

Abstract
Background
Tissue plasminogen activator (tPA) regulates fibrinolysis and is routinely used as ischemic stroke pharmacotherapy. We hypothesized that brain microvascular tPA expression and barrier properties of endothelial cells are substantially related.
Methods
Human brain microvascular endothelial cells were incubated with two agents known to modify cAMP pathways: forskolin and rolipram. We analyzed development of endothelial barrier properties, i.e., trans-endothelial electrical resistance (TEER), permeability of endothelial cell monolayer, expression of influx transporter glut-1 and endothelial tight junction molecules occludin and claudin-5, tPA antigen release, and levels of endothelial tPA mRNA.
Results
Forskolin plus rolipram-treated endothelial cells showed increased TEER compared to controls (174±20% of control at day six, p<0.01), while permeability to albumin and 70kDa dextran was reduced (21±6.8% of control and 3.8±0.3% of control, respectively, p<0.001). In addition, occludin and claudin-5 protein were up-regulated, occludin mRNA was increased to 206±60% of control (p<0.05), glut-1 mRNA was increased to 196±68% of control (p<0.05), levels of tPA protein were reduced to 35±7.0% of control (p<0.001) after six days, and tPA mRNA was reduced to 32±7.7% of control (p<0.01). TPA and occludin mRNA levels were inversely associated (r=−0.68, p<0.05). Conclusions: In this in vitro model, barrier properties were strongly linked (by inverse association) with tPA expression of brain microvascular endothelial cells.
Key Words: Endothelium, Tissue plasminogen activator, Stroke, Microvasculature, Fibrinolysis
Introduction
Tissue plasminogen activator (tPA), a serine protease regulator of fibrinolysis, is largely endothelial-derived and processes plasminogen into proteolytically active plasmin [1]. Reduced fibrinolytic capacity increases risk of thrombosis, and tPA as pharmacotherapy remains the only proven treatment of acute ischemic stroke [2]. Prior cell culture investigation using brain microvascular endothelial cells and astrocytes has shown that endothelial tPA expression is negatively regulated by astrocytes [3, 4]. Astrocyte regulation of tPA was inversely related to barrier properties of brain microvascular endothelial cells [3, 4] and was mediated by TGF-beta [5].
In the current study, we further investigated the relationship between barrier properties of brain microvascular endothelial cells and expression of tPA. Prior work has demonstrated importance of organ-specific regulation of thrombosis and hemostasis [6]. We wished to analyze tPA expression using a monoculture system, thereby avoiding potential confounding effects of a cellular milieu involving multiple cell types. We hypothesized that brain microvascular tPA expression and barrier properties of endothelial cells are substantially related. We tested this hypothesis using human brain microvascular endothelial cells (HBECs) and two cAMP agents (forskolin and rolipram) known to enhance barrier characteristics in vitro [7, 8].
Materials and Methods
Cell Culture
HBECs (Applied Cell Biology Research Institute, Kirkland, WA) were cultured in Medium 131 (Cascade Biologics, Portland, OR) containing microvascular growth supplement and 5% fetal bovine serum and characterized by immunoreactivity for von Willebrand factor (Dako, Carpinteria, CA) and uptake of acetylated LDL labeled with 1-1'-dioctacecyl-1-1-3-3-3'-3'-tetamethyl-indocarbocyanine perchlorate (Biomedical Technologies, Stoughton, MA). HBEC passages 5-10 were used for experiments. HBECs were plated at density of 1×105 cells/cm2.
Measurement of Trans-Endothelial Electrical Resistance
We measured trans-endothelial electrical resistance (TEER) using two different systems. With the first system (“ECIS method”), continuous changes in TEER were monitored in real-time by electric cell-substrate impedance sensing (ECIS) using ECIS Model 1600R instrument (Applied BioPhysics, Troy, NY), as previously described [8]. HBECs were plated into 8-well arrays (8W10E), each well containing ten gold microelectrodes, 72 hours prior to addition of 50 uM forskolin (Sigma, St Louis, MO) and/or 10 uM rolipram (A.G. Scientific, San Diego, CA). Long-term resistance changes were monitored at 400 Hz. Data were automatically and continuously collected and recorded by computer. Experiments were performed after cells reached confluence, with basal TEER values > 1500 Ω. Resistance was calculated by subtracting resistance readings of cell-free preparations from that of HBEC preparations and are shown as percentage change, as previously described [9-19]. Conditioned media were collected and frozen at -80° C prior to enzyme immunoassay. On day six, cells were harvested and used for quantitative PCR analysis. In separate experiments conducted as above, we added 250 ng/ml tPA (American Diagnostica Inc., Stamford, CT) to cell culture preparations at the same time as the addition of forskolin and/or rolipram.
The second method (“transwell system”) measured TEER using a EVOM Voltohmmeter (World Precision Instrument, Sarasota, FL). HBECs were plated into 12-well transwells at 105 cells/cm2 72 hours prior to addition of 50 uM forskolin and 10 uM rolipram. Resistance was measured at day six. Conditioned media were collected at day six and frozen at -80° C prior to enzyme immunoassay.
Enzyme Immunoassay for tPA
Enzyme immunoassay was performed using human tPA ELISA kit purchased from American Diagnostica Inc. 20 µl of tPA standards and 20 µl of test samples were added to the wells. After 1 hour incubation, 50 µl conjugate was added and incubated for an additional 30 minutes. The plates were washed prior to the addition of substrate. After 15 minutes, the reaction was terminated and absorbance was read at 490 nm.
Quantitative Real-Time Polymerase Chain Reaction (PCR; Taqman)
RNeasy Mini kits (Qiagen, Valencia, CA) were used for RNA extraction. RNAase-Free DNase digestion (Qiagen, Valencia, CA) was used to remove genomic DNA from the RNA preparations. Taqman kits (Applied Biosystems, Foster City, CA) were used for reverse transcription. 200 ng total mRNA was added in 20 µl reverse-transcription reaction and the mixture was incubated 5 minutes at 25°C, 30 minutes at 48°C, then 5 minutes at 95°C. cDNAs were stored at -20°C for further use.
