

Abstract
Protein S-glutathionylation is one of the important cysteine oxidations that regulate various redox-mediated biological processes. Despite several existing methods, there are few proteomic approaches to identify and quantify specific cysteine residues susceptible to S-glutathionylation. Previously, we have developed a clickable glutathione approach that labels intracellular glutathione with azido-Ala by using a mutant of glutathione synthetase. In this report, we developed a quantification strategy with clickable glutathione by using isotopically-labelled heavy and light derivatives of azido-Ala, which provides the relative quantification of glutathionylated peptides in the mass spectrometry-based proteomic analysis. We have applied isotopically-labelled clickable glutathione to HL-1 cardiomyocytes, quantifying relative levels of 1,398 glutathionylated peptides upon addition of hydrogen peroxide. Importantly, we highlight elevated levels of glutathionylation on sarcomere-associated muscle proteins while validating glutathionylation of two structural proteins, α-actinin and desmin. Our report provides a chemical proteomic strategy to quantify specific glutathionylated cysteines.
Keywords: Proteomics, Protein modification, S-glutathionylation, clickable glutathione, quantification, hydrogen peroxide
Graphical Abstract
Identification of protein S-glutathionylation is important to understand the redox-mediated biological processes regulated by the reactive oxygen species. Isotopically-labelled clickable glutathione was developed and applied for relative quantification of glutathionylated peptides upon addition of hydrogen peroxide to a cardiomyocyte cell line
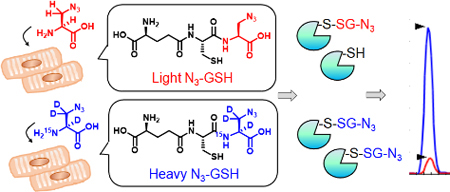
Introduction
Hydrogen peroxide (H2O2) is an important signaling molecule in redox signaling while its overproduction is associated with oxidative stress in various diseases, including cardiovascular diseases.[1–2] The importance of H2O2 as a regulatory molecule or pathological contributor to the cardiovascular diseases continues to grow.[3–4] Notably, cysteine is one of the unique amino acids that has high susceptibility to oxidation by H2O2, and cysteine oxidation is emerging as an important molecular event in redox signaling or oxidative stress that contributes to biological phenotype of H2O2.[5–6] Cysteine can be oxidized to various oxoforms by H2O2, including S-sulfenylation, S-glutathionylation, and disulfide formation, many of which form via distinct chemical mechanisms and are likely to confer different functional alterations on proteins due to varying sizes and electronic properties of individual modifications.[7] Among various oxoforms, S-glutathionylation is one of the major oxoforms that involves addition of bulky glutathione to protein cysteine residues via disulfide formation.[8] The biological or functional significance of S-glutathionylation, especially in the cardiovascular system, has been demonstrated with many examples of redox-active proteins, including titin, SERCA, and eNOS.[9–13]
With growing biological significance of protein S-glutathionylation, many biochemical approaches have been developed to investigate glutathionylated proteins,[14–18] especially with proteomic identification of glutathionylated peptides to find specific cysteines susceptible to glutathionylation.[18–20] One common proteomic approach includes a biotin-switch method or its derivatives in which glutathionylated cysteines are reduced by glutaredoxin and re-alkylated by biotin or enriched by resins.[15–16, 21] The biotin-switch method has been also coupled with isobaric tag to quantify the level of glutathionylation.[16, 22–23] Alternatively, we have previously developed a chemoselective method, namely clickable glutathione, to directly detect and identify glutathionylated proteins.[17, 24–25] In this approach, we used a mutant of glutathione synthetase (GS M4) that utilizes azido-Ala as a substrate in place of Gly during synthesis of glutathione (γGlu-Cys-Gly), which results in biosynthesis of clickable glutathione (γGlu-Cys-azido-Ala, N3-GSH, azido-glutathione, Figure 1) in situ in cells.[17] After glutathionylation by clickable glutathione, the subsequent click reaction enables to identify and characterize glutathionylated proteins.[26] We recently demonstrated the mass spectrometry-based identification of specific glutathionylated cysteines, especially by using biotin-alkyne with an acid-labile DADPS-cleavable linker, finding over 1,700 specific cysteine sites for glutathionylation in cardiomyocytes.[20]
Figure 1. Isotopic-labeling strategy to identify and quantify glutathionylated cysteines.

