

Abstract
A comparative study of the impact of small, medium-sized, and bulky α,β-dehydroamino acids (ΔAAs) on the structure and stability of Balaram’s incipient 310-helical peptide (1) is reported. Replacement of the N-terminal Aib residue of 1 with a ΔAA afforded peptides 2a–c that maintained the 310-helical shape of 1. In contrast, installation of a ΔAA in place of Aib-3 yielded peptides 3a–c that preferred a β-sheet-like conformation. The impact of the ΔAA on peptide structure was independent of size, with small (ΔAla), medium-sized (Z-ΔAbu), and bulky (ΔVal) ΔAAs exerting similar effects. The proteolytic stabilities of 1 and its analogs were determined by incubation with Pronase. Z-ΔAbu and ΔVal increased the resistance of peptides to proteolysis when incorporated at the 3-position and had negligible impact on stability when placed at the 1-position, whereas ΔAla-containing peptides degraded rapidly regardless of position. Exposure of peptides 2a–c and 3a–c to the reactive thiol cysteamine revealed that ΔAla-containing peptides underwent conjugate addition at room temperature, while Z-ΔAbu- and ΔVal-containing peptides were inert even at elevated temperatures. These results suggest that both bulky and more accessible medium-sized ΔAAs should be valuable tools for bestowing rigidity and proteolytic stability on bioactive peptides.
Graphical Abstract
INTRODUCTION
The great potential of peptides as therapeutic agents1 continues to inspire the discovery of new methods for increasing their short half-lives, which are caused by facile proteolytic degradation and rapid renal clearance. A host of structural motifs including peptoids,2 β and α/β peptides,3 stapled peptides,4 hydrogen bond surrogates,5 γAA-peptides,6 d-peptides,7 α,α-disubstituted amino acids,8 N-amino peptides,9 and β-turn mimics10 can decrease their susceptibility to proteolysis. Additionally, techniques such as PEGylation,11 conjugation to albumin or antibody fragments,12 and anchoring to plasma membranes13 are effective at slowing renal clearance and/or proteolysis. In spite of these successes, only a modest number of peptide drugs is currently reaching the market.14 Thus, more strategies for increasing the half-lives of these potential therapeutics are urgently required.
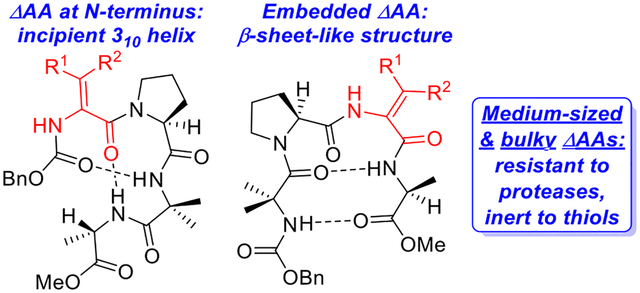
In seminal work conducted 35–40 years ago, Stammer and co-workers established that α,β-dehydroamino acids (ΔAAs) such as ΔAla, Z-ΔPhe, and Z-ΔLeu could improve the proteolytic stability of the enkephalin peptide hormones.15 In regards to ΔPhe and ΔLeu, this phenomenon can be attributed to the A1,3 strain caused by the alkene, which limits the conformational freedom of these residues and increases the rigidity of peptides that contain them. This rigidity favors folded states instead of random coil conformations and leads to greater stability since folded peptides undergo proteolysis more slowly than unfolded peptides.16 Subsequent to Stammer’s pioneering investigations, Chauhan17,18 and others19 studied the impact of ΔAAs containing trisubstituted alkenes (e.g., ΔPhe, ΔLeu) on peptide structure. In general, these residues promote the formation of β-turns or 310 helices (Figure 1). In fact, Dong and co-workers recently exploited the turn-inducing properties of ΔPhe as an aid to cyclizing pentapeptides.20 This ΔAA and its analogs have been inserted into the backbones of bioactive peptides with promising results.21 ΔPhe and other ΔAAs that incorporate trisubstituted alkenes are typically encountered as the more stable Z-isomers due to challenges in constructing the E-isomers22 and E-to-Z isomerization that often occurs during peptide coupling reactions.23
Figure 1.

Two types of dehydroamino acids.
Despite their presence in bioactive peptides including the yaku’amides,24 phomopsins,25 and myxovalargin A,26 bulky dehydroamino acids such as ΔVal and ΔIle that contain tetrasubstituted alkenes have been less studied than their counterparts with trisubstituted Z-alkenes (Figure 1).27,28 We hypothesized that the elevated A1,3 strain characteristic of bulky ΔAAs would render them more effective than smaller ΔAAs at rigidifying peptides and increasing their proteolytic stability. However, this strain also makes them more difficult to incorporate into peptides than their smaller relatives. A few years ago, we developed a protocol for synthesizing peptides containing ΔVal and its bis-homolog dehydroethylnorvaline (ΔEnv) via solid-phase peptide synthesis.29 We applied this method to the construction of β-hairpins and demonstrated that inserting bulky ΔAAs into these peptides leads to an increased preference for the folded state as well as substantially higher proteolytic stability.30 The synthetic challenges that we faced during this endeavor, which included construction of congested azlactone dipeptides and sluggish couplings of these species, caused us to wonder how the effect of bulky ΔAAs on peptide structure and stability compared to that of medium-sized ΔAAs with trisubstituted alkenes. Specifically, we were unsure if the extra effort required to furnish peptides containing bulky ΔAAs was worthwhile. Comparisons of these two types of ΔAAs in the literature have been limited to studies of individual amino acids27f–h or dipeptides27a that are too small to exhibit secondary structure. Accordingly, we resolved to examine the impact of small (i.e., ΔAla), medium-sized (i.e., dehydro-2-aminobutyric acid or ΔAbu), and bulky ΔAAs (i.e., ΔVal) on the structure and stability of incipient 310-helical peptides.31 Herein, we report the results of this investigation.
RESULTS AND DISCUSSION
In 1979, Balaram and co-workers synthesized tetrapeptide 1 (Figure 2) and studied its structure using X-ray crystallography and NMR spectroscopy. They determined that 1 forms two overlapping β-turns in solution and in the solid state, representing an incipient 310 helix due to the presence of consecutive (i → i + 3) hydrogen bonds.31 This compound is well-suited as a system for studying the impacts of different ΔAAs on peptide structure and stability, as the turn-inducing properties of these residues should cause subtle yet measurable perturbations to its secondary structure. Moreover, the short and simple sequence of 1 should render its ΔAA-containing variants synthetically accessible and relatively straightforward to study via NMR spectroscopy. Thus, we targeted peptides 2 (Figure 2) in which the Aib-1 residue is replaced by ΔVal (2a), ΔAla (2b), or Z-ΔAbu (2c) in order to probe the impact of these ΔAAs when they are inserted at the (i + 1) position of one of the β-turns contained in the incipient 310 helix. Similarly, peptides 3 would permit evaluation of the effect of ΔAAs when placed at the (i + 2) position.
Figure 2.
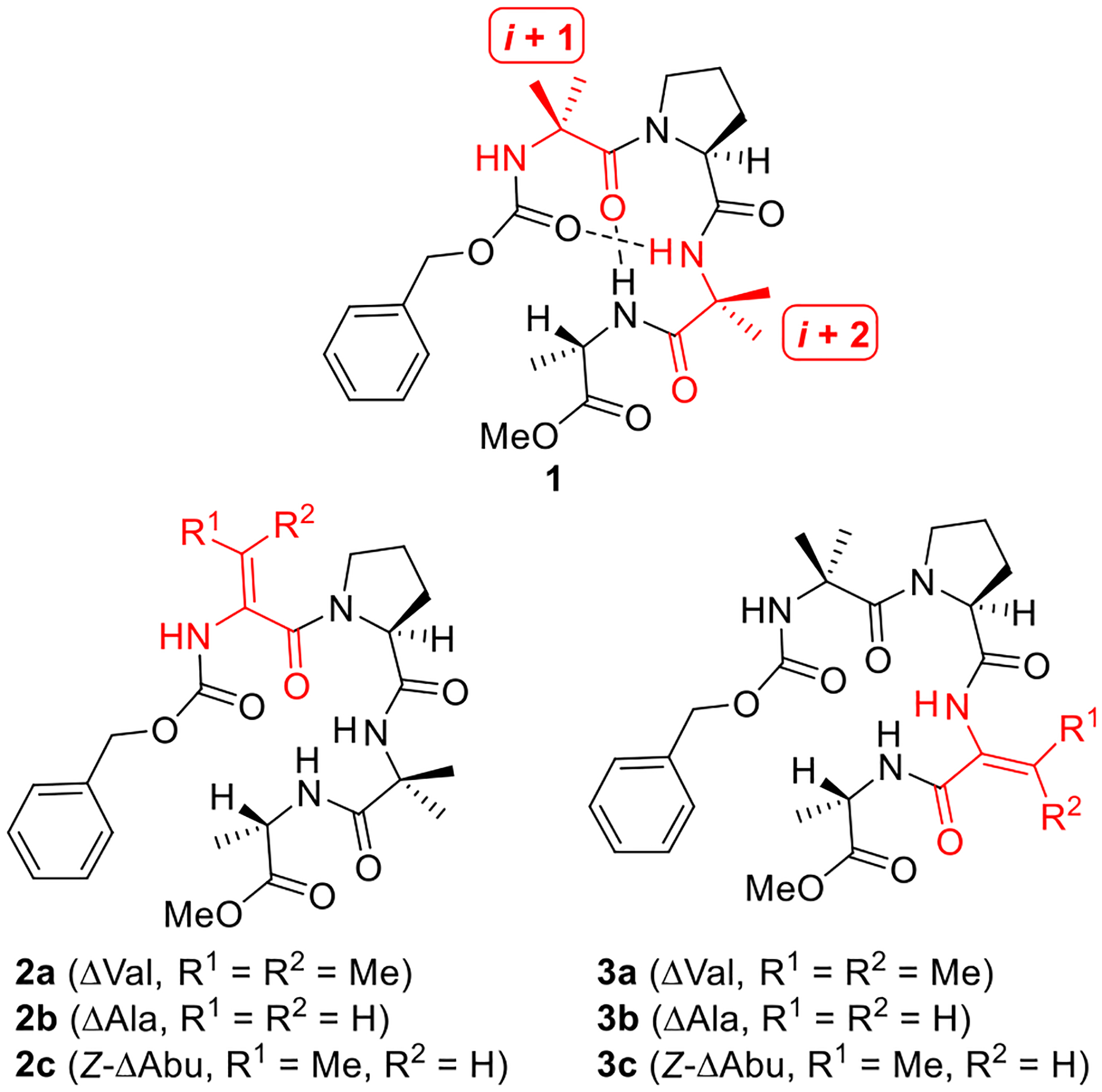
Balaram’s incipient 310 helical peptide (1) and proposed ΔAA-containing variants (2 and 3).
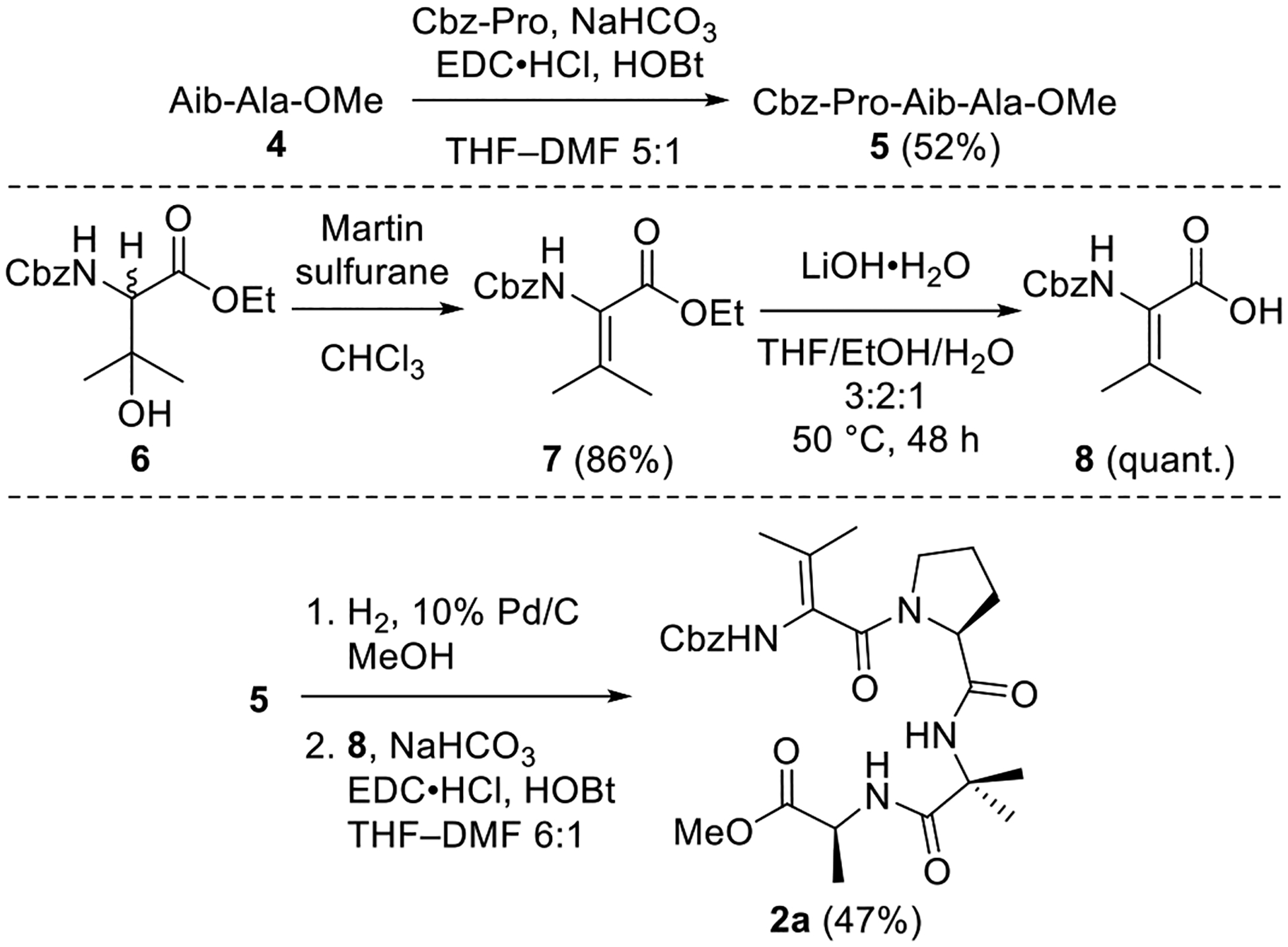
The synthesis of analog 2a incorporating a bulky ΔAA in place of the Aib-1 residue in 1 commenced with the construction of tripeptide 5 via coupling of Aib-Ala-OMe (4) and Cbz-Pro (Scheme 1). The moderate yield is presumably due to the steric hindrance of the two coupling partners. ΔVal derivative 7 was generated in good yield by using Martin sulfurane to dehydrate β-OHVal derivative 6, which is readily available via OsO4-catalyzed aminohydroxylation.29,32 Saponification of 7 was sluggish owing to the A1,3 strain caused by the β-methyl groups, with two days required to obtain complete conversion to acid 8. Coupling of this acid with the amine derived from hydrogenolysis of tripeptide 5 furnished the targeted tetrapeptide 2a in reasonable yield considering bulkiness of the reactants.
Scheme 1. Synthesis of ΔVal Analog 2a.
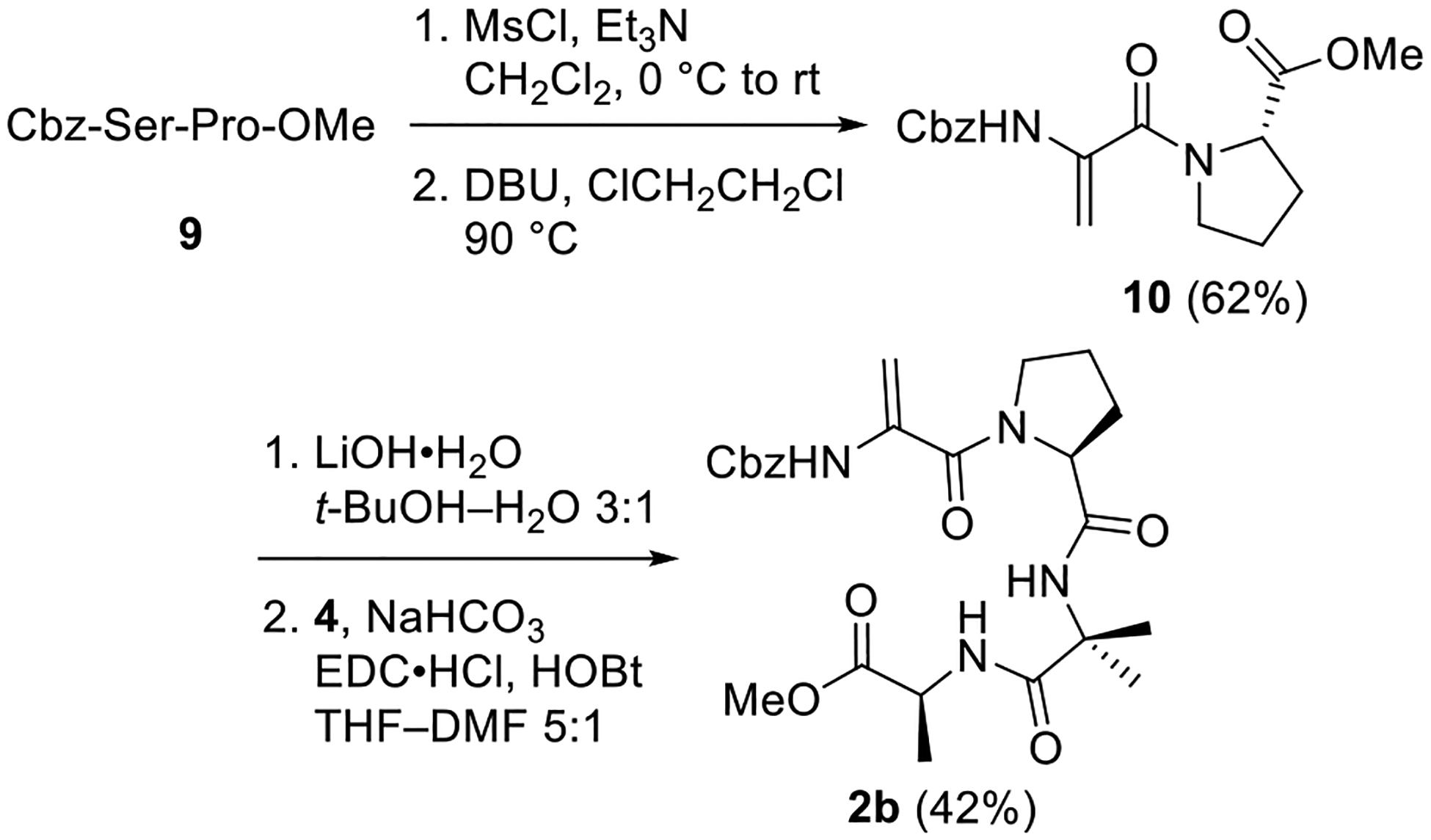
Preparation of ΔAla-containing analog 2b proceeded smoothly as outlined in Scheme 2. Dehydration of the dipeptide Cbz-Ser-Pro-OMe (9) was accomplished by mesylation of the primary alcohol followed by exposure to DBU and heat.33 No racemization of the Pro residue was detected under these conditions. Saponification of the resulting ΔAla-containing dipeptide and coupling with dipeptide 4 delivered 2b in an acceptable yield considering the hindered nature of the reactants.
Scheme 2. Synthesis of ΔAla Analog 2b.
The synthesis of variant 2c possessing the medium-sized trisubstituted ΔAA Z-ΔAbu required coupling of dipeptides 4 and 11, which was a challenging reaction (Scheme 3). Brief attempts at optimization revealed that HBTU could promote the coupling in low but consistent yields. While it is possible that either protecting the secondary alcohol of 11 or inverting the order of the dehydration and coupling steps could have resulted in better yields, we elected not to explore these alternatives since we were able to generate the required amounts of 2c using the short sequence shown in Scheme 3. This peptide was produced in modest yield by dehydrating tetrapeptide 12 with the Martin sulfurane.22a
Scheme 3. Synthesis of Z-ΔAbu Analog 2c.
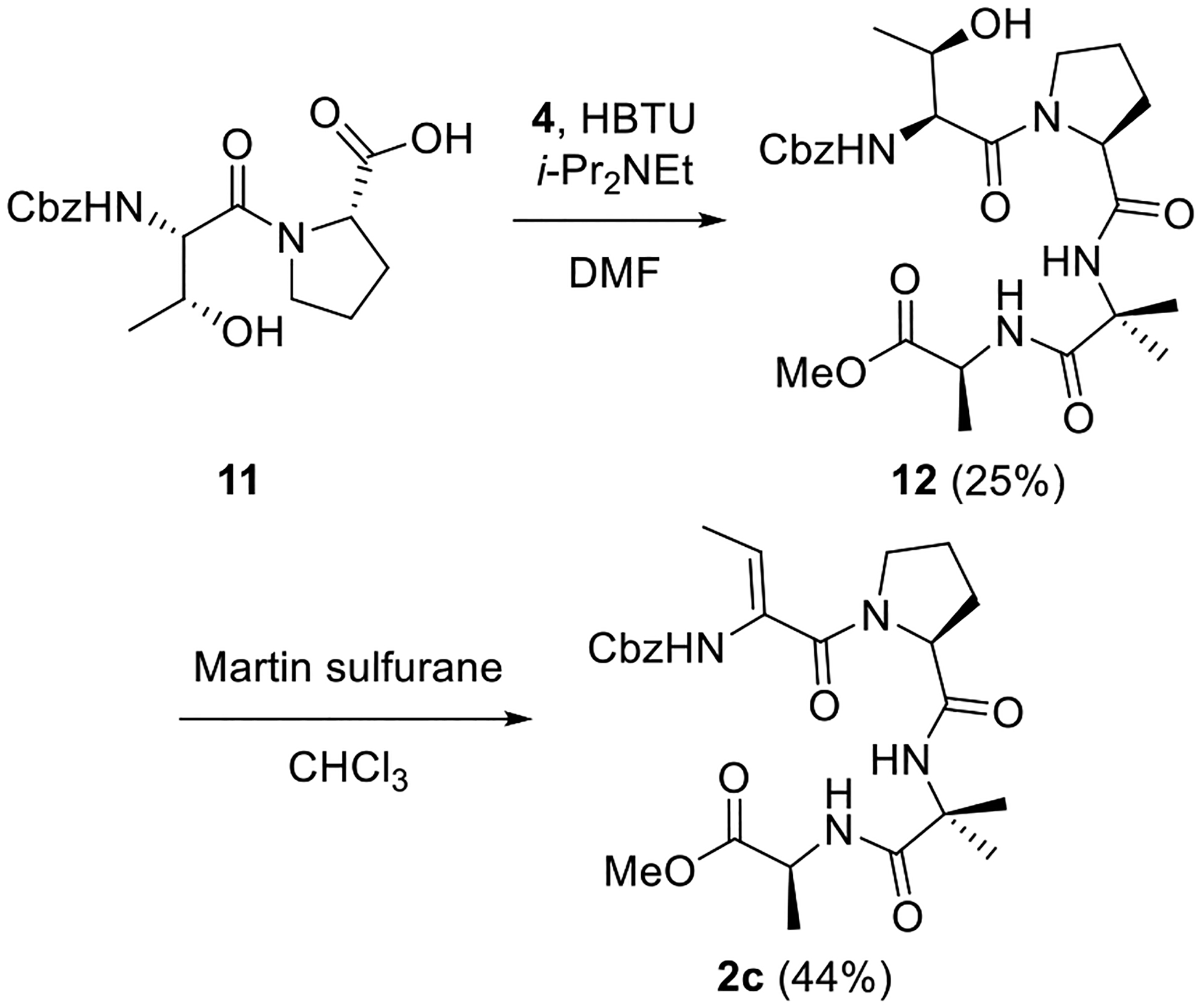
Analog 3a, which contains ΔVal instead of the Aib-3 residue of Balaram’s peptide, was synthesized as shown in Scheme 4. The dipeptide Cbz-Aib-Pro was coupled with racemic β-OH-Val derivative 14 to afford tripeptide 15 in good yield. Saponification of this peptide was followed by dehydration and azlactone formation utilizing NaOAc and Ac2O in THF.30 Ring-opening and coupling of the hindered azlactone intermediate was difficult. A brief survey of conditions revealed that serviceable yields of tetrapeptide 3a could be obtained by first hydrolyzing the azlactone through treatment with NaOH and then employing PyBOP to mediate coupling with Ala-OMe.34
Scheme 4. Synthesis of ΔVal Analog 3a.
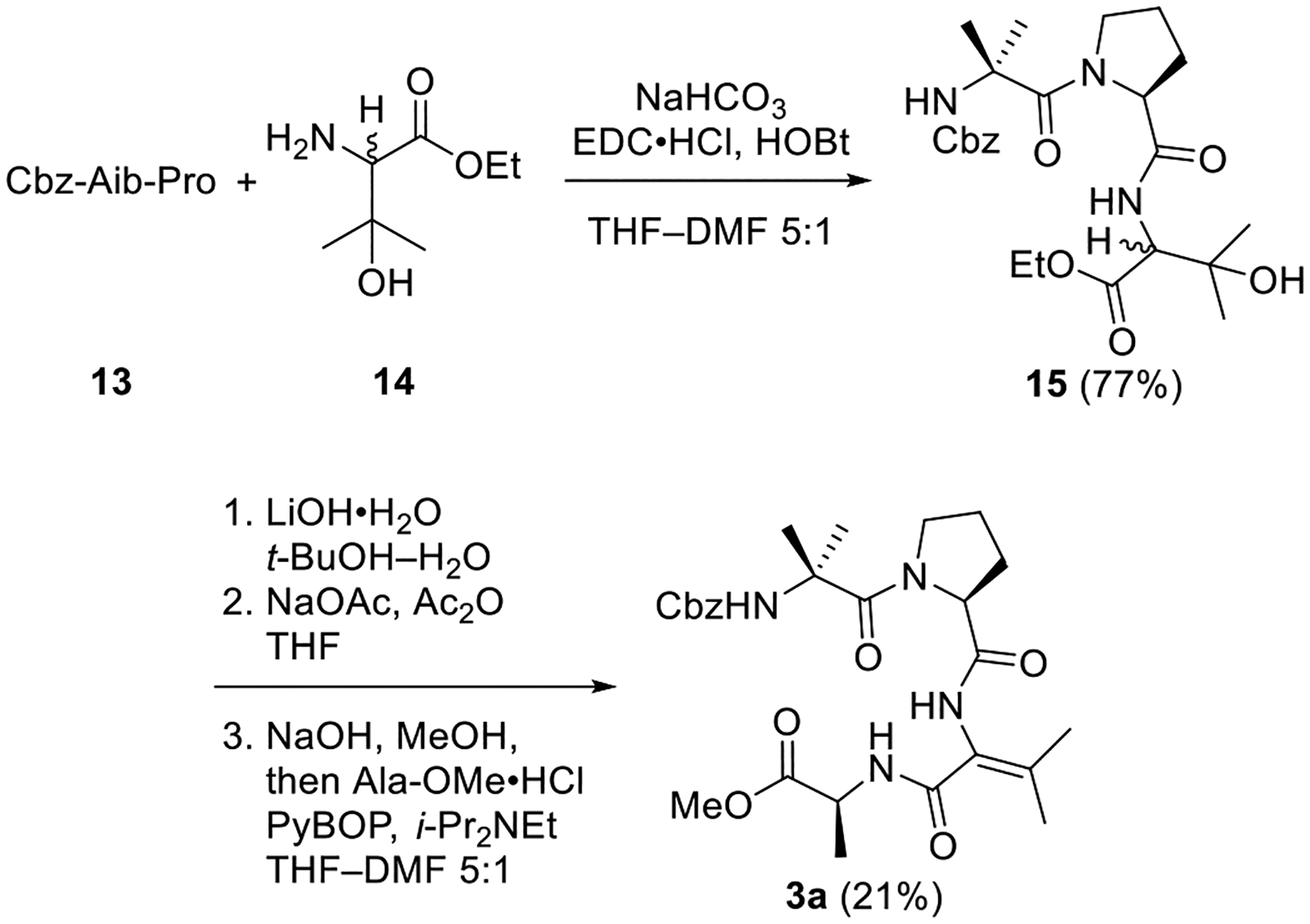
Construction of variant 3b wherein the Aib-3 residue of 1 is replaced by ΔAla is summarized in Scheme 5. Hydrogenolysis of the dipeptide Cbz-Ser-Ala-OMe (16) was followed by coupling with dipeptide 13, furnishing tetrapeptide 17 in moderate yield. As in the coupling of 11, protection of the alcohol moiety in 16 was eschewed in favor of a short and direct synthetic route to 3b. Mesylation and elimination of the primary alcohol in 17 was lower-yielding than the analogous transformation of dipeptide 9 (see Scheme 2), as 3b was obtained in 36% yield.
Scheme 5. Synthesis of ΔAla Analog 3b.
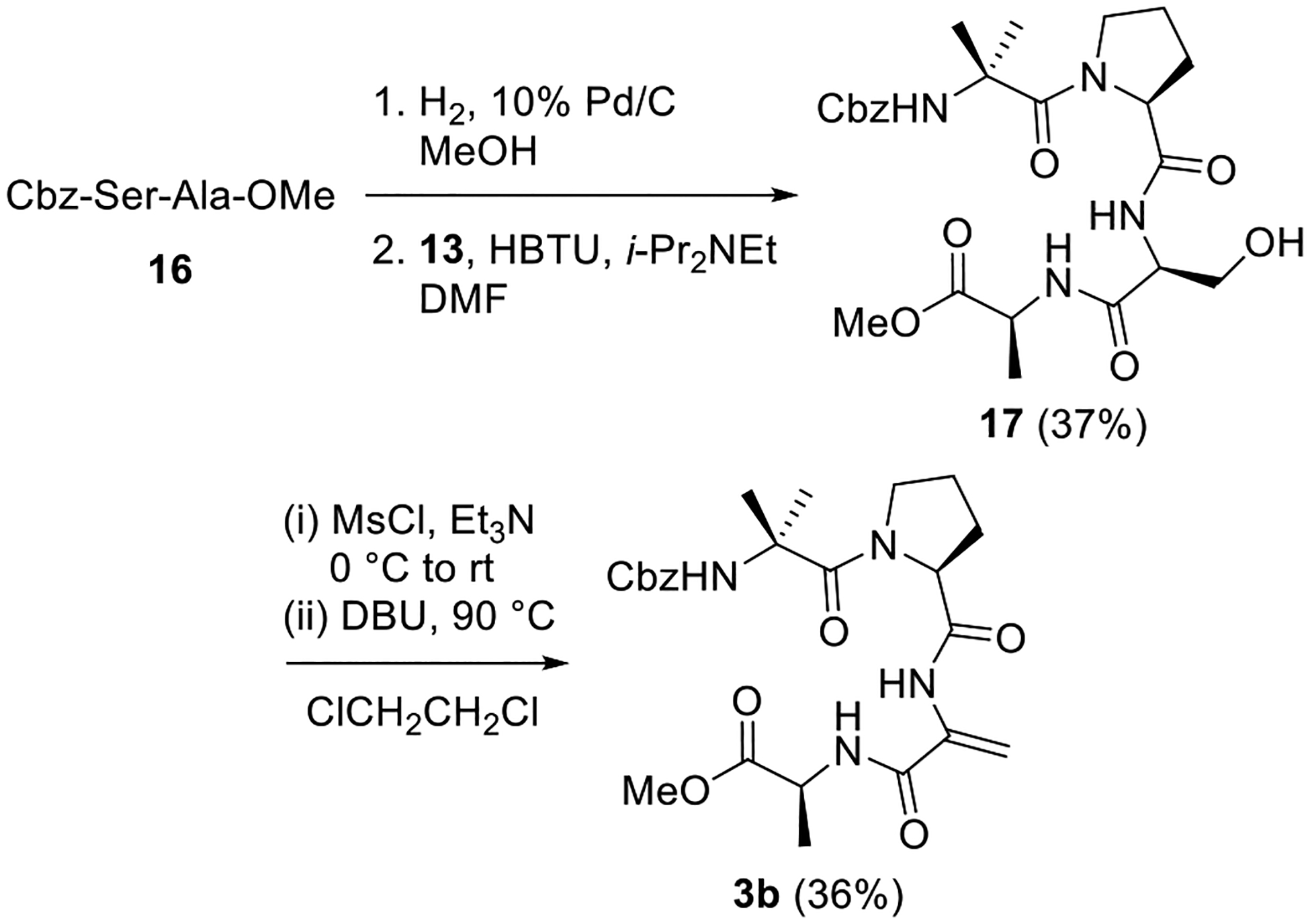
Preparation of analog 3c possessing Z-ΔAbu in place of Aib-3 commenced with coupling of 13 to dipeptide 18 (Scheme 6). We suspect that steric hindrance is the primary reason for the low yields of this reaction. Exposure of the resulting tetrapeptide 19 to the Martin sulfurane afforded 3c. The poor yields that we encountered while synthesizing some of the analogs of 1 are indicative of limits in current methods for constructing hindered peptides containing ΔAAs. While our concise routes enabled us to overcome these problems and generate sufficient quantities of the small target peptides 2 and 3, we plan to address these limitations in future studies.
Scheme 6. Synthesis of Z-ΔAbu Analog 3c.
The 1H NMR spectra of 2a–c and 3a–c in CDCl3 solutions provided evidence of a rigid and well-defined major conformation for each peptide. The peptides in each series exhibit similar spectral characteristics, suggesting that the position of the ΔAA in the chain has a larger impact on peptide structure than the nature of the ΔAA (i.e., small, medium-sized, or bulky). Monitoring of the NH moieties of 2a–c in CDCl3 solutions doped with D2O revealed rapid H–D exchange of the ΔAA carbamate NH and negligible H–D exchange of the Aib and Ala amide NH groups. These observations are consistent with the 310-helical pattern of intramolecular hydrogen bonds shown in Figure 3. In contrast, analogous experiments conducted with 3a–c showed rapid H–D exchange of the ΔAA amide NH and minimal H–D exchange of the Aib carbamate NH and the Ala amide NH groups. These data fit the β-sheet-like pattern of intramolecular hydrogen bonds depicted in Figure 3. Interestingly, in CDCl3 solutions the ΔAA residues do not participate in intramolecular hydrogen bonding regardless of their position in the chain.
Figure 3.
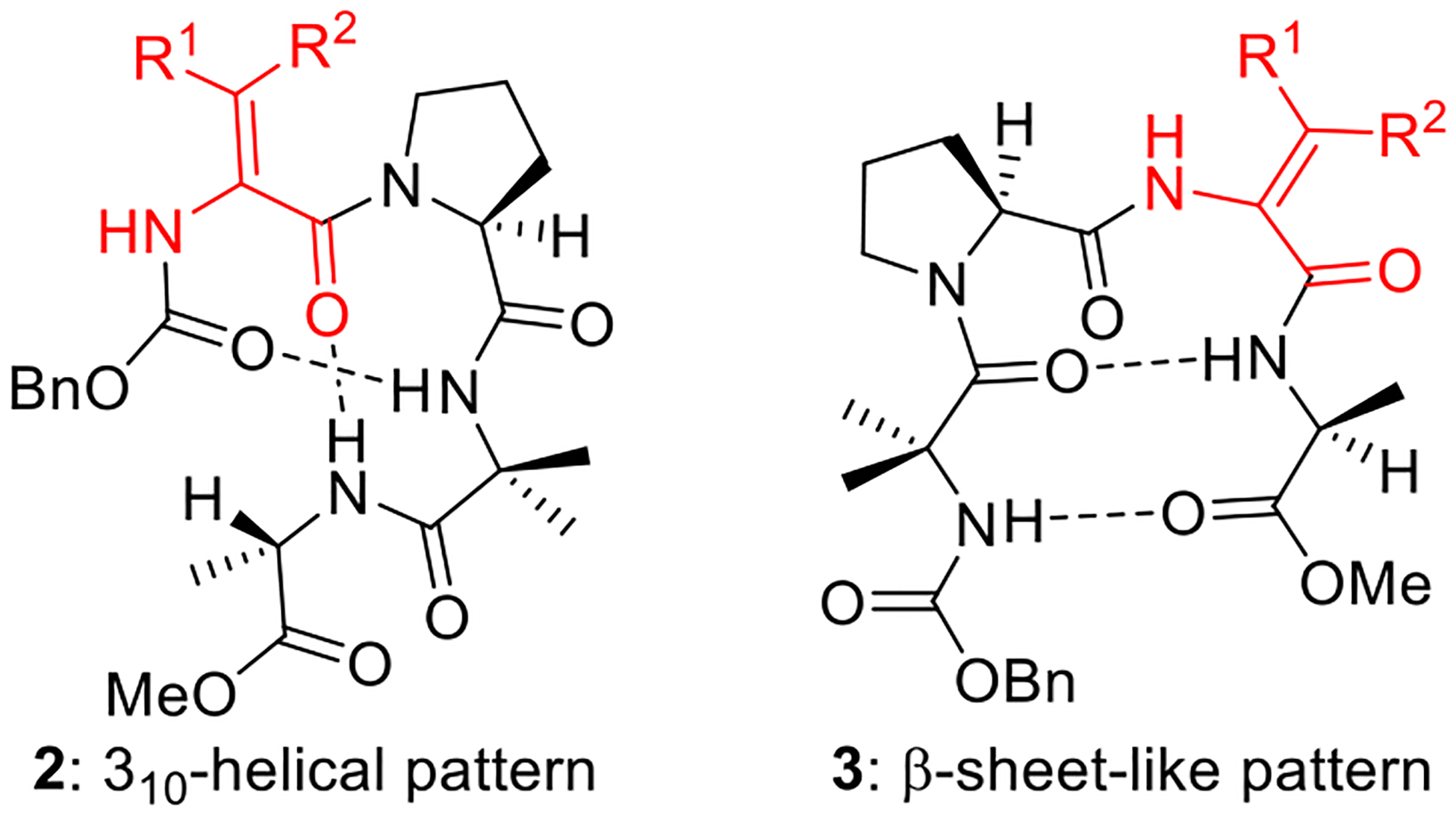
310-Helical and β-sheet-like conformations.
In an attempt to determine the impact of a polar environment on the hydrogen bonding patterns of 2 and 3, we conducted variable-temperature 1H NMR experiments with solutions of these peptides in DMSO-d6. In this solvent, only the Ala amide NH groups of 2a–c, 3a, and 3c participate in intramolecular hydrogen bonding. Apparently, the intramolecular hydrogen bonds involving the Aib amide or carbamate NH groups that are present in the less polar CDCl3 solvent are weak enough to be disrupted by interactions with the highly polar DMSO-d6 solvent molecules. Surprisingly, only the ΔAla amide NH of peptide 3b forms an intramolecular hydrogen bond in DMSO-d6 solution. Perhaps the absence of A1,3 strain in the small ΔAla residue allows this peptide to adopt a conformation in a strongly polar solvent that is unavailable to the other peptides with larger ΔAA residues and intrinsic A1,3 strain.
NOE-restrained structural calculations using the program CYANA 2.135 provided insights into the solution structures of 1 and its analogs. First, ROESY spectra were acquired for each compound with a mixing time of 200 ms. The resulting cross-peaks were then converted to upper-distance constraints. A standard simulated annealing calculation was then performed for 100 conformers of each peptide with the upper-limit file and experimentally determined hydrogen bonding pattern as input. The ten structures with the lowest objective target function were then superimposed to create a final ensemble for each peptide, which was then analyzed based on structure and RMSD values using the molecular visualization software VMD. These ensembles are shown in Figure 4. The parent peptide 1 exhibits the highest RMSD value of 1.154 Å, suggesting that it has more conformational freedom than its analogs. As anticipated from the intramolecular hydrogen bonding data, the peptides with a ΔAA in place of Aib-1 (i.e., 2a–c) form incipient 310-helical structures and the peptides with a ΔAA in place of Aib-3 (i.e., 3a–c) resemble β-turns. These structural data demonstrate that the position of the ΔAA plays an important role in determining the conformation preferred by each analog.
Figure 4.
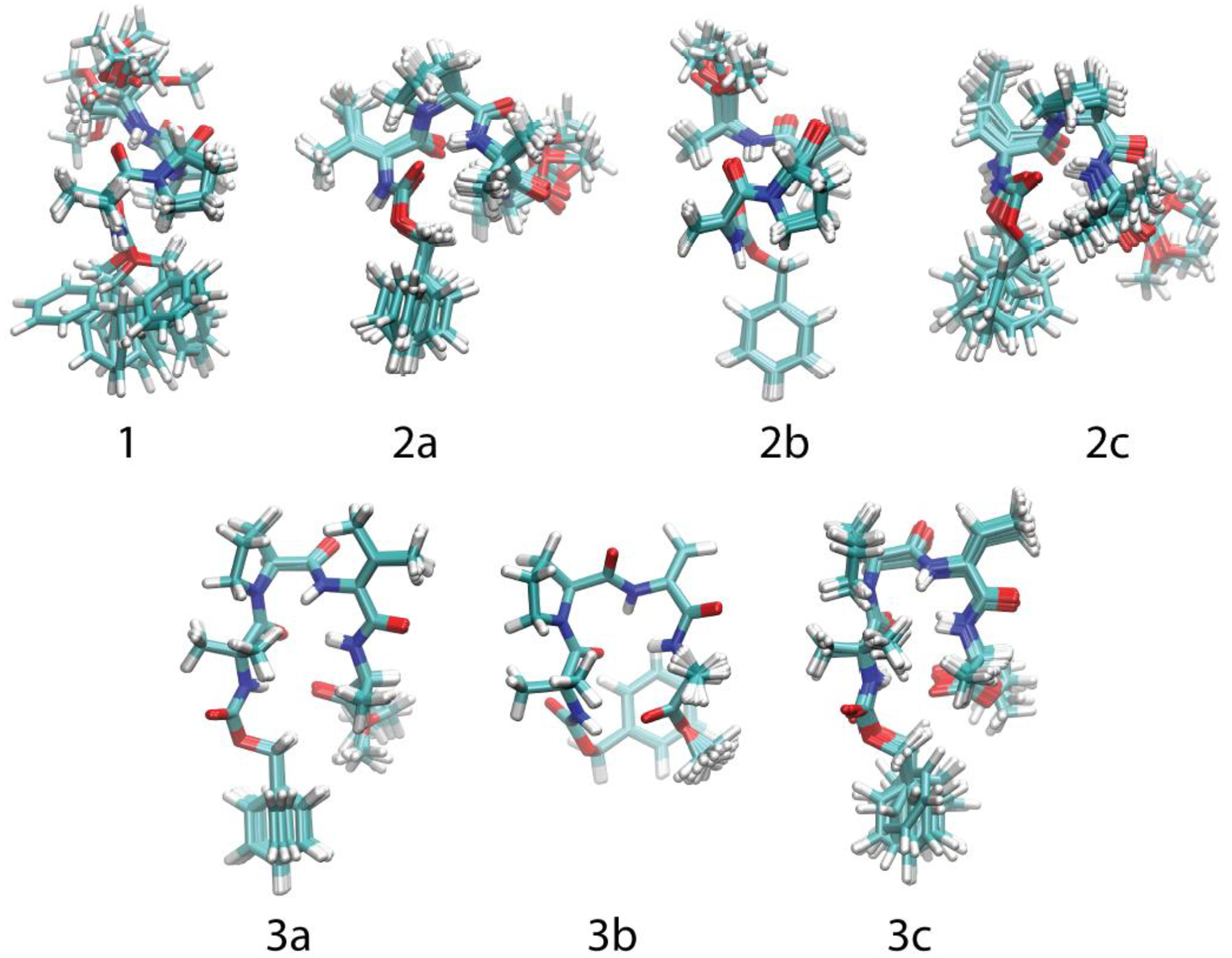
Solution-phase structures of 1, 2a–c, and 3a–c as calculated using NOE distance restraints.
Peptides 3a–c exhibit lower RMSD values than peptides 2a–c, indicating that placement of a ΔAA at the Aib-3 position leads to a greater increase in rigidity than does placement at the Aib-1 position. Interestingly, the conformations of the ΔVal and Z-ΔAbu residues in peptides of the same series are quite similar (i.e., 2a and 2c, 3a and 3c). However, the Z-ΔAbu-containing analogs 2c and 3c exhibit higher RMSD values than the corresponding ΔVal-containing analogs 2a and 3a. While both residues apparently prefer similar low-energy conformations, it appears that the additional A1,3 strain inherent in a tetrasubstituted alkene relative to a trisubstituted alkene destabilizes the high-energy conformations of ΔVal to a greater degree than those of Z-ΔAbu. Thus, our data suggest that medium-sized and bulky ΔAAs exert similar effects on the structures of peptides, but that bulky ΔAAs impart more rigidity.
The stability of 1 and its analogs to proteolytic degradation was determined by treating the peptides with Pronase, a cocktail of proteases from Streptomyces griseus that aggressively cleaves peptide bonds in a nonspecific fashion.36 Since 1 is relatively stable to proteolysis due to its two Aib residues, a high concentration of Pronase was required for it to degrade on a reasonable timescale. As a result, the kinetics of peptide degradation were affected by self-proteolysis of the enzymes. Although this prevented us from measuring half-lives, we were able to compare the amount of each peptide that remained after 30 minutes of incubation with Pronase as recorded in Table 1. Peptides 2b and 3b containing ΔAla were significantly more labile than Balaram’s peptide, as the relative abundance of the former peptides after 30 minutes of exposure to Pronase was ca. 10–30% that of the latter. Thus, it is clear that Aib is more effective than ΔAla at protecting peptides from proteolysis. Interestingly, the effects of Z-ΔAbu and ΔVal were dependent on their location in the peptide chain. Analogs 2a and 2c possessing a ΔAA at the N-terminus were equally or slightly less stable than 1, respectively. In contrast, variants 3a and 3c in which the ΔAA replaces Aib-3 were more stable than 1, as approximately twice as much of each remained after exposure to Pronase for 30 minutes. While the ΔVal-containing peptides 2a and 3a were slightly more stable than the Z-ΔAbu-containing peptides 2b and 3b, the differences in performance were not dramatic. Although a more detailed and comprehensive study would be required to generate firm conclusions, these data suggest that Z-ΔAbu and ΔVal might confer comparable increases in proteolytic stability upon peptides that contain them. Importantly, our results show that in some cases the stabilizing impact of these two ΔAAs can exceed that of the α,α-disubstituted residue Aib.
Table 1.
Proteolysis of 1 and Analogs by Pronasea
| peptide | amount remaining after 30 min (%) |
|---|---|
| 1 | 31 |
| 2a | 31 |
| 2b | 9 |
| 2c | 21 |
| 3a | 61 |
| 3b | 3 |
| 3c | 58 |
Solutions of peptides (0.5 mM, 500 μL) in 1X PBS buffer (10 mM sodium phosphate, 137 mM NaCl, 2.7 mM KCl buffer, pH 7.4) were each treated with a solution of Pronase (0.88 mg/mL, 200 μL). Degradation of peptides was monitored using HPLC.
Conventional wisdom indicates that ΔAAs are reactive electrophiles that undergo conjugate additions by biological nucleophiles such as thiols.37 In fact, enzyme-catalyzed conjugate additions of thiols to ΔAla and ΔAbu are involved in the biosynthesis of lantibiotics.38 However, a close look at the literature reveals that uncatalyzed conjugate additions of thiols to ΔAAs larger than ΔAla typically require large excesses of thiol to proceed.39 Alternatively, placement of the ΔAA at the C-terminus of a peptide instead of embedded within the backbone can increase its electrophilicity.40 Based on these prior results, we postulated that steric hindrance at the β-carbon atom would significantly attenuate the reactivity of medium-sized and bulky ΔAAs to thiols in the absence of enzymes. We probed this hypothesis by exposing solutions of 2a–c and 3a–c in DMSO-d6 to ca. 2 equiv of cysteamine, a highly nucleophilic thiol.41 As anticipated, ΔAla-containing peptides 2b and 3b were steadily consumed under these conditions. The decomposition of cysteamine in solution complicated our efforts to calculate half-lives for these reactions. Nonetheless, it was clear that 3b was consumed more rapidly than 2b, as 65% of the former and 93% of the latter remained after 40 minutes of incubation at room temperature.
In contrast, ΔVal-containing peptides 2a and 3a as well as Z-ΔAbu-containing peptides 2c and 3c were inert to cysteamine at temperatures up to 100 °C, at which point the thiol decomposed completely. Thus, neither of these residues is reactive to thiols when embedded within a peptide (i.e., not located at the C-terminus). While further investigation is warranted, these results suggest that both medium-sized and bulky ΔAAs can be incorporated into bioactive peptides without fear of in vivo degradation due to conjugate addition. Interestingly, subjection of α-methylene-γ-butyrolactone to cysteamine caused complete consumption of the electrophile within 5 minutes at room temperature. This result shows that ΔAla-containing peptides 2b and 3b are only modestly reactive to cysteamine by comparison, implying that peptides including this residue might also be useful for some biological applications.
CONCLUSIONS
In an effort to compare the impacts of small (i.e., ΔAla), medium-sized (i.e., Z-ΔAbu), and bulky ΔAAs (i.e., ΔVal) on peptide structure and stability, we synthesized ΔAA-containing analogs 2a–c and 3a–c of Balaram’s incipient 310-helical peptide 1. The different types of ΔAAs exerted similar effects on conformation in CDCl3 solution according to NOE-restrained structural calculations. Variants 2a–c possessing a ΔAA in place of Aib-1 all preferred the 310-helical shape of the original peptide, whereas analogs 3a–c with a ΔAA replacing Aib-3 all adopted a β-sheet-like orientation instead. Thus, the location of the ΔAA in the backbone played a larger role than the size of the residue in determining the structures of the peptides. The proteolytic stabilities of 1 and its analogs were evaluated by exposure to Pronase. ΔAla-containing analogs 2b and 3b were degraded rapidly, demonstrating that this small ΔAA does not protect peptides from proteolysis. Z-ΔAbu and ΔVal increased proteolytic stability to similar degrees, with the most pronounced effect (ca. twice as stable as 1) occurring when the ΔAA replaced Aib-3. Intriguingly, peptides containing Z-ΔAbu and ΔVal were completely unreactive to conjugate addition by the potent nucleophile cysteamine at temperatures up to 100 °C, whereas ΔAla-containing peptides reacted at room temperature. The comparable impacts of Z-ΔAbu and ΔVal on proteolytic stability suggests that the former residue is likely to be preferred to the latter due to its relative ease of synthesis, except in cases where the increased rigidity caused by the elevated A1,3-strain in ΔVal is desired. The inertness of both of these residues to thiols suggests that they can be incorporated into bioactive peptides without fear of degradation by biological nucleophiles. While we hope that these conclusions will be valid with peptides larger than 2 and 3, further experimentation is required to confirm this premise. When viewed as a whole, our results provide indications of the promise of medium-sized and bulky ΔAAs as tools for imparting rigidity and proteolytic stability to peptides. Future studies will focus on the impact of ΔAAs on other secondary structures as well as the synthesis and study of additional ΔAAs including medium-sized ΔAAs that are larger than ΔAbu (e.g., ΔPhe and ΔLeu).42
EXPERIMENTAL SECTION
General Details.
Dimethylformamide and tetrahydrofuran were dried by passage through cylinders of activated alumina. Chloroform was dried over 4 Å molecular sieves and Na2SO4 under Ar overnight. An oil bath was used to heat reactions that required elevated temperatures to proceed. Flash chromatography was conducted using 60–230 mesh silica gel. 1H NMR spectra were acquired on a 500 MHz spectrometer with chloroform (7.27 ppm) as internal reference. Signals are reported as follows: s (singlet), d (doublet), t (triplet), q (quartet), quint (quintet), dd (doublet of doublets), br s (broad singlet), m (multiplet). 13C NMR spectra were acquired on a spectrometer operating at 125 MHz with chloroform (77.23 ppm) as internal reference. Infrared spectra were obtained on an FT-IR spectrometer. MS data were obtained using ESI techniques.
Methyl (2-amino-2-methylpropanoyl)-l-alaninate (4).
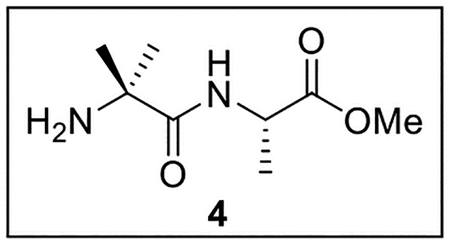
A solution of Z-Aib-OH (1.5034 g, 6.34 mmol) in anhydrous THF–DMF (5:1, 48 mL) at 0 °C under Ar was treated sequentially with Ala-OMe (944.5 mg, 6.77 mmol, 1.1 equiv), HOBt (ca. 20% H2O content, 1.2988 g, 7.69 mmol, 1.2 equiv), EDC·HCl (1.7356 g, 9.05 mmol, 1.4 equiv), and NaHCO3 (1.2811 g, 15.25 mmol, 2.4 equiv). The resulting mixture was slowly warmed to rt and stirred for 24 h. The reaction was quenched by the addition of sat aq NaHCO3 (25 mL), diluted with H2O (30 mL), and extracted with EtOAc (5 × 40 mL). The combined organic layers were dried (Na2SO4) and concentrated in vacuo. Flash chromatography (200 mL of SiO2, 0–30% EtOAc in hexanes gradient elution) afforded Z-Aib-Ala-OMe (1.936 g, 6.01 mmol, 95%) as a white solid: [α]25D −3.3 (c 1.2, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 7.38–7.31 (m, 5H), 6.80 (br s, 1H), 5.29 (br s, 1H), 5.11 (s, 2H), 4.59–4.52 (m, 1H), 3.75 (s, 3H), 1.56 (s, 3H), 1.54 (s, 3H), 1.38 (br s, 3H); 13C{1H} NMR (CDCl3, 125 MHz) δ 173.8, 173.4, 155.0, 136.2, 128.6 (2C), 128.2, 128.1 (2C), 66.8, 56.9, 52.4, 48.2, 25.7, 25.2, 18.2; IR (film) υmax 3327, 2998, 1725, 1660, 1522, 1454, 1255, 1074 cm−1; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C16H22N2O5H 323.1607; Found 323.1600.
A solution of Z-Aib-Ala-OMe (1.001 g, 3.11 mmol) in CH3OH (10.0 mL) was treated with 10% Pd/C (501.1 mg, 0.5 wt equiv) at rt under Ar. The resulting mixture was stirred at rt under H2 (500 psi) for 72 h, then diluted with H2O (10 mL) and extracted with EtOAc (4 × 6 mL). The combined organic layers were dried (Na2SO4) and concentrated in vacuo. The crude Aib-Ala-OMe (4, 559.6 mg, 2.97 mmol, 96%) was used directly in subsequent coupling reactions without further purification.
Benzyl (S)-2-((1-(((S)-1-methoxy-1-oxopropan-2-yl)amino)-2-methyl-1-oxopropan-2-yl)carbamoyl)pyrrolidine-1-carboxylate (5).
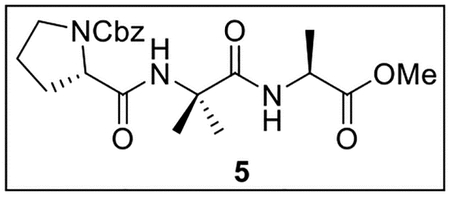
A solution of Z-Pro-OH (44.8 mg, 0.180 mmol) in anhydrous THF–DMF (5:1, 6 mL) at 0 °C under Ar was treated sequentially with 4 (35.4 mg, 0.188 mmol, 1.0 equiv), HOBt (ca. 20% H2O content, 38.3 mg, 0.227 mmol, 1.3 equiv), EDC·HCl (50.5 mg, 0.263 mmol, 1.5 equiv), and NaHCO3 (38.3 mg, 0.456 mmol, 2.5 equiv). The resulting mixture was slowly warmed to rt and stirred for 24 h. The reaction was quenched by the addition of sat aq NaHCO3 (3 mL), diluted with H2O (6 mL), and extracted with EtOAc (5 × 10 mL). The combined organic layers were dried (Na2SO4) and concentrated in vacuo. Flash chromatography (100 mL of SiO2, 0–80% EtOAc in hexanes gradient elution) afforded 5 (39.3 mg, 0.0937 mmol, 52%) as a white film: [α]25D −55 (c 0.78, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 7.38–7.30 (m, 5H), 7.22 (br s, 1H), 6.67 (br s, 1H), 5.18 (d, J = 12.2 Hz, 1H), 5.12 (d, J = 12.4 Hz, 1H), 4.52 (quint, J = 7.2 Hz, 1H), 4.20–4.15 (m, 1H), 3.73 (s, 3H), 3.60–3.54 (m, 1H), 3.52–3.45 (m, 1H), 2.25–2.14 (m, 1H), 2.08–2.00 (m, 1H), 1.95–1.87 (m, 2H), 1.55 (s, 3H), 1.49 (s, 3H), 1.37 (d, J = 7.2 Hz, 3H); 13C{1H} NMR (CDCl3, 125 MHz) δ 173.6, 173.5, 171.4, 156.0, 136.3, 128.6 (2C), 128.2, 127.8 (2C), 67.4, 61.2, 57.3, 52.2, 48.3, 47.1, 28.9, 26.5, 24.7, 24.4, 17.8; IR (film) υmax 3319, 2997, 1751, 1685, 1519, 1421, 1357, 1174, 1110 cm−1; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C21H29N3O6H 420.2135; Found 420.2127.
Ethyl 2-(((Benzyloxy)carbonyl)amino)-3-methylbut-2-enoate (7).
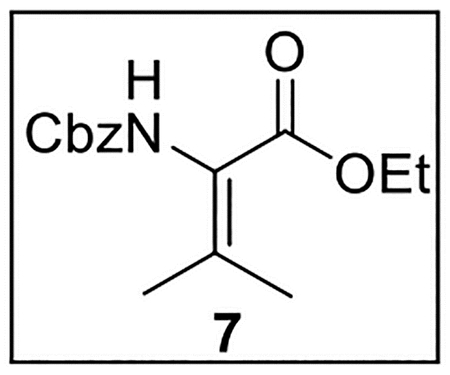
A solution of 632 (172 mg, 0.582 mmol) in anhydrous CHCl3 (3 mL) at 0 °C was treated with Martin sulfurane (783.2 mg, 1.16 mmol, 2.0 equiv) and stirred at 0 °C to rt for 1 h. The mixture was then dried (Na2SO4) and concentrated in vacuo. Flash chromatography (150 mL of SiO2, 0–1.5% MeOH in CH2Cl2 gradient elution) afforded 7 (139.4 mg, 0.503 mmol, 86%) as a yellow oil: 1H NMR (CDCl3, 500 MHz) δ 7.41–7.30 (m, 5H), 5.99 (br s, 1H), 5.16 (s, 2H), 4.26–4.19 (m, 2H), 2.19 (s, 3H), 1.91 (s, 3H), 1.31–1.25 (m, 3H); 13C{1H} NMR (CDCl3, 125 MHz) δ 164.9, 159.9, 145.6, 136.3, 128.5 (2C), 128.2, 128.1 (2C), 126.2, 67.1, 60.8, 22.5, 21.3, 14.2; IR (film) υmax 3324, 2924, 1715, 1499, 1454, 1373, 1308, 1249, 1090 cm−1; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C15H19NO4H 278.1392; Found 278.1383.
2-(((Benzyloxy)carbonyl)amino)-3-methylbut-2-enoic acid (8).
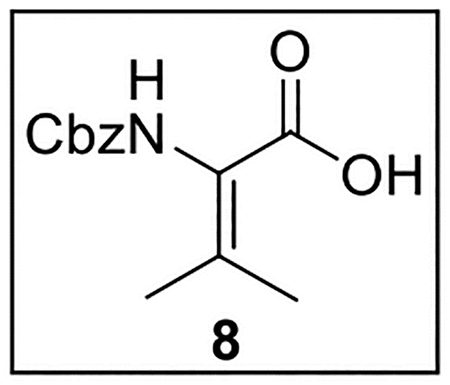
A solution of 7 (22.0 mg, 0.0793 mmol) in THF–EtOH–H2O (3:2:1, 3 mL) at rt was treated with LiOH·H2O (15.2 mg, 0.362 mmol, 4.6 equiv). The resulting mixture was stirred at 50 °C for 48 h. The reaction was acidified using 1N HCl until pH ~2, and the mixture was then extracted with EtOAc (4 × 10 mL). The combined organic layers were washed with brine (8 mL), dried (Na2SO4), and concentrated in vacuo. The crude acid 8 (20 mg, 0.079 mmol, quant.), was obtained as a yellow oil and used in subsequent reactions without further purification.
Methyl (2-((S)-1-(2-(((Benzyloxy)carbonyl)amino)-3-methylbut-2-enoyl)pyrrolidine-2-carboxamido)-2-methylpropanoyl)-l-alaninate (2a).
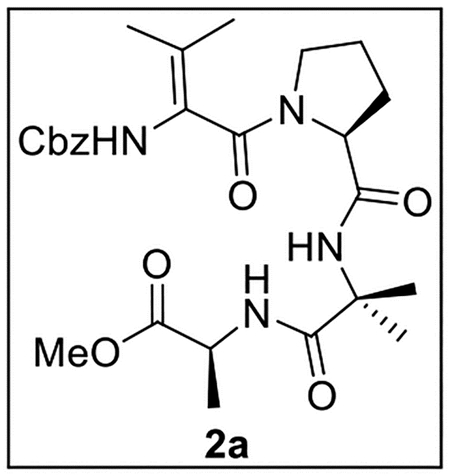
A solution of 5 (34.3 g, 0.0818 mmol) in MeOH (4 mL) at rt under Ar was treated with 10% Pd/C (21.5 mg, 0.63 wt equiv). The resulting mixture was stirred at rt under H2 (500 psi) for 72 h, then filtered through Celite, dried (Na2SO4), and concentrated in vacuo. The crude Pro-Aib-Ala-OMe product (18.5 mg, 0.0648 mmol, 79%) was used directly in subsequent coupling reactions without further purification.
A solution of acid 8 (20.0 mg, 0.0802 mmol, 1.2 equiv) in anhydrous THF–DMF (6:1, 3.5 mL) at 0 °C under Ar was treated sequentially with Pro-Aib-Ala-OMe (18.5 mg, 0.0648 mmol, 1.0 equiv), HOBt (ca. 20% H2O content, 17.0 mg, 0.101 mmol, 1.6 equiv), EDC·HCl (21.7 mg, 0.113 mmol, 1.7 equiv), and NaHCO3 (14.6 mg, 0.174 mmol, 2.7 equiv). The resulting mixture was stirred at 0 °C to rt for 48 h. The reaction was quenched by the addition of sat aq NaHCO3 (3 mL) and diluted with H2O (6 mL), and the mixture was then extracted with EtOAc (5 × 10 mL). The combined organic layers were dried (Na2SO4) and concentrated in vacuo. Flash chromatography (100 mL of SiO2, 0–80% EtOAc in hexanes gradient elution) afforded 2a (15.8 mg, 0.0306 mmol, 47%) as a white solid: [α]25D −16 (c 0.9, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 7.41 (br s, 1H), 7.40–7.34 (m, 5H), 7.33 (br s, 1H), 6.35 (s, 1H), 5.14 (d, J = 12.2 Hz, 1H), 5.06 (d, J = 12.2 Hz, 1H), 4.53–4.48 (m, 2H), 3.69 (s, 3H), 3.66–3.61 (m, 1H), 3.55–3.49 (m, 1H), 2.35–2.28 (m, 1H), 2.18–2.09 (m, 1H), 2.04–1.91 (m, 2H), 1.76 (s, 3H), 1.73 (s, 3H), 1.55 (s, 3H), 1.51 (s, 3H), 1.37 (d, J = 7.2 Hz, 3H); 13C{1H} NMR (CDCl3, 125 MHz) δ 174.4, 173.4, 170.6, 166.7, 153.9, 135.4, 128.7 (2C), 128.6, 128.1 (2C), 124.6, 122.7, 67.6, 61.0, 57.3, 52.1, 48.4, 48.3, 29.0, 25.6, 25.4, 24.5, 19.8, 18.3, 17.6; IR (film) υmax 3322, 2924, 1640, 1518, 1445, 1240, 1261 cm−1; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C26H36N4O7H 517.2662; Found 517.2677.
Methyl ((Benzyloxy)carbonyl)-l-serylprolinate (9).
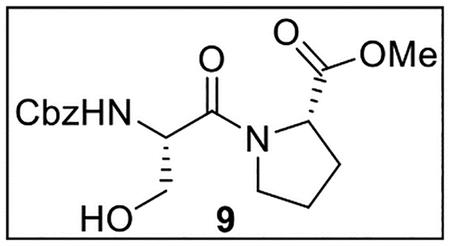
A solution of Z-Ser-OH (500.5 mg, 2.09 mmol) in anhydrous THF–DMF (5:1, 18 mL) under Ar at 0 °C was treated sequentially with Pro-OMe·HCl (363.4 mg, 2.19 mmol, 1.05 equiv), HOBt (ca. 20% H2O content, 458.8 mg, 2.72 mmol, 1.3 equiv), EDC·HCl (680.9 mg, 3.55 mmol, 1.7 equiv), and NaHCO3 (403.7 mg, 4.81 mmol, 2.3 equiv). The resulting mixture was slowly warmed to rt and stirred for 24 h. The reaction was quenched by the addition of sat aq NaHCO3 (15 mL), diluted with H2O (25 mL), and extracted with EtOAc (4 × 25 mL). The combined organic layers were dried (Na2SO4) and concentrated in vacuo. Flash chromatography (185 mL of SiO2, 0–5% MeOH in CH2Cl2 gradient elution) afforded 9 (597.3 mg, 1.70 mmol, 81%) as a white solid: [α]25D −25 (c 0.6, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 7.36–7.29 (m, 5H), 5.95–5.89 (m, 1H), 5.09 (s, 2H), 5.07 (br s, 1H), 4.70–4.64 (m, 1H), 4.59–4.55 (m, 1H), 3.92–3.88 (m, 1H), 3.86–3.79 (m, 1H), 3.76–3.70 (m, 1H), 3.73 (s, 3H), 3.41–3.39 (m, J = 7.2 Hz, 1H), 2.27–2.15 (m, 2H), 2.03–1.94 (m, 2H); 13C{1H} NMR (CDCl3, 125 MHz) δ 172.9, 169.9, 156.2, 136.2, 128.5 (2C), 128.1, 128.0 (2C), 67.0, 63.8, 58.9, 53.8, 52.7, 47.3, 28.9, 24.8; IR (film) υmax 3315, 2953, 2348, 1720, 1639, 1529, 1449, 1216, 1061 cm−1; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C17H22N2O6H 351.1556; Found 351.1549.
Methyl (2-(((Benzyloxy)carbonyl)amino)acryloyl)-l-prolinate (10).
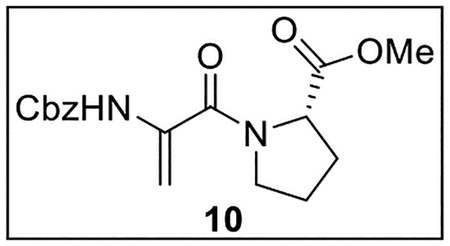
A solution of 9 (380.7 mg, 1.09 mmol) in 1,2-dichloroethane (15 mL) at 0 °C was treated with Et3N (380 μL, 276 mg, 2.73 mmol, 2.5 equiv), then stirred at 0 °C for 10 min. The resulting mixture was treated dropwise with MsCl (170 μL, 252 mg, 2.20 mmol, 2.0 equiv) and stirred at 0 °C to rt for 1 h. It was then treated with DBU (810 μL, 825 mg, 5.42 mmol, 5.0 equiv) and stirred at 90 °C for 2 h. The mixture was concentrated in vacuo, and the residue was dissolved in EtOAc (15 mL) and washed with 10% aq citric acid (10 mL), sat aq NaHCO3 (2 × 8 mL), and brine (10 mL). The combined organic layers were dried (Na2SO4) and concentrated in vacuo. Flash chromatography (110 mL of SiO2, 0–13% MeOH in CH2Cl2 gradient elution) afforded 10 (223.2 mg, 0.672 mmol, 62%) as a white solid: [α]25D −43 (c 0.6, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 7.38–7.30 (m, 5H), 7.23 (br s, 1H), 6.13 (s, 1H), 5.14 (s, 3H), 4.55–4.48 (m, 1H), 3.83–3.77 (m, 2H), 3.74 (s, 3H), 2.30–2.21 (m, 1H), 2.10–2.01 (m, 1H), 2.00–1.84 (m, 2H); 13C{1H} NMR (CDCl3, 125 MHz) δ 172.3, 165.0, 153.3, 136.0, 134.8, 128.5 (2C), 128.3, 128.1 (2C), 102.6, 66.8, 59.8, 52.3, 50.4, 28.9, 25.4; IR (film) υmax 3303, 2953, 2881, 1738, 1622, 1511, 1438, 1219, 1088 cm−1; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C17H20N2O5H 333.1445; Found 333.1438.
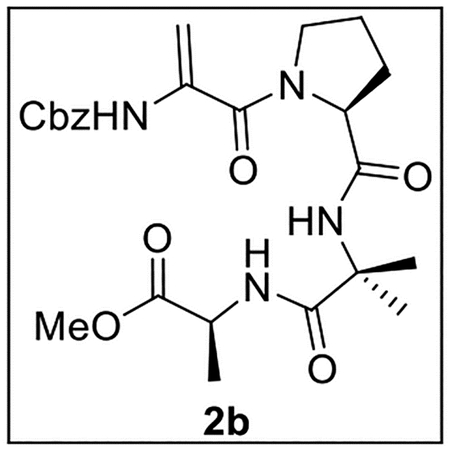
Methyl (2-(1-(2-(((Benzyloxy)carbonyl)amino)acryloyl)pyrrolidine-2-carboxamido)-2-methylpropanoyl)-l-alaninate (2b).
A solution of 10 (150.6 mg, 0.453 mmol) in t-BuOH–H2O (3:1, 10 mL) at 0 °C was treated with LiOH·H2O (95.1 mg, 2.27 mmol, 5.0 equiv) and stirred at rt for 4 h. The reaction was quenched by the addition of 1N HCl until pH ~4, and the mixture was extracted with EtOAc (4 × 10 mL). The combined organic layers were washed with brine (10 mL), dried (Na2SO4), and concentrated in vacuo. Crude acid Z-ΔAla-Pro-OH (105.2 mg, 0.330 mmol, 73%) was obtained as a colorless oil and was used in subsequent reactions without further purification.
A solution of Z-ΔAla-Pro-OH (105 mg, 0.330 mmol) in anhydrous THF–DMF (5:1, 18 mL) at 0 °C under Ar was treated sequentially with Aib-Ala-OMe (4, 65.2 mg, 0.346 mmol, 1.05 equiv), HOBt (ca. 20% H2O content, 71.7 mg, 0.425 mmol, 1.3 equiv), EDC·HCl (88.5 mg, 0.462 mmol, 1.4 equiv), and NaHCO3 (66.5 mg, 0.792 mmol, 2.4 equiv). The resulting mixture was slowly warmed to rt and stirred for 24 h. The reaction was quenched by the addition of sat aq NaHCO3 (5 mL), diluted with H2O (5 mL), and extracted with EtOAc (4 × 8 mL). The combined organic layers were dried (Na2SO4) and concentrated in vacuo. Flash chromatography (60 mL of SiO2, 0–3% MeOH in CH2Cl2 gradient elution) afforded 2b (93.7 mg, 0.192 mmol, 58%, 42% from 10) as a white solid: [α]25D −14 (c 0.5, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 7.40–7.31 (m, 5H), 7.27 (br s, 1H), 7.17 (br s, 1H), 6.96 (br s, 1H), 5.60 (s, 1H), 5.15 (d, J = 12.3 Hz, 1H), 5.12 (d, J = 12.4 Hz, 1H), 5.01 (s, 1H), 4.51 (quint, J = 7.2 Hz, 1H), 4.42–4.36 (m, 1H), 3.75–3.67 (m, 2H), 3.66 (s, 3H), 2.22–2.12 (m, 2H), 2.11–2.03 (m, 1H), 1.93–1.86 (m, 1H), 1.56 (s, 3H), 1.52 (s, 3H), 1.33 (d, J = 7.2 Hz, 3H); 13C{1H} NMR (CDCl3, 125 MHz) δ 174.0, 173.6, 170.7, 165.7, 153.4, 135.8, 135.6, 128.6 (2C), 128.5, 128.2 (2C), 102.6, 67.4, 61.7, 57.3, 52.2, 50.2, 48.3, 28.7, 26.3, 25.3, 24.6, 17.7; IR (film) υmax 3324, 2951, 1715, 1649, 1523, 1450, 1243, 1090 cm−1; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C24H32N4O7H 489.2349; Found 489.2357.
((Benzyloxy)carbonyl)-l-threonyl-l-proline (11).
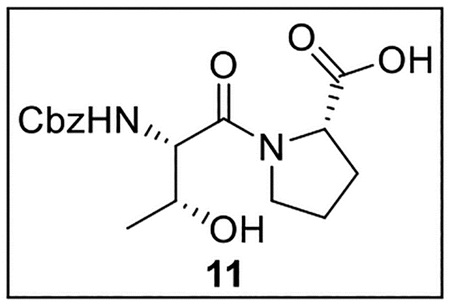
A solution of Z-Thr-OH (1.004 g, 3.96 mmol) in anhydrous THF–DMF (5:1, 24 mL) under Ar at 0 °C was treated sequentially with Pro-OMe·HCl (707 mg, 4.27 mmol, 1.1 equiv), HOBt (ca. 20% H2O content, 811.5 mg, 4.80 mmol, 1.2 equiv), EDC·HCl (1.085 g, 5.66 mmol, 1.4 equiv), and NaHCO3 (798.1 mg, 9.50 mmol, 2.4 equiv). The resulting mixture was allowed to warm to rt and was stirred for 24 h. The reaction was quenched by the addition of sat aq NaHCO3 (25 mL), diluted with H2O (30 mL), and extracted with EtOAc (5 × 40 mL). The combined organic layers were dried (Na2SO4) and concentrated in vacuo. Flash chromatography (200 mL of SiO2, 0–30% EtOAc in hexanes gradient elution) afforded Z-Thr-Pro-OMe (1.3584 g, 3.73 mmol, 94%) as a white solid: [α]25D −88 (c 0.49, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 7.40–7.31 (m, 5H), 5.70 (d, J = 9.0 Hz, 1H), 5.14 (d, J = 12.2 Hz, 1H), 5.10 (d, J = 12.4 Hz, 1H), 4.57–4.53 (m, 1H), 4.48 (dd, J = 9.2, 2.0 Hz, 1H), 4.24–4.17 (m, 1H), 3.86–3.80 (m, 1H), 3.78–3.73 (m, 1H), 3.75 (s, 3H), 3.38 (br s, 1H), 2.30–2.21 (m, 1H), 2.10–1.97 (m, 3H), 1.24 (d, J = 6.4 Hz, 3H); 13C{1H} NMR (CDCl3, 125 MHz) δ 172.5, 170.6, 156.6, 136.2, 128.5 (2C), 128.2, 128.0 (2C), 67.3, 67.1, 58.8, 56.0, 52.5, 47.3, 28.9, 24.9, 18.5; IR (film) υmax 3399, 2977, 1744, 1720, 1634, 1524, 1439, 1218, 1176, 1027 cm−1; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C18H24N2O6H 365.17126; Found 365.17005.
A solution of Z-Thr-Pro-OMe (800.0 mg, 2.20 mmol) in t-BuOH–H2O (3:1, 12 mL) at 0 °C was treated with LiOH·H2O (480.5 mg, 11.45 mmol, 5.2 equiv) and stirred for 4 h. The reaction was quenched by the addition of 1 N HCl until pH ~2, and the mixture was extracted with EtOAc (4 × 15 mL). The combined organic layers were washed with brine (10 mL), dried (Na2SO4), and concentrated in vacuo. Crude acid 11 (616.7 mg, 1.76 mmol, 80%) was obtained as a colorless oil and was used in subsequent reactions without further purification.
Methyl (2-((S)-1-(((Benzyloxy)carbonyl)-l-threonyl)pyrrolidine-2-carboxamido)-2-methylpropanoyl)-l-alaninate (12).
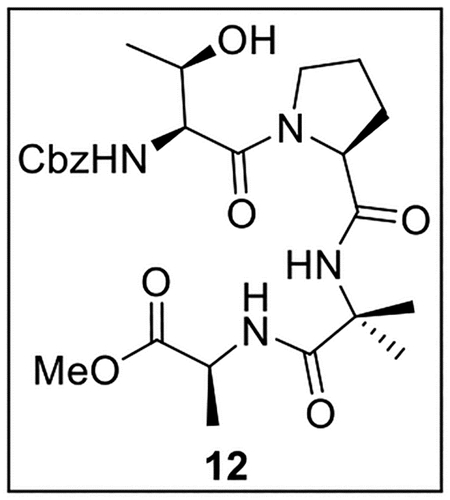
A solution of acid 11 (240.4 mg, 0.686 mmol) in anhydrous DMF (10 mL) at 0 °C under Ar was treated sequentially with HBTU (262.3 mg, 0.692 mmol, 1.0 equiv), i-Pr2NEt (360 μL, 267 mg, 2.07 mmol, 3.0 equiv), and amine 4 (159.9 mg, 0.850 mmol, 1.2 equiv). The resulting mixture was warmed to rt and stirred for 2 h, then diluted with EtOAc (15 mL) and washed with sat aq NH4Cl (3 × 15 mL), sat aq NaHCO3 (2 × 15 mL), and brine (15 mL). The organic layer was dried (Na2SO4) and concentrated in vacuo. Flash chromatography (100 mL of SiO2, 2–4.5% MeOH in CH2Cl2 gradient elution) afforded 12 (89.5 mg, 0.172 mmol, 25%) as a white solid: [α]25D −34 (c 0.34, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 7.37–7.32 (m, 6H), 7.00 (br s, 1H), 6.94 (d, J = 7.0 Hz, 1H), 5.75 (d, J = 8.7 Hz, 1H), 5.14 (d, J = 12.2 Hz, 1H), 5.10 (d, J = 12.2 Hz, 1H), 4.56–4.52 (m, 2H), 4.50–4.48 (m, 1H), 4.26–4.21 (m, 1H), 3.74 (s, 3H), 3.73–3.70 (m, 2H), 2.27–2.18 (m, 1H), 2.13–2.07 (m, 1H), 2.06–1.96 (m, 2H), 1.59 (s, 3H), 1.46 (s, 3H), 1.37 (d, J = 7.2 Hz, 3H), 1.26 (d, J = 4.0 Hz, 3H); 13C{1H} NMR (CDCl3, 125 MHz) δ 173.9, 173.8, 171.1, 170.7, 156.6, 136.2, 128.5 (2C), 128.2, 128.0 (2C), 67.2, 67.1, 60.5, 57.1, 56.5, 52.5, 48.3, 47.7, 27.3, 26.8, 25.2, 23.9, 19.0, 18.1; IR (film) υmax 3312, 2923, 1735, 1633, 1529, 1454, 1256 cm−1; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C25H36N4O8H 521.2611; Found 521.2602.
Methyl (2-((S)-1-((Z)-2-(((Benzyloxy)carbonyl)amino)but-2-enoyl)pyrrolidine-2-carboxamido)-2-methylpropanoyl)-l-alaninate (2c).
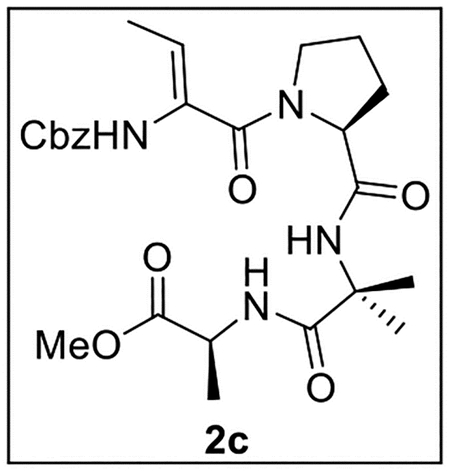
A solution of tetrapeptide 12 (55.0 mg, 0.106 mmol) in anhydrous CHCl3 (3 mL) at 0 °C was treated with Martin sulfurane (152 mg, 0.226 mmol, 2.1 equiv). The resulting mixture was stirred at 0 °C to rt for 1 h, dried (Na2SO4), and concentrated in vacuo. Flash chromatography (150 mL of SiO2, 2–4.5% MeOH in CH2Cl2 gradient elution) afforded 2c (23.1 mg, 0.0460 mmol, 44%) as a white film: [α]25D −32 (c 0.85, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 7.40–7.31 (m, 7H), 6.49 (br s, 1H), 5.52 (q, J = 6.8 Hz, 1H), 5.16 (d, J = 12.1 Hz, 1H), 5.10 (d, J = 12.1 Hz, 1H), 4.54–4.45 (m, 2H), 3.70 (s, 3H), 3.63–3.50 (m, 2H), 2.24–2.12 (m, 2H), 1.98–1.85 (m, 2H), 1.73 (d, J = 7.0 Hz, 3H), 1.55 (s, 3H), 1.53 (s, 3H), 1.37 (d, J = 7.2 Hz, 3H);13C{1H} NMR (CDCl3, 125 MHz) δ 174.4, 173.5, 170.6, 166.6, 153.7, 135.4, 130.7, 128.7 (2C), 128.6, 128.3 (2C), 117.1, 67.8, 61.4, 57.3, 52.1, 49.6, 48.3, 29.0, 25.6, 25.4, 24.9, 17.6, 11.7; IR (film) υmax 3320, 2924, 2359, 1669, 1521, 1456, 1255 cm−1; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C25H34N4O7H 503.2506; Found 503.2513.
(2-(((Benzyloxy)carbonyl)amino)-2-methylpropanoyl)-l-proline (13).
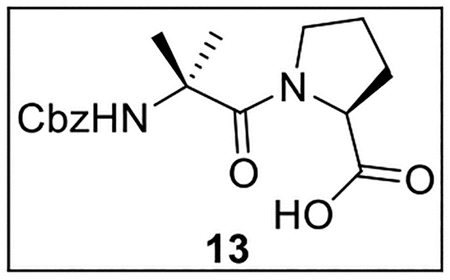
A solution of Z-Aib-OH (750.6 mg, 3.16 mmol) in anhydrous THF–DMF (5:1, 24 mL) at 0 °C under Ar was treated sequentially with Pro-OMe·HCl (557.6 mg, 3.37 mmol, 1.1 equiv), HOBt (ca. 20% H2O content, 645.1 mg, 3.82 mmol, 1.2 equiv), EDC·HCl (861.2 mg, 4.49 mmol, 1.4 equiv), and NaHCO3 (641.7 mg, 7.64 mmol, 2.4 equiv). The resulting mixture was allowed to warm to rt and was stirred for 24 h. The reaction was quenched by the addition of sat aq NaHCO3 (15 mL), diluted with H2O (30 mL), and extracted with EtOAc (5 × 30 mL). The combined organic layers were dried (Na2SO4) and concentrated in vacuo. Flash chromatography (150 mL of SiO2, 0–30% EtOAc in hexanes gradient elution) afforded Z-Aib-Pro-OMe (750.7 mg, 2.15 mmol, 68%) as a white solid: [α]25D −52 (c 2.64, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 7.36–7.29 (m, 5H), 5.65 (br s, 1H), 5.11 (d, J = 11.8 Hz, 1H), 5.03 (d, J = 11.6 Hz, 1H), 4.58–4.52 (m, 1H), 3.70 (s, 3H), 3.50–3.42 (m, 1H), 3.38–3.29 (m, 1H), 2.08–1.94 (m, 2H), 1.91–1.80 (m, 2H), 1.59 (s, 3H), 1.54 (s, 3H); 13C{1H} NMR (CDCl3, 125 MHz) δ 173.0, 172.1, 154.2, 136.6, 128.4 (2C), 128.2, 128.1 (2C), 66.4, 60.8, 56.7, 52.1, 47.9, 27.7, 25.8, 24.7, 24.3; IR (film) υmax 3303, 2986, 1720, 1623, 1525, 1411, 1364, 1258, 1168, 1074 cm−1; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C18H24N2O5H 349.1763; Found 349.1757.
A solution of Z-Aib-Pro-OMe (56.2 mg, 0.161 mmol) in t-BuOH (1.0 mL) and H2O (0.4 mL) was treated with LiOH·H2O (34.1 mg, 0.813 mmol, 5.0 equiv), then stirred at rt for 4 h. The resulting mixture was acidified to pH 4~5 by the addition of 1 N HCl, diluted with H2O (2 mL), and extracted with EtOAc (3 × 5 mL). The combined organic layers were dried (Na2SO4) and concentrated in vacuo. The crude carboxylic acid 13 was used directly in subsequent coupling reactions without further purification.
Ethyl (S)-2-((S)-1-(2-(((Benzyloxy)carbonyl)amino)-2-methylpropanoyl)pyrrolidine-2-carboxamido)-3-hydroxy-3-methylbutanoate (15).
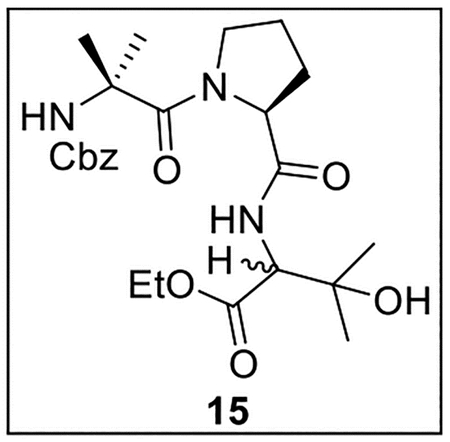
A solution of 13 (506.8 mg, 1.52 mmol) in anhydrous THF–DMF (5:1, 12 mL) at 0 °C under Ar was treated with 1432 (248.4 mg, 1.54 mmol, 1.0 equiv), HOBt (ca. 20% H2O content, 322.3 mg, 1.91 mmol, 1.3 equiv), EDC·HCl (413.5 mg, 2.16 mmol, 1.4 equiv), and NaHCO3 (314.0 mg, 3.74 mmol, 2.5 equiv). The resulting mixture was warmed to rt and stirred for 24 h. The reaction was quenched by the addition of sat aq NaHCO3 (15 mL), diluted with H2O (30 mL), and extracted with EtOAc (5 × 40 mL). The combined organic layers were dried (Na2SO4) and concentrated in vacuo. Flash chromatography (150 mL of SiO2, 0–60% EtOAc in hexanes gradient elution) afforded 15 (558.4 mg, 1.17 mmol, 77%) as a colorless oil that was a 1:1 mixture of diastereomers: 1H NMR (CDCl3, 500 MHz) δ 7.68–7.60 (m, 1H), 7.42–7.32 (m, 5H), 5.22–5.02 (m, 3H), 4.70–4.55 (m, 1H), 4.40–4.15 (m, 3H), 4.06 and 3.85 (2 br s, 1H), 3.72–3.60 (m, 1H), 3.467–3.35 (m, 1H), 2.21–2.12 (m, 1H), 2.02–1.80 (m, 3H), 1.61 (s, 6H), 1.48 and 1.45 (2s, 3H), 1.44 and 1.36 (2s, 3H), 1.33–1.29 (m, 3H); 13C{1H} NMR (CDCl3, 125 MHz) δ 172.4, 171.6 and 171.5, 170.7, 155.7 and 154.9, 135.8 and 135.4, 128.9 and 128.7 (2C), 128.6, 128.5 (2C), 72.2 and 71.9, 68.2, 67.3, 63.0 and 62.2, 61.0 and 60.6, 57.2, 48.2 and 48.0, 28.8, 27.6, 27.1 and 26.6, 26.3 and 26.2, 25.7, 24.7, 14.2; IR (film) υmax 3294, 2980, 1725, 1651, 1536, 1456, 1400, 1274, 1196, 1089 cm−1; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C24H35N3O7H 478.2553; Found 478.2546.
Methyl (2-((S)-1-(2-(((Benzyloxy)carbonyl)amino)-2-methylpropanoyl)pyrrolidine-2-carboxamido)-3-methylbut-2-enoyl)-l-alaninate (3a).
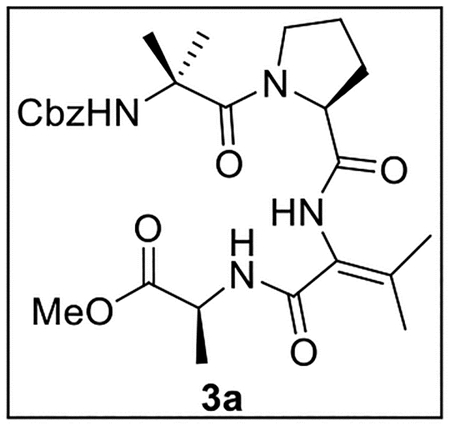
A solution of tripeptide 15 (75.8 mg, 0.159 mmol) in t-BuOH–H2O (3:1, 8 mL), was treated with LiOH·H2O (36.0 mg, 0.858 mmol, 5.4 equiv). The mixture was stirred at rt for 4 h, and the reaction was quenched by the addition of 1N HCl until pH ~2. The mixture was then extracted with EtOAc (4 × 15 mL), and the combined organic layers were washed with brine (10 mL), dried (Na2SO4), and concentrated in vacuo. The crude acid was then dissolved in anhydrous THF (8 mL) and treated with NaOAc (19.3 mg, 0.235 mmol, 1.5 equiv) and Ac2O (75 μL, 81.2 mg, 0.795 mmol, 5.0 equiv). The resulting mixture was stirred at rt under Ar for 12 h, then diluted with MeOH (10 mL), stirred for an additional 30 min, and diluted with H2O (10 mL). The mixture was extracted with EtOAc (4 × 15 mL), and the combined organic layers were washed with brine (10 mL), dried (Na2SO4), and concentrated in vacuo.
The crude azlactone (40.2 mg, 0.0972 mmol) was then dissolved in MeOH (2 mL) and treated dropwise with 1 N NaOH (970 μL, 0.97 mmol, 10 equiv). The resulting mixture was stirred at rt for 24 h, then washed with sat aq NaHCO3 (2 × 5 mL) and acidified by the addition of 1 N HCl to pH ~2. The mixture was extracted with EtOAc (3 × 5 mL), and the combined organic layers were dried (Na2SO4) and concentrated in vacuo. The crude C-terminal dehydroamino acid was dissolved in anhydrous THF–DMF (5:1, 3 mL) and treated sequentially with Ala-OMe (42.6 mg, 0.305 mmol, 3.1 equiv), PyBOP (152.4 mg, 0.293 mmol, 3.0 equiv), and i-Pr2NEt (100 μL, 74.2 mg, 0.574 mmol, 3.6 equiv). The resulting mixture was stirred at rt under Ar for 24 h, then extracted with EtOAc (5 × 5 mL). The combined organic layers were dried (Na2SO4) and concentrated in vacuo. Flash chromatography (25 mL of SiO2, 2–4.5% MeOH in CH2Cl2 gradient elution) afforded 3a (17.0 mg, 0.0329 mmol, 34% from azlactone and 21% from 15) as a white film: [α]25D +12.7 (c 0.41, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 8.14 (s, 1H), 7.39–7.30 (m, 6H), 5.30 (s, 1H), 5.16 (d, J = 12.1 Hz, 1H), 4.96 (d, J = 12.1 Hz, 1H), 4.59–4.51 (m, 2H), 3.71 (s, 3H), 3.70–3.65 (m, 1H), 3.26–3.19 (m, 1H), 2.30–2.23 (m, 1H), 2.23 (s, 3H), 1.90–1.78 (m, 3H), 1.78 (s, 3H), 1.55 (s, 3H) 1.46 (d, J = 7.2 Hz, 3H), 1.44 (s, 3H); 13C{1H} NMR (CDCl3, 125 MHz) δ 173.5, 172.4, 171.0, 165.4, 155.3, 144.6, 136.0, 128.6 (2C), 128.5, 128.4 (2C), 122.3, 67.3, 63.2, 57.0, 52.1, 48.5, 48.3, 28.7, 26.5, 26.0, 24.6, 22.1, 21.1, 17.6; IR (film) υmax 3306, 2987, 1666, 1625, 1535, 1454, 1273, 1077 cm−1; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C26H36N4O7H 517.2662; Found 517.2670.
Methyl ((Benzyloxy)carbonyl)-l-seryl-l-alaninate (16).
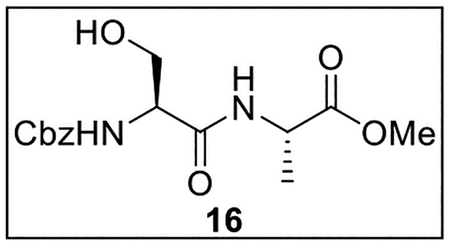
A solution of Z-Ser-OH (500.2 mg, 2.09 mmol) in anhydrous THF–DMF (5:1, 18 mL) under Ar at 0 °C was treated sequentially with Ala-OMe·HCl (291.7 mg, 2.09 mmol, 1.0 equiv), HOBt (ca. 20% H2O content, 423.6 mg, 2.51 mmol, 1.2 equiv), EDC·HCl (520.8 mg, 2.72 mmol, 1.3 equiv), and NaHCO3 (403.8 mg, 4.81 mmol, 2.3 equiv). The resulting mixture was slowly warmed to rt and stirred for 24 h. The reaction was quenched by the addition of sat aq NaHCO3 (20 mL), diluted with H2O (25 mL), and extracted with EtOAc (4 × 25 mL). The combined organic layers were dried (Na2SO4) and concentrated in vacuo. Flash chromatography (170 mL of SiO2, 0–5% MeOH in DCM gradient elution) afforded 16 (489.3 mg, 1.51 mmol, 72%) as a white solid: [α]25D −11 (c 0.6, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 7.39–7.31 (m, 5H), 7.13–7.01 (m, 1H), 5.92–5.83 (m, 1H), 5.13 (s, 2H), 5.10 (br s, 1H), 4.57 (quint, J = 7.3 Hz, 1H), 4.33–4.28 (m, 1H), 4.08–4.00 (m, 1H), 3.76 (s, 3H), 3.68 (dd, J = 11.2, 6.2 Hz, 1H), 1.41 (d, J = 7.1 Hz, 3H); 13C{1H} NMR (CDCl3, 125 MHz) δ 173.4, 170.6, 156.5, 136.0, 128.6 (2C), 128.3, 128.1 (2C), 67.3, 63.0, 55.4, 52.7, 48.3, 17.7; IR (film) υmax 3314, 1736, 1665, 1562, 1547, 1460, 1217, 1058 cm−1; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C15H20N2O6H 325.1400; Found 325.1408.
Methyl (2-(((Benzyloxy)carbonyl)amino)-2-methylpropanoyl)-l-prolyl-l-seryl-l-alaninate (17).
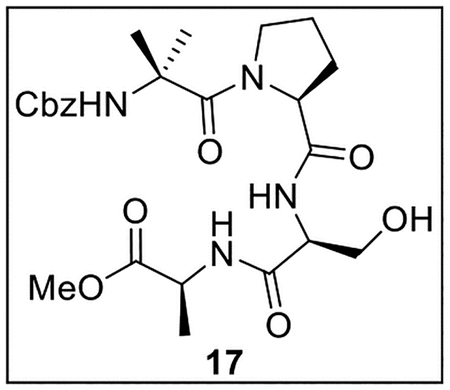
A solution of 16 (106.7 mg, 0.329 mmol) in MeOH (6.0 mL) was treated with 10% Pd/C (70.2 mg, 0.66 wt equiv) at rt under Ar. The resulting mixture was stirred at rt under H2 (500 psi) for 48 h, then diluted with H2O (10 mL) and extracted with EtOAc (4 × 6 mL). The combined organic layers were dried (Na2SO4) and concentrated in vacuo. The crude Ser-Ala-OMe (54.4 mg, 0.286 mmol, 87%) was used directly in subsequent couplings without further purification.
A solution of crude carboxylic acid 13 (94.6 mg, 0.283 mmol) in anhydrous DMF (3.0 mL) at 0 °C under Ar was treated with HBTU (107.4 mg, 0.282 mmol, 1.0 equiv) and i-Pr2NEt (51.0 μL, 37.8 mg, 0.293 mmol, 1.0 equiv). The resulting mixture was treated dropwise with a solution of Ser-Ala-OMe (54.4 mg, 0.286 mmol, 1.0 equiv) in anhydrous DMF (1 mL) and stirred at 0 °C under Ar for 2 h. It was then diluted with H2O (8 mL) and extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with sat aq NH4Cl (3 × 5 mL), sat aq NaHCO3 (2 × 4 mL), and brine (6 mL), dried (Na2SO4), and concentrated in vacuo. Flash chromatography (110 mL of SiO2, 0–10% MeOH in CH2Cl2 gradient elution) afforded 17 (61.6 mg, 0.122 mmol, 37% from 16) as a white solid: [α]25D + 22 (c 0.6, CHCl3); 1H NMR (CDCl3, 500 MHz, ca. 1.2:1 mixture of rotamers) δ 7.63 and 7.57 (2d, J = 8.8 Hz and J = 7.4 Hz, 1H), 7.38–7.32 (m, 5H), 7.21 (br s, 1H), 5.75 (br s, 1H), 5.34 (br s, 1H), 5.23–5.19 and 5.17 (m and d, J = 11.8 Hz, 1H), 5.00 and 4.99 (2d, J = 12.2 and 12.1 Hz, 1H), 4.60–4.50 (m, 2H), 4.06–4.00 (m, 1H), 3.83 (dd, J = 11.4, 4.7 Hz, 1H), 3.71 (s, 3H), 3.67–3.60 (m, 1H), 3.18–3.09 (m, 2H), 2.33–2.27 (m, 1H), 2.17–2.10 and 1.92–1.85 (2m, 1H), 1.80–1.70 (m, 2H), 1.59 and 1.54 (2s, 3H), 1.46 (d, J = 7.4 Hz, 3H) 1.45 and 1.42 (2s, 3H); 13C{1H} NMR (CDCl3, 125 MHz) δ 174.9, 172.9 and 172.8, 171.4, 170.9, 155.9 and 155.3, 135.9 and 135.6, 128.7 (2C), 128.6, 128.3 (2C), 67.7 and 67.3, 63.3 and 63.0, 62.4, 57.2, 54.7, 52.3, 48.7, 48.3 and 48.1, 28.6 and 28.5, 26.7 and 26.5, 26.0 and 25.7, 24.7 and 24.3, 17.4; IR (film) υmax 3306, 2987, 1666, 1625, 1535, 1454, 1273, 1077 cm−1; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C24H34N4O8H 507.2455; Found 507.2461.
Methyl (2-((S)-1-(2-(((Benzyloxy)carbonyl)amino)-2-methylpropanoyl)pyrrolidine-2-carboxamido)acryloyl)-l-alaninate (3b).
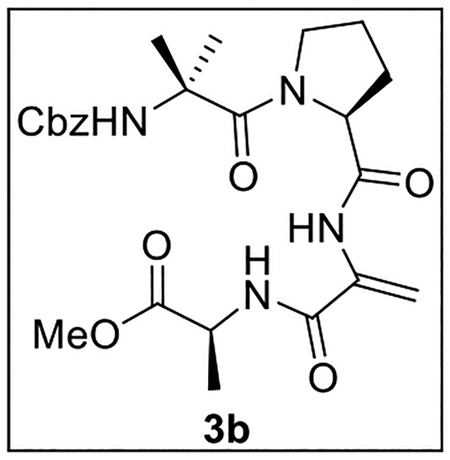
A solution of 17 (40.4 mg, 0.0798 mmol) in 1,2-dichloroethane (6 mL) at 0 °C was treated with Et3N (28 μL, 20.3 mg, 0.20 mmol, 2.5 equiv), then stirred at 0 °C for 10 min. The resulting mixture was treated dropwise with MsCl (12 μL, 17.8 mg, 0.155 mmol, 1.9 equiv), warmed to rt, and stirred for 1 h. It was then treated with DBU (60 μL, 61.1 mg, 0.401 mmol, 5.0 equiv) and stirred at 90 °C for 16 h. The mixture was concentrated in vacuo, and the residue was dissolved in EtOAc (10 mL) and washed with 10% aq citric acid (6 mL), sat aq NaHCO3 (2 × 5 mL), and brine (10 mL). The combined organic layers were dried (Na2SO4) and concentrated in vacuo. Flash chromatography (50 mL of SiO2, 0–10% MeOH in CH2Cl2 gradient elution) afforded 3b (14.2 mg, 0.0291 mmol, 36%) as a white solid: [α]25D +6.1 (c 0.6, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 8.45 (s, 1H), 7.42–7.30 (m, 5H), 7.10 (br s, 1H), 5.90 (s, 1H), 5.88 (s, 1H), 5.29 (s, 1H), 5.13 (d, J = 11.6 Hz, 1H), 5.00 (d, J = 12.0 Hz, 1H), 4.63 (quint, J = 7.1 Hz, 1H), 4.59–4.53 (m, 1H), 3.76 (s, 3H), 3.62–3.55 (m, 1H), 3.30–3.22 (m, 1H), 2.17–2.09 (m, 1H), 1.96–1.90 (m, 1H), 1.88–1.75 (m, 2H), 1.57 (s, 3H), 1.50 (s, 3H), 1.46 (d, J = 7.2 Hz, 3H); 13C{1H} NMR (CDCl3, 125 MHz) δ 176.7, 173.0, 172.6, 171.0, 156.0, 136.1, 128.7 (2C), 128.6 (2C), 128.5, 128.1, 111.9, 67.2, 66.9, 63.2, 57.0, 52.5, 48.6, 29.4, 26.7, 26.0, 24.6, 18.0; IR (film) υmax 3301, 2950, 1668, 1532, 1455, 1263, 1170, 1075 cm−1; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C24H32N4O7H 489.2349; Found 489.2357.
Methyl l-Threonyl-l-alaninate (18).
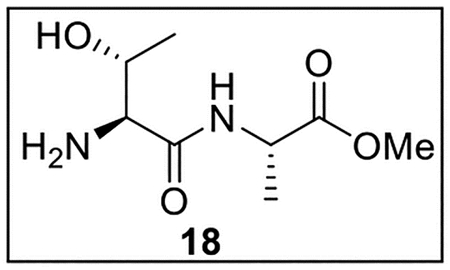
A solution of Z-Thr-OH (1.0021 g, 3.96 mmol) in anhydrous THF–DMF (5:1, 24 mL) under Ar at 0 °C was treated sequentially with Ala-OMe·HCl (594.0 mg, 4.26 mmol, 1.1 equiv), HOBt (ca. 20% H2O content, 800.5 mg, 4.74 mmol, 1.2 equiv), EDC·HCl (1.0621 g, 5.54 mmol, 1.4 equiv), and NaHCO3 (801.4 mg, 9.54 mmol, 2.4 equiv). The resulting mixture was allowed to warm to rt and was stirred for 24 h. The reaction was quenched by the addition of sat aq NaHCO3 (25 mL), diluted with H2O (30 mL), and extracted with EtOAc (5 × 40 mL). The combined organic layers were dried (Na2SO4) and concentrated in vacuo. Flash chromatography (200 mL of SiO2, 0–30% EtOAc in hexanes gradient elution) afforded Z-Thr-Ala-OMe (1.2778 g, 3.78 mmol, 95%) as a white solid: [α]25D −23 (c 0.50, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 7.39–7.32 (m, 5H), 6.96 (d, J = 6.7 Hz, 1H), 5.77 (d, J = 7.6 Hz, 1H), 5.17 (d, J = 12.2 Hz, 1H), 5.12 (d, J = 12.2 Hz, 1H), 4.56 (quint, J = 7.3 Hz, 1H), 4.38–4.32 (m, 1H), 4.19 (dd, J = 7.8, 2.0 Hz, 1H), 3.76 (s, 3H), 3.17 (br s, 1H), 1.40 (d, J = 7.2 Hz, 3H), 1.20 (d, J = 6.5 Hz, 3H); 13C{1H} NMR (CDCl3, 125 MHz) δ 173.1, 170.5, 156.8, 136.0, 128.6 (2C), 128.3, 128.0 (2C), 67.3, 66.9, 58.2, 52.6, 48.1, 18.0, 17.9; IR (film) υmax 3324, 3066, 3035, 2956, 1742, 1661, 1538, 1455, 1384, 1293, 1219, 1150, 1051 cm−1; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C16H22N2O6H 339.15561; Found 339.15492.
A solution of Z-Thr-Ala-OMe (1.200 g, 3.55 mmol) in MeOH (10.0 mL) was treated with 10% Pd/C (602.5 mg, 0.5 wt equiv) at rt under Ar. The suspension was then pressurized under hydrogen gas (500 psig) and stirred for 72 h. The reaction was filtered through Celite, dried (Na2SO4), and concentrated in vacuo. Amine 18 (534.1 mg, 2.62 mmol, 74%) was obtained as a white solid and was used in subsequent reactions without further purification.
Methyl (2-(((Benzyloxy)carbonyl)amino)-2-methylpropanoyl)-l-prolyl-l-threonyl-l-alaninate (19).
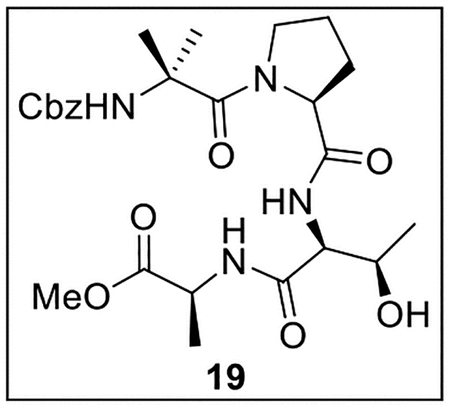
A solution of acid 13 (500.0 mg, 1.50 mmol, 1.0 equiv) in anhydrous THF–DMF (5:1, 12 mL) at 0 °C under Ar was treated with amine 18 (301.1 mg, 1.48 mmol), HOBt (ca. 20% H2O content, 310.6 mg, 1.84 mmol, 1.2 equiv), EDC·HCl (396.0 mg, 2.07 mmol, 1.4 equiv), and NaHCO3 (304.1 mg, 3.62 mmol, 2.5 equiv). The resulting mixture was warmed to rt, then stirred for 48 h. The reaction was quenched by the addition of sat aq NaHCO3 (15 mL), diluted with H2O (30 mL), and extracted with EtOAc (5 × 40 mL). The combined organic layers were dried (Na2SO4) and concentrated in vacuo. Flash chromatography (150 mL of SiO2, 2–4.5% MeOH in CH2Cl2 gradient elution) afforded 19 (167.6 mg, 0.322 mmol, 22%) as a white solid: [α]25D −8.1 (c 0.23, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 7.44 (d, J = 9.2 Hz, 1H), 7.40–7.34 (m, 7H), 5.17 (s, 1H), 5.15 (d, J = 12.4 Hz, 1H), 5.05 (d, J = 12.1 Hz, 1H), 4.61–4.53 (m, 4H), 3.78–3.73 (m, 1H), 3.73 (s, 3H), 3.32–3.27 (m, 1H), 2.36–2.31 (m, 1H), 1.89–1.83 (m, 3H), 1.55 (s, 3H), 1.47–1.44 (m, 3H), 1.46 (s, 3H), 1.21 (d, J = 6.4 Hz, 3H); 13C{1H} NMR (CDCl3, 125 MHz) δ 173.0, 172.5, 171.8, 170.6, 155.5, 135.4, 128.7 (2C), 128.7, 128.4 (2C), 67.8, 67.1, 63.5, 58.3, 57.2, 52.2, 48.6, 48.4, 28.9, 26.7, 26.0, 24.6, 19.5, 17.5; IR (film) υmax 3317, 2925, 1744, 1656, 1542, 1454, 1410, 1275, 1211, 1156, 1089 cm−1; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C25H36N4O8H+ 521.2611; Found 521.2602.
Methyl ((Z)-2-((S)-1-(2-(((Benzyloxy)carbonyl)amino)-2-methylpropanoyl)pyrrolidine-2-carboxamido)but-2-enoyl)-l-alaninate (3c).
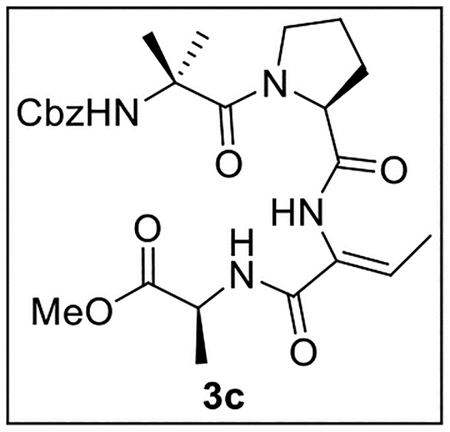
A solution of tetrapeptide 19 (100 mg, 0.192 mmol) in anhydrous CHCl3 (3 mL) at 0 °C was treated with Martin sulfurane (251.1 mg, 0.373 mmol, 1.9 equiv) and stirred at 0 °C to rt for 1 h. The mixture was dried (Na2SO4) and concentrated in vacuo. Flash chromatography (150 mL of SiO2, 2–4.5% MeOH in CH2Cl2 gradient elution) afforded 3c (35.0 mg, 0.0696 mmol, 36%) as a white film: [α]25D +18 (c 2.4, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 8.13 (s, 1H), 7.36–7.29 (m, 6H), 6.96 (q, J = 7.0 Hz, 1H), 5.41 (s, 1H), 5.16 (d, J = 12.1 Hz, 1H), 4.90 (d, J = 12.1 Hz, 1H), 4.61 (quint, J = 7.2 Hz, 1H), 4.55–4.50 (m, 1H), 3.70 (s, 3H), 3.69–3.65 (m, 1H), 3.18–3.10 (m, 1H), 2.32–2.27 (m, 1H), 1.85–1.74 (m, 3H), 1.72 (d, J = 7.1 Hz, 3H), 1.54 (s, 3H), 1.46 (d, J = 7.2 Hz, 3H), 1.43 (s, 3H); 13C{1H} NMR (CDCl3, 125 MHz) δ 173.2, 172.7, 170.7, 164.1, 155.5, 136.0, 133.8, 128.62 (2C), 128.57 (2C), 128.5 (2C), 67.3, 63.4, 57.0, 52.2 (2C), 48.6, 29.0, 26.7, 26.0, 24.4, 17.6, 13.4; IR (film) υmax 3274, 2925, 1625, 1536, 1270, 1076 cm−1; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C25H34N4O7H 503.2506; Found 503.2512.
Summary of NOE distance-restrained calculations.
Manual NMR structural calculations were performed to generate conformational ensembles of all the peptides that were studied. The ensembles were generated using the simulated annealing algorithm CYANA35 in conjunction with ROESY chemical shift and peak data for the peptides 1, 2a–c, and 3a–c. Dihedral restraints were not used due to the presence of multiple nonstandard amino acids, which lack sufficient secondary chemical shift analysis data. The final structural ensemble for each peptide consists of the 10 lowest-energy conformations as determined by the CYANA objective-function from 100 preliminary structures.
Procedure for proteolysis assays of 1, 2a–c, and 3a–c.
Pronase E from Streptomyces griseus (EC 232–909-5) was purchased from Sigma-Aldrich. This mixture of enzymes was dissolved in 1X PBS buffer (10 mM sodium phosphate, 137 mM NaCl, 2.7 mM KCl buffer, pH 7.4) at a concentration of 0.881 mg/mL. Then, solutions of each peptide (1, 2a, 2b, 2c, 3a, 3b, and 3c) in 1X PBS buffer (0.50 mM, 0.5 mL) at 38 °C were treated with an aliquot (200 μL) of the Pronase E solution. Aliquots (50 μL) were removed after 0, 15, 30, 45, 60, 90, and 120 min. The aliquots were quenched with glacial acetic acid (20 μL), diluted to 75 μL with 1X PBS buffer, and analyzed by HPLC (Phenomenex Jupiter C18, 5 μm particle size, 300 Å pore size, 4.6 ´ 250 mm, 75 μL injection volume, 40%– 90% CH3CN in H2O gradient over 30 min, then 95% CH3CN in H2O for 10 min, flow rate: 1 mL/min).
General procedure for thiol reactivity assays.
A solution of ΔAA-containing peptide 2 or 3 (typically 0.010–0.015 mmol) in DMSO-d6 (600 μL) containing a small amount (<1 mg) of 1,3,5-trimethoxybenzene as an internal standard was treated with 2 equiv of a freshly prepared 0.5 M solution of cysteamine in DMSO-d6 (typically 40–60 μL). 1H NMR spectra were acquired at various times, and disappearance of the starting peptide was quantified by comparing the integration values of diagnostic signals to the signals of the internal standard. In cases where the peptide was unreactive at room temperature, the solution was heated at successively higher temperatures (60 °C, 80 °C, and 100 °C) and 1H NMR spectra were acquired intermittently. As cysteamine degraded slowly, experiments were halted once it was completely absent from the solution according to 1H NMR.
Supplementary Material
Acknowledgment.
We thank the National Institutes of Health (1R15GM114789-01A1 and 3R15GM114789-01A1S1) and Brigham Young University (Undergraduate Research Awards to M.A.L. and D.W.K.) for support. Funding for S.T.M. and G.D. was provided by the National Science Foundation Chemistry and Biochemistry REU Site to Prepare Students for Graduate School and an Industrial Career under award CHE-1757627.
Footnotes
Publisher's Disclaimer: This document is confidential and is proprietary to the American Chemical Society and its authors. Do not copy or disclose without written permission. If you have received this item in error, notify the sender and delete all copies.
Supporting Information Available.
Data from proteolysis assays and peptide HPLC traces, tabulated 1D and 2D NMR data of peptides 1, 2a–c, and 3a–c, tabulated restraints and parameters used in structural calculations, structures of peptides 1, 2a–c, and 3a–c calculated using NMR data, ROESY spectrum snapshots, and copies of 1H and 13C NMR spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).(a) Henninot A; Collins JC; Nuss JM The Current State of Peptide Drug Discovery: Back to the Future? J. Med. Chem 2018, 61, 1382–1414. [DOI] [PubMed] [Google Scholar]; (b) Tsomaia N Peptide Therapeutics: Targeting the Undruggable Space. Eur. J. Med. Chem 2015, 94, 459–470. [DOI] [PubMed] [Google Scholar]; (c) Fosgerau K; Hoffmann T Peptide Therapeutics: Current Status and Future Directions. Drug Discovery Today 2015, 20, 122–128. [DOI] [PubMed] [Google Scholar]; (d) Kaspar AA; Reichert JM Future Directions for Peptide Therapeutics Development. Drug Discovery Today 2013, 18, 807–817. [DOI] [PubMed] [Google Scholar]; (e) Craik DJ; Fairlie DP; Liras S; Price D The Future of Peptide-Based Drugs. Chem. Biol. Drug Des 2013, 81, 136–147. [DOI] [PubMed] [Google Scholar]
- (2).(a) Webster AM; Cobb SL Recent Advances in the Synthesis of Peptoid Macrocycles. Chem. - Eur. J 2018, 24, 7560–7573. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sun J; Zuckermann RN Peptoid Polymers: A Highly Designable Bioinspired Material. ACS Nano 2013, 7, 4715–4732. [DOI] [PubMed] [Google Scholar]; (c) Yoo B; Shin SBY; Huang ML; Kirshenbaum K Peptoid Macrocycles: Making the Rounds with Peptidomimetic Oligomers. Chem. - Eur. J 2010, 16, 5528–5537. [DOI] [PubMed] [Google Scholar]; (d) Fowler SA; Blackwell HE Structure–Function Relationships in Peptoids: Recent Advances Toward Deciphering the Structural Requirements for Biological Function. Org. Biomol. Chem 2009, 7, 1508–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) Cabrele C; Martinek TA; Reiser O; Berlicki Ł Peptides Containing β-Amino Acid Patterns: Challenges and Successes in Medicinal Chemistry. J. Med. Chem 2014, 57, 9718–9739. [DOI] [PubMed] [Google Scholar]; (b) Horne WS; Gellman SH Foldamers with Heterogeneous Backbones. Acc. Chem. Res 2008, 41, 1399–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Seebach D; Gardiner J β-Peptidic Peptidomimetics. Acc. Chem. Res 2008, 41, 1366–1375. [DOI] [PubMed] [Google Scholar]; (d) Cheng RP; Gellman SH; DeGrado WF β-Peptides: From Structure to Function. Chem. Rev 2001, 101, 3219–3252. [DOI] [PubMed] [Google Scholar]
- (4).(a) Reguera L; Rivera DG Multicomponent Reaction Toolbox for Peptide Macrocyclization and Stapling. Chem. Rev 2019, 119, 9836–9860. [DOI] [PubMed] [Google Scholar]; (b) Lau YH; de Andrade P; Wu Y; Spring DR Peptide Stapling Techniques Based on Different Macrocyclisation Chemistries. Chem. Soc. Rev 2015, 44, 91–102. [DOI] [PubMed] [Google Scholar]; (c) Cromm PM; Spiegel J; Grossmann TN Hydrocarbon Stapled Peptides as Modulators of Biological Function. ACS Chem. Biol 2015, 10, 1362–1375. [DOI] [PubMed] [Google Scholar]; (d) Walensky LD; Bird GH Hydrocarbon-Stapled Peptides: Principles, Practice, and Progress. J. Med. Chem 2014, 57, 6275–6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patgiri A; Jochim AL; Arora PS A Hydrogen Bond Surrogate Approach for Stabilization of Short Peptide Sequences in α-Helical Conformation. Acc. Chem. Res 2008, 41, 1289–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Teng P; Shi Y; Sang P; Cai J γ-AAPeptides as a New Class of Peptidomimetics. Chem. - Eur. J 2016, 22, 5458–5466. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shi Y; Teng P; Sang P; She F; Wei L; Cai J γ-AAPeptides: Design, Structure, and Applications. Acc. Chem. Res 2016, 49, 428–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weinstock MT; Francis JN; Redman JS; Kay MS Protease-Resistant Peptide Design—Empowering Nature’s Fragile Warriors Against HIV. Biopolymers (Pept. Sci.) 2012, 98, 431–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Toniolo C; Formaggio F; Kaptein B; Broxterman QB You Are Sitting on a Gold Mine! Synlett 2006, 1295–1310. [Google Scholar]
- 9.Sarnowski MP; Kang CW; Elbatrawi YM; Wojtas L; Del Valle JR Peptide N-Amination Supports β-Sheet Conformations. Angew. Chem., Int. Ed 2017, 56, 2083–2086. [DOI] [PubMed] [Google Scholar]
- (10).(a) Lubell WD; Khashper A Design, Synthesis, Conformational Analysis and Application of Indolizidin-2-one Dipeptide Mimics. Org. Biomol. Chem 2014, 12, 5052–5070. [DOI] [PubMed] [Google Scholar]; (b) MacDonald M; Aubé J Approaches to Cyclic Peptide β-Turn Mimics. Curr. Org. Chem 2001, 5, 417–438. [Google Scholar]
- (11).(a) Nischan N; Hackenberger CPR Site-specific PEGylation of Proteins: Recent Developments. J. Org. Chem 2014, 79, 10727–10733. [DOI] [PubMed] [Google Scholar]; (b) Jevševar S; Kunstelj M; Porekar VG PEGylation of Therapeutic Proteins. Biotechnol J 2010, 5, 113–128. [DOI] [PubMed] [Google Scholar]; (c) Harris JM; Chess RB Effect of PEGylation on Pharmaceuticals. Nat. Rev. Drug Discovery 2003, 2, 214–221. [DOI] [PubMed] [Google Scholar]
- (12).(a) Liu Z; Chen X Simple Bioconjugate Chemistry Serves Great Clinical Advances: Albumin as a Versatile Platform for Diagnosis and Precision Therapy. Chem. Soc. Rev 2016, 45, 1432–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Elsadek B; Kratz F Impact of Albumin on Drug Delivery – New Applications on the Horizon. J. Controlled Release 2012, 157, 4–28. [DOI] [PubMed] [Google Scholar]; (c) Pollaro L; Heinis C Strategies to Prolong the Plasma Residence Time of Peptide Drugs. Med. Chem. Commun 2010, 1, 319–324. [Google Scholar]
- 13.O’Banion CP; Nguyen LT; Wang Q; Priestman MA; Holly SP; Parise LV; Lawrence DS The Plasma Membrane as a Reservoir, Protective Shield, and Light-Triggered Launch Pad for Peptide Therapeutics. Angew. Chem., Int. Ed 2016, 55, 950–954. [DOI] [PubMed] [Google Scholar]
- 14. https://www.fda.gov/drugs/development-approval-process-drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products.
- (15).(a) English ML; Stammer CH The Enzyme Stability of Dehydropeptides. Biochem. Biophys. Res. Commun 1978, 83, 1464–1467. [DOI] [PubMed] [Google Scholar]; (b) English ML; Stammer CH d-Ala2, ΔZPhe4-Methionine Enkephalin Amide, a Dehydropeptide Hormone. Biochem. Biophys. Res. Commun 1978, 85, 780–782. [DOI] [PubMed] [Google Scholar]; (c) Shimohigashi Y; Chen H-C; Stammer CH The Enzyme Stability of Dehydro-Enkephalins. Peptides 1982, 3, 985–987. [DOI] [PubMed] [Google Scholar]; (d) Shimohigashi Y; Stammer CH Dehydro-enkephalins. Part 7. A Potent Dehydroleucine-enkephalin Resistant to Carboxypeptidase. J. Chem. Soc., Perkin Trans 1 1983, 803–808. [Google Scholar]
- (16).(a) Daniel RM; Cowan DA; Morgan HW; Curran MP A Correlation Between Protein Thermostability and Resistance to Proteolysis. Biochem. J 1982, 207, 641–644. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Imoto T; Yamada H; Ueda T Unfolding Rates of Globular Proteins Determined by Kinetics of Proteolysis. J. Mol. Biol 1986, 190, 647–649. [DOI] [PubMed] [Google Scholar]; (c) Parsell DA; Sauer RT The Structural Stability of a Protein Is an Important Determinant of Its Proteolytic Susceptibility in Escherichia coli. J. Biol. Chem 1989, 264, 7590–7595. [PubMed] [Google Scholar]; (d) Klink TA; Raines RT Conformational Stability Is a Determinant of Ribonuclease A Cytotoxicity. J. Biol. Chem 2000, 275, 17463–17467. [DOI] [PubMed] [Google Scholar]; (e) Ahmad S; Kumar V; Ramanand KB; Rao NM Probing Protein Stability and Proteolytic Resistance by Loop Scanning: A Comprehensive Mutational Analysis. Protein Sci 2012, 21, 433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).(a) Rajashankar KR; Ramakumar S; Chauhan VS Design of a Helical Motif Using α,β-Dehydrophenylalanine Residues: Crystal Structure of Boc-Val-ΔPhe-Phe-Ala-Phe-ΔPhe-Val-ΔPHe-Gly-OCH3, a 310-Helical Nonapeptide. J. Am. Chem. Soc 1992, 114, 9225–9226. [Google Scholar]; (b) Bhandary KK; Chauhan VS Peptide Design 310-Helical Conformation of a Linear Pentapeptide Containing Two Dehydrophenylalanines, Boc-Gly-ΔZ-Phe-Leu-ΔZPhe-Ala-NHCH3. Biopolymers 1993, 33, 209–217. [DOI] [PubMed] [Google Scholar]; (c) Pieroni O; Fissi A; Jain RM; Chauhan VS Solution Structure of Dehydropeptides: A CD Investigation. Biopolymers 1996, 38, 97–108. [DOI] [PubMed] [Google Scholar]; (d) Jain RM; Rajashankar KR; Ramakumar S; Chauhan VS First Observation of Left-Handed Helical Conformation in a Dehydro Peptide Containing Two l-Val Residues. Crystal and Solution Structure of Boc-l-Val-ΔPhe-ΔPhe-ΔPhe-l-Val-OMe. J. Am. Chem. Soc 1997, 119, 3205–3211. [Google Scholar]
- (18).(a) Jain R; Chauhan VS Conformational Characteristics of Peptides Containing α,β-Dehydroamino Acid Residues. Biopolymers (Pept. Sci.) 1996, 40, 105–119. [DOI] [PubMed] [Google Scholar]; (b) Mathur P; Ramakumar S; Chauhan VS Peptide Design Using α,β-Dehydroamino Acids: From β-Turns to Helical Hairpins. Biopolymers (Pept. Sci.) 2004, 76, 150–161. [DOI] [PubMed] [Google Scholar]; (c) Gupta M; Chauhan VS De Novo Design of α,β-Didehydrophenylalanine Containing Peptides: From Models to Applications. Biopolymers 2011, 95, 161–173. [DOI] [PubMed] [Google Scholar]
- (19).(a) Singh TP; Narula P; Patel HC α,β-Dehydro Residues in the Design of Peptide and Protein Structures. Acta Crystallogr 1990, B46, 539–545. [DOI] [PubMed] [Google Scholar]; (b) Zecchini GP; Paradisi MP; Torrini I; Lucente G; Gavuzzo E; Mazza F; Pochetti G; Paci M; Sette M; Di Nola A; Veglia G; Traniello S; Spisani S Synthesis, Conformation, and Activity of HCO-Met-ΔZLeu-Phe-Ome, an Active Analogue of Chemotactic N-Formyltripeptides. Biopolymers 1993, 33, 437–451. [DOI] [PubMed] [Google Scholar]; (c) Inai Y; Oshikawa T; Yamashita M; Hirabayashi T; Hirako T Structural and Conformational Properties of (Z)-β-(1-Naphthyl)-dehydroalanine Residue. Biopolymers 2001, 58, 9–19. [DOI] [PubMed] [Google Scholar]; (d) Inai Y; Hirabayashi T A Helical Arrangement of β-Substituents of Dehydropeptides: Synthesis and Conformational Study of Sequential Nona- and Dodecapeptides Possessing (Z)-β-(1-Naphthyl)-dehydroalanine Residues. Biopolymers 2001, 59, 356–369. [DOI] [PubMed] [Google Scholar]; (e) Buczek A; Siodłak D; Bujak M; Broda MA Effects of Side-Chain Orientation on the Backbone Conformation of the Dehydrophenylalanine Residue. Theoretical and X-ray Study. J. Phys. Chem. B 2011, 115, 4295–4306. [DOI] [PubMed] [Google Scholar]; (f) Jewginski M; Latajka R; Krezel A; Haremza K; Makowski M; Kafarski P Influence of Solvents on Conformation of Dehydropeptides. J. Mol. Struct 2013, 1035, 129–139. [Google Scholar]; (g) Buczek A; Makowski M; Jewgiński M; Latajka R; Kupka T; Broda MA Toward Engineering Efficient Peptidomimetics. Screening Conformational Landscape of Two Modified Dehydroaminoacids. Biopolymers 2014, 101, 28–40. [DOI] [PubMed] [Google Scholar]
- 20.Le DN; Riedel J; Kozlyuk N; Martin RW; Dong VM Cyclizing Pentapeptides: Mechanism and Application of Dehydrophenylalanine as a Traceless Turn-Inducer. Org. Lett 2017, 19, 114–117. [DOI] [PubMed] [Google Scholar]
- (21).(a) Yamada K; Shinoda S.-s.; Oku H; Komagoe K; Katsu T; Katakai R Synthesis of Low-Hemolytic Antimicrobial Dehydropeptides Based on Gramicidin S. J. Med. Chem 2006, 49, 7592–7595. [DOI] [PubMed] [Google Scholar]; (b) Yamada K; Kodaira M; Shinoda S.-s.; Komagoe K; Oku H; Katakai R; Katsu T; Matsuo I Structure–Activity Relationships of Gramicidin S Analogs Containing (β−3-Pyridyl)-α,β-dehydroalanine Residues on Membrane Permeability. Med. Chem. Commun 2011, 2, 644–649. [Google Scholar]; (c) Grigoryan HA; Hambardzumyan AA; Mkrtchyan MV; Topuzyan VO; Halebyan GP; Asatryan RS α,β-Dehydrophenylalanine Choline Esters, a New Class of Reversible Inhibitors of Human Acetylcholinesterase and Butyrylcholinesterase. Chem.-Biol. Interact 2008, 171, 108–116. [DOI] [PubMed] [Google Scholar]; (d) Pathak S; Chauhan VS Rationale-Based, De Novo Design of Dehydrophenylalanine-Containing Antibiotic Peptides and Systematic Modification in Sequence for Enhanced Potency. Antimicrob. Agents Chemother 2011, 55, 2178–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Mishra A; Misra A; Vaishnavi TS; Thota C; Gupta M; Ramakumar S; Chauhan VS Conformationally Restricted Short Peptides Inhibit Human Islet Amyloid Polypeptide (hIAPP) Fibrillization. Chem. Commun 2013, 49, 2688–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Giordano C; Punzi P; Lori C; Chiaraluce R; Consalvi V β-Sheet Breaker Peptides Containing α,β-Dehydrophenylalanine: Synthesis and In Vitro Activity Studies. ChemPlusChem 2014, 79, 1036–1043. [Google Scholar]
- (22).(a) Yokokawa F; Shioiri T Novel Stereospecific Dehydration of β-Hydroxy-α-amino Acids Using Martin’s Sulfurane. Tetrahedron Lett 2002, 43, 8679–8682. [Google Scholar]; (b) Sai H; Ogiku T; Ohmizu H Stereoselective Syntheses of (E)-α,β-Dehydroamino Acids and (E)-α,β-Dehydropeptides by Stereospecific Dehydration with 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC). Synthesis 2003, 201–204. [Google Scholar]
- (23).(a) Yamashita T; Kuranaga T; Inoue M Solid-Phase Total Synthesis of Bogorol A: Stereocontrolled Construction of Thermodynamically Unfavored (E)-2-Amino-2-butenamide. Org. Lett 2015, 17, 2170–2173. [DOI] [PubMed] [Google Scholar]; (b) Butler E; Florentino L; Cornut D; Gomez-Campillos G; Liu H; Regan AC; Thomas EJ Synthesis of Macrocyclic Precursors of the Vioprolides. Org. Biomol. Chem 2018, 16, 6935–6960. [DOI] [PubMed] [Google Scholar]
- 24.Ueoka R; Ise Y; Ohtsuka S; Okada S; Yamori T; Matsunaga S Yaku’amides A and B, Cytotoxic Linear Peptides Rich in Dehydroamino Acids from the Marine Sponge Ceratopsion sp. J. Am. Chem. Soc 2010, 132, 17692–17694. [DOI] [PubMed] [Google Scholar]
- 25.Culvenor CCJ; Edgar JA; Mackay MF; Gorst-Allman CP; Marasas WFO; Steyn PS; Vleggaar R; Wessels PL Structure Elucidation and Absolute Configuration of Phomopsin A, a Hexapeptide Mycotoxin Produced by Phomopsis Leptostromiformis. Tetrahedron 1989, 45, 2351–2372. [Google Scholar]
- (26).(a) Irschik H, Gerth K, Kemmer T, Steinmetz H, Reichenbach H The Myxovalargins, New Peptide Antibiotics from Myxococcus Fulvus (Myxobacterales). I. Cultivation, Isolation and Some Chemical and Biological Properties. J. Antibiot 1983, 36, 6–12. [DOI] [PubMed] [Google Scholar]; (b) Irschik H, Reichenbach H The Mechanism of Action of Myxovalargin A, a Peptide Antibiotic from Myxococcus Fulvus. J. Antibiot 1985, 38, 1237–1245. [DOI] [PubMed] [Google Scholar]; (c) Steinmetz H; Irschik H; Reichenbach H; Höfle G Structure Elucidation of the Peptide Antibiotics Myxovalargin A–D. Chem. Pept. Proteins 1989, 4, 13–18. [Google Scholar]
- (27).(a) Pietrzyński G; Rzeszotarska B; Kubica Z Conformational Investigation of α,β-Dehydropeptides. Part IV. β-Turn in Saturated and α,β-Unsaturated Peptides Ac-Pro-Xaa-NHCH3: NMR and IR Studies. Int. J. Peptide Protein Res 1992, 40, 524–531. [PubMed] [Google Scholar]; (b) Vijayaraghavan R; Kumar P; Dey S; Singh TP Design of Peptides with α,β-Dehydro Residues: A Dipeptide with a Branched β-Carbon Dehydro Residue at the (i+1) Position, Methyl N-(Benzyloxycarbonyl)-α,β-didehydrovalyl-l-tryptophanate. Acta Crystallogr., Sect. C 2001, 57, 1220–1221. [DOI] [PubMed] [Google Scholar]; (c) Vijayaraghavan R; Kumar P; Dey S; Singh TP Design of Peptides with Branched β-Carbon Dehydro-residues: Syntheses, Crystal Structures and Molecular Conformations of Two Peptides, (I) N-Carbobenzoxy-ΔVal-Ala-Leu-OCH3 and (II) N-Carbobenzoxy-ΔIle-Ala-Leu-OCH3. J. Pept. Res 2003, 62, 63–69. [DOI] [PubMed] [Google Scholar]; (d) Makker J; Dey S; Mukherjee S; Vijayaraghavan R; Kumar P; Singh TP Design of Peptides with α,β-Dehydro-residues: Synthesis, Crystal Structure and Molecular Conformation of a Tetrapeptide Z-ΔVal-Val-ΔPhe-Ile-Ome. J. Mol. Struct 2003, 654, 119–124. [Google Scholar]; (e) Siodłak D; Rzeszotarska B; Broda MA; Kozioł AE; Kołodziejczyk E Conformational Investigation of α,β-Dehydropeptides. N-Acetyl-α,β-dehydrovaline-N’,N’-dimethylamide. Acta Biochim. Pol 2004, 51, 145–152. [PubMed] [Google Scholar]; (f) Broda MA; Siodłak D; Rzeszotarska B Conformational Investigation of α,β-Dehydropeptides. XV: N-Acetyl-α,β-dehydroamino Acid N’N’-Dimethylamides: Conformational Properties from Infrared and Thereorectical Studies. J. Pept. Sci 2005, 11, 546–555. [DOI] [PubMed] [Google Scholar]; (g) Siodłak D; Grondys J; Lis T; Bujak M; Broda MA; Rzeszotarska B The Conformational Properties of Dehydrobutyrine and Dehydrovaline: Theoretical and Solid-State Conformational Studies. J. Pept. Sci 2010, 16, 496–505. [DOI] [PubMed] [Google Scholar]; (h) Siodłak D; Bujak M; Staś M Intra- and Intermolecular Forces Dependent Main Chain Conformations of Esters of α,β-Dehydroamino Acids. J. Mol. Struct 2013, 1047, 229–236. [Google Scholar]
- 28.For a review on bulky ΔAAs, see:; Jiang J; Ma Z; Castle SL Bulky α,β-Dehydroamino Acids: Their Occurrence in Nature, Synthesis, and Applications. Tetrahedron 2015, 71, 5431–5451. [Google Scholar]
- 29.Jiang J; Luo S; Castle SL Solid-Phase Synthesis of Peptides Containing Bulky Dehydroamino Acids. Tetrahedron Lett 2015, 56, 3311–3313. [Google Scholar]
- 30.Jalan A; Kastner DW; Webber KGI; Smith MS; Price JL; Castle SL Bulky Dehydroamino Acids Enhance Proteolytic Stability and Folding in β-Hairpin Peptides. Org. Lett 2017, 19, 5190–5193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nagaraj R; Shamala N; Balaram P Stereochemically Constrained Linear Peptides. Conformations of Peptides Containing α-Aminoisobutyric Acid. J. Am. Chem. Soc 1979, 101, 16–20. [Google Scholar]
- 32.Ma Z; Naylor BC; Loertscher BM; Hafen DD; Li JM; Castle SL Regioselective Base-Free Intermolecular Aminohydroxylations of Hindered and Functionalized Alkenes. J. Org. Chem 2012, 77, 1208–1214. [DOI] [PubMed] [Google Scholar]
- 33.Ross AC; Liu H; Pattabiraman VR; Vederas JC Synthesis of the Lantibiotic Lactocin S Using Peptide Cyclizations on Solid Phase. J. Am. Chem. Soc 2010, 132, 462–463. [DOI] [PubMed] [Google Scholar]
- 34.Wołczański G; Lisowski M A General Method for Preparation of N-Boc-Protected or N-Fmoc-Protected α,β-Didehydropeptide Building Blocks and Their Use in the Solid-Phase Peptide Synthesis. J. Pept. Sci 2018, 24, e3091. [DOI] [PubMed] [Google Scholar]
- (35).(a) Güntert P; Mumenthaler C; Wüthrich K Torsion Angle Dynamics for NMR Structure Calculation with the New Program DYANA. J. Mol. Biol 1997, 273, 283–298. [DOI] [PubMed] [Google Scholar]; (b) Yilmaz EM; Güntert P NMR Structure Calculation for All Small Molecule Ligands and Non-Standard Residues from the PDB Chemical Component Dictionary. J. Biomol. NMR 2015, 63, 21–37. [DOI] [PubMed] [Google Scholar]
- 36.Cline LL; Waters ML The Structure of Well-Folded β-Hairpin Peptides Promotes Resistance to Peptidase Degradation. Biopolymers (Pept. Sci.) 2009, 92, 502–507. [DOI] [PubMed] [Google Scholar]
- 37.Yang B; Wang N; Schnier PD; Zheng F; Zhu H; Polizzi NF; Ittuveetil A; Saikam V; DeGrado WF; Wang Q; Wang PG; Wang L Genetically Introducing Biochemically Reactive Amino Acids Dehydroalanine and Dehydrobutyrine in Proteins. J. Am. Chem. Soc 2019, 141, 7698–7703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chatterjee C; Paul M; Xie L; van der Donk WA Biosynthesis and Mode of Action of Lantibiotics. Chem. Rev 2005, 105, 633–683. [DOI] [PubMed] [Google Scholar]
- (39).(a) Gutiérrez A; Osante I; Cativiela C Synthesis of cis- and trans-(±)-3-Mercaptoproline and Pipecolic Acid Derivatives via thio-Michael Addition. Tetrahedron Lett 2018, 59, 1661–1665. [Google Scholar]; (b) Villadsen NL; Hansen BK; Svenningsen EB; Jørgensen KH; Tørring T; Poulsen TB Stendomycin and Pantomycin Are Identical Natural Products: Preparation of a Functionalized Bioactive Analogue. J. Org. Chem 2018, 83, 7303–7308. [DOI] [PubMed] [Google Scholar]
- 40.Aydillo C; Avenoza A; Busto JH; Jiménez-Osés G; Peregrina JM; Zurbano MM A Biomimetic Approach to Lanthionines. Org. Lett 2012, 14, 334–337. [DOI] [PubMed] [Google Scholar]
- 41.Avonto C; Taglialatela-Scafati O; Pollastro F; Minassi A; Di Marzo V; De Petrocellis L; Appendino G An NMR Spectroscopic Method to Identify and Classify Thio-Trapping Agents: Revival of Michael Acceptors for Drug Discovery? Angew. Chem., Int. Ed 2011, 50, 467–471. [DOI] [PubMed] [Google Scholar]
- 42.A preliminary version of this manuscript was deposited on the ChemRxiv preprint server:; Joaquin D; Lee MA; Kastner DW; Singh J; Morrill ST; Damstedt G; Castle SL Impact of Dehydroamino Acids on the Structure and Stability of Incipient 310-Helical Peptides. ChemRxiv 2019, 10.26434/chemrxiv.9968375.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.