

Summary
It has been well documented that the ER responds to cellular stresses through the unfolded protein response (UPR), but it is unknown how the Golgi responds to similar stresses. In this study, we treated HeLa cells with ER stress inducers, thapsigargin (TG), tunicamycin (Tm), and dithiothreitol (DTT), and found that only TG treatment resulted in Golgi fragmentation. TG induced Golgi fragmentation at a low dose and short time when UPR was undetectable, indicating that Golgi fragmentation occurs independently of ER stress. Further experiments demonstrated that TG induces Golgi fragmentation through elevating intracellular Ca2+ and protein kinase Cα (PKCα) activity, which phosphorylates the Golgi stacking protein GRASP55. Significantly, activation of PKCα with other activating or inflammatory agents, including phorbol 12-myristate 13-acetate and histamine, modulates Golgi structure in a similar fashion. Hence, our study revealed a novel mechanism through which increased cytosolic Ca2+ modulates Golgi structure and function.
Subject Areas: Biological Sciences, Cell Biology, Functional Aspects of Cell Biology
Graphical Abstract
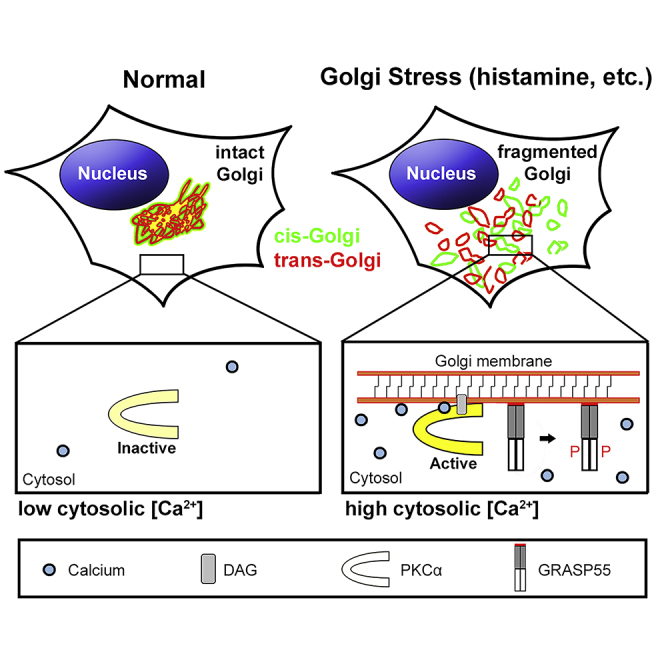
Highlights
-
•
Thapsigargin (TG) treatment leads to Golgi fragmentation independent of ER stress
-
•
TG induces Golgi fragmentation through elevated cytosolic Ca2+
-
•
TG-induced cytosolic Ca2+ spikes activate PKCα that phosphorylates GRASP55
-
•
Histamine modulates the Golgi structure and function by a similar mechanism
Biological Sciences; Cell Biology; Functional Aspects of Cell Biology
Introduction
In mammalian cells, the Golgi apparatus is characterized by a multilayer stacked structure of ∼5–7 flattened cisternal membranes, and stacks are often laterally linked to form a ribbon located in the perinuclear region of the cell (Tang and Wang, 2013, Wang and Seemann, 2011). The exact mechanism of Golgi stack formation is not fully understood, but it has been shown that the Golgi re-assembly stacking protein of 55 kDa (GRASP55, also called GORASP2) and its homolog GRASP65 (GORASP1) play essential roles in Golgi stacking (Wang et al., 2003, Zhang and Wang, 2015). Both GRASPs are peripheral membrane proteins that share similar domain structures and overlapping functions (Wang and Seemann, 2011). GRASP65 is predominantly concentrated in the cis Golgi, whereas GRASP55 is localized on medial-trans cisternae. Both GRASPs form trans-oligomers through their N-terminal GRASP domains that “glue” adjacent Golgi cisternae together into stacks (Wang et al., 2003, Xiang and Wang, 2010) and ribbons (Feinstein and Linstedt, 2008, Puthenveedu et al., 2006). GRASP oligomerization is regulated by phosphorylation; mitotic phosphorylation of GRASP55 and GRASP65 at the C-terminal serine/proline-rich (SPR) domain inhibits oligomerization and results in Golgi cisternal unstacking and disassembly (Tang et al., 2012, Wang et al., 2005, Xiang and Wang, 2010).
The Golgi exhibits different morphology in different cell types and tissues as well as under different conditions. For example, in many secretory cells such as Brunner's gland of platypus, the Golgi forms large, well-formed stacks (Krause, 2000), whereas electron micrographs show reorganization of Golgi membranes in prolactin cells of female rats upon cessation of a sucking stimulus (Rambourg et al., 1993). In neurons, increased neuronal activity causes dispersal of the Golgi at the resolution of light microscopy (Thayer et al., 2013). In Alzheimer disease, the Golgi membranes are dispersed and fragmented in neurons from human brain and mouse models (Joshi et al., 2015). Golgi fragmentation is also observed in other neurodegenerative diseases, including Parkinson (Mizuno et al., 2001) and Huntington (Hilditch-Maguire et al., 2000) diseases and amyotrophic lateral sclerosis (ALS) (Fujita and Okamoto, 2005, Gonatas et al., 1998, Mourelatos et al., 1996). In addition, the Golgi has also been shown to be fragmented in lung, prostate, and breast cancers (Petrosyan et al., 2014, Sewell et al., 2006, Tan et al., 2016). A plausible hypothesis is that the Golgi adjusts its structure and function in response to different physiological and pathological conditions; however, the molecular mechanisms that control Golgi structure and function under disease conditions are so far not well understood.
The Golgi structure can be modulated experimentally such as by molecular manipulations of GRASP55 and GRASP65. Microinjection of antibodies against GRASP55 or GRASP65 into cells inhibits post-mitotic stacking of newly formed Golgi cisternae (Wang et al., 2003, Wang et al., 2008). Knockdown (KD, by siRNA) or knockout (KO, by CRISPR/Cas9) of either GRASP reduces the number of cisternae per stack (Sutterlin et al., 2005, Tang et al., 2010), whereas simultaneous depletion of both GRASPs causes fragmentation of the entire Golgi stack (Bekier et al., 2017, Xiang and Wang, 2010). Expression of non-phosphorylatable GRASP65 mutants enhances Golgi stacking in interphase and inhibits Golgi disassembly in mitosis (Tang et al., 2010). Because GRASPs play critical roles in Golgi structure formation, it is reasonable to speculate that physiological and pathological cues may trigger Golgi fragmentation through GRASP55/65 modification, such as phosphorylation (Ahat et al., 2019a, Li et al., 2019a). Using GRASPs as tools to manipulate Golgi stack formation, it has been demonstrated that Golgi cisternal unstacking accelerates protein trafficking but impairs accurate glycosylation and sorting (Bekier et al., 2017, Xiang et al., 2013). In addition, GRASP depletion also affects other cellular activities such as cell attachment, migration, growth, and autophagy (Ahat et al., 2019b, Zhang et al., 2018, Zhang et al., 2019).
Protein kinase C (PKC) is a large family of multifunctional serine/threonine kinases that are activated by signals such as increases in the concentration of diacylglycerol (DAG) and/or intracellular calcium ions (Ca2+). In cells, PKCs are mainly cytosolic, but transiently localize to membranes such as endosomes and Golgi upon activation (Chen et al., 2004, El Homasany et al., 2005). Membrane association of PKC is via a C1 domain that interacts with DAG in the membrane. Conventional PKCs (cPKCs) also contain a C2 domain that binds Ca2+ ions, which further enhances their membrane association and activity (Nishizuka, 1995). Knockdown of atypical PKCs (aPKCs) using siRNA causes a reduction in peripheral ERGIC-53 clusters without affecting the Golgi morphology (Farhan et al., 2010). In addition, increased PKC activity has been implicated in cancer (Cooke et al., 2017, Kim et al., 2013), but the mechanism by which PKC may contribute to invasion, inflammation, tumorigenesis, and metastasis is not fully understood (Griner and Kazanietz, 2007).
