

Graphical abstract

Abbreviations: PKN, protein kinase N; Abl, Abelson murine leukemia viral oncogene; IC50, half maximal inhibitory concentration; AGC, protein kinase A/G/C families; PKC, protein kinase C; PRK, protein kinase C-related kinase; PAK2, p21 activated kinase 2; PRO2, glutamate 5-kinase Pro2; STK, serine/threonine kinase; PDB, protein databank; PARP, poly(ADP-ribose) polymerase; ChEMBL, European Molecular Biology Laboratory Chemical database; CLK, CDC2-like kinase; SAR, structure activity relationship; CDI, 1,1′-carbonyldiimidazole; TR-FRET, time resolved fluorescence resonance energy transfer; THF, tetrahydrofuran; EtOH, ethanol; HATU, hexafluorophosphate azabenzotriazole tetramethyl uronium; DIPEA, N,N-diisopropylethylamine; DCM, dichloromethane; AcOH, acetic acid; DMF, N,N-dimethyl-formamide; KD, dissociation constant; Ki, inhibitor constant; NMR, nuclear magnetic resonance; DMSO, dimethyl sulfoxide; MeOH, methanol; GST, glutathione S-transferase; DNA, deoxyribonucleic acid; SFM, scanning force microscopy; HEPES, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; TCEP, tris(2-carboxyethyl)phosphine; EDTA, ethylenediaminetetraacetic acid; SDS, sodium dodecyl sulfate; PAGE, polyacrylamide gel electrophoresis; ATP, adenosine triphosphate; EGTA, egtazic acid; CV, column volumes
Keywords: Kinases, Cancer, Heart failure, Inflammation, AGC kinase, PKN2, PRK2, Protein kinase N2, Benzimidazole, Chemical probe, Chemical tool
Abstract
Kinases are signalling proteins which have proven to be successful targets for the treatment of a variety of diseases, predominantly in cancers. However, only a small proportion of kinases (<20%) have been investigated for their therapeutic viability, likely due to the lack of available chemical tools across the kinome. In this work we describe initial efforts in the development of a selective chemical tool for protein kinase N2 (PKN2), a relatively unexplored kinase of interest in several types of cancer. The most successful compound, 5, has a measured IC50 of 0.064 μM against PKN2, with ca. 17-fold selectivity over close homologue, PKN1.
Chemical tools/probes are drug-like compounds used to answer biological questions. They need not possess all the properties of a drug candidate, which can be dialled in at a later point in the drug development process. These compounds only need to be sufficiently stable, potent and selective towards their particular target.1, 2
Historically, the approval of imatinib3 as an effective Abl kinase inhibitor for treating chronic myeloid leukaemia stimulated efforts to better understand the 518 human protein kinases and their role in disease. Trends in research4 suggest that less than 20% of the human kinome has been well-studied,5 and selective inhibitors are only available for an even smaller fraction of those kinases.
Protein kinase N2 (PKN2) (Fig. 1) is one of these understudied kinases. It is an AGC-type serine/threonine protein kinase. There are more than 60 AGC protein kinases in the human genome with 14 further classifications. PKN2 falls into the PKN sub-family, closely related to the PKC sub-family, and is one of three homologues (PKN1/2/3). It has a number of pseudonyms which include protein kinase C-related kinase 2 (PRK2), PKNγ, PAK2, PRO2, and STK7.6
Fig. 1.

PKN2 and its domain organisation. The structure organisation contains three repeats of ACC domain (anti-parallel coiled-coil) in the N-termini region (pink/orange), a C2 calcium binding-like domain and in the C-terminal, the Ser/Thr kinase domain.
PKNs have a fairly conserved primary sequence and they share the same architecture. The catalytic domain of PKN2 has 87% percent identity with PKN1; 70% with PKN3; and 50% with PKC kinases, while the N-termini regions are less conserved, sharing only 48% and 40% between PKN1/2 and PKN2/3, respectively.7, 8
PKNs have been linked to various cellular roles, including cytoskeleton regulation,9 transport,10 cell adhesion,11 nutrient signalling,12 and cell cycle,13 as well as being a target of interest in colon,14 breast,15 renal,16 head,17 neck,17 and prostate cancers.18 They are also reportedly involved in inflammation19, 20 and heart failure.21 So far, there is one X-ray crystal structure of PKN2 publicly available in the Protein Data Bank (PDB ID: 4CRS) (Fig. 2).