TaqMan real-time quantitative PCR was performed in a GeneAmp 5700 sequence detector (Applied Biosystems, Foster City, CA). The oligonucleotide primers, 5' fluorescent dye-labeled (FAM) reporter probes, and 3' fluorescent dye-labeled (TAMRA) quencher probes were designed using Primer Express software (Applied Biosystems, Foster City, CA). The PCR reaction, performed in a 20 µl total volume, contained 10 µl TaqMan Universal PCR Master Mix (Applied Biosystems, Foster City, CA) and 2 µl (0.5 µM) cDNA, primers, and probes. The PCR reaction was carried out at 50°C for 2 minutes to activate uracil-N-glycosylase (UNG), 95°C for 10 minutes to activate Taq polymerase, followed by 40 cycles of denaturation at 95oC for 15 seconds and primer annealing-extension at 60°C for 1 minute. A standard curve, generated from three-fold serial dilutions of HBEC cDNA, was used to determine tPA and β-actin concentrations. Quantity of tPA was adjusted to β-actin levels (ie, dividing by quantity of β-actin). Sequences of TaqMan primers and probes for real-time PCR are listed in Table 1.
Table 1.
Sequences of TaqMan primers and probes for real-time PCR.
| Gene* | Sequence | Reaction concentration [nM] |
|---|---|---|
| tPA | Forward: 5'-GGCCTTGTCTCCTTTCTATTCG -3' | 900 |
| Reverse: 5'-GCGGCTGGATGGGTACAGT -3' | 900 | |
| Probe: FAM-TGACATGAGCCTCCTTCAGCCGCT-TAMRA | 50 | |
| β-actin | Forward: 5'-CCTGGCACCCAGCACAAT -3' | 300 |
| Reverse: 5'-GCCGATCCACACGGAGTACT -3' | 300 | |
| Probe: FAM-ATCAAGATCATTGCTCCTCCTGAGCGC -TAMRA | 100 | |
| TM | Forward: 5' -CGAAGACACAGACTGCGATTTG -3' | 900 |
| Reverse: 5'-GGCTGCCGACCAATAACG -3' | 900 | |
| Probe: FAM-CCAGGTCCTCACTACCGGGCGC-TAMRA | 100 | |
| PAI | Forward: 5'-GCCCCACTTCTTCAAGCTGTT-3' | 900 |
| Reverse: 5'-TGGCTCTCTCCACCTCTGAAA-3' | 900 | |
| Probe: FAM-TCCACTTGCTTGACCGTGCTCCG-TAMRA | 200 | |
| Occludin | Forward: 5'- AAGGTCAAAGAGAACAGAGCAAGATC -3' | 900 |
| Reverse: 5'- GTCCTCCTCCAGCTCATCACA -3' | 900 | |
| Probe: FAM- AGACAGACTACACAACTGGCGGCGAGTCC -TAMRA | 50 | |
| Glut-1 | Forward: 5'- GCTCATCAACCGCAACGAG -3' | 300 |
| Reverse: 5'- TCATGGGTCACGTCAGCTGT -3' | 300 | |
| Probe: FAM-AGAACCGGGCCAAGAGTGTGCTAAAGAA-TAMRA | 100 |
Barrier Permeability Assay
HBECs were cultured on 12 mm diameter transwell insert (3 µm pore size) (Corning, Corning NY). After six days culture, media were replaced and fluorescein isothiocyanate (FITC)-labeled dextran (1 mg/ml, molecular size: 70kDa, Invitrogen, Carlsbad, CA) was added to the upper chambers. Fluorescence intensity was monitored using Chameleon Mikrowin-2000 microplate fluorescent reader (Bioscan, Washington, DC). Upper chamber was sampled at the beginning; bottom chamber was sampled every 15 minutes for one hour. Permeability (P, cm/h) was calculated using the following equation: P= [V/(A×C0)] × [dC/dt], where V is the receiver volume (the volume of bottom chamber, 1 ml), A is the surface area of the endothelial monolayer, C0 is the concentration of the donor solution (the initial concentration of the upper chamber), and dC/dt is the rate of concentration change on the receiver side [20]. Permeability of endothelial cell monolayer (Pe) was calculated using the following equation: 1/P=1/Pe+1/Pf., where Pf is the permeability of the transwell [21].
Western blot
To analyze expression of occludin protein, cell pellets were lysed in RIPA buffer (58 mM NaCl, 5 mM EDTA, 10 mM Tris/HCl, pH 7.2, 0.1% SDS, 1% sodium deoxycholate, 1% Triton X-100, 10 µg/ml PMSF, 2 µg/ml aprotinin, 2 µg/ml leupeptin and 1 µg/ml pepstatin A). Samples were applied to a 10% polyacrylamide gel and then subjected to electrophoresis. After transferring to PVDF (Polyvinylidene Difluoride) membrane (Millipore, Bedford, MA), the membrane was incubated with tris-buffered saline containing 5% milk for 1 hour, then incubated with antibodies to occludin (BD Bioscience, San Diego, CA), claudin-5 (BD Bioscience, San Diego, CA) or actin (Santa Cruz, Santa Cruz, CA). The membrane was washed with 0.1% Tween 20 in TBS for 30 minutes. Horseradish peroxidase-labeled secondary antibody was then incubated for 1 hour. The washed membranes were processed with West Pico chemiluminescent detection system (Pierce, Rockford, IL). A Kodak Image Station 2000mm (Kodak, Rochester, NY) was used to acquire images.
Statistical Analysis
Data are presented as percent of mean control value at day 0. Differences among the means of the treatment groups were tested using analysis of variance and Dunnett's two-tailed t-test, testing if any treatments are significantly different from the control group. Differences were considered statistically significant if p<0.05.
