitzman JO, Gullingsrud J, Hause RJ, Fowler DM, Parvin JD, Shendure J, Fields S. Massively parallel functional analysis of BRCA1 RING domain variants. Genetics. 2015;200:413–422. doi: 10.1534/genetics.115.175802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Findlay Gregory M., Daza Riza M., Martin Beth, Zhang Melissa D., Leith Anh P., Gasperini Molly, Janizek Joseph D., Huang Xingfan, Starita Lea M., Shendure Jay. Accurate classification of BRCA1 variants with saturation genome editing. Nature. 2018;562(7726):217–222. doi: 10.1038/s41586-018-0461-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Melnikov A, Rogov P, Wang L, Gnirke A, Mikkelsen TS. Comprehensive mutational scanning of a kinase in vivo reveals substrate-dependent fitness landscapes. Nucleic Acids Res. 2014;42:1–8. doi: 10.1093/nar/gku511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matreyek KA, Starita LM, Stephany JJ, Martin B, Chiasson MA, Gray VE, Kircher M, Khechaduri A, Dines JN, Hause RJ, et al. Multiplex assessment of protein variant abundance by massively parallel sequencing. Nat Genet. 2018;50:874–882. doi: 10.1038/s41588-018-0122-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Calabrese R, Capriotti E, Fariselli P, Martelli PL, Casadio R. Functional annotations improve the predictive score of human disease-related mutations in proteins. Human Mutat. 2009;30:1237–1244. doi: 10.1002/humu.21047. [DOI] [PubMed] [Google Scholar]
- 34.Capriotti E, Calabrese R, Fariselli P, Martelli PL, Altman RB, Casadio R. WS-SNPs&GO: a web server for predicting the deleterious effect of human protein variants using functional annotation. BMC genomics. 2013;14(Suppl 3). [DOI] [PMC free article] [PubMed]
- 35.Rentzsch Philipp, Witten Daniela, Cooper Gregory M, Shendure Jay, Kircher Martin. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Research. 2018;47(D1):D886–D894. doi: 10.1093/nar/gky1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yates CM, Filippis I, Kelley LA, Sternberg MJE. SuSPect: Enhanced prediction of single amino acid variant (SAV) phenotype using network features. J Mol Biol. 2014;426:2692–2701. doi: 10.1016/j.jmb.2014.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adzhubei Ivan A, Schmidt Steffen, Peshkin Leonid, Ramensky Vasily E, Gerasimova Anna, Bork Peer, Kondrashov Alexey S, Sunyaev Shamil R. A method and server for predicting damaging missense mutations. Nature Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hecht M, Bromberg Y, Rost B. Better prediction of functional effects for sequence variants. BMC Genomics. 2015;16:S1. doi: 10.1186/1471-2164-16-S8-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012;40:452–457. doi: 10.1093/nar/gks539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, Musolf A, Li Q, Holzinger E, Karyadi D, et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am J Hum Genet. 2016;99:877–885. doi: 10.1016/j.ajhg.2016.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carter H, Douville C, Stenson PD, Cooper DN, Karchin R. Identifying Mendelian disease genes with the variant effect scoring tool. BMC genomics. 2013;14(Suppl 3):S3. doi: 10.1186/1471-2164-14-S3-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Amberger JS, Bocchini CA, Scott AF, Hamosh A. OMIM.org: leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res. 2019;47:D1038–D1043. doi: 10.1093/nar/gky1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kawabata T, Ota M, Nishikawa K. The protein mutant database. Nucleic Acids Res. 1999;27:355–357. doi: 10.1093/nar/27.1.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Consortium TU. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47:D506–D515. doi: 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44:D862–D868. doi: 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reeb J, Hecht M, Mahlich Y, Bromberg Y, Rost B. Predicted molecular effects of sequence variants link to system level of disease. PLoS Comput Biol. 2016;12:e1005047. doi: 10.1371/journal.pcbi.1005047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pejaver V, Babbi G, Casadio R, Folkman L, Katsonis P, Kundu K, Lichtarge O, Martelli PL, Miller M, Moult J, et al. Assessment of methods for predicting the effects of PTEN and TPMT protein variants. Human Mutat. 2019;40:1495–1506. doi: 10.1002/humu.23838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Livesay B, Marsh JA. Using deep mutational scanning data to benchmark computational phenotype predictors and identify pathogenic missense mutations. bioRxiv. 2019.