Fig. 2.

Crystallographic structure of PKN2 bound to ATP-γS (PDB ID: 4CRS).
These previous studies have elucidated functions for PKN2 using molecular and cell biology techniques, and the conclusions would be greatly supported by validation through the use of small molecule inhibitors, especially to evaluate PKN2′s potential as a cancer drug target. Potent inhibitors are known for several AGC kinase family members, including ROCK22, 23, 24, 25 and PKC,26 but currently there are no sufficiently selective inhibitors for PKN2.12
This work describes an initial effort to develop such compounds based around a benzimidazole core. Compound 5 was previously developed as a PARP inhibitor27, 28, 29 but exhibited higher potency towards PKN2 than its desired target. Benzimidazoles are N-containing heterocycles that are prevalent in medicinal chemistry.30 The compound was found as part of a screen of the Abbott chemical library31 via the ChEMBL database when searching for PKN2 inhibitors. It had a reported Ki of 0.040 μM against PKN2 while only inhibiting two out of 137 other kinases (PKN1 and CLK4) with potencies lower than 0.100 μM.31 This was deemed a good starting point for repurposing the compound as a PKN2 inhibitor. We report the synthesis of that compound and subsequent SAR studies to determine its viability as a chemical tool for establishing the potential of PKN2 as a therapeutic target.
Compound 5 was successfully synthesised via a four step synthesis (Scheme 1). 2-Amino-3-nitro-benzoic acid (1) was treated with ammonia and CDI-coupling conditions32 to form amide 2. The 3-nitro group was reduced to aniline 3 with sodium dithionate,33 followed by the coupling of isonicotinic acid to the 3-position aniline to form amide 4,34 which was then heated in acetic acid to form benzimidazole 5.35
Scheme 1.

The scope of this chemistry enabled the synthesis of 14 analogues using commercially available nitroanilines and di-anilines. Additional alkylation conditions allowed the capping of the benzimidazole N-H36 (6) and alternative amide coupling conditions were used for preparing compound 1137 and the penultimate amide intermediate used to make compound 19.38
The potencies and selectivities of these compounds were tested using a TR-FRET binding-displacement assay in which the IC50 values were measured (Table 1). Calculation of Ki values using the Cheng-Prusoff equation and the KD of the tracer (previously determined) allowed the affinity of the inhibitors for PKN2 and PKN1 to be compared (Table 1).
Table 1.

| # | R | R1 | R2 | X | Ar | PKN2 |
PKN1 |
||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50 (µM) | Standard Deviation (µM) | Ki (µM) | IC50 (µM) | Standard Deviation (µM) | Ki (µM) | ||||||
| 5 | CONH2 | H | H | H | 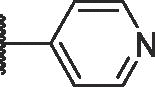 |
0.064 | 0.001 | 0.03 | 1.10 | 0.11 | 0.54 |
| 6 | CONH2 | H | H | CH2COOMe | 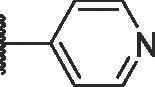 |
5.40 | 0.48 | 2.69 | 52.84 | 1.74 | 25.94 |
| 7 | CONH2 | H | H | H | 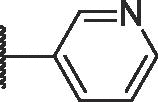 |
16.38 | 3.86 | 8.19 | 66.29 | 38.04 | 32.54 |
| 8 | CONH2 | H | H | H |  |
47.66 | 4.06 | 23.83 | 137.92 | 100.95 | 67.71 |
| 9 | CONH2 | H | H | H | 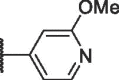 |
13.79 | 1.18 | 6.90 | 61.59 | 11.95 | 30.23 |
| 10 | CONHMe | H | H | H | 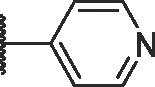 |