Results
We examined TEER during the experiments conducted over a six day period, using ECIS and transwell systems. Using ECIS system, TEER of endothelial cells incubated with forskolin plus rolipram increased to 220±26% (p<0.001) of their baseline values and 174±20% of control values at day six (p<0.01) (Fig. 1a). Addition of 250 ng/ml tPA to cell culture preparations had no significant effect on TEER, and endothelial cell count among the four groups did not differ significantly (data not shown). Using transwell system, TEER of endothelial cells incubated with forskolin and rolipram increased to 85±1.5Ω*cm2 (178±3% of control) at day six (p<0.001, Fig. 1c).
Fig. 1.
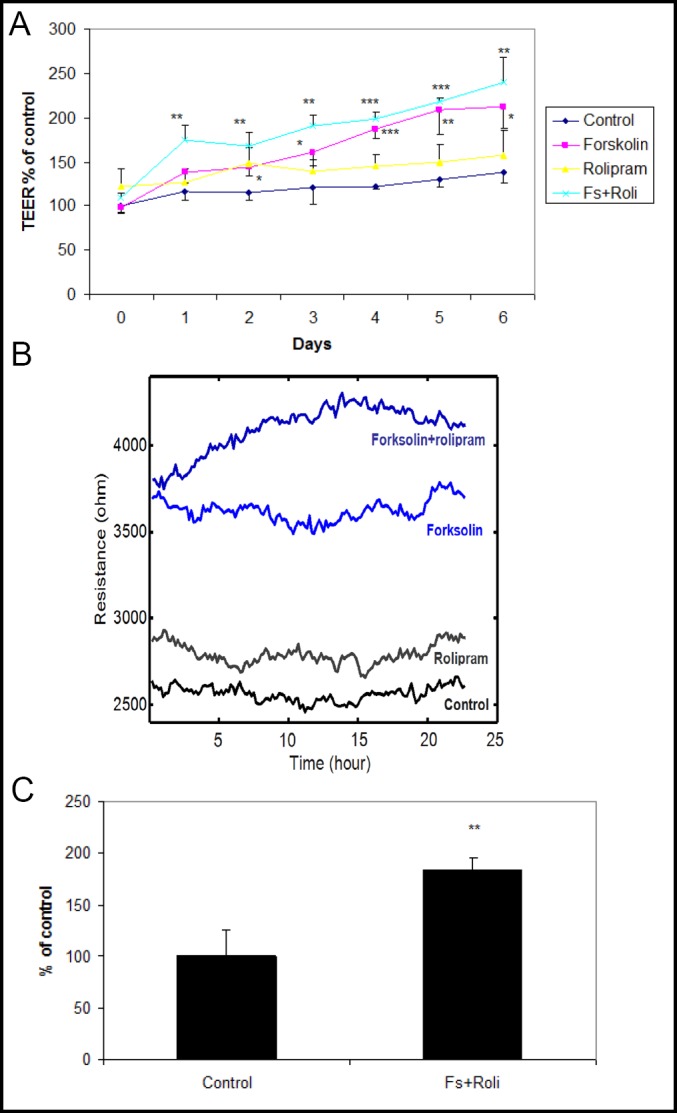
a: Continuous measurement of TEER using ECIS system. During six days of culture, human brain endothelial cells incubated with forskolin and rolipram demonstrated significantly increased TEER at all time points. Data from one of three independent experiments, presented as mean ± SD. (*p<0.05, **p<0.01, ***p<0.001 compared to control). b: ECIS tracing showing mean TEER values during the final day of a six day experiment. c: Measurement of TEER using transwell system. At day six, human brain endothelial cells incubated with forskolin and rolipram demonstrated significantly increased TEER. Data from one of three independent experiments, presented as mean ± SD. (**p<0.01 compared to control).
We measured several variables related to endothelial barrier properties at day six. Occludin mRNA increased significantly in forskolin plus rolipram-treated endothelial cells (206±60% of control, p<0.05) (Fig. 2a). Occludin mRNA and TEER were strongly correlated (r=0.72, p<0.01, Fig. 2b). Expression of occludin and claudin-5 protein also increased in forskolin plus rolipram-treated endothelial cells compared to control (Fig. 2c) as did glut1 mRNA (196±68% of control p<0.05) (Fig. 3). Permeability of 70kDa dextran at day six was 2.8×10-5 cm/s for control endothelial cells. It was significantly decreased to 1.4×10-5 cm/s in rolipram-treated endothelial cells (51±14% of control, p<0.05), to 1.7×10-6 cm/s in forskolin-treated cells (6.1±1.6% of control, p<0.001), and to 1.1×10-6 cm/s in forskolin plus rolipram-treated cells (3.8±0.3% of control, p<0.001) (Fig. 4).
Fig. 2.