(A) A scheme for quantification of glutathionylated peptides. A mutant of glutathione synthetase (GS M4) in HL-1 cells uses light or heavy azido-Ala to synthesize isotopically-labelled clickable glutathione. Upon addition of stimulus, two cohorts of lysates containing glutathionylated proteins with a light or heavy label were combined and subjected to click reaction with biotin-DADPS-alkyne, pull-down by streptavidin-beads, and tryptic digestion. Glutathionylated peptides were eluted by acidic cleavage of a DADPS linker and analyzed by LC-MS/MS, which provides a MS1-peak area ratio (RH/L) of heavy- to light-labelled peptides. (B) The structure of biotin-DADPS-alkyne with its cleavage site (dot line).
In this report, we have developed a quantification strategy with clickable glutathione, which measures the relative levels of glutathionylated peptides in mass spectrometric analysis by using isotopically-labelled clickable glutathione. We have designed and synthesized isotopically-labelled heavy and light derivatives of azido-Ala, demonstrated their identical labeling of clickable glutathione, and identified and quantified glutathionylated cysteines with isotopic mass difference upon addition of H2O2. Specifically, we have applied an isotopic clickable glutathione approach to HL-1 cardiomyocytes and identified 1,398 glutathionylated cysteines, whose relative levels of glutathionylation were quantified after addition of H2O2. From proteomic analysis, we highlight a list of muscle-relevant or sarcomeric proteins with their elevated levels of glutathionylation in response to H2O2. In addition, we validated glutathionylation of specific cysteines in two important structural proteins in cardiomyocytes, such as α-actinin and desmin.
Result and Discussion
Design and synthesis of isotopically-labelled azido-Ala
In our approach, clickable glutathione is synthesized by GS M4 that catalyzes coupling of intracellular γGlu-Cys with L-azido-Ala exogenously added to cells. We envisioned that addition of isotopic light and heavy derivatives of azido-Ala will produce the corresponding clickable glutathione with isotopic mass difference (Figure 1A), which can be combined and analyzed for quantification in the subsequent mass spectrometric analysis after click reaction, enrichment, and elution of glutathionylated peptides, as outlined in Figure 1A and Figure S1 in the Supporting Information. Therefore, we have designed a heavy derivative of L-azido-Ala that contains three deuterium (D) atoms and one nitrogen-15 (15N) atom, which has +4 Da mass higher than light azido-Ala (Figure 1). Heavy L-azido-Ala was synthesized starting from 2,3,3-D3,15N-L-serine by simple 6-step reactions (Scheme S1), which resulted in overall 15% yield.
Examination of isotopically-labelled clickable glutathione to detect S-glutathionylation
First, light and heavy azido-Ala derivatives were compared for their efficiency to synthesize clickable glutathione by GS M4. In-vitro kinetic analysis showed that both light and heavy azido-Ala derivatives are used as substrates of GS M4 with the same enzymatic rates (Figure 2A). Subsequently, LC-MS analysis of in-vitro enzymatic products showed that the corresponding light and heavy derivatives of azido-glutathione were synthesized in equal amounts (Figure S2). Similarly, when light or heavy azido-Ala was incubated to HEK293 cells stably expressing GS M4 (HEK293/GS M4)[26], the same amounts of respective azido-glutathione derivatives were detected in lysates by LC-MS analysis (Figure 2B), confirming that light and heavy azido-Ala derivatives are used identical by GS M4 to synthesize the corresponding clickable glutathione.
Figure 2. Evaluation of light and heavy derivatives of azido-Ala to identify glutathionylated peptides with isotopic mass difference.

(A) Light and heavy azido-Ala derivatives are used equally by GS M4 in vitro. GS M4 enzyme activity was measured with γ-Glu-Cys and ATP in the presence of two different concentrations (0.2 and 1 mM) of light azido-Ala (red) or heavy azido-Ala (blue), showing identical enzymatic rates. (B) Light and heavy azido-Ala derivatives produce the equal amount of respective azido-glutathione in cells. HEK293 expressing GS M4 (HEK293/GS M4) were incubated with light or heavy azido-Ala. Lysates were analyzed by LC-MS to detect the mass of light or heavy-labelled azido-glutathione. (C) Light and heavy azido-Ala derivatives detect the identical pattern of glutathionylation. HEK293/GS M4 cells incubated with light or heavy azido-Ala were treated with H2O2 (1 mM, 15 min). Lysates were then subjected to click reaction with Cy5-alkyne for fluorescence detection or Coomassie stains. (D) Light and heavy azido-Ala derivatives identify glutathionylated peptides with isotopic mass difference. Purified GSTO1 was glutathionylated by addition of diamide (0.2 mM, 30 min) in the presence of equal amount of light- and heavy-labelled azido-glutathione derivatives (both 1 mM). Glutathionylated GSTO1 was processed for click reaction with biotin-DDE-alkyne, tryptic digestion, pull-down, elution, and MALDI-MS analysis.[12, 20] Data are representative of 3 independent experiments.