In this study, we performed high-resolution microscopy and biochemistry experiments to determine how the Golgi responds to cellular stresses such as ER stress. Although not all ER stress inducers caused Golgi fragmentation, treatment of cells with the Ca2+-ATPase inhibitor thapsigargin (TG) resulted in Golgi fragmentation with a low dose and short time in which ER stress was undetectable, indicating that Golgi fragmentation occurs independently of ER stress. Further experiments demonstrated that TG-induced cytosolic Ca2+ spikes activate PKC that phosphorylates GRASP55. Interestingly, inflammatory factors such as histamine modulate the Golgi structure through a similar mechanism. Thus, we have uncovered a novel pathway through which cytosolic Ca2+ modulates the Golgi structure and function.
Results
TG Induces Golgi Fragmentation and UPR
It has been hypothesized that ER stress and the unfolded protein response (UPR) cause Golgi fragmentation and dysfunction through overloading misfolded proteins into the Golgi (Oku et al., 2011). To test this hypothesis, we performed a time course treatment of HeLa cells with a well-known UPR inducer, TG, which specifically blocks the sarcoendoplasmic reticulum Ca2+ transport ATPase (SERCA) (Xu et al., 2004) and causes Ca2+ dysregulation (Ito et al., 2015). We assessed the Golgi morphology by co-staining the cells for GM130, a cis-Golgi marker, and TGN46, a protein in the trans-Golgi network. As shown in Figures 1A and 1B, the Golgi became fragmented after TG treatment, and the response was linear over time (Figures 1A, 1B, and S1A). Although Golgi fragmentation was more obvious after a longer treatment, it became detectable in shorter treatments such as 10 min. More careful examination of the Golgi morphology by super-resolution fluorescence microscopy demonstrated that the Golgi ribbon was broken down, as the Golgi appeared as disconnected puncta. The stacks were also defective, as indicated by the separation of GM130 and TGN46 signals (Figures 1C and 1D).
Figure 1.

TG Induces Golgi Fragmentation and UPR
(A) Short-term TG treatment causes Golgi fragmentation. HeLa cells were treated with 250 nM TG, fixed at the indicated time points, and stained for GM130 (cis-Golgi) and TGN46 (trans-Golgi). Scale bar, 20 μm.
(B) Quantitation of (A) for cells with fragmented Golgi using GM130 as the Golgi marker.
(C and D) Super-resolution images of DMSO- (C) and TG-treated (D) HeLa cells. Cells were treated with 2 μM TG for 1 h, stained as in (A), and imaged with a Leica SP8 STED microscope. Indicated areas are enlarged and shown on the right as merged GM130 (green) and TGN46 (red). To quantify Golgi unstacking in these images, relative fluorescence intensity was plotted along a random line through the Golgi region. Note the agreement in peaks in control (C) and relative disagreement in peaks in the TG-treated cell (D). Scale bar in main images, 5 μm; in inserts, 1 μm.
(E) Longer term TG treatment results in ER stress. Cells treated as in (A) were analyzed by Western blot of indicated proteins. Note that TG treatment increases the levels of p-eIF2α, Bip, and CHOP.
(F–G) Quantitation of the ratio of p-eIF2α/eIF2α and the Bip levels from (E), with the no-treatment control normalized to 1.
All quantitation results are shown as mean ± SEM from at least three independent experiments; statistical analyses were performed using two-tailed Student's t-tests (∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001).
To correlate Golgi fragmentation with UPR, we performed Western blot of TG-treated cells to assess the levels of several UPR markers, including phosphorylated eukaryotic translation initiation factor 2A eIF2α (p-eIF2α), the ER chaperone binding of immunoglobulin protein (Bip), and the CCAAT-enhancer-binding protein homologous protein (CHOP). As shown in Figures 1E–1G, longer term TG treatment, for example, 2 h or longer, caused UPR, as indicated by the increase of all three markers. When the treatment was reduced to 30 min, only the p-eIF2α level increased, whereas Bip and CHOP did not change. This indicates that the minimal time for UPR to occur is ∼30 min under our experimental conditions. Consistently, no significant increase in the level of any of these UPR markers was detected when the treatment was reduced to below 30 min. Interestingly, the Golgi in a significant proportion of cells was fragmented at this time. Golgi fragmentation was obvious with 10-min TG treatment when UPR was undetectable and became more prevalent at 30-min treatment (Figures 1A and 1B). The fact that Golgi fragmentation occurs earlier than UPR indicates that Golgi fragmentation is unlikely a downstream effect of ER stress, but rather occurs independently of UPR.
It is worth mentioning that TG treatment did not affect the level of key Golgi structural proteins, including the Golgi stacking proteins GRASP55 and GRASP65, the Golgi tethering protein GM130, and the Golgi SNARE Gos28 (Figure 1E), indicating that TG induces Golgi fragmentation likely through modification rather than degradation of Golgi structural proteins. In addition, TG-induced Golgi fragmentation is reversible; when TG was washed out, the Golgi structure gradually returned to its normal shape (Figures S1B–S1D). Consistently, TG-treatment did not induce apoptosis as shown by Annexin V staining. In contrast, staurosporine treatment, which is known to induce apoptosis, increased Annexin V cell surface staining (Figures S1D and S1E). In addition, TG treatment did not seem to affect the organization of the actin and microtubule cytoskeleton (Figures S1F and S1G).
Tunicamycin or Dithiothreitol Treatment Induces UPR but Not Golgi Fragmentation
To test whether the hypothesis that Golgi fragmentation occurs independently of UPR applies only to TG treatment or also to other ER stress inducers, we repeated the same set of experiments by treating cells with tunicamycin (Tm), an antibiotic that induces ER stress by inhibiting N-glycosylation and the accumulation of misfolded proteins in the ER lumen. Tm treatment did not affect the Golgi morphology after 360 min, as indicated by the GM130 and TGN46 signals (Figures S2A–S2C). Further analysis of Tm-treated cells by electron microscopy (EM) also did not reveal any significant changes in the Golgi structure (Figure S2D). The treatment indeed induced UPR, as indicated by the robust increase in the p-eIF2α, Bip, and CHOP levels, in particular after 120 min (Figures S2E–S2G). Six-hour Tm treatment increased the width of the ER cisternae and caused ER fragmentation (Figure S2H). As Tm, dithiothreitol (DTT) treatment also did not cause Golgi fragmentation, although Bip and CHOP levels increased significantly after 120 min of treatment (Figures S3A–S3D). We also performed super-resolution microscopy to examine the Golgi structure in parallel after Tm, DTT, or TG treatment. Similar to that observed in control cells, the Golgi structure is intact in Tm- and DTT-treated cells, with extensive overlap between cis- and trans-Golgi markers, whereas TG treatment caused not only fragmentation of the Golgi structure but also separation of cis- and trans-Golgi markers (Figures S3E and S3F). Taken together, these results indicate that ER stress is unlikely a direct cause of Golgi fragmentation.
TG Induces Golgi Fragmentation Prior to UPR Through Elevated Cytosolic Ca2+
We next sought to decouple the Golgi stress response from UPR after TG treatment. As a complementary approach to the timecourse experiment shown in Figure 1, we titrated TG (1–250 nM) in the treatment. Here we treated cells for 20 min, a time point prior to UPR becoming detectable when cells were treated with 250 nM TG (Figure 1). The results showed that Golgi fragmentation increased linearly in response to the increasing TG concentration, and importantly, TG at low doses (1–250 nM) effectively caused Golgi fragmentation (Figures 2A and 2B). For comparison, we also assessed UPR in the same cells. As shown in Figures 2C–2E, treatment of cells with up to 250 nM TG for 20 min did not cause UPR as indicated by the p-eIF2α and Bip levels. These results indicate that TG triggers Golgi fragmentation independent of ER stress. Furthermore, we carried out similar experiments in normal rat kidney (NRK) cells and RAW 264.7 murine macrophages and obtained similar results (Figure S4), indicating that the effect of TG treatment on the Golgi structure is not cell-type specific.
Figure 2.

Low Concentration of TG Induces Golgi Fragmentation Prior to UPR Through Elevated Cytosolic Ca2+
(A) HeLa cells were treated with the indicated concentrations of TG for 20 min and stained for GM130. Scale bar, 20 μm.
