

Abstract
Exposure to arsenic in contaminated drinking water is an emerging public health problem that impacts more than 200 million people worldwide. Accumulating lines of evidence from epidemiological studies revealed that chronic exposure to arsenic can result in various human diseases including cancer, type 2 diabetes, and neurodegenerative disorders. Arsenic is also classified as a Group I human carcinogen. In this review, we survey extensively different modes of action for arsenic-induced carcinogenesis, with focus being placed on arsenic-mediated impairment of DNA repair pathways. Inorganic arsenic can be bioactivated by methylation, and the ensuing products are highly genotoxic. Bioactivation of arsenicals also elicits the production of reactive oxygen and nitrogen species (ROS and RNS), which can directly damage DNA and modify cysteine residues in proteins. Results from recent studies suggest zinc finger proteins as crucial molecular targets for direct binding to As3+ or for modifications by arsenic-induced ROS/RNS, which may constitute a common mechanism underlying arsenic-induced perturbations of DNA repair.
Graphical Abstract
1. INTRODUCTION
Being the 20th most abundant element in the Earth’s crust, arsenic (As) is widely distributed in the environment.1 Arsenic pollution incidents have been documented around the world, including the United States, influencing approximately 200 million people in over 70 countries primarily through contaminated drinking water.2 For instance, the levels of arsenic in groundwater in most regions in Bangladesh exceed the level set by the U.S. EPA (i.e., 10 ppb).3,4 Arsenic has been one of the most widely studied metals/metalloids in the last 20 years, and it has been placed on the top of the hazardous substance priority list by the U.S. Agency for Toxic Substances and Disease Registry (ATSDR) for over 15 years.
Aquatic arsenic is the most significant source of arsenic contamination in groundwater due to its solubility.5 Inorganic arsenite (As3+) and arsenate (As5+) are the predominant forms of arsenic in water,6 and they also confer higher toxicity and display higher mobility in the environment when compared to organic forms of arsenic.7 Naturally occurring inorganic arsenic (iAs) is present mainly in its sulfide form within complex minerals containing silver, lead, copper, nickel, antimony, cobalt, and iron.1 Arsenic can be mobilized by a variety of natural and anthropogenic activities, where natural occurrences, such as volcano eruptions and weathering of rocks and soils, are only secondary mobilizers in comparison to anthropogenic activities.1 Mining, smelting of nonferrous metals, burning of fossil fuels, and agricultural irrigation using contaminated groundwater are the primary routes through which arsenic species are released into the environment, though historically the application of arsenic-containing pesticides also released a significant amount of arsenic to agricultural soil.1
Arsenic is classified as a Group I human carcinogen by the International Agency for Research on Cancer.1,8 Over the last few decades, an increasing body of evidence from numerous epidemiological and animal studies has documented a strong association between arsenic exposure and tumor progression in skin, lung, bladder, kidney, and liver.1,4,9–16
Carcinogenesis is a multistep process encompassing cancer initiation, promotion, and malignant progression.17,18 Cancer can arise after accumulation of mutations in cellular DNA, which could emanate from impaired capacity in DNA repair. Metabolic and other endogenous cellular processes constantly generate ROS and reactive metabolites, which can result in DNA damage.19 The ensuing DNA adducts need to be efficiently removed by the DNA repair machinery; otherwise, they may elicit nucleotide misincorporation during DNA replication, thereby inducing mutations in DNA.20 A mutated proto-oncogene could be activated, whereas a mutated tumor suppressor gene may not function properly, both scenarios favoring carcinogenesis.
There are a number of proposed mechanisms for arsenic-elicited carcinogenesis, including, but not limited to, elevated oxidative stress, diminished DNA repair, dysregulated cell proliferation and apoptosis, and aberrant DNA methylation and histone post-translational modifications.21 In the following sections, we review briefly the biotransformation of arsenic species and discuss these different modes of action of arsenic carcinogenesis, with the focus being placed on the molecular mechanisms through which arsenic exposure leads to compromised DNA repair.
2. BIOTRANSFORMATION OF INORGANIC ARSENIC
Toxicity of inorganic arsenic (As3+ and As5+) in humans depends largely on their metabolism. Approximately 90% of ingested inorganic arsenic (As3+ or As5+) is absorbed by the gastrointestinal tract.22 Inorganic As5+ subsequently undergoes a sequential process, that is, glutathione (GSH)-mediated two-electron reduction to As3+, and oxidative methylation of As3+ by arsenite methyltransferase (As3MT) to pentavalent organic arsenic species (e.g., MMAV and DMAV) in the liver (Figure 1).21,23 DMAV was previously shown to be a teratogen, a nephrotoxin, a tumor promoter, and a complete carcinogen in mammals.24–27 In this biotransformation process, iAs can also be methylated to yield trivalent arsenic compounds such as MMAIII and DMAIII (Figure 1), which exhibit higher potency in being cytotoxic/genotoxic agents and enzyme inhibitors over iAs3+.23 Therefore, it is important to consider both inorganic arsenic and their trivalent methylated arsenic species when discussing arsenic toxicity.28–30
Figure 1.

Inorganic arsenic and its metabolism. In liver, absorbed As5+ is reduced to As3+ by GSH as an electron donor, and As3+ undergoes sequential methylation and reduction with SAM and GSH as the donors of methyl group and electron, respectively, to generate MMAV, MMAIII DMAV, and DMAIII.
3. ARSENIC EXPOSURE AND OXIDATIVE STRESS
Arsenic-induced oxidative stress has been widely studied and may constitute a major factor contributing to arsenic carcinogenesis (Figure 2). Inorganic and methylated trivalent arsenic species have been shown to induce the generation of reactive oxygen species (ROS) and oxidative stress in mammalian cells.31–33
Figure 2.

iAs-elicited oxidative stress enhances carcinogenesis through impairing DNA repair pathway to induce mutations in DNA. As3+ can induce the overproduction of ROS and RNS through mitochondria dysfunction, cellular antioxidant imbalance, and impairment of ROS-scavenging enzymes. Hence, iAs-elicited oxidative stress induces oxidative DNA damage, disturbs PTMs of DNA repair enzymes, and disrupts protein tyrosine phosphorylation, thereby enhancing DNA mutations to promote carcinogenesis.
Apart from direct generation of ROS from arsenic and its metabolites, arsenic exposure can result in antioxidant imbalance, mitochondrial dysfunction, and impairment of ROS-scavenging enzymes, which together result in arsenic-induced oxidative stress, as noted previously.34 Because glutathione (GSH) serves as an electron donor in arsenic metabolism,21,23 the intracellular GSH pool is heavily depleted upon chronic arsenic exposure and becomes unavailable for scavenging ROS as a cellular defense mechanism against oxidative stress. In addition, arsenic-mediated disruption of the mitochondrial electron transport chain exacerbates oxidative stress, because mitochondrion constitutes a major source of intracellular ROS.35–37 Moreover, arsenic-induced oxidative stress emanates from impaired activities of ROS-scavenging enzymes such as superoxide dismutase, catalase, glutathione peroxidase, glutathione S-transferase, and glutathione reductase.34,38
Arsenic carcinogenesis may arise, in part, from the genotoxicity of ROS and RNS. Along this line, increasing lines of evidence demonstrated that iAs and its methylated metabolites damage DNA indirectly through the induction of free radicals.33,39–42 For instance, hydroxyl radicals generated from arsenite exposure are believed to react with nucleobases in DNA to yield DNA lesions, for example, 8-oxo-7,8-dihydroguanine, 5-hydroxycytosine, and 5-hydroxyuracil.43 In addition, arsenite-induced ROS and RNS can elicit cross-links between DNA and proteins or other molecules in cells.44 Moreover, in vitro studies with cultured human cells indicated that arsenite-induced oxidative stress causes persistent telomere attrition, DNA strand breaks, chromosomal aberrations, and sister chromatid exchanges.45–48
Aside from oxidative DNA damage, arsenite-induced ROS may perturb the cellular functions of ROS as secondary messengers.33,34 Last but not least, ROS/RNS generated from arsenite exposure can directly impair important cysteine-containing proteins involved in DNA repair and DNA damage response (DDR) signaling. For example, inhibition of PARP1 by peroxynitrite through S-nitrosylation of its zinc finger cysteines was shown to compromise DNA repair.49,50
4. INHIBITION OF DNA REPAIR
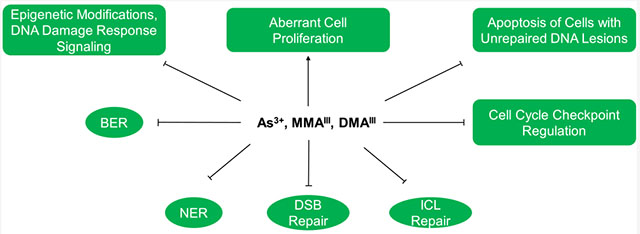
Arsenite alone is a weak mutagenic agent, but it is known to enhance the mutagenicity of other carcinogens. For instance, arsenite was found to enhance the mutagenicity of X-rays, UV light, methylmethanesulfonate (MMS), and diepoxybutane in mammalian cells.51–53 Arsenic’s role in augmenting the mutagenicity of other carcinogens perhaps can be attributed to its ability in inhibiting the repair of DNA lesions induced by these carcinogens. In this section, we review the previous work regarding the perturbation of various cellular DNA repair and DDR pathways after exposure to iAs (Figure 3).
Figure 3.

Major events governing the disruption of DNA repair pathways by iAs and its trivalent metabolites. Arsenite and its metabolites induce cell proliferation while inhibiting BER, NER, DSB repair, ICL repair, DDR signaling, cell cycle checkpoint regulation, and apoptosis of damaged cells. These together diminish the capacity of DNA repair and impair genetic integrity.
4.1. Excision Repair.
A number of studies revealed that arsenite can target several key molecular players in base excision repair (BER) and nucleotide excision repair (NER) pathways through perturbing the expression levels of DNA repair genes or catalytic activities of DNA repair proteins.
4.1.1. BER.
BER is a crucial DNA repair pathway mainly responsible for the removal of oxidatively generated and alkylated nucleobase lesions, apurinic/apyrimidinic (AP) sites, and strand breaks.54 For instance, 8-oxoG is one of the most abundant oxidatively generated DNA lesions,55–57 and 8-oxoguanine DNA glycosylase-1 (OGG1) is the main glycosylase responsible for the excision of 8-oxoG from DNA in mammals.57–59 A previous study revealed a dose-dependent decline in mRNA level and enzymatic activity of OGG1 in A549 human lung epithelial cells after exposure to micromolar concentrations of arsenite and its metabolites.60
Aside from OGG1, AP endonuclease 1 (APE1), the major endonuclease responsible for the excision of apurinic/apyrimidinic (AP) sites in eukaryotic cells, was shown to be diminished at the mRNA and protein levels after exposure to As3+.61 Additionally, DNA polymerase β, an important enzyme for DNA repair synthesis during BER,62 exhibited decreased expression at both the mRNA and protein levels after exposure to As3+ at concentrations that are ≥5 μM.61 Moreover, Osmond et al.63 observed a dose-dependent decrease in mRNA levels of APE1, DNA ligase I (LIGI), OGG1, PARP1, and DNA polymerase β (DNA Polβ) in 24-week old mice subchronically (2 weeks) exposed to arsenite-contaminated drinking water, further substantiating that arsenic exposure can impair the BER pathway.
4.1.2. NER.
NER is a critical and versatile DNA repair pathway for the removal of bulky DNA adducts and helix-distorting lesions induced by environmental carcinogens (e.g., UV-induced dimeric DNA photoproducts and adducts generated from metabolites of polycyclic aromatic hydro-carbons).64
A number of prior studies revealed that arsenic mainly interferes with NER by disrupting the gene expression levels and activities of crucial NER players. For instance, exposure to arsenic in drinking water was found to be correlated with reductions in mRNA levels of ERCC1, XPB, and XPF genes in lymphocytes,65 and the mRNA and protein expression levels of ERCC1 were also shown to be diminished upon arsenic exposure in a follow-up study.66 Additionally, a dose-dependent decline in mRNA levels of ERCC1 gene was observed in human cardiomyocytes following a 72-h exposure to arsenite.67
An earlier large-scale microarray analysis revealed that the mRNA expression levels of a number of DNA repair genes, encompassing XPC, DDB2, and TP53, were significantly down-regulated in human epidermal keratinocytes after exposure to submicromolar concentrations of arsenite.68 Another microarray study showed that the mRNA expression levels of XPD, PCNA, APE1, RFC, XPC, and DNA ligase I were reduced by at least 1.5-fold following a 4-h exposure to 5 μM arsenite.69 Moreover, treatment of human skin fibroblast cells with arsenite and MMAIII lowered, in a dose-dependent manner, the mRNA levels of XPC and DDB2 as well as the protein level of XPC.70 Furthermore, treatment of IMR-90 human lung fibroblasts with arsenite reduced the protein level of XPC, partially through proteasomal degradation, as well as reducing the mRNA levels of several NER genes, including XPA, XPC, and DDB2.71 A recent Bru-seq study showed that a 1-h acute exposure to 5 μM arsenite led to diminished transcription of RAD23B and DDB2 genes.72 Impairment of NER by arsenic was also observed for DNA lesions induced by cisplatin, a clinically used chemotherapeutic agent for treating human cancers through the generation of Pt-d(GpG) intrastrand cross-link lesions in DNA.73 In particular, exposure to arsenite prevented the induction of XPC after treatment of mice with cisplatin.74
Interestingly, arsenic exposure has been shown not to affect the protein level of XPA,71 which is essential for recognition of damaged DNA and subsequent recruitment of other NER components, especially RPA70 and TFIIH.75,76 The XPA protein contains a Cys4 (C4)-type zinc finger that is involved in binding with damaged DNA and RPA70. Biochemical studies indicated that mutations in any of the four zinc-coordinating cysteines result in an unfolded protein.77 Arsenite has been demonstrated to interact with zinc finger proteins by substituting for the zinc ion,78,79 and several zinc finger proteins involved in DNA repair, for example, XPA and poly(ADP-ribose) polymerase 1 (PARP-1), have been shown to be direct molecular targets for binding with iAs3+ and MMAIII.80–83
Lastly, arsenite exposure has been proposed to interfere with and inhibit NER activity through NO-mediated nitrosylation of DNA repair proteins.49,50,84,85 All the above studies together support that arsenite and its trivalent metabolites may perturb NER by perturbing the central NER players at both transcript and protein levels.
4.2. DNA Ligation.
DNA ligases assume important roles in various DNA metabolic processes including DNA replication, repair, and recombination, and arsenite has been shown to inhibit the DNA ligation process. It was reported that the levels of mRNA, protein and enzymatic activities of DNA ligase I and DNA ligase III are significantly diminished in mammalian cells after exposure to iAs3+ and MMAIII.61,86 It was also shown that arsenite inhibits DNA ligation by interacting with the vicinal cysteines in DNA ligase III, thereby retarding DNA break rejoining in MMS-treated hamster cells.87 In addition, XRCC1 plays an indispensable role in recruiting and stabilizing ligase IIIα in the DNA ligation step of excision repair by acting as a scaffolding protein,88–91 where down-regulation of the XRCC1 protein by iAs exposure also contributes to the impairment of the DNA ligation step of the excision repair pathways.92 Because the inhibition of DNA ligation by arsenic exposure prevents the completion of DNA repair, it may lead to accumulation of damaged intermediates including single- and double-strand breaks, ultimately contributing to genome instability.