Next, we began to compare detection of glutathionylation by light and heavy clickable glutathione derivatives. After incubation of light or heavy azido-Ala to HEK293/GS M4 cells, glutathionylation was induced by addition of H2O2 (1 mM, 15 min). Subsequently, lysates were subjected to click reaction with Cy5-alkyne. The following in-gel fluorescence analysis showed identical patterns and intensities of global glutathionylation from both cells treated with light or heavy azido-Ala (lane 2 vs. 4, Figure 2C), demonstrating identical detection of glutathionylated proteins by isotopically-labelled clickable glutathione. In addition, isotopic mass difference of a glutathionylated peptide was examined with purified GSTO1 in vitro (Figure 2D).[12, 20] Purified GSTO1 was subjected to glutathionylation in the presence of equal amounts of light and heavy azido-glutathione derivatives in vitro. After click reaction with biotin-alkyne, GSTO1 was digested by trypsin. Biotinylated peptides were enriched by streptavidin-beads. MALDI analysis of eluted samples found the similar ion intensity of two peaks (m/z 1284.68 and 1288.71) with 4 Da mass difference that correspond to the molecular weights of glutathionylated peptides (FC32*PFAER) labelled by light or heavy azido-glutathione (the median intensity ratio of heavy- to light-labelled peptide = 0.88 ± 0.09, n = 3) (Figure 2D). The identified Cys32 is a redox-sensitive active-site cysteine of GSTO1 involved in glutathionylation.[27] These biochemical data demonstrate that light and heavy azido-Ala derivatives enable to detect glutathionylation in the same manner while providing the mass separation of identical glutathionylated peptides.
Proteomic analysis of S-glutathionylation by isotopically-labelled clickable glutathione
To evaluate light and heavy azido-Ala derivatives for proteomic identification of glutathionylated cysteines, HL-1 cardiomyocyte cell line was selected as a model system because of the importance of redox signaling and oxidative stress in cardiomyocytes.[1] Also, we have recently used a clickable glutathione approach with HL-1 cells, confirming its feasibility to identify glutathionylated proteins in this cell line.[20] First, HL-1 cells expressing a GS M4 mutant were either incubated with heavy or light azido-Ala (Figure 1A). Subsequently, two cohorts of cells with heavy or light azido-Ala were incubated with equal amounts of H2O2 (both 1 mM) (E1, Figure 3A). Lysates treated with heavy or light azido-Ala were then combined and processed for mass identification of glutathionylated peptides: glutathionylated proteins were conjugated with biotin-DADPS-alkyne by click reaction and enriched by streptavidin-beads. After on-bead trypsin digestion, glutathionylated peptides were eluted by acidic cleavage of the DADPS linker (Figure 1 and Figure S1).
Figure 3. Identification and quantification of glutathionylated peptides by isotopically-labelled clickable glutathione.

(A) Experimental conditions to identify glutathionylated peptides by isotopic azido-Ala, and the number of quantified glutathionylated peptides under indicated condition by LC-MS. (B) The RH/L values and the coefficient of variation (CV) of glutathionylated peptides. The box plots of RH/L values (left) and the CV percentage (right) with the median value (line), box (25–75%), and whiskers (10–90%). (C) Distribution of RH/L values of individual glutathionylated peptides. The RH/L value of 4 is indicated by a dotted line. Examples of sarcomeric proteins with cysteine sites are indicated by arrow and colors (yellow in E1 and cyan in E2). (D) Graphical display of MS1-peaks that shows relative quantification of heavy (blue)- to light (red)-labelled peptides. Selected sarcomeric or muscle-relevant proteins are shown. Retention times are indicated with MS1-peaks. (E-F) Validation of glutathionylated cysteines by Western blots. HL-1 cells expressing GS M4 were used without transfection of α-actinin or desmin. HEK293/GS M4 cells were transfected with WT or Cys mutants of α-actinin or desmin. After addition of H2O2 (15 min), glutathionylated proteins were subjected to click reaction with biotin-alkyne and pull-down with streptavidin-beads, and detected by Western blotting with individual antibodies, including α-actinin, desmin, and FLAG. Data are representative of 2 independent experiments.