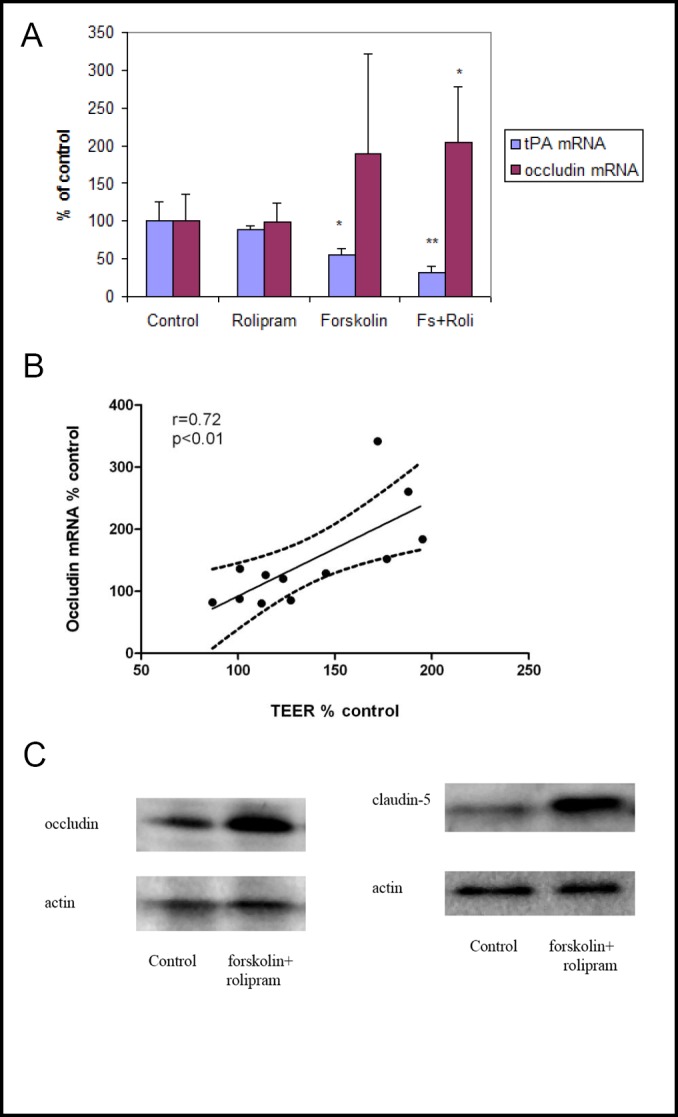
a: Measurement of tPA and occludin mRNA at day six using quantitative RT-PCR. Human brain endothelial cells incubated with forskolin, with or without rolipram, demonstrated reduced tPA mRNA expression; HBEC incubated with forskolin and rolipram demonstrated increased occludin mRNA. Data from three independent experiments combined, presented as mean ± SD. (*p<0.05, **p<0.01 compared to control). b: TEER and occludin mRNA expression were highly correlated (r=0.72). Data from three independent experiments combined. c: Measurement of occludin and claudin-5 protein levels by Western blot. Forskolin plus rolipram increased occludin protein compared to control. Representative Western blot from three independent experiments.
Fig. 3.
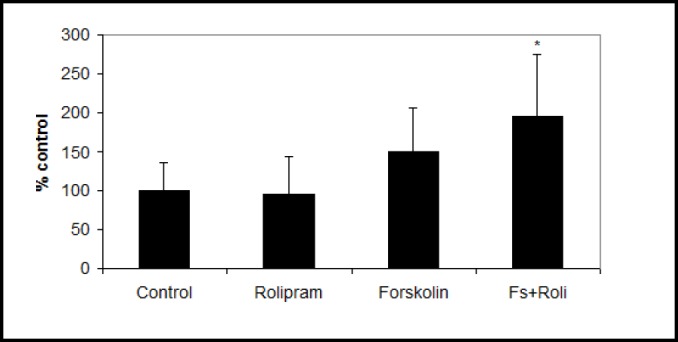
After six days culture, glut1 mRNA was significantly increased in human brain endothelial cells incubated with forskolin plus rolipram. Data from three independent experiments combined, presented as mean ± SD. (*p<0.05 compared to control).
Fig. 4.
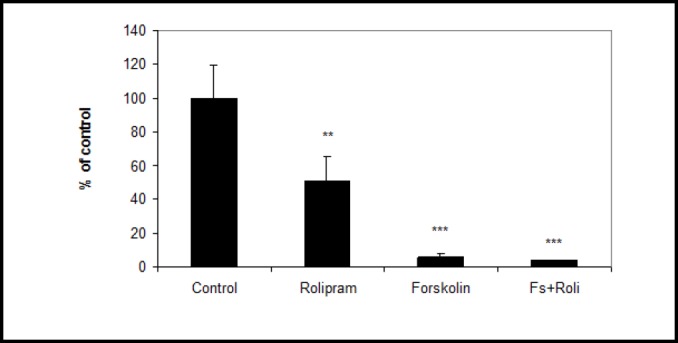
Treatment reduced permeability to 70kDa dextran for human brain endothelial cell monolayer. Data from one of three independent experiments combined, presented as mean ± SD. (**p<0.01, ***p<0.001 compared to control).
We examined release of tPA protein with HBEC grown on ECIS slides (Fig. 5a). At baseline (day zero) tPA protein level for control was 42±5 ng/ml. At day two, tPA protein was significantly reduced in forskolin alone (61±14% of control, p<0.001) and forskolin plus rolipram groups (62±15% of control, p<0.001). By day four, tPA protein was further reduced in both forskolin alone (37±9.3% of control, p<0.001) and forskolin plus rolipram groups (32±11% of control, p<0.001). By day six, tPA protein was significantly reduced by rolipram alone (87±7.5% of control, p<0.01), forskolin alone (42±9.0% of control, p<0.001), and forskolin plus rolipram (35±7.0% of control, p<0.001). Using the transwell system, tPA levels at day six were significantly reduced in forskolin plus rolipram treated cells (35±3.2%, p<0.001) (Fig. 5b). We compared tPA levels in top and bottom transwell chambers (ie, above and below endothelial cell monolayers). Forskolin plus rolipram treatment produced similar tPA levels in both chambers, which were both reduced (p<0.05) compared to controls (data not shown).
Fig. 5.
