

Abstract
Objective
To investigate β‐amyloid and tau depositions using Pittsburgh compound B (PiB) positron emission tomography (PET) and AV1451 tau PET imaging in aging multiple sclerosis (MS) patients.
Methods
Patients with MS (n = 16) and controls (n = 80) matched for age, sex, and APOE ε4 status from the population‐based Mayo Clinic Study of Aging who underwent PiB PET imaging were studied. Of these individuals, 12 patients with MS and 60 matching controls also underwent AV1451 tau PET. Cortical PiB and AV1451 standard uptake value ratios (SUVrs) from the entire cortex and previously determined Alzheimer disease (AD) signature regions in the same population were calculated for group comparisons and testing for associations with age.
Results
AD signature PiB SUVr (odds ratio [OR] [95% confidence interval (CI)] = 0.52 [0.27–0.98], p = 0.044), total cortical PiB SUVr (OR [95% CI] = 0.52 [0.28–0.99], p = 0.048), and the frequency of abnormal PiB SUVrs (OR [95% CI] = 0.10 [0.01–0.90], p = 0.040) were lower in MS than controls. Although AD‐signature and total cortical AV1451 SUVrs were not different between the groups, the frequency of abnormal AV1451 SUVrs was higher (OR [95% CI] = 10.65 [1.10–103.35], p = 0.041) in MS than controls. The association of AD signature PiB SUVr with age was steeper in the controls compared to patients with MS (estimate [95% CI] = −0.14 [−0.023 to −0.006], p = 0.002). Similarly, the association of total cortical PiB SUVr with age was steeper in the controls compared to patients with MS (estimate [95% CI] = −0.13 [−0.021 to −0.005], p = 0.002). There was no difference in the association of AV1451 SUVr findings with age between the MS patients and controls.
Interpretation
Although both β‐amyloid and tau are biomarkers of cognitive aging and AD, cortical β‐amyloid deposition was lower in MS than age‐matched controls, suggesting that some aspect of MS pathobiology retards the accumulation of β‐amyloid but not the accumulation of tau. ANN NEUROL 2020;87:556–567
Extracellular amyloid plaques and intraneuronal neurofibrillary tangles are the 2 hallmarks of Alzheimer disease (AD) pathology. Pittsburgh compound B (PiB) positron emission tomography (PET) is a biomarker of β‐amyloid load in plaques,1 and AV1451 tau PET is a biomarker of post‐translationally modified tau protein accumulation associated with AD.2, 3 Although β‐amyloid and tau PET imaging are widely studied in cognitive aging and AD, little is known about findings in aging multiple sclerosis (MS) patients.4 Aging is a critical factor in developing neurodegenerative clinical and pathological phenotypes of MS,5, 6, 7 and β‐amyloid and tau biomarkers may be utilized to understand the influence of AD pathophysiology on cognitive aging in MS patients.
Evidence from animal models indicates that inflammatory demyelination‐induced microglial activation may influence β‐amyloid deposition. Immunization with antiamyloid antibodies in AD mouse models and in humans is an established strategy for reducing AD‐related β‐amyloid.8, 9, 10, 11 However, antibody‐independent activation of microglia in an experimental autoimmune encephalomyelitis model of inflammatory demyelination also reduced cortical β‐amyloid deposition,12 suggesting that autoimmune encephalomyelitis may modify cortical β‐amyloid deposition through antibody‐independent mechanisms. On the contrary, in an autopsy cohort of MS patients, the incidence of AD pathology in patients older than 64 years was similar to a normal aging cohort.13 Coexistence of MS and AD pathology has been mentioned in several historic case reports14, 15 and case series,13, 16, 17 as recently reviewed.18 However, none of these studies have been in population‐based cohorts, nor were they compared to a matched control group from the same population.
In this prospective population‐based study with a matched case–control design, our first objective was to determine β‐amyloid and tau depositions with PiB PET and AV1451 tau PET imaging in patients with MS compared to age‐, sex‐, and APOE ε4 status–matched participants without MS. Our second objective was to determine the associations between β‐amyloid and tau depositions and their relationship with age in MS patients compared to controls.
Patients and Methods
Study Population
Individuals enrolled in the Mayo Clinic Study of Aging (MCSA), a prospective population‐based study of cognitive aging,19 were included. Among 5,988 participants enrolled in the MCSA between 2004 to 2019, 32 were previously diagnosed with MS. Their diagnoses were retrospectively confirmed using the most recent diagnostic criteria20, 21 by a neurologist (B.Z.) and an MS specialist (O.H.K.). The current frequency of MS (0.5%) in the MCSA was concordant with the prevalence of MS in Olmsted County, Minnesota, population (0.2%).22 Female‐to‐male ratio was similar at 2:1, and 38% of our study population had progressive MS, which was less than that of the Olmsted County population‐based MS cohort,5 where 48% had progressive MS, likely reflecting a slightly more benign nature of the patients who decided to participate in an aging study later in life. PiB PET imaging was added to the MCSA protocol in 2008. Of the 32 patients with MS, 16 underwent PiB PET imaging. The 16 patients with MS who did not have PET imaging either declined imaging consent from the beginning during enrollment or were no longer interested in imaging during the course of the study; 6 were deceased during the study period and did not undergo PET. The characteristics including age (p = 0.15), sex (p = 0.71), APOE ε4 carrier status (p = 0.91), education (p = 0.25), and global cognitive function (p = 0.06) were not different between the 16 patients who had PET imaging and the 16 patients who did not. Of the 16 patients who participated in the PiB PET study, 12 also underwent AV1451 tau PET imaging. Fewer patients underwent AV1451 tau PET because AV1451 tau PET was added to the MCSA protocol in 2015. Each patient with MS was matched to 5 cognitively unimpaired controls without a demyelinating disease from the same cohort for age, sex, and APOE ε4 carrier status as well as availability of AV1451 PET scans. Three of the participants with MS were classified as having mild cognitive impairment (MCI); therefore, these patients were additionally matched to controls with MCI for a secondary analysis.
The study protocol was approved by the Mayo Clinic and Olmsted Medical Center institutional review boards, and each participant signed informed consent.