After LC-MS/MS analysis (triplicate, E1), individual pairs of glutathionylated peptides with isotopic +4 Da mass difference were identified (at least 2 out of 3 replicates) and quantified to give a ratio (RH/L) by Skyline software,[28] which provides a MS1 peak integration ratio of heavy- to light-labeled peptides. In the E1 experiment (1 / 1 mM H2O2 for light / heavy), 1,398 glutathionylated peptides with both light and heavy labels were identified and assigned with RH/L values (Figure 3A and Table S1). Importantly, the median RH/L value in E1 is 0.94 (Figure 3B, left), which is close to 1 (92.6% of RH/L in a range of 0.6–1.4, Figure 3C and Figure S3), supporting relatively identical quantification of glutathionylated peptides when equal amounts of H2O2 are added to cells. The median value of the coefficient of variation (CV) in E1 (13.5%) indicates the relatively consistent quantification among triplicate experiments (Figure 3B, right). Previously, we have identified glutathionylated cysteines from sarcomere-associated proteins or the mitochondrial electron transport chains by proteomic analysis,[20] many of which were also consistently found in E1 (Figure 3C and Table S2). The MS1 peaks for individual identified peptides confirmed that their RH/L values are close to 1, as seen with ACTN4 C352, ACTN1 C480, and DES C332 (1.07, 0.84, and 0.88, respectively) and others (Figure 3D and Figure S4). These experiments demonstrate identification and quantification of glutathionylated peptides by isotopically-labelled clickable glutathione.
Quantitative analysis of glutathionylated peptides upon addition of H2O2
Next, we began to quantify different levels of glutathionylated peptides upon addition of H2O2. To maximize the number of quantifiable cysteines, we have compared levels of glutathionylation without and with a relatively high amount (1 mM) of H2O2: two cohorts of cells incubated with light or heavy azido-Ala were treated without and with H2O2 (0 / 1 mM H2O2 for light / heavy, E2, Figure 3A). After the same processes shown in Figure 1A, mass spectrometric quantification analysis identified 797 pairs of glutathionylated peptides with light and heavy labels (at least 2 out of 3 replicates) (Figure 3A). The RH/L values of 797 cysteines are widely distributed with the median value of 16.3 (3.7 < RH/L < 70.2, 80%, 637 out of 797 cysteines) (Figure 3B, left). The median CV value (46.7%) for E2 is relatively high, as opposed to CV (14.6%) in E1 (Figure 3B, right), which likely results from difficulty of quantifying low levels of light-labeled peptides in E2. Cysteines with high RH/L values in E2 indicate that their levels of glutathionylation increase upon addition of H2O2: 89% of identified glutathionylated peptides (707 out of 797) have their RH/L values higher than 4 (Figure 3C), reflecting significant increases of global glutathionylation upon addition of H2O2 (Figure 2C). To emphasize potential implication of glutathionylation in cardiac function, we searched and summarized sarcomeric proteins (identified from NCBI GO annotation[29]) or muscle-relevant proteins with their RH/L values (Figure 3C–D and Table S2). These examples include myofilamental proteins [TTN C19461 (RH/L 32.1), MYL7 C43 (RH/L 19.8), MYBPC3 C619 (RH/L 82.0)], structural proteins [ACTN1 C480 (RH/L 24.0), ACTN4 C500 (RH/L 18.2), DES C332 (RH/L 30.5), FLNC C1067 (RH/L 108.7)], adaptor or regulatory proteins [CSRP3 C25 (RH/L 18.0), PDLIM5 C73 (RH/L 22.4), PDLIM3 C77 (RH/L 46.2)], sarcomeric chaperons [UNC45b C636 (RH/L 106.4), BAG3 C185 (RH/L 12.8), OBSL1 C1719 (RH/L 7.1)], respiratory complex [UQCRC1 C454 (RH/L 158.7)], showing their relatively high RH/L values (Figure 3D and Table S2).