2.12 | 0.27 | 1.06 | 10.59 | 1.70 | 5.20 |
| 11 | CONMe2 | H | H | H |  |
38.84 | 9.57 | 19.42 | 56.66 | 25.96 | 27.81 |
| 12 | H | CONH2 | H | H | 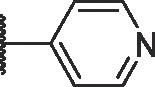 |
16.23 | 3.79 | 8.11 | 99.41 | 34.63 | 48.80 |
| 13 | H | H | H | H | 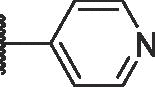 |
7.71 | 0.21 | 3.85 | 45.54 | 17.58 | 22.36 |
| 14 | NO2 | H | H | H |  |
2.50 | 0.06 | 1.25 | 58.94 | 8.33 | 28.93 |
| 15 | COOMe | H | H | H | 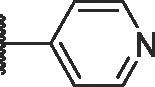 |
31.29 | 5.20 | 15.64 | 38.81 | 13.48 | 19.05 |
| 16 | H | COOEt | H | H | 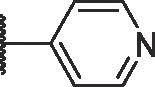 |
42.09 | 23.86 | 21.05 | 56.69 | 1.61 | 27.83 |
| 17 | H | C N | H | H | 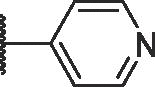 |
5.02 | 0.79 | 2.51 | 38.54 | 2.26 | 18.92 |
| 18 | H | NO2 | H | H | 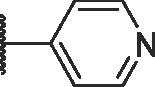 |
25.85 | 0.58 | 12.92 | 109.30 | 33.23 | 53.66 |
| 19 | CONH2 | H | Br | H | 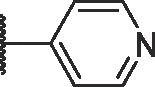 |
0.17 | 0.01 | 0.08 | 4.45 | 0.39 | 2.19 |
Compound 5 was validated as a PKN2 inhibitor (Ki = 0.032 μM) with 17-fold selectivity over PKN1 (Ki = 0.500 μM) which was not previously included in the Abbott library screen used in the Metz et al. study.31
The benzimidazole N—H was capped using chemistry described by Tsukamoto et al.36 While the alkylation conditions given were said to be applicable to methylation of the benzimidazole using the corresponding methyl halide, this proved unsuccessful; a dimethylated product formed instead, thought to be due to the susceptibility for the 4′-pyridyl to also alkylate after the benzimidazole N—H. Repeating the specific reaction conditions used by the authors incorporated a methyl acetate ester at the 1-position (6) which led to loss of binding to PKN2.
Moving the 4′-pyridyl nitrogen in 7 and 8 resulted in loss of activity, as did introducing an electron-donating methoxy group at the 3′-position (9). This suggests the 4′-pyridyl ring acts as the hinge binder. Attempts to make the 2′-pyridyl and 4′-pyrimidine analogues were unsuccessful (Scheme 2).
Scheme 2.

Compounds 21 and 23 could not be synthesised from intermediates 20 and 22
Capping the amide with one (10) or two (11) methyl groups led to increasing loss of activity respectively. Potency was lost when the amide was moved to the 5-position of the benzimidazole ring (12), Removing the amide completely (13) or exchanging the 4- or 5-position for another functional group (14–18) also led to loss of activity.
Introduction of a bromine at the 6-position (19) was hoped to provide a useful handle for incorporating various alkyl/aryl groups at that position using Suzuki coupling chemistry.39, 40 This reaction was attempted at multiple stages of the synthetic route but was unsuccessful. Compound 19 was active against PKN2 but was nearly three times less potent than compound 5. Despite this reduction in potency, compound 19 is 26-fold selective over PKN1.
The SAR exploration around 5 confirms that the primary amide at the 4-position, 4'-pyridyl and free N—H at the 1-position are necessary for the compound’s activity against PKN2. Subsequent analogues prepared for this series did not improve potency for the target within the PKN family but did result in a slight improvement in selectivity over PKN1 in compound 19.
