

Abstract
The oxidation of methionine is an important post-translational modification of proteins with numerous roles in physiology and pathology. However, the quantitative analysis of methionine oxidation on a proteome-wide scale has been hampered by technical limitations. Methionine is readily oxidized in vitro during sample preparation and analysis. In addition, there is a lack of enrichment protocols for peptides that contain an oxidized methionine residue, making the accurate quantification of methionine oxidation difficult to achieve on a global scale. Herein, we report a methodology to circumvent these issues by isotopically labeling unoxidized methionines with 18O-labeled hydrogen peroxide and quantifying the relative ratios of 18O- and 16O-oxidized methionines. We validate our methodology using artificially oxidized proteomes made to mimic varying degrees of methionine oxidation. Using this method, we identify and quantify a number of novel sites of in vivo methionine oxidation in an unstressed human cell line.
Keywords: methionine oxidation, redox chemistry, hydrogen peroxide, oxidative protein damage, quantitative proteomics
Graphical Abstract
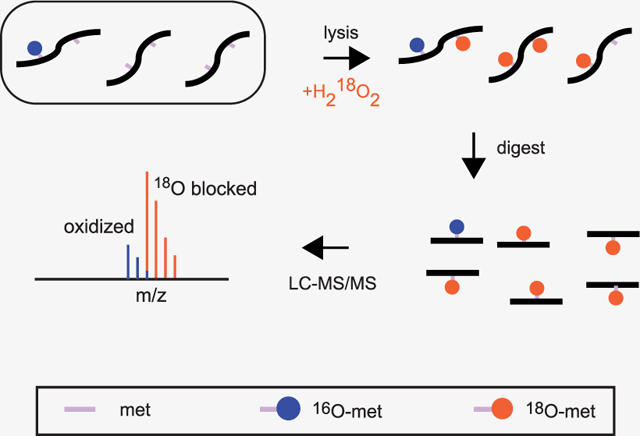
INTRODUCTION
The oxidation of methionine side chains has recently emerged as an important pathway for the post-translational modification of proteins. Methionine is normally a hydrophobic amino acid with an oxidatively labile thioether group.1 When oxidized, the chemical and physical properties of methionine are altered, causing alterations in the protein structure and stability.2–4 In this context, the oxidation of methionine has traditionally been thought of as a form of spontaneous protein damage. However, recent studies have shown that the regulated oxidation of methionine can also serve as a mechanism for modulating protein function.
A notable example of regulatory methionine oxidation is carried out by a family of enzymes known as MICALs (molecule interacting with CasL) that enzymatically oxidize specific methionine residues on actin.5–7 The oxidation of these methionine residues causes the formation of “fragile” F-actin filaments with an accelerated rate of depolymerization that has been shown to be important for the clearance of F-actin during cytokinesis and cell abscission.7,8 Furthermore, it has been shown that the conformation-specific oxidation of methionine residues in calmodulin results in a reduced association between calmodulin and the plasma membrane and an increase in calmodulin degradation by the proteasome.9,10 More recently, it has been shown that the liquid–liquid phase separation of the Ataxin-2 protein in Saccharomyces cerevisiae is regulated by the redox status of methionines in its low complexity domain.11 In the case of F-actin, the oxidation of methionine is enzymatic, whereas in the latter two cases, it is believed to be chemical. Furthermore, methionine sulfoxides can be reduced by the action of specialized methionine sulfoxide reductase (Msr) enzymes, providing another potential mode of regulation.12–14
Despite its importance to protein structure and function, the large-scale investigation of methionine oxidation in a complex matrix, such as the cellular proteome, is hampered by technical limitations. Methionine oxidation has been shown to spuriously accumulate during the upstream stages of a typical bottom-up proteomics workflow. In particular, methionine oxidation has been shown to increase with the length of trypsin digestion as well as the strength of ionization energy during electrospray ionization (ESI).15,16 These observations make it difficult to distinguish methionines that are oxidized in vivo from those that are artifactually oxidized in vitro during the course of sample preparation and mass spectrometric analysis. In addition, the bias of data-dependent acquisition (DDA) for more abundant peptides and a lack of adequate enrichment protocols complicate the identification and quantification of methionines that are oxidized at low levels. Furthermore, oxidation of methionine residues results in significant changes in retention times and ionization propensities, making it difficult to accurately quantify fractional oxidation by directly comparing the relative intensities of oxidized and unoxidized spectra of methionine-containing peptides.
Several recent methods for the quantification of methionine oxidation have been developed with the aim of circumventing these technical limitations. Ghesquierè et al. demonstrated a method termed COFRADIC (combined fractional diagonal chromatography) proteomics.17 This procedure isolates peptides that contain oxidized methionines by taking advantage of chromatographic shifts in identical reverse-phase high-performance liquid chromatography (RP-HPLC) runs of peptides before and after the reduction of methionine sulfoxides by purified Msr enzymes. Using this methodology, the authors were able to isolate and identify a large set of oxidized methionine residues in a hydrogen peroxide-stressed proteome from human Jurkat cells. This method was successful in increasing the number of methionine sulfoxide containing peptides that were detected compared to traditional bottom-up proteomic methods.18 However, the COFRADIC approach requires multiple additional sample preparation steps prior to proteomic analysis as well as the production of an isotopically labeled reference proteome.
Liu et al. and Shipman et al. independently developed a strategy for the quantification of methionine oxidation that relies on the isotopic labeling of unoxidized methionine residues with H2 18O2 during the early stages of sample preparation and prior to liquid chromatography–mass spectrometry (LC–MS/MS) analysis.19,20 This strategy results in the conversion of all unoxidized methionines to an 18O-labeled version of the oxidized peptide. Conversely, peptides that are already oxidized in vivo retain their 16O modifications. The 2 Da mass difference between the 16O- and 18O-labeled methionine-containing peptides is then used to distinguish between peptides that were unoxidized from those that were oxidized in vivo. The authors of these studies demonstrate that this strategy allows for the accurate quantification of methionine residues in a single protein.
Here, we report a modified version of the H2 18O2 blocking methodology and extend the quantification of 16O/18O-labeled methionine pairs to a proteome-wide level. Our strategy relies on the spectral identifications and MS1 annotations of the 18O-labeled peptides, which are then used to identify, deconvolute, and quantify the relative population of in vivo oxidized (16O-modified) peptides. We demonstrate the feasibility of this experimental approach and use it to measure in vivo methionine oxidation levels in unstressed human cells. Our data identifies a number of novel in vivo methionine oxidation sites while indicating that, as a whole, methionine oxidation is rare within the proteome of unstressed cells.
EXPERIMENTAL PROCEDURES
Cell Culture, Lysis, and H2 18O2 Treatment
Wild-type human epidermal fibroblast (MJT) cells were grown to confluency in Dulbecco’s modified Eagle medium (DMEM), supplemented with 15% fetal bovine serum (FBS) and 1% penicillin–streptomycin (Invitrogen), and harvested by trypsinization. Cells were lysed in 50 mM triethylammonium bicarbonate (TEAB) (Fischer Scientific) and 5% sodium dodecyl sulfate (SDS) by high-energy sonication and clarified of cell debris by centrifugation at 16 000×g for 10 min. Following lysis, protein concentration was quantified by bicinchoninic acid (BCA) assay and immediately diluted (1:1) to a final protein concentration of 0.5 mg/mL with either 18O heavy (Cambridge Isotope Laboratories) or 16O light (Fisher) H2O2 to a final H2O2 concentration of 1.25%. The oxidation reaction was allowed to continue for 1 h at room temperature. Disulfide bonds were reduced by adding 2 mM dithiothreitol (DTT) (Fisher), and protein alkylation was performed with 10 mM iodoacetamide (IAA) (Sigma). Samples were acidified by adding phosphoric acid to a final concentration of 1.2% and subsequently diluted 7-fold with 90% methanol in 100 mM TEAB. The samples were added to an S-trap column (Protofi), and the column was washed twice with 90% methanol in 100 mM TEAB. Trypsin (Pierce) was added to the S-trap column at a ratio of 1:25 (trypsin/protein), and the digest reaction was allowed to continue overnight at 37 °C. Peptides were eluted in 80 μL of 50 mM TEAB followed by 80 μL of 0.1% trifluoroacetic acid (TFA) (Pierce) in water and 80 μL of 50/50 acetonitrile/water in 0.1% TFA. Titration experiments were prepared by mixing light (16O)- and heavy (18O)-labeled proteomes in the specified ratio to a final protein amount of 50 μg. To increase proteome coverage, high-pH fractionation was conducted on extracts prior to LC–MS/MS. Extracts were fractionated using homemade C18 spin columns. Eight different elution buffers were made in 100 mM ammonium formate (pH 10) with 5, 7.5, 10, 12.5, 15, 17.5, 20, and 50% acetonitrile added. All fractions were then lyophilized and resuspended in 25 μL of 0.1% TFA.