To validate our identification and quantification analysis, we attempted to confirm glutathionylation of identified cysteines in α-actinin and desmin, which are important proteins for maintaining the sarcomere structure and function in muscle.[30–31] Mouse α-actinin 1 (ACTN1) has 11 cysteines, among which 3 cysteines (C370, C480, C690) were identified for glutathionylation (Figure 3C–D and Table S2). Notably, α-actinin 1 C480 is conserved in all α-actinin isoforms (ACTN1–4). Desmin has only one cysteine, C332, which was found glutathionylated (Figure 3C–D and Table S2). To confirm glutathionylation of α-actinin and desmin, HL-1 cells expressing GS M4 were treated with H2O2 (1 mM). Glutathionylated proteins were enriched by streptavidin-beads after click reaction with biotin-alkyne. Western blot analysis of eluted samples detected glutathionylation of both α-actinin (lane 1 vs. 2, Figure 3E) and desmin (lane 1 vs. 2, Figure 3F) upon addition of H2O2. Next, to determine glutathionylation at the specific cysteines, cysteine mutants of α-actinin 1 (ACTN1 C480S) and desmin (DES C332S), together with their wild-types (WT) were produced and expressed to HEK293/GS M4. Addition of H2O2 induced glutathionylation of both α-actinin 1 WT (lane 4 vs. 5, Figure 3E) and desmin WT (lane 4 vs. 5, Figure 3F). In contrast, levels of glutathionylation were comparatively reduced in α-actinin C480S (lane 5 vs. 8, Figure 3E) and desmin C332S (lane 5 vs 8, Figure 3F) after incubation of H2O2, supporting glutathionylation of their specific cysteines. The relatively strong signal of glutathionylation in α-actinin 1 C480S upon addition of H2O2 (lane 8 in Figure 3E) indicate glutathionylation at other cysteines of α-actinin 1 in addition to C480. Due to the importance of ACTN2 in muscle, we also examined glutathionylation of sarcomeric α-actinin 2 (ACTN2) WT and its cysteine mutant, C487S, which corresponds to C480S of α-actinin 1 (ACTN1). Notably, α-actinin 2 WT was also found glutathionylated upon addition of H2O2 (lane 10 vs. 11, Figure 3E). In contrast, α-actinin 2 C487S showed a weak signal of glutathionylation (lane 11 vs. 14, Figure 3E), supporting glutathionylation of α-actinin 2 at C487. These biochemical data support valid identification and quantification of glutathionylated cysteines by isotopically-labelled clickable glutathione.
Conclusion
Protein S-glutathionylation is an important molecular event that regulates various redox-mediated biological processes.[8] The chemical proteomic quantification of protein modifications has made significant advances toward understanding biological processes as well as developing new pharmacological agents.[32–33] Although a few approaches exist to characterize protein S-glutathionylation, our approach uses an engineered glutathione synthetase mutant (GS M4) that selectively labels intracellular glutathione with a chemical azide-functionality, thereby directly detecting or enriching glutathionylated proteins.[17, 24] In this report, we have devised biosynthesis of isotopically-labelled clickable glutathione by feeding heavy and light derivatives of azido-Ala to cells, which enable to quantify glutathionylated peptides with isotopic mass difference. While SILAC[34] or isobaric tagging[35] could be used instead of isotopic clickable glutathione, our approach may hold several advantages, such as eliminating a need to extensively label a whole proteome as in SILAC[34] or introducing an isotopic-labeling step at the beginning of experiment, as opposed to isobaric tagging.[35] We have demonstrated the use of light and heavy derivatives of azido-Ala for proteomic identification and quantification of glutathionylated peptides upon addition of H2O2, especially estimating RH/L values of 797 glutathionylated cysteines. We also highlighted glutathionylation of muscle-relevant proteins, providing the opportunity to investigate the functional impact of glutathionylation on specific cysteines in cardiomyocytes. Lastly, in recent years, cysteine reactivity or oxidation profiling has been valuable to predict and annotate hyper-reactive or functional cysteines.[32, 36–37] Our quantification strategy developed here could be applicable to profile glutathionylation-susceptibility in the future.
Experimental Section
Cell culture and induction of glutathionylation.
HL-1 cells (Sigma, SCC065) were cultured in Claycomb medium (Sigma, 51800C) supplemented with 10% FBS (GE HyClone), penicillin (100 units/mL), streptomycin (100 μg/mL), L-glutamine (2 mM), and norepinephrine (0.1 mM) in fibronectin-gelatin-coated flasks. HL-1 cells at 80% confluency were infected with adenovirus-expressing GS M4 (Ad/GS M4, VectorBiolab). After 24 h, cells were incubated with azido-Ala (heavy or light) (0.6 mM) in Claycomb medium for additional 20 h. Cells were then treated with H2O2 for 15 min. Cells were washed with cold PBS and lysed with a lysis solution containing 1% SDS, 100 mM LiCl, 100 mM HEPES (pH 7.6), 50 mM N-ethylmaleimide (NEM), and protease inhibitor cocktail (Fisher). The solution was allowed to rotate at 4°C for 30 min and passed through a 26-gauge needle 10 times. Lysates collected were analyzed for protein concentration by Bradford assay.