4.3. Fanconi Anemia (FA)/BRCA Pathway for DNA Interstrand Cross-Link and DNA–Protein Cross-Link Repair.
DNA interstrand cross-links (ICLs) can arise from endogenous metabolism or from exposure to therapeutic cross-linking agents such as mitomycin C (MMC).93 ICLs are extremely cytotoxic because covalent linkage of the two complementary strands of DNA blocks essential DNA metabolic processes including replication and transcription.93 FA/BRCA pathway, which encompasses three stages of DNA repair processes, that is, nucleolytic incision, translesion synthesis (TLS) and homologous recombination (HR), is indispensable for the repair of DNA ICLs.93,94 As3+ was shown to disrupt the FA/BRCA pathway-mediated repair of DNA ICLs.95
In the FA/BRCA pathway, monoubiquitination of FANCD2 is essential for the recruitment of SLX4/FANCP, an endonuclease protein complex required for unhooking the DNA cross-link and for the downstream TLS and HR steps of the ICL repair pathway, to DNA damage sites.93,96 Monoubiquitination of FANCD2, catalyzed by the E3 ubiquitin ligase FANCL,97,98 is also necessary for the relocalization of the Fanconi-associated nuclease 1 into nuclear DNA repair foci for recovery of stalled replication forks during ICL repair.99 Recently, arsenite was shown to inhibit the repair of DNA ICLs induced by MMC through diminishing monoubiquitination and compromising the access of FANCD2 to DNA damage sites in chromatin in cultured human cells.100 This occurs through inhibition of the E3 ubiquitin ligase activity of FANCL via direct binding of arsenite to its RING finger domain.100 We reason that, apart from FANCL, arsenite may also bind to RING finger-containing SUMO E3 ligases PIAS1 and RNF4, which may inhibit the SUMOylation, polyubiquitination, and degradation of FANCA, thereby perturbing FA/BRCA pathway.101
Unhooking of an ICL by XPF-ERCC1 is necessary for the stable localization of FANCD2 onto chromatin and its subsequent HR-mediated repair of DNA DSBs, as manifested by the failure to repair ICL-induced DSBs in XPF-ERCC1-deficient human cells.102–104 Decreases in the mRNA levels of XPF and ERCC1,65 and in the protein level of ERCC1,66 in individuals exposed to arsenite in drinking water suggest that arsenite may impair ICL repair through suppressing mRNA and protein expression and disrupting the proper functions of the XPF-ERCC1 complex.
BRCA1 has been proposed to be crucial for homologous recombination-independent repair of DNA ICLs by promoting the recruitment of FANCD2 to DNA damage sites.105,106 In particular, BRCA1 was shown to antagonize the inhibitory effect of the Ku70-Ku80 heterodimer on FANCD2 foci formation105 and promote the unloading of the CMG helicase from stalled replication forks during ICL repair.107 Additionally, BRCA1 is believed to amplify the FA/BRCA pathway by regulating FANCD2’s interaction with other proteins.108 It has been hypothesized that BRCA1 is involved in homology-based DNA repair through its E3 ubiquitin ligase activity; the RING domain of the ligase is indispensable for its interaction with BRCA1-associated RING domain (BARD1) protein, forming a heterodimeric complex to modulate the stability and enzymatic activity of BRCA1.107,109 Exposure to arsenite was recently found to diminish the recruitment of BRCA1 to DNA DSB sites,110 suggesting that arsenite might also interfere with ICL repair through binding and inhibiting the E3 ubiquitin ligase activity of the BRCA1-BARD1 complex.
4.4. DNA Double-Strand Break Repair.
Double-strand breaks (DSBs) are among the most deleterious types of DNA lesions, which can lead to mutations, loss of heterozygosity, and chromosomal rearrangement; if not properly repaired, they can lead to cell death and cancer.111,112 In mammalian cells, DSB repair proceeds through two different pathways, namely HR and nonhomologous end-joining (NHEJ).113 Exposure to arsenic was shown to induce DSBs and ultimately lead to chromosomal aberrations and sister chromatid exchanges.114,115 Exposure to arsenic was also found to inhibit DNA DSB repair and influence the DNA DSB repair pathway choice by favoring error-prone NHEJ repair while inhibiting the error-free HR pathway, leading to mis-repair of DSBs and genome instability.116
Since DSB repair occurs on DNA substrates that are localized in chromatin, the efficiency in DSB repair depends mainly on how accessible the site of damage is, which is largely determined by the compactness of the local chromatin.117 Generation of open chromatin involves the actions of multisubunit chromatin-remodeling complexes and post-translational modifications of core histone proteins. In the latter regard, acetylation of lysine 16 in histone H4 (H4K16Ac) and monoubiquitination of lysine 120 in histone H2B (H2BK120ub) represent those histone epigenetic marks that promote the formation of biochemically accessible chromatin at or near DNA DSB sites.110,118,119 Recently, it was reported that arsenite inhibits H4K16Ac by binding to the zinc finger motif of two MYST family histone acetyltransferases TIP60 and hMOF,120,121 and arsenite was also shown to inhibit H2BK120ub catalyzed by RNF20-RNF40 histone E3 ubiquitin ligase in a similar fashion, thereby diminishing the recruitment of BRCA1 and RAD51 to DSB sites for repair.110 Therefore, arsenite could disrupt DSB repair by inhibiting histone epigenetic modifications, which leads to compact chromatin structures unfavorable for DNA DSB repair.
In addition to the aforementioned histone modifications that regulate the accessibility of DNA lesions in chromatin, a myriad of zinc finger proteins are involved in post-translational modifications (PTMs) of proteins that control DDR and transcription of DNA repair genes. For instance, the RING finger E3 ubiquitin ligases RNF8 and RNF168 are essential for DDR in response to DSB formation through ubiquitination of H2A/H2AX surrounding DNA DSB sites,122 where RNF168 interacts with PALB2-containing protein complex to DSB-induced H2A ubiquitination, thereby promoting DSB repair.123 In addition, DNA DSB repair pathway choice is modulated by deubiquitinating enzymes (DUBs) and other E3 ubiquitin ligases (e.g., RNF169 and RNF126).124–127 In this vein, being a negative regulator of the ubiquitin-dependent DDR signaling, RNF169 directly recognizes RNF168-mediated ubiquitination near DNA DSB sites and competes with other ligases for nonproteolytic ubiquitination at DSB sites to limit the deposition of 53BP1 and RAP80, thereby fine-tuning the DSB repair pathway choice.125,126,128 Moreover, after being recruited to DSB sites in a RNF8-dependent manner, RNF126 directly interacts with and ubiquitinates RNF168 to negatively regulate the RNF168-mediated H2AX ubiquitination and favor the HR-mediated repair of DSBs.127
Within seconds after DSB induction, poly(ADP-ribose) polymerases, including PARP-1, sense, recognize, and bind to DSBs to catalyze global protein poly(ADP-ribosyl)ation (PARylation).129–131 Global PARylation around DSB sites serves as a docking platform for rapid recruitment of various DNA repair factors including MRE11, NBS1, BARD1, CHFR, and RNF146 to chromatin.130,132 Meanwhile, PARP-1 can PARylate different proteins globally, including BRCA1, DNA-dependent protein kinase catalytic subunit (DNA-PKcs), and core histones, to promote DNA DSB repair.132–134 In addition, arsenite was shown to interfere with DNA damage-elicited global PARylation in human cells by inhibiting PARP1 activity through displacement of zinc ions from its zinc finger motifs.80,135–137
PARP1 perhaps can be viewed as a typical paradigm among DNA repair proteins, many of which contain redox-sensitive cysteine residues within zinc finger domains.34,138,139 Given that arsenic exposure can stimulate the generation of ROS/RNS,33,49,140 arsenite-induced oxidative stress can result in modification of thiol groups on the cysteine residues in zinc finger motifs of these DNA repair proteins, leading to the loss of their enzymatic functions.82,85,141,142 This has been demonstrated for PARP1, which could be inhibited by peroxynitrite-mediated S-nitrosation of its zinc finger cysteine(s).49,50,80 Global PARylation, a PTM predominantly mediated by PARP1 and critical for immediate initiation of DDR to maintain genomic stability, was shown to be markedly inhibited upon an 18-h exposure to 0.01 μM arsenite.143 Therefore, apart from direct As3+ binding, arsenic-induced oxidative stress also contributes, in part, to diminished DNA repair arising from arsenic exposure (Figure 4).84,144
Figure 4.

Modes of action of inorganic arsenic and iAs-induced ROS/RNS in impairing the enzymatic activity of zinc finger (ZnF) proteins. iAs and ROS/RNS can target vicinal cysteines within the zinc coordination spheres of zinc finger proteins: (i) As3+ directly binds to these cysteines more strongly than Zn2+; (ii) ROS oxidizes these cysteines to form a series of oxidization products, such as –SOH and –S–S–; (iii) RNS, especially peroxynitrite, can S-nitrosylate these cysteines. In all these cases, Zn2+ bound within zinc finger motifs is released through its displacement by As3+, which alters the conformation of zinc finger proteins and hence their enzymatic activities.
Recently, CTCF, a versatile 11-zinc finger transcription regulator with well-established roles in three-dimensional genome organization and transcriptional regulation, was found to facilitate DNA DSB repair by enhancing HR.145,146 CTCF is recruited to DSB sites through its zinc finger domains independently of PARylation.145 Therefore, substitution of zinc ions within those 11 zinc finger domains of CTCF by iAs3+ can diminish its DNA binding capability. Additionally, a recent study demonstrated that CTCF binds to MRE11 and CtIP through its zinc finger domains, which enables robust CtIP recruitment for 5′-end DNA resection, thereby promoting HR while suppressing NHEJ pathway of DNA DSB repair.147 Hence, the binding of iAs3+ with CTCF might explain, in part, how arsenite disrupts the outcome of this DSB repair pathway.
4.5. Disruption of DNA Damage Response Signaling.
Arsenite exposure has been shown to impair DDR signaling, especially through dysregulation of protein PARylation, ubiquitination, and SUMOylation,148,149 as reviewed recently.72 DDR is a tightly regulated temporal- and spatial-sensitive chromatin-associated process important for sensing DNA damage, recruiting DNA repair machinery to damage sites, and intertwining DNA repair with other DNA-transacting activities.150 For example, H2AX phosphorylation, PARylation, and histone acetylation mediated by ATM, PARP1, and TIP60, respectively, are among the earliest events in DNA damage response; they are activated by DNA damage and involve early and rapid detection of DNA lesions and chromatin decompaction, thereby providing better accessibility for DNA repair machinery to DNA damage sites.150 Reversible ubiquitination and SUMOylation of DDR proteins are crucial for effective DDR signaling and repair of DNA DSBs,151 where zinc finger-containing ubiquitin ligases and SUMO-conjugating enzymes can be disrupted by arsenic exposure, with examples of RAD18, MORC2, RNF4, and RNF111 being briefly discussed below.72
During replication stress, the E3 ubiquitin ligase RAD18 induces monoubiquitination of PCNA, which is in turn recognized and bound by Spartan for its subsequent recruitment of Pol η (i.e., a TLS polymerase important for bypassing UV-induced DNA lesions).152–154 The monoubiquitinated PCNA also promotes efficient monoubiquitination and chromatin localization of FANCD2,155,156 and this ubiquitination is indispensable for recruiting SNM1A to DNA repair complexes assembled at MMC- and UV-induced DNA lesions to promote ICL repair.157
PARylation is also important in DDR. To achieve DNA damage-induced PARylation and PAR-dependent recruitment of DNA repair proteins to DNA damage sites, PARP1 recruits chromatin remodeling enzyme MORC2 to DNA damage sites and catalyzes PARylation of its CW-type zinc finger domain, thereby activating its ATPase and chromatin remodeling activities. Meanwhile, PARylated MORC2 stabilizes PARP1 through enhancing the NAT10-mediated acetylation of lysine 949 in PARP1, which is no longer subjected to CHFR ubiquitination and the subsequent proteasomal degradation.132,158 This illustrates that the crosstalk between different DNA repair enzymes is important for the dynamic PARylation in DDR.
SUMOylation and ubiquitination are also necessary for robust DDR. To favor DDR with coordinated SUMOylation and ubiquitination, the SUMO E3 ligases PIAS1 and PIAS4 are recruited to DSB sites and lead to accumulation of SUMO1/2/3 at DSB sites, which leads to the recruitment of RNF4 to DNA damage sites.159,160 RNF4 subsequently ubiquitinates and facilitates the degradation of polySUMOylated MDC1 and RPA, thus promoting efficient DSB repair.148,161–164
Similar to RNF4, RNF111 promotes nonproteolytic ubiquitination of SUMOylated XPC, which is in turn recruited to UV-damaged DNA.165 Given the extensive involvement of zinc finger-harboring ubiquitin and SUMO E3 ligases in DDR signaling (e.g., RAD18, RNF4), arsenite exposure may hamper ubiquitination or SUMOylation through direct binding or inducing oxidative modifications of cysteine residues in their zinc finger motifs, thereby disrupting DDR.