Neuropsychological Testing
Four cognitive domains of memory, language, visuospatial skills, and attention/executive function were assessed by the neuropsychological battery completed by all MCSA participants.19 A global cognitive function score was calculated from the average of individual domain scores. Participants were classified based upon a consensus diagnosis by physicians, neuropsychologists, and study coordinators.
Magnetic Resonance Imaging Methods
The magnetic resonance imaging (MRI) protocol was performed on 3.0 T scanners (GE Healthcare, Waukesha, WI; and Siemens, Malvern, PA)23 and included a T2‐weighted fluid‐attenuated inversion recovery sequence and a T1‐weighted 3‐dimensional (3D) high‐resolution magnetization–prepared rapid acquisition gradient‐echo (MPRAGE) sequence for anatomical segmentation and labeling.
PET Methods
The PET imaging protocol was performed on a PET/CT scanner (Discovery; GE Healthcare) operating in 3D mode using 11C‐PiB tracer for β‐amyloid PET and 18F‐AV1451 tracer for tau PET. PiB PET scans were acquired in a 20‐minute scan of four 5‐minute dynamic frames after an injection of 11C‐PiB (555MBq; range, 292–729MBq) and an uptake period of 40 minutes. AV1451 PET scans were acquired in a 20‐minute scan of four 5‐minute dynamic frames after an injection of 18F‐AV1451 (370MBq; range, 333–407MBq) and an uptake period of 80 minutes. The dynamic frame images were averaged to create a single static PET image. An iterative reconstruction algorithm was applied. A 5mm Gaussian postfilter was used, and attenuation, scatter, radioactive decay, and random coincidences were corrected.24
PET images were analyzed using an automated imaging processing pipeline. For gray matter and white matter segmentation, PET images were registered to each participant's own T1‐weighted 3D MPRAGE using SPM12 with the Mayo Clinic Adult Lifespan Template (https://www.nitrc.org/projects/mcalt/).25 From the PiB and AV1451 PET images, standard uptake value ratios (SUVrs) were calculated using cerebellar crus gray matter as the reference region. We analyzed AD signature PiB and AV1451 SUVrs as well as total cortical PiB and AV1451 SUVrs in both groups. The AD signature PiB SUVr was obtained from the bilateral prefrontal, orbitofrontal, temporal, anterior cingulate, and parietal (including precuneus and posterior cingulate) gray matter regions as previously described.26 The (median) AD signature AV1451 SUVr was obtained from the entorhinal, amygdala, parahippocampal, fusiform, inferior, and middle temporal gray matter regions as previously described.27, 28 AD signature PiB SUVr was used to classify participants as PiB positive or negative using a cutoff point of 1.48, and AD signature AV1451 SUVr was used to classify participants as AV1451 positive or negative using a cutoff point of 1.25 as previously operationalized.27, 29 We also investigated the cortical PiB and AV1451 SUVrs obtained from the entire cortex from 41 regions of interest (ROIs).
Statistical Analysis
Demographic, clinical, and imaging characteristics of controls and patients with MS were described using means and standard deviations for continuous variables and counts and percentages for categorical variables. To compare the control and MS groups, conditional logistic regression models were used to account for matching. Due to the skewness in data distribution, PiB SUVr data were log transformed. Linear regressions, adjusting for the matched sets through blocks, were used to describe the associations of PiB or AV1451 SUVr with age and the associations of PiB SUVr with AV1451 SUVr among controls and patients with MS. We included main effects for group to incorporate shifts in the regression lines and interaction terms to test for differences in slopes between the 2 groups.
Results
Demographics and Participants’ Characteristics
In total, 16 patients with MS and 80 cognitively unimpaired controls without MS were matched, of which 69% were women and 31% were APOE ε4 positive. Age at imaging was (mean ± standard deviation [SD]) 63.9 ± 9.5 years in the MS patients and 64.0 ± 9.1 years in controls. The Short Test of Mental Status scores,30 memory, language, attention/executive, visuospatial, and global z scores were not different between the MS and control groups (Table 1).
Table 1.
Characteristics of Patients with MS and Age‐, Sex‐, APOE ε4‐Status Matched Cognitively Unimpaired Controls from a Population‐Based Cohort
| Characteristic | Controls, n = 80 | MS, n = 16 | Odds Ratio (95% CI) | p |
|---|---|---|---|---|
| Women, n (%) | 55 (69) | 11 (69) | — | 1.00 |
| Age, yr | 64.0 (9.1) | 63.9 (9.5) | 0.70 (0.25–1.98) | 0.50 |
| APOE ε4 carrier status, n (%) | 25 (31) | 5 (31) | — | 1.00 |
| Education, yr | 15.1 (2.1) | 14.7 (2.3) | 0.91 (0.71–1.17) | 0.47 |
| Cognitive tests | ||||
| Short Test of Mental Status30 | 36.2 (2.0) | 35.6 (2.2) | 0.87 (0.68–1.10) | 0.24 |
| Global z score | 0.62 (0.87) | 0.31 (0.93) | 0.70 (0.37–1.30) | 0.26 |
| Memory z score | 0.65 (0.88) | 0.25 (1.05) | 0.62 (0.35–1.12) | 0.11 |
| Attention/executive z score | 0.51 (0.87) | 0.03 (1.52) | 0.65 (0.40–1.07) | 0.09 |
| Language z score | 0.40 (0.88) | −0.02 (0.74) | 0.56 (0.30–1.05) | 0.07 |
| Visuospatial z score | 0.46 (0.95) | 0.19 (1.09) | 0.69 (0.37–1.29) | 0.25 |
| PET imaginga | ||||
| AD signature PiB SUVr | 1.54 (0.41) | 1.35 (0.09) | 0.52 (0.27–0.98) | 0.044 |
| Total cortical PiB SUVr | 1.53 (0.36) | 1.37 (0.09) | 0.52 (0.28–0.99) | 0.048 |
| AD signature AV1451 SUVr | 1.17 (0.07) | 1.20 (0.07) | 1.95 (0.72–5.29) | 0.192 |
| Total cortical AV1451 SUVr | 1.12 (0.07) | 1.15 (0.09) | 1.61 (0.64–4.04) | 0.312 |
| Abnormal PiB PET, n (%) | 26 (32) | 1 (6) | 0.10 (0.01–0.90) | 0.040 |
| Abnormal AV1451 PET, n (%) | 6 (10) | 4 (33) | 10.65 (1.10–103.35) | 0.041 |
Mean (standard deviation) is listed for the continuous variables and count (%) for the categorical variables. P values and odds ratios between groups come from a conditional logistic regression accounting for the matching. A log transformation was done for the PiB SUVr due to skewness.
