

Abstract
Emerging evidence indicates that macrophage functional polarization is critically involved in the development of atherosclerosis (AS). Here, we examined the role of 5-aminolaevulinic acid (ALA)-mediated non-lethal sonodynamic therapy (NL-SDT) in macrophage-subset polarization and atherosclerotic lesion stability and explored the potential underlying mechanisms. Using Western diet-fed apolipoprotein E (apoE)−/− and green fluorescent protein (GFP)-positive bone marrow (BM) chimeric mouse models, we demonstrated that NL-SDT promoted phenotypic switching of both BM-derived and resident macrophages from M1 to M2 and significantly inhibited AS progression. Further mechanistic studies indicated that NL-SDT enhanced macrophage differentiation toward the M2 phenotype by activating the reactive oxygen species (ROS)–5′ AMP-activated protein kinase (AMPK)–mammalian target of rapamycin complex 1 (mTORC1)–autophagy signaling pathway in murine BM-derived M1 macrophages (BMDM1s). Moreover, NL-SDT drastically reduced lipid droplets, mainly by promoting apoAI-mediated cholesterol efflux in vitro. Specifically, administration of pharmacological inhibitors to the animal model showed a reciprocal effect on NL-SDT-induced macrophage polarization. These findings indicate that NL-SDT engages a virtuous cycle that enhances M1-to-M2 polarization, cholesterol efflux, and anti-inflammatory reactions in advanced plaque in vivo and in BMDM1s in vitro by activating the ROS–AMPK–mTORC1–autophagy pathway. This discovery might help elucidate the mechanism underlying NL-SDT as a potential treatment to prevent atherothrombotic events.
Keywords: Atherosclerosis, Non-lethal sonodynamic therapy, Macrophage polarization, AMPK, mTORC1, Autophagy
Graphical abstract
Highlights
-
•
NL-SDT enhances M1-to-M2 shift and significantly inhibits atherosclerosis progression in a mouse model.
-
•
NL-SDT induces autophagy by activating the AMPK signaling pathway.
-
•
M2-like macrophages promoted by NL-SDT facilitate cholesterol efflux and attenuate intracellular cholesterol deposition.
-
•
The ROS–AMPK–mTORC1–autophagy pathway is critical for NL-SDT-mediated effects on M2 macrophage polarization.
Non-standard abbreviations and acronyms
- 3-MA
3-methyladenine
- ABC
ATP-binding cassette
- ACC
acetyl-CoA carboxylase
- ALA
5-aminolaevulinic acid
- AMPK
5′ AMP-activated protein kinase
- apoE
apolipoprotein E
- ARG
arginase
- AS
atherosclerosis
- ATG5
autophagy related 5
- BMDM1
bone marrow-derived M1 macrophage
- BMT
bone marrow transplantation
- EGFPBM
enhanced green fluorescent protein-bone marrow
- HDL
high-density lipoprotein
- IFNγ
interferon-gamma
- IL
interleukin
- iNOS
induced nitric oxide synthase
- LC3
light chain 3
- LDL
low-density lipoprotein
- LPS
lipopolysaccharide
- LSCM
laser-scanning confocal microscopy
- LXRα
liver X receptor alpha
- MGL
macrophage galactose-type lectin
- mTORC1
mammalian target of rapamycin complex 1
- NAC
N-acetyl-l-cysteine
- NF-κB
nuclear factor kappaB
- NL-SDT
non-lethal sonodynamic therapy
- NO
nitric oxide
- PPAR
peroxisome proliferator-activated receptor
- PpIX
protoporphyrin IX
- RNAi
RNA interference
- ROS
reactive oxygen species
- RT-PCR
reverse transcription polymerase chain reaction
- siRNA
small-interfering RNA
- SMC
smooth-muscle cell
- STAT6
signal transducer and activator of transcription 6
- TC
total cholesterol
- TGF
transforming growth factor
- TEM
transmission electron microscopy
- TNF
tumor necrosis factor
1. Introduction
Atherosclerosis (AS) is the leading cause of vascular disease and morbidity and mortality worldwide [1]. Acute rupture of burgeoning atherosclerotic plaques triggers local thrombosis, ultimately resulting in partial or complete occlusion of the affected artery and manifesting as potentially lethal myocardial infarction or stroke [2]. Macrophages recognize and uptake lipids and function decisively during atherogenesis as heterogeneous and versatile cells that respond to microenvironment signals, with subsets of activated macrophages exhibiting distinct pathologic roles in atherogenesis [3,4]. According to signature stimuli, activated macrophages are mainly classified into proinflammatory (M1) or anti-inflammatory (M2) subsets. Classically activated M1 macrophages, are present mainly in unstable plaques of symptomatic patients, predominate rupture-prone shoulder regions, and boost the production of pro-atherogenic inflammatory mediators and tissue-degrading enzymes, such as matrix metalloproteinases, thereby contributing to sustained inflammation and plaque vulnerability [5,6]. Conversely, alternatively activated M2 macrophages, primarily generated by interleukin (IL)-4 and IL-13, are expressed mostly in asymptomatic plaques, resistant to cholesterol loading, and exhibit increased ability to store esterified cholesterol and facilitate fibrous cap formation by enhancing collagen production [7]. The anti-inflammatory and profibrotic M2 phenotypes actively counteract M1-derived deleterious effects to stabilize plaques. Consequently, equilibrium between these two phenotypes is likely related to plaque outcome and subsequent thromboembolic complications. Accordingly, therapeutic modulation of macrophage polarization has emerged as a novel therapeutic strategy for AS management.
Autophagy is a key catabolic recycling pathway involved in the degradation of different substrates, such as misfolded proteins, lipids, or damaged organelles, and that contributes to cellular recovery and plays vital roles in AS development [8]. During early AS, autophagy at a basal level is efficient and atheroprotective; however, it becomes defective in advanced lesions. Notably, impaired macrophage autophagy results in decreased lesional efferocytosis and hyperactivation of inflammasomes, thereby exacerbating atherosclerotic plaque development [9,10]. Moreover, accumulating evidence demonstrates that autophagy represents a crucial mechanism for regulating macrophage polarization. Because moderate activation of autophagy suppresses M1 polarization, promotes M2 polarization, and alleviates inflammatory reactions, the need for a method to therapeutically regulate autophagy appears to be clinically relevant [11,12].
5′ AMP-activated protein kinase (AMPK) is a well-conserved, energy-sensing enzyme comprising a catalytic α subunit and regulatory β and γ subunits [13,14]. Recent advances show that AMPK not only functions as a key nutrient-sensitive kinase but also as an oxidative-stress sensor and redox regulator [15]. Remarkably, AMPK can be activated by mitochondria-derived oxidant challenge [16], and upon activation, directly phosphorylate tuberous sclerosis complex 2 (TSC2), thereby inhibiting mammalian target of rapamycin complex 1 (mTORC1) activity associated with autophagy [17]. Accumulating evidence demonstrates that the AMPK and mTORC1 signaling pathways are associated with macrophage polarization, with M2 polarization mediated by macrophage-specific microRNA-33 dependent on AMPK activation [18], and decreased M2 polarization in TSC2-defective macrophages harboring constitutively activated mTORC1 [19]. Moreover, examination of atherosclerotic plaques in mice harboring genetic ablation of mTORC1 signaling in macrophages revealed a noteworthy decrease in M1-associated chemokine expression [20], suggesting the importance of both AMPK and mTORC1 signaling in the processes of macrophage polarization and autophagy.