- 49.Gray VE, Hause RJ, Luebeck J, Shendure J, Fowler DM. Quantitative Missense Variant Effect Prediction Using Large-Scale Mutagenesis Data. Cell Systems. 2018;6:116–124. doi: 10.1016/j.cels.2017.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adkar BV, Tripathi A, Sahoo A, Bajaj K, Goswami D, Chakrabarti P, Swarnkar MK, Gokhale RS, Varadarajan R. Protein model discrimination using mutational sensitivity derived from deep sequencing. Structure. 2012;20:371–381. doi: 10.1016/j.str.2011.11.021. [DOI] [PubMed] [Google Scholar]
- 51.Araya CL, Fowler DM, Chen W, Muniez I, Kelly JW, Fields S. A fundamental protein property, thermodynamic stability, revealed solely from large-scale measurements of protein function. Proc Natl Acad Sci. 2012;109:16858–16863. doi: 10.1073/pnas.1209751109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brenan L, Andreev A, Cohen O, Pantel S, Kamburov A, Cacchiarelli D, Persky NS, Zhu C, Bagul M, Goetz EM, et al. Phenotypic characterization of a comprehensive set of MAPK1/ERK2 missense mutants. Cell Rep. 2016;17:1171–1183. doi: 10.1016/j.celrep.2016.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Heredia JD, Park J, Brubaker RJ, Szymanski SK, Gill KS, Procko E. Mapping interaction sites on human chemokine receptors by deep mutational scanning. J Immunol. 2018;200:3825–3839. doi: 10.4049/jimmunol.1800343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hietpas RT, Jensen JD, Bolon DNA. Experimental illumination of a fitness landscape. Proc Natl Acad Sci. 2011;108:7896–7901. doi: 10.1073/pnas.1016024108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hietpas RT, Bank C, Jensen JD, Bolon DNA. Shifting fitness landscapes in response to altered environments. Evolution. 2013;67:3512–3522. doi: 10.1111/evo.12207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jiang Li, Mishra Parul, Hietpas Ryan T., Zeldovich Konstantin B., Bolon Daniel N. A. Latent Effects of Hsp90 Mutants Revealed at Reduced Expression Levels. PLoS Genetics. 2013;9(6):e1003600. doi: 10.1371/journal.pgen.1003600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kitzman JO, Starita LM, Lo RS, Fields S, Shendure J. Massively parallel single amino acid mutagenesis. Nat Methods. 2014;44:3516–3521. doi: 10.1038/nmeth.3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Klesmith JR, Bacik JP, Michalczyk R, Whitehead TA. Comprehensive sequence-flux mapping of a Levoglucosan utilization pathway in E. coli. ACS Synth Biol. 2015;4:1235–1243. doi: 10.1021/acssynbio.5b00131. [DOI] [PubMed] [Google Scholar]
- 59.Majithia AR, Tsuda B, Agostini M, Gnanapradeepan K, Rice R, Peloso G, Patel KA, Zhang X, Broekema MF, Patterson N, et al. Prospective functional classification of all possible missense variants in PPARG. Nat Genet. 2016;48:1570–1575. doi: 10.1038/ng.3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rockah-Shmuel L, Tóth-Petróczy Á, Tawfik DS. Systematic mapping of protein mutational space by prolonged drift reveals the deleterious effects of seemingly neutral mutations. PLoS Comput Biol. 2015;11:1–28. doi: 10.1371/journal.pcbi.1004421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Romero PA, Tran TM, Abate AR. Dissecting enzyme function with microfluidic-based deep mutational scanning. Proc Natl Acad Sci. 2015;112:7159–7164. doi: 10.1073/pnas.1422285112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sarkisyan KS, Bolotin DA, Meer MV, Usmanova DR, Mishin AS, Sharonov GV, Ivankov DN, Bozhanova NG, Baranov MS, Soylemez O, et al. Local fitness landscape of the green fluorescent protein. Nature. 