LC–MS/MS Analysis
Fractionated peptides were injected onto a homemade 30 cm C18 column with 1.8 μm beads (Sepax), with an Easy nLC-1200 HPLC (Thermo Fisher), connected to a Fusion Lumos Tribrid mass spectrometer (Thermo Fisher). Solvent A was 0.1% formic acid in water, while solvent B was 0.1% formic acid in 80% acetonitrile. Ions were introduced to the mass spectrometer using a Nanospray Flex source operating at 2 kV. The gradient began at 3% B and held for 2 min, increased to 10% B over 5 min, increased to 38% B over 68 min, then ramped up to 90% B in 3 min and was held for 3 min, before returning to starting conditions in 2 min and re-equilibrating for 7 min, for a total run time of 90 min. The Fusion Lumos was operated in data-dependent mode, with MS1 scans acquired in the Orbitrap and MS2 scans acquired in the ion trap. The cycle time was set to 1.5 s to ensure that there were enough scans across the peak. Monoisotopic precursor selection (MIPS) was set to Peptide. The full scan was collected over a range of 375–1400 m/z, with a resolution of 120 000 at an m/z of 200, an AGC target of 4e5, and a maximum injection time of 50 ms. Peptides with charge states between 2 and 5 were selected for fragmentation. Precursor ions were fragmented by collision-induced dissociation (CID) using a collision energy of 30% with an isolation width of 1.1 m/z. The ion trap scan rate was set to rapid, with a maximum injection time of 35 ms and an AGC target of 1e4. Dynamic exclusion was set to 20 s.
MaxQuant Analysis and Data Conversion
Raw files for all samples were searched against the Homo sapiens UniProt database (downloaded 8/7/2017) using the integrated Andromeda search engine with the MaxQuant software.21 Peptide and protein quantifications were performed with MaxQuant using the default parameter settings. 18O Methionine sulfoxide, 16O methionine sulfoxide, 18O methionine sulfone, and N-terminal acetylation were set as variable modifications, and carbamidomethyl cysteine was set as a fixed modification. Raw files were converted to the mzXML format with the ProteoWizard’s MSConvert software22 using the vendor-supplied peak picking algorithm and the threshold peak filter set to the top 1500 peaks in each scan. The MaxQuant-supplied evidence file and the mzXML file were used as the input into a custom algorithm described below.
When analyzing the prevalence of other H2O2-induced modifications (Figure 2B), MaxQuant searches were conducted for unmodified peptides (except for carbamidomethylation of cysteines as a constant modification). The total measured intensities of all unmodified peptide spectral matches (PSMs) containing specific residues were measured as fractions of total intensities of all PSMs.
Figure 2.
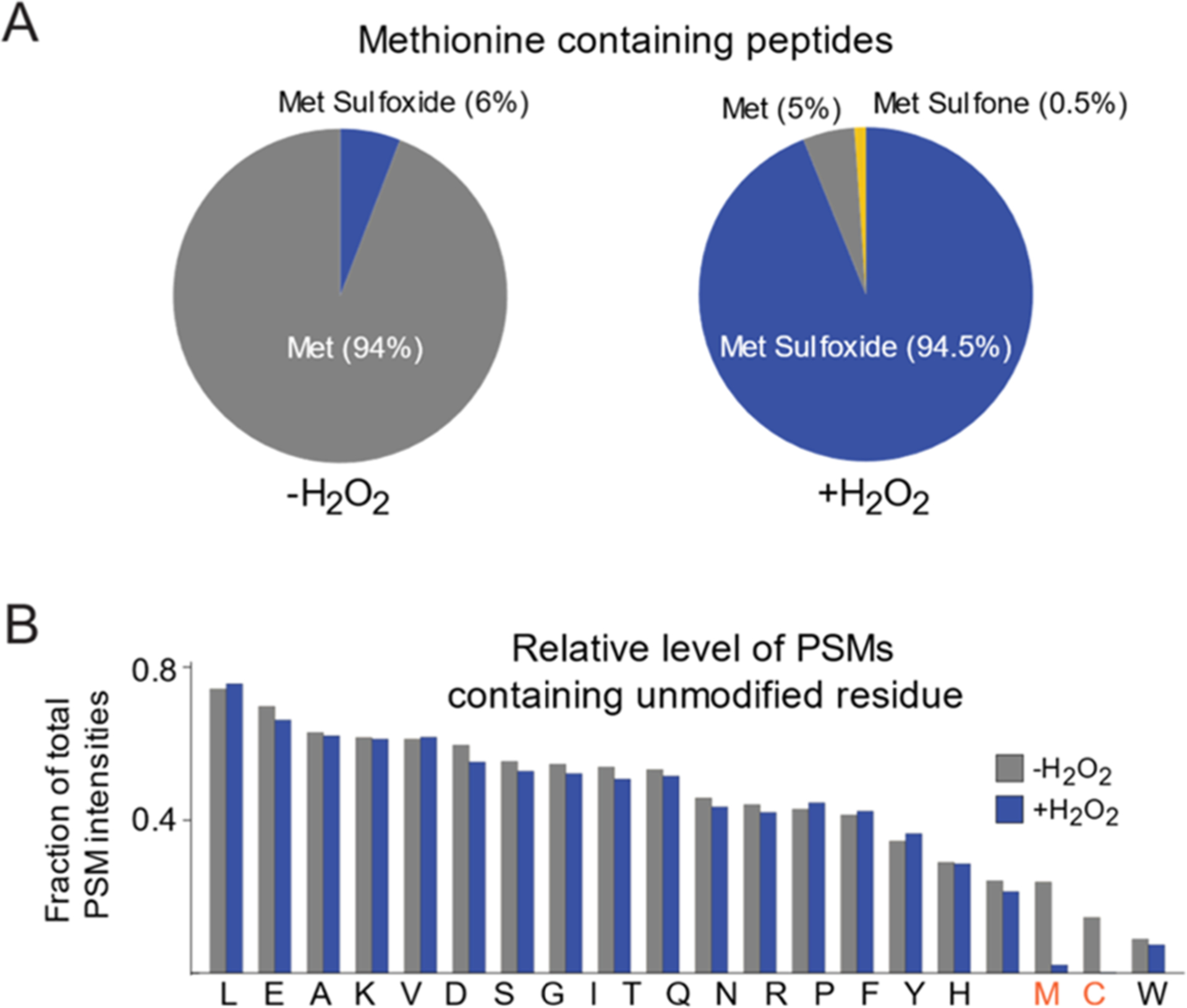
Validation of oxidation conditions. (A) The total spectral intensity of single methionine-containing peptides with no cysteines in unblocked samples (left) is predominately composed of peptides with unoxidized methionines with a small fraction of peptides containing methionine sulfoxides. This trend is reversed in blocked samples (right) where the predominant variants are methionine sulfoxide containing peptides with a small fraction of unoxidzed peptides and peptides with doubly oxidized methionine sulfones. (B) Comparison of relative numbers of PSMs found for unmodified peptides containing at least one of each amino acid before and after blocking with hydrogen peroxide. Amino acids that are found to be significantly modified by hydrogen peroxide treatment are shown in red.