Sonodynamic therapy (SDT) is an emerging medical technique that utilizes low-intensity ultrasound to locally activate a preloaded sonosensitizer [21]. Activation of the sonosensitizer by ultrasound generates ROS, a key factor in mediating the biological effects induced by SDT. Because of its non-invasive, efficacious, repeatable, strong tissue-penetrating, and region-focusing properties in the absence of obvious energy attenuation, SDT application as a novel treatment for AS has recently been investigated by us and other research groups. Our group previously reported that 5-aminolevulinic acid (ALA)-derived protoporphyrin IX (PpIX) could selectively accumulate in the mitochondria of macrophages, but not smooth muscle cells in advanced plaque [22]. Additionally, we found that ALA-mediated SDT (ALA-SDT) ameliorated AS in atherosclerotic rabbit and apolipoprotein E (apoE)-deficient mouse models [22,23]. Furthermore, in our pilot clinical trial involving patients with atherosclerotic peripheral artery disease, combined treatment with SDT and atorvastatin significantly inhibited AS progression within 4 weeks accompanied by a long-lasting effect for at least 40 weeks [23]. Therefore, in this study, we further delineated the underlying mechanism of ALA-mediated non-lethal SDT (NL-SDT) treatment on macrophage polarization and plaque stability due to the critical role of macrophages in atherosclerotic plaque development [24].
2. Methods
2.1. Establishment of the mouse AS model
Male apoE−/− (B6·129P2-Apoe tm1Unc/J; Vital River Laboratory Animal Technology Co., Ltd., Beijing, China) mice (8-weeks old) were fed a Western-type diet (21% fat, 0.15% cholesterol; MD12015; Medicience Ltd., Jiangsu, China) for 12 weeks to develop advanced atherosclerotic plaques and then randomly separated into different treatment groups (n = 16/group; Fig. S1A). For the NL-SDT group, the thorax area was exposed to 0.4 W/cm2 intensity ultrasonic radiation for 15 min at 2 h post-injection of ALA (60 mg/kg) into the caudal vein [25]. After NL-SDT, the mice were fed a normal diet. During the entire process and 1 day after the treatment, the mice were kept from light. All animal studies were performed according to regulations of the Harbin Medical University Human Ethics Committee, and the care and use of the animals conformed to the US National Institutes of Health Guide for Care and Use of Laboratory Animals.
2.2. Bone marrow transplantation (BMT) and the chimeric enhanced green fluorescent protein (EGFP)BMapoE−/− mouse model
BMT was performed as previously described, with modifications [26]. BM cells were harvested from male 6-week-old EGFP-transgenic mice (B6 ACTb-EGFP; Cyagen Biosciences Inc.) by flushing the femur and tibias with sterile phosphate-buffered saline (PBS). After irradiation with a single dose of 8 Gy, the suspended EGFP+ BM cells (1 × 107 cells) were injected via the caudal vein into male 6-week-old apoE−/− recipient mice. The chimeric EGFPBM apoE−/− mice were housed in autoclaved cages and fed normal chow for 2 weeks after BMT to allow for BM reconstitution and then switched to the Western-type diet for an additional 12 weeks to induce lesion formation.
2.3. Culture and activation of murine BM-derived macrophages
BM cells were harvested from male 6-week-old C57BL/6 J wild-type mice (Vital River Laboratory Animal Technology Co., Ltd.) by flushing the femur and tibias using a syringe. The isolated cells were then suspended in culture medium containing 10 ng/mL recombinant mouse granulocyte macrophage-colony stimulating factor at a density of 2 × 106 cells/mL, seeded in 35-mm Petri dishes, and maintained at 37 °C in a humidified atmosphere containing 5% CO2. Fresh media was changed on days 3 and 6, and adherent cells were polarization into M1 macrophages by stimulation with 10 ng/mL lipopolysaccharide plus 10 ng/mL interferon-γ for 24 h. M1 macrophages were subsequently transformed into foam cells by incubating with 100 μg/mL oxidized low-density lipoprotein (oxLDL) in serum-free RPMI-1640 medium containing 0.3% BSA for 24 h.
2.4. Application of specific inhibitors in vivo
The AMPK inhibitor compound C (10 mg/kg) and the autophagy inhibitor lys05 (10 mg/kg) were dissolved in sterile PBS and administrated intraperitoneally to apoE−/− mice 2 h before ultrasonic radiation, according to previous studies [27,28]. The mTOR inhibitor rapamycin (4 mg/kg) was dissolved using a dilution of 0.25% Tween-80 and 0.2% carboxymethylcellulose [29]. Control and NL-SDT mice received sterile PBS.
2.5. Sonication and treatment protocol
Cells were exposed to NL-SDT, as described previously [25]. The ultrasonic generator, transducer, and power amplifier used in this study were designed and assembled by Harbin Institute of Technology (Harbin, China). A 35-mm Petri dish was placed in a degassed water bath 30 cm away from the customized ultrasonic transducer (diameter: 3.5 cm; resonance frequency: 1.0 MHz; duty factor: 10%; and repetition frequency: 100 Hz). The exposure time was 5 min, and the ultrasonic intensity was 0.1 W/cm2. During NL-SDT, the temperature of the solution inside the Petri dishes varied by < 0.1 °C, as detected by a digital thermometer. M1 macrophages were incubated with 1 mM ALA in serum-free RPMI-1640 medium for different time intervals at 37 °C in the dark to determine the intracellular metabolic kinetics of ALA-PpIX. After ALA incubation, cells were co-stained with 200 nM Mito-tracker Green for 30 min and 1 μg/mL Hoechst 33,342 for 15 min in order to observe the subcellular localization of the sensitizer. The differentiated cells were pre-treated with the autophagy inhibitor 3-methyladenine (3-MA; 5 mM) or the lysosomal inhibitor bafilomycin A1 (50 nM) for 2 h, and 5 mM of the reactive oxygen species (ROS) inhibitor N-acetyl-l-cysteine (NAC) was added 1 h before NL-SDT.
2.6. Flow cytometric analysis
Aortas were collected from chimeric EGFPBM apoE−/− mice and digested by enzymes, as previously described [30]. Macrophages detached from 35-mm Petri dishes and cells within the mouse aorta were stained with conjugated antibodies against macrophage-subset markers for 1 h on ice in the dark, after which the samples were determined with a FACSCalibur flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA), and the data were analyzed with FlowJo software (FlowJo, LLC, Ashland, OR, USA).
2.7. Measurement of nitric oxide (NO) production and arginase (ARG) activity
NO levels in the supernatant were estimated as nitrite using the Griess reagent, as described previously [31]. ARG activity was measured according to the formation of urea after incubation of lysates from activated macrophages with arginine, as described elsewhere [31].