2016;533:397–401. doi: 10.1038/nature17995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Starita LM, Pruneda JN, Lo RS, Fowler DM, Kim HJ, Hiatt JB, Shendure J, Brzovic PS, Fields S, Klevit RE. Activity-enhancing mutations in an E3 ubiquitin ligase identified by high-throughput mutagenesis. Proc Natl Acad Sci. 2013;110:E1263–E1272. doi: 10.1073/pnas.1303309110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stiffler Michael A, Hekstra Doeke R, Ranganathan R. Evolvability as a function of purifying selection in TEM-1 β-lactamase. Cell. 2015;160:882–892. doi: 10.1016/j.cell.2015.01.035. [DOI] [PubMed] [Google Scholar]
- 65.Traxlmayr MW, Hasenhindl C, Hackl M, Stadlmayr G, Rybka JD, Borth N, Grillari J, Rüker F, Obinger C. Construction of a stability landscape of the CH3 domain of human IgG1 by combining directed evolution with high throughput sequencing. J Mol Biol. 2012;423:397–412. doi: 10.1016/j.jmb.2012.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bromberg Y, Kahn PC, Rost B. Neutral and weakly nonneutral sequence variants may define individuality. Proc Natl Acad Sci U S A. 2013;110:14255–14260. doi: 10.1073/pnas.1216613110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bromberg Y, Rost B. SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res. 2007;35:3823–3835. doi: 10.1093/nar/gkm238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–423. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Niroula A, Vihinen M. How good are pathogenicity predictors in detecting benign variants ? PLoS Comput Biol. 2019;15:1–17. doi: 10.1371/journal.pcbi.1006481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Andersen LL, Terczyńska-Dyla E, Mørk N, Scavenius C, Enghild JJ, Höning K, Hornung V, Christiansen M, Mogensen TH, Hartmann R. Frequently used bioinformatics tools overestimate the damaging effect of allelic variants. Genes Immun. 2017;20:10–22. doi: 10.1038/s41435-017-0002-z. [DOI] [PubMed] [Google Scholar]
- 71.Anderson D, Lassmann T. A phenotype centric benchmark of variant prioritisation tools. Genomic Medicine. 2018;3. [DOI] [PMC free article] [PubMed]
- 72.Grimm DG, Azencott CA, Aicheler F, Gieraths U, Macarthur DG, Samocha KE, Cooper DN, Stenson PD, Daly MJ, Smoller JW, et al. The evaluation of tools used to predict the impact of missense variants is hindered by two types of circularity. Hum Mutat. 2015;36:513–523. doi: 10.1002/humu.22768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Miller M, Bromberg Y, Swint-Kruse L. Computational predictors fail to identify amino acid substitution effects at rheostat positions. Sci Rep. 2017;7:41329. doi: 10.1038/srep41329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cline MS, Babbi G, Bonache S, Cao Y, Casadio R, Cruz X, Díez O, Gutiérrez-Enríquez S, Katsonis P, Lai C, et al. Assessment of blind predictions of the clinical significance of BRCA1 and BRCA2 variants. Human Mutat. 2019;40:1546–1556. doi: 10.1002/humu.23861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stein Amelie, Fowler Douglas M., Hartmann-Petersen Rasmus, Lindorff-Larsen Kresten. Biophysical and Mechanistic Models for Disease-Causing Protein Variants. Trends in Biochemical Sciences. 2019;44(7):575–588. doi: 10.1016/j.tibs.2019.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Riesselman AJ, Ingraham JB, Marks DS. Deep generative models of genetic variation capture the effects of mutations. Nature Methods. 