(B) Quantitation of cells with fragmented Golgi in (A).
(C) Cells treated with TG as in (A) were analyzed by Western blots.
(D and E) Quantitation of p-eIF2α/eIF2α and Bip in (C) from five independent experiments.
(F) BAPTA-AM inhibits TG-induced Golgi fragmentation. HeLa cells treated with 100 nM TG for indicated times with or without 60 μM BAPTA-AM (B/AM) and stained for GM130. Scale bar, 20 μm.
(G) Quantitation of (F) from three independent experiments. Statistical analyses were performed using two-tailed Student's t tests (∗, p ≤ 0.05; ∗∗, p ≤ 0.01; ∗∗∗, p ≤ 0.001).
(H) Electron micrographs of Golgi profiles in HeLa cells treated with 250 nM TG and B/AM for 20 min. Note that the Golgi is comprised of bulbous saccules in the TG-treatment, whereas in the B/AM pretreated cells the cisternae appear straight and well-stacked. Asterisks (∗) indicate nuclei. Scale bar, 0.5 μm.
(I) Quantitation of the morphological features of Golgi stacks on the EM images in (H). For statistics, B/AM and TG were compared with DMSO treatment, whereas TG + B/AM was compared with TG treatment.
We next asked how TG treatment induces Golgi fragmentation. Knowing that TG increases cytosolic Ca2+ (Jones and Sharpe, 1994), we employed the membrane permeable Ca2+ chelator BAPTA-AM to test whether TG induces Golgi fragmentation through cytosolic Ca2+. We pre-treated cells with BAPTA-AM alone (60 μM) for 30 min and then with or without TG (100 nM) for 0, 15, 30, and 60 min (Figures 2F and 2G). The result showed that BAPTA-AM significantly prevented TG-induced Golgi fragmentation, whereas BAPTA-AM alone did not affect the Golgi morphology. Subsequent EM analysis confirmed TG-induced Golgi fragmentation and its rescue by BAPTA-AM (Figures 2H and 2I). TG treatment reduced the number of cisternae per stack and the length of cisternae but increased the number of vesicles surrounding each stack. These effects were largely abolished by the addition of BAPTA-AM. These results demonstrated that cytosolic Ca2+ is required for TG-induced Golgi fragmentation. Consistent with this notion, treatment of cells with a Ca2+ ionophore, ionomycin (Io), also caused Golgi fragmentation (Figures S5A and S5B). Therefore, the driving force behind TG-induced Golgi fragmentation is the elevated cytosolic Ca2+.
TG-Induced Golgi Fragmentation Increases Protein Trafficking in the Golgi
As GRASP-depletion-mediated Golgi destruction affects Golgi functions such as protein trafficking (Ahat et al., 2019b, Xiang et al., 2013), we examined the effect of TG treatment on the trafficking of the vesicular stomatitis virus glycoprotein (VSV-G) using the well-established RUSH system (Boncompain et al., 2012). Cells were transfected with a plasmid that encodes both the invariant chain of the major histocompatibility complex (Ii, an ER protein) fused to core streptavidin and VSV-G fused to streptavidin-binding peptide (SBP). Under growth conditions without biotin, the interaction between streptavidin and SBP retains VSV-G in the ER. Upon the addition of biotin, this interaction is disrupted, resulting in synchronous release of the VSV-G reporter from the ER to the Golgi. Because VSV-G is a glycoprotein, we used endoglycosidase H (EndoH) to distinguish its core (ER and cis Golgi) and complex (trans Golgi and post-Golgi) glycosylation forms as an indicator of trafficking. As shown in Figure 3A, TG treatment first slightly decreased VSV-G trafficking at 15-min release but then increased VSV-G trafficking at 60 and 90 min compared with DMSO control. Our previous studies showed that VSV-G reaches the cis Golgi at 15–20 min and trans Golgi at ∼90 min (Bekier et al., 2017, Li et al., 2019b). These results suggest that TG treatment may delay VSV-G release possibly by slowing down its folding; but once it reaches the cis Golgi, VSV-G trafficking across the Golgi stack is significantly accelerated. Monensin (Mo) is known to disrupt the Golgi structure and blocks TGN exit (Fliesler and Basinger, 1987) and thus was used as a control. As expected, monensin treatment resulted in VSV-G accumulation in the Golgi (Figures 3A–3C).
Figure 3.

TG-Induced Golgi Fragmentation Has Minor Effect on Protein Trafficking
(A) Cells were transfected with the Str-li_VSVG wt-SBP-EGFP plasmid for 16 h followed by a 30-min treatment with DMSO, 250 nM TG, or 10 μM monensin (Mo) at 37°C. Cells were then incubated with complete medium containing 40 μM biotin (chase) for the indicated times, lysed and treated with (+) or without (−) EndoH, and analyzed by Western blot for GFP.
(B) Quantification of (A) for the percentage of EndoH-resistant VSV-G from three independent experiments. Quantitation results are shown as Mean ± SEM. Statistical analyses were performed using two-tailed Student's t-tests by comparing with the control (∗p ≤ 0.05).
(C) Representative images of (A) showing the subcellular localization of VSVG-EGFP at indicated time points after biotin chase. Scale bar, 20 μm.
(D) Fluorescent images showing the subcellular localization of ManII-GFP in cells treated with DMSO (Ctrl) or 250 nM TG at the indicated time points of BFA washout. ManII-GFP appears in the Golgi area beginning at 60 min, whereas this takes longer in control cells. Scale bar, 20 μm.
To confirm these results using an alternative approach, we treated cells with Brefeldin A (BFA) to accumulate ManII-GFP in the ER. We then washed out BFA and analyzed ManII-GFP in ER-to-Golgi trafficking. The results showed that ManII-GFP started to accumulate in the Golgi at 60 min of BFA washout in the presence of TG, whereas the same observation occurred at 90 min in the control (Figure 3D).
TG Induces Golgi Fragmentation Through PKCα Activation
Given that phosphorylation of Golgi structural proteins has been shown to cause Golgi fragmentation in physiological conditions such as in mitosis (Tang et al., 2010, Wang et al., 2003, Xiang and Wang, 2010), as well as in pathological conditions such as in Alzheimer disease (Joshi et al., 2014), we explored the possibility that phosphorylation of Golgi structural proteins may play a role in TG-induced Golgi fragmentation. We treated cells with staurosporine, a non-selective kinase inhibitor, and a number of specific inhibitors of calcium-related kinases such as protein kinase Cs (PKCs) and Ca2+/calmodulin-dependent protein kinases (CAMKs). As shown in Figures S5C and S5D, staurosporine significantly reduced Golgi fragmentation in TG-treated cells. In addition, Bisindolylmaleimide I (BIM1), a selective PKC inhibitor, and KN-93, an inhibitor of CAMKII, also partially reduced Golgi fragmentation in TG-treated cells, whereas the myosin light-chain kinase inhibitor ML-7 and the protein kinase A (PKA) inhibitors H-89 and PKI had no such effects (Figures 4A, 4B, S5C, and S5D). These results suggest that either PKC and/or CAMKII is involved in TG-induced Golgi fragmentation. Because both BIM1 and KN-93 inhibitors have pleiotropic effects, we selected two alternative drugs, Gö6976 and KN-62, to inhibit PKC and CAMKII, respectively. Although Gö6976 inhibited TG-induced Golgi fragmentation effectively, KN-62 had no effect (Figures 4A and 4B), suggesting a major role of PKC in TG-induced Golgi fragmentation.
Figure 4.

TG induces Golgi Fragmentation through PKCα
(A) Inhibition of PKC reduces TG-induced Golgi fragmentation. HeLa cells were pre-treated with DMSO, BAPTA-AM (B/AM, 60 μM for 10 min), staurosporine (STS, general kinase inhibitor, 2 μM for 10 min), KN-62 or KN-93 (CAMKII inhibitors, 10 μM and 5 μM, respectively, 10 min), or BIM1 or Gӧ6976 (PKC inhibitors, 2 μM and 4 μM, respectively, 10 min), and then with 250 nM TG for 20 min followed by immunostaining of GM130 and TGN46. Scale bar, 20 μm.
(B) Quantitation of cells in (A) with fragmented Golgi based on the GM130 pattern in the cell.