2.8. Histopathology and terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) staining
At the end of the experiments, the animals were deeply anesthetized and euthanized with an intraperitoneal injection of pentobarbital sodium (100 mg/kg). After transcardial heparinized saline perfusion, the mouse heart and aorta were carefully removed using a dissecting microscope. The top half of the heart was excised, fixed with 4% paraformaldehyde, dehydrated, and serially cross-sectioned according to routine procedures. For histopathologic assays, tissue samples were embedded in paraffin (n = 8/group), and for TUNEL, Oil red O, or immunofluorescence staining, aortic roots were embedded in optimal cutting temperature compound (n = 8/group). DNA fragmentation in apoptotic cells was determined by an in situ cell death-detection kit according to manufacturer instructions.
2.9. Statistical analysis
All experiments were preformed independently at least three times. Quantitative data are presented as the mean ± standard deviation (SD) values, and Student's two-tailed t-tests or one-way analysis of variance (ANOVA), followed by the Student–Newman–Keuls test was applied, as appropriate. Statistical analyses were performed using SAS software (v.9.2; SAS institute, Cary, NC, USA). A P < 0.05 was considered significant.
An expanded Methods section is available in the online-only Data Supplement.
3. Results
3.1. NL-SDT reduces plaque size and alters the composition of atherosclerotic plaques in apoE−/− mice
Autophagy is anti-apoptotic and maintains cellular homeostasis in an adverse environment. Western blot results demonstrated that compared with those of control mice, more than two rounds of repeated NL-SDT in apoE−/− mice significantly upregulated protein levels of the microtubule-associated protein light chain 3 (LC3)II, as well as the LC3II:LC3I ratio, which are hallmarks of autophagosome formation (Fig. S1B). These findings prompted us to evaluate the impact of three rounds of NL-SDT. As shown in Fig. 1A and B, NL-SDT significantly inhibited AS progression, with histologic analysis showing a significant decrease in lipids and increase in collagen content in NL-SDT-treated lesions. Interestingly, NL-SDT did not significantly alter the number of macrophages, smooth-muscle cells (SMCs), apoptotic cells, body weight, or serum lipid profile (Fig. 1C–J and Supplementary Table 1).
Fig. 1.

NL-SDT reduces plaque size and alters the composition of atherosclerotic plaques in Western diet-fed apoE−/−mice. (A) Representative histopathologic images of atherosclerotic lesions and quantification of the plaque area in different groups. Scale bar: 500 μm. (B) Atherosclerotic lesions with Oil red O staining and statistical analysis of the percentage of en face plaque area after NL-SDT. (C–H) Representative histopathologic images of atherosclerotic lesions and quantification of the percentage of corresponding composition in murine plaques. Scale bar: 200 μm. (I and J) Immunofluorescence of terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL)-stained plaque lesions and quantification of TUNEL-positive apoptotic cells per mm2 plaque area. Scale bar: 200 μm. (A and C–I): n = ~7–8; and (B): n = 5. ***P < 0.001 vs. control from Student's two-tailed t-test. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.2. NL-SDT promotes a shift in M1/M2 equilibrium toward M2 in vivo
To determine how NL-SDT alters the composition of atherosclerotic plaques without affecting cell apoptosis and total macrophage number, we assessed the proportion of macrophage subsets using double-immunofluorescence experiments. The results showed that F4/80+ macrophages from NL-SDT-treated plaques exhibited decreased levels of the M1 marker induced NO synthase (iNOS; 52.5 ± 12.5% vs. 14.9 ± 5.5%; P < 0.001) and increased levels of M2 markers, including ARG1 (21.3 ± 4.9% vs. 40.8 ± 4.8%; P < 0.001), CD206 (23.5 ± 7.4% vs. 50.8 ± 8.1%; P < 0.001), and CD163 (17.9 ± 3.7% vs. 38.7 ± 4.6%; P < 0.001) (Fig. 2A–D). Moreover, levels of the inflammatory cytokines tumor necrosis factor (TNF)α and IL-6 were markedly reduced, and levels of the anti-inflammatory cytokines transforming growth factor (TGF)-β and IL-10 were elevated in NL-SDT-treated plaques, indicating amelioration of NL-SDT-induced inflammation (Fig. 2E–H).
Fig. 2.

NL-SDT facilitates a shift in M1/M2 equilibrium toward M2 in vivo. (A–D) Immunofluorescence of plaque lesions double-stained for F4/80 and iNOS, ARG1, CD163, or CD206 and quantification of corresponding double-positive cells in murine plaques. Scale bar: 200 μm. (E–H) Quantitative analysis of TNFα, IL-6, IL-10, or TGFβ-positive areas showing the effects of NL-SDT on inflammation within the atheroma of apoE−/− mouse aortic roots. (I and J) Representative dot plots and quantification of retrieved aortic cells exhibiting the effects of NL-SDT on macrophage polarization within the atheroma of the Western diet-fed chimeric EGFPBMapoE−/− mouse aortic roots. (A–H): n = ~7–8; and (I and J): n = 5. Student's two-tailed t-test was used for statistical analyses. **P < 0.01, ***P < 0.001 vs. control.
To further analyze the effect on macrophage polarization in atherosclerotic plaques, we generated chimeric EGFPBM apoE−/− mice by transplantation of BM cells from EGFP transgenic mice after sublethal irradiation of apoE−/− mice (Fig. S2A). The mean percentage of EGFP+ cells among peripheral nucleated blood cells was 90.5 ± 3.3% in chimeric EGFPBM apoE−/− mice, which was close to that in donor EGFP transgenic mice (93.2 ± 2.4%; Fig. S2B). The retrieved aortic cells were first divided into donor-origin BM-derived GFP+ and tissue-origin GFP− populations then further analyzed based on iNOS and CD206 levels (Fig. 2I and J). The results showed that NL-SDT decreased the proportion of iNOS+CD206− cells from 27.3 ± 6.9% to 4.9 ± 2.4% and from 9.5 ± 2.7% to 2.1 ± 0.9% and increased iNOS−CD206+ cells from 9.7 ± 2.7% to 21.5 ± 3.8% and from 1.9 ± 0.7% to 5.5 ± 1.3% for the GFP+ and GFP− populations, respectively. The M1/M2 rate was defined as the ratio of iNOS+ population to the CD206+ population. Interestingly, NL-SDT clearly decreased the M1/M2 rate by 17% and 9% for the GFP+ and GFP− populations, respectively. These results indicated that NL-SDT promoted phenotypic switching of both BM-derived and resident macrophages from M1 to M2 in the aorta of chimeric EGFPBM apoE−/− mice.
3.3. NL-SDT activates autophagy in vitro
To determine the optimal NL-SDT-treatment parameters, we treated murine BM-derived M1 (BMDM1) macrophages (Figs. S2C–E) with NL-SDT in vitro. Cell viability decreased markedly from 81.5 ± 6.5% (2 mM) to 50.9 ± 4.3% (10 mM) as compared with that of the ALA-untreated group (Fig. S3A). Additionally, the fluorescence intensity of 1 mM ALA-PpIX measured using a multimode microplate reader increased over time, peaked at 6 h, and then faded over time in BMDM1s, which mimicked the pattern observed previously in foam cells (Fig. S3B) [32]. Laser-scanning confocal microscopy (LSCM) images showed that the red PpIX fluorescence was almost exclusively distributed in the mitochondria at peak fluorescence intensity (Fig. S3C). Moreover, there was no significant change in apoptotic or necrotic ratio at ultrasonic intensities of 0.1 W/cm2 or 0.2 W/cm2 (Figs. S3D–F). Therefore, 1 mM ALA and 0.1 W/cm2 ultrasonic intensity were applied as the optimal NL-SDT-treatment parameters in vitro.