2018;15:816–822. doi: 10.1038/s41592-018-0138-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim Young-Eun, Ki Chang-Seok, Jang Mi-Ae. Challenges and Considerations in Sequence Variant Interpretation for Mendelian Disorders. Annals of Laboratory Medicine. 2019;39(5):421. doi: 10.3343/alm.2019.39.5.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sherry ST, Ward M-H, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rives A, Goyal S, Meier J, Guo D, Ott M, Zitnick CL, Ma J, Fergus R. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. bioRxiv preprint. 2019. [DOI] [PMC free article] [PubMed]
- 80.Heinzinger M, Elnaggar A, Wang Y, Dallago C, Nechaev D, Matthes F, Rost B. Modeling aspects of the language of life through transfer-learning protein sequences. BMC Bioinformatics. 2019;20(1):723. 10.1186/s12859-019-3220-8. [DOI] [PMC free article] [PubMed]
- 81.Pundir S, Martin MJ, O'Donovan C. UniProt tools. Curr Protoc Bioinformatics. 2016;53:1.29.21–21.29.15. doi: 10.1002/0471250953.bi0129s53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Esposito D, Weile J, Shendure J, Starita LM, Papenfuss AT, Roth FP, Fowler DM, Rubin AF. MaveDB: an open-source platform to distribute and interpret data from multiplexed assays of variant effect. Genome Biol. 2019;20(1):223. 10.1186/s13059-019-1845-6. [DOI] [PMC free article] [PubMed]
- 83.Firnberg E, Labonte JW, Gray JJ, Ostermeier M. A comprehensive, high-resolution map of a Gene's fitness landscape. Mol Biol Evol. 2014;31:1581–1592. doi: 10.1093/molbev/msu081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fabian P, Michel V, Grisel O, Blondel M, Prettenhofer P, Weiss R, Vanderplas J, Cournapeau D, Pedregosa F, Varoquaux G, et al. Scikit-learn: machine learning in python. J Mach Learn Res. 2011;12:2825–2830. [Google Scholar]
- 85.Virtanen Pauli, Gommers Ralf, Oliphant Travis E., Haberland Matt, Reddy Tyler, Cournapeau David, Burovski Evgeni, Peterson Pearu, Weckesser Warren, Bright Jonathan, van der Walt Stéfan J., Brett Matthew, Wilson Joshua, Millman K. Jarrod, Mayorov Nikolay, Nelson Andrew R. J., Jones Eric, Kern Robert, Larson Eric, Carey C J, Polat İlhan, Feng Yu, Moore Eric W., VanderPlas Jake, Laxalde Denis, Perktold Josef, Cimrman Robert, Henriksen Ian, Quintero E. A., Harris Charles R., Archibald Anne M., Ribeiro Antônio H., Pedregosa Fabian, van Mulbregt Paul. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nature Methods. 2020;17(3):261–272. doi: 10.1038/s41592-019-0686-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wilcox RR. Comparing dependent robust correlations. Br J Math Stat Psychol. 2016;69:215–224. doi: 10.1111/bmsp.12069. [DOI] [PubMed] [Google Scholar]
- 87.Turck N, Vutskits L, Sanchez-Pena P, Robin X, Hainard A, Gex-Fabry M, Fouda C, Bassem H, Mueller M, Lisacek F, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics. 2011;8:12–77. doi: 10.1186/1471-2105-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.R Core Team. R: a language and environment for statistical computing. In: R Foundation for Statistical Computing; 2018.
- 89.PolyPhen2 Webserver.http://genetics.bwh.harvard.edu/pph2/bgi.shtml Accessed: 15 Apr 2019.
- 90.Envision webserver. https://envision.gs.washington.edu/shiny/envision_new/ Accessed: 15 Apr 2019.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Supporting Online Material (SOM) containing additional figures, tables and notes.
Data Availability Statement
The datasets generated and code for their analysis are available on Mendeley Data (10.17632/2rwrkp7mfk.1).