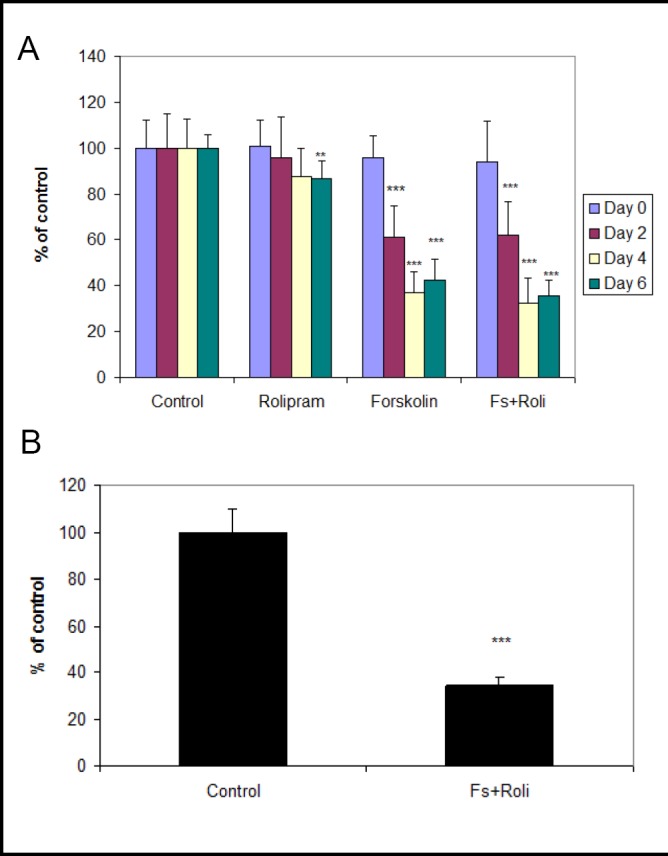
a: Measurement of tPA protein at day 0, 2, 4 and 6 using ECIS system. Conditioned media of human brain endothelial cells incubated with forskolin, with or without rolipram, showed reduced tPA antigen beginning at day two and became more prominent at days four and six. Data from three independent experiments combined, presented as mean ± SD. (**p<0.01, *** p<0.001 compared to control). b: Measurement of tPA protein at day six using transwell system. Conditioned media of human brain endothelial cells incubated with forskolin plus rolipram showed reduced tPA antigen. Data from one of three independent experiments, presented as mean ± SD. (***p<0.001 compared to control).
Analysis of tPA mRNA at day six showed significant reductions in forskolin-treated endothelial cells (55±8.9% of control, p<0.05) and in the forskolin plus rolipram-treated cells (32±7.7% of control, p<0.01) (Fig. 2a). Moreover, tPA mRNA and occludin mRNA were highly and negatively correlated (r=−0.68, p<0.05) (Fig. 6). tPA mRNA and TEER were also inversely associated (r=−0.88, p<.01). Thus, tPA expression and release were reduced as TEER and occludin and glut1 mRNA expression increased and as permeability decreased.
Fig. 6.
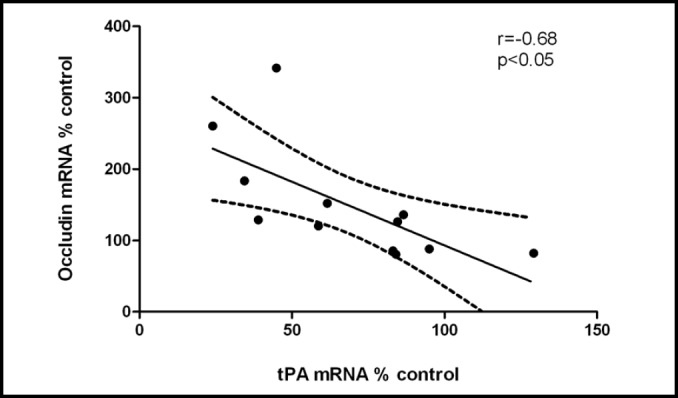
tPA and occludin mRNA expression were highly and inversely correlated (r=−0.68). Data from three independent experiments combined.
Prior work has shown regulation of thrombomodulin (endothelial co-activator of circulating protein C) and plasminogen activator inhibitor-1 (PAI-1, principal inhibitor of tPA) by astrocytes [3, 22]. In the current study, we measured mRNA expression of thrombomodulin and PAI-1 after six days culture. HBEC incubated with forskolin plus rolipram demonstrated significantly decreased thrombomodulin mRNA (73±11% of control, p<0.01) (Fig. 7a). PAI-1 mRNA levels tended to increase in HBECs incubated with forkskolin, with or without rolipram (Fig. 7b).
Fig. 7.
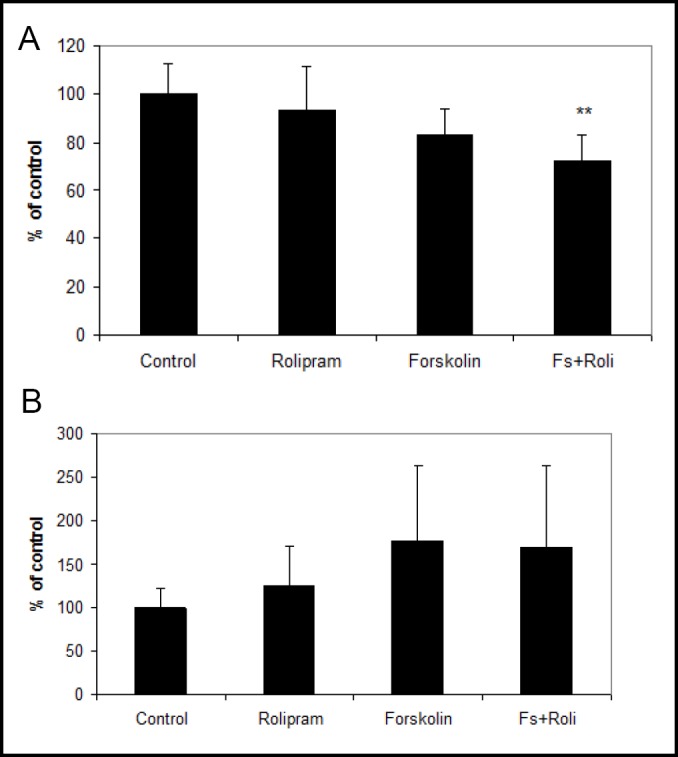
a: Measurement of thrombomodulin mRNA at day 6 using quantitative RT-PCR. Endothelial cells incubated with both forskolin and rolipram showed a significant reduction of thrombomodulin mRNA. Data from six independent experiments combined, presented as mean ± SD. (**p<0.01 compared to control). b: During the same time period, PAI-1 expression tended to increase in HBEC incubated with forskolin (177±87% of control, p=0.2) and with forskolin plus rolipram (170±94% of control, p=0.3). Data from three independent experiments combined, presented as mean ± SD.