Western blot results demonstrated that LC3II levels and the LC3II: LC3I ratio were markedly elevated in 0.1 W/cm2 SDT and 0.2 W/cm2 SDT groups (Fig. 3A), and that administration of the lysosomal inhibitor bafilomycin A1 further increased LC3-II levels in NL-SDT-treated BMDM1s, demonstrating enhanced autophagic flux (Fig. 3B). The relative levels of ROS increased significantly in the NL-SDT group (Fig. S4), and the increased ROS upon NL-SDT is mitochondrial derived as revealed by MitoSOX fluorescence assay (Fig. S4C). Electron micrography of autophagic vesicles revealed an increase number of autophagosomes in NL-SDT-treated cells (Fig. 3C). Additionally, pretreatment with the ROS scavenger NAC reversed this effect, as confirmed by the lower number of autophagic vesicles in the cells (Fig. 3D). We subsequently performed the GFP-LC3 cleavage assay to determine autophagic flux, finding that the elevated levels of free GFP induced by NL-SDT were also inhibited by NAC pre-treatment (Fig. 3E–G). Moreover, LSCM results showed that NL-SDT stimulated LC3 protein clustering, and that the increase in fluorescent puncta was mostly suppressed by NAC pre-treatment (Fig. 3H and I). Collectively, these results demonstrated that ROS contributed to the activation of NL-SDT-induced autophagy.
Fig. 3.

NL-SDT activates autophagy in vitro. (A) Protein level of LC3B in different groups according to western blot. U indicates ultrasound; S, NL-SDT. (B) Western blot analysis of LC3B in the presence or absence of bafilomycin A1 (Baf-A1; 50 nM). Baf-A1 pretreatment significantly increased NL-SDT-induced LC3 conversion. (C and D) Electron micrograph showing the ultrastructural changes in NL-SDT-treated cells ± NAC. Red arrows indicate autophagosomes. Scale bar: 2 μm. (E–G) Detection of autophagic flow in each group by western blot using the GFP-LC3 fusion protein. (H and I) Representative LSCM images and quantification of punctate spots of LC3 induced by NL-SDT. Scale bar: 10 μm. GFP-LC3 puncta-containing cells were quantified by counting GFP-positive cells. We used >100 cells per experiment in (I). (A–G): n = ~4–5, and data were representative of three independent experiments. ANOVA followed by the Student–Newman–Keuls test was applied. ***P < 0.001 vs. control; ###P < 0.001 vs. the NL-SDT group. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.4. NL-SDT activates autophagy via the ROS–AMPK–mTORC1 signaling pathway in vitro
To investigate the molecular mechanism underlying NL-SDT induction of autophagy, we performed semi-quantitative detection of 18 phosphorylated mouse proteins (mouse AKT pathway phosphorylation array; RayBiotech, Norcross, GA, USA) in BMDM1 cells, with the results suggesting that NL-SDT might affect the AMPK–mTORC1 signaling cascade (n = 4; Fig. S5A). We then examined pathway activation using western blot, finding increased levels of phosphorylated AMPKα (Thr172) and acetyl-CoA carboxylase (ACC), whereas levels of phosphorylated mTOR (Ser2448), p70 S6 kinase 1, and 4E-binding protein 1 were decreased following NL-SDT (Fig. 4A). Moreover, NAC successfully suppressed the increased phosphorylation of AMPK and ACC and the decreased phosphorylation of mTOR regulated by NL-SDT (Fig. 4B). Furthermore, small-interfering (si)RNA-mediated downregulation of AMPK prevented NL-SDT-mediated repression of mTORC1, suggesting that AMPK mediates mTORC1 repression (Fig. 4C). Similarly, LC3BII levels in NL-SDT-treated cells increased following downregulation of mTOR levels (Fig. 4D), with enhancement of NL-SDT-induced autophagy via AMPK–mTORC1 signaling further supported by TEM and LSCM results (Fig. 4F). These results indicated that mTORC1 downregulation by AMPK was required for NL-SDT-mediated activation of autophagy.
Fig. 4.

NL-SDT activates autophagy via the ROS–AMPK–mTORC1 signaling pathway in vitro. (A and B) Levels of proteins associated with AMPK–mTORC1–autophagy signaling in each group detected by western blot. Quantification of protein levels (right). (C–F) The effects of AMPK, mTOR, or ATG5 knockdown by siRNA on NL-SDT-induced autophagy in BMDM1s detected by western blot, electron micrography, and LSCM. Quantification of protein levels (left bottom). Scale bar (black): 1 μm. Scale bar (white): 10 μm. For all experiments, n = ~4–5, and experiments were performed independently in triplicate. ANOVA followed by the Student–Newman–Keuls test was used for statistical analyses. **P < 0.01, ***P < 0.001 vs. control; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. the NL-SDT group.
3.5. NL-SDT promotes transition toward the M2 phenotype in vitro
Emerging evidence indicates that autophagy is critically involved in macrophage activation and function; therefore, we examined the effects of NL-SDT on macrophage polarization in vitro by analyzing levels of macrophage-phenotype biomarker genes by reverse transcription polymerase chain reaction (RT-PCR). As shown in Fig. S6, NL-SDT significantly attenuated mRNA levels of M1-specific genes, including Arg2, C–C motif chemokine ligand 5, Il6, Nos2, and Tnfa, but markedly upregulated mRNA levels of M2-specific genes, including Arg1, chitinase-like protein 3, Il10, macrophage galactose-type lectin (Mgl)1, Mgl2, resistin-like molecule alpha, and Tgfb. Mechanistically, these effects were blocked by administration of NAC or the autophagy inhibitor 3-MA and confirmed by western blot and flow cytometry (Figs. S7A–C). Interestingly, NL-SDT did not alter levels of M2-associated phosphorylated signal transducer and activator of transcription 6 (STAT6; Fig. S8). Additionally, NL-SDT-treated cells displayed elevated ARG1 activity, significantly decreased NO levels, and secreted greater amounts of anti-inflammatory cytokines, including IL-10 and TGFβ while releasing lower levels of proinflammatory cytokines, including IL-6 and TNFα (Figs. S7D–I), relative to untreated cells, with these effects also attenuated by NAC or 3-MA administration. These data suggested that NL-SDT-mediated autophagy promoted the polarization of M1 macrophages toward the M2 phenotype.
To confirm the involvement of the AMPK–mTORC1–autophagy pathway in NL-SDT-mediated M1-to-M2 transition, we employed RNA interference (RNAi) and found that downregulation of this pathway prevented the NL-SDT-induced M1-to-M2 transition (Fig. 5 and Fig. S9). Collectively, these results demonstrated that the ROS–AMPK–mTORC1–autophagy pathway critically mediated the effects of NL-SDT on macrophage polarization.
Fig. 5.

The AMPK–mTOR–autophagy pathway critically mediates the effects of NL-SDT on macrophage polarization. AMPKα, mTOR, or ATG5 levels were knocked down by RNAi in BMDM1s before exposing cells to NL-SDT. (A) Western blot of cell lysates using antibodies against NF-κB, iNOS, ARG1, CD163, CD206, and actin. (B and C) Representative flow cytograms and quantification of cell markers for M1 (iNOS) and M2 (CD206) macrophages after NL-SDT (n = 4). S, NL-SDT. (D) Quantitative measurements of the urea level in cells from different treatment groups (n = 6). (E) Quantitative measurements of the amounts of NO in the supernatant produced by experimental samples (n = ~6–8). (F–I) Quantification of cytokine levels in the supernatant of M1 macrophages in different treatment groups detected by enzyme-linked immunosorbent assay (n = 4). Data are shown as the means ± SD and representative of three independent experiments. ANOVA followed by the Student–Newman–Keuls test was used for statistical analyses. ***P < 0.001 vs. control; ##P < 0.01, ###P < 0.001 vs. the NL-SDT group.
3.6. NL-SDT decreases foam-cell formation in vitro by enhancing cholesterol efflux
Ouimet et al. [33] reported that autophagy attenuates foam-cell formation and AS development. In the present study, we showed that NL-SDT significantly decreased lipid content in advanced lesions (Fig. 1B–D). Accordingly, Oil red O staining of BMDM1 derived foam cells showed that NL-SDT substantially reduced the number of lipid droplets (Fig. 6A). Furthermore, results of a quantitative intracellular cholesterol assay indicated that NL-SDT markedly decreased both free and total cholesterol (TC) contents as compared with those in the control group (Fig. 6C). Additionally, NL-SDT enhanced mRNA level of lectin-like oxLDL receptor 1 expression (Fig. 6F), whereas that of Cd36 and macrophage scavenger receptor1 was unaffected (Fig. S10). Therefore, NL-SDT maintained cholesterol homeostasis by modulating the balance between cholesterol influx and efflux. Moreover, we observed significant increases in the expression of cholesterol-efflux mediators peroxisome proliferator-activated receptor gamma, liver X receptor alpha, ATP-binding cassette (Abc)a1, and Abcg1 in lipid-loaded M1 macrophages post-NL-SDT (Fig. 6G–N), suggesting that the decreased lipid retention was likely due to the promotion of cholesterol efflux by NL-SDT. Indeed, fluorometric assays to evaluate cholesterol efflux revealed that the apoAI-mediated cholesterol efflux was markedly stimulated in NL-SDT-treated cells (Fig. 6O and P). By contrast, these effects were abolished by NAC or 3-MA administration or down-regulation of AMPK and autophagy related 5 (ATG5) levels, whereas downregulated mTOR levels showed cooperative effects, implying that the ROS–AMPK–mTOR–autophagy cascade was involved in promoting NL-SDT-induced cholesterol efflux (Fig. 6).
Fig. 6.

NL-SDT decreases foam-cell formation in vitro. (A, B, and D) Representative microscopic photographs of cells stained with Oil Red O. Scale bar: 10 μm. (C and E) Quantification of TC and free cholesterol contents in each group (n = 4). (F–N) The effects of NL-SDT on the transcription of genes associated with macrophage lipid metabolism in vitro (n = ~4–6). (O and P) Promotion of cholesterol efflux induced by NL-SDT and assessed by fluorometric assays measuring cholesterol efflux (n = ~4–5). Data are presented as means ± SD, and experiments were independently performed in triplicate. ANOVA followed by the Student–Newman–Keuls test was used for statistical analyses. ***P < 0.001 vs. control group; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. the NL-SDT group. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.7. NL-SDT-mediated autophagy contributes to M2 polarization in advanced plaques
Consistent with our in vitro observation that NL-SDT improves autophagy via AMPK–mTORC1 signaling, the percentages of phosphorylated-AMPKα- and LC3B-positive areas increased, whereas the percentage of phosphorylated-mTOR-positive areas decreased in NL-SDT-treated lesions (Fig. 7A–C), with these effects supported by western blot (Fig. 7D). We then systemically administered the specific AMPK inhibitor compound C, the mTOR inhibitor rapamycin, and the autophagy inhibitor lys05 to Western-diet-fed apoE−/− mice, respectively. Quantitative image analysis indicated activation of AMPK–mTOR–autophagy signaling in NL-SDT-treated plaques (Fig. 7E–G). Application of the different inhibitors did not cause any remarkable change in the body weights or serum lipid profiles, including TC, triglyceride, LDL, and high-density lipoprotein levels (Supplementary Table 2). However, the specific blockade of AMPK or autophagy significantly diminished NL-SDT-mediated reductions in plaque area and lipid content, as well as elevations in collagen content, while exerting no significant effects on macrophage or SMC content (Fig. 7H–M). Moreover, double-immunofluorescence experiments demonstrated that inhibitor administration of compound C or lys05 markedly suppressed NL-SDT-induced M1-to-M2 polarization in advanced plaques (Fig. 7N–Q). Although the parameters affected by rapamycin treatment displayed similar trends relative to those in NL-SDT mice, the differences were not statistically significant. Therefore, we postulated that the discrepancy could be due to the fewer mice used in the experiment. A novel experiment with a higher number of mice might be necessary in future studies in order to confirm the role of mTOR in NL-SDT-mediated effects in vivo (Fig. 7G–Q). Collectively, these results showed that the activation of AMPK and autophagy by NL-SDT is pivotal for M2 polarization and subsequent stabilization of advanced atherosclerotic plaques.
Fig. 7.

NL-SDT-mediated autophagy contributes to M2 phenotype polarization in advanced plaques. (A–C and E–G) Quantitative measurements of corresponding protein levels in murine plaques in each group. (D) Levels of proteins associated with AMPK–mTOR–autophagy signaling detected by western blot. (H–M) Quantification of the percentage of corresponding composition in murine plaques. (N–Q) Quantification of corresponding double-positive cells in murine plaques of each group. For the analysis of significant differences between two groups, Student's two-tailed t-test was applied. The differences among three or more groups were analyzed via ANOVA followed by the Student–Newman–Keuls test. n = ~7–8. **P < 0.01, ***P < 0.001 vs. control group; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. the NL-SDT group.
4. Discussion
The central role of macrophages in AS development and related pathological complications is well documented [24]; however, the underlying mechanism and therapeutic solutions remain a riddle. Understanding the mechanisms involved in macrophage heterogeneity provides new insights into macrophage-centered diagnostic and therapeutic strategies for multiple diseases, including AS. Although we preliminarily observed the effects of SDT treatment on macrophage autophagy according to in vitro experiments [34,35], we definitely need to obtain validation of the effects of NL-SDT by in vivo studies for preclinical application.