5. DISRUPTION OF CELL CYCLE CHECKPOINTS, PROMOTION OF CELL PROLIFERATION, AND SUPPRESSION OF APOPTOSIS
In arsenic-induced carcinogenesis, iAs has been shown to interfere with cell cycle regulation, promote cell proliferation, and suppress apoptosis, which indirectly inhibit DNA repair (i.e., by not providing enough time) and allow cells with DNA damage to propagate166–168
Cell cycle checkpoints, including DNA damage checkpoints at the G1/S and G2/M boundaries as well as in the S phase, tightly regulate cell cycle progression by accurately assessing mitogenic signals and properly repairing DNA damage while avoiding further propagation of damaged genomes through promoting apoptosis of the severely damaged cells.169–172 This regulation is executed by checkpoint proteins, which comprise cyclins, cell cycle-dependent kinases, and phosphatases.170,171
A recent study demonstrated that a 48-h exposure of acute promyelocytic leukemia (APL) cells to 2 μM iAs3+ increased the mRNA expression of several cell cycle-associated genes, including CCND1 (encodes for cyclin D1 protein), CCNE1 (cyclin E1 protein), and GADD45A, but reduced those of CCNF (cyclin F) and CDKN1A (p21), resulting in a transition of cell populations from G1/S phases to G2/M phases and arresting cell cycle progression. This result suggests that acute exposure to iAs3+ disturbs cell cycle checkpoints, leading to uncontrolled cell cycle progression and proliferation of APL cells.168 Notably, the DNA damage checkpoint at the G1/S boundary was bypassed by iAs3+-mediated alterations in expression of checkpoint proteins, especially cyclin D1.173–175
Another recent study demonstrated that a 1-month exposure of human BEAS-2B cells and keratinocytes to 0.5 μM arsenite delayed the transition from mitosis by compromising mitotic checkpoint through the attenuation of anaphase promoting complex-mediated cyclin B1 degradation.167 In this vein, long-term arsenite exposure up-regulates Polo-like kinase 1 via acting on Akt in the PI3K/Akt pathway, thereby potentiating mitotic catastrophe and genetic instability.167,176
Arsenite-elicited up-regulation of p53 protein expression and abrogation of p53-dependent increase in p21 expression together unleash the checkpoint restraints at the G1/S and G2/M boundaries as well as in the S phase.174,177,178 Chronic low-dose (e.g., 14 days, 0.1 μM) and acute noncytotoxic-level (e.g., 24 h, 1 μM) of arsenite exposure, as well as acute low-level (e.g., 24 h, 1 μM) of MMAIII exposure, were found to induce p53 protein expression in normal human fibroblast cells.70,174,177 In addition, arsenite-elicited up-regulation of Hdm2 and the ensuing ubiquitination of p53 promote nuclear export of p53, thereby disrupting its ability to transcriptionally activate its target genes, including p21 and NER genes.179–181 These may give rise to unimpeded cell cycle progression and accrual of mutations from unrepaired DNA lesions.178,181
Arsenite has been reported to promote the proliferation of human cells.33,68,182–187 Arsenite is thought to achieve this through stimulating pathways for cell proliferation and survival (e.g., Erk, EGFR, MAPK pathways) while inhibiting pathways involved in cell death (e.g., JNK signaling) via modulation of a myriad of transcription factors (e.g., AP-1 and NF-κB).187 Exposure to 5 μM arsenite was found to increase the proliferation of SH-SY5Y human neuroblastoma cells via activation of ERK in VEGF signaling, which might favor tumor progression.184 Arsenite-induced cell proliferation was shown to arise from elevated levels of epidermal growth factor receptor (EGFR) ligand, heparin-binding EGF, and its subsequent activation of EGFR phosphorylation that induces pERK and cyclin D1 expression in human cells.188 Arsenite-elicited ERK signaling is required for arsenic-induced transactivation of NF-κB,189 which might be mediated by arsenic-stimulated oxidative stress.190–192 Additionally, low-dose arsenite treatment has been documented to activate ERK, as well as transcription factors E2F1 and Activating Protein 1 (AP-1), enhance the DNA binding activities of AP-1 and NF-κB, and elevate the expression of a number of positive cell growth-related genes including FOS, JUN, MYC, and EGR-1.185,190,193–197
The major cell growth and ROS-mediated pathways are regulated by protein tyrosine phosphorylation, which itself is controlled by tyrosine kinases and protein tyrosine phosphatases. Therefore, arsenite exposure is believed to inactivate protein tyrosine phosphatases by ROS/RNS-induced modifications of redox-sensitive cysteines at their active sites, thereby augmenting the total cellular tyrosine phosphorylation.34,198–203 Combined, arsenite and arsenite-induced ROS/RNS can stimulate a phosphorylated state of EGFR, and activate ERK, transcription factor AP-1 complex, and its downstream target genes JUN, FOS, and MYC, thereby increasing cyclin D1 expression.190,204 Together with arsenite-activated E2F transcription factors and their modulation of cyclin E levels, arsenite exposure elicits unchecked cell cycle progression and uncontrolled cell proliferation.194,204–206
Chronic exposure to arsenic has been shown to increase cell survival and elevate levels of DNA damage in cultured human cells.42 PARP1 inhibition by low concentrations of arsenic has been proposed to enhance the survival of cells with unrepaired DNA lesions including a population of “initiated carcinogenic cells” that represents the first step of the multistage carcinogenesis process.18 Chronic arsenic exposure was also shown to decrease p53 at the post-translational level via arsenic-induced PARylation as well as the mRNA expression level of Bax.42 This arsenite-elicited inhibition of apoptotic mediators was also found to impair the XPC-mediated global-genome NER, resulting in mutation accrual and neoplastic transformation of DNA damage-containing cells.42
Together, arsenite-induced positive cell proliferation and suppression of apoptosis confer insufficient time for efficient DNA repair before replication of damaged DNA and/or allow cells with damaged DNA to propagate, which may give rise to mutations and genome instability.
6. EPIGENETIC DYSREGULATION ASSOCIATED WITH ARSENIC-INDUCED CARCINOGENESIS
Arsenic-elicited carcinogenesis is believed to stem, in part, from its disruption of epigenetic signaling by alterations of histone PTMs and DNA methylation patterns (Figure 5). Histone PTMs and DNA methylation tightly regulate the chromatin dynamics to modulate the inheritable expression patterns of different genes.207 Therefore, arsenic can induce carcinogenesis by epigenetic silencing of tumor suppressor genes or activation of oncogenes. Here, we review the current evidence about the role of arsenic exposure in modulating the epigenetic pathway of gene regulation.
Figure 5.

Major events through which inorganic arsenite and its trivalent metabolites disrupt epigenetic integrity via inhibition of epigenetic regulators and chromatin modifiers. As3+, MMAIII, and DMAIII can impair the enzymatic activities of DNA epigenetic regulators (e.g., DNMTs, Tet, and CTCF) and chromatin-modifying enzymes (e.g., hMOF, TIP60, and PARP1), which subsequently perturb DNA methylation and histone PTMs, respectively, thereby disrupting epigenetic integrity.
6.1. Alterations of Histone PTMs.
In the nucleus, DNA is packaged into chromatin, where the nucleosome core consists of stretches of DNA (~147 bp) wrapping around a histone octamer consisting of two copies each of core histones H2A, H2B, H3, and H4.208 Hence, nucleosomes form linear 11 nm beads-on-a-string structures that further compact into 30 nm fibers and other higher-order chromatin states.209 The N-terminal histone tails extending from nucleosomes are subjected to a range of PTMs, including methylation, acetylation, phosphorylation, ubiquitination, SUMOylation, ADP ribosylation, deimination, and proline isomerization,210 which in turn modify the chromatin compaction and recruitment of nonhistone proteins, including gene regulatory factors and DNA repair enzymes, to chromatin. The formation of open, relaxed chromatin conformation is required for DNA repair machinery to gain access to the spatially confined region surrounding DNA damage sites, as described in the “access-repair-restore” model.211,212
Inorganic arsenic and its metabolites have been documented to disrupt histone PTMs, including but not limited to H2AX phosphorylation, H2AX ubiquitination, H2B ubiquitination, H3 methylation, and H4K16 acetylation,110,120,121,213,214 which were reviewed elsewhere.207,215,216 Here, we focus on the effect of arsenic exposure on those histone PTMs that are closely associated with DNA repair and genomic stability.
A number of previous in vitro studies have demonstrated that arsenic exposure elicits alterations in a variety of global histone PTMs, which include loss of H4K16Ac, H3K27me3, and ubiquitination of H2B, as well as gain of H3K4me2, H3K4me3, H3K9me2, H3K9Ac, H3K14Ac, and phosphorylation of H3S10 and H2AX (γH2AX).118,217–222 For example, the PBMC from the participants of the folic acid and creatine supplementation trial (FACT) study exposed to 50–500 μg/L arsenite in drinking water exhibited a decrease in H3K9me3 and H3K9ac, and a gain in H3K9me2.217,220 In addition, A549 human lung carcinoma cells displayed a global loss of H3K4me1 and a global gain of H3K4me2 and H3K4me3 following a 24-h exposure to 1 μM arsenite, where H3K4me3 remained elevated even at 1 week after arsenite withdrawal.219 In another study, a 24-h exposure of A549 cells to arsenite led to elevated levels of gene-silencing marks, H3K9me2 and H3K27me3, while also augmenting the global level of H3K4me3, a gene-activating mark.218
In contrast to transcriptional activators, elevated levels of H3K9me2 mediated by increased mRNA and protein levels of histone methyltransferase G9a are correlated with transcriptional repression,218,223 which has been shown to be involved in the silencing of tumor suppressor genes in cultured cancer cells.224,225 H3K27me3 is frequently accompanied by inactive promoters and gene silencing, and it labels chromatin by polycomb repressive complex 1 (PRC1) via H2AK119 ubiquitination to facilitate chromatin compaction.226 Exposure of human cells to 0.5 μM arsenic trioxide (ATO) induces the expression of components PRC2 protein complex, consisting of SUZ12, EZH2, and BMI1, resulting in elevated H3K27me3 and the ensuing diminished expression of tumor suppressors p16INK4a and p14ARF.227 Arsenite-induced H3K27me3 in chromatin silences tumor suppressor genes, for example, HOXB7 and CDKN2A, which are involved in DNA repair.228–230 Therefore, histone H3 PTMs are among the targets of arsenic exposure and aid in the subsequent disruption of DNA repair.
As briefly discussed above, histone H2BK120ub and histone H4K16Ac also play a significant role in the generation of relaxed chromatin environment that is conducive for the access of DNA repair enzymes. Histone H2BK120ub is crucial for decompacting the 30 nm chromatin fiber,231 while H4K16Ac also decondenses chromatin,232 thereby facilitating DSB repair by increasing the accessibility of chromatin to DNA repair machinery.233–235 H2BK120ub is mediated by the E3 ubiquitin ligase composed of the RNF20-RNF40 heterodimer,233 whereas H4K16Ac is modulated by both hMOF and TIP60 MYST-family of histone acetyltransferases.236 In UROtsa human bladder epithelial cells, global H4K16Ac levels were reduced in a dose- and time-dependent manner upon exposure to As3+ and MMAIII.118 Arsenite exposure was also documented to diminish H2BK120ub and H4K16Ac by inhibiting the above-mentioned zinc finger-containing proteins.110,120,121 Moreover, the PARylation of lysine residues of the core histone tails mediated by PARP1, including H2AK13, H2BK30, H3K27, H3K37, and H4K16, can result in a rapid decondensation of chromatin around DNA damage sites and facilitate DNA repair.133,134,237 Therefore, arsenic exposure could result in a compact chromatin structure by interfering with these histone-modifying enzymes and by diminishing the chromatin-decompacting histone PTMs, limiting the access of DNA repair proteins to chromatin.
Finally, phosphorylation of H2AX, which is mediated by ATM and DNA-PKcs following DNA damage,238 contributes to the initiation of DNA damage response.214,239 Phosphorylation of H2AX at different sites triggers distinct downstream cellular processes. For instance, phosphorylation of Tyr142 in H2AX stimulates XPD-dependent apoptosis and enhances DDR.240 Recently, it was reported that a 24-h exposure to 4 μM ATO significantly stimulated levels of phosphorylated H2AX in mouse embryonic fibroblasts (MEFs), possibly via inhibition of de novo dTMP biosynthesis through inducing SUMOylation, ubiquitination, and subsequent degradation of MTHFD1.213 Since iAs has been documented to elicit ATR and DNA-PKcs in vivo and in vitro,241–243 this increase in H2AX phosphorylation is thought to originate from the activation of ATR and DNA-PKcs and might also be modulated by TOPK.244
Studies have suggested that arsenic-induced phosphorylation of histone H3 might be responsible for the up-regulation of caspase 10, a proto-apoptotic factor,222 and the proto-oncogenes FOS and JUN,245 which can lead to transformation of human fibroblast cells and the induction of tumors in animals.246 Additionally, since arsenite could induce FOS and JUN via activation of JNKs and p38/MAPK2 kinases, and promote H3S10 phosphorylation,221,247 therefore, the impact of the arsenic-induced H3 phosphorylation on DNA repair may be regulated by JNK/MAPK pathway, which was recently shown to be linked to DNA damage response.248
6.2. DNA Methylation.
DNA methylation constitutes another important epigenetic mechanism of gene regulation. Depending on the type of regulatory elements where the methylation occurs, the effect of DNA methylation on gene expression varies. Under normal circumstances, DNA methylation events in the promoter and gene body are associated with gene repression and activation, respectively.249 Alterations in DNA methylation are known to play roles in carcinogenesis partly through inactivation of tumor suppressor genes or activation of oncogenes.250
Several potential mechanisms have been proposed to account for the arsenite-induced alterations in DNA methylation including SAM deficiency, diminished expression of DNMT genes, inhibition of Tet proteins, and aberrant occupancy of CTCF binding on promoters of DNMT and TET genes (Figure 6). As noted above, biotransformation of inorganic arsenic depletes SAM, which is also utilized in DNA methylation catalyzed by DNA cytosine-5-methyltransferases (DNMTs).251 In addition, arsenic exposure was found to repress, in a dose-dependent manner, the mRNA expression levels and activities of DNA methyltransferases DNMT1, DNMT3A, and DNMT3B, thereby resulting in a loss of global DNA methylation.251–253 Interestingly, arsenic exposure was shown to reduce CTCF expression and inhibit the binding of CTCF to DNA, which diminishes the occupancy of CTCF in the promoters of DNMT1, DNMT3A, and DNMT3B genes; this may explain the observation of arsenite-induced diminished expressions of DNMT genes.254
Figure 6.

Arsenite disrupts DNA methylation. Methylation events in promoters repress gene expression, whereas those in gene bodies activate gene expression. Metabolism of iAs induces SAM deficiency, which results in global DNA hypomethylation. iAs exposure leads to decreased expressions of DNMT1, DNMT3A, DNMT3B, thus diminishing global DNA methylation. Additionally, iAs selectively inhibits the binding of CTCF to promoters of genes (e.g., DNMTs), leading to repression of tumor suppressor genes. Meanwhile, iAs inhibits Tet proteins, thus reducing the level of 5-hmC, which can be inhibited by weakened occupancy of CTCF in the promoters of TET genes. Combined together, iAs can repress tumor suppressors and activate proto-oncogenes, thereby impairing DNA repair and genome integrity.
Arsenic exposure has been documented to result in global DNA hypomethylation,175,255–257 which is a hallmark of various human cancers.258–260 It was reported that chronic exposure of cultured rat liver cells to a low dose (0.5 μM) of arsenite resulted in global DNA hypomethylation.257 DNA hypomethylation was also observed in leukocytes of human populations who were exposed to arsenic and developed skin cancers.261 Chronic exposure of mice to 45 ppm arsenite for 48 weeks induced hepatic global DNA hypomethylation as well as promoter hypomethylation of the ESR1 gene, which encodes for estrogen receptor α.175 Promoter hypomethylation is believed to stimulate the expression of ESR1 gene, which in turn can induce cell cycle-dependent DSBs and contribute to initiation of breast cancer.262,263 Arsenic-induced promoter hypomethylation of the ESR1 gene is consistent with the observation of frequent mutations of DDR and DNA repair proteins in estrogen-dependent breast cancers, suggesting that ER signaling converges to inhibit effective DNA repair and apoptosis, thereby favoring proliferation.175,263
Although arsenic exposure causes global DNA hypomethylation, it also leads to promoter hypermethylation and repression of specific tumor suppressor genes. For instance, the DNA repair gene MLH1 displays significant promoter hypermethylation in whole blood obtained from humans chronically exposed to arsenic.264 Additionally, significant promoter hypermethylation of NER genes (ERCC2, RPA1, POLD3, POLE2) was observed in human hepatocytes exposed to 0.2 μM ATO for 3 months.265 Recently, hypermethylation of NER genes ERCC1 and ERCC2, and suppression of their expression in human cells, was also correlated with chronic arsenic exposure.266
5-Methylcytosine (5-mC) in DNA can be oxidized by the ten-eleven translocation (Tet) family enzymes to 5-hydroxymethylcytosine (5-hmC), which may convey regulatory epigenetic functions by binding to specific proteins to confer active gene transcription.267–270 In addition, diminished levels of 5-hmC in DNA are hallmarks of human cancers.268 Tet enzymes were found to prevent DNA damage-induced chromosome mis-segregation, indicating that 5-hmC was pivotal in promoting DNA repair and maintenance of genome integrity.271,272 Arsenite was shown to bind directly with the zinc finger motifs of Tet proteins and inhibit the Tet-mediated oxidation of 5-mC to 5-hmC in HEK293T cells with ectopic expression of Tet proteins and in mouse embryonic stem cells following an acute 24-h exposure to 2–5 μM arsenite.267 On the other hand, global hyper-hydroxymethylation of cytosine was observed in arsenite-transformed BEAS-2B cells (after a 8-week exposure to 0.5 μM arsenite),273 which was accompanied by elevated expression of Tet enzymes and was attributed to arsenic-elicited selective inhibition of CTCF binding on the proximal, weaker CTCF binding sites in the promoters of TET genes.273 The differences in these observations are likely attributed to the uses of different cell lines and exposure conditions, where long-term exposure to arsenite may induce adaptive response in cells. Future studies in laboratory animals and human subjects are needed to determine how arsenic exposure affects the levels of 5-hmC in vivo.
Combined together, chromatin compaction around damaged DNA and disturbed methylation pattern in DNA upon arsenite exposure perturb the sophisticated epigenetic network of DNA repair machinery, which may compromise genome stability and result in arsenic-elicited carcinogenesis.
7. CONCLUSIONS AND PERSPECTIVES
In this review, distinct modes of action for arsenic-induced impairment of DNA repair pathways are extensively discussed. Unlike conventional carcinogens (e.g., alkylating agents, UV light), which can damage DNA directly, arsenic’s role in carcinogenesis perhaps resides on its ability to compromise the repair of DNA lesions induced by DNA damaging agents that are formed from endogenous metabolism or arise from other environmental exposure. Multiple mechanisms are likely at play, and they encompass diminished expression of DNA repair genes, functional disruption of DNA repair proteins, induction of a chromatin environment that is not conducive for DNA repair, and aberrant cell cycle regulation. Hence, chronic arsenic exposure can result in a progressive decline in DNA repair capacity, which may represent a crucial pathway through which arsenic contributes to carcinogenesis. In addition, elevated production of ROS/RNS from arsenic exposure and ensuing accumulation of DNA lesions from these species may also contribute, in part, to arsenic carcinogenesis.
The findings cited in this review also suggest that zinc finger proteins, which are encoded by genes constituting approximately 10% of the human genome and play significant roles in DNA repair as well as epigenetic regulation, constitute important molecular targets for arsenic binding (Figure 7).274 iAs exposure induces ROS/RNS, which can target the redox-active nucleophilic sulfhydryl group on cysteine residues located within the zinc finger motifs of these proteins. The resulting modification products, especially S-nitrosylation, disturb the native Zn2+ coordination sphere of the zinc finger proteins and alter their structure and functions.49,50,275–279 Thus, direct iAs3+ binding, perhaps in conjunction with oxidation and nitrosylation of cysteine sulfhydryl groups of zinc finger motifs of proteins involved in DNA repair and epigenetic regulation of gene expression, may represent important molecular mechanisms underlying the modes of action for the exacerbated DNA repair capacity in arsenic-induced carcinogenesis.
Figure 7.

Arsenite and iAs-induced oxidative stress enhance DNA damage through disrupting the functions of zinc finger proteins. iAs-induced oxidative stress and As3+ itself can disrupt the zinc finger-containing epigenetic regulators and DNA repair enzymes, thereby impairing DNA repair. Simultaneously, oxidative stress generates oxidative DNA damage. These together may result in tumorigenesis.
It is important to note that, while substantial progress has been made in this area of research, much remains to be done to further illustrate the above-mentioned mechanisms. First, most of the previously published studies were conducted with the use of cell-based systems. It will be important to examine whether the findings made from the cell-based assays can be extended to laboratory animals and human subjects exposed to environmentally relevant levels of arsenic species.
Second, RING finger proteins make up a large family of E3 ubiquitin ligases,280 and it will be important to systematically investigate, at the proteome-wide scale, how exposure to arsenite modulates the ubiquitination of proteins, especially those that are involved in DNA repair and DNA damage response signaling.
Third, the C3H-type zinc-finger proteins represent the second largest group of RNA-binding proteins in mammals.281 It remains unclear how arsenic exposure affects the functions of these RNA-binding proteins and influences the metabolisms of RNA (i.e., stability, translation efficiency, and alternative splicing of RNA), especially those mRNA species that encode DNA repair proteins and epigenetic modifiers or those noncoding RNAs that modulate chromatin structure.
Lastly, if diminished DNA repair and elevated oxidative DNA damage constitute the major mechanisms of arsenic carcinogenesis, we would expect to observe elevated rates of mutations in arsenic-exposed laboratory animals and human subjects. In this vein, Hei et al.282 demonstrated, by employing AL cell assay, that arsenite could induce, in a dose-dependent manner, mutations (mostly large deletions) in mammalian cells. They also observed that the mutagenicity of arsenite could be diminished markedly by cotreating cells with a radical scavenger, dimethyl sulfoxide.282 It will be important to unveil the degree to which arsenic exposure leads to mutagenesis. Recent advances in next-generation sequencing, especially exome sequencing,283 render it possible to unveil how arsenic exposure leads to mutagenesis in laboratory animals or human subjects.
ACKNOWLEDGMENTS
The authors would like to thank the National Institutes of Health for supporting this research (R01 ES029749). N.P. was supported in part by an NRSA T32 Institutional Training Grant (T32 ES018827).
Biographies
Biographies
Lok Ming Tam received his B.S. degree in Cell and Molecular Biology from Chinese University of Hong Kong (Hong Kong SAR) in 2014. After participating in an exchange program at the University of California Davis (2012–2013), he became fascinated with environmental toxicology. He subsequently joined the graduate program in Environmental Toxicology at the University of California Riverside. He worked under the guidance of Prof. Yinsheng Wang and earned his Ph.D. degree in late 2019. His research interests include protein quality control and etiology of human diseases, with a special focus on the arsenic impairment of protein quality control machinery.
Nathan Price obtained his Ph.D. degree in Chemical Biology from the University of Missouri Columbia in 2014, where he worked with Prof. Kent S. Gates on chemistry of abasic site-derived interstrand cross-link lesions. He subsequently joined the laboratory of Yinsheng Wang at the University of California Riverside, and his main research projects involve the development and application of shuttle vector methods for studying how structurally defined DNA lesions affect DNA replication in human cells. He also developed a strong interest in scientific and medical writing.
Yinsheng Wang is currently a Distinguished Professor in the Chemistry Department and the Environmental Toxicology graduate program at the University of California Riverside. His research interest encompasses DNA damage and mutagenesis, proteomics, and epigenetics. In recent years, he also developed a strong interest in understanding the molecular mechanisms of arsenic toxicity and carcinogenicity. His group members employ a multidisciplinary approach, including mass spectrometry-based bioanalytical chemistry, synthetic organic chemistry, molecular biology, genetics, and genomics, to tackle their research projects.
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.chemrestox.9b00464
The authors declare no competing financial interest.
Contributor Information
Lok Ming Tam, Environmental Toxicology Graduate Program, University of California, Riverside, California 92521, United States.
Nathan E. Price, Department of Chemistry, University of California, Riverside, California 92521, United States
Yinsheng Wang, Environmental Toxicology Graduate Program and Department of Chemistry, University of California, Riverside, California 92521, United States.
REFERENCES
- (1).International Agency for Research on Cancer (IARC). (2012) Arsenic, Metals, Fibres, and Dusts, Vol. 100C, The International Agency for Research on Cancer; https://www.iarc.fr/. [Google Scholar]
- (2).Ravenscroft P, Brammer H, and Richards K (2009) Arsenic Pollution: A Global Synthesis, Wiley-Blackwell, UK. [Google Scholar]
- (3).Kemper K, and Minnatullah K (2005) Towards a More Effective Operational Response. World Bank; I, 1–68. [Google Scholar]
- (4).Hossain MF (2006) Arsenic contamination in Bangladesh - An overview. Agric., Ecosyst. Environ 113, 1–16. [Google Scholar]
- (5).Chung JY, Yu SD, and Hong YS (2014) Environmental source of arsenic exposure. J. Prev. Med. Public Heal 47, 253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Nicomel NR, Leus K, Folens K, Van Der Voort P, and Du Laing G (2016) Technologies for arsenic removal from water: Current status and future perspectives. Int. J. Environ. Res. Public Health 13, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Wang S, and Mulligan CN (2006) Occurrence of arsenic contamination in Canada: Sources, behavior and distribution. Sci. Total Environ 366, 701–721. [DOI] [PubMed] [Google Scholar]
- (8).US Environmental Protection Agency. (1988) Special Report on Ingested Inorganic Arsenic: Skin Cancer; Nutritional Essentiality EPA/625/3–87/-13, EPA. [Google Scholar]
- (9).Rossman TG, Uddin AN, and Burns FJ (2004) Evidence that arsenite acts as a cocarcinogen in skin cancer. Toxicol. Appl Pharmacol 198, 394–404. [DOI] [PubMed] [Google Scholar]
- (10).Tseng WP, Chu HM, How SW, Fong JM, Lin CS, and Yeh S (1968) Prevalence of skin cancer in an endemic area of chronic arsenicism in taiwan. J. Natl. Cancer Inst 40, 453–463. [PubMed] [Google Scholar]
- (11).Smith AH, Goycolea M, Haque R, and Biggs M (1998) Lou. Marked Increase in Bladder and Lung Cancer Mortality in a Region of Northern Chile due to Arsenic in Drinking Water. Am. J. Epidemiol 147, 660–669. [DOI] [PubMed] [Google Scholar]
- (12).Marshall G, Ferreccio C, Yuan Y, Bates MN, Steinmaus C , Selvin S, Liaw J, and Smith AH (2007) Fifty-Year study of lung and bladder cancer mortality in Chile related to arsenic in drinking water. J. Natl. Cancer Inst 99, 920–928. [DOI] [PubMed] [Google Scholar]
- (13).Ferreccio C, González C, Milosavjlevic V, Marshall G, Sancha AM, and Smith AH (2000) Lung cancer and arsenic concentrations in drinking water in Chile. Epidemiology 11 , 673–679. [DOI] [PubMed] [Google Scholar]
- (14).Hopenhayn-Rich C, Biggs ML, Fuchs A, Bergoglio R, Tello EE, Nicolli H, and Smith AH (1996) Bladder Cancer Mortality Associated with Arsenic in Drinking Water in Argentina. Epidemiology 7, 117–124. [DOI] [PubMed] [Google Scholar]
- (15).Hopenhayn-Rich C, Biggs ML, and Smith AH (1998) Lung and kidney cancer mortality associated with arsenic in drinking water in Cordoba, Argentina. Int. J. Epidemiol 27, 561–569. [DOI] [PubMed] [Google Scholar]
- (16).Cebrian ME, Albores A, Aguilar M, and Blakely E (1983) Chronic arsenic poisoning in the North of Mexico. Hum. Toxicol 2, 121–133. [DOI] [PubMed] [Google Scholar]
- (17).Basu AK (2018) DNA damage, mutagenesis and cancer. Int. J. Mol. Sci 19, 970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Weston A, and Harris CC (2003) Multistage Carcinogenesis. Holland-Frei Cancer Medicine (Kufe DW, Pollock RE, and Weichselbaum RR, Eds.) 6th ed., BC Decker, Hamilton (ON) https://www.ncbi.nlm.nih.gov/books/NBK13982/. [Google Scholar]
- (19).De Bont R (2004) Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis 19, 169–185. [DOI] [PubMed] [Google Scholar]
- (20).Marnett LJ, and Burcham PC (1993) Endogenous DNA Adducts: Potential and Paradox. Chem. Res. Toxicol 6, 771–785. [DOI] [PubMed] [Google Scholar]
- (21).Hughes MF (2002) Arsenic toxicity and potential mechanisms of action. Toxicol. Lett 133, 1–16. [DOI] [PubMed] [Google Scholar]
- (22).Vahter M, and Norin H (1980) Metabolism of 74As-labeled trivalent and pentavalent inorganic arsenic in mice. Environ. Res 21 , 446–457. [DOI] [PubMed] [Google Scholar]
- (23).Drobna Z, Styblo M, and Thomas DJ (2009) An Overview of Arsenic Metabolism and Toxicity. Curr. Protoc. Toxicol 42, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Rogers EH, Chernoff N, and Kavlock RJ (1981) The teratogenic potential of cacodylic acid in the rat and mouse. Drug Chem. Toxicol 4, 49–61. [DOI] [PubMed] [Google Scholar]
- (25).Murai T, Iwata H, Otoshi T, Endo G, Horiguchi S, and Fukushima S (1993) Renal lesions induced in F344/DuCrj rats by 4-weeks oral administration of dimethylarsinic acid. Toxicol. Lett 66, 53–61. [DOI] [PubMed] [Google Scholar]
- (26).Yamamoto S, Konishi Y, Matsuda T, Murai T, Shibata M, Matsui-Yuasa I, Otani S, Kuroda K, Endo G, and Fukushima S (1995) Cancer Induction by an Organic Arsenic Compound, Dimethylarsinic Acid (Cacodylic Acid), in F344/DuCrj Rats after Pretreatment with Five Carcinogens. Cancer Res. 55, 1271–1276. [PubMed] [Google Scholar]
- (27).Wei M, Wanibuchi H, Morimura K, Iwai S, Yoshida K, Endo G, Nakae D, and Fukushima S (2002) Carcinogenicity of dimethylarsinic acid in male F344 rats and genetic alterations in induced urinary bladder tumors. Carcinogenesis 23, 1387–1397. [DOI] [PubMed] [Google Scholar]
- (28).Trouba KJ, Wauson EM, and Vorce RL (2000) Sodium arsenite-induced dysregulation of proteins involved in proliferative signaling. Toxicol. Appl. Pharmacol 164, 161–170. [DOI] [PubMed] [Google Scholar]
- (29).Cholanians AB, Phan AV, Ditzel EJ, Camenisch TD, Lau SS, and Monks TJ (2016) Arsenic induces accumulation of α-synuclein: Implications for synucleinopathies and neurodegeneration. Toxicol. Sci 153, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Liu S, Guo X, Wu B, Yu H, Zhang X, and Li M (2015) Arsenic induces diabetic effects through beta-cell dysfunction and increased gluconeogenesis in mice. Sci. Rep 4, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Yamanaka K, Hoshino M, Okamoto M, Sawamura R, Hasegawa A, and Okada S (1990) Induction of DNA Damage by Dimethylarsine, A Metabolite of Inorganic Arsenics, is for the Major Part Likly Due to Its Peroxyl Radical. Biochem. Biophys. Res. Commun 168, 58–64. [DOI] [PubMed] [Google Scholar]
- (32).Liu SX, Athar M, Lippai I, Waldren C, and Hei TK (2001) Induction of oxyradicals by arsenic: Implication for mechanism of genotoxicity. Proc. Natl. Acad. Sci. U. S. A 98, 1643–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Ruiz-Ramos R, Lopez-Carrillo L, Rios-Perez AD, De Vizcaya-Ruíz A, and Cebrian ME (2009) Sodium arsenite induces ROS generation, DNA oxidative damage, HO-1 and c-Myc proteins, NF-κB activation and cell proliferation in human breast cancer MCF-7 cells. Mutat. Res., Genet. Toxicol. Environ. Mutagen 674, 109–115. [DOI] [PubMed] [Google Scholar]
- (34).Flora SJS (2011) Arsenic-induced oxidative stress and its reversibility. Free Radical Biol. Med 51, 257–281. [DOI] [PubMed] [Google Scholar]
- (35).Muller FL, Liu Y, and Van Remmen H (2004) Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem 279, 49064–49073. [DOI] [PubMed] [Google Scholar]
- (36).Liu SX, Davidson MM, Tang X, Walker WF, Athar M, Ivanov V, and Hei TK (2005) Mitochondrial damage mediates genotoxicity of arsenic in mammalian cells. Cancer Res. 65, 3236–3242. [DOI] [PubMed] [Google Scholar]
- (37).Gadelha FR, Thomson L, Fagian MM, Costa ADT, Radi R, and Vercesi AE (1997) Ca2+-independent permeabilization of the inner mitochondrial membrane by peroxynitrite is mediated by membrane protein thiol cross-linking and lipid peroxidation. Arch. Biochem. Biophys 345, 243–250. [DOI] [PubMed] [Google Scholar]
- (38).Nordenson I, and Beckman L (1991) Is the genotoxic effect of arsenic mediated by oxygen free radicals? Hum. Hered 41, 71–73. [DOI] [PubMed] [Google Scholar]
- (39).Hei TK, Liu SUX, and Waldren C (1998) Mutagenicity of arsenic in mammalian cells: Role of reactive oxygen species. Proc. Natl. Acad. Sci. U. S. A 95, 8103–8107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Shi H, Shi X, and Liu KJ (2004) Oxidative mechanism of arsenic toxicity and carcinogenesis. Mol. Cell. Biochem 255, 67–78. [DOI] [PubMed] [Google Scholar]
- (41).Kligerman AD, Doerr CL, Tennant AH, Harrington-Brock K, Allen JW, Winkfield E, Poorman-Allen P, Kundu B, Funasaka K, Roop BC, Mass MJ, and DeMarini DM (2003) Methylated Trivalent Arsenicals as Candidate Ultimate Genotoxic Forms of Arsenic: Induction of Chromosomal Mutations but Not Gene Mutations. Environ. Mol. Mutagen 42, 192–205. [DOI] [PubMed] [Google Scholar]
- (42).Singh KP, Kumari R, Treas J, and Dumond JW (2011) Chronic exposure to arsenic causes increased cell survival, DNA damage, and increased expression of mitochondrial transcription factor A (mtTFA) in human prostate epithelial cells. Chem. Res. Toxicol 24, 340–349. [DOI] [PubMed] [Google Scholar]
- (43).Huang C, Ke Q, Costa M, and Shi X (2004) Molecular mechanisms of arsenic carcinogenesis. Mol. Cell. Biochem 255, 57–66. [DOI] [PubMed] [Google Scholar]
- (44).Bau DT, Wang TS, Chung CH, Wang ASS, and Jan KY (2002) Oxidative DNA adducts and DNA-protein cross-links are the major DNA lesions induced by arsenite. Environ. Health Perspect 110, 753–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Helleday T, Nilsson R, and Jenssen D (2000) Arsenic[III] and heavy metal ions induce intrachromosomal homologous recombination in the hprt gene of V79 Chinese hamster cells. Environ. Mol. Mutagen 35, 114–122. [DOI] [PubMed] [Google Scholar]
- (46).Coluzzi E, Colamartino M, Cozzi R, Leone S, Meneghini C , O’Callaghan N, and Sgura A (2014) Oxidative stress induces persistent telomeric DNA damage responsible for nuclear morphology change in mammalian cells. PLoS One 9, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Liu L, Trimarchi JR, Navarro P, Blasco MA, and Keefe DL (2003) Oxidative stress contributes to arsenic-induced telomere attrition, chromosome instability, and apoptosis. J. Biol. Chem 278, 31998–32004. [DOI] [PubMed] [Google Scholar]
- (48).Klein CB, Leszczynska J, Hickey C, and Rossman TG (2007) Further evidence against a direct genotoxic mode of action for arsenic-induced cancer. Toxicol. Appl. Pharmacol 222, 289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Zhou X, Ding X, Shen J, Yang D, Hudson LG, and Liu KJ (2019) Peroxynitrite contributes to arsenic-induced PARP-1 inhibition through ROS/RNS generation. Toxicol. Appl. Pharmacol 378, 114602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Zhou X, Cooper KL, Huestis J, Xu H, Burchiel SW, Hudson LG, and Liu KJ (2016) S-nitrosation on zinc finger motif of PARP-1 as a mechanism of DNA repair inhibition by arsenite. Oncotarget 7, 80482–80492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Jha AN, Noditi M, Nilsson R, and Natarajan AT (1992) Genotoxic effects of sodium arsenite on human cells. Mutat. Res. Fundam. Mol. Mech. Mutagen 284, 215–221. [DOI] [PubMed] [Google Scholar]
- (52).Lee-Chen SF, Gurr JR, Lin IB, and Jan KY (1993) Arsenite enhances DNA double-strand breaks and cell killing of methyl methanesulfonate-treated cells by inhibiting the excision of alkali-labile sites. Mutat. Res. DNA Repair 294, 21–28. [DOI] [PubMed] [Google Scholar]
- (53).Wiencke JK, and Yager JW (1992) Specificity of arsenite in potentiating cytogenetic damage induced by the DNA crosslinking agent diepoxybutane. Environ. Mol. Mutagen 19, 195–200. [DOI] [PubMed] [Google Scholar]
- (54).Hang B (2007) Base excision repair. DNA Repair Genet. Instab. Cancer, 23–64. [Google Scholar]
- (55).Cooke MS, Evans MD, Dizdaroglu M, and Lunec J (2003) Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 17, 1195–1214. [DOI] [PubMed] [Google Scholar]
- (56).Ziech D, Franco R, Georgakilas AG, Georgakila S, Malamou-Mitsi V, Schoneveld O, Pappa A, and Panayiotidis MI (2010) The role of reactive oxygen species and oxidative stress in environmental carcinogenesis and biomarker development. Chem.- Biol. Interact 188, 334–339. [DOI] [PubMed] [Google Scholar]
- (57).Grollman AP, and Moriya M (1993) Mutagenesis by 8-oxoguanine: an enemy within. Trends Genet. 9, 246–249. [DOI] [PubMed] [Google Scholar]
- (58).Shockley AH, Doo DW, Rodriguez GP, and Crouse GF (2013) Oxidative damage and mutagenesis in Saccharomyces cerevisiae: Genetic studies of pathways affecting replication fidelity of 8-oxoguanine. Genetics 195, 359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Ba X, and Boldogh I (2018) 8-Oxoguanine DNA glycosylase 1: Beyond repair of the oxidatively modified base lesions. Redox Biol. 14, 669–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Ebert F, Weiss A, Bültemeyer M, Hamann I, Hartwig A, and Schwerdtle T (2011) Arsenicals affect base excision repair by several mechanisms. Mutat. Res. Fundam. Mol. Mech. Mutagen 715, 32–41. [DOI] [PubMed] [Google Scholar]
- (61).Sykora P, and Snow ET (2008) Modulation of DNA polymerase beta-dependent base excision repair in cultured human cells after low dose exposure to arsenite. Toxicol. Appl. Pharmacol 228, 385–394. [DOI] [PubMed] [Google Scholar]
- (62).Idriss HT, Al-Assar O, and Wilson SH (2002) DNA polymerase β. Int. J. Biochem. Cell Biol 34, 321–324. [DOI] [PubMed] [Google Scholar]
- (63).Osmond MJ, Kunz BA, and Snow ET (2010) Age and exposure to arsenic alter base excision repair transcript levels in mice. Mutagenesis 25, 517–522. [DOI] [PubMed] [Google Scholar]
- (64).Scharer OD (2013) Nucleotide Excision Repair in Eukaryotes. Cold Spring Harbor Perspect. Biol 5, No. a012609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Andrew AS, Karagas MR, and Hamilton JW (2003) Decreased DNA repair gene expression among individuals exposed to arsenic in United States drinking water. Int. J. Cancer 104, 263–268. [DOI] [PubMed] [Google Scholar]
- (66).Andrew AS, Burgess JL, Meza MM, Demidenko E, Waugh MG, Hamilton JW, and Karagas MR (2006) Arsenic exposure is associated with decreased DNA repair in vitro and in individuals exposed to drinking water arsenic. Environ. Health Perspect 114, 1193–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Mo J, Xia Y, Wade TJ, DeMarini DM, Davidson M, and Mumford J (2011) Altered gene expression by low-dose arsenic exposure in humans and cultured cardiomyocytes: Assessment by real-time PCR arrays. Int. J. Environ. Res. Public Health 8, 2090–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Hamadeh HK, Trouba KJ, Amin RP, Afshari CA, and Germolec D (2002) Coordination of altered DNA repair and damage pathways in arsenite-exposed keratinocytes. Toxicol. Sci 69, 306–316. [DOI] [PubMed] [Google Scholar]
- (69).Andrew AS, Warren AJ, Barchowsky A, Temple KA, Klei L, Soucy NV, O’Hara KA, and Hamilton JW (2003) Genomic and Proteomic Profiling of Responses to Toxic Metals in Human Lung Cells. Environ. Health Perspect 111, 825–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Nollen M, Ebert F, Moser J, Mullenders LHF, Hartwig A, and Schwerdtle T (2009) Impact of arsenic on nucleotide excision repair: XPC function, protein level, and gene expression. Mol. Nutr. Food Res 53, 572–582. [DOI] [PubMed] [Google Scholar]
- (71).Holcomb N, Goswami M, Han SG, Scott T, D’Orazio J, Orren DK, Gairola CG, and Mellon I (2017) Inorganic arsenic inhibits the nucleotide excision repair pathway and reduces the expression of XPC. DNA Repair 52, 70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Muenyi CS, Ljungman M, and States JC (2015) Arsenic disruption of DNA damage responses—potential role in carcinogenesis and chemotherapy. Biomolecules 5, 2184–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Yimit A, Adebali O, Sancar A, and Jiang Y (2019) Differential damage and repair of DNA-adducts induced by anti-cancer drug cisplatin across mouse organs. Nat. Commun 10, 309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Muenyi CS, States VA, Masters JH, Fan TW, Helm CW, and States JC (2011) Sodium arsenite and hyperthermia modulate cisplatin-DNA damage responses and enhance platinum accumulation in murine metastatic ovarian cancer xenograft after hyperthermic intraperitoneal chemotherapy (HIPEC). J. Ovarian Res 4,9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Saijo M, Takedachi A, and Tanaka K (2011) Nucleotide excision repair by mutant xeroderma pigmentosum group A (XPA) proteins with deficiency in interaction with RPA. J. Biol. Chem 286, 5476–5483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Sugitani N, Sivley RM, Perry KE, Capra JA, and Chazin WJ (2016) XPA: A key scaffold for human nucleotide excision repair. DNA Repair 44, 123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Morita EH, Ohkubo T, Kuraoka I, Shirakawa M, Tanaka K , and Morikawa K (1996) Implications of the zinc-finger motif found in the DNA-binding domain of the human XPA protein. Genes Cells 1, 437–442. [DOI] [PubMed] [Google Scholar]
- (78).Quintal SM, Depaula QA, and Farrell NP (2011) Zinc finger proteins as templates for metal ion exchange and ligand reactivity. Chemical and biological consequences. Metallomics 3, 121–139. [DOI] [PubMed] [Google Scholar]
- (79).Jiang J, Tam LM, Wang P, and Wang Y (2018) Arsenite Targets the RING Finger Domain of Rbx1 E3 Ubiquitin Ligase to Inhibit Proteasome-Mediated Degradation of Nrf2. Chem. Res. Toxicol 31, 380–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Ding W, Liu W, Cooper KL, Qin X-J, de Souza Bergo PL , Hudson LG, and Liu KJ (2009) Inhibition of poly(ADP-ribose) Polymerase-1 by Arsenite interferes with repair of oxidative DNA damage. J. Biol. Chem 284, 6809–6817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Asmuss M (2000) Differential effects of toxic metal compounds on the activities of Fpg and XPA, two zinc finger proteins involved in DNA repair. Carcinogenesis 21, 2097–2104. [DOI] [PubMed] [Google Scholar]
- (82).Hartwig A, Blessing H, Schwerdtle T, and Walter I (2003) Modulation of DNA repair processes by arsenic and selenium compounds. Toxicology 193, 161–167. [DOI] [PubMed] [Google Scholar]
- (83).Qin XJ, Hudson LG, Liu W, Timmins GS, and Liu KJ (2008) Low concentration of arsenite exacerbates UVR-induced DNA strand breaks by inhibiting PARP-1 activity. Toxicol. Appl. Pharmacol 232, 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Bau DT, Gurr JR, and Jan KY (2001) Nitric oxide is involved in arsenite inhibition of pyrimidine dimer excision. Carcinogenesis 22, 709–716. [DOI] [PubMed] [Google Scholar]
- (85).Davis KL, Martin E, Turko IV, and Murad F (2001) Novel Effects of Nitric Oxide. Annu. Rev. Pharmacol. Toxicol 41 , 203–236. [DOI] [PubMed] [Google Scholar]
- (86).Snow ET, Hu Y, Klein CB, McCluskey KL, Schuliga M , and Sykora P (2003) Regulation of redox and DNA repair genes by arsenic: low dose protection against oxidative stress? Arsenic Exposure and Health Effects V., 305–319. [Google Scholar]
- (87).Lynn S, Lai HT, Gurr JR, and Jan KY (1997) Arsenite retards DNA break rejoining by inhibiting DNA ligation. Mutagenesis 12, 353–358. [DOI] [PubMed] [Google Scholar]
- (88).Ellenberger T, and Tomkinson AE (2008) Eukaryotic DNA Ligases: Structural and Functional Insights. Annu. Rev. Biochem 77, 313–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Cappelli E, Taylor R, Cevasco M, Abbondandolo A, Caldecott K, and Frosina G (1997) Involvement of XRCC1 and DNA ligase III gene products in DNA base excision repair. J. Biol. Chem 272, 23970–23975. [DOI] [PubMed] [Google Scholar]
- (90).Cannan WJ, Rashid I, Tomkinson AE, Wallace SS, and Pederson DS (2017) The human ligase IIIa-XRCC1 protein complex performs DNA nick repair after transient unwrapping of nucleosomal DNA. J. Biol. Chem 292, 5227–5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Caldecott KW, McKeown CK, Tucker JD, Ljungquist S, and Thompson LH (1994) An interaction between the mammalian DNA repair protein XRCC1 and DNA ligase III. Mol. Cell. Biol 14, 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Li J-H, and Rossman TG (1989) Inhibition of DNA ligase activity by arsenite: A possible mechanism of its comutagenesis. Mol. Toxicol 2, 1–9. [PubMed] [Google Scholar]
- (93).Kim H, and D’Andrea AD (2012) Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 26, 1393–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Wang W (2007) Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat. Rev. Genet 8, 735–748. [DOI] [PubMed] [Google Scholar]
- (95).Peremartí J, Ramos F, Marcos R, and Hernández, A (2014) Arsenic exposure disrupts the normal function of the FA/BRCA repair pathway. Toxicol. Sci 142, 93–104. [DOI] [PubMed] [Google Scholar]
- (96).Yamamoto KN, Kobayashi S, Tsuda M, Kurumizaka H, Takata M, Kono K, Jiricny J, Takeda S, and Hirota K (2011) Involvement of SLX4 in interstrand cross-link repair is regulated by the Fanconi anemia pathway. Proc. Natl. Acad. Sci. U. S. A 108, 6492–6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Alpi AF, Pace PE, Babu MM, and Patel KJ (2008) Mechanistic Insight into Site-Restricted Monoubiquitination of FANCD2 by Ube2t, FANCL, and FANCI. Mol. Cell 32, 767–777. [DOI] [PubMed] [Google Scholar]
- (98).Meetei AR, De Winter JP, Medhurst AL, Wallisch M, Waisfisz Q, van de Vrugt HJ, Oostra AB, Yan Z, Ling C, Bishop CE, Hoatlin ME, Joenje H, and Wang W (2003) A novel ubiquitin ligase is deficient in Fanconi anemia. Nat. Genet 35, 165–170. [DOI] [PubMed] [Google Scholar]
- (99).Chaudhury I, Stroik DR, and Sobeck A (2014) FANCD2-Controlled Chromatin Access of the Fanconi-Associated Nuclease FAN1 Is Crucial for the Recovery of Stalled Replication Forks. Mol. Cell. Biol 34, 3939–3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Jiang J, Bellani M, Li L, Wang P, Seidman MM, and Wang Y (2017) Arsenite Binds to the RING Finger Domain of FANCL E3 Ubiquitin Ligase and Inhibits DNA Interstrand Crosslink Repair. ACS Chem. Biol 12, 1858–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Xie J, Kim H, Moreau LA, Puhalla S, Garber J, Abo MA, Takeda S, and D’Andrea AD (2015) RNF4-mediated polyubiquitination regulates the Fanconi anemia/BRCA pathway. J. Clin. Invest 125, 1523–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Bhagwat N, Olsen AL, Wang AT, Hanada K, Stuckert P, Kanaar R, D’Andrea A, Niedernhofer LJ, and McHugh PJ (2009) XPF-ERCC1 Participates in the Fanconi Anemia Pathway of Cross-Link Repair. Mol. Cell. Biol 29, 6427–6437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Rahn JJ, Adair GM, and Nairn RS (2010) Multiple roles of ERCC1-XPF in Mammalian Interstrand Crosslink Repair. Environ. Mol. Mutagen 51, 567–581. [DOI] [PubMed] [Google Scholar]
- (104).Niedernhofer LJ, Odijk H, Budzowska M, van Drunen E, Maas A, Theil AF, de Wit J, Jaspers NGJ, Beverloo HB, Hoeijmakers JHJ, and Kanaar R (2004) The Structure-Specific Endonuclease Ercc1-Xpf Is Required To Resolve DNA Interstrand Cross-Link-Induced Double-Strand Breaks. Mol. Cell. Biol 24, 5776–5787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Long DT, and Walter JC (2012) A Novel Function for BRCA1 in Crosslink Repair? Mol. Cell 46, 111–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Bunting SF, Callen E, Kozak ML, Kim JM, Wong N, Lopez-Contreras AJ, Ludwig T, Baer R, Faryabi RB, Malhowski A, Chen H-T, Fernandez-Capetillo O, D’Andrea A, and Nussenzweig A (2012) BRCA1 Functions Independently of Homologous Recombination in DNA Interstrand Crosslink Repair. Mol. Cell 46, 125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Long DT, Joukov V, Budzowska M, and Walter JC (2014) BRCA1 promotes unloading of the CMG helicase from a stalled DNA replication fork. Mol. Cell 56, 174–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).D’Andrea AD (2013) BRCA1: A Missing Link in the Fanconi Anemia/BRCA Pathway. Cancer Discovery 3, 376–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Rosen EM (2013) BRCA1 in the DNA damage response and at telomeres. Front. Genet 4, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Zhang F, Paramasivam M, Cai Q, Dai X, Wang P, Lin K, Song J, Seidman MM, and Wang Y (2014) Arsenite Binds to the RING Finger Domains of RNF20-RNF40 Histone E3 Ubiquitin Ligase and Inhibits DNA Double-Strand Break Repair. J. Am. Chem. Soc 136, 12884–12887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Hoeijmakers JHJ (2001) Genome maintenance mechanisms for preventing cancer. Nature 411, 366–374. [DOI] [PubMed] [Google Scholar]
- (112).Lindahl T, and Wood RD (1999) Quality control by DNA repair. Science 286, 1897–1905. [DOI] [PubMed] [Google Scholar]
- (113).Symington LS, and Gautier J (2011) Double-Strand Break End Resection and Repair Pathway Choice. Annu. Rev. Genet 45, 247–271. [DOI] [PubMed] [Google Scholar]
- (114).Dong JT, and Luo XM (1993) Arsenic-induced DNA-strand breaks associated with DNA-protein crosslinks in human fetal lung fibroblasts. Mutat. Res. Lett 302, 97–102. [DOI] [PubMed] [Google Scholar]
- (115).Jimenez-Villarreal J, Rivas-Armendariz DI, Pineda-Belmontes CP, Betancourt-Martinez ND, Macias-Corral MA, Guerra-Alanis AJ, Nino-Castaneda MS, and Moran-Martinez J (2017) Detection of damage on single- or double-stranded DNA in a population exposed to arsenic in drinking water. GMR, Genet. Mol. Res 16, 16. [DOI] [PubMed] [Google Scholar]
- (116).Morales ME, Derbes RS, Ade CM, Ortego JC, Stark J, Deininger PL, and Roy-Engel AM (2016) Heavy Metal Exposure Influences Double Strand Break DNA Repair Outcomes. PLoS One 11, No. e0151367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Price BD, and D’Andrea AD (2013) Chromatin remodeling at DNA double-strand breaks. Cell 152, 1344–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Jo WJ, Ren X, Chu F, Aleshin M, Wintz H, Burlingame A, Smith MT, Vulpe CD, and Zhang L (2009) Acetylated H4K16 by MYST1 Protects UROtsa Cells from the Carcinogen Arsenic and is Decreased Following Chronic Arsenic Exposure. Toxicol. Appl. Pharmacol 241, 294–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).Chu F, Ren X, Chasse A, Hickman T, Zhang L, Yuh J, Smith MT, and Burlingame AL (2011) Quantitative mass spectrometry reveals the epigenome as a target of arsenic. Chem.-Biol. Interact 192, 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Tam LM, Jiang J, Wang P, Li L, Miao W, Dong X, and Wang Y (2017) Arsenite binds to the zinc finger motif of TIP60 histone acetyltransferase and induces its degradation via the 26S proteasome. Chem. Res. Toxicol 30, 1685–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Liu D, Wu D, Zhao L, Yang Y, Ding J, Dong L, Hu L, Wang F, Zhao X, Cai Y, and Jin J (2015) Arsenic trioxide reduces global histone H4 acetylation at lysine 16 through direct binding to histone acetyltransferase hMOF in human cells. PLoS One 10, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Mattiroli F, Vissers JHA, Van Dijk WJ, Ikpa P, Citterio E, Vermeulen W, Marteijn JA, and Sixma TK (2012) RNF168 ubiquitinates K13–15 on H2A/H2AX to drive DNA damage signaling. Cell 150, 1182–1195. [DOI] [PubMed] [Google Scholar]
- (123).Luijsterburg MS, Typas D, Caron MC, Wiegant WW, van den Heuvel D, Boonen RA, Couturier AM, Mullenders LH, Masson J-Y, and van Attikum H (2017) A PALB2-interacting domain in RNF168 couples homologous recombination to DNA break-induced chromatin ubiquitylation. eLife 6, 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Nakada S (2016) Opposing roles of RNF8/RNF168 and deubiquitinating enzymes in ubiquitination-dependent DNA double-strand break response signaling and DNA-repair pathway choice. J. Radiat. Res 57, i33–i40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Chen J, Feng W, Jiang J, Deng Y, and Huen MSY (2012) Ring finger protein RNF169 antagonizes the ubiquitin-dependent signaling cascade at sites of DNA damage. J. Biol. Chem 287, 27715–27722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Poulsen M, Lukas C, Lukas J, Bekker-Jensen S, and Mailand N (2012) Human RNF169 is a negative regulator of the ubiquitin-dependent response to DNA double-strand breaks. J. Cell Biol 197, 189–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Zhang L, Wang Z, Shi R, Zhu X, Zhou J, Peng B, and Xu X (2018) RNF126 Quenches RNF168 Function in the DNA Damage Response. Genomics, Proteomics Bioinf. 16, 428–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).An L, Dong C, Li J, Chen J, Yuan J, Huang J, Chan KM, Yu C, and Huen MSY (2018) RNF169 limits 53BP1 deposition at DSBs to stimulate single-strand annealing repair. Proc. Natl. Acad. Sci U. S. A 115, E8286–E8295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Wei H, and Yu X (2016) Functions of PARylation in DNA Damage Repair Pathways. Genomics, Proteomics Bioinf. 14, 131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Liu C, Vyas A, Kassab MA, Singh AK, and Yu X (2017) The role of poly ADP-ribosylation in the first wave of DNA damage response. Nucleic Acids Res. 45, 8129–8141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (131).Beck C, Robert I, Reina-San-Martin B, Schreiber V, and Dantzer F (2014) Poly(ADP-ribose) polymerases in double-strand break repair: Focus on PARP1, PARP2 and PARP3. Exp. Cell Res 329, 18–25. [DOI] [PubMed] [Google Scholar]
- (132).Chaudhuri AR, and Nussenzweig A (2017) The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol 18, 610–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Poirier GG, de Murcia G, Jongstra-Bilen J, Niedergang C, and Mandel P (1982) Poly (ADP-ribosyl)ation of polynucleosomes causes relaxation of chromatin structure. Proc. Natl. Acad. Sci. U. S. A 79, 3423–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Messner S, Altmeyer M, Zhao H, Pozivil A, Roschitzki B, Gehrig P, Rutishauser D, Huang D, Caflisch A, and Hottiger MO (2010) PARP1 ADP-ribosylates lysine residues of the core histone tails. Nucleic Acids Res. 38, 6350–6362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Qin XJ, Liu W, Li YN, Sun X, Hai CX, Hudson LG, and Liu KJ (2012) Poly(ADP-ribose) polymerase-1 inhibition by arsenite promotes the survival of cells with unrepaired DNA lesions induced by UV exposure. Toxicol. Sci 127, 120–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Zhou X, Sun X, Cooper KL, Wang F, Liu KJ, and Hudson LG (2011) Arsenite interacts selectively with zinc finger proteins containing C3H1 or C4 motifs. J. Biol. Chem 286, 22855–22863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (137).Ding X, Zhou X, Cooper KL, Huestis J, Hudson LG, and Liu KJ (2017) Differential sensitivities of cellular XPA and PARP-1 to arsenite inhibition and zinc rescue. Toxicol. Appl. Pharmacol 331, 108–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Brandes N, Schmitt S, and Jakob U (2009) Thiol-based redox switches in eukaryotic proteins. Antioxid. Redox Signaling 11, 997–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Vilas CK, Emery LE, Denchi EL, and Miller KM (2018) Caught with One’s Zinc Fingers in the Genome Integrity Cookie Jar. Trends Genet. 34, 313–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Shi H, Hudson LG, and Liu KJ (2004) Oxidative stress and apoptosis in metal ion-induced carcinogenesis. Free Radical Biol. Med 37, 582–593. [DOI] [PubMed] [Google Scholar]
- (141).Witkiewicz-Kucharczyk A, and Bal W (2006) Damage of zinc fingers in DNA repair proteins, a novel molecular mechanism in carcinogenesis. Toxicol. Lett 162, 29–42. [DOI] [PubMed] [Google Scholar]
- (142).Phoa N, and Epe B (2002) Influence of nitric oxide on the generation and repair of oxidative DNA damage in mammalian cells. Carcinogenesis 23, 469–475. [DOI] [PubMed] [Google Scholar]
- (143).Hartwig A, Pelzer A, Asmuss M, and Burkle A (2003) Very Low Concentrations of Arsenite Suppress Poly(ADP-Ribosyl)ation in Mammalian Cells. Int. J. Cancer 104, 1–6. [DOI] [PubMed] [Google Scholar]
- (144).Jaiswal M, LaRusso NF, Shapiro RA, Billiar TR, and Gores GJ (2001) Nitric oxide-mediated inhibition of DNA repair potentiates oxidative DNA damage in cholangiocytes. Gastroenterology 120, 190–199. [DOI] [PubMed] [Google Scholar]
- (145).Hilmi K, Jangal M, Marques M, Zhao T, Saad A, Zhang C, Luo VM, Syme A, Rejon C, Yu Z, Krum A, Fabian MR, Richard S, Alaoui-Jamali M, Orthwein A, McCaffrey L, and Witcher M (2017) CTCF facilitates DNA double-strand break repair by enhancing homologous recombination repair. Sci. Adv 3, No. e1601898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Lang F, Li X, Zheng W, Li Z, Lu D, Chen G, Gong D, Yang L, Fu J, Shi P, and Zhou J (2017) CTCF prevents genomic instability by promoting homologous recombination-directed DNA. Proc. Natl. Acad. Sci. U. S. A 114, 10912–10917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Hwang SY, Kang MA, Baik CJ, Lee Y, Hang NT, Kim BG, Han JS, Jeong JH, Park D, Myung K, and Lee JS (2019) CTCF cooperates with CtIP to drive homologous recombination repair of double-strand breaks. Nucleic Acids Res. 47, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Galanty Y, Belotserkovskaya R, Coates J, and Jackson SP (2012) RNF4, a SUMO-targeted ubiquitin E3 ligase, promotes DNA double-strand break repair. Genes Dev. 26, 1179–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Pinder JB, Attwood KM, and Dellaire G (2013) Reading, writing, and repair: The role of ubiquitin and the ubiquitin-like proteins in DNA damage signaling and repair. Front. Genet 4, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (150).Giglia-Mari G, Zotter A, and Vermeulen W (2011) DNA damage response. Cold Spring Harbor Perspect. Biol 3, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Jackson SP, and Durocher D (2013) Regulation of DNA Damage Responses by Ubiquitin and SUMO. Mol. Cell 49, 795–807. [DOI] [PubMed] [Google Scholar]
- (152).Centore RC, Yazinski SA, Tse A, and Zou L (2012) Spartan/C1orf124, a Reader of PCNA Ubiquitylation and a Regulator of UV-Induced DNA Damage Response. Mol. Cell 46, 625–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (153).Ting L, Jun H, and Junjie C (2010) RAD18 lives a double life: Its implication in DNA double-strand break repair. DNA Repair 9, 1241–1248. [DOI] [PubMed] [Google Scholar]
- (154).Vaziri C, Tateishi S, Mutter-Rottmayer E, and Gao Y (2016) Roles of RAD18 in DNA Replication and Postreplication Repair Genome Stability: From Virus to Human Application, pp 257–273, Elsevier Inc. [Google Scholar]
- (155).Ceccaldi R, Sarangi P, and D’Andrea AD (2016) The Fanconi anaemia pathway: New players and new functions. Nat. Rev. Mol. Cell Biol 17, 337–349. [DOI] [PubMed] [Google Scholar]
- (156).Williams SA, Longerich S, Sung P, Vaziri C, and Kupfer GM (2011) The E3 ubiquitin ligase RAD18 regulates ubiquitylation and chromatin loading of FANCD2 and FANCI. Blood 117, 5078–5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (157).Yang K, Moldovan GL, and D’Andrea AD (2010) RAD18-dependent recruitment of SNM1A to DNA repair complexes by a ubiquitin-binding zinc finger. J. Biol. Chem 285, 19085–19091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (158).Zhang L, and Li D-Q (2019) MORC2 regulates DNA damage response through a PARP1-dependent pathway. Nucleic Acids Res. 47, 8502–8520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Galanty Y, Belotserkovskaya R, Coates J, Polo S, Miller KM, and Jackson SP (2009) Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks. Nature 462, 935–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (160).Hendriks IA, and Vertegaal ACO (2015) Sumo in the DNA damage response. Oncotarget 6, 15734–15735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).Luo K, Zhang H, Wang L, Yuan J, and Lou Z (2012) Sumoylation of MDC1 is important for proper DNA damage response. EMBO J. 31, 3008–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (162).Chung I, and Zhao X (2015) Dna break-induced sumoylation is enabled by collaboration between a sumo ligase and the ssdnabinding complex rpa. Genes Dev. 29, 1593–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Dou H, Huang C, Singh M, Carpenter PB, and Yeh ETH (2010) Regulation of DNA Repair through DeSUMOylation and SUMOylation of Replication Protein A Complex. Mol. Cell 39, 333–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).Dhingra N, Wei L, and Zhao X (2019) Replication protein A (RPA) sumoylation positively influences the DNA damage checkpoint response in yeast. J. Biol. Chem 294, 2690–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (165).Poulsen SL, Hansen RK, Wagner SA, van Cuijk L, van Belle GJ, Streicher W, Wikstrom M, Choudhary C, Houtsmuller AB, Marteijn JA, Bekker-Jensen S, and Mailand N (2013) RNF111/Arkadia is a SUMO-targeted ubiquitin ligase that facilitates the DNA damage response. J. Cell Biol 201, 787–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Luo Q, Li Y, Deng J, and Zhang Z (2015) PARP-1 inhibitor sensitizes arsenic trioxide in hepatocellular carcinoma cells via abrogation of G2/M checkpoint and suppression of DNA damage repair. Chem.-Biol. Interact 226, 12–22. [DOI] [PubMed] [Google Scholar]