The odds ratio for the PET SUVr represents a 0.1 unit SUVr change for tau and with PiB being a 0.1 unit log SUVr change.
There were 16 MS patients who had PiB PET imaging, and 12 of these 16 MS patients had AV1451 PET imaging. Each MS patient was matched to 5 cognitively unimpaired controls for age, sex, and APOE ε4 carrier status as well as availability of AV1451 PET imaging.
AD = Alzheimer disease; CI = confidence interval; MS = multiple sclerosis; PET = positron emission tomography; PiB = Pittsburgh compound B; SUVr = standardized uptake value ratio.
Of the 16 patients with MS, 10 were in the relapsing–remitting phase, and 6 (38%) were in the progressive phase at the time of imaging. The frequency of patients in progressive phase was also 38% among 16 patients with MS who did not participate in PET imaging. There were 5 patients who had used disease‐modifying treatments (n = 4 interferon beta‐1a, n = 1 dimethyl fumarate), with a median treatment duration of 11 years (range = 4–15 years). This finding is expected given the older age of MS patients in our study because their active MS phase predated evolution of modern disease‐modifying treatments. AD signature and total cortical PiB SUVr values of these 5 individuals were all within the range of the PiB SUVr values observed in MS patients who did not use disease‐modifying treatments (Table 2).
Table 2.
Individual MS Patient Characteristics
| Patient | Sex | APOE ε4 Status | MS Phase | MS Onset Age, yr | DMT | PET Imaging Age, yr | MCI | PiB SUVra | AV1451 SUVra |
|---|---|---|---|---|---|---|---|---|---|
| 1 | F | + | Progressive | 29 | − | 80 | − | 1.39 | − |
| 2 | F | − | Relapsing | 33 | − | 77 | + | 1.23 | 1.32 |
| 3 | F | − | Progressive | 40 | − | 75 | − | 1.33 | 1.17 |
| 4 | F | + | Relapsing | 46 | − | 74 | − | 1.42 | − |
| 5 | F | − | Relapsing | 47 | − | 69 | + | 1.38 | 1.26 |
| 6 | F | − | Progressive | 66 | − | 67 | − | 1.35 | 1.06 |
| 7 | M | − | Progressive | 49 | IFNb 1a | 64 | − | 1.42 | 1.20 |
| 8 | M | − | Progressive | 51 | − | 63 | − | 1.38 | − |
| 9 | M | + | Progressive | 53 | − | 63 | + | 1.23 | 1.17 |
| 10 | F | − | Relapsing | 32 | − | 63 | − | 1.31 | 1.27 |
| 11 | F | − | Relapsing | 35 | − | 58 | − | 1.55 | 1.18 |
| 12 | F | − | Relapsing | 52 | DMF | 57 | − | 1.40 | 1.29 |
| 13 | F | + | Relapsing | 43 | IFNb 1a | 57 | − | 1.39 | 1.23 |
| 14 | M | + | Relapsing | 46 | IFNb 1a | 57 | − | 1.24 | 1.16 |
| 15 | M | − | Relapsing | 42 | IFNb 1a | 53 | − | 1.33 | − |
| 16 | F | − | Relapsing | 33 | − | 45 | − | 1.25 | 1.14 |
Alzheimer disease signature PiB and AV1451 SUVrs are displayed in the table.
DMF = dimethyl fumarate; DMT = disease‐modifying treatment; F = female; IFNb 1a = interferon beta 1a; M = male; MCI = mild cognitive impairment; MS = multiple sclerosis; PiB = Pittsburgh compound B; SUVr = standardized uptake value ratio.
PiB PET Imaging Findings
The AD signature PiB SUVr in the MS patients (mean ± SD = 1.35 ± 0.09; odds ratio [OR] [95% confidence interval (CI)] = 0.52 [0.27–0.98], p = 0.04) was lower than in the controls (mean ± SD = 1.54 ± 0.41). Similarly, the total cortical PiB SUVr in the MS patients (mean ± SD = 1.37 ± 0.09; OR [95% CI] = 0.52 [0.28–0.99], p = 0.048) was lower than in the controls (mean ± SD = 1.53 ± 0.36). Using the previously operationalized cutoff of 1.48 in this population,27 the frequency of abnormal AD signature PiB SUVr was lower (OR [95% CI] = 0.10 [0.01–0.90], p = 0.04) in the MS patients (n = 1, 6%) compared to controls (n = 26, 32%; see Table 1).
Findings in all 41 ROIs that were studied and ranked by ORs for distinguishing patients with MS and controls are shown in Figure 1A. Cortical PiB SUVrs were lower in patients with MS than in controls in all 41 ROIs, and in 21 of 41 ROIs the difference was statistically significant (p < 0.05).
Figure 1.
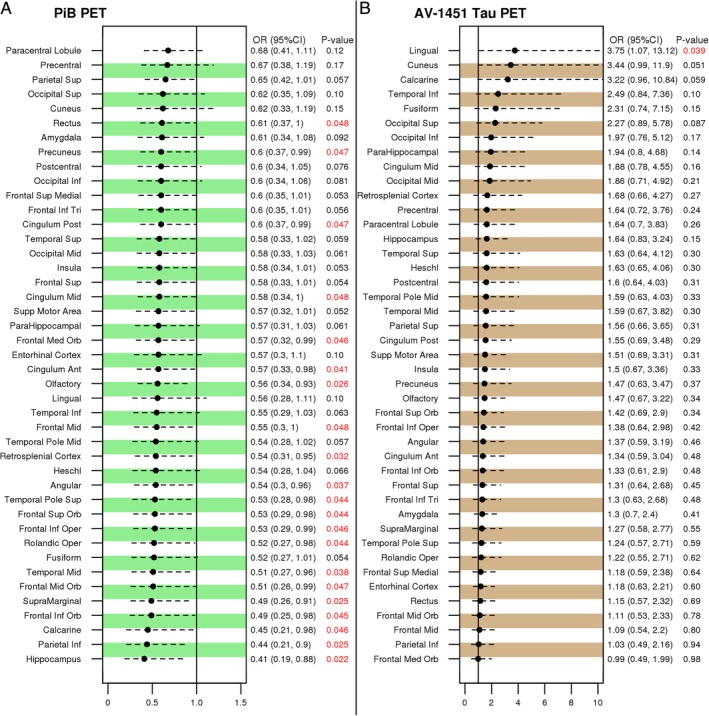
Regional positron emission tomography (PET) findings comparing patients with multiple sclerosis (MS) to controls. The differences in 41 regions of interest are displayed and ranked from top to bottom by the odds ratios (OR). The statistically significant p values are labeled in red for the differences in Pittsburgh compound B (PiB) standard uptake value ratios (SUVrs; A) and AV1451 tau SUVr (B) comparing patients with MS to controls. Ant = anterior; CI = confidence interval; Inf = inferior; Med = medial; Mid = middle; Oper = opercular; Orb = orbital; Post = posterior; Sup = superior; Tri = triangular.