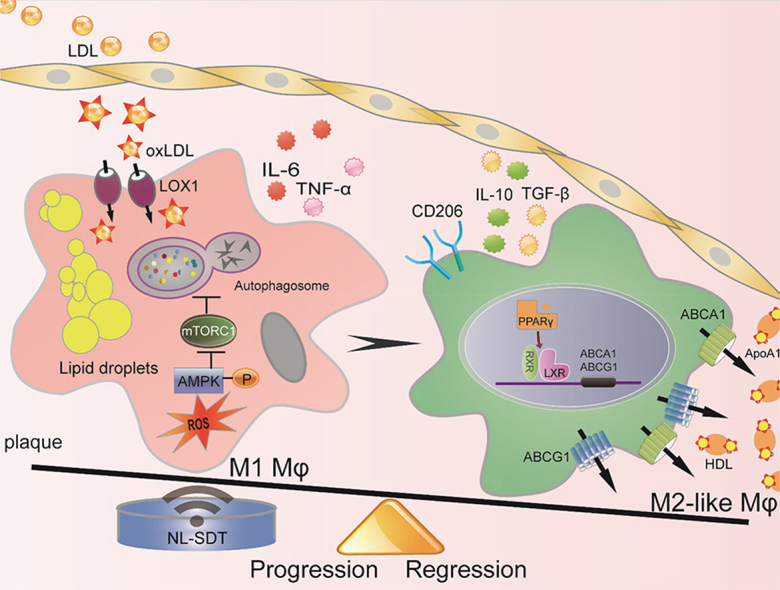
In this study, we focused on macrophage response to NL-SDT treatment in atherosclerotic plaque and demonstrated that NL-SDT activated AMPK–mTORC1–dependent autophagy, as well as promoted a shifting of both BM-derived and resident macrophages toward an M2-like phenotype and eventual amelioration of AS (an overview is shown in Fig. 8). These findings suggest NL-SDT as a potential and effective treatment for advanced atherosclerotic lesions based on its selective modulation of macrophage polarization within the atheroma.
Fig. 8.

Schematic of the anti-AS mechanism of NL-SDT. ROS-dependent activation of autophagy through the AMPK–mTORC1 signaling pathway is induced by NL-SDT, which enhances M1-to-M2 shift, cholesterol efflux, and anti-inflammatory reactions.
Recent studies indicate that the AMPK–mTORC1 signaling pathway acts as a “master switch” of macrophage functional polarization, with AMPKα stimulation resulting in decreased expression of M1-related genes and promoting an M2 phenotype, whereas attenuated AMPKα levels dramatically elevate levels of M1-related transcripts and facilitate transition to an M1 phenotype [36,37]. Moreover, M2 polarization is defective in macrophages harboring constitutively activated mTORC1 both in vivo and in vitro [19,20]. The present study showed for the first time the direct effects of NL-SDT on macrophage polarization, and that stimulation of M2-predominant responses by NL-SDT was due to activation of AMPK–mTOR–autophagy signaling (Fig. 5, Fig. 7N–Q). Indeed, NL-SDT dramatically suppressed inflammatory reactions and reduced the expression of the major inflammation-associated regulator nuclear factor-kappaB (NF-κB) in vitro (Fig. 5A and F–I) in a process dependent on AMPK–mTOR–autophagy signaling, and consistent with previous findings [19,38,39]. These findings highlight the AMPK–mTOR cascade as a central determinant of macrophage polarization and a therapeutic pathway in inhibition of AS development.
The role of the M1 versus M2 phenotype in AS pathogenesis has been thoroughly investigated; however, their respective function in cholesterol deposition remains contentious and confusing. Recent studies indicated that M2 macrophages are more susceptible to foam-cell formation than M1 macrophages via accelerated cholesterol uptake [40,41], suggesting that shifting differentiated macrophages to M2 might not be an ideal therapeutic strategy. In human atherosclerotic plaques, alternative macrophages display lower cholesterol efflux capacities [7]. Intriguingly, we found that apoAI-mediated cholesterol efflux in NL-SDT-treated cells was substantially enhanced (Fig. 6). This observation that NL-SDT promoted the alternative phenotype but enhanced cholesterol efflux is presumably due to the activation of AMPK–mTOR–autophagy signaling. A previous study reported that administration of the AMPK agonist 5-aminoimidazole-4-carboxamide ribonucleotide restrains hypercholesterolemia-induced endothelial dysfunction through cholesterol efflux from vascular endothelial cells, whereas inhibition of AMPK with compound C has reciprocal effects [42]. Additionally, a novel AMPK activator, WS070117, attenuates intracellular ABCA1 degradation, resulting in enhanced cholesterol efflux [43]. Moreover, the mTOR inhibitor rapamycin and its semi-derivatives decrease lipid accumulation and activate cholesterol efflux via autophagy [44]. Furthermore, recent intriguing results show that the autophagic machinery degrades lipid droplets to transfer free cholesterol for efflux in an ABCA1-dependent process [45]. Therefore, our findings suggest that the AMPK–mTOR–autophagy pathway represents a new target for cholesterol-efflux regulation, and that NL-SDT, in particular, is capable of both alleviating inflammation and maintaining cholesterol homeostasis in a novel mechanism through which NL-SDT protects against AS.
We previously demonstrated that ALA-SDT induces foam-cell apoptosis, enhances cholesterol efflux, and ameliorates AS [22]. In the present study, we demonstrated that NL-SDT also exerted a protective function, interestingly not via apoptosis but rather by activating autophagy. Different intensities of SDT exert identically beneficial effects locally without systemic off-target toxicity via divergent mechanisms, which might be a finding of great significance for future translational applications. Theoretically, lower-intensity SDT makes the treatment of severer plaques safer, which might be due to a reduced susceptibility to complications from acute plaque rupture.
Because lysosomes mediate the degradation of diverse cargos through processes, such as autophagosome processing, deficiency of plaque macrophages is a critical factor associated with dysfunctional autophagy [46]. Therefore, a major limitation of the present study is the lack of an exploration of autophagosome-lysosome biogenesis, including effects on the autophagy chaperone p62. We systemically administrated specific inhibitors for in vivo experiments; however, this approach can have many off-target effects. In subsequent studies, we aim to broaden our understanding of the role of autophagy in M2 polarization in AS by collecting more convincing in vivo data from experimental animals, such as mouse models of macrophage-specific gene modulation.
In summary, our findings revealed a novel mechanism by which NL-SDT protects against AS, with NL-SDT locally displaying beneficial effects on lesion sites and suggesting its potential efficacy as an appealing regimen for managing AS.
Funding
This work was supported by the Major Research Equipment Development Project of National Natural Science of China (81727809) and the State Key Project of National Natural Science of China (81530052).
Declaration of competing interest
None declared.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2020.101501.