Chemical tools are needed to facilitate the exploration of lesser understood kinases such as PKN2 for its roles in healthy and cancerous cells. Benzimidazole 5 was validated as an inhibitor of PKN2 with IC50 0.064 μM and with ca. 17-fold selectivity over PKN1 with reported high selectivity across the wider kinome31. Our efforts to develop a new compound to inhibit PKN2 resulted in compound 19 which was 26-fold selective for PKN2 over PKN1 despite having a near three-fold reduction in potency compared to compound 5.
Acknowledgments
Acknowledgements
This work was supported by a Continuing Excellence Fund from the Genome Damage and Stability Centre, University of Sussex. Thanks also to additional funding from the Wellcome Trust for initial assay experiments.
This work was also supported by the Brazilian agencies FAPESP (Fundação de Amparo à Pesquisa do Estado de São Paulo) (2013/50724-5 and 2014/50897-0) and CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico) (465651/2014-3). The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck KGaA Darmstadt Germany, MSD, Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, Takeda, and Wellcome [106169/ZZ14/Z]. We thank the staff of the Life Sciences Core Facility (LaCTAD) from State University of Campinas (UNICAMP), for the proteomics analysis. AMF received a CAPES fellowship (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior- 88887.136437/2017-00).
To the best of our knowledge there are no competing interests with involved parties.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bmcl.2020.127040.
Contributor Information
Fiona Scott, Email: f.scott@sussex.ac.uk.
Angela M. Fala, Email: angelafala@gmail.com.
Lewis E. Pennicott, Email: l.e.pennicott@sussex.ac.uk.
Tristan D. Reuillon, Email: treuillo@its.jnj.com.
Katlin B. Massirer, Email: kmassire@unicamp.br.
Jonathan M. Elkins, Email: jon.elkins@sgc.ox.ac.uk.
Simon E. Ward, Email: wards10@cardiff.ac.uk.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Blagg J., Workman P. Choose and use your chemical probe wisely to explore cancer biology. Cancer Cell. 2017;32(1):9–25. doi: 10.1016/j.ccell.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arrowsmith C.H., Audia J.E., Austin C., et al. The promise and peril of chemical probes. Nat Chem Biol. 2015;11(8):536–541. doi: 10.1038/nchembio.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gleevec: the Breakthrough in Cancer Treatment. http://www.nature.com/scitable/topicpage/gleevec-the-breakthrough-in-cancer-treatment-565. Accessed March 17, 2016.
- 4.Fedorov O., Müller S., Knapp S. The (un)targeted cancer kinome. Nat Chem Biol. 2010;6(3):166–169. doi: 10.1038/nchembio.297. [DOI] [PubMed] [Google Scholar]
- 5.Fabbro D., Cowan-Jacob S.W., Moebitz H. Ten things you should know about protein kinases: IUPHAR Review 14. Br J Pharmacol. 2015;172(11):2675–2700. doi: 10.1111/bph.13096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arencibia J.M., Pastor-Flores D., Bauer A.F., Schulze J.O., Biondi R.M. AGC protein kinases: from structural mechanism of regulation to allosteric drug development for the treatment of human diseases. Biochim Biophys Acta – Proteins Proteomics. 2013;1834(7):1302–1321. doi: 10.1016/j.bbapap.2013.03.010. [DOI] [PubMed] [Google Scholar]
- 7.Lim W., Tan B., Zhu Y., et al. The very C-terminus of PRK1/PKN is essential for its activation by RhoA and downstream signaling. Cell Signal. 2006;18(9):1473–1481. doi: 10.1016/j.cellsig.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 8.Bauer A.F., Sonzogni S., Meyer L., et al. Regulation of protein kinase C-related protein kinase 2 (PRK2) by an intermolecular PRK2-PRK2 interaction mediated by Its N-terminal domain. J Biol Chem. 2012;287(24):20590–20602. doi: 10.1074/jbc.M111.327437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amano M., Nakayama M., Kaibuchi K. Rho-kinase/ROCK: a key regulator of the cytoskeleton and cell polarity. Cytoskeleton. 2010;67:545–554. doi: 10.1002/cm.20472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leroux A.E., Schulze J.O., Biondi R.M. AGC kinases, mechanisms of regulation and innovative drug development. Semin Cancer Biol. 2018;48:1–17. doi: 10.1016/j.semcancer.2017.05.011. [DOI] [PubMed] [Google Scholar]