- (167).Ganapathy S, Liu J, Xiong R, Yu T, Makriyannis A, and Chen C (2018) Chronic low dose arsenic exposure preferentially perturbs mitotic phase of the cell cycle. Genes and Cancer. 10, 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (168).Hassani S, Khaleghian A, Ahmadian S, Alizadeh S, Alimoghaddam K, Ghavamzadeh A, and Ghaffari SH (2018) Redistribution of cell cycle by arsenic trioxide is associated with demethylation and expression changes of cell cycle related genes in acute promyelocytic leukemia cell line (NB4). Ann. Hematol 97, 83–93. [DOI] [PubMed] [Google Scholar]
- (169).Zhou BS, and Elledge SJ (2000) The DNA damage response: putting checkpoints in Perspective. Nature 408, 433–439. [DOI] [PubMed] [Google Scholar]
- (170).Nyberg KA, Michelson RJ, Putnam CW, and Weinert TA (2002) Toward Maintaining the Genome: DNA Damage and Replication Checkpoints. Annu. Rev. Genet 36, 617–656. [DOI] [PubMed] [Google Scholar]
- (171).Barnum KJ, and O’Connell MJ (2014) Cell Cycle Regulation by Checkpoints. Methods Mol. Biol 1170, 29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (172).Shaltiel IA, Krenning L, Bruinsma W, and Medema RH (2015) The same, only different - DNA damage checkpoints and their reversal throughout the cell cycle. J. Cell Sci 128, 607–620. [DOI] [PubMed] [Google Scholar]
- (173).Beezhold K, Klei LR, and Barchowsky A (2017) Regulation of cyclin D1 by arsenic and microRNA inhibits adipogenesis. Toxicol. Lett 265, 147–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (174).Vogt BL, and Rossman TG (2001) Effects of arsenite on p53, p21 and cyclin D expression in normal human fibroblasts - A possible mechanism for arsenite’s comutagenicity. Mutat. Res. Fundam. Mol. Mech. Mutagen 478, 159–168. [DOI] [PubMed] [Google Scholar]
- (175).Chen H, Li SF, Liu J, Diwan BA, Barrett JC, and Waalkes MP (2004) Chronic inorganic arsenic exposure induces hepatic global and individual gene hypomethylation: Implications for arsenic hepatocarcinogenesis. Carcinogenesis 25, 1779–1786. [DOI] [PubMed] [Google Scholar]
- (176).Chen YJ, Lai KC, Kuo HH, Chow LP, Yih LH, and Lee TC (2014) HSP70 colocalizes with PLK1 at the centrosome and disturbs spindle dynamics in cells arrested in mitosis by arsenic trioxide. Arch. Toxicol 88, 1711–1723. [DOI] [PubMed] [Google Scholar]
- (177).Salazar AM, Ostrosky-Wegman P, Menéndez D, Miranda E , García-Carrancá A, and Rojas E (1997) Induction of p53 protein expression by sodium arsenite. Mutat. Res. Fundam. Mol. Mech. Mutagen 381, 259–265. [DOI] [PubMed] [Google Scholar]
- (178).Karimian A, Ahmadi Y, and Yousefi B (2016) Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair 42, 63–71. [DOI] [PubMed] [Google Scholar]
- (179).Huang Y, Zhang J, Mchenry KT, Kim MM, Zeng W, Lopez-Pajares V, Dibble CC, Mizgerd JP, and Yuan ZM (2008) Induction of Cytoplasmic Accumulation ofp53: A Mechanism for Low Levels of Arsenic Exposure to Predispose Cells for Malignant Transformation. Cancer Res. 68, 9131–9136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (180).Tang F, Liu G, He Z, Ma W-Y, Bode AM, and Dong Z (2006) Arsenite Inhibits p53 Phosphorylation, DNA Binding Activity, and p53 Target Gene p21 Expression in Mouse Epidermal JB6 Cells. Mol. Carcinog 45, 861–870. [DOI] [PubMed] [Google Scholar]
- (181).Shen S, Wang C, Weinfeld M, and Le XC (2013) Inhibition of nucleotide excision repair by arsenic. Chin. Sci. Bull 58, 214–221. [Google Scholar]
- (182).Germolec DR, Yoshida T, Gaido K, Wilmer JL, Simeonova PP, Kayama F, Burleson F, Dong W, Lange RW, and Luster MI (1996) Arsenic induces overexpression of growth factors in human keratinocytes. Toxicol. Appl Pharmacol 141, 308–318. [DOI] [PubMed] [Google Scholar]
- (183).Simeonova PP, and Luster MI (2000) Mechanisms of arsenic carcinogenicity: genetic or epigenetic mechanisms? J. Env. Pathol. Toxicol. Oncol 19, 281–286. [PubMed] [Google Scholar]
- (184).Watcharasit P, Visitnonthachai D, Suntararuks S, Thiantanawat A, and Satayavivad J (2012) Low arsenite concentrations induce cell proliferation via activation of VEGF signaling in human neuroblastoma SH-SY5Y cells. Environ. Toxicol. Pharmacol 33, 53–59. [DOI] [PubMed] [Google Scholar]
- (185).Simeonova PP, Wang S, Toriuma W, Kommineni V, Matheson J, Unimye N, Kayama F, Harki D, Ding M, Vallyathan V, and Luster MI (2000) Arsenic mediates cell proliferation and gene expression in the bladder epithelium association with activating protein-1 transactivation. Cancer Res. 60, 3445–3453. [PubMed] [Google Scholar]
- (186).Jeon BG, Kumar BM, Kang EJ, Maeng GH, Lee YM, Hah YS, Ock SA, Kwack DO, Park BW, and Rho GJ (2011) Differential cytotoxic effects of sodium meta-arsenite on human cancer cells, dental papilla stem cells and somatic cells correlate with telomeric properties and gene expression. Anticancer Res. 31, 4315–4328. [PubMed] [Google Scholar]
- (187).Dreval K, Tryndyak V, Kindrat I, Twaddle NC, Orisakwe OE, Mudalige TK, Beland FA, Doerge DR, et al. (2018) and Pogribny IP Cellular and molecular effects of prolonged low-level sodium arsenite exposure on human hepatic HepaRG cells. Toxicol. Sci 162, 676–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (188).Andrew AS, Mason RA, Memoli V, and Duell EJ (2009) Arsenic activates EGFR pathway signaling in the lung. Toxicol. Sci 109, 350–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (189).Huang C, Li J, Ding M, Wang L, Shi X, Castranova V, Vallyathan V, Gong J, and Costa M (2001) Arsenic-induced NFKB transactivation through Erks-and JNKs-dependent pathways in mouse epidermal JB6 cells. Mol. Cell. Biochem 222, 29–34. [PubMed] [Google Scholar]
- (190).Chowdhury R, Chatterjee R, Giri AK, Mandal C, and Chaudhuri K (2010) Arsenic-induced cell proliferation is associated with enhanced ROS generation, Erk signaling and CyclinA expression. Toxicol. Lett 198, 263–271. [DOI] [PubMed] [Google Scholar]
- (191).Gupta A, Rosenberger SF, and Bowden GT (1999) Increased ROS levels contribute to elevated transcription factor and MAP kinase activities in malignantly progressed mouse keratinocyte cell lines. Carcinogenesis 20, 2063–2073. [DOI] [PubMed] [Google Scholar]
- (192).Morgan MJ, and Liu ZG (2011) Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 21, 103–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (193).Liu Y, Guyton KZ, Gorospe M, Xu Q, Lee JC, and Holbrook NJ (1996) Differential activation of ERK, JNK/SAPK and P38/CSBP/RK map kinase family members during the cellular response to arsenite. Free Radical Biol. Med 21, 771–781. [DOI] [PubMed] [Google Scholar]
- (194).Kao YT, Wu CH, Wu SY, Lan SH, Liu HS, and Tseng YS (2017) Arsenic treatment increase Aurora-A overexpression through E2F1 activation in bladder cells. BMC Cancer 17, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (195).Wijeweera JB, Gandolfi AJ, Parrish A, and Lantz RC (2001) Sodium arsenite enhances AP-1 and NFκB DNA binding and induces stress protein expression in precision-cut rat lung slices. Toxicol. Sci 61, 283–294. [DOI] [PubMed] [Google Scholar]
- (196).Lau ATY, Li M, Xie R, He QY, and Chiu JF (2004) Opposed arsenite-induced signaling pathways cell proliferation or apoptosis in cultured lung cells. Carcinogenesis 25, 21–28. [DOI] [PubMed] [Google Scholar]
- (197).Chen H, Liu J, Zhao CQ, Diwan BA, Merrick BA, and Waalkes MP (2001) Association of c-myc overexpression and hyperproliferation with arsenite-induced malignant transformation. Toxicol. Appl. Pharmacol 175, 260–268. [DOI] [PubMed] [Google Scholar]
- (198).Natarajan V, Scribner WM, Al-Hassani M, and Vepa S (1998) Reactive oxygen species signaling through regulation of protein tyrosine phosphorylation in endothelial cells. Environ. Health Perspect 106, 1205–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (199).Hamann I, and Klotz LO (2013) Arsenite-induced stress signaling: Modulation of the phosphoinositide 3/-kinase/Akt/FoxO signaling cascade. Redox Biol. 1, 104–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (200).Rodríguez-Gabriel MA, and Russell P (2005) Distinct signaling pathways respond to arsenite and reactive oxygen species in Schizosaccharomyces pombe. Eukaryotic Cell 4, 1396–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (201).Souza K, Maddock DA, Zhang Q, Chen J, Chiu C, Mehta S, and Wan Y (2001) Arsenite activation of PI3K/AKT cell survival pathway is mediated by p38 in cultured human keratinocytes. Mol. Med 7, 767–772. [PMC free article] [PubMed] [Google Scholar]
- (202).Wetzler M, Brady MT, Tracy E, Li ZR, Donohue KA, O’Loughlin KL, Cheng Y, Mortazavi A, McDonald AA, Kunapuli P, Wallace PK, Baer MR, Cowell JK, and Baumann H (2006) Arsenic Trioxide Affects Signal Transducer and Activator of Transcription Proteins through Alteration of Protein Tyrosine Kinase Phosphorylation. Clin. Cancer Res 12, 6817–6825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (203).Hunter T (1998) The phosphorylation of proteins on tyrosine: Its role in cell growth and disease. Philos. Trans. R. Soc., B 353, 583–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (204).Wee P, and Wang Z (2017) Epidermal growth factor receptor cell proliferation signaling pathways. Cancers 9, 1–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (205).Muller H, and Helin K (2000) The E2F transcription factors: Key regulators of cell proliferation. Biochim. Biophys. Acta, Rev. Cancer 1470, M1. [DOI] [PubMed] [Google Scholar]
- (206).Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA, and Dynlacht BD (2002) E2F integrates cell cycle progression with DNA repair, replication, and G 2 /M checkpoints. Genes Dev 16, 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (207).Eckstein M, Eleazer R, Rea M, and Fondufe-Mittendorf Y (2017) Epigenomic reprogramming in inorganic arsenic-mediated gene expression patterns during carcinogenesis. Rev. Environ. Health 32, 93–103. [DOI] [PubMed] [Google Scholar]
- (208).Luger K, Mader AW, Richmond RK, Sargent DF, and Richmond TJ (1997) Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389, 251–260. [DOI] [PubMed] [Google Scholar]
- (209).Mcginty RK, and Tan S (2015) Nucleosome structure and function. Chem. Rev 115, 2255–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (210).Kouzarides T (2007) Chromatin Modifications and Their Function. Cell 128, 693–705. [DOI] [PubMed] [Google Scholar]
- (211).Smerdon MJ (1991) DNA repair and the role of chromatin structure. Curr. Opin. Cell Biol 3, 422–428. [DOI] [PubMed] [Google Scholar]
- (212).Soria G, Polo SE, and Almouzni G (2012) Prime, Repair, Restore: The Active Role of Chromatin in the DNA Damage Response. Mol. Cell 46, 722–734. [DOI] [PubMed] [Google Scholar]
- (213).Kamynina EA, Lachenauer ER, DiRisio AC, Liebenthal RP, Field MS, and Stover PJ (2017) Arsenic trioxide targets MTHFD1 and SUMO-dependent nuclear de novo thymidylate biosynthesis. Proc. Natl. Acad. Sci. U. S. A 114, E2319–E2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (214).Zhu F, Zykova TA, Peng C, Zhang J, Cho YY, Zheng D, Yao K, Ma WY, Lau ATY, Bode AM, and Dong Z (2011) Phosphorylation of H2AX at Ser139 and a new phosphorylation site Ser16 by RSK2 decreases H2AX ubiquitination and inhibits cell transformation. Cancer Res. 71, 393–403. [DOI] [PubMed] [Google Scholar]
- (215).Howe CG, and Gamble MV (2016) Influence of Arsenic on Global Levels of Histone Posttranslational Modifications: a Review of the Literature and Challenges in the Field. Curr. Environ. Heal. reports 3, 225–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (216).Ren X, Mchale CM, Skibola CF, Smith AH, Smith MT, and Zhang L (2011) An emerging role for epigenetic dysregulation in arsenic toxicity and carcinogenesis. Environ. Health Perspect 119, 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (217).Chervona Y, Hall MN, Arita A, Wu F, Sun H, Tseng HC, Ali E, Uddin MN, Liu X, Zoroddu MA, Gamble MV, and Costa M (2012) Associations between arsenic exposure and global posttranslational histone modifications among adults in Bangladesh. Cancer Epidemiol., Biomarkers Prev 21 , 2252–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (218).Zhou X, Sun H, Ellen TP, Chen H, and Costa M (2008) Arsenite alters global histone H3 methylation. Carcinogenesis 29, 1831–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (219).Zhou X, Li Q, Arita A, Sun H, and Costa M (2009) Effects of Nickel, Chromate, and Arsenite on Histone 3 Lysine Methylation. Toxicol. Appl. Pharmacol 236, 78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (220).Pournara A, Kippler M, Holmlund T, Ceder R, Grafstrom R, Vahter M, Broberg K, and Wallberg AE (2016) Arsenic alters global histone modifications in lymphocytes in vitro and in vivo. Cell Biol. Toxicol 32, 275–284. [DOI] [PubMed] [Google Scholar]
- (221).Ray PD, Huang B-W, and Tsuji Y (2015) Coordinated Regulation of Nrf2 and Histone H3 Serine 10 Phosphorylation in Arsenite-activated Transcription of the Human Heme Oxygenase-1 Gene. Biochim. Biophys. Acta, Gene Regul. Mech 1849, 1277–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (222).Li J, Chen P, Sinogeeva N, Gorospe M, Wersto RP, Chrest FJ, Barnes J, and Liu Y (2002) Arsenic trioxide promotes histone H3 phosphoacetylation at the chromatin of CASPASE-10 in acute promyelocytic leukemia cells. J. Biol. Chem 277, 49504–49510. [DOI] [PubMed] [Google Scholar]
- (223).Peterson CL, and Laniel MA (2004) Histones and histone modifications. Curr. Biol 14, 546–551. [DOI] [PubMed] [Google Scholar]
- (224).Estève PO, Hang GC, and Pradhan S (2007) Molecular mechanisms of transactivation and doxorubicin-mediated repression of survivin gene in cancer cells. J. Biol. Chem 282, 2615–2625. [DOI] [PubMed] [Google Scholar]
- (225).McGarvey KM, Fahrner JA, Greene E, Martens J, Jenuwein T, and Baylin SB (2006) Silenced tumor suppressor genes reactivated by DNA demethylation do not return to a fully euchromatic chromatin state. Cancer Res. 66, 3541–3549. [DOI] [PubMed] [Google Scholar]
- (226).Steffen PA, and Ringrose L (2014) What are memories made of? How polycomb and trithorax proteins mediate epigenetic memory. Nat. Rev. Mol. Cell Biol 15, 340–356. [DOI] [PubMed] [Google Scholar]
- (227).Kim HG, Kim DJ, Li S, Lee KY, Li X, Bode AM, and Dong Z (2012) Polycomb (PcG) proteins, BMI1 and SUZ12, regulate arsenic-induced cell transformation. J. Biol. Chem 287, 31920–31928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (228).Rubin E, Wu X, Zhu T, Cheung JCY, Chen H, Lorincz A, Pandita RK, Sharma GG, Ha HC, Gasson J, Hanakahi LA, Pandita TK, and Sukumar S (2007) A role for the HOXB7 homeodomain protein in DNA repair. Cancer Res. 67, 1527–1535. [DOI] [PubMed] [Google Scholar]
- (229).Li J, Poi MJ, and Tsai M-D (2011) The Regulatory Mechanisms of Tumor Supressor p16INK4 and Relevance to cancer. Biochemistry 50, 5566–5582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (230).Weber HO, Samuel T, Rauch P, and Funk JO (2002) Human p14arf-mediated cell cycle arrest strictly depends on intact p53 signaling pathways. Oncogene 21, 3207–3212. [DOI] [PubMed] [Google Scholar]
- (231).Fierz B, Chatterjee C, Mcginty RK, Bar-Dagan M, Daniel P, and Muir TW (2011) Histone H2B ubiquitylation disrupts local and higher order chromatin compaction. Nat. Chem. Biol 7, 113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (232).Shogren-Knaak M, Ishii H, Sun J-M, Pazin MJ, Davie JR, and Peterson CL (2006) Histone H4-K16 Acetylation Controls Chromatin Structure and Protein Interactions. Science 311, 844–847. [DOI] [PubMed] [Google Scholar]
- (233).Moyal L, Lerenthal Y, Gana-Weisz M, Mass G, So S, Wang SY, Eppink B, Chung YM, Shalev G, Shema E, Shkedy D, Smorodinsky NI, van Vliet N, Kuster B, Mann M, Ciechanover A, Dahm-Daphi J, Kanaar R, Hu MCT, Chen DJ, Oren M, and Shilon Y (2011) Requirement of ATM-Dependent Monoubiquitylation of Histone H2B for Timely Repair of DNA Double-Strand Breaks. Mol. Cell 41, 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (234).Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, and Lukas J (2007) RNF8 Ubiquitylates Histones at DNA Double-Strand Breaks and Promotes Assembly of Repair Proteins. Cell 131 , 887–900. [DOI] [PubMed] [Google Scholar]
- (235).Zhang R, Erler J, and Langowski J (2017) Histone Acetylation Regulates Chromatin Accessibility: Role of H4K16 in Inter-nucleosome Interaction. Biophys. J 112, 450–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (236).Sapountzi V, and Cote J (2011) MYST-family histone acetyltransferases: Beyond chromatin. Cell. Mol. Life Sci 68, 1147–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (237).Tulin AV (2017) Poly (ADP-Ribose) Polymerase (Methods and Protocols) (Tulin AV, Ed.) 2nd ed., Humana Press, Totowa, NJ. [Google Scholar]
- (238).Ding D, Zhang Y, Wang J, Zhang X, Gao Y, Yin L, Li Q, Li J, and Chen H (2016) Induction and inhibition of the pan-nuclear gamma-H2AX response in resting human peripheral blood lymphocytes after X-ray irradiation. Cell Death Discovery 2, 16011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (239).Podhorecka M, Skladanowski A, and Bozko P (2010) H2AX Phosphorylation: Its Role in DNA Damage Response and Cancer Therapy. J. Nucleic Acids 2010, 920161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (240).Kaushik Tiwari M, and Rogers FA (2013) XPD-dependent activation of apoptosis in response to triplex-induced DNA damage. Nucleic Acids Res. 41, 8979–8994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (241).Joe YS, Jeong JH, Yang S, Kang H, Motoyama N, Pandolfi PP, Chung JH, and Kim MK (2006) ATR, PML, and CHK2 play a role in arsenic trioxide-induced apoptosis. J. Biol. Chem 281, 28764–28771. [DOI] [PubMed] [Google Scholar]
- (242).Watanabe R, Unuma K, Noritake K, Funakoshi T, Aki T, and Uemura K (2017) Ataxia telangiectasia and rad3 related (ATR)-promyelocytic leukemia protein (PML) pathway of the DNA damage response in the brain of rats administered arsenic trioxide. J. Toxicol. Pathol 30, 333–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (243).Li Y, Zhao Y, Jiang R, Xu Y, Ling M, Pang Y, Shen L, Zhou Y, Zhang J, Zhou J, Wang X, and Liu Q (2012) DNA-PKcs-mediated stabilization of p53 by JNK2 is involved in arsenite-induced DNA damage and apoptosis in human embryo lung fibroblast cells. Toxicol. Lett 210, 302–310. [DOI] [PubMed] [Google Scholar]
- (244).Zykova TA, Zhu F, Lu C, Higgins L, Tatsumi Y, Abe Y, Bode AM, and Dong Z (2006) Lymphokine-activated killer T-cell-originated protein kinase phosphorylation of histone H2AX prevents arsenite-induced apoptosis in RPMI7951 melanoma cells. Clin. Cancer Res 12, 6884–6893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (245).Li J, Gorospe M, Barnes J, and Liu Y (2003) Tumor promoter arsenite stimulates histone H3 phosphoacetylation of proto-oncogenes c-fos and c-jun chromatin in human diploid fibroblasts. J. Biol. Chem 278, 13183–13191. [DOI] [PubMed] [Google Scholar]
- (246).Xanthoudakis S, Miao G, Wang F, Pan YC, and Curran T (1992) Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J. 11, 3323–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (247).Cavigelli M, Li WW, Lin A, Su B, Yoshioka K, and Karin M (1996) The tumor promoter arsenite stimulates AP-1 activity by inhibiting a JNK phosphatase. EMBO J. 15, 6269–6279. [PMC free article] [PubMed] [Google Scholar]
- (248).Picco V, and Pagès G (2013) Linking JNK Activity to the DNA Damage Response. Genes Cancer 4, 360–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (249).Jjingo D, Conley AB, Yi SV, Lunyak VV, and King Jordan I (2012) On the presence and role of human gene-body DNA methylation. Oncotarget 3, 462–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (250).Jones PA, and Gonzalgo ML (1997) Altered DNA methylation and genome instability: A new pathway to cancer? Proc. Natl. Acad. Sci. U. S. A 94, 2103–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (251).Reichard JF, Schnekenburger M, and Puga A (2007) Long term low-dose arsenic exposure induces loss of DNA methylation. Biochem. Biophys. Res. Commun 352, 188–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (252).Cui X, Wakai T, Shirai Y, Yokoyama N, Hatakeyama K, and Hirano S (2006) Arsenic trioxide inhibits DNA methyltransferase and restores methylation-silenced genes in human liver cancer cells. Hum. Pathol 37, 298–311. [DOI] [PubMed] [Google Scholar]
- (253).Ahlborn GJ, Nelson GM, Ward WO, Knapp G, Allen JW, Ouyang M, Roop BC, Chen Y, O’Brien T, Kitchin KT, and Delker DA (2008) Dose response evaluation of gene expression profiles in the skin of K6/ODC mice exposed to sodium arsenite. Toxicol. Appl. Pharmacol 227, 400–416. [DOI] [PubMed] [Google Scholar]
- (254).Rea M, Eckstein M, Eleazer R, Smith C, and Fondufe-Mittendorf YN (2017) Genome-wide DNA methylation reprogramming in response to inorganic arsenic links inhibition of CTCF binding, DNMT expression and cellular transformation. Sci. Rep 7, 41474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (255).Goering PL, Aposhian HV, Mass MJ, Cebrián M, Beck BD, and Waalkes MP (1999) The enigma of arsenic carcinogenesis: Role of metabolism. Toxicol. Sci 49, 5–14. [DOI] [PubMed] [Google Scholar]
- (256).Coppin JF, Qu W, and Waalkes MP (2008) Interplay between cellular methyl metabolism and adaptive efflux during oncogenic transformation from chronic arsenic exposure in human cells. J. Biol. Chem 283, 19342–19350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (257).Zhao CQ, Young MR, Diwan BA, Coogan TP, and Waalkes MP (1997) Association of arsenic-induced malignant transformation with DNA hypomethylation and aberrant gene expression. Proc. Natl. Acad. Sci. U. S. A 94, 10907–10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (258).Pfeifer GP (2018) Defining driver DNA methylation changes in human cancer. Int. J. Mol. Sci 19, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (259).Hon GC, Hawkins RD, Caballero OL, Lo C, Lister R, Pelizzola M, Valsesia A, Ye Z, Kuan S, Edsall LE, Camargo AA, Steveson BJ, Ecker JR, Baina V, Strausberg RL, Simpson AJ, and Ren B (2012) Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res. 22, 246–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (260).Esteller M, Fraga MF, Guo M, Garcia-Foncillas J, Hedenfalk I, Godwin AK, Trojan J, Vaurs-Barriere C, Bignon YJ, Ramus S, Benitez J, Caldes T, Akiyama Y, Yuasa Y, Launonen V , Canal MJ, Rodriguez R, Capella G, Peinado MA, Borg A, Aaltonen LA, Ponder BA, Baylin SB, and Herman JG (2001) DNA methylation patterns in hereditary human cancers mimic sporadic tumorigenesis. Hum. Mol. Genet 10, 3001–3007. [DOI] [PubMed] [Google Scholar]
- (261).Pilsner JR, Liu X, Ahsan H, Ilievski V, Slavkovich V, Levy D, Factor-Litvak P, Graziano JH, and Gamble MV (2009) Folate deficiency, hyperhomocysteinemia, low university creatinine, and hypomethylation of leukocyte DNA are risk factors for arsenic-induced skin lesions. Environ. Health Perspect 117, 254–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (262).Williamson LM, and Lees-Miller SP (2011) Estrogen receptor α-mediated transcription induces cell cycle-dependent DNA double-strand breaks. Carcinogenesis 32, 279–285. [DOI] [PubMed] [Google Scholar]
- (263).Caldon CE (2014) Estrogen Signaling and the DNA Damage Response in Hormone Dependent Breast Cancers. Front. Oncol 4, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (264).Hossain MB, Vahter M, Concha G, and Broberg K (2012) Environmental arsenic exposure and DNA methylation of the tumor suppressor gene p16 and the DNA repair gene MLH1: Effect of arsenic metabolism and genotype. Metallomics 4, 1167–1175. [DOI] [PubMed] [Google Scholar]
- (265).Miao Z, Wu L, Lu M, Meng X, Gao B, Qiao X, Zhang W , and Xue D (2015) Analysis of the transcriptional regulation of cancer-related genes by aberrant DNA methylation of the cis-regulation sites in the promoter region during hepatocyte carcinogenesis caused by arsenic. Oncotarget 6, 21493–21506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (266).Zhang A, Li H, Xiao Y, Chen L, Zhu X, Li J, Ma L, Pan X, Chen W, and He Z (2017) Aberrant methylation of nucleotide excision repair genes is associated with chronic arsenic poisoning. Biomarkers 22, 429–438. [DOI] [PubMed] [Google Scholar]
- (267).Liu S, Jiang J, Li L, Amato NJ, Wang Z, and Wang Y (2015) Arsenite Targets the Zinc Finger Domains of Tet Proteins and Inhibits Tet-Mediated Oxidation of 5-Methylcytosine. Environ. Sci. Technol 49, 11923–11931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (268).Bachman M, Uribe-Lewis S, Yang X, Williams M, Murrell A, and Balasubramanian S (2014) 5-Hydroxymethylcytosine is a predominantly stable DNA modification. Nat. Chem 6, 1049–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (269).Mellen M, Ayata P, Dewell S, Kriaucionis S, and Heintz N (2012) MeCP2 Binds to 5hmC Enriched within Active Genes and Accessible Chromatin in the Nervous System. Cell 151, 1417–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (270).Williams K, Christensen J, Pedersen MT, Johansen JV, Cloos PAC, Rappsilber J, and Helin K (2011) TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature 473, 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (271).Kafer GR, Li X, Horii T, Suetake I, Tajima S, Hatada I, and Carlton PM (2016) 5-Hydroxymethylcytosine Marks Sites of DNA Damage and Promotes Genome Stability. Cell Rep. 14, 1283–1292. [DOI] [PubMed] [Google Scholar]
- (272).Kantidze OL, and Razin SV (2017) 5-hydroxymethylcytosine in DNA repair: A new player or a red herring? Cell Cycle 16, 1499–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (273).Rea M, Gripshover T, and Fondufe-Mittendorf Y (2018) Selective inhibition of CTCF binding by iAs directs TET-mediated reprogramming of 5-hydroxymethylation patterns in iAs-transformed cells. Toxicol. Appl. Pharmacol 338, 124–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (274).Maret W (2013) Zinc Biochemistry: From a Single Zinc Enzyme to a key element of life. Adv. Nutr 4, 82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (275).Dickinson BC, and Chang CJ (2011) Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat. Chem. Biol 7, 504–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (276).Lange M, Ok K, Shimberg GD, Bursac B, Marko L, Ivanovic-Burmazovic I, Michel SLJ, and Filipovic MR (2019) Direct Zinc Finger Protein Persulfidation by H2S Is Facilitated by Zn2+. Angew. Chem., Int. Ed 58, 7997–8001. [DOI] [PubMed] [Google Scholar]
- (277).Lee SJ, and Michel SLJ (2010) Cysteine oxidation enhanced by iron in tristetraprolin, a zinc finger peptide. Inorg. Chem 49, 1211–1219. [DOI] [PubMed] [Google Scholar]
- (278).Kluska K, Adamczyk J, and Krezel A (2018) Metal binding properties, stability and reactivity of zinc fingers. Coord. Chem. Rev 367, 18–64. [Google Scholar]
- (279).Zhou X, Cooper KL, Sun X, Liu KJ, and Hudson LG (2015) Selective sensitization of zinc finger protein oxidation by reactive oxygen species through arsenic binding. J. Biol. Chem 290, 18361–18369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (280).Lipkowitz S, and Weissman AM (2011) RINGs of good and evil: RING finger ubiquitin ligases at the crossroads of tumour suppression and oncogenesis. Nat. Rev. Cancer 11 , 629–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (281).Cook KB, Kazan H, Zuberi K, Morris Q, and Hughes TR (2011) RBPDB: A database of RNA-binding specificities. Nucleic Acids Res. 39, 301–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (282).Hei TK, Liu SX, and Waldren C (1998) Mutagenicity of arsenic in mammalian cells: Role of reactive oxygen species. Proc. Natl. Acad. Sci. U. S. A 95, 8103–8107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (283).The International Cancer Genome Consortium (2010) International network of cancer genome projects. Nature 464, 993–998. [DOI] [PMC free article] [PubMed] [Google Scholar]