All raw and processed data are available at ProteomeX-change Consortium via the PRIDE database (accession number PXD014629).
Custom Search and Quantification
For each MS1 peptide feature that was identified by MaxQuant, a theoretical isotope cluster was generated using the atomic composition of the peptide and the known abundance of natural isotopes. Next, for each peptide feature, the associated retention time range identified by MaxQuant was used to subset a swath of the MS1 spectra predicted to contain the associated feature. The swath was then filtered for peaks with a centroided mass to charge ratio within a 14 ppm window centered on the predicted mass to charge ratios for the identified peptide feature. Assembled peaks were then summed across the retention time range and projected onto two separate two-dimensional (2D) planes: the m/z-intensity plane and the retention time–intensity plane. Isotopic peaks were sequentially connected along the m/z axis only if they improved the fit to the theoretical isotope cluster along the m/z-intensity plane.
The spectra were then mined for the differentially labeled peptide within the same retention time window. If the identified peptide was unlabeled, the search defaulted to searching for the light (−2 Da) modification within the retention time window. Using the atomic composition of the peptide, theoretical models were generated for each peptide with theoretical ratios between the light- and heavy-labeled peptide ranging between 0 and 1 with a step size of 0.01. The theoretical model with the best fit to the observed data along the m/z-intensity plane was taken as the measured light/heavy ratio for that peptide.
| (1) |
where In is the summed intensity of each isotopic peak and n is the number of isotopic peaks assembled into the model. A minimum of three isotopic peaks was required to assemble a model with values ranging from 3 to 9 and a mean of 6.8 across all experiments. Intensities were expressed as values ranging between 0 and 1, normalized by the most intense isotopic peak. A maximum mean squared error (MSE) value of 5.00e–3 was allowed for a peptide to be quantified. The range of MSE values for peptides quantified across all experiments was 5.51e–06 to 4.99e–03 with a mean of 8.42e–04.
Three measures of model quality were used to assess each model and assign statistical confidence. On the retention time–intensity plane, a Spearman correlation in the retention time profiles between the light- and heavy-labeled peptide was measured (referred to as the RT Spearman correlation in Figure 3). In addition, a percent shift in the center of masses of the elution profiles between the heavy- and light-labeled peptide was measured (referred to as RT shift in Figure 3).
| (2) |
where In is the intensity of a datapoint expressed as a value ranging between 0 and 1 normalized by the maximum intensity and RTn is the associated retention time, RTS is the retention time start, and RTE is the retention time end, such that (RTE − RTS) is the retention time length. Next, the accumulated error along the m/z axis of the individual data points used to create the model was measured as the average difference between the theoretical mass to charge ratio and the centroided mass to charge ratio of the peaks assembled into the model (referred to as the m/z error in Figure 3).
| (3) |
| (4) |
where n is equal to the number of isotopic peaks assembled into the model and is identical to n in eq 1. Within each isotopic peak, the negative and positive m/z errors were allowed to cancel each other to maximize sensitivity to mass drift. However, between isotope clusters, the absolute value of the error is averaged.
Figure 3.
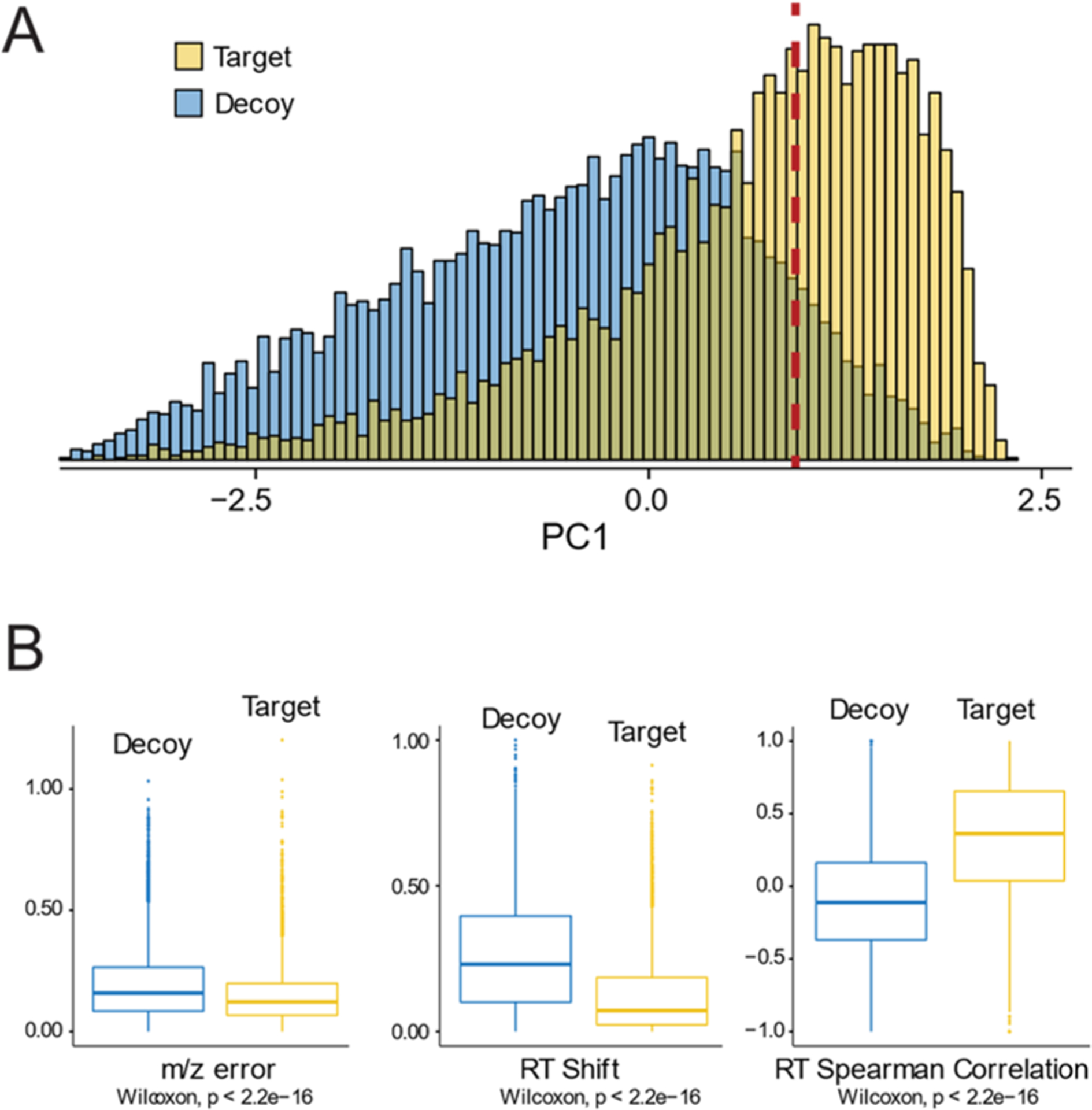
Quality control scoring results. (A) Principal component analysis (PCA) of the quality parameters shown in (B) was used to calculate a quality score for each peptide. Methionine-containing peptides (target) for which a true −2 Da modification is expected to exist have better scores than non-methionine-containing peptides (decoy) for which no −2 Da modification is expected to exist. The red dotted line shows the principal component value at which there is a global 10% false positive rate (FPR). All subsequent data analyses are done on peptides with principal component scores that are better than the cutoff value represented by the red dotted line. (B) The three quality parameters used to calculate the principal component score (see Experimental Procedures) show a significant distinction between target and decoy peptides. For both (A) and (B), results from the baseline titration point (0.0 18O:1.0 16O) are shown as a representative example.
Each peptide feature has a uniquely quantified model; however, most peptides are represented by multiple peptide features. As a final step, peptide features were aggregated together according to their sequence and modification status. Measurements taken along the m/z axis (fraction oxidized and m/z error) were reported as the intensity-weighted mean of each feature associated with the peptide.
| (5) |
where In is equal to the total intensity of the assembled feature and Xn is equal to the fraction oxidized or m/z error associated with the feature. Measurements taken along the retention time axis (Spearman correlation, relative RT shift) were reported as the cumulative result after consolidating all features associated with a peptide into a single feature. As a final step, the measured fraction oxidized for each peptide was corrected by the labeling efficiency of that peptide. Labeling efficiencies were calculated by comparing the assembled intensity of the oxidized peptide to the unoxidized version.
| (6) |
All subsequent data analyses were restricted to peptides with a 90% labeling efficiency or greater.