- 11.Calautti E., Grossi M., Mammucari C., et al. Fyn tyrosine kinase is a downstream mediator of Rho/PRK2 function in keratinocyte cell–cell adhesion. J Cell Biol. 2002;156(1):137–148. doi: 10.1083/jcb.200105140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wallroth A., Koch P.A., Marat A.L., Krause E., Haucke V. Protein kinase N controls a lysosomal lipid switch to facilitate nutrient signalling via mTORC1. Nat Cell Biol. 2019;21(9):1093–1101. doi: 10.1038/s41556-019-0377-3. [DOI] [PubMed] [Google Scholar]
- 13.Schmidt A., Durgan J., Magalhaes A., Hall A. Rho GTPases regulate PRK2/PKN2 to control entry into mitosis and exit from cytokinesis. EMBO J. 2007;26(6):1624–1636. doi: 10.1038/sj.emboj.7601637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng Y., Zhu Y., Xu J., et al. PKN2 in colon cancer cells inhibits M2 phenotype polarization of tumor-associated macrophages via regulating DUSP6-Erk1/2 pathway. Mol Cancer. 2018;17:13. doi: 10.1186/s12943-017-0747-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin W., Huang J., Yuan Z., Feng S., Xie Y., Ma W. Protein kinase C inhibitor chelerythrine selectively inhibits proliferation of triple-negative breast cancer cells. Sci Rep. 2017;7(1):2022. doi: 10.1038/s41598-017-02222-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hopkins S.R., Mcgregor G.A., Murray J.M., Downs J.A., Savic V. Novel synthetic lethality screening method identifies TIP60-dependent radiation sensitivity in the absence of BAF180. DNA Repair (Amst) 2016;46:47–54. doi: 10.1016/j.dnarep.2016.05.030. [DOI] [PubMed] [Google Scholar]
- 17.Rajagopalan P., Nanjappa V., Patel K., et al. Role of protein kinase N2 (PKN2) in cigarette smoke-mediated oncogenic transformation of oral cells. J Cell Commun Signal. 2018;12(4):709–721. doi: 10.1007/s12079-017-0442-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Sullivan A.G., Mulvaney E.P., Hyland P.B., Kinsella B.T. Protein kinase C-related kinase 1 and 2 play an essential role in thromboxane-mediated neoplastic responses in prostate cancer. Oncotarget. 2015;6(28):26437–26456. doi: 10.18632/oncotarget.4664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park Y.H., Wood G., Kastner D.L., Chae J.J. Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat Immunol. 2016;17(8):914–921. doi: 10.1038/ni.3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schnappauf O., Chae J.J., Kastner D.L., Aksentijevich I. The pyrin inflammasome in health and disease. Front Immunol. 2019;10:1745. doi: 10.3389/fimmu.2019.01745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakaguchi T., Takefuji M., Wettschureck N., et al. Protein kinase N promotes stress-induced cardiac dysfunction through phosphorylation of myocardin-related transcription factor A and disruption of its interaction with actin. Circulation. 2019 doi: 10.1161/CIRCULATIONAHA.119.041019. [DOI] [PubMed] [Google Scholar]
- 22.Shaw D., Hollingworth G., Soldermann N., et al. Novel ROCK inhibitors for the treatment of pulmonary arterial hypertension. Bioorg Med Chem Lett. 2014;24(20):4812–4817. doi: 10.1016/j.bmcl.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 23.Pan J., Yin Y., Zhao L., Feng Y. Discovery of (S)-6-methoxy-chroman-3-carboxylic acid (4-pyridin-4-yl-phenyl)-amide as potent and isoform selective ROCK2 inhibitors. Bioorg Med Chem. 2019;27(7):1382–1390. doi: 10.1016/J.BMC.2019.02.047. [DOI] [PubMed] [Google Scholar]
- 24.Doe C., Bentley R., Behm D.J., et al. Novel Rho kinase inhibitors with anti-inflammatory and vasodilatory activities. J Pharmacol Exp Ther. 2007;320(1):89–98. doi: 10.1124/jpet.106.110635. [DOI] [PubMed] [Google Scholar]
- 25.Goodman K.B., Cui H., Dowdell S.E., et al. Development of dihydropyridone indazole amides as selective Rho-kinase inhibitors. J Med Chem. 2007;50(1):6–9. doi: 10.1021/jm0609014. [DOI] [PubMed] [Google Scholar]
- 26.Arencibia J.M., Fröhner W., Krupa M., et al. An allosteric inhibitor scaffold targeting the PIF-pocket of atypical protein kinase C isoforms. ACS Chem Biol. 2017;12(2):564–573. doi: 10.1021/acschembio.6b00827. [DOI] [PubMed] [Google Scholar]
- 27.Kock M, Lubisch W, Jentzsch A, Use of Parp Inhibitors in Cosmetic Preparations. WO/2001/082877A2 2001.