In the secondary analysis, when patients with MS were matched to controls for their MCI status in addition to age, sex, and APOE ε4, the AD signature and total cortical PiB SUVrs remained lower in the MS patients than in controls (p = 0.05). The frequency of abnormal AD signature PiB SUVrs also remained lower (p = 0.027) in the MS patients (n = 1, 6%) compared to controls (n = 26, 32%).
The association of age and AD signature PiB SUVr was steeper in the controls compared to patients with MS (estimate [95% CI] = −0.14 [−0.023 to −0.006], p = 0.002). Similarly, the association of age and total cortical PiB SUVr was steeper in the controls compared to patients with MS (estimate [95%CI] = −0.13 [−0.021 to −0.005], p = 0.002; Fig 2).
Figure 2.
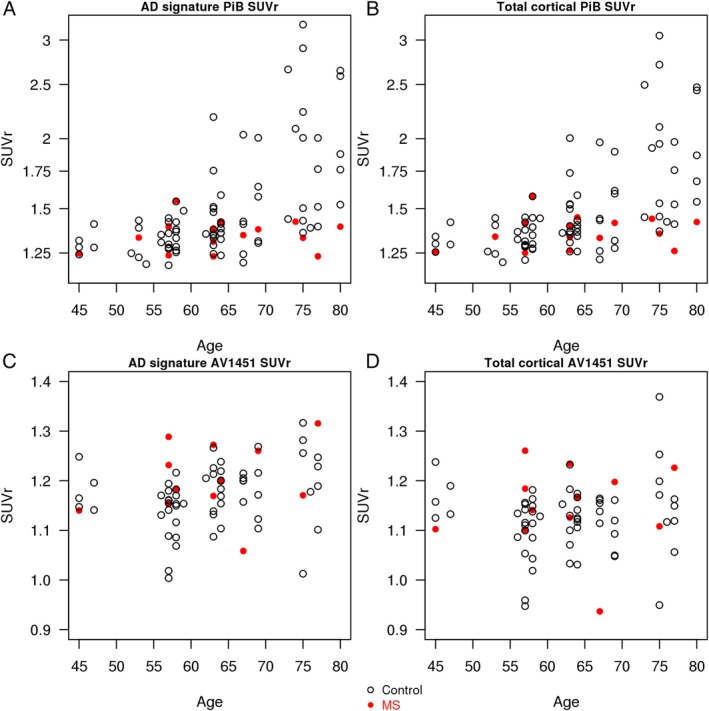
Association of Pittsburgh compound B (PiB) standard uptake value ratios (SUVrs) and AV1451 SUVr with age. Association between Alzheimer disease (AD) signature PiB SUVr and age (A) and association between total cortical PiB SUVr and age (B) in controls and multiple sclerosis (MS) patients. The associations of age with PiB SUVrs were steeper in the controls compared to the MS patients. Association between AD signature AV1451 SUVr and age (C) and association between total cortical AV1451 SUVr and age (D) in controls and MS patients. There was no difference in slopes of associations of age with AV1451 SUVrs between the 2 groups.
AV1451 TAU PET Imaging Findings
The AD signature AV1451 SUVr did not differ (OR [95% CI] = 1.95 [0.72–5.29], p = 0.19) between the MS patients (n = 12, mean ± SD = 1.20 ± 0.07) and the matched controls (n = 60, mean ± SD = 1.17 ± 0.07). Similarly, the total cortical AV1451 SUVr in the MS patients (mean ± SD = 1.15 ± 0.09) did not differ (OR [95% CI] = 1.61 [0.64–4.04], p = 0.31) from the matched controls (mean ± SD = 1.12 ± 0.07). Using the previously operationalized cutoff of 1.25 in this population,29 the frequency of elevated AD signature AV1451 SUVr was different (OR [95% CI] = 10.65 [1.10–103.35], p = 0.04) between patients with MS (n = 4, 33%) and controls (n = 6, 10%; see Table 1).
Findings in all 41 ROIs that were studied and ranked by ORs for distinguishing patients with MS and controls are shown in Figure 1B. Overall, regional cortical AV‐1451 SUVrs were higher in patients with MS compared to controls, but this did not reach statistical significance in any of the regions except for the lingual gyrus ROI (p = 0.039).
In the secondary analysis, when patients with MS were matched to controls for their MCI status in addition to age, sex, and APOE ε4, the AD signature and total cortical AV1451 SUVrs did not differ between the groups. Similarly, the frequency of abnormal AD signature AV‐1451 SUVr was not different (OR [95% CI] = 4.31 [0.71–26.27], p = 0.11) between patients with MS (n = 4, 33%) and controls (n = 9, 15%) in this secondary analysis that included patients with MCI.
There was no difference in slopes of associations of age and AD signature AV1451 SUVr (estimate [95% CI] = −0.000 [−0.006 to 0.005], p = 0.88) or age and total cortical AV1451 SUVr (estimate [95% CI] = −0.000 [−0.006 to 0.006], p = 0.96) between the 2 groups (see Fig 2).