Assigning Statistical Confidence
Unlabeled peptides are not expected to have a −2 Da modification, and the model assembled for those peptides is an experimental approximation of the null distribution that is measured as a function of the background alone. All (−) methionine, (−) cysteine-containing peptides are unlabeled and assigned to a decoy dataset, whereas all (+) methionine, (−) cysteine are assigned to a target dataset
Quality parameters discussed above are reduced to a single score by principal component analysis. The distribution of principal component scores within the decoy dataset (unlabeled peptides) is then used to calculate a false positive rate (FPR) for each score.
| (7) |
Gene Ontology Enrichments
Gene ontologies (GO) were searched using the Database for Annotation, Visualization, and Integrated Discovery (DAVID).23,24 Proteins that contained at least one methionine that were found to be significantly oxidized between two biological replicates were used as the target dataset (N = 30), and proteins that were quantified between two biological replicates but not found to contain any significantly oxidized methionines were used as the background dataset (N = 941). A cutoff of 0.05 for the Bonferroni-corrected p-values was used to determine significantly enriched terms.
RESULTS
18O Blocking Methodology
We developed a stable isotope labeling strategy that allows for the accurate quantification of in vivo methionine oxidation while preventing artifactual in vitro methionine oxidation that typically occurs during the course of a bottom-up proteomic workflow. An overview of the experimental strategy is illustrated in Figure 1. The spontaneous in vitro oxidation of methionine during sample preparation may result in an overestimation of in vivo oxidation levels. We circumvent this problem by forced oxidation of methionines with 18O-labeled hydrogen peroxide (H2 18O2) at the time of cell lysis. Hence, relative intensities of 16O- and 18O-modified methionine-containing peptide provide a measure of in vivo fractional oxidation prior to lysis.
Figure 1.
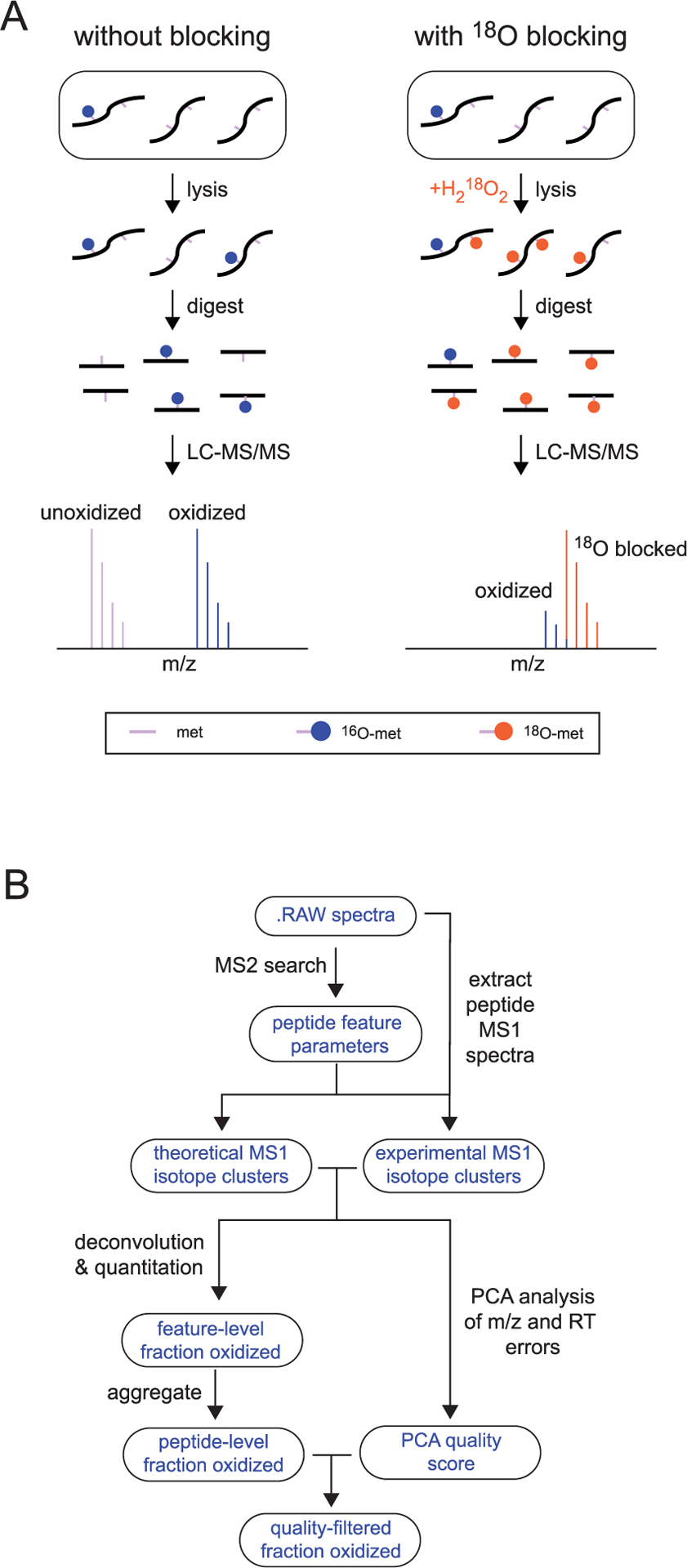
Schematic overview of 18O blocking methodology. (A) A comparison of strategies for quantifying methionine oxidation without (left) or with (right) blocking unoxidized methionines with a heavy-labeled oxidizing agent. Unlike blocked samples, unblocked samples accumulate oxidized methionines during sample preparation, resulting in an overestimation of oxidation levels. (B) A general overview of the computational strategy for finding, deconvoluting, and quantifying the relative ratios of light- and heavy-labeled peptides in blocked samples (see Experimental Procedures for additional details).
To rapidly block in vivo unoxidized methionines with 18O, cells were lysed in a denaturing buffer and subsequently exposed to excess levels of 18O-labeled hydrogen peroxide. Oxidation was allowed to proceed for 1 h at room temperature. These oxidation conditions allow for the near-complete labeling of all methionine-containing peptides (Figure 2A). In oxidized extracts, only ~5% of the total methionine-containing peptide intensities were due to unlabeled (unoxidized) peptides, <1% were due to doubly oxidized methionine sulfones, and the remaining ~95% were due to light-or heavy-labeled methionine sufloxides (Figure 2A). Independently conducted searches showed a low prevalence of other oxidized residues with the exception of cysteines (Figure 2B). Because of this potential complication, peptides containing both methionines and cysteines (~1% of all methionine-containing peptides) were excluded from the analysis.
Quantitation of Fractional Oxidation
A full description of the data analysis pipeline is provided in Experimental Procedures, and a brief graphical overview is shown in Figure 1B. Since in vivo 16O-oxidized methionines were rare within the unstressed MJT cells (see below), 18O-modified forms of methionine-containing peptides were typically identified by mass spectrometric data-dependent acquisition (DDA) of 18O-oxidized samples. The measured retention time (RT) and mass to charge ratios (m/z) of the 18O-labeled peptides were then used to conduct a model-dependent search to identify the entire isotope cluster and measure the relative ratios of 16O- and 18O-modified variants of the peptide. Since 16O- and 18O-modified peptides differ in mass by only 2 Da, quantitation of their relative levels requires deconvolution of isotopic clusters. In rare cases where the 16O-labeled version of methionine-containing peptides was identified by DDA, the quantitation was reversed to maximize coverage.