Click reaction and fluorescence detection of glutathionylated proteins.
Proteins (100 μg) in lysates were precipitated by addition of cold acetone (4 times volume). After centrifugation at 13,000 RPM for 5 min, the supernatant was removed. The pellet was air-dried for 5 min and resuspended in a suspension buffer (40 μL) containing 1% SDS (5 μL), 10 X PBS (5 μL), DMSO (5 μL), and water (25 μL). The mixture was sonicated until the pellet was completely dissolved. To the mixture were added 10 mM Cy5-alkyne in DMSO (0.5 μL), followed by a click solution (10 μL) [pre-made with 20 mM CuBr in DMSO/tBuOH (3:1 vol/vol) (5 μL) and 20 mM THPTA (5 μL)]. The mixture was incubated at room temperature for 1 h in the dark. After separation by SDS-PAGE, proteins in gels were analyzed by FluorChem Q imaging system (Biorad) or Coomassie stains.
Pull-downs and Western blot analysis of glutathionylated proteins.
After click reaction with biotin-alkyne, as described above, proteins were precipitated with ice-cold acetone (4 times volume). The resulting pellet was resuspended in PBS containing 1.2% SDS with sonication. The resuspended proteins were added to PBS containing streptavidin-agarose (Pierce). After incubation overnight at 4°C, beads were washed with PBS containing 0.2% SDS and PBS only, and eluted by SDS-loading dye. Eluted proteins were resolved on SDS-PAGE, transferred to PVDF membrane. The membrane was blocked and incubated with primary antibodies diluted in a blocking buffer at 4°C overnight, including α-actinin (Abcam, Cat# ab9465) (1:500), desmin (Abcam, Cat# ab32362) (1:1000) and FLAG antibody (Sigma, Cat# F1804) (1:1000). Protein levels were visualized by chemiluminescence using appropriate HRP-conjugated secondary antibodies.
Proteomic sample preparation.
Two different lysates made with heavy azido-Ala or light azido-Ala (5 mg proteins per each lysate) were combined, and processed for click reactions, as described above, with biotin-DADPS-alkyne (Click chemistry tools). After click reaction, proteins were precipitated with ice-cold acetone (4 times volume). After centrifugation at 14,000 RPM for 15 min at 4°C, the pellet was resuspended in PBS (1 mL) containing 1.2% SDS with sonication. The resuspended solution was diluted with PBS (5 mL) containing streptavidin-agarose (100 μL bead volume) (Pierce) and incubated overnight at 4°C. Proteins on beads were washed with PBS containing 0.2% SDS and PBS only, and incubated in a denaturation PBS solution (0.5 mL) containing 6 M urea at 37°C for 45 min. Protein on beads were then incubated with a digestion PBS buffer (0.2 mL) containing 2 M urea, 1 mM CaCl2, and Trypsin/Lys-C (Promega) (5 μg) overnight at 37°C. After digestion, beads were washed with PBS containing 0.2% SDS (3 × 1 mL), PBS (3 × 1 mL), and water (3 × 1 mL). Peptides on beads were then eluted by adding 10% aq. formic acid (100 μL x 2) for 30 min, followed by a wash (100 μL). Eluates were combined, lyophilized, and analyzed by LC-MS/MS.
LC MS/MS analysis.
Dried peptides were resuspended in 0.1% formic acid, 0.005% trifluoroacetic acid and 5% acetonitrile and separated by UHPLC reverse phase chromatography using PepMap RSLC C18 columns and an EASY-nLC system before introduction into an Orbitrap Fusion mass spectrometer (Thermo Fisher). Settings for MS1 included a scan range of 375–1600 m/z at 240,000 resolution. For data-dependent MS2 scans detected in the ion trap, peptides with +2 and +3 charges were fragmented by collision induced dissociation (CID, at 32% collision energy) and peptides with charges +3 to +7 were fragmented by electron transfer dissociation (ETD, with calibrated charge-dependent parameters). Cycle time was set at 2.5 seconds over the 60 min gradient, and Dynamic Exclusion was turned on (exclude for 20 s).