Association of PiB PET with AV1451 PET Findings
The AD signature PiB SUVr increased more with increasing AD signature AV1451 SUVr in controls than in patients with MS (estimate [95% CI] = −1.32 [−2.55 to −0.097], p = 0.03), but there was no difference in slopes for total cortical PiB SUVr with total cortical AV1451 SUVr (estimate [95% CI] = −0.77 [−1.77 to 0.23], p = 0.13; Fig 3). In both cases, PiB SUVr was positively associated with AV1451 SUVr.
Figure 3.
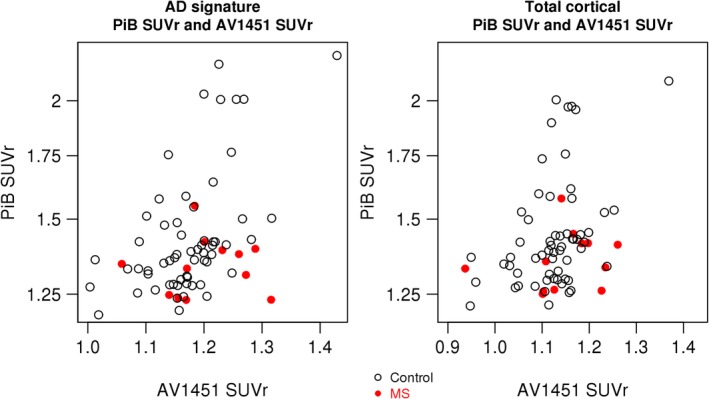
Association of Alzheimer disease (AD) signature and total cortical Pittsburgh compound B (PiB) standard uptake value ratios (SUVrs) with AV1451 SUVr. Association of AD signature PiB with AV1451 SUVrs (left) and association of total cortical AD signature PiB with AV1451 SUVrs (right) in controls and multiple sclerosis (MS) patients. The AD signature PiB SUVr increased more with increasing AD signature AV1451 SUVr in controls than in patients with MS, but there was no difference in slopes for total cortical PiB SUVr with total cortical AV1451 SUVr.
Association of PiB PET and AV1451 PET Findings with MS Disease Duration
For an explanatory analysis, the association of MS disease duration with AV1451 and PiB uptake was studied. Longer disease duration associated with higher cortical AV151 SUVr (AD signature AV1451 SUVr r = 0.42; p = 0.19 and total cortical AV1451 SUVr r = 0.46; p = 0.15 after adjusting for age); however, the findings did not reach statistical significance. On the other hand, such trends were not observed for the association of MS disease duration and PiB SUVr (AD signature PiB SUVr r = 0.10; p = 0.72 and total cortical PiB SUVr r = 0.17; p = 0.53 after adjusting for age).
Pathology
A woman with MS had a pathological evaluation. She was an APOE ε4 carrier. The MS disease onset was at age 29 years, and she had secondary progressive MS since the age of 67 years. She did not use any disease‐modifying treatments for MS. Her cognitive function z scores at the time of imaging at age 81 years were within normal limits. She had diffuse generalized cerebral and cerebellar atrophy and extensive white matter changes with T2 hyperintensities in the white matter. The AD signature PiB SUVr was 1.39, below the cutoff for β‐amyloid positivity (<1.48), 7 years before death. Neuropathological evaluation at age 88 years revealed extensive chronic demyelinating lesions involving subcortical and periventricular regions of the brain, cerebellar white matter, brainstem, and spinal cord. Evaluation also revealed moderate neocortical diffuse plaques and sparse neuritic plaques. Her Consortium to Establish a Registry of Alzheimer's Disease (CERAD) plaque score was “A,” and Thal β‐amyloid phase was 2 with neocortical and entorhinal β‐amyloid deposits, consistent with a negative PiB PET SUVr.31 The Braak neurofibrillary tangle stage was I–II with sparse entorhinal neurofibrillary tangles and moderate pretangles, which classified her as possible primary age‐related taupathy.31, 32 Hippocampal sclerosis was present with associated TAR DNA‐binding protein 43 immunoreactive lesions (Fig 4).31
Figure 4.
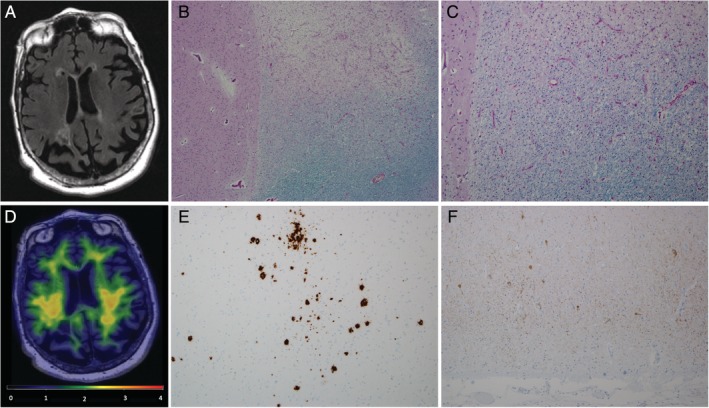
Imaging and pathologic evaluation of a multiple sclerosis (MS) patient, a female 81‐year‐old APOE ε4 carrier. (A) Brain magnetic resonance imaging showed generalized cerebral and cerebellar atrophy and extensive white matter changes with T2 hyperintensities in the white matter, particularly in the periventricular regions. (B, C) Anterior basal ganglia (level of head of caudate nucleus) with adjacent white matter chronic demyelinating lesion (luxol fast blue/periodic acid Schiff). Original magnification, (B) × 40 and (C) × 100. (D) Pittsburgh compound B (PiB) positron emission tomography (PET) was negative with an Alzheimer disease signature PiB SUVr of 1.39. (E) Middle frontal gyrus with sparse neuritic plaques and moderate diffuse plaques (β‐amyloid immunostain). (F) Tau immunostain of entorhinal cortex with sparse neurofibrillary tangles. (E, F) Original magnification, ×100.
Discussion
In this prospective population‐based study of β‐amyloid and AV‐1451 tau PET imaging in MS patients, we made several observations as compared to controls from the same population, matched for age, sex, and APOE ε4 status: (1) there was a paucity of cortical β‐amyloid deposition in patients with MS compared to controls, but there was no difference in cortical tau deposition; (2) the association of older age with higher cortical β‐amyloid deposition was steeper in controls than MS patients; (3) the relationship between older age and higher cortical tau levels was not different between MS patients and controls; and (4) previous studies have shown that higher cortical PiB SUVr is strongly associated with higher cortical AV1451 SUVr,33, 34, 35, 36 and our results were consistent with this pattern in controls, but this relationship was attenuated in MS patients.