Discussion
We studied the response of HBECs to treatment designed to enhance endothelial barrier properties. Treatment consisted of two cAMP-modifying agents, forskolin and rolipram. Over six days treatment, we induced increased TEER, increased expression of endothelial tight junction proteins occludin and claudin-5, reduced endothelial permeability to dextran, and increased expression of influx transporter Glut-1. These four elements are characteristic of increased endothelial barrier properties. As these elements became manifest, expression of endothelial tPA declined - both the mRNA and protein levels. Addition of exogenous tPA to cell culture preparations did not alter the results, suggesting that low tPA is a consequence rather than a cause of increased barrier properties. These findings support the contention that brain endothelial tPA expression is restricted with presence of enhanced barrier properties.
The current findings are consistent with prior work demonstrating distinctive characteristics of brain expression of tPA. In vitro studies demonstrated systemic, but not brain, endothelial cells release tPA in response to α-thrombin [23, 24]. Rodent studies showed dependence of endothelial tPA expression on size and location of vessel [25]. In an investigation of tPA expression in primate brain, capillary tPA expression was largely absent; capillaries comprised nearly 40% of all microvessels, yet more than 95% of capillaries showed no immunoreactivity for tPA [26]. Work from our laboratory showed astrocyte regulation of brain microvascular endothelial tPA expression [3, 4]. Thrombomodulin, known to co-activate protein C with subsequent inhibition of PAI-1 [27], exhibited reduced expression in the current study, as was seen in prior work using astrocyte-endothelial co-cultures [22, 5]. Moreover, the principal tPA inhibitor PAI-1 tended to have increased expression in the current study, consistent with work done in astrocyte-endothelial co-cultures [3].
Taken together, these data indicate restricted expression of tPA with enhanced barrier properties. For our model, we utilized HBECs and two cAMP-modifying agents, forskolin and rolipram. Elevated cAMP plays a key role in generating increased endothelial barrier properties in vitro [28, 29]. Two cAMP-dependent signal pathways, protein kinase A and Epac, are involved in blood-brain barrier (BBB) formation. cAMP-dependent protein kinase A regulates cytoskeleton rearrangement [30]. Epac pathway modulates VE-cadherin junction integrity and actin reorganization, resulting in decreased endothelial permeability [31] and also mediates redistribution, to sites of cell-cell contact, of tight junction and adherens junction molecules [32]. Nevertheless, these findings are not indicative of a cAMP-specfic effect. Note that prior work has demonstrated that astrocytes enhance brain endothelial barrier properties concurrent with reduced tPA [3, 4]; TGF-beta also induces similar findings [5]. Moreover, enhanced endothelial glut1 is consistent with upregulation of gamma glutamyl transpeptidase reported in prior similar work [3, 4].
In the current study, forskolin with rolipram increased TEER, increased expression of tight junction constituents occludin and claudin-5 and influx transporter glut1, and decreased endothelial permeability to both albumin and 70kDa dextran. Tight junctions, a principal determinant of TEER [33], have multiple associated molecules including occludin [34, 35], which has a sealant and regulatory role for tight junctions [34]. Claudin-5 also contributes to elevated TEER [36] and is one of the most prominent tight junction proteins induced in BBB models [37]. Our model demonstrated the expected strong association between TEER and occludin expression. We also demonstrated reduced endothelial permeability to 70kDa FITC-dextran (a measure of paracellular permability [38]) and to albumin. Albumin permeability is generally viewed as indicative of trans-endothelial permeability, probably via vesicles [39].
We observed that cAMP elevation agents forskolin and rolipram decreased tPA release from HBECs. The role of cAMP in regulation of tPA differs between in vitro systems. cAMP stimulated the acute release of tPA from passage one human umbilical vein endothelial cells at 30 minutes [40]. Forskolin tended to decrease tPA protein and mRNA in human thyroid follicular cells at approximately 7 days [41]. We measured relatively long-term, rather than acute, tPA release from passaged cells. The in vitro nature of our investigation, using later passage endothelial cells, represents a significant limitation of our study and extrapolations to in vivo phenomena require caution.
In conclusion, we show restricted expression of brain microvascular tPA when increased barrier properties of endothelial cells become manifest. This is consistent with multiple lines of evidence showing tight regulation of fibrinolysis factors in the brain microvasculature. These findings indicate close linkage between expression of brain microvascular tPA and endothelial barrier properties in vitro.
Acknowledgments
This research was supported by the National Institutes of Health research grant NS20989. Dr. Fisher has received support from Boehringer Ingelheim (research support, speakers' bureau, honoraria) and Otsuka Pharmaceutical Company (research support, honoraria).