Protein identification and quantification:
RAW files were searched with MaxQuant (version 1.6.2.10) against the Uniprot mouse complete database downloaded 2017.07.14 (16844 entries) plus a contaminant database. N-terminal acetylation and methionine oxidation were variable modifications. S-Glutathionylation of cysteine with light azido-glutathione (addition of C16H24N6O7S, 444.14272) or heavy azido-glutathione (addition of C16H212H3N515NO7S, 448.15858) was used for modifications. All other parameters were left at default values. Peptide spectra matches were accepted at a 1% false discovery rate as determined by a reversed database search. Peptide quantifications were analyzed with Skyline software (version 4.2.0), as reported previously.[38] Spectral libraries were built by importing all msms.txt files from MaxQuant to Skyline. FASTA database (Uniprot mouse 2017.07.14) was imported to Skyline. Raw files were imported to Skyline for peak picking. Peptide list was refined to remove peptides without glutathionylation. In MS1-filtering, mass accuracy was set to 10 ppm, and a retention time window (± 2.0 min) was used to find the corresponding peptide peaks in all runs lacking MS/MS identification. For proper peak picking, individual peptide peaks were manually inspected for top three isotopic peaks in the chromatographic traces, and peptides with isotope dot product (idotp) scores lower than 0.8 were manually removed. When necessary, manual integration was applied to have idotp values higher than 0.8. The ratios of peptide areas of heavy to light labels were calculated automatically. From three replicates, peptides identified 2 or 3 times with idotp score higher than 0.8 were assigned with the median RH/L values.
Supplementary Material
Acknowledgements
This work was supported by National Institute of Health Grant R01 HL131740 (Y.-H.A) and Wayne State University Research Award (M. Y. A). The Wayne State University Proteomics Core was supported through the NIH Center Grant P30 ES 020957, the NIH Cancer Center Support Grant P30 CA 02253 and the NIH Shared Instrumentation Grant S10 OD 010700. We thank Dr. Joseph Caruso, Dr. Nicholas Carruthers, and Dr. Paul Stemmer in WSU proteomic center for analysis.
Footnotes
DATA AVAILABILITY
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE [1] partner repository with the dataset identifier PXD014937.
Conflict of Interest
The authors declare no conflict of interest
References
- [1].Burgoyne JR, Mongue-Din H, Eaton P, Shah AM, Circ. Res 2012, 111, 1091–1106. [DOI] [PubMed] [Google Scholar]
- [2].Chen YR, Zweier JL, Circ. Res 2014, 114, 524–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Eaton P, Shah AM, J. Mol. Cell. Cardiol 2014, 73, 1–1. [DOI] [PubMed] [Google Scholar]
- [4].Steinhorn B, Sorrentino A, Badole S, Bogdanova Y, Belousov V, Michel T, Nat. Commun 2018, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Finkel T, J. Biol. Chem 2012, 287, 4434–4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Janssen-Heininger YMW, Mossman BT, Heintz NH, Forman HJ, Kalyanaraman B, Finkel T, Stamler JS, Rhee SG, van der Vliet A, Free Radic. Biol. Med 2008, 45, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Paulsen CE, Carroll KS, Chem. Rev 2013, 113, 4633–4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Xiong Y, Uys JD, Tew KD, Townsend DM, Antioxid. Redox Signal 2011, 15, 233–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS, Schoneich C, Cohen RA, Nat. Med 2004, 10, 1200–1207. [DOI] [PubMed] [Google Scholar]
- [10].Alegre-Cebollada J, Kosuri P, Giganti D, Eckels E, Rivas-Pardo JA, Hamdani N, Warren CM, Solaro RJ, Linke WA, Fernandez JM, Cell 2014, 156, 1235–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sakai J, Li JY, Subramanian KK, Mondal S, Bajrami B, Hattori H, Jia YH, Dickinson BC, Zhong J, Ye KQ, Chang CJ, Ho YS, Zhou J, Luo HBR, Immunity 2012, 37, 1037–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Munkanatta Godage DNP, VanHecke GC, Samarasinghe KTG, Feng HZ, Hiske M, Holcomb J, Yang Z, Jin JP, Chung CS, Ahn YH, Nat. Commun 2018, 9, 4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chen CA, Wang TY, Varadharaj S, Reyes LA, Hemann C, Talukder MAH, Chen YR, Druhan LJ, Zweier JL, Nature 2010, 468, 1115–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sullivan DM, Wehr NB, Fergusson MM, Levine RL, Finkel T, Biochemistry 2000, 39, 11121–11128. [DOI] [PubMed] [Google Scholar]