- 28.Lubisch W, Kock M et al. Heterocyclically substituted benzimidazoles, the production and application thereof. US6696437. 2004.
- 29.Takayama K, Koga Y et al. Benzimidazole Derivatives. WO/2001/021615. 2001.
- 30.Velík J., Baliharová V., Fink-Gremmels J., Bull S., Lamka J., Skálová L. Benzimidazole drugs and modulation of biotransformation enzymes. Res Vet Sci. 2004;76(2):95–108. doi: 10.1016/J.RVSC.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 31.Metz J.T., Johnson E.F., Soni N.B., Merta P.J., Kifle L., Hajduk P.J. Navigating the kinome. Nat Chem Biol. 2011;7(4):200–202. doi: 10.1038/nchembio.530. [DOI] [PubMed] [Google Scholar]
- 32.Francis C. Rix, Smita Kacker, Sudhin Datta, Rul Zhao VRE. Olefin polymerization catalyst system and process for use thereof. US7601666. 2005.
- 33.Schmidt A., Shilabin A.G., Nieger M., Mariand M., Levillain P., Sense J.M. On benzo[b][1,4]diazepinium-olates, -thiolates and -carboxylates as anti-Hückel mesomeric betaines. Org Biomol Chem. 2003;1(23):4342–4350. doi: 10.1039/B308412D. [DOI] [PubMed] [Google Scholar]
- 34.Van Steijvoort B.F., Kaval N., Kulago A.A., Maes B.U.W. Remote functionalization: palladium-catalyzed C5(sp3)-H arylation of 1-Boc-3-aminopiperidine through the use of a bidentate directing group. ACS Catal. 2016;6(7):4486–4490. doi: 10.1021/acscatal.6b00841. [DOI] [Google Scholar]
- 35.White A.W., Almassy R., Calvert A.H., et al. Resistance-modifying agents. 9. 1 synthesis and biological properties of benzimidazole inhibitors of the DNA repair enzyme poly(ADP-ribose) polymerase. J Med Chem. 2000;43(22):4084–4097. doi: 10.1021/jm000950v. [DOI] [PubMed] [Google Scholar]
- 36.Tsukamoto G., Yoshino K., Kohno T., Ohtaka H., Kagaya H., Ito K. Synthesis and antiinflammatory activity of some 2-(substituted-pyridinyl)benzimidazoles. J Med Chem. 1980;23(7):734–738. doi: 10.1021/jm00181a007. [DOI] [PubMed] [Google Scholar]
- 37.Tanaka H, Bohno A, et al. Azole-Substituted Pyridine Compound. WO/2019/031618. 2019.
- 38.Yoshikawa M., Motoshima K., Fujimoto K., Tai A., Kakuta H., Sasaki K. Pyridinium cationic-dimer antimalarials, unlike chloroquine, act selectively between the schizont stage and the ring stage of Plasmodium falciparum. Bioorg Med Chem. 2008;16(11):6027–6033. doi: 10.1016/J.BMC.2008.04.051. [DOI] [PubMed] [Google Scholar]
- 39.Hong S., Kim J., Yun S.-M., et al. Discovery of new benzothiazole-based inhibitors of breakