

Summary
Hepatocellular carcinoma (HCC) is the most common type of liver cancer with limited treatments. Asia has the highest HCC incidence rates; China accounts for over 50% of all HCC cases worldwide. T‐cell receptor (TCR) ‐engineered T‐cell immunotherapies specific for human leukocyte antigen (HLA) ‐A*02:01‐restricted α‐fetoprotein (AFP) peptide have shown encouraging results in clinics. HLA‐A*24:02 is more common than HLA‐A*02:01 in Asian countries, including China. Here we identified a novel HLA‐A*24:02‐restricted peptide KWVESIFLIF (AFP2–11) located in AFP signal peptide domain by mass spectrometric analysis of HLA‐bound peptides from HepG2 cells. A TCR (KWV3.1) specific for AFP2–11‐HLA‐A*24:02 was isolated from peripheral blood mononuclear cells of a healthy donor. The binding affinity of soluble KWV3.1 to its antigen was determined to be ~55 μm, within the affinity range of native TCRs for self‐antigens. KWV3.1‐transfected T cells could specifically activate and kill AFP2–11 pulsed T2‐A24 cells and AFP+ HLA‐A*24:02+ tumor cell lines, demonstrating that AFP2–11 can be naturally presented on the surface of AFP+ tumor cell lines. The newly identified antigenic peptide can provide a novel target for immunotherapeutic strategies for patients with AFP+ HLA‐A*24:02+ HCC.
Keywords: α‐fetoprotein, hepatocellular carcinoma, HLA‐A*24:02, mass spectrometry, T‐cell immunotherapy
We identified a novel HLA‐A*24:02‐restricted antigen KWVESIFLIF (AFP2–11) derived from the α‐fetoprotein (AFP) signal peptide by mass spectrometric analysis of immune‐precipitated hepatocellular carcinoma (HCC) cell lysate and demonstrated its natural presentation with AFP+ tumor cell lines. The immunogenicity of AFP2–11 was confirmed by isolation of a specific TCR from an HLA‐A*24:02+ healthy donor, which conferred T cells by gene transduction with capability to kill tumor cells. This study offers a new target for the development of immunotherapy for HCC patients.

Abbreviations
- AFP
α‐fetoprotein
- FBS
fetal bovine serum
- HCC
hepatocellular carcinoma
- HLA
human leukocyte antigen
- HPLC
high‐performance liquid chromatography
- IFN‐γ
interferon‐γ
- IL‐2
interleukin‐2
- MHC
major histocompatibility complex
- MS
mass spectrometry
- PBMC
peripheral blood mononuclear cell
- SP
signal peptide
- TAP
antigen peptide transporter
- TCR
T‐cell receptor
- TCR‐T
T‐cell receptor engineered T cell
Introduction
Being the sixth most common cancer and the fourth most common cause of cancer‐related deaths globally,1 liver cancer is a major threat to health. Hepatocellular carcinoma (HCC) is the most prevalent type of primary liver cancer predominantly occurring in patients with chronic liver diseases as a result of hepatitis virus infection or alcohol abuse.2, 3, 4 HCC is particularly common in China, where it accounts for over 50% of global incidence and deaths each year.3, 4, 5 With very limited systemic therapies, HCC in advanced stages has poor prognosis and poor survival rates.3, 4, 6 Therefore, novel cancer treatment modalities are urgently needed for HCC patients.
Immunotherapy has emerged as a new promising strategy for HCC patients.7, 8 The immune checkpoint inhibitor nivolumab has been approved by the US Food and Drug Administration for the treatment of advanced HCC. However, it is unlikely to benefit the majority of patients with HCC because of the low response rate (20%) and treatment‐related adverse effects.9 T‐cell receptor‐modified T‐cell (TCR‐T) therapy is capable of targeting a wide variety of antigenic peptides presented by major histocompatibility complexes (pMHCs) and has demonstrated significant clinical efficacy in solid tumors,10, 11, 12 so is likely to provide new strategies for treating HCC.
α‐Fetoprotein (AFP) is expressed during embryonic development13, 14 and transcriptionally repressed immediately after birth.14, 15, 16 It was considered that AFP was not expressed, or was expressed at almost undetectable levels in healthy tissues.17 However, AFP becomes transcriptionally reactivated and is highly expressed in approximately 50%–80% of HCC patients.18, 19, 20 As a result, AFP is an attractive target for TCR‐T therapy.
Human leukocyte antigen (HLA)‐A*02:01 (hereinafter referred to as HLA‐A2) ‐restricted AFP peptides15 are the most widely studied AFP epitopes. AFP peptide‐based immunization has shown only modest clinical responses in patients.15, 21, 22 Several groups have developed TCRs17, 23 or TCR‐like antibodies24 specific for HLA‐A2/AFP158–166. T cells transduced with the TCRs or chimeric antigen receptor could specifically recognize and kill AFP+ tumor cells in vitro and eradicate tumors in murine models. Adoptive transfer of the engineered T cells has shown safety and early efficacy in HCC patients in an on‐going clinical trial.25 Hence, adoptive T‐cell immunotherapy is a promising new avenue for HCC treatment.
Not only the incidence rates for HCC show geographic variations worldwide, but also the HLA allele frequencies vary in different countries. Therefore, it is highly desirable to develop AFP epitopes restricted by HLA alleles that are more common in countries where the burden of HCC is high. HLA‐A2 is the most common HLA‐A allele in Europe and North America, where the HCC incidence is low, but not in Asia, where the burden of HCC is much higher.26, 27 HLA‐A*24:02 (hereinafter referred to as HLA‐A24), on the other hand, is much more common in Asia, such as in China and Japan.28 Several HLA‐A24‐restricted AFP peptides have been identified29 and used as anti‐tumor vaccines,30 but none have demonstrated potential in TCR‐T therapy.
Here, we report on a novel HLA‐A24‐restricted AFP2–11 peptide (KWVESIFLIF) discovered using mass spectrometry (MS) analysis. Moreover, an anti‐AFP2–11 TCR (KWV3.1) was identified from an AFP‐specific T‐cell clone isolated from peripheral blood mononuclear cells (PBMCs) of an HLA‐A24+ healthy donor. T cells transfected with KWV3.1 mediated specific cytokine release and lysis of target cells when co‐cultured with AFP+ cell lines. We demonstrated that AFP2–11 could be naturally processed and presented by HLA‐A24 on tumor cells. Therefore AFP2–11 is a promising epitope target for HLA‐A24 HCC patients and KWV3.1 has the therapeutic potential to be developed into an HLA‐A24‐restricted TCR‐T therapy that may cover a larger portion of the HCC patients compared with HLA‐A2‐restricted therapies.
Materials and methods
Collection of HLA‐A24‐restricted peptides on the cell surface
HepG2 cells (ATCC, Manassas, VA) were lyzed with lyzing buffer (20 mm Tris–HCl (pH7·5), 150 mm NaCl, 0·5% Triton X‐100) and centrifuged at 8000 g. The supernatant was collected and further filtered through a vacuum pump (Welch, Mt Prospect, IL) and incubated with the anti‐HLA‐A11/A24 antibody (Clone A11.1M, produced in‐house) at 4° overnight, and then immunoprecipitated with rProtein A Sepharose beads (GE Life Sciences, Chalfont St Giles, UK) at 4° for 4 hr. The pellet was washed with washing buffer (20 mm Tris–HCl, pH 7·5). A 10% (vol/vol) acetic acid/water (Acros Organics, Franklin Lakes, NJ) elution buffer was used to dissociate the bound peptide–HLA from the antibody–rProtein A Sepharose beads. To collect peptides bound to HLA, the eluate was incubated at 95° for 5 min, centrifuged through an Amicon Ultra 10k device (Millipore, Billerica, MA) and concentrated by vacuum centrifugation (Labconco, Kansas City, MO).
Analysis of peptides using nano high‐performance liquid chromatography and MS
The peptides were separated by high‐performance liquid chromatography (HPLC). Briefly, the sample containing the peptides was injected into an Agilent 1260 HPLC system, separated by gradient elution from a C18 reverse‐phase column (ZORBAX 300SB‐C18, 1·0 × 150 mm, 3·5 µm), with a binary gradient of 30%–70% solvent B (0·1%/2%/98%, vol/vol/vol, formic acid/water/acetonitrile) within 5 min, and fractionated in 100 µl aliquots.
The peptide fractions were analyzed by an Ultimate 3000 Nano LC (Dionex, Sunnyvale, CA) coupled to a Q Exactive Plus (ThermoFisher Scientific, Franklin Lakes, NJ). The LC method included a C18 precolumn (ThermoFisher Scientific; Acclaim PepMap®100, 100 µm × 2 cm, nanoViper, 5 µm, 100 Å) for concentrating and desalting peptides before loading onto an analytical column (ThermoFisher Scientific; Acclaim PepMap®100, 75 µm × 15 cm, nanoViper, 3 µm, 100 Å). The sample was also analyzed on a Nano Ultra LC (Eksigent, Dublin, CA) with an analytical column (Eksigent, 75 µm × 15 cm, 3 µm, 120 Å) configured with a trap column (Eksigent, 350 µm × 0·5 mm, 3 µm, 120 Å) coupled to a Triple TOF 5600+ Mass Spectrometer (Sciex, Warrington, UK).
Database search and analysis
The acquired MS raw data were processed in PEAKS studio 8.0 (Bioinformatics Solutions Inc., Waterloo, ON, Canada) using the Uniprot FASTA database for human data (https://www.uniprot.org/). The candidate peptides were determined based on b ions and y ions of the MS/MS spectra and confirmed by comparison with the spectra of the corresponding synthetic peptides.
Synthetic peptides
Peptides were synthesized using GenScript. The quality of peptides, including purity, amino acid sequence and molecular weight, was evaluated by MS coupled with HPLC.
T cells and tumor cell lines
The PBMCs were isolated from buffy coats (Guangzhou Blood Center, Guangzhou, China) using density centrifugation. CD8+ T cells were freshly isolated from PBMCs using the EasySep™ Human CD8+ T‐Cell Isolation Kit (StemCell Technologies, Vancouver, BC, Canada). All tumor cell lines were obtained from ATCC. The T2‐A24 cell line was generated from its parental T2 cell line by stable transduction of a lentiviral vector encoding the HLA‐A24 heavy chain and further analyzed using anti‐HLA‐A24 antibody (One Lambda, West Hills, CA) and anti‐mouse IgG Fab2 antibody (Cell Signaling, Danvers, MA). SNU398‐AFP cell line was derived from its parental cell line, SNU398 (AFP− HLA‐A24+), through stable transduction of a lentiviral vector encoding AFP. T2‐A24, SNU398, SNU398‐AFP cell lines were cultured in RPMI‐1640 (Invitrogen Life Technologies, Carlsbad, CA) supplemented with 10% heat‐inactivated fetal bovine serum (FBS) (Life Technologies). The HepG2 cell line (AFP+ HLA‐A24+) was cultured in minimal essential medium (Life Technologies) supplemented with 10% heat‐inactivated FBS. The human B‐lymphoblastoid cell lines were established by Epstein–Barr virus exogenous transformation of peripheral blood B cells and cultured in RPMI‐1640 supplemented with 10% heat‐inactivated FBS.
Isolation of antigen‐specific CD8+ T‐cell clones
CD8+ T cells were stained by phycoerythrin‐labeled pMHC tetramers (produced in‐house) for 30 min at 4°, followed by allophycocyanin‐labeled anti‐CD8 antibody (BioLegend, San Diego, CA) for 15 min at 4°. The tetramer‐positive CD8+ T cells were sorted by a FACS Aria III sorter (Becton Dickinson, Franklin Lakes, NJ) and cultured in TexMACS GMP medium (Miltenyi Biotec, Bergisch Gladbach, Germany) supplemented with 10% (vol/vol) human serum (Gemini), 100 units/ml penicillin (Gibco, Grand Island, NY), 100 μg/ml streptomycin (Gibco), 10 IU/ml interleukin‐2 (IL‐2), 10 ng/ml IL‐7 and 30 ng/ml IL‐21 (all from PeproTech, Rocky Hill, NJ) along with 5 × 104 irradiated human B‐lymphoblastoid cell lines pulsed with specific peptide in a U‐bottom 96‐well plate (Corning, Corning, NY). The expanded T cells were cloned by limiting dilution in a U‐bottomed 96‐well plate and further analyzed by flow cytometry and ELISpot assays.
Cloning of TCR α‐chain and β‐chain genes
The total RNA of the T‐cell clones was isolated using the Quick‐RNA MicroPrep Kit (Zymo, Irvine, CA) and reverse‐transcribed into cDNA library using the 5′ SMARTer RACE cDNA Amplification Kit (Clontech, Mountain View, CA). The TCR Vα and Vβ genes were amplified from the cDNA library using UPM‐Mix‐Forward primers (supplied by RACE cDNA Amplification Kit) and TCR‐αβ reverse primers (TCR‐α reverse: 5′‐GAGTCTCTCAGCTGGTACACGGCAGGGT‐3′, TCR‐β reverse: 5′‐TTCTGATGGCTCAAACACAGCGACCT‐3′). Both TCR‐α and TCR‐β PCR products were subcloned into the pMD19‐T vector (Takara Bio, Mountain View, CA), and then sequenced.
Preparation of soluble TCR, peptide‐HLA and tetramer
Disulfide bond‐linked soluble TCR was produced as previously described.31 Peptide bound‐HLA molecules and tetramer were prepared as described elsewhere.32
Surface plasmon resonance
The binding of soluble TCR to pMHC was determined using surface plasmon resonance on a Biacore 4000 (GE) as described previously.32
mRNA preparation
TCR‐α and TCR‐β genes were cloned into pGEM vector (Promega, Madison, WI) and linearized by AvrII at 37° for 3 hr, followed by gel extraction using the QIAquick PCR Purification Kit (Qiagen, Hilden, Germany). The extracted linearized products were used as templates for mRNA transcription using the mMESSAGE mMACHINE® T7 Ultra Kit (Life Technologies) according to standard instructions. The transcribed mRNA was polyadenylated by poly(A) polymerase using the RNeasy Mini Kit (Qiagen), and finally purified and stored at −80°.
Electroporation
Before electroporation, the concentration of CD8+ T cells was adjusted to 5 × 105 cells/ml and stimulated by human anti‐CD3/CD28 dynabeads (Gibco) at a 2 : 1 ratio for 48 hr at 37° in complete RPMI‐1640 medium supplemented with 10% FBS, 100 U/ml penicillin, 100 g/ml streptomycin and 30 IU/ml IL‐2. After activation, anti‐CD3/CD28 dynabeads were removed by the magnetic separation rack. Approximately 1 × 106 activated CD8+ T cells were gently mixed with 1·5 μg TCR‐α mRNA and 1·5 μg TCR‐β mRNA using the P3 Primary Cell 4D‐Nucleofector X Kit (Lonza, Basel, Switzerland). The mixture was immediately electroporated using the Amaxa 4D Nucleofector (Lonza) and immediately cultured in a flat‐bottom 96‐well plate (Corning) at 37° for approximately 16 hr. After transduction, the expression level of TCR on T‐cell surface was determined by flow cytometry.
Enzyme‐linked immunospot and cytotoxicity assay
The interferon‐γ (IFN‐γ) release was determined by the enzyme‐linked immunospot (ELISpot) assay. Briefly, a total of 2 × 103 T cells were co‐cultured with 2 × 104 target cells in an ELISpot plate overnight at 37°. For the antibody blocking experiments, the target cells were incubated with anti‐HLA‐A24 mAb (One Lambda) or IgG mAb (Abcam, Cambridge, UK) for 1 hr at room temperature before co‐culturing. The IFN‐γ release was determined using the ELISpot assay kit (BD Biosciences, San Jose, CA) according to the manufacturer's instructions. The spots were counted by the AID iSpot Reader Spectrum (AID GmbH). The data were analyzed using graphpad prism 5 (GraphPad, San Diego, CA). The cytotoxicity activity of T cells was determined by lactate dehydrogenase release assay. Briefly, T cells were co‐cultured with the target cells at the indicated effector : target ratios in a flat‐bottomed 96‐well plate at 37° for 18 hr in phenol red‐free RPMI‐1640 medium supplemented with 5% FBS. After incubation, the release level of lactate dehydrogenase was determined using the CytoTox 96 Non‐Radioactive Cytotoxicity Assay kit (Promega) according to the manufacturer's instructions.
Results
Identification and analysis of AFP2–11 peptide
We employed the MS‐based approach33 to discover AFP peptides for immunotherapy. HepG2 is a well‐studied HLA‐A2+ and HLA‐A24+ HCC cell line highly expressing AFP, so is well‐suited for identifying both HLA‐A2+ and HLA‐A24+ restricted immunogenic peptides. A large amount of HepG2 cells were collected and lyzed, and the surface peptide–HLA molecules were immunoprecipitated by anti‐HLA‐A24 antibodies. The peptides bound to HLA were released, and the peptide sequence was determined and analyzed by MS. An AFP2–11 peptide KWVESIFLIF was identified from the peptidomics data generated from the HepG2 cell lysate (Fig. 1a). To verify the identity of the peptide, we compared the MS spectrum with that of synthesized reference peptide (Fig. 1b), and found that they were coincident with each other. The predicted binding affinity of the peptide to HLA‐A24 was high according to the NetMHC 4.0 prediction.34 Moreover, soluble AFP2–11–HLA‐A24 can be produced with high purity (see Supplementary material, Figure S1), which is in an agreement with the peptide–HLA binding affinity prediction. Therefore, AFP2–11 is a potential HLA‐A24‐restricted peptide epitope.
Figure 1.
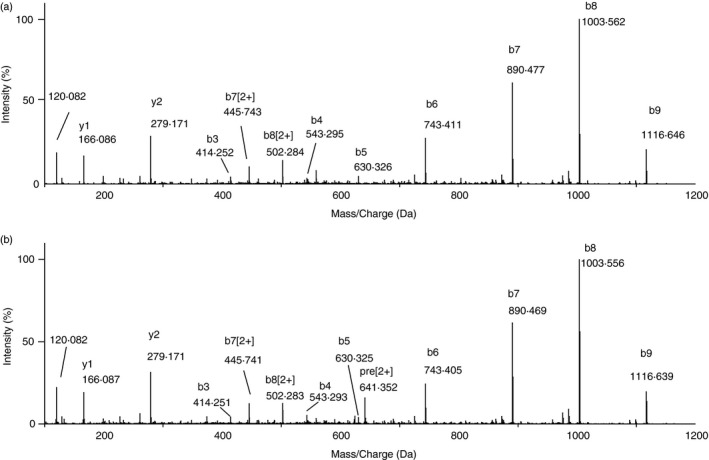
Identification of AFP2–11 peptide (KWVESIFLIF) by mass spectrometry (MS). (a) MS/MS spectrum of the AFP2–11 peptide identified from HepG2 cells. (b) MS/MS spectrum of the synthesized reference peptide
Isolation of AFP2–11–HLA‐24‐specific T‐cell clones
To verify the immunogenicity of the AFP2–11 peptide, AFP2–11–HLA‐A24‐specific CD8+ T cells were isolated from PBMCs of HLA‐A24+ healthy donors and cloned by limiting dilution at a density of c.1–2 cells/well in a 96‐well plate. One high avidity T‐cell clone was identified (Fig. 2). The majority of the cells of the clone can specifically bind to AFP2–11–HLA‐A24 tetramer and a minor tetramer‐negative population (~15%) was also noticeable (Fig. 2a). The tetramer‐negative cells were probably non‐AFP2–11‐specific T cells mixed in the clone during limiting dilution cloning. This minor population did not affect the subsequent experiments, so was not pursued further. The AFP2–11‐specific T‐cell clone could mediate IFN‐γ release when co‐cultured with T2‐A24 cells [transporter of antigen peptide (TAP) ‐deficient T2 cells transduced with HLA‐A24, see Supplementary material, Figure S2] pulsed with the specific peptide AFP2–11, but not with non‐specific control peptide in ELISpot assays (Fig. 2b). To determine whether the peptide can be naturally presented on the cell surface, we tested the activation of the clone by AFP‐expressing tumor cell lines. The clone could mediate IFN‐γ release when co‐cultured with the AFP‐positive cell line HepG2 (HLA‐A24+ AFP+), but not with the AFP‐negative cell line SNU398 (HLA‐A24+ AFP−). However, overexpressing AFP in SNU398 (SNU398‐AFP) enabled the cell line to activate the T‐cell clone (Fig. 2b). Our data suggested that AFP2–11 is a peptide epitope that can be naturally processed and presented by HLA‐A24 on tumor cells.
Figure 2.
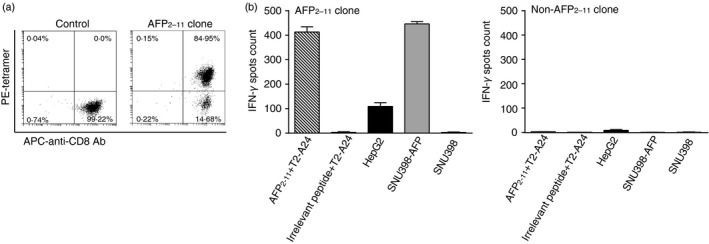
Characterization of the AFP2–11‐specific T‐cell clone. (a) The AFP2–11‐specific clone (right) and a non‐specific control clone (left) were stained with anti‐CD8 antibody and AFP2–11/HLA‐A24 tetramer and analyzed by flow cytometry. (b) Interferon‐γ (IFN‐γ) release mediated by the AFP2–11 clone against T2‐A24 pulsed with AFP2–11 (10−6 m) or an irrelevant peptide (10−6 m) or tumor cell lines: HepG2 (AFP+ HLA‐A24+), SNU398 (AFP− HLA‐A24+) and SNU398‐AFP (AFP overexpressed, HLA‐A24+). The non‐AFP2–11‐specific clone was used as a negative control. IFN‐γ release was determined by enzyme‐linked immune absorbent spot (elispot) assay. Error bars indicate standard deviation of triplicate measurements
Binding affinity of KWV3.1
To further explore the antigen‐specificity of the T‐cell clone, the TCR‐α and TCR‐β chain genes from the T‐cell clone were isolated and sequenced. The TCR‐α and TCR‐β genes (hereinafter referred to as KWV3.1) were found to be TRAV3 and TRBV7‐9, respectively. KWV3.1 was produced by refolding of Escherichia coli expressed inclusion bodies and purified using anion exchange and size exclusion chromatography (see Supplementary material, Figure S3). The binding affinity of soluble KWV3.1 for its cognate pMHC was determined using surface plasmon resonance. The equilibrium binding constant (KD) of KWV3.1 was revealed as 54·8 μm (Fig. 3a,b), coincident with the affinity range of wild‐type TCR to self‐antigens.35, 36 Moreover, no non‐specific binding of KWV3.1 to a panel of irrelevant pMHCs was detected (see Supplementary material, Table S1). These results suggested that KWV3.1 is specific for AFP2–11–HLA‐A24 with a modest affinity.
Figure 3.

Surface plasmon resonance analysis of the binding between soluble KWV3.1 and its cognate ligand. Biotinylated AFP2–11–HLA‐A24 was immobilized on a streptavidin‐coated bicore chip. Soluble KWV3.1 with different concentrations of 9·37, 18·75, 37·50, 75·00 and 150·00 μm were sequentially injected. The equilibrium binding constant (KD) of KWV3.1 was determined to be 54·8 μm. Binding kinetics was analyzed on a Biacore 4000. KD were obtained by Scatchard analysis
Biological functions of KWV3.1‐transfected T cells
To test whether KWV3.1 is capable of mediating cytokine release by T cells, mRNA encoding KWV3.1 containing mouse constant domains (for reducing the ratio of paring with endogenous TCR)37 was electroporated in CD8+ T cells activated by CD3/CD28 beads. High level of KWV3.1 expression could be detected on the cell surface as determined by HLA‐A24+/AFP2–11 tetramer and anti‐murine TCR‐β antibody staining (Fig. 4a). T cells expressing KWV3.1 can mediate IFN‐γ released when co‐cultured with T2‐A24 cells loaded with AFP2–11 peptide in a concentration‐dependent manner (Fig. 4b), but did not response to T2‐A24 cells loaded with irrelevant peptide or no peptide. In addition, T cells expressing KWV3.1 can be activated by HepG2 cells (HLA‐A24+ AFP+), but not by the AFP‐negative cell line SNU398 (HLA‐A24+ AFP−) (Fig. 4c). Overexpression of AFP in SNU398 (SNU398‐AFP) enabled the cells to activate KWV3.1‐transfected T cells (Fig. 4c), suggesting that KWV3.1 is AFP‐specific.
Figure 4.

Functional analysis of KWV3.1‐transfected T cells. (a) Flow cytometric analysis of KWV3.1‐transfected T cells. The mRNA encoding KWV3.1 α‐ and β‐chains or buffer alone (control) were electroporated in T cells activated by CD3/CD28 beads. The electroporated T cells were stained with anti‐mouse T‐cell receptor‐β chain (mTRBC) antibody or AFP2–11/HLA‐A24 tetramer and analyzed on a flow cytometer. (b) Specific activation of KWV3.1‐transfected T cells against T2‐A24 cells pulsed with various concentrations of AFP2–11. Interferon‐γ (IFN‐γ) release was determined by elispot assay. The irrelevant peptide‐pulsed T2‐A24 and the unpulsed T2‐A24 were used as negative controls. (c) Specific activation of KWV3.1‐transfected T cells co‐cultured with tumor cell lines. KWV3.1‐transfected T cells were specifically activated by HepG2 (AFP+ HLA‐A24+) and SNU398‐AFP (AFP overexpressed, HLA‐A24+), but not by SNU398 (AFP− HLA‐A24+). IFN‐γ release was determined by elispot assay. A6 was used as a negative TCR control. (d) Cytotoxicity of KWV3.1‐transfected T cells against T2‐A24 loaded with varied concentrations of AFP2–11 peptide. Lactate dehydrogenase (LDH) assay was implemented at an effector : target ratio of 5 : 1. The A6‐transfected T cells incubated with T2‐A24 loaded with 10−6 m AFP2–11 were used as negative control. (e) The cytotoxic activity of KWV3.1‐transfected T cells against AFP‐expressing tumor cell lines at effector : target ratios of 1 : 1, 3 : 1, 5 : 1 and 10 : 1. A6 was used as a negative TCR control. Error bars indicate standard deviation of triplicate measurements
We further examine whether KWV3.1 could mediate specific cytotoxicity. T cells expressing KWV3.1 were able to lyze T2‐A24 cells pulsed with AFP2–11 in a concentration‐dependent manner (Fig. 4d) and AFP‐positive tumor cell lines (HepG2 and SNU398‐AFP) at various effector : target cell ratios (Fig. 4e), but did not affect T2‐A24 cells loaded with an irrelevant peptide (Fig. 4d) or the AFP‐negative cell line (SNU398, Fig. 4e). The assay with AFP2–11 pulsed T2‐A24 showed that KWV3.1‐expressing T cells were able to specifically kill targets at relatively low antigen densities when the cells were pulsed with 10−10 m to 10−12 m AFP2–11 (Fig. 4d), although the corresponding IFN‐γ released was merely detectable (Fig. 4b) within that density range. The reason behind these results is outside the scope of this investigation, but will be studied in the future. Taken together, these results suggest that KWV3.1 has the therapeutic potential for developing a TCR‐T therapy for patients with HLA‐A24+ HCC in future studies.
HLA‐A24 restriction of KWV3.1
To further explore the antigen specificity of KWV3.1, we conducted the antibody blocking assay. The level of IFN‐γ released by T cells expressing KWV3.1 co‐cultured with SNU398‐AFP was significantly inhibited by anti‐HLA‐A24 antibody blocking, but was not affected by the control IgG antibody (Fig. 5), suggesting that the activation of T cells was mediated by the interaction between KWV3.1 and HLA‐A24+/AFP2–11.
Figure 5.
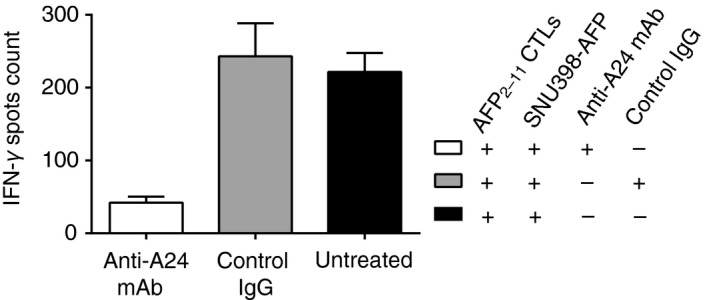
Inhibition of specific recognition of KWV3.1‐transfected T cells by anti‐HLA‐A24 antibody. Before incubation with KWV3.1‐transduced T cells, SNU398‐AFP cells were pre‐incubated with anti‐HLA‐A24 monoclonal antibody (mAb) or IgG control for 1 hr at room temperature. The production of interferon‐γ (IFN‐γ) by KWV3.1‐transfected T cells when incubated with SNU398‐AFP cells was significantly inhibited by anti‐HLA‐A24 mAb in comparison with the untreated SNU398‐AFP cells, but not influenced by the control IgG. IFN‐γ release was determined by elispot assay at an effector : target ratio of 1 : 10. Error bars indicate standard deviation of triplicate measurements
Discussion
The discovery of tumor targets, such as programmed cell death protein 1 (PD‐1) for checkpoint inhibitors and CD19 for chimeric antigen receptor–T‐cell therapies, plays a critical role in recent immunotherapy breakthroughs. The targets for conventional antibody‐based therapies are usually limited to membrane proteins, whereas secreted or intracellular proteins, which are common sources of tumor‐specific antigens, are considered not targetable. TCR‐based therapies, on the other hand, do not have such limitation, and so provide broader opportunities. However, TCR‐T therapies are only applicable to patients carrying certain HLA alleles because of the HLA restriction of the antigens. HLA‐A2‐restricted epitopes are the focus of the majority of current researches. Therefore, to make TCR‐based therapies amenable for a broader group of patients, identification and verification of peptide epitopes restricted by other HLAs is needed.
The discovery of tumor antigen‐derived peptides is non‐trivial given the complexity of antigen presentation.38 In silico prediction and MS are both effective methods.10, 33 As only a small portion of peptides are actually presented on the cell surface,38 the majority of predicted epitopes may not be valid targets for clinical applications. Mass spectrometry has been considered to be an unbiased methodology to comprehensively interrogate the naturally presented repertoire of HLA‐binding peptides. Several HLA‐A24‐restricted AFP peptides, but not including AFP2–11, have been reported based on prediction algorithms29 and some of them have been shown to induce AFP‐specific cytotoxic T lymphocytes.29, 30 However, previous studies39 showed that the human telomerase catalytic subunit‐derived peptide 540–548 predicted by algorithms was not effectively processed and presented on the cell surface, although it could induce human telomerase catalytic subunit‐specific cytotoxic T lymphocytes.40 In this regard, those predicted epitopes may need to be further characterized. On the other hand, we not only applied the MS‐based approach to identify AFP2–11 that has not been reported by the prediction based method, but also demonstrated that AFP2–11 could be a target for TCR‐T immunotherapy. Our result suggests that the two discovery approaches are complementary, and that peptides detected by MS can prove to be straightforward as naturally processed and presented targets for immunotherapy.
It is interesting to note that AFP2–11 is located at the signal peptide (SP) domain of AFP and this location may have important implications. First, it has been shown that SP‐derived epitopes can be processed and presented through TAP‐independent pathways.41, 42 If this is the case with AFP2–11, which is yet to be experimentally confirmed, TCR‐T cells targeting this peptide may overcome tumor escape as a result of impaired TAP function.43 Second, the SP domains have been proposed as better vaccine candidates,44 because of the efficient stimulation for T‐cell immune responses in both mouse and human studies. Our findings support the use of AFP SP as a vaccine for HLA‐A24 patients because of an epitope within the sequence.
In general, TCRs obtained from the peripheral T‐cell repertoire exhibit low affinity for tumor‐associated antigen‐derived peptides because of negative selection in the thymus. The functional avidity of a TCR, within a certain range, is directly correlated with its ligand binding affinity17, 45, 46 and therefore T cells expressing low‐affinity TCRs have limited clinical efficacy. Optimizing TCR affinity, while avoiding non‐specificity, becomes essential for the development of TCR‐T therapy.17 KWV3.1 reported here has a relatively low affinity of ~54·8 μm. Although T cells expressing KWV3.1 can mediate IFN‐γ release and specific killing of target cell lines, the functional avidity may need to be improved for better efficacy at tumor microenvironment and hence affinity enhancement of KWV3.1 is required to achieve optimal clinical efficacy in TCR‐T therapy. We are in the process of optimizing KWV3.1 affinity by phage display technology, which has been demonstrated to be highly effective in generating high‐affinity TCRs,47 and have observed significantly enhanced functional avidity for some KWV3.1 mutants (unpublished data).
Although adoptive transfer of TCR‐T cells has shown significant clinical response in solid tumors, such as melanoma and synovial cell sarcoma,48, 49 the success of TCR‐T therapy for HCC is still challenging. The strong immune suppressive microenvironment of HCC, characterized by the presence of immunosuppressive cells and elevated expression of immune checkpoint molecules,50, 51 poses a major barrier to clinical efficacy in immunotherapy. Therefore, TCR‐T therapy in combination with methods for modulating the tumor microenvironment, such as checkpoint blockage,9 may represent a favorable strategy for HCC immunotherapy. Interestingly, unlike most tumor cells that evade immune surveillance by down‐regulation of HLA molecules. HCC cells are frequently associated with HLA up‐regulation,52 which can be beneficial to TCR‐based therapy. Another concern is toxicity associated with TCR‐T therapy. AFP is not strictly restricted in tumor cells: a low level of AFP expression can be detected in non‐malignant liver cells and other normal tissues.17 Careful adjustment of the antigen sensitivity of AFP‐specific TCRs might be required to avoid autoreactivity. Moreover, off‐target toxicity arises when the TCR cross‐reacts with self‐antigen‐derived peptides.53 Strategic screening techniques54 can be applied to minimize this type of risk.
In this study, we identified and characterized a novel HLA‐A24‐restricted AFP‐derived peptide AFP2–11, and demonstrated the potential application of the AFP2–11‐specific TCR KWV3.1 in adoptive T‐cell immunotherapy. Further studies revolving around affinity enhancement, cross‐reactivity screening and HCC immunosuppression may transform KWV3.1 into an effective TCR‐T therapy for HCC.
Disclosures
All authors are employees of Guangdong Xiangxue Life Sciences, Ltd.
Supporting information
Figure S1. SDS‐PAGE analysis of biotinylated soluble AFP2‐11–HLA‐A24.
Figure S2. (Wanli Wu et al unpublished). Flow cytometric analysis of HLA‐A24 transfected T2 cells.
Figure S3. SDS‐PAGE analysis of soluble KWV3.1.
Table S1. Non‐specific binding of KWV3.1 to irrelevant ligands.
Acknowledgements
YL, SZ and ZJL planned the project, designed the experiments, and analyzed and interpreted the data. ZJL, QPL, JTC, YYM and YLC performed the experiments and collected data. HPG established the platform for mass spectrometry‐based epitope identification. HPG, XHY, HJZ and WLW contributed expertise. ZJL, SZ and YL wrote and reviewed the paper. YL conceived the concept and approved the final version submitted for publication. This work was supported by the National key R&D Program (grant number 2016YFC1303404); the Science and Technology Program of Guangzhou (grant number 201704020220); the Frontier Program of Guangzhou Regenerative Medicine and Health Guangdong Laboratory (grant number 2018GZR110105010); and the State Key Laboratory of Respiratory Disease (grant numbers SKLRD‐QN‐201915; SKLRD‐MS‐201902).
Zhenjuan Li and Haiping Gong contributed equally to this study.
Contributor Information
Shi Zhong, Email: zhongsh@xphcn.com.
Yi Li, Email: li_yi@gibh.ac.cn.
References
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018; 68:394–424. [DOI] [PubMed] [Google Scholar]
- 2. Akinyemiju T, Abera S, Ahmed M, Alam N, Alemayohu MA, Allen C et al The burden of primary liver cancer and underlying etiologies from 1990 to 2015 at the global, regional, and national level: results from the global burden of disease study 2015. JAMA Oncol 2017; 3:1683–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Llovet JM, Montal R, Villanueva A. Randomized trials and endpoints in advanced HCC: role of PFS as a surrogate of survival. J Hepatol 2019; 70:1262–77. [DOI] [PubMed] [Google Scholar]
- 4. Llovet JM, Villanueva A. Liver cancer: effect of HCV clearance with direct‐acting antiviral agents on HCC. Nat Rev Gastroenterol Hepatol 2016; 13:561–2. [DOI] [PubMed] [Google Scholar]
- 5. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019; 69:7–34. [DOI] [PubMed] [Google Scholar]
- 6. Zhang S, Yue M, Shu R, Cheng H, Hu P. Recent advances in the management of hepatocellular carcinoma. J BUON 2016; 21:307–11. [PubMed] [Google Scholar]
- 7. El‐Khoueiry A. The promise of immunotherapy in the treatment of hepatocellular carcinoma. Am Soc Clin Oncol Educ Book 2017; 37:311–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Buonaguro L, Mauriello A, Cavalluzzo B, Petrizzo A, Tagliamonte M. Immunotherapy in hepatocellular carcinoma. Ann Hepatol 2019; 18:291–7. [DOI] [PubMed] [Google Scholar]
- 9. El‐Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C et al Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open‐label, non‐comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017; 389:2492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Desrichard A, Snyder A, Chan TA. Cancer neoantigens and applications for immunotherapy. Clin Cancer Res 2016; 22:807–12. [DOI] [PubMed] [Google Scholar]
- 11. Ikeda H. T‐cell adoptive immunotherapy using tumor‐infiltrating T cells and genetically engineered TCR‐T cells. Int Immunol 2016; 28:349–53. [DOI] [PubMed] [Google Scholar]
- 12. Ikeda H. Cancer immunotherapy utilizing T cell receptor gene engineering. Gan To Kagaku Ryoho 2017; 44:273–7. [PubMed] [Google Scholar]
- 13. Abelev GI, Eraiser TL. Cellular aspects of α‐fetoprotein reexpression in tumors. Semin Cancer Biol 1999; 9:95–107. [DOI] [PubMed] [Google Scholar]
- 14. Abelev GI. α‐Fetoprotein in ontogenesis and its association with malignant tumors. Adv Cancer Res 1971; 14:295–358. [DOI] [PubMed] [Google Scholar]
- 15. Butterfield LH, Meng WS, Koh A, Vollmer CM, Ribas A, Dissette VB et al T cell responses to HLA‐A*0201‐restricted peptides derived from human α fetoprotein. J Immunol 2001; 166:5300–308. [DOI] [PubMed] [Google Scholar]
- 16. Mizejewski GJ. Biological role of α‐fetoprotein in cancer: prospects for anticancer therapy. Expert Rev Anticancer Ther 2002; 2:709–35. [DOI] [PubMed] [Google Scholar]
- 17. Docta RY, Ferronha T, Sanderson JP, Weissensteiner T, Pope GR, Bennett AD et al Tuning T‐cell receptor affinity to optimize clinical risk‐benefit when targeting α‐fetoprotein‐positive liver cancer. Hepatology 2019; 69:2061–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kubota M, Yagi M, Kanada S, Yamazaki S, Tanaka S, Asami K et al Effect of postoperative chemotherapy on the serum α‐fetoprotein level in hepatoblastoma. J Pediatr Surg 2004; 39:1775–8. [DOI] [PubMed] [Google Scholar]
- 19. Nakagawara A, Ikeda K, Tsuneyoshi M, Daimaru Y, Enjoji M, Watanabe I et al Hepatoblastoma producing both α‐fetoprotein and human chorionic gonadotropin. Clinicopathologic analysis of four cases and a review of the literature. Cancer 1985; 56:1636–42. [DOI] [PubMed] [Google Scholar]
- 20. Johnson PJ. Role of α‐fetoprotein in the diagnosis and management of hepatocellular carcinoma. J Gastroenterol Hepatol 1999; 14(Suppl):S32–36. [DOI] [PubMed] [Google Scholar]
- 21. Butterfield LH, Ribas A, Meng WS, Dissette VB, Amarnani S, Vu HT et al T‐cell responses to HLA‐A*0201 immunodominant peptides derived from α‐fetoprotein in patients with hepatocellular cancer. Clin Cancer Res 2003; 9(16 Pt 1):5902–8. [PubMed] [Google Scholar]
- 22. Liu Y, Daley S, Evdokimova VN, Zdobinski DD, Potter DM, Butterfield LH. Hierarchy of α fetoprotein (AFP)‐specific T cell responses in subjects with AFP‐positive hepatocellular cancer. J Immunol 2006; 177:712–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhu W, Peng Y, Wang L, Hong Y, Jiang X, Li Q et al Identification of α‐fetoprotein‐specific T‐cell receptors for hepatocellular carcinoma immunotherapy. Hepatology 2018; 68:574–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu H, Xu Y, Xiang J, Long L, Green S, Yang Z et al Targeting α‐fetoprotein (AFP)‐MHC complex with CAR T‐cell therapy for liver cancer. Clin Cancer Res 2017; 23:478–88. [DOI] [PubMed] [Google Scholar]
- 25. Goyal L, Meyer T, Frigault M, Feun LG, Bruix J, El‐Khoueiry A et al Abstract 3183: Initial safety of AFP SPEAR T‐cells in patients with advanced hepatocellular carcinoma. Cancer Res 2019; 79:3183. [Google Scholar]
- 26. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F et al Cancer statistics in China, 2015. CA Cancer J Clin 2016; 66:115–32. [DOI] [PubMed] [Google Scholar]
- 27. Teo EK, Fock KM. Hepatocellular carcinoma: an Asian perspective. Dig Dis 2001; 19:263–8. [DOI] [PubMed] [Google Scholar]
- 28. Nakaoka H, Inoue I. Distribution of HLA haplotypes across Japanese Archipelago: similarity, difference and admixture. J Hum Genet 2015; 60:683–90. [DOI] [PubMed] [Google Scholar]
- 29. Mizukoshi E, Nakamoto Y, Tsuji H, Yamashita T, Kaneko S. Identification of α‐fetoprotein‐derived peptides recognized by cytotoxic T lymphocytes in HLA‐A24+ patients with hepatocellular carcinoma. Int J Cancer 2006; 118:1194–1204. [DOI] [PubMed] [Google Scholar]
- 30. Nakagawa H, Mizukoshi E, Kobayashi E, Tamai T, Hamana H, Ozawa T et al Association between high‐avidity T‐cell receptors, induced by α‐fetoprotein‐derived peptides, and anti‐tumor effects in patients with hepatocellular carcinoma. Gastroenterology 2017; 152:1395–1406.e1310. [DOI] [PubMed] [Google Scholar]
- 31. Boulter JM, Glick M, Todorov PT, Baston E, Sami M, Rizkallah P et al Stable, soluble T‐cell receptor molecules for crystallization and therapeutics. Protein Eng 2003; 16:707–11. [DOI] [PubMed] [Google Scholar]
- 32. Zhang H, Zhang J, Chen L, Weng Z, Tian Y, Zhao H et al Targeting naturally occurring epitope variants of hepatitis C virus with high‐affinity T‐cell receptors. J Gen Virol 2017; 98:374–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bassani‐Sternberg M, Coukos G. Mass spectrometry‐based antigen discovery for cancer immunotherapy. Curr Opin Immunol 2016; 41:9–17. [DOI] [PubMed] [Google Scholar]
- 34. Nielsen M, Andreatta M. NetMHCpan‐3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med 2016; 8:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Aleksic M, Liddy N, Molloy PE, Pumphrey N, Vuidepot A, Chang KM et al Different affinity windows for virus and cancer‐specific T‐cell receptors: implications for therapeutic strategies. Eur J Immunol 2012; 42:3174–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stone JD, Harris DT, Kranz DM. TCR affinity for p/MHC formed by tumor antigens that are self‐proteins: impact on efficacy and toxicity. Curr Opin Immunol 2015; 33:16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, Morgan RA. Enhanced antitumor activity of murine‐human hybrid T‐cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res 2006; 66:8878–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Neefjes J, Jongsma ML, Paul P, Bakke O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol 2011; 11:823–36. [DOI] [PubMed] [Google Scholar]
- 39. Ayyoub M, Migliaccio M, Guillaume P, Liénard D, Cerottini J‐C, Romero P et al Lack of tumor recognition by hTERT peptide 540–548‐specific CD8+ T cells from melanoma patients reveals inefficient antigen processing. Eur J Immunol 2001; 31:2642–51. [DOI] [PubMed] [Google Scholar]
- 40. Purbhoo MA, Li Y, Sutton DH, Brewer JE, Gostick E, Bossi G et al The HLA A*0201‐restricted hTERT(540–548) peptide is not detected on tumor cells by a CTL clone or a high‐affinity T‐cell receptor. Mol Cancer Ther 2007; 6:2081–91. [DOI] [PubMed] [Google Scholar]
- 41. Wolfel C, Drexler I, Van Pel A, Thres T, Leister N, Herr W et al Transporter (TAP)‐ and proteasome‐independent presentation of a melanoma‐associated tyrosinase epitope. Int J Cancer 2000; 88:432–8. [PubMed] [Google Scholar]
- 42. Oliveira CC, van Hall T. Alternative antigen processing for MHC class I: multiple roads lead to Rome. Front Immunol 2015; 6:298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. van Hall T, Wolpert EZ, van Veelen P, Laban S, van der Veer M, Roseboom M et al Selective cytotoxic T‐lymphocyte targeting of tumor immune escape variants. Nat Med 2006; 12:417–24. [DOI] [PubMed] [Google Scholar]
- 44. Kovjazin R, Carmon L. The use of signal peptide domains as vaccine candidates. Hum Vaccin Immunother 2014; 10:2733–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hebeisen M, Allard M, Gannon PO, Schmidt J, Speiser DE, Rufer N. Identifying individual T cell receptors of optimal avidity for tumor antigens. Front Immunol 2015; 6:582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhong S, Malecek K, Johnson LA, Yu Z, Vega‐Saenz de Miera E, Darvishian F et al T‐cell receptor affinity and avidity defines antitumor response and autoimmunity in T‐cell immunotherapy. Proc Natl Acad Sci USA 2013; 110:6973–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li Y, Moysey R, Molloy PE, Vuidepot AL, Mahon T, Baston E et al Directed evolution of human T‐cell receptors with picomolar affinities by phage display. Nat Biotechnol 2005; 23:349–54. [DOI] [PubMed] [Google Scholar]
- 48. Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME et al Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY‐ESO‐1. J Clin Oncol 2011; 29:917–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS et al Gene therapy with human and mouse T‐cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 2009; 114:535–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lu C, Rong D, Zhang B, Zheng W, Wang X, Chen Z et al Current perspectives on the immunosuppressive tumor microenvironment in hepatocellular carcinoma: challenges and opportunities. Mol Cancer 2019; 18:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mizukoshi E, Kaneko S. Immune cell therapy for hepatocellular carcinoma. J Hematol Oncol 2019; 12:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shen Y, Xia M, Zhang J, Xu L, Yang J, Chen A et al IRF‐1 and p65 mediate upregulation of constitutive HLA‐A antigen expression by hepatocellular carcinoma cells. Mol Immunol 2009; 46:2045–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC et al Identification of a Titin‐derived HLA‐A1‐presented peptide as a cross‐reactive target for engineered MAGE A3‐directed T cells. Sci Transl Med 2013; 5:197ra103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bijen HM, van der Steen DM, Hagedoorn RS, Wouters AK, Wooldridge L, Falkenburg JHF et al Preclinical strategies to identify off‐target toxicity of high‐affinity TCRs. Mol Ther 2018; 26:1206–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. SDS‐PAGE analysis of biotinylated soluble AFP2‐11–HLA‐A24.
Figure S2. (Wanli Wu et al unpublished). Flow cytometric analysis of HLA‐A24 transfected T2 cells.
Figure S3. SDS‐PAGE analysis of soluble KWV3.1.
Table S1. Non‐specific binding of KWV3.1 to irrelevant ligands.