Transparency document related to this article can be found online at https://doi.org/10.1016/j.redox.2020.101501.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Transparency document
References
- 1.Roth G.A., Forouzanfar M.H., Moran A.E., Barber R., Nguyen G., Feigin V.L., Naghavi M., Mensah G.A., Murray C.J. Demographic and epidemiologic drivers of global cardiovascular mortality. N. Engl. J. Med. 2015;372:1333–1341. doi: 10.1056/NEJMoa1406656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bentzon J.F., Otsuka F., Virmani R., Falk E. Mechanisms of plaque formation and rupture. Circ. Res. 2014;114:1852–1866. doi: 10.1161/CIRCRESAHA.114.302721. [DOI] [PubMed] [Google Scholar]
- 3.Martinez F.O., Helming L., Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu. Rev. Immunol. 2009;27:451–483. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- 4.Cochain C., Zernecke A. Macrophages and immune cells in atherosclerosis: recent advances and novel concepts. Basic Res. Cardiol. 2015;110:34. doi: 10.1007/s00395-015-0491-8. [DOI] [PubMed] [Google Scholar]
- 5.Stoger J.L., Gijbels M.J., van der Velden S., Manca M., van der Loos C.M., Biessen E.A., Daemen M.J., Lutgens E., de Winther M.P. Distribution of macrophage polarization markers in human atherosclerosis. Atherosclerosis. 2012;225:461–468. doi: 10.1016/j.atherosclerosis.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 6.Lee C.W., Hwang I., Park C.S., Lee H., Park D.W., Kang S.J., Lee S.W., Kim Y.H., Park S.W., Park S.J. Macrophage heterogeneity of culprit coronary plaques in patients with acute myocardial infarction or stable angina. Am. J. Clin. Pathol. 2013;139:317–322. doi: 10.1309/AJCP7KEYGN3OBGQX. [DOI] [PubMed] [Google Scholar]
- 7.Chinetti-Gbaguidi G., Baron M., Bouhlel M.A., Vanhoutte J., Copin C., Sebti Y., Derudas B., Mayi T., Bories G., Tailleux A., Haulon S., Zawadzki C., Jude B., Staels B. Human atherosclerotic plaque alternative macrophages display low cholesterol handling but high phagocytosis because of distinct activities of the PPARgamma and LXRalpha pathways. Circ. Res. 2011;108:985–995. doi: 10.1161/CIRCRESAHA.110.233775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shao B.Z., Han B.Z., Zeng Y.X., Su D.F., Liu C. The roles of macrophage autophagy in atherosclerosis. Acta Pharmacol. Sin. 2016;37:150–156. doi: 10.1038/aps.2015.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liao X., Sluimer J.C., Wang Y., Subramanian M., Brown K., Pattison J.S., Robbins J., Martinez J., Tabas I. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metabol. 2012;15:545–553. doi: 10.1016/j.cmet.2012.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nussenzweig S.C., Verma S., Finkel T. The role of autophagy in vascular biology. Circ. Res. 2015;116:480–488. doi: 10.1161/CIRCRESAHA.116.303805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu K., Zhao E., Ilyas G., Lalazar G., Lin Y., Haseeb M., Tanaka K.E., Czaja M.J. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy. 2015;11:271–284. doi: 10.1080/15548627.2015.1009787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matta S.K., Kumar D. AKT mediated glycolytic shift regulates autophagy in classically activated macrophages. Int. J. Biochem. Cell Biol. 2015;66:121–133. doi: 10.1016/j.biocel.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 13.Towler M.C., Hardie D.G. AMP-activated protein kinase in metabolic control and insulin signaling. Circ. Res. 2007;100:328–341. doi: 10.1161/01.RES.0000256090.42690.05. [DOI] [PubMed] [Google Scholar]
- 14.Salt I.P., Hardie D.G. AMP-activated protein kinase: an ubiquitous signaling pathway with key roles in the cardiovascular system. Circ. Res. 2017;120:1825–1841. doi: 10.1161/CIRCRESAHA.117.309633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang S., Song P., Zou M.H. AMP-activated protein kinase, stress responses and cardiovascular diseases. Clin. Sci. (Lond.) 2012;122:555–573. doi: 10.1042/CS20110625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lamberts R.R., Onderwater G., Hamdani N., Vreden M.J., Steenhuisen J., Eringa E.C., Loer S.A., Stienen G.J., Bouwman R.A. Reactive oxygen species-induced stimulation of 5'AMP-activated protein kinase mediates sevoflurane-induced cardioprotection. Circulation. 2009;120:S10–S15. doi: 10.1161/CIRCULATIONAHA.108.828426. [DOI] [PubMed] [Google Scholar]
- 17.Inoki K., Zhu T., Guan K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 18.Ouimet M., Ediriweera H.N., Gundra U.M., Sheedy F.J., Ramkhelawon B., Hutchison S.B., Rinehold K., van Solingen C., Fullerton M.D., Cecchini K., Rayner K.J., Steinberg G.R., Zamore P.D., Fisher E.A., Loke P., Moore K.J. MicroRNA-33-dependent regulation of macrophage metabolism directs immune cell polarization in atherosclerosis. J. Clin. Invest. 2015;125:4334–4348. doi: 10.1172/JCI81676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Byles V., Covarrubias A.J., Ben-Sahra I., Lamming D.W., Sabatini D.M., Manning B.D., Horng T. The TSC-mTOR pathway regulates macrophage polarization. Nat. Commun. 2013;4:2834. doi: 10.1038/ncomms3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ai D., Jiang H., Westerterp M., Murphy A.J., Wang M., Ganda A., Abramowicz S., Welch C., Almazan F., Zhu Y., Miller Y.I., Tall A.R. Disruption of mammalian target of rapamycin complex 1 in macrophages decreases chemokine gene expression and atherosclerosis. Circ. Res. 2014;114:1576–1584. doi: 10.1161/CIRCRESAHA.114.302313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang X., Jia Y., Wang P., Liu Q., Zheng H. Current status and future perspectives of sonodynamic therapy in glioma treatment. Ultrason. Sonochem. 2017;37:592–599. doi: 10.1016/j.ultsonch.2017.02.020. [DOI] [PubMed] [Google Scholar]
- 22.Wang H., Yang Y., Sun X., Tian F., Guo S., Wang W., Tian Z., Jin H., Zhang Z., Tian Y. Sonodynamic therapy-induced foam cells apoptosis activates the phagocytic PPARgamma-LXRalpha-ABCA1/ABCG1 pathway and promotes cholesterol efflux in advanced plaque. Theranostics. 2018;8:4969–4984. doi: 10.7150/thno.26193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun X., Guo S., Yao J., Wang H., Peng C., Li B., Wang Y., Jiang Y., Wang T., Yang Y., Cheng J., Wang W., Cao Z., Zhao X., Li X., Sun J., Yang J., Tian F., Chen X., Li Q., Gao W., Shen J., Zhou Q., Wang P., Li Z., Tian Z., Zhang Z., Cao W., Li M., Tian Y. Rapid inhibition of atherosclerotic plaque progression by sonodynamic therapy. Cardiovasc. Res. 2019;115:190–203. doi: 10.1093/cvr/cvy139. [DOI] [PubMed] [Google Scholar]
- 24.Moore K.J., Sheedy F.J., Fisher E.A. Macrophages in atherosclerosis: a dynamic balance. Nat. Rev. Immunol. 2013;13:709–721. doi: 10.1038/nri3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y., Wang W., Xu H., Sun Y., Sun J., Jiang Y., Yao J., Tian Y. Non-lethal sonodynamic therapy inhibits atherosclerotic plaque progression in ApoE-/- mice and attenuates ox-LDL-mediated macrophage impairment by inducing heme oxygenase-1. Cell. Physiol. Biochem. 2017;41:2432–2446. doi: 10.1159/000475913. [DOI] [PubMed] [Google Scholar]