MS1 spectra of crude extracts are generally complex, and light 16O-labeled peptides commonly have low abundance relative to background peptides. We were, therefore, concerned that some of our assembled isotope cluster models may be dominated by the background signal coming from overlapping peptide features. To distinguish models representing true 16O-labeled peptides from the background, we calculated a quality score for each model that was dependent on the chromatographic correlation between the 16O- and 18O-labeled peptides as well as the m/z error of individual peaks assembled into the model. To assess the efficacy of our scoring function to distinguish true hits from the background, we conducted a decoy search composed of unlabeled peptide spectra for which we did not expect a true −2 Da modification (e.g., nonmethionine-containing peptides). Figure 3 shows a comparison of the distribution of scores calculated for labeled vs unlabeled peptides in our baseline titration point. The calculated scores show strong correlations with the different measures of the model error (Figure 3B), and labeled peptides have a significantly higher distribution of scores than unlabeled peptides (Figure 3A). This suggests that our scoring strategy can be used to distinguish true hits from the background. Using the distribution of scores in the unlabeled dataset, a false positive rate (FPR) was calculated for each score. All subsequent data analyses are restricted to peptides with scores associated with a FPR of <10%. An additional filter is applied to the fit of the theoretical isotope cluster with a hard cutoff of 5.0e–3. The filter applied to the fit to the theoretical isotope cluster is a user set parameter and was adjusted to balance accuracy and sensitivity. Together, the data in Figure 3 indicate that our scoring strategy effectively limits the level of background noise in our dataset and allows us to identify highly confident models.
Quantitative Validation of Fractional Oxidation Measurements
As a first application, we sought to validate our quantitative approach by conducting a spike-in titration experiment. At varying ratios, fully light (16O)-labeled proteome prepared from MJT cells was spiked into a background of a heavy (18O)-labeled proteome from the identical cell extract. Four titration samples were prepared with spike-in ratios of 0:1.0, 0.1:0.9, 0.25:0.75, and 0.4:0.6. As expected, the measured distribution of light/heavy ratios increases proportionally in accordance to spike-in ratios (Figure 4 and Table S1). Importantly, the measured light/heavy ratios for the decoy dataset do not increase with the amount of labeled spike-in, suggesting that they represent a true measurement of the background (Figure 4B and Table S1).
Figure 4.
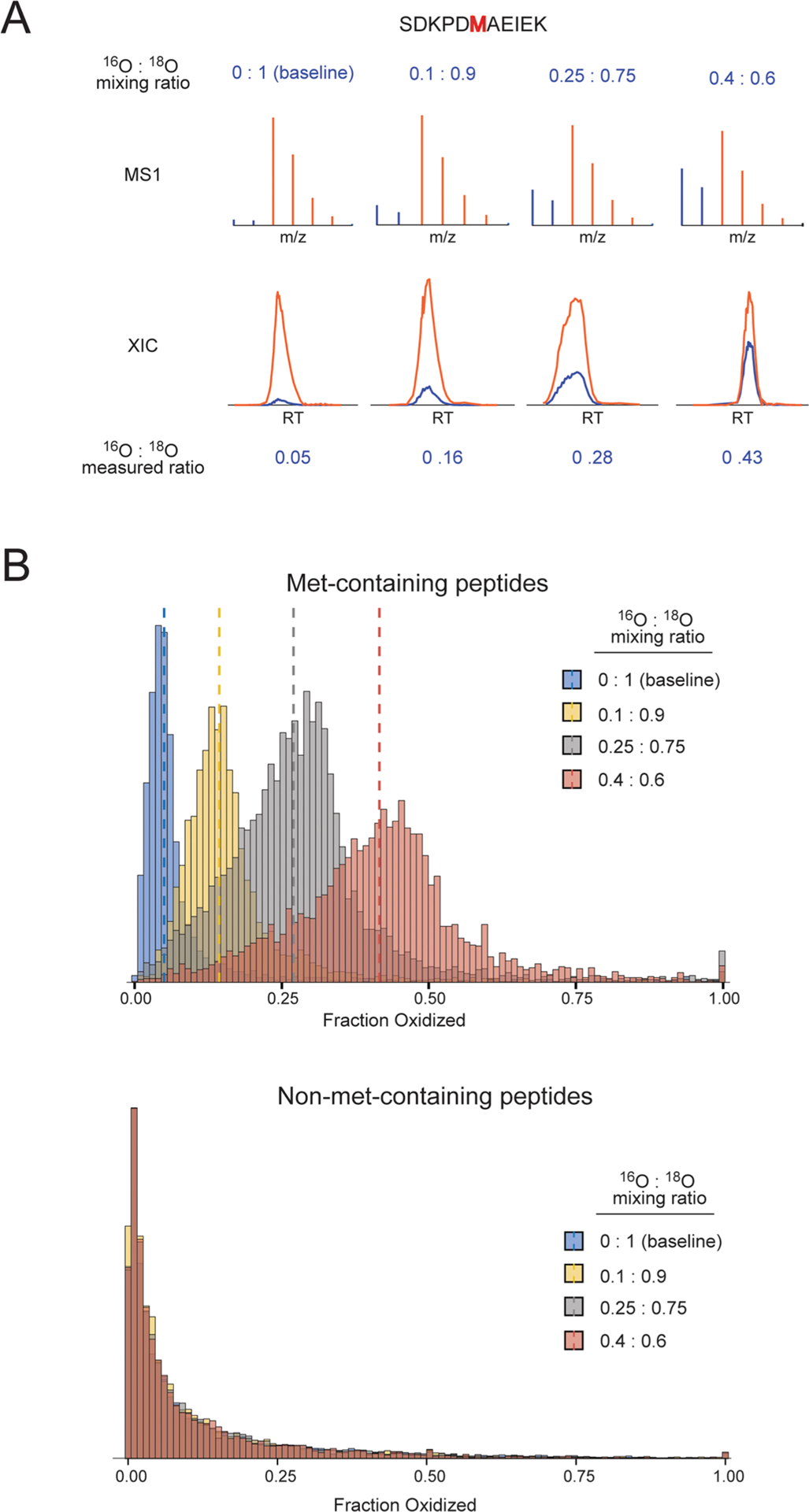
Spike-in titration results and validation of methodology. (A) A peptide detected in all titration points is shown as a representative example. The MS1 spectra of one feature representing the peptide are shown at the top and the total XIC of all features associated with the peptide is shown at the bottom. Red and blue colors represent the peaks and intensities corresponding to 18O and 16O peptides, respectively. The top and bottom rows of numbers indicate the mixing ratio and the measured fractional oxidation, respectively. (B) The distribution of measured fractional oxidation levels of all single methionine-containing peptides with no cysteines that passed the quality score filter is shown on top. Dotted lines indicate the respective median values. The null distribution of fractional oxidation levels for all non-methionine-containing peptides is shown at the bottom.
As tabulated in Table 1, the measured medians of fractional oxidation for the four spike-in experiments were 0.050, 0.140, 0.280, and 0.421. It is important to note that although the spike-in sample can be assumed to be fully light (16O) labeled, the same cannot be assumed for the heavy-labeled sample. The heavy-labeled sample was not cleared of all in vivo (16O) methionine oxidation prior to labeling, and the H 182O2 used as the labeling reagent contains some 16O isotopic impurity, thus the heavy-labeled background sample contains a mixture of 16O- and 18O-labeled methionine-containing peptides. Under the assumption that the majority of methionine-containing peptides are not oxidized under unstressed conditions, the global median of the light/heavy ratio for all methionine-containing peptides is representative of the isotopic impurity of the 18O-labeled H2O2 stock. We, therefore, calculated the expected median for each titration point using a dilution formula that takes into account the measured global median of the baseline titration point (0.0 18O:1.0 16O) as a value representative of the amount of isotopic impurity present in the background prior to spike-in.
| (8) |
where median0 is equal to the measured median of the light/heavy ratio for the background prior to spike-in, (1 − Fx) is equal to the fraction of the sample that is the heavy-labeled background by volume and Fx is the fraction of the sample that is light labeled by volume. The protein concentrations of the heavy- and light-labeled samples were standardized prior to mixing. Medianx is equal to the median of the light/heavy ratio for the light-labeled sample (the light-labeled sample contains no heavy-labeled peptides) and is equal to 1.0 for all calculations. Table 1 lists the measured medians and the expected medians. The difference between the expected median and the measured median did not exceed a maximum value of 0.9%, indicating that our experimental approach is capable of measuring the global levels of methionine fractional oxidation with a high degree of accuracy.
Table 1.
Global Statistics of Methionine Oxidation
| experiment | total met-containing peptides | Met-containing peptides analyzed | measured median fraction oxidized | expected median fraction oxidized |
|---|---|---|---|---|
| 0.0:1.0 baseline (replicate 1) | 12 981 | 2789 | 0.050 | NA |
| 0.0:1.0 baseline (replicate 2) | 11 767 | 2081 | 0.035 | NA |
| 0.10:0.90 | 12144 | 3879 | 0.140 | 0.145 |
| 0.25:0.75 | 10 765 | 4910 | 0.280 | 0.288 |
| 0.40:0.60 | 8433 | 3783 | 0.421 | 0.430 |
Levels of in Vivo Methionine Oxidation
The distribution of methionine oxidation levels in an unstressed human proteome appears to be significantly lower than previous estimates made on oxidatively stressed human proteomes.17 Within our baseline measurement of methionine oxidation in an unstressed human proteome, 39.8% of methionine-containing peptides had levels of in vivo oxidized (light-labeled) peptides that were below the limit of detection. The levels of light-labeled peptides that were below the limit of detection decreased as the amount of light-labeled spike-in increased (Table 1). For those peptides that had detectable levels of methionine oxidation, the median value measured for fractional oxidation was 5.0%, a value that is within the limit of isotopic impurity of H2 18O2 provided by the manufacturer (Figure 5).
Figure 5.
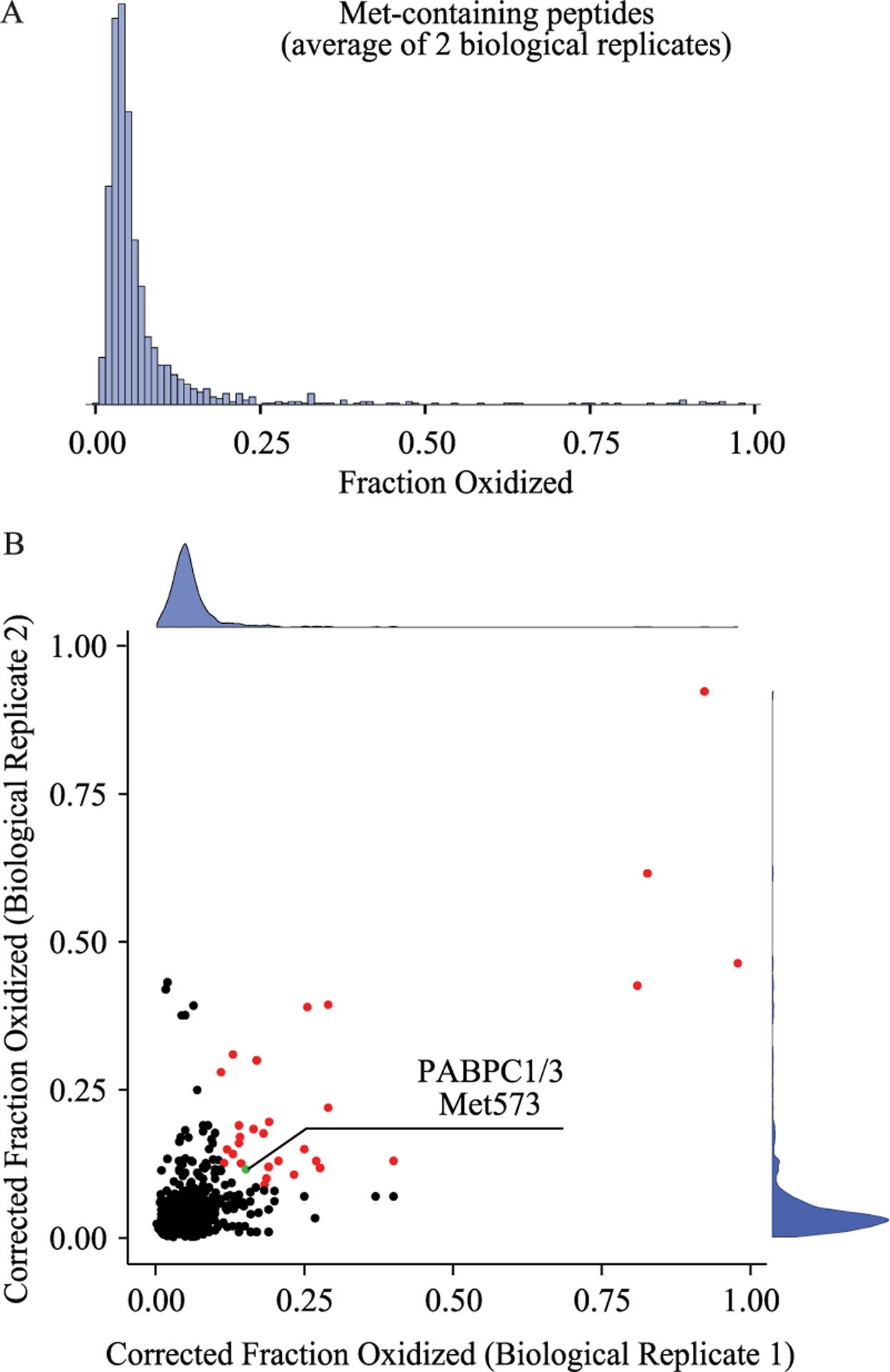
Distribution of in vivo methionine oxidation levels in unstressed MJT cells. (A) Measurements of methionine oxidation levels were made in two biological replicates and the distribution of the averaged results is shown. (B) A comparison of peptide level measurements of methionine oxidation between two biological replicates. The distributions of each replicate are shown on the respective axes. Dots are colored red if they are found to be highly and reproducibly oxidized in both replicates and have a labeling efficiency of at least 80% in both replicates. Met573 from PABCP1/3 (discussed below) is shown in green at the indicated position. Peptides represented by the red colored data points are tabulated in Table S2.
Measurements conducted on a biological replicate con-firmed the observation that the levels of methionine oxidation in an unstressed human proteome are low and within the limit of isotopic impurity with a median value of 3.5% (Figure 5 and Table 1). We observed a slight shift in observed methionine oxidation levels between the biological replicates. However, biological replicates were prepared using separate batches of 18O-labeled hydrogen peroxide, making it difficult to ascribe the observed shift to a biological source and highlighting the importance of batch effects in our assay. Nonetheless, we observed good agreement in the measured levels of methionine oxidation between replicates on a peptide to peptide level and were able to qualitatively detect a subset of peptides that were significantly oxidized in both replicates (Figure 5B).
For a subset of the isotope clusters assembled and used to quantify methionine oxidation, there is direct overlapping MS/MS evidence that both labeling variants of the peptide are present in the model (Table S2). Despite the observation that global levels of methionine oxidation are generally low, we observed a subpopulation of methionine-containing peptides that are observed to have reproducibly high levels of methionine oxidation in vivo (Table S2). A representative example of a verified and highly oxidized peptide is shown in Figure 6 and discussed below.
Figure 6.