(C) HeLa cells were treated with 100 nM PMA for 1 h; 4-alpha-PMA of the same concentration was used as a control. Scale bar, 20 µm.
(D) Quantitation of cells with fragmented Golgi in (C).
(E) Cells treated as in (C) were analyzed by Western blot to show that PMA treatment does not change the PKCα expression level.
(F) Activation of ectopically expressed PKCα triggers its Golgi localization (indicated by “→”) and Golgi fragmentation (∗). Cells transfected with GFP or PKCα-GFP were treated with 100 nM PMA or 4-alpha for 1 h. Note the fragmented Golgi in these cells upon PMA treatment. Scale bar, 20 μm.
(G) Quantitation of cells in (F) with fragmented Golgi.
(H) Western blot of cells from (F) showing that ectopic PKC expression does not alter the endogenous PKCα expression level.
(I) HeLa cells were transfected with control (Ctrl-i) or PKC-specific siRNA for 48 h and then treated with 250 nM TG for 20 min. Cells were fixed and stained for GM130 (green) to show the Golgi structure. Scale bar, 20 μm.
(J) Quantitation of Golgi fragmentation of cells in (I).
(K) Cells in (I) were blotted for endogenous PKCα to evaluate the siRNA knockdown efficiency.
All quantitation results are shown as Mean ± SEM from three independent experiments. Statistical analyses were performed using two-tailed Student's t-tests (∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001; NS, non-significant).
To further confirm that PKC activation causes Golgi fragmentation, we treated cells with phorbol 12-myristate 13-acetate (PMA), a widely used PKC activator, and its inactive enantiomer, 4-alpha-phorbol myristate acetate (4-alpha). The results showed that 4-alpha had no effect on the Golgi structure, whereas PMA treatment caused Golgi fragmentation (Figures 4C and 4D), although 4-alpha and PMA had no effect on the level of PKC expression (Figure 4E). In addition, expression of CAMKIIβ had no effect on the Golgi morphology (Figures S5E and S5F). There have been reports that activation of MAPK/ERK or PKD signaling causes Golgi fragmentation (Jamora et al., 1999, Jesch et al., 2001), we therefore inhibited these two kinases with U0126 or H-89, respectively. Pre-treatment of cells with these kinase inhibitors did not prevent Golgi fragmentation upon the addition of TG (Figures S5G and S5H). Taken together, these results indicate that TG induces Golgi fragmentation through PKC activation.
PKC has multiple isoforms including α, βI, βII, γ, δ, ϵ, η, ζ, and ι (Kajimoto et al., 2001). To identify the PKC isoform responsible for TG-induced Golgi fragmentation, we expressed GFP-tagged PKC isoforms, including all four known classical PKC (cPKC) isoforms (α, βI, βII, γ) that respond to Ca2+ stimuli, one from the non-calcium responsive novel PKC (nPKC, δ) and one from the atypical PKC (nPKC, ζ) subfamily (Figures S6A and S6B). To enhance the activity of expressed PKC, we also treated cells with PMA, using 4-alpha as a control. The results showed that expression of PKCα and treatment of cells with PMA increased Golgi fragmentation (Figures 4F–4H, S6A, and S6B). Interestingly, in addition to the localization to the plasma membrane as previously reported (Becker and Hannun, 2003), wild-type PKCα-GFP was also concentrated on the Golgi upon PMA treatment, as indicated by the colocalization with GM130 (Figures 4F and S6B), whereas other PKC isoforms, or the inactive PKCα K368R mutant (Baier-Bitterlich et al., 1996), did not show the same phenotype (Figures 4F and S6B–S6D). To further specify that PKCα mediates TG-induced Golgi fragmentation, we knocked down PKCα in cells with siRNA. The results showed that PKCα depletion reduces Golgi fragmentation after TG treatment (Figures 4I–4K). Taken together, these results demonstrate that PKCα activation causes Golgi fragmentation.
PKCα Induces Golgi Fragmentation Through GRASP55 Phosphorylation
Because activated PKCα localizes to the Golgi, we thought it might phosphorylate Golgi structural proteins. To identify potential PKCα targets on the Golgi, we performed gel mobility shift assays on a number of Golgi structural proteins, tethering factors, and SNARE proteins after TG treatment (Figure S6E). To ensure that the band shift was caused by phosphorylation, we also applied staurosporine (2 μM for 10 min prior to TG treatment) to TG-treated cells to broadly inhibit phosphorylation. Among the proteins tested, GRASP55 and GRASP65 showed a smear above the main bands (Figure S6E), indicating a partial phosphorylation of the proteins. To increase the resolution of phosphorylated proteins we utilized phos-tag gels, which showed GRASP55, but not GRASP65, to be significantly shifted up after TG treatment (250 nM, 1 h) (Figure S6F). TG-induced mobility shift of GRASP55 was not seen upon Tm treatment (Figure 5A, lanes 2 vs. 3) and was less dramatic than that by nocodazole (Noc) treatment that blocks cells in mitosis when GRASP55 is fully phosphorylated (Figure 5A, lanes 3 vs. 4) (Xiang and Wang, 2010), indicating that TG induced partial phosphorylation of GRASP55. The mobility shift of GRASP55 triggered by TG treatment was abolished by the addition of staurosporine (Figure 5C, lanes 4 vs. 3; Figure S6F, lanes 3 vs. 2), validating the mobility shift by phosphorylation. In addition, incubation of purified recombinant GRASP55 with purified PKCα caused GRASP55 phosphorylation, confirming that PKCα can directly phosphorylate GRASP55 (Figure 5D). In this experiment, PKCα was also autophosphorylated (Figure 5D). Taken together, these results demonstrate that TG treatment activates PKCα, which subsequently phosphorylates GRASP55.
Figure 5.

TG Induces Golgi Fragmentation through GRASP55 Phosphorylation
(A) GRASP55 is phosphorylated upon TG treatment. HeLa cells treated with Tm, TG, or nocodazole (Noc) were analyzed by phos-tag gels and Western blot. Note the mobility shift of GRASP55 upon TG treatment compared with control (Ctrl). Nocodazole-arrested mitotic cells were used as a positive control for GRASP55 phosphorylation.
(B) TG-induced GRASP55 mobility shift is abolished by kinase inhibition. Cells were pre-treated with 2 μM staurosporine (STS) for 10 min and then with 250 nM TG for 1 h followed by analysis with phos-tag gels and Western blot.
(C) Quantitation of GRASP55 phosphorylation in TG-treated cells. The intensity of the phosphorylated (upper) band was quantified by densitometry analysis and plotted relative to the intensities of the full length (lower) band. Shown are the results of relative phosphorylation of GRASP55 from three independent experiments.
(D) PKCα phosphorylates GRASP55 in vitro. Purified PKCα and GRASP55 were incubated in a kinase buffer with or without ATP as indicated and analyzed by phos-tag gels and Western blot for GRASP55 and PKCα.
(E) Mapping the phosphorylation site on GRASP55 by expressing GRASP55 truncation mutants. Indicated GFP-tagged GRASP55 constructs were expressed in HeLa cells. After TG treatment, GRASP55 was analyzed by mobility shift as in (A). Note the mobility shift in lanes 8, 10, and 12.
(F) Quantitation of GRASP55 phosphorylation in TG-treated cells from (E).
(G) TG-induced Golgi fragmentation is rescued by the expression of non-phosphorylatable GRASP55 proteins. Cells expressing the indicated GRASP55 constructs were stained for giantin. Scale bar, 20 µm.
(H) Quantitation of cells in (G) with fragmented Golgi. Results are shown as Mean ± SEM.
Statistical analyses were performed using two-tailed Student's t-tests (∗p ≤ 0.05; ∗∗p ≤ 0.01).