β‐amyloid accumulation in the cortex occurs with advancing age, and around 20% of the cognitively unimpaired adults over the age of 65 years have an abnormal PiB PET scan.27 On the contrary, patients with MS had significantly lower cortical β‐amyloid deposition than their matched controls. The association of age with AD signature and total cortical PiB SUVr was steeper in controls compared to MS patients, indicating that β‐amyloid deposition does not follow the expected increase with aging in MS patients. Based on our data, MS patients appear to be protected from β‐amyloid deposition defined by β‐amyloid PET, after accounting for potential confounders such as age, sex, and APOE ε4 status. When patients were matched to controls also for the MCI status, the cortical β‐amyloid deposition remained lower in MS patients.
The coexistence of MS and AD pathology has been reported in case reports and case series,14, 15, 16, 17, 18 but there have been no studies investigating biomarkers of AD‐related pathology in vivo. In an autopsy cohort of 23 MS patients who died after age 64 years, the incidence of AD pathology in the temporal cortex, characterized by β‐amyloid plaques and neurofibrillary tangles, was similar to an independent normal aging population.13 AV1451 tau PET findings of the current study are consistent with those of the autopsy study. However, we observed lower β‐amyloid deposition on PiB PET in MS patients than in controls, and none of the 6 patients with MS older than 64 years had an abnormal PiB PET scan. Several differences exist between the autopsy study by Dal Bianco et al13 and our study. In our study, MS patients and matched cognitively unimpaired controls were ascertained from the same population and underwent the same clinical and imaging protocols, whereas in the autopsy study the control group was from an independent cohort.37 Therefore, due to differences in control populations, it is difficult to compare the studies based on differences between patients with MS and controls. Potentially, if patients with MS from both studies were directly compared, it is possible that levels of amyloid pathology could be similar. In fact, our patient who came to autopsy had low levels of amyloid pathology (Thal stage 2) and did not reach PiB positivity (PiB SUVr >1.48).38 Confirming our findings in an autopsy study with patients and controls drawn from the same cohort could resolve the inconsistency between the 2 studies. Moreover, we were able to quantify the entire cortical β‐amyloid deposition with PiB PET imaging in vivo, whereas the autopsy study13 had limited sampling of brain regions including the hippocampus, the entorhinal cortex, and the temporal lobe without specifying which other regions of temporal lobe were assessed. In fact, in our study the difference in PiB SUVr between patients with MS and controls was statistically significant in the hippocampus, but in some of the other temporal lobe regions such as the entorhinal cortex, inferior temporal, and fusiform gyri this difference was attenuated (see Fig 1A). Therefore, due to potential sampling from regions with smaller differences, it is also possible that the autopsy study may have failed to capture the extent of our findings.
When inflammatory demyelination was induced in an AD mouse model, β‐amyloid deposition was reduced.12 Our finding of lower cortical β‐amyloid levels in a central nervous system (CNS) inflammatory disorder such as MS is consistent with this observation. Furthermore, trafficking of monocytes into the CNS may also contribute to the β‐amyloid clearance as previously shown.39 Although microglia have an important role in MS pathogenesis, the influence of chronic microglia activation on β‐amyloid deposition and clearance in MS patients may potentially be varied.13, 40 Inflammation may be neuroprotective in earlier stages of AD as microglia promotes β‐amyloid clearance,41 but as the disease progresses, microglia develop a toxic and deleterious phenotype.42 Profound microglia activation is also observed in the progressive phase of MS,43 and brain‐wide microglial density is related to MS risk variants.44
In addition to the suggested potential role of microglia, other yet to be determined disease‐specific inflammatory mechanisms may be leading to the low levels of cortical β‐amyloid deposition in MS. In line with this, recent clinical trials have failed to show any positive effect of nonsteroidal anti‐inflammatory drugs (NSAIDs) in individuals at risk for AD.45, 46 Therefore, the reduced risk of AD among long‐term NSAID users with chronic inflammatory diseases in previous observational studies47, 48 would suggest a possible positive impact of the specific inflammatory mechanisms (eg, microglia activation) due to the inflammatory disease itself rather than the treatment. Alternatively, suppression of certain aspects of the inflammatory cascade associated with the disease and allowing other aspects to be active could have impacted β‐amyloid deposition. These possibilities need further study and may point to a common immune‐activation pathway responsible for dampened amyloid deposition or increased β‐amyloid clearance.
Although there are a few observations on β‐amyloid deposition in inflammatory demyelinating diseases, there is little information on tau deposition in MS.13 In the current study, AD signature and total cortical AV1451 uptake and the associations of AV1451 uptake and age were not different between MS patients and controls. According to the β‐amyloid cascade hypothesis, β‐amyloid deposition initiates a downstream sequence of events promoting tauopathy and neurodegeneration.49 In the current study, although controls were cognitively unimpaired and cognitive function domain scores were not different between the groups, 3 participants in the MS group had MCI. None of these 3 patients had β‐amyloid positivity on PiB PET, but 2 had elevated tau on AV1451 PET. Furthermore, there was a modest relationship between longer MS disease duration and higher AV1451 uptake, but this did not reach statistical significance, likely due to small sample size. This relationship should be studied in larger studies. Our findings suggest that, in MS, tau pathology appears to evolve independent of β‐amyloid deposition but may still be associated with cognitive impairment. Therefore, tau pathology in MS does not seem to follow the β‐amyloid cascade hypothesis.
Normal β‐amyloid biomarkers with abnormal tau and/or neurodegeneration suggest neuropathologic processes other than AD, which can be classified as “suspected non‐Alzheimer pathophysiology (SNAP).”50 As in all SNAP cases, determining the etiology of cognitive impairment or dementia in elderly MS patients who have neurodegeneration due to MS may be challenging, such as in a recent case report on a 70‐year‐old woman who had an almost 30‐year history of MS.51 Although this case was clinically diagnosed with AD dementia, the pathologic evaluation revealed that she did not have any neuropathological features of AD and that her dementia was due to MS.51 Hence, in vivo diagnostic accuracy may be facilitated through the use of imaging biomarkers.
A strength of our study is that the MS patients were drawn from a population‐based cohort and compared to the age‐, sex‐, and APOE ε4‐matched controls from the same population. Furthermore, MS patients that enrolled in the MCSA were similar to the MS patients in the Olmsted County population, and there was no clear difference between patients who agreed to undergo PET versus those who did not. Future work confirming the current findings in a larger independent cohort of MS patients would be valuable.
In conclusion, our results suggest that a chronic CNS‐specific disorder, characterized by episodic inflammatory CNS insults followed by endogenous activation of reparative mechanisms, could be protective of β‐amyloid pathology. Because the pathologic evidence in this imaging study is limited to only 1 patient, we note that low cortical PiB SUVr in MS patients may also be due to other yet to be understood reasons related to MS disease pathophysiology.
Author Contributions
K.K., O.H.K., and B.Z. contributed to the conception and design of the study. All authors contributed to the data acquisition and analyses. B.Z., R.R.R., K.K., and O.H.K. contributed to drafting the manuscript, tables, and figures.
Potential Conflicts of Interest
V.J.L.: research support from GE Healthcare, Siemens Molecular Imaging, Avid Radiopharmaceuticals. R.C.P.: scientific advisory board of GE Healthcare. K.K.: research support from Avid Radiopharmaceuticals. The other authors have nothing to report.
Acknowledgment
This study was funded by the NIH (NIA: P50 AG016574, U01 AG006786, R01 AG011378, R01 AG041851, R01 AG034676, R01 AG040042, RF1 AG57547, RF1 AG55151, U54 AG44170; NINDS: R01 NS097495; NCRR: C06 RR018898), the Elsie and Marvin Dekelboum Family Foundation, the Schuler Foundation, the Liston Award, the GHR Foundation, the Alexander Family Alzheimer's Disease Research Professorship of the Mayo Clinic, and the Mayo Foundation for Medical Education and Research. The funding sources had no role in study design, collection, analysis, interpretation, or decision to submit this paper.
We thank Avid Radiopharmaceuticals for their support in supplying AV‐1451 precursor, chemistry production advice and oversight, and the US Food and Drug Administration regulatory cross‐filing permission and documentation needed for this work.
References
- 1. Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound‐B. Ann Neurol 2004;55:306–319. [DOI] [PubMed] [Google Scholar]
- 2. Lowe VJ, Curran G, Fang P, et al. An autoradiographic evaluation of AV‐1451 tau PET in dementia. Acta Neuropathol Commun 2016;4:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chien DT, Bahri S, Szardenings AK, et al. Early clinical PET imaging results with the novel PHF‐tau radioligand [F‐18]‐T807. J Alzheimers Dis 2013;34:457–468. [DOI] [PubMed] [Google Scholar]
- 4. Zeydan B, Lowe VJ, Schwarz CG, et al. Pittsburgh compound‐B PET white matter imaging and cognitive function in late multiple sclerosis. Mult Scler 2018;24:739–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tutuncu M, Tang J, Zeid NA, et al. Onset of progressive phase is an age‐dependent clinical milestone in multiple sclerosis. Mult Scler 2013;19:188–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Frischer JM, Weigand SD, Guo Y, et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann Neurol 2015;78:710–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zeydan B, Kantarci OH. MS progression is predominantly driven by age‐related mechanisms—commentary. Mult Scler 2019;25:906–908. [DOI] [PubMed] [Google Scholar]
- 8. Schenk D, Barbour R, Dunn W, et al. Immunization with amyloid‐beta attenuates Alzheimer‐disease‐like pathology in the PDAPP mouse. Nature 1999;400:173–177. [DOI] [PubMed] [Google Scholar]
- 9. Weiner HL, Lemere CA, Maron R, et al. Nasal administration of amyloid‐beta peptide decreases cerebral amyloid burden in a mouse model of Alzheimer's disease. Ann Neurol 2000;48:567–579. [PubMed] [Google Scholar]
- 10. Wilcock DM, Munireddy SK, Rosenthal A, et al. Microglial activation facilitates Abeta plaque removal following intracranial anti‐Abeta antibody administration. Neurobiol Dis 2004;15:11–20. [DOI] [PubMed] [Google Scholar]
- 11. Nicoll JAR, Buckland GR, Harrison CH, et al. Persistent neuropathological effects 14 years following amyloid‐beta immunization in Alzheimer's disease. Brain 2019;142:2113–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Frenkel D, Maron R, Burt DS, Weiner HL. Nasal vaccination with a proteosome‐based adjuvant and glatiramer acetate clears beta‐amyloid in a mouse model of Alzheimer disease. J Clin Invest 2005;115:2423–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dal Bianco A, Bradl M, Frischer J, et al. Multiple sclerosis and Alzheimer's disease. Ann Neurol 2008;63:174–183. [DOI] [PubMed] [Google Scholar]
- 14. Weber W, Ulrich J. Multiple sclerosis, diffuse lymphoplasmocytic encephalitis and Alzheimer's disease in a patient with progressive dementia. Eur Neurol 1976;14:266–274. [DOI] [PubMed] [Google Scholar]
- 15. Barkhof F, Scheltens P, Kamphorst W. Pre‐and post‐mortem MR imaging of unsuspected multiple sclerosis in a patient with Alzheimer's disease. J Neurol Sci 1993;117:175–178. [DOI] [PubMed] [Google Scholar]
- 16. Frischer JM, Bramow S, Dal‐Bianco A, et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 2009;132:1175–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Flanagan EP, Knopman DS, Keegan BM. Dementia in MS complicated by coexistent Alzheimer disease: diagnosis premortem and postmortem. Neurol Clin Pract 2014;4:226–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Luczynski P, Laule C, Hsiung GR, et al. Coexistence of multiple sclerosis and Alzheimer's disease: a review. Mult Scler Relat Disord 2019;27:232–238. [DOI] [PubMed] [Google Scholar]
- 19. Roberts RO, Geda YE, Knopman DS, et al. The Mayo Clinic Study of Aging: design and sampling, participation, baseline measures and sample characteristics. Neuroepidemiology 2008;30:58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol 2011;69:292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol 2018;17:162–173. [DOI] [PubMed] [Google Scholar]