- 26.Endo J., Sano M., Fujita J., Hayashida K., Yuasa S., Aoyama N., Takehara Y., Kato O., Makino S., Ogawa S., Fukuda K. Bone marrow derived cells are involved in the pathogenesis of cardiac hypertrophy in response to pressure overload. Circulation. 2007;116:1176–1184. doi: 10.1161/CIRCULATIONAHA.106.650903. [DOI] [PubMed] [Google Scholar]
- 27.Li J., Zeng Z., Viollet B., Ronnett G.V., McCullough L.D. Neuroprotective effects of adenosine monophosphate-activated protein kinase inhibition and gene deletion in stroke. Stroke. 2007;38:2992–2999. doi: 10.1161/STROKEAHA.107.490904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ndoye A., Budina-Kolomets A., Kugel C.H., 3rd, Webster M.R., Kaur A., Behera R., Rebecca V.W., Li L., Brafford P.A., Liu Q., Gopal Y.N.V., Davies M.A., Mills G.B., Xu X., Wu H., Herlyn M., Nicastri M.C., Winkler J.D., Soengas M.S., Amaravadi R.K., Murphy M.E., Weeraratna A.T. ATG5 mediates a positive feedback loop between wnt signaling and autophagy in melanoma. Canc. Res. 2017;77:5873–5885. doi: 10.1158/0008-5472.CAN-17-0907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Phung T.L., Ziv K., Dabydeen D., Eyiah-Mensah G., Riveros M., Perruzzi C., Sun J., Monahan-Earley R.A., Shiojima I., Nagy J.A., Lin M.I., Walsh K., Dvorak A.M., Briscoe D.M., Neeman M., Sessa W.C., Dvorak H.F., Benjamin L.E. Pathological angiogenesis is induced by sustained Akt signaling and inhibited by rapamycin. Canc. Cell. 2006;10:159–170. doi: 10.1016/j.ccr.2006.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Galkina E., Kadl A., Sanders J., Varughese D., Sarembock I.J., Ley K. Lymphocyte recruitment into the aortic wall before and during development of atherosclerosis is partially L-selectin dependent. J. Exp. Med. 2006;203:1273–1282. doi: 10.1084/jem.20052205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Classen A., Lloberas J., Celada A. Macrophage activation: classical versus alternative. Methods Mol. Biol. 2009;531:29–43. doi: 10.1007/978-1-59745-396-7_3. [DOI] [PubMed] [Google Scholar]
- 32.Wang H., Yang Y., Chen H., Dan J., Cheng J., Guo S., Sun X., Wang W., Ai Y., Li S., Li Z., Peng L., Tian Z., Yang L., Wu J., Zhong X., Zhou Q., Wang P., Zhang Z., Cao W., Tian Y. The predominant pathway of apoptosis in THP-1 macrophage-derived foam cells induced by 5-aminolevulinic acid-mediated sonodynamic therapy is the mitochondria-caspase pathway despite the participation of endoplasmic reticulum stress. Cell. Physiol. Biochem. 2014;33:1789–1801. doi: 10.1159/000362958. [DOI] [PubMed] [Google Scholar]
- 33.Ouimet M., Franklin V., Mak E., Liao X., Tabas I., Marcel Y.L. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metabol. 2011;13:655–667. doi: 10.1016/j.cmet.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang Y., Kou J., Han X., Li X., Zhong Z., Liu Z., Zheng Y., Tian Y., Yang L. ROS-dependent activation of autophagy through the PI3K/Akt/mTOR pathway is induced by hydroxysafflor yellow A-sonodynamic therapy in THP-1 macrophages. Oxid Med Cell Longev. 2017 doi: 10.1155/2017/8519169. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li X., Zhang X., Zheng L., Kou J., Zhong Z., Jiang Y., Wang W., Dong Z., Liu Z., Han X., Li J., Tian Y., Zhao Y., Yang L. Hypericin-mediated sonodynamic therapy induces autophagy and decreases lipids in THP-1 macrophage by promoting ROS-dependent nuclear translocation of TFEB. Cell Death Dis. 2016;7:e2527. doi: 10.1038/cddis.2016.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sag D., Carling D., Stout R.D., Suttles J. Adenosine 5'-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J. Immunol. 2008;181:8633–8641. doi: 10.4049/jimmunol.181.12.8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Namgaladze D., Snodgrass R.G., Angioni C., Grossmann N., Dehne N., Geisslinger G., Brune B. AMP-activated protein kinase suppresses arachidonate 15-lipoxygenase expression in interleukin 4-polarized human macrophages. J. Biol. Chem. 2015;290:24484–24494. doi: 10.1074/jbc.M115.678243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Salminen A., Hyttinen J.M., Kaarniranta K. AMP-activated protein kinase inhibits NF-kappaB signaling and inflammation: impact on healthspan and lifespan. J. Mol. Med. (Berl.) 2011;89:667–676. doi: 10.1007/s00109-011-0748-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumase F., Takeuchi K., Morizane Y., Suzuki J., Matsumoto H., Kataoka K., Al-Moujahed A., Maidana D.E., Miller J.W., Vavvas D.G. AMPK-activated protein kinase suppresses Ccr2 expression by inhibiting the NF-kappaB pathway in RAW264.7 macrophages. PloS One. 2016;11 doi: 10.1371/journal.pone.0147279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van Tits L.J., Stienstra R., van Lent P.L., Netea M.G., Joosten L.A., Stalenhoef A.F. Oxidized LDL enhances pro-inflammatory responses of alternatively activated M2 macrophages: a crucial role for Kruppel-like factor 2. Atherosclerosis. 2011;214:345–349. doi: 10.1016/j.atherosclerosis.2010.11.018. [DOI] [PubMed] [Google Scholar]
- 41.Oh J., Riek A.E., Weng S., Petty M., Kim D., Colonna M., Cella M., Bernal-Mizrachi C. Endoplasmic reticulum stress controls M2 macrophage differentiation and foam cell formation. J. Biol. Chem. 2012;287:11629–11641. doi: 10.1074/jbc.M111.338673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li D., Zhang Y., Ma J., Ling W., Xia M. Adenosine monophosphate activated protein kinase regulates ABCG1-mediated oxysterol efflux from endothelial cells and protects against hypercholesterolemia-induced endothelial dysfunction. Arterioscler. Thromb. Vasc. Biol. 2010;30:1354–1362. doi: 10.1161/ATVBAHA.110.204230. [DOI] [PubMed] [Google Scholar]
- 43.Huang L., Fan B., Ma A., Shaul P.W., Zhu H. Inhibition of ABCA1 protein degradation promotes HDL cholesterol efflux capacity and RCT and reduces atherosclerosis in mice. J. Lipid Res. 2015;56:986–997. doi: 10.1194/jlr.M054742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kurdi A., Martinet W., De Meyer G.R.Y. mTOR inhibition and cardiovascular diseases: dyslipidemia and atherosclerosis. Transplantation. 2018;102:S44–S46. doi: 10.1097/TP.0000000000001693. [DOI] [PubMed] [Google Scholar]
- 45.Ouimet M., Marcel Y.L. Regulation of lipid droplet cholesterol efflux from macrophage foam cells. Arterioscler. Thromb. Vasc. Biol. 2012;32:575–581. doi: 10.1161/ATVBAHA.111.240705. [DOI] [PubMed] [Google Scholar]
- 46.Sergin I., Evans T.D., Zhang X., Bhattacharya S., Stokes C.J., Song E., Ali S., Dehestani B., Holloway K.B., Micevych P.S., Javaheri A., Crowley J.R., Ballabio A., Schilling J.D., Epelman S., Weihl C.C., Diwan A., Fan D., Zayed M.A., Razani B. Exploiting macrophage autophagy-lysosomal biogenesis as a therapy for atherosclerosis. Nat. Commun. 2017;8:15750. doi: 10.1038/ncomms15750. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.