GRASP55 contains an N-terminal GRASP domain that forms dimers and oligomers and a C-terminal SPR domain with multiple phosphorylation sites (Xiang and Wang, 2010, Zhang and Wang, 2015). To map the PKCα phosphorylation site on GRASP55, we expressed GFP-tagged GRASP55 truncation mutants (Zhang et al., 2018), treated the cells with TG, and determined their phosphorylation. A visible mobility shift of the GRASP55 variants was observed on the mutants possessing amino acids (aa)251-300 but not the truncated forms shorter than aa250 (Figures 5E and 5F). To further determine the functional consequence of GRASP55 phosphorylation, we expressed these constructs and treated cells with or without TG. The exogenously expressed GRASP55 truncation mutants were targeted to the Golgi as indicated by giantin as a Golgi marker but had no impact on the Golgi structure (Figure S6G). However, when cells were treated with TG, expression of the N-terminal aa250 or shorter reduced TG-induced Golgi fragmentation, whereas expression of N-terminal aa300 or longer had no significant effect (Figures 5G and 5H). These results demonstrated that phosphorylation of GRASP55 within aa251-300 is important for TG-induced Golgi fragmentation.
Histamine Modulates the Golgi Structure Through the Same Pathway as TG Treatment
It is known that histamine activates Ca2+-dependent PKC isoforms and upregulates cytokine secretion via the release of calcium from the ER into the cytosol (Matsubara et al., 2005). It has also been shown that histamine triggers protein secretion and Golgi fragmentation (Saini et al., 2010), but the underlying mechanism has not been revealed. Therefore, we treated HeLa cells with histamine and determined the effect on Golgi morphology. As shown in Figures 6A and 6B, histamine treatment induced Golgi fragmentation in a dose- and time-dependent manner. More than 40% of cells possessed fragmented Golgi after 100 μM histamine treatment for 1 h, a concentration and time often used in previous studies (Sahoo et al., 2017, Xie et al., 2018). Subsequent EM analysis confirmed that histamine treatment induced alterations in the Golgi structure, including fewer cisternae per stack, shorter cisternae, and an increased number of Golgi-associated vesicles (Figures 6C, 6D, and S7A).
Figure 6.

Histamine Induces Golgi Fragmentation through PKC Activation
(A) HeLa cells were treated with indicated concentrations of histamine for 1 h, stained for GM130, and quantified for the percentage of cells with fragmented Golgi. Shown are the quantitation results.
(B) HeLa cells were treated with 100 μM histamine for the indicated times and analyzed as in (A). Note that histamine induced Golgi fragmentation within 2 h.
(C) Electron micrographs of Golgi profiles in HeLa cells treated with DMSO control (Ctrl, left panel) or 100 μM histamine for 1 h (right panel). Note the reduced size of the Golgi in histamine-treated cells. Scale bar, 0.5 μm.
(D) Quantitation of the morphological features of the Golgi stacks in (C). For statistics, histamine treated cells were compared with control cells.
(E) Cells were pre-treated with (+) or without (−) the PKC inhibitor Gö6976 at 37°C for 10 min followed by the addition of 100 μM histamine for 60 min. Scale bar, 20 μm.
(F) Quantitation of cells with fragmented Golgi in (E).
(G) Expression of the GRASP55-GCaMP7 Golgi Ca2+ sensor. HeLa cells transfected with indicated proteins were analyzed by Western blot for GFP.
(H) Histamine treatment increases the GRASP55-GCaMP7 signal. HeLa cells were co-transfected with mCherry-GM130 (red) and GRASP55-GCAMP7 (green). Shown are still frames before (left panel) or after (right) histamine was added. Scale bar, 5 µm.
(I) TG and ionomycin treatments increase the GRASP55-GCaMP7 signal. HeLa cells expressing GRASP55-GCaMP7 were treated with 100 μM histamine (Hist), 250 nM TG, or 1 μM ionomycin (Io) for 1 h. Shown are the quantitation of the fluorescence intensity before and after the drug was added.
All quantitation results are shown as Mean ± SEM. Statistical analyses were performed using two-tailed Student's t-tests (∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001).
It has been previously shown that histamine activates Gβγ, which causes TGN fragmentation (Saini et al., 2010). Because HeLa cells do not express Gβγ, and histamine treatment triggers fragmentation of the entire Golgi stack (Figures 6C and 6D), the Golgi fragmentation observed in our study may occur through a different mechanism. Indeed, as TG, histamine-induced Golgi fragmentation also depended on PKC, as the addition of the PKC inhibitor Gö6976 reduced histamine-induced Golgi fragmentation (Figures 6E and 6F). To investigate the functional consequence of histamine treatment on Golgi function, VSV-G trafficking experiments were performed as above. As shown in Figures S7B and S7C, histamine treatment slightly decreased VSV-G trafficking within 45-min release but then began to increase the trafficking speed in the Golgi at later time points. These results demonstrate that histamine treatment affects Golgi structure and function through a similar mechanism as TG.
Although it has been well documented that TG or histamine treatment elevates Ca2+ level in the cytosol, whether this is also true for the Ca2+ level in the Golgi region has not been reported. To test the effect of TG or histamine treatment on the Ca2+ level in the Golgi region in real time, we fused the Ca2+ probe GCaMP (Muto et al., 2013) to GRASP55, expressed the GRASP55-GCaMP construct in cells (Figure 6G), and performed live cell imaging. Treatment of cells with 100 μM histamine caused a robust calcium spike (Figure 6H; Video S1). Subsequent experiments using this novel Golgi-localized Ca2+ probe demonstrated that treatment of cells with TG and Io also significantly elevated the Ca2+ level in the Golgi (Figure 6I). Taken together, our results demonstrated that histamine or TG treatment elevates the Ca2+ level in the Golgi region, which subsequently activates PKCα, leading to GRASP55 phosphorylation and Golgi fragmentation. Thus, this study revealed a novel mechanism of how histamine, and perhaps other drugs, modulates Golgi structure and function.
Live cell imaging of HeLa cells expressing both mCherry-GM130 (red) and GRASP55-GCaMP(green) treated with 100 μM histamine. The time when histamine was added is indicated in the video. Note the elevated green signal when histamine was added.
Discussion
In this study, by comparing Golgi fragmentation with ER stress in response to TG, Tm, and DTT treatments, we uncovered a novel signaling pathway through which increased cytosolic Ca2+ triggers Golgi fragmentation through PKCα activation and GRASP55 phosphorylation. Significantly, we also demonstrated that histamine modulates the Golgi structure and function via a similar mechanism, which opens a new window through which we can better understand the effect of histamine on cell physiology.
One possible model of Golgi stress is that the expanding capacity of the ER during cellular stress leads to the failure of the Golgi as it is over-burdened with misfolded or improperly folded proteins, affecting its functions such as glycosylation (Oku et al., 2011). However, our results do not support this hypothesis for two reasons. First, although three ER stress inducers, TG, Tm, and DTT, all induce ER stress, only TG treatment causes Golgi fragmentation. Second, TG induces Golgi fragmentation at a low dose and time when UPR is undetectable. These results demonstrate that Golgi fragmentation occurs independently of ER stress, perhaps via the modification of pre-existing cellular materials. Therefore, the Golgi may possess its own mechanism to sense and respond to stress alongside or completely separate from the ER. Furthermore, our study revealed a novel mechanism that coordinates Golgi structure and perhaps function: TG treatment increases cytoplasmic Ca2+, which activates PKCα, which subsequently phosphorylates GRASP55, impairing its function in Golgi structure formation. GRASP55 therefore provides the conceptual link between an extracellular cue and Golgi morphological change during stress.
GRASP55 is comprised of an N-terminal GRASP domain (aa1-212) that forms dimers and oligomers and functions as a membrane tether to maintain an intact Golgi structure and an SPR domain (aa212-454) that undergoes post-translational modifications and functions as the regulatory domain of the protein (Xiang and Wang, 2010, Zhang and Wang, 2015). Originally, GRASP55 was found to be phosphorylated by ERK2 at T225 and T222 (Jesch et al., 2001). Subsequently, additional sites, such as S245 and T249, were identified to be phosphorylated in mitosis, which is required for mitotic Golgi disassembly (Xiang and Wang, 2010). Recently, GRASP55 was discovered to be de-O-GlcNAcylated upon energy or nutrient deprivation and regulates autophagosome maturation (Zhang et al., 2018, Zhang et al., 2019). These results indicate that GRASP55 is an excellent candidate to function as both a sensor and effector of cellular stresses. Thus, GRASP55 is likely a master regulator of Golgi structure formation, function, and stress responses.
Our in vitro kinase assay demonstrated that PKCα can directly phosphorylate GRASP55 likely on more than one site. The mobility shift of GRASP55 observed in cells consisted of only one obvious band, whereas the in vitro experiment showed two clear bands, suggesting that phosphorylation of GRASP55 in cells might occur less frequently. Using GRASP55 truncation mutants we mapped the site(s) of PKC-mediated phosphorylation to the aa251-300 region. Expression of truncation mutants of GRASP55 that lack this region significantly reduced TG-induced Golgi fragmentation. Previously, it has been shown by mass spectrometry that GRASP55 is phosphorylated on S441 after TG treatment, but the kinase mediating this phosphorylation is unknown (Gee et al., 2011). Although our results are consistent with this previous study, the exact phosphorylation site(s) need further investigation.
Histamine is a neuroendocrine hormone involved in the regulation of stomach acid secretion, brain function, and immune response; many of these functions involve secretion (Karpati et al., 2018, Sahoo et al., 2017, Xie et al., 2018). The role of histamine in immune response is often through the activation of the downstream kinase PKCα. For example, histamine enhances the secretion of granulocyte-macrophage colony stimulation factor (GM-CSF) and nerve growth factor (NGF) in different cell types, both through a PKCα-dependent mechanism (Sohen et al., 2001). Interestingly, histamine promotes HeLa cell proliferation and growth and has been shown to be elevated in cancers where Golgi is fragmented and secretion is enhanced. In our experiments, histamine induced a clear Golgi fragmentation phenotype, confirming a link between histamine and Golgi fragmentation. Additionally, expression of PKCα, but not other PKC isoforms, along with a stimulation with PMA, exhibited an additive Golgi fragmentation effect. Consistent with prior work showing that disassembly of Golgi stacks accelerates protein trafficking (Xiang et al., 2013), our findings therefore offer a mechanism for how histamine increases secretion of inflammatory factors.
How Ca2+ controls membrane trafficking at the plasma membrane has been well documented in regulated secretion in specific cell types such as neurons, neuroendocrine cells, and mast cells, whereas its role in other cell types is less well known. Ca2+ dynamic at the Golgi as well as its role in membrane trafficking at the Golgi is still an understudied area. There are EF-hand Ca2+ binding proteins associated in the Golgi. For example, Cab45 is located in the Golgi lumen, whereas Calnuc is found in both cell cytosol and membrane fractions (Lin et al., 1998). At the cis-Golgi, Calnuc binds Gαi and Gαs, which is thought to be important for vesicular trafficking (Lin et al., 2000). There are also P-Type ATPases (SPCAs) such as SPCA1 located in the Golgi that regulate Ca2+ homeostasis in the Golgi and control neural polarity (Sepulveda et al., 2009, Vanoevelen et al., 2007). Our study provided a novel link between thapsigargin and histamine treatment, elevation of Ca2+ concentration in the Golgi region, activation of PKC and phosphorylation of Golgi GRASP55, and modification of Golgi structure and function.
Our study revealed that TG induces Golgi fragmentation through increasing cytosolic Ca2+ and GRASP55 phosphorylation. A similar case has been described previously in Alzheimer disease, where cytosolic calcium increases by Aβ treatment triggers activation of a cytosolic protease, calpain, which cleaves p35 to generate p25 and activate Cdk5, a cytoplasmic kinase that is highly expressed in neurons (Lew et al., 1994). Subsequently, activated Cdk5 phosphorylates GRASP65 and perhaps other Golgi structural proteins, leading to Golgi fragmentation (Joshi et al., 2014, Joshi et al., 2015). Although PKC and GRASP55 were not the focus in this study, expression of a phosphorylation-deficient mutant of GRASP55 significantly reduced Golgi fragmentation as well as Aβ production. Taken together, our studies indicate that the Golgi is sensitive to cellular stimuli and stresses as in disease conditions,and responds to signaling cues to adjust its structure and function through increasing cytosolic Ca2+ and GRASP55 modification. Future studies defining the detailed mechanisms may help understand disease pathologies with Golgi and trafficking defects.
Limitations of the Study
Phosphorylation occurs more frequently in vitro than in cells. The exact phosphorylation site(s) on GRASP55 needs further investigation.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Drs. Mohammed Akaaboune, Peter Arvan, Hesham El-Shewy, Yusuf Hannun, Isabel Martinez Peña, Franck Perez, Joachim Seemann, Haoxing Xu, and Kezhong Zhang for reagents; Drs. Peter Blumberg, John Kuwada, Edward Stuenkel, and Haoxing Xu for technical assistance; Gregg Sobocinski, Dana Holcomb, Miles McKenna, and Edwin De Feijter for microscope and equipment expertise. This work was supported by the National Institutes of Health (Grants GM112786, GM105920, and GM130331), MCubed and the Fastforward Protein Folding Disease Initiative of the University of Michigan to Y.W.. S.I. is a graduate student who has been partially supported by the Mary Sue and Kenneth Coleman Endowed Fellowship Fund from the University of Michigan. Many thanks to past and current members of the Wang lab, including Erpan Ahat, Michael Bekier, Shijiao Huang, Courtney Killeen, Haoran Huang, and Ron Benyair.
Author Contributions
Conception and design: S. R., S. I., and Y. W.
Development of methodology: S. I., S. R., X. Z., J. Z., J. L., and Y. W.
Acquisition of data: S. I., S. R., M. F., X. Z., J. Z., and D. E.
Analysis and interpretation of data: S. I., S. R., M. F., and Y. W.
Writing, review, and/or revision of the manuscript: S. I., and Y. W.
Administrative, technical, or material support: S. I., and Y. W.
Study supervision: Y. W.
Declaration of Interests
The authors declare no competing financial interests.
Published: March 27, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.100952.
Data and CodeAvailability
All data from this study is available upon request.