References
- 1.Oliver JJ, Webb DJ, Newby DE. Stimulated tissue plasminogen activator release as a marker of endothelial function in humans. Arterioscler Thromb Vasc Biol. 2005;25:2470–2479. doi: 10.1161/01.ATV.0000189309.05924.88. [DOI] [PubMed] [Google Scholar]
- 2.The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group Tissue plasminogen activator for acute ischemic stroke. N Engl J Med. 1995;333:1581–1587. doi: 10.1056/NEJM199512143332401. [DOI] [PubMed] [Google Scholar]
- 3.Tran ND, Schreiber SS, Fisher M. Astrocyte regulation of endothelial tissue plasminogen activator in a blood-brain barrier model. J Cereb Blood Flow Metab. 1998;18:1316–1324. doi: 10.1097/00004647-199812000-00006. [DOI] [PubMed] [Google Scholar]
- 4.Kim JA, Wang SJ, Fisher MJ. Astrocyte regulation of human brain capillary endothelial fibrinolysis. Thromb Res. 2003;112:159–165. doi: 10.1016/j.thromres.2003.10.021. [DOI] [PubMed] [Google Scholar]
- 5.Tran ND, Correale J, Schreiber SS, Fisher M. Transforming growth factor-beta mediates astrocyte-specific regulation of brain endothelial anticoagulant factors. Stroke. 1999;30:1671–1678. doi: 10.1161/01.str.30.8.1671. [DOI] [PubMed] [Google Scholar]
- 6.Rosenberg RD, Aird WC. Vascular-bed-specific hemostasis and hypercoagulable states. N Engl J Med. 1999;340:1555–1564. doi: 10.1056/NEJM199905203402007. [DOI] [PubMed] [Google Scholar]
- 7.Baumer Y, Spindler V, Werthmann RC, Bunemann M, Waschke J. Role of Rac 1 and cAMP in endothelial barrier stabilization and thrombin-induced barrier breakdown. J Cell Physiol. 2009;220:716–726. doi: 10.1002/jcp.21819. [DOI] [PubMed] [Google Scholar]
- 8.Bruckener KE, el Baya A, Galla HJ, Schmidt MA. Permeabilization in a cerebral endothelial barrier model by pertussis toxin involves the PKC effector pathway and is abolished by elevated levels of cAMP. J Cell Sci. 2003;116:1837–1846. doi: 10.1242/jcs.00378. [DOI] [PubMed] [Google Scholar]
- 9.Di Lorenzo A, Fernandez-Hernando C, Cirino G, Sessa WC. Akt1 is critical for acute inflammation and histamine-mediated vascular leakage. Proc Natl Acad Sci U S A. 2009;106:14552–14557. doi: 10.1073/pnas.0904073106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tripathi AK, Sullivan DJ, Stins MF. Plasmodium falciparum-infected erythrocytes decrease the integrity of human blood-brain barrier endothelial cell monolayers. J Infect Dis. 2007;195:942–950. doi: 10.1086/512083. [DOI] [PubMed] [Google Scholar]
- 11.Grab DJ, Garcia-Garcia JC, Nikolskaia OV, Kim YV, Brown A, Pardo CA, Zhang Y, Becker KG, Wilson BA, de A Lima AP, Scharfstein J, Dumler JS. Protease activated receptor signaling is required for african trypanosome traversal of human brain microvascular endothelial cells. PLoS Negl Trop Dis. 2009;3:e479. doi: 10.1371/journal.pntd.0000479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grab DJ, Nyarko E, Nikolskaia OV, Kim YV, Dumler JS. Human brain microvascular endothelial cell traversal by borrelia burgdorferi requires calcium signaling. Clin Microbiol Infect. 2009;15:422–426. doi: 10.1111/j.1469-0691.2009.02869.x. [DOI] [PubMed] [Google Scholar]
- 13.Adam AP, Sharenko AL, Pumiglia K, Vincent PA. Src-induced tyrosine phosphorylation of VE-cadherin is not sufficient to decrease barrier function of endothelial monolayers. J Biol Chem. 2010;285:7045–7055. doi: 10.1074/jbc.M109.079277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Birukova AA, Burdette D, Moldobaeva N, Xing J, Fu P, Birukov KG. Rac GTPase is a hub for protein kinase A and Epac signaling in endothelial barrier protection by cAMP. Microvasc Res. 2010;79:128–138. doi: 10.1016/j.mvr.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huppert J, Closhen D, Croxford A, White R, Kulig P, Pietrowski E, Bechmann I, Becher B, Luhmann HJ, Waisman A, Kuhlmann CR. Cellular mechanisms of IL-17-induced blood-brain barrier disruption. FASEB J. 2010;24:1023–1034. doi: 10.1096/fj.09-141978. [DOI] [PubMed] [Google Scholar]
- 16.Sidhaye VK, Chau E, Breysse PN, King LS. Septin-2 mediates airway epithelial barrier function in physiologic and pathologic conditions. Am J Respir Cell Mol Biol. 2010;45:120–126. doi: 10.1165/rcmb.2010-0235OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Umapathy NS, Zemskov EA, Gonzales J, Gorshkov BA, Sridhar S, Chakraborty T, Lucas R, Verin AD. Extracellular betanicotinamide adenine dinucleotide (beta-NAD) promotes the endothelial cell barrier integrity via PKA- and EPAC1/RAC1-dependent actin cytoskeleton rearrangement. J Cell Physiol. 2010;223:215–223. doi: 10.1002/jcp.22029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Z, Ginnan R, Abdullaev IF, Trebak M, Vincent PA, Singer HA. Calcium/calmodulin-dependent protein kinase II delta 6 (CaMKIIdelta6) and RhoA involvement in thrombin-induced endothelial barrier dysfunction. J Biol Chem. 2010;285:21303–21312. doi: 10.1074/jbc.M110.120790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao Y, Gorshkova IA, Berdyshev E, He D, Fu P, Ma W, Su Y, Usatyuk PV, Pendyala S, Oskouian B, Saba JD, Garcia JG, Natarajan V. Protection of LPS-induced murine acute lung injury by sphingosine-1-phosphate lyase suppression. Am J Respir Cell Mol Biol. 2011;45:426–435. doi: 10.1165/rcmb.2010-0422OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eddy EP, Maleef BE, Hart TK, Smith PL. In vitro models to predict blood-brain barrier permeability. Adv Drug Deliv Rev. 1997;23:185–198. [Google Scholar]
- 21.Deli MA, Abraham CS, Kataoka Y, Niwa M. Permeability studies on in vitro blood-brain barrier models: Physiology, pathology, and pharmacology. Cell Mol Neurobiol. 2005;25:59–127. doi: 10.1007/s10571-004-1377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tran ND, Wong VL, Schreiber SS, Bready JV, Fisher M. Regulation of brain capillary endothelial thrombomodulin mRNA expression. Stroke. 1996;27:2304–2310. doi: 10.1161/01.str.27.12.2304. [DOI] [PubMed] [Google Scholar]
- 23.Shatos MA, Orfeo T, Doherty JM, Penar PL, Collen D, Mann KG. Alpha-thrombin stimulates urokinase production and DNA synthesis in cultured human cerebral microvascular endothelial cells. Arterioscler Thromb Vasc Biol. 1995;15:903–911. doi: 10.1161/01.atv.15.7.903. [DOI] [PubMed] [Google Scholar]
- 24.Grau GE, de Moerloose P, Bulla O, Lou J, Lei Z, Reber G, Mili N, Ricou B, Morel DR, Suter PM. Haemostatic properties of human pulmonary and cerebral microvascular endothelial cells. Thromb Haemost. 1997;77:585–590. [PubMed] [Google Scholar]
- 25.Levin EG, Santell L, Osborn KG. The expression of endothelial tissue plasminogen activator in vivo: A function defined by vessel size and anatomic location. J Cell Sci. 1997;110:139–148. doi: 10.1242/jcs.110.2.139. [DOI] [PubMed] [Google Scholar]
- 26.Levin EG, del Zoppo GJ. Localization of tissue plasminogen activator in the endothelium of a limited number of vessels. Am J Pathol. 1994;144:855–861. [PMC free article] [PubMed] [Google Scholar]
- 27.Nesheim M, Wang W, Boffa M, Nagashima M, Morser J, Bajzar L. Thrombin, thrombomodulin and TAFI in the molecular link between coagulation and fibrinolysis. Thromb Haemost. 1997;78:386–391. [PubMed] [Google Scholar]
- 28.Rubin LL, Hall DE, Porter S, Barbu K, Cannon C, Horner HC, Janatpour M, Liaw CW, Manning K, Morales J, et al. A cell culture model of the blood-brain barrier. J Cell Biol. 1991;115:1725–1735. doi: 10.1083/jcb.115.6.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolburg H, Neuhaus J, Kniesel U, Krauss B, Schmid EM, Ocalan M, Farrell C, Risau W. Modulation of tight junction structure in blood-brain barrier endothelial cells. Effects of, tissue culture second messengers and cocultured astrocytes. J Cell Sci. 1994;107:1347–1357. doi: 10.1242/jcs.107.5.1347. [DOI] [PubMed] [Google Scholar]
- 30.Liu F, Verin AD, Borbiev T, Garcia JG. Role of cAMP-dependent protein kinase A activity in endothelial cell cytoskeleton rearrangement. Am J Physiol Lung Cell Mol Physiol. 2001;280:L1309–1317. doi: 10.1152/ajplung.2001.280.6.L1309. [DOI] [PubMed] [Google Scholar]
- 31.Kooistra MR, Corada M, Dejana E, Bos JL. Epac1 regulates integrity of endothelial cell junctions through VE-cadherin. FEBS Lett. 2005;579:4966–4972. doi: 10.1016/j.febslet.2005.07.080. [DOI] [PubMed] [Google Scholar]
- 32.Cullere X, Shaw SK, Andersson L, Hirahashi J, Luscinskas FW, Mayadas TN. Regulation of vascular endothelial barrier function, by Epac a cAMP-activated exchange factor for Rap GTPase. Blood. 2005;105:1950–1955. doi: 10.1182/blood-2004-05-1987. [DOI] [PubMed] [Google Scholar]
- 33.Gaillard PJ, de Boer AG. Relationship between permeability status of the blood-brain barrier and in vitro permeability coefficient of a drug. Eur J Pharm Sci. 2000;12:95–102. doi: 10.1016/s0928-0987(00)00152-4. [DOI] [PubMed] [Google Scholar]
- 34.Wolburg H, Lippoldt A. Tight junctions of the blood-brain barrier: Development, composition and regulation. Vascul Pharmacol. 2002;38:323–337. doi: 10.1016/s1537-1891(02)00200-8. [DOI] [PubMed] [Google Scholar]
- 35.Feldman GJ, Mullin JM, Ryan MP. Occludin: Structure, function and regulation. Adv Drug Deliv Rev. 2005;57:883–917. doi: 10.1016/j.addr.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 36.Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 37.Perriere N, Yousif S, Cazaubon S, Chaverot N, Bourasset F, Cisternino S, Decleves X, Hori S, Terasaki T, Deli M, Scherrmann JM, Temsamani J, Roux F, Couraud PO. A functional in vitro model of rat blood-brain barrier for molecular analysis of efflux transporters. Brain Res. 2007;1150:1–13. doi: 10.1016/j.brainres.2007.02.091. [DOI] [PubMed] [Google Scholar]
- 38.Tai LM, Holloway KA, Male DK, Loughlin AJ, Romero IA. Amyloid-beta-induced occludin down-regulation and increased permeability in human brain endothelial cells is mediated by MAPK activation. J Cell Mol Med. 2009;14:1101–1112. doi: 10.1111/j.1582-4934.2009.00717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Plateel M, Teissier E, Cecchelli R. Hypoxia dramatically increases the nonspecific transport of blood-borne proteins to the brain. J Neurochem. 1997;68:874–877. doi: 10.1046/j.1471-4159.1997.68020874.x. [DOI] [PubMed] [Google Scholar]
- 40.Hegeman RJ, van den Eijnden-Schrauwen Y, Emeis JJ. Adenosine 3':5'-cyclic monophosphate induces regulated secretion of tissue-type plasminogen activator and von Willebrand factor from cultured human endothelial cells. Thromb Haemost. 1998;79:853–858. [PubMed] [Google Scholar]
- 41.Susarla R, Watkinson JC, Eggo MC. Regulation of plasminogen activators in human thyroid follicular cells and their relationship to differentiated function. J Cell Physiol. 2007;212:643–654. doi: 10.1002/jcp.21060. [DOI] [PubMed] [Google Scholar]