- [15].Lind C, Gerdes R, Hamnell Y, Schuppe-Koistinen I, von Lowenhielm HB, Holmgren A, Cotgreave IA, Arch. Biochem. Biophys 2002, 406, 229–240. [DOI] [PubMed] [Google Scholar]
- [16].Su D, Gaffrey MJ, Guo J, Hatchell KE, Chu RK, Clauss TRW, Aldrich JT, Wu S, Purvine S, Camp DG, Smith RD, Thrall BD, Qian WJ, Free Radic. Biol. Med 2014, 67, 460–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Samarasinghe KTG, Godage DNPM, VanHecke GC, Ahn YH, J. Am. Chem. Soc 2014, 136, 11566–11569. [DOI] [PubMed] [Google Scholar]
- [18].Chiang BY, Chou CC, Hsieh FT, Gao SJ, Lin JCY, Lin SH, Chen TC, Khoo KH, Lin CH, Angew. Chem. Int. Ed. Engl 2012, 51, 5871–5875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Guo J, Gaffrey MJ, Su D, Liu T, Camp DG, Smith RD, Qian WJ, Nat. Protoc 2014, 9, 64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].VanHecke GC, Abeywardana MY, Ahn YH, Proteome Res J. 2019, 18, 1806–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Aesif SW, Janssen-Heininger YMW, Reynaert NL, Methods Enzymol. 2010, 474, 289–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Duan JC, Kodali VK, Gaffrey MJ, Guo J, Chu RK, Camp DG, Smith RD, Thrall BD, Qian WJ, ACS Nano 2016, 10, 524–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kramer PA, Duan J, Gaffrey MJ, Shukla AK, Wang L, Bammler TK, Qian WJ, Marcinek DJ, Redox Biol. 2018, 17, 367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kekulandara DN, Samarasinghe KTG, Godage DNPM, Ahn YH, Org. Biomol. Chem 2016, 14, 10886–10893. [DOI] [PubMed] [Google Scholar]
- [25].Samarasinghe KTG, Ahn YH, Synlett 2015, 26, 285–293. [Google Scholar]
- [26].Samarasinghe KTG, Godage DNPM, Zhou YN, Ndombera FT, Weerapana E, Ahn YH, Mol. Biosyst 2016, 12, 2471–2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Menon D, Board PG, J. Biol. Chem 2013, 288, 25769–25779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ, Bioinformatics 2010, 26, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, Foulger R, Eilbeck K, Lewis S, Marshall B, Mungall C, Richter J, Rubin GM, Blake JA, Bult C, Dolan M, Drabkin H, Eppig JT, Hill DP, Ni L, Ringwald M, et al. , Nucleic Acids Res. 2004, 32, D258–D261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ribeiro ED, Pinotsis N, Ghisleni A, Salmazo A, Konarev PV, Kostan J, Sjoblom B, Schreiner C, Polyansky AA, Gkougkoulia EA, Holt MR, Aachmann FL, Zagrovic B, Bordignon E, Pirker KF, Svergun DI, Gautel M, Djinovic-Carugo K, Cell 2014, 159, 1447–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Clemen CS, Herrmann H, Strelkov SV, Schroder R, Acta Neuropathol. 2013, 125, 47–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MBD, Bachovchin DA, Mowen K, Baker D, Cravatt BF, Nature 2010, 468, 790–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Moellering RE, Cravatt BF, Chem. Biol 2012, 19, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Harsha HC, Molina H, Pandey A, Nat. Protoc 2008, 3, 505–516. [DOI] [PubMed] [Google Scholar]
- [35].Rauniyar N, Yates JR, Proteome Res J. 2014, 13, 5293–5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Fu L, Liu KK, Sun MG, Tian CP, Sun R, Betanzos CM, Tallman KA, Porter NA, Yang Y, Guo DJ, Liebler DC, Yang J, Mol. Cell. Proteomics 2017, 16, 1815–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].van der Reest J, Lilla S, Zheng L, Zanivan S, Gottlieb E, Nat. Commun 2018, 9, 1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Yang J, Gupta V, Carroll KS, Liebler DC, Nat. Commun 2014, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.