Supplemental Information
References
- Ahat E., Li J., Wang Y. New insights into the Golgi stacking proteins. Front. Cell Dev. Biol. 2019;7:131. doi: 10.3389/fcell.2019.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahat E., Xiang Y., Zhang X., Bekier M.E., Wang Y. GRASP depletion-mediated Golgi destruction decreases cell adhesion and migration via the reduction of alpha5beta1 integrin. Mol. Biol. Cell. 2019;30:766–777. doi: 10.1091/mbc.E18-07-0462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baier-Bitterlich G., Uberall F., Bauer B., Fresser F., Wachter H., Grunicke H., Utermann G., Altman A., Baier G. Protein kinase C-theta isoenzyme selective stimulation of the transcription factor complex AP-1 in T lymphocytes. Mol. Cell Biol. 1996;16:1842–1850. doi: 10.1128/mcb.16.4.1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker K.P., Hannun Y.A. cPKC-dependent sequestration of membrane-recycling components in a subset of recycling endosomes. J. Biol. Chem. 2003;278:52747–52754. doi: 10.1074/jbc.M305228200. [DOI] [PubMed] [Google Scholar]
- Bekier M.E., 2nd, Wang L., Li J., Huang H., Tang D., Zhang X., Wang Y. Knockout of the Golgi stacking proteins GRASP55 and GRASP65 impairs Golgi structure and function. Mol. Biol.Cell. 2017;28:2833–2842. doi: 10.1091/mbc.E17-02-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boncompain G., Divoux S., Gareil N., de Forges H., Lescure A., Latreche L., Mercanti V., Jollivet F., Raposo G., Perez F. Synchronization of secretory protein traffic in populations of cells. Nat. Methods. 2012;9:493–498. doi: 10.1038/nmeth.1928. [DOI] [PubMed] [Google Scholar]
- Chen D., Purohit A., Halilovic E., Doxsey S.J., Newton A.C. Centrosomal anchoring of protein kinase C betaII by pericentrin controls microtubule organization, spindle function, and cytokinesis. J. Biol. Chem. 2004;279:4829–4839. doi: 10.1074/jbc.M311196200. [DOI] [PubMed] [Google Scholar]
- Cooke M., Magimaidas A., Casado-Medrano V., Kazanietz M.G. Protein kinase C in cancer: the top five unanswered questions. Mol. Carcinog. 2017;56:1531–1542. doi: 10.1002/mc.22617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Homasany B.S.E.D., Volkov Y., Takahashi M., Ono Y., Keryer G., Delouvee A., Looby E., Long A., Kelleher D. The scaffolding protein CG-NAP/AKAP450 is a critical integrating component of the LFA-1-induced signaling complex in migratory T cells. J. Immunol. 2005;175:7811–7818. doi: 10.4049/jimmunol.175.12.7811. [DOI] [PubMed] [Google Scholar]
- Farhan H., Wendeler M.W., Mitrovic S., Fava E., Silberberg Y., Sharan R., Zerial M., Hauri H.P. MAPK signaling to the early secretory pathway revealed by kinase/phosphatase functional screening. J. Cell Biol. 2010;189:997–1011. doi: 10.1083/jcb.200912082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinstein T.N., Linstedt A.D. GRASP55 regulates Golgi ribbon formation. Mol. Biol.Cell. 2008;19:2696–2707. doi: 10.1091/mbc.E07-11-1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fliesler S.J., Basinger S.F. Monensin stimulates glycerolipid incorporation into rod outer segment membranes. J. Biol. Chem. 1987;262:17516–17523. [PubMed] [Google Scholar]
- Fujita Y., Okamoto K. Golgi apparatus of the motor neurons in patients with amyotrophic lateral sclerosis and in mice models of amyotrophic lateral sclerosis. Neuropathology. 2005;25:388–394. doi: 10.1111/j.1440-1789.2005.00616.x. [DOI] [PubMed] [Google Scholar]
- Gee H.Y., Noh S.H., Tang B.L., Kim K.H., Lee M.G. Rescue of DeltaF508-CFTR trafficking via a GRASP-dependent unconventional secretion pathway. Cell. 2011;146:746–760. doi: 10.1016/j.cell.2011.07.021. [DOI] [PubMed] [Google Scholar]
- Gonatas N.K., Gonatas J.O., Stieber A. The involvement of the Golgi apparatus in the pathogenesis of amyotrophic lateral sclerosis, Alzheimer's disease, and ricin intoxication. Histochem. Cell Biol. 1998;109:591–600. doi: 10.1007/s004180050257. [DOI] [PubMed] [Google Scholar]
- Griner E.M., Kazanietz M.G. Protein kinase C and other diacylglycerol effectors in cancer. Nat. Rev. Cancer. 2007;7:281–294. doi: 10.1038/nrc2110. [DOI] [PubMed] [Google Scholar]
- Hilditch-Maguire P., Trettel F., Passani L.A., Auerbach A., Persichetti F., MacDonald M.E. Huntingtin: an iron-regulated protein essential for normal nuclear and perinuclear organelles. Hum. Mol. Genet. 2000;9:2789–2797. doi: 10.1093/hmg/9.19.2789. [DOI] [PubMed] [Google Scholar]
- Ito Y., Takeda Y., Seko A., Izumi M., Kajihara Y. Functional analysis of endoplasmic reticulum glucosyltransferase (UGGT): synthetic chemistry’s initiative in glycobiology. Semin. Cell Dev. Biol. 2015;41:90–98. doi: 10.1016/j.semcdb.2014.11.011. [DOI] [PubMed] [Google Scholar]
- Jamora C., Yamanouye N., Van Lint J., Laudenslager J., Vandenheede J.R., Faulkner D.J., Malhotra V. Gbetagamma-mediated regulation of Golgi organization is through the direct activation of protein kinase D. Cell. 1999;98:59–68. doi: 10.1016/S0092-8674(00)80606-6. [DOI] [PubMed] [Google Scholar]
- Jesch S.A., Lewis T.S., Ahn N.G., Linstedt A.D. Mitotic phosphorylation of Golgi reassembly stacking protein 55 by mitogen-activated protein kinase ERK2. Mol. Biol. Cell. 2001;12:1811–1817. doi: 10.1091/mbc.12.6.1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones K.T., Sharpe G.R. Thapsigargin raises intracellular free calcium levels in human keratinocytes and inhibits the coordinated expression of differentiation markers. Exp. Cell Res. 1994;210:71–76. doi: 10.1006/excr.1994.1011. [DOI] [PubMed] [Google Scholar]
- Joshi G., Bekier M.E., 2nd, Wang Y. Golgi fragmentation in Alzheimer’s disease. Front. Neurosci. 2015;9:340. doi: 10.3389/fnins.2015.00340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi G., Chi Y., Huang Z., Wang Y. Abeta-induced Golgi fragmentation in Alzheimer's disease enhances Abeta production. Proc. Natl. Acad. Sci. U S A. 2014;111:E1230–E1239. doi: 10.1073/pnas.1320192111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimoto T., Ohmori S., Shirai Y., Sakai N., Saito N. Subtype-specific translocation of the delta subtype of protein kinase C and its activation by tyrosine phosphorylation induced by ceramide in HeLa cells. Mol. Cell Biol. 2001;21:1769–1783. doi: 10.1128/MCB.21.5.1769-1783.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpati A., Yoshikawa T., Nakamura T., Iida T., Matsuzawa T., Kitano H., Harada R., Yanai K. Histamine elicits glutamate release from cultured astrocytes. J. Pharmacol. Sci. 2018;137:122–128. doi: 10.1016/j.jphs.2018.05.002. [DOI] [PubMed] [Google Scholar]
- Kim H., Zamel R., Bai X.H., Liu M. PKC activation induces inflammatory response and cell death in human bronchial epithelial cells. PLoS One. 2013;8:e64182. doi: 10.1371/journal.pone.0064182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause W.J. Brunner's glands: a Structural,Histochemical and pathological profile. Prog. Histochem. Cytochem. 2000;35:255–367. [PubMed] [Google Scholar]
- Lew J., Huang Q.Q., Qi Z., Winkfein R.J., Aebersold R., Hunt T., Wang J.H. A brain-specific activator of cyclin-dependent kinase 5. Nature. 1994;371:423–426. doi: 10.1038/371423a0. [DOI] [PubMed] [Google Scholar]
- Li J., Ahat E., Wang Y. Springer Nature; 2019. Golgi Structure and Function in Health, Stress and Diseases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Tang D., Ireland S.C., Wang Y. DjA1 maintains Golgi integrity via interaction with GRASP65. Mol. Biol. Cell. 2019;30:478–490. doi: 10.1091/mbc.E18-10-0613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin P., Fischer T., Weiss T., Farquhar M.G. Calnuc, an EF-hand Ca(2+) binding protein, specifically interacts with the C-terminal alpha5-helix of G(alpha)i3. Proc. Natl. Acad. Sci. U S A. 2000;97:674–679. doi: 10.1073/pnas.97.2.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin P., Le-Niculescu H., Hofmeister R., McCaffery J.M., Jin M., Hennemann H., McQuistan T., De Vries L., Farquhar M.G. The mammalian calcium-binding protein, nucleobindin (CALNUC), is a Golgi resident protein. J. Cell Biol. 1998;141:1515–1527. doi: 10.1083/jcb.141.7.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara M., Tamura T., Ohmori K., Hasegawa K. Histamine H1 receptor antagonist blocks histamine-induced proinflammatory cytokine production through inhibition of Ca2+-dependent protein kinase C, Raf/MEK/ERK and IKK/I kappa B/NF-kappa B signal cascades. Biochem. Pharmacol. 2005;69:433–449. doi: 10.1016/j.bcp.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Mizuno Y., Hattori N., Kitada T., Matsumine H., Mori H., Shimura H., Kubo S., Kobayashi H., Asakawa S., Minoshima S. Familial Parkinson's disease. Alpha-synuclein and parkin. Adv. Neurol. 2001;86:13–21. [PubMed] [Google Scholar]