- 22. Mayr WT, Pittock SJ, McClelland RL, et al. Incidence and prevalence of multiple sclerosis in Olmsted County, Minnesota, 1985–2000. Neurology 2003;61:1373–1377. [DOI] [PubMed] [Google Scholar]
- 23. Schwarz CG, Wiste HJ, Gunter JL, et al. Variability in MRI and PET measurements introduced by change in MRI vendor. Alzheimers Dement 2019;15:P104–P105. [Google Scholar]
- 24. Schwarz CG, Senjem ML, Gunter JL, et al. Optimizing PiB‐PET SUVR change‐over‐time measurement by a large‐scale analysis of longitudinal reliability, plausibility, separability, and correlation with MMSE. Neuroimage 2017;144:113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schwarz CG, Gunter JL, Ward CP, et al. The Mayo Clinic Adult Life Span Template: better quantification across the life span. Alzheimers Dement 2017;13(suppl):P93–P94. [Google Scholar]
- 26. Jack CR Jr, Lowe VJ, Senjem ML, et al. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer's disease and amnestic mild cognitive impairment. Brain 2008;131(pt 3):665–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jack CR Jr, Wiste HJ, Weigand SD, et al. Defining imaging biomarker cut points for brain aging and Alzheimer's disease. Alzheimers Dement 2017;13:205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lowe VJ, Wiste HJ, Senjem ML, et al. Widespread brain tau and its association with ageing, Braak stage and Alzheimer's dementia. Brain 2018;141:271–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lowe VJ, Bruinsma TJ, Wiste HJ, et al. Cross‐sectional associations of tau‐PET signal with cognition in cognitively unimpaired adults. Neurology 2019;93:e29–e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kokmen E, Naessens JM, Offord KP. A short test of mental status: description and preliminary results. Mayo Clin Proc 1987;62:281–288. [DOI] [PubMed] [Google Scholar]
- 31. Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging–Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol 2012;123:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Crary JF, Trojanowski JQ, Schneider JA, et al. Primary age‐related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 2014;128:755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kantarci K, Lowe VJ, Boeve BF, et al. AV‐1451 tau and beta‐amyloid positron emission tomography imaging in dementia with Lewy bodies. Ann Neurol 2017;81:58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cho H, Choi JY, Hwang MS, et al. In vivo cortical spreading pattern of tau and amyloid in the Alzheimer disease spectrum. Ann Neurol 2016;80:247–258. [DOI] [PubMed] [Google Scholar]
- 35. Brier MR, Gordon B, Friedrichsen K, et al. Tau and Abeta imaging, CSF measures, and cognition in Alzheimer's disease. Sci Transl Med 2016;8:338ra66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Marks SM, Lockhart SN, Baker SL, Jagust WJ. Tau and beta‐amyloid are associated with medial temporal lobe structure, function, and memory encoding in normal aging. J Neurosci 2017;37:3192–3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Giannakopoulos P, Hof PR, Mottier S, et al. Neuropathological changes in the cerebral cortex of 1258 cases from a geriatric hospital: retrospective clinicopathological evaluation of a 10‐year autopsy population. Acta Neuropathol 1994;87:456–468. [DOI] [PubMed] [Google Scholar]
- 38. Murray ME, Lowe VJ, Graff‐Radford NR, et al. Clinicopathologic and 11C‐Pittsburgh compound B implications of Thal amyloid phase across the Alzheimer's disease spectrum. Brain 2015;138(pt 5):1370–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Frenkel D, Wilkinson K, Zhao L, et al. Scara1 deficiency impairs clearance of soluble amyloid‐beta by mononuclear phagocytes and accelerates Alzheimer's‐like disease progression. Nat Commun 2013;4:2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lassmann H. Mechanisms of neurodegeneration shared between multiple sclerosis and Alzheimer's disease. J Neural Transm (Vienna) 2011;118:747–752. [DOI] [PubMed] [Google Scholar]
- 41. Heneka MT, Carson MJ, El Khoury J, et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol 2015;14:388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Prokop S, Miller KR, Heppner FL. Microglia actions in Alzheimer's disease. Acta Neuropathol 2013;126:461–477. [DOI] [PubMed] [Google Scholar]
- 43. Kutzelnigg A, Lucchinetti CF, Stadelmann C, et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 2005;128(pt 11):2705–2712. [DOI] [PubMed] [Google Scholar]
- 44. Felsky D, Patrick E, Schneider JA, et al. Polygenic analysis of inflammatory disease variants and effects on microglia in the aging brain. Mol Neurodegener 2018;13:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Group AR , Lyketsos CG, Breitner JC, et al. Naproxen and celecoxib do not prevent AD in early results from a randomized controlled trial. Neurology 2007;68:1800–1808. [DOI] [PubMed] [Google Scholar]
- 46. Meyer PF, Tremblay‐Mercier J, Leoutsakos J, et al. INTREPAD: a randomized trial of naproxen to slow progress of presymptomatic Alzheimer disease. Neurology 2019;92:e2070–e2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cote S, Carmichael PH, Verreault R, et al. Nonsteroidal anti‐inflammatory drug use and the risk of cognitive impairment and Alzheimer's disease. Alzheimers Dement 2012;8:219–226. [DOI] [PubMed] [Google Scholar]
- 48. Chang KH, Hsu YC, Hsu CC, et al. Prolong exposure of NSAID in patients with RA will decrease the risk of dementia: a nationwide population‐based cohort study. Medicine (Baltimore) 2016;95:e3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002;297:353–356. [DOI] [PubMed] [Google Scholar]
- 50. Jack CR Jr, Knopman DS, Weigand SD, et al. An operational approach to National Institute on Aging–Alzheimer's Association criteria for preclinical Alzheimer disease. Ann Neurol 2012;71:765–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tobin WO, Popescu BF, Lowe V, et al. Multiple sclerosis masquerading as Alzheimer‐type dementia: clinical, radiological and pathological findings. Mult Scler 2016;22:698‐704. [DOI] [PMC free article] [PubMed] [Google Scholar]