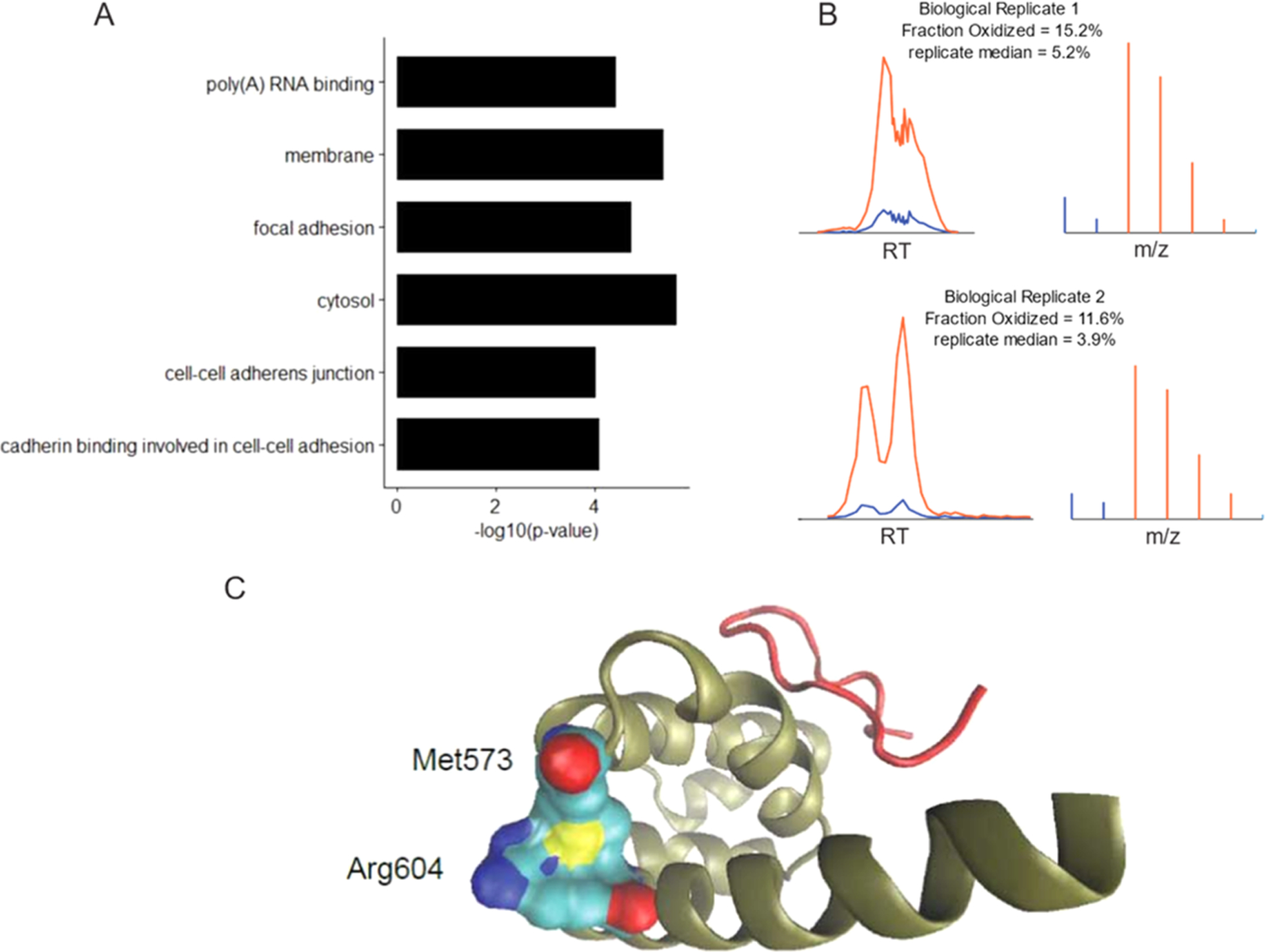
Gene ontology enrichment and a representative example of an oxidized protein (PABCP1/3). (A) Gene ontology terms associated with peptides found to be significantly oxidized between two biological replicates (FDR-corrected p-value < 0.05). (B) Models used to calculate the fraction oxidized for the peptide LFPLIQAMHPTLAGK within PABCP1/3. The total XIC of all features associated with the peptide is shown on the left and the summed MS1 spectra of the best-associated feature are shown on the right. Red and blue colors represent the peaks and intensities corresponding to 18O and 16O peptides. The “replicate median” values indicate the median fraction oxidized measurements for all methionine-containing peptides in the replicate experiment. (C) The structure of the C-terminal peptide-binding domain of PABPC1 (PDB:3KTP).25 The interacting partner GW182 is shown in red, and the main chain of PABPC1 is shown in gold. Sulfurs are colored yellow, carbons are colored cyan, oxygens are colored, red and nitrogens are colored dark blue.
DISCUSSION
We report a stable isotope labeling strategy that allows for quantification of in vivo levels of methionine oxidation on a proteome-wide scale. We used this approach to provide accurate measurements of methionine oxidation in an unstressed human proteome. This methodology relies on labeling and blocking of all in vivo unoxidized methionines with a heavy oxygen label immediately following cell lysis. As seen in Figure 2, the labeling and blocking efficiency of methionines was approximately 95%, thereby mitigating the accumulation of in vitro methionine oxidation that typically hampers the quantitative analysis of in vivo methionine oxidation.
Previous descriptions of the biological significance of methionine oxidation have stemmed from the observation that methionines in proteins are readily oxidized by reactive oxygen species such as hydrogen peroxide. For this reason, methionine oxidation has traditionally been thought of as a form of protein damage that occurs stochastically in response to oxidative stress. This notion, as well as previous measurements of methionine oxidation conducted on oxidatively stressed proteomes, suggested that global levels of methionine oxidation may be moderately high and broadly distributed.17 However, previous attempts at quantifying global levels of in vivo methionine oxidation in unstressed cells have relied on the use of reporter molecules and antibodies with low specificity.26,27 These approaches may not provide an accurate census of levels of methionine oxidation and do not allow for the quantification of the distribution of methionine oxidation across a diverse array of methionine-containing protein sequences. Here, we were able to quantify >3800 chemically distinct methionine-containing peptides in their native context across two biological replicates with ~1000 peptides shared between them. As seen in Figure 5, baseline levels of in vivo methionine oxidation in an unstressed human proteome are tightly distributed around median values of 5.0 and 3.5% in biological replicates 1 and 2, respectively, which are within the limit of isotopic impurity of 18O-labeled hydrogen peroxide used as a labeling reagent. In addition, 39.8 and 44.3% of all methionine-containing peptides analyzed had levels of oxidation that are below the limit of detection in replicates 1 and 2, respectively. These observations suggest that the levels of methionine oxidation in unstressed cells are maintained at minimal levels and only a small subset of methionine-containing peptides are significantly oxidized.
Although the global levels of methionine oxidation were observed to be low, our screen was able to detect a list of peptide sequences having reproducibly high levels of methionine oxidation with high confidence (Figure 5B and Table S2). Significantly oxidized peptides were defined using a nonparametric Wilcoxon test comparing the distance from the median for each peptide to the average distance from the median for all peptides. Gene ontology analysis of significantly oxidized peptides shows an enrichment for methionine oxidation found in the cytosol and cell membranes as well as proteins involved in cell adhesion and/or poly(A) RNA binding.
A representative example of a highly confident model that contains direct MS/MS evidence for both the heavy and light peptides is shown in Figure 6. Met573 from the C-terminal peptide-binding domain of polyA binding protein (PABPC1/3) is observed to be approximately 15.2 and 11.2% oxidized in vivo in replicates 1 and 2, respectively (p-value = 0.023). As can be seen in Figure 6, the sulfur atom of met573 appears to pack tightly against the backbone of Arg604, mediating a spatial interaction between the boundary region of two α-helices. Oxidation of met573 may disrupt this interaction and alter the peptide-binding properties of PABPC1/3. Interestingly, it has recently been shown that in S. cerevisiae, an interacting partner of PABPC1/3, Ataxin-2, is released from phase-separated granules in a methionine oxidation-dependent manner in response to mitochondrial stress where it subsequently binds to TORC to modulate cellular metabolism and dampen translational activity.28 Under conditions of mitochondrial stress, the further oxidation of met573 in the peptide-binding domain of PABCP1/3 may increase the propensity for Ataxin-2 to interact with TORC by disrupting its interaction with PABPC1/3.
In conclusion, we report a methodology that is able to accurately quantify levels of in vivo methionine oxidation across a large subset of methionine-containing peptides. The methodology is robustly sensitive toward detecting shifts in the global levels of methionine oxidation and can potentially be used in future studies to monitor changes in the distribution of methionine oxidation levels under various cellular conditions.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank members of the Ghaemmaghami lab at the University of Rochester for helpful discussions and suggestions. This work was supported by grants from the National Institutes of Health (R35 GM119502, 1S100D021486-01, and 1S10OD025242-01).
ABBREVIATIONS
- AGC
automatic gain control
- BCA
bicinchoninic acid
- CID
collision-induced dissociation
- COFRADIC
combined fractional diagonal chromatography
- DDA
data-dependent acquisition
- DTT
dithiothreitol
- DMEM
Dulbecco’s modified Eagle medium
- ESI
electrospray ionization
- FBS
fetal bovine serum
- FDR
false discovery rate
- HCD
higher-energy collisional dissociation
- IAA
iodoacetamide
- MICAL
molecule interacting with CasL
- m/z
mass to charge ratio
- PSM
peptide-spectrum match
- RP-HPLC
reverse-phase high-performance liquid chromatography
- RT
retention time
- SDS
sodium dodecyl sulfate
- TCEP
Tris(2-carboxyethyl)-phosphine
- TEAB
triethylammonium bicarbonate
- TFA
trifluoroacetic acid
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.9b00505.
Peptide level measured oxidation levels, quality control parameters, and score (XLSX)
Table of highly oxidized peptides (XLSX)
The authors declare no competing financial interest.
REFERENCES
- 1.Glaser CB; Li CH Reaction of bovine growth hormone with hydrogen peroxide. Biochemistry 1974, 13, 1044–1047. [DOI] [PubMed] [Google Scholar]
- 2.Chao CC; Ma YS; Stadtman ER Modification of protein surface hydrophobicity and methionine oxidation by oxidative systems. Proc. Natl. Acad. Sci. U.S.A 1997, 94, 2969–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Samson AL; Knaupp AS; Kass I; Kleifeld O; Marijanovic EM; Hughes VA; Lupton CJ; Buckle AM; Bottomley SP; Medcalf RL Oxidation of an exposed methionine instigates the aggregation of glyceraldehyde-3-phosphate dehydrogenase. J. Biol. Chem 2014, 289, 26922–26936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hsu YR; Narhi LO; Spahr C; Langley KE; Lu HS In vitro methionine oxidation of Escherichia coli-derived human stem cell factor: effects on the molecular structure, biological activity, and dimerization. Protein Sci 1996, 5, 1165–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grintsevich EE; Ge P; Sawaya MR; Yesilyurt HG; Terman JR; Zhou ZH; Reisler E Catastrophic disassembly of actin filaments via Mical-mediated oxidation. Nat. Commun 2017, 8, No. 2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hung RJ; Pak CW; Terman JR Direct redox regulation of F-actin assembly and disassembly by Mical. Science 2011, 334, 1710–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee BC; Peterfi Z; Hoffmann FW; Moore RE; Kaya A; Avanesov A; Tarrago L; Zhou Y; Weerapana E; Fomenko DE; Hoffmann PR; Gladyshev VN MsrB1 and MICALs regulate actin assembly and macrophage function via reversible stereoselective methionine oxidation. Mol. Cell 2013, 51, 397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frémont S; Hammich H; Bai J; Wioland H; Klinkert K; Rocancourt M; Kikuti C; Stroebel D; Romet-Lemonne G; Pylypenko O; Houdusse A; Echard A Oxidation of F-actin controls the terminal steps of cytokinesis. Nat. Commun 2017, 8, No. 14528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Balog EM; Lockamy EL; Thomas DD; Ferrington DA Site-specific methionine oxidation initiates calmodulin degradation by the 20S proteasome. Biochemistry 2009, 48, 3005–3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCarthy MR; Thompson AR; Nitu F; Moen RJ; Olenek MJ; Klein JC; Thomas DD Impact of methionine oxidation on calmodulin structural dynamics. Biochem. Biophys. Res. Commun 2015, 456, 567–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kato M; Yang YS; Sutter BM; Wang Y; McKnight SL; Tu BP Redox State Controls Phase Separation of the Yeast Ataxin-2 Protein via Reversible Oxidation of Its Methionine-Rich Low-Complexity Domain. Cell 2019, 177, 711–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Antoine M; Boschi-Muller S; Branlant G Kinetic characterization of the chemical steps involved in the catalytic mechanism of methionine sulfoxide reductase A from Neisseria meningitidis. J. Biol. Chem 2003, 278, 45352–45357. [DOI] [PubMed] [Google Scholar]
- 13.Olry A; Boschi-Muller S; Branlant G Kinetic characterization of the catalytic mechanism of methionine sulfoxide reductase B from Neisseria meningitidis. Biochemistry 2004, 43, 11616–11622. [DOI] [PubMed] [Google Scholar]
- 14.Moskovitz J; Poston JM; Berlett BS; Nosworthy NJ; Szczepanowski R; Stadtman ER Identification and characterization of a putative active site for peptide methionine sulfoxide reductase (MsrA) and its substrate stereospecificity. J. Biol. Chem 2000, 275, 14167–14172. [DOI] [PubMed] [Google Scholar]
- 15.Zang L; Carlage T; Murphy D; Frenkel R; Bryngelson P; Madsen M; Lyubarskaya Y Residual metals cause variability in methionine oxidation measurements in protein pharmaceuticals using LC-UV/MS peptide mapping. J. Chromatogr. B 2012, 895–896, 71–76. [DOI] [PubMed] [Google Scholar]
- 16.Chen M; Cook KD Oxidation artifacts in the electrospray mass spectrometry of Abeta Peptide. Anal. Chem 2007, 79, 2031–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghesquière B; Jonckheere V; Colaert N; Van Durme J; Timmerman E; Goethals M; Schymkowitz J; Rousseau F; Vandekerckhove J; Gevaert K Redox proteomics of protein-bound methionine oxidation. Mol. Cell. Proteomics 2011, 10, No. M110.006866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosen H; Klebanoff SJ; Wang Y; Brot N; Heinecke JW; Fu X Methionine oxidation contributes to bacterial killing by the myeloperoxidase system of neutrophils. Proc. Natl. Acad. Sci. U.S.A 2009, 106, 18686–18691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu H; Ponniah G; Neill A; Patel R; Andrien B Accurate determination of protein methionine oxidation by stable isotope labeling and LC-MS analysis. Anal. Chem 2013, 85, 11705–11709. [DOI] [PubMed] [Google Scholar]
- 20.Shipman JT; Go EP; Desaire H Method for Quantifying Oxidized Methionines and Application to HIV-1 Env. J. Am. Soc. Mass Spectrom 2018, 29, 2041–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cox J; Mann M MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 2008, 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- 22.Kessner D; Chambers M; Burke R; Agus D; Mallick P ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 2008, 24, 2534–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang DW; Sherman BT; Lempicki RA Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 2009, 4, 44–57. [DOI] [PubMed] [Google Scholar]
- 24.Huang DW; Sherman BT; Lempicki RA Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kozlov G; Safaee N; Rosenauer A; Gehring K Structural basis of binding of P-body-associated proteins GW182 and ataxin-2 by the Mlle domain of poly(A)-binding protein. J. Biol. Chem 2010, 285, 13599–13606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Péterfi Z; Tarrago L; Gladyshev VN Practical guide for dynamic monitoring of protein oxidation using genetically encoded ratiometric fluorescent biosensors of methionine sulfoxide. Methods 2016, 109, 149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Le DT; Liang X; Fomenko DE; Raza AS; Chong CK; Carlson BA; Hatfield DL; Gladyshev VN Analysis of methionine/selenomethionine oxidation and methionine sulfoxide reductase function using methionine-rich proteins and antibodies against their oxidized forms. Biochemistry 2008, 47, 6685–6694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang Y-S; Kato M; Wu X; Litsios A; Sutter BM; Wang Y; Hsu C-H; Wood NE; Lemoff A; Mirzaei H; Heinemann M; Tu BP Yeast Ataxin-2 Forms an Intracellular Condensate Required for the Inhibition of TORC1 Signaling during Respiratory Growth. Cell 2019, 177, 697–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.