- Mourelatos Z., Gonatas N.K., Stieber A., Gurney M.E., Dal Canto M.C. The Golgi apparatus of spinal cord motor neurons in transgenic mice expressing mutant Cu,Zn superoxide dismutase becomes fragmented in early, preclinical stages of the disease. Proc. Natl. Acad. Sci. U S A. 1996;93:5472–5477. doi: 10.1073/pnas.93.11.5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muto A., Ohkura M., Abe G., Nakai J., Kawakami K. Real-time visualization of neuronal activity during perception. Curr. Biol. 2013;23:307–311. doi: 10.1016/j.cub.2012.12.040. [DOI] [PubMed] [Google Scholar]
- Nishizuka Y. Protein kinase C and lipid signaling for sustained cellular responses. FASEB J. 1995;9:484–496. [PubMed] [Google Scholar]
- Oku M., Tanakura S., Uemura A., Sohda M., Misumi Y., Taniguchi M., Wakabayashi S., Yoshida H. Novel cis-acting element GASE regulates transcriptional induction by the Golgi stress response. Cell Struct. Funct. 2011;36:1–12. doi: 10.1247/csf.10014. [DOI] [PubMed] [Google Scholar]
- Petrosyan A., Holzapfel M.S., Muirhead D.E., Cheng P.W. Restoration of compact Golgi morphology in advanced prostate cancer enhances susceptibility to galectin-1-induced apoptosis by modifying mucin O-glycan synthesis. Mol. Cancer Res. 2014;12:1704–1716. doi: 10.1158/1541-7786.MCR-14-0291-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthenveedu M.A., Bachert C., Puri S., Lanni F., Linstedt A.D. GM130 and GRASP65-dependent lateral cisternal fusion allows uniform Golgi-enzyme distribution. Nat. Cell Biol. 2006;8:238–248. doi: 10.1038/ncb1366. [DOI] [PubMed] [Google Scholar]
- Rambourg A., Clermont Y., Chretien M., Olivier L. Modulation of the Golgi apparatus in stimulated and nonstimulated prolactin cells of female rats. Anat. Rec. 1993;235:353–362. doi: 10.1002/ar.1092350304. [DOI] [PubMed] [Google Scholar]
- Sahoo N., Gu M., Zhang X., Raval N., Yang J., Bekier M., Calvo R., Patnaik S., Wang W., King G. Gastric acid secretion from parietal cells is mediated by a Ca(2+)efflux channel in the tubulovesicle. Dev.Cell. 2017;41:262–273 e266. doi: 10.1016/j.devcel.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini D.K., Karunarathne W.K., Angaswamy N., Saini D., Cho J.H., Kalyanaraman V., Gautam N. Regulation of Golgi structure and secretion by receptor-induced G protein betagamma complex translocation. Proc. Natl. Acad. Sci. U S A. 2010;107:11417–11422. doi: 10.1073/pnas.1003042107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepulveda M.R., Vanoevelen J., Raeymaekers L., Mata A.M., Wuytack F. Silencing the SPCA1 (secretory pathway Ca2+-ATPase isoform 1) impairs Ca2+ homeostasis in the Golgi and disturbs neural polarity. J. Neurosci. 2009;29:12174–12182. doi: 10.1523/JNEUROSCI.2014-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sewell R., Backstrom M., Dalziel M., Gschmeissner S., Karlsson H., Noll T., Gatgens J., Clausen H., Hansson G.C., Burchell J. The ST6GalNAc-I sialyltransferase localizes throughout the Golgi and Is responsible for the synthesis of the tumor-associated sialyl-Tn O-glycan in human breast cancer. J. Biol. Chem. 2006;281:3586–3594. doi: 10.1074/jbc.M511826200. [DOI] [PubMed] [Google Scholar]
- Sohen S., Ooe H., Hashima M., Nonaka T., Fukuda K., Hamanishi C. Activation of histamine H1 receptor results in enhanced proteoglycan synthesis by human articular chondrocyte: involvement of protein kinase C and intracellular Ca(2+) Pathophysiology. 2001;8:93–98. doi: 10.1016/s0928-4680(01)00066-9. [DOI] [PubMed] [Google Scholar]
- Sutterlin C., Polishchuk R., Pecot M., Malhotra V. The Golgi-associated protein GRASP65 regulates spindle dynamics and is essential for cell division. Mol. Biol.Cell. 2005;16:3211–3222. doi: 10.1091/mbc.E04-12-1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan X., Banerjee P., Guo H.F., Ireland S., Pankova D., Ahn Y.H., Nikolaidis I.M., Liu X., Zhao Y., Xue Y. Epithelial-to-mesenchymal transition drives a pro-metastatic Golgi compaction process through scaffolding protein PAQR11. J. Clin. Invest. 2016;127:117–131. doi: 10.1172/JCI88736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D., Wang Y. Cell cycle regulation of Golgi membrane dynamics. Trends Cell Biol. 2013;23:296–304. doi: 10.1016/j.tcb.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D., Yuan H., Wang Y. The role of GRASP65 in Golgi cisternal stacking and cell cycle progression. Traffic. 2010;11:827–842. doi: 10.1111/j.1600-0854.2010.01055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D., Yuan H., Vielemeyer O., Perez F., Wang Y. Sequential phosphorylation of GRASP65 during mitotic Golgi disassembly. Biol. Open. 2012;1:1204–1214. doi: 10.1242/bio.20122659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thayer D.A., Jan Y.N., Jan L.Y. Increased neuronal activity fragments the Golgi complex. Proc. Natl. Acad. Sci. U S A. 2013;110:1482–1487. doi: 10.1073/pnas.1220978110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanoevelen J., Dode L., Raeymaekers L., Wuytack F., Missiaen L. Diseases involving the Golgi calcium pump. Subcell Biochem. 2007;45:385–404. doi: 10.1007/978-1-4020-6191-2_14. [DOI] [PubMed] [Google Scholar]
- Wang Y., Satoh A., Warren G. Mapping the functional domains of the Golgi stacking factor GRASP65. J. Biol. Chem. 2005;280:4921–4928. doi: 10.1074/jbc.M412407200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Seemann J. Golgi biogenesis. Cold Spring Harb. Perspect. Biol. 2011;3:a005330. doi: 10.1101/cshperspect.a005330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Seemann J., Pypaert M., Shorter J., Warren G. A direct role for GRASP65 as a mitotically regulated Golgi stacking factor. EMBO J. 2003;22:3279–3290. doi: 10.1093/emboj/cdg317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Wei J.H., Bisel B., Tang D., Seemann J. Golgi cisternal unstacking stimulates COPI vesicle budding and protein transport. PLoS One. 2008;3:e1647. doi: 10.1371/journal.pone.0001647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y., Wang Y. GRASP55 and GRASP65 play complementary and essential roles in Golgi cisternal stacking. J.Cell Biol. 2010;188:237–251. doi: 10.1083/jcb.200907132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y., Zhang X., Nix D.B., Katoh T., Aoki K., Tiemeyer M., Wang Y. Regulation of protein glycosylation and sorting by the Golgi matrix proteins GRASP55/65. Nat. Commun. 2013;4:1659. doi: 10.1038/ncomms2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie G., Wang F., Peng X., Liang Y., Yang H., Li L. Modulation of mast cell toll-like receptor 3 expression and cytokines release by histamine. Cell Physiol. Biochem. 2018;46:2401–2411. doi: 10.1159/000489646. [DOI] [PubMed] [Google Scholar]
- Xu C., Ma H., Inesi G., Al-Shawi M.K., Toyoshima C. Specific structural requirements for the inhibitory effect of thapsigargin on the Ca2+ ATPase SERCA. J. Biol. Chem. 2004;279:17973–17979. doi: 10.1074/jbc.M313263200. [DOI] [PubMed] [Google Scholar]
- Zhang X., Wang L., Ireland S.C., Ahat E., Li J., Bekier Ii M.E., Zhang Z., Wang Y. GORASP2/GRASP55 collaborates with the PtdIns3K UVRAG complex to facilitate autophagosome-lysosome fusion. Autophagy. 2019;15:1–14. doi: 10.1080/15548627.2019.1596480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Wang L., Lak B., Li J., Jokitalo E., Wang Y. GRASP55 senses glucose deprivation through O-GlcNAcylation to promote autophagosome-lysosome fusion. Dev.Cell. 2018;45:245–261.e6. doi: 10.1016/j.devcel.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Wang Y. GRASPs in Golgi structure and function. Front. Cell Dev. Biol. 2015;3:84. doi: 10.3389/fcell.2015.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Live cell imaging of HeLa cells expressing both mCherry-GM130 (red) and GRASP55-GCaMP(green) treated with 100 μM histamine. The time when histamine was added is indicated in the video. Note the elevated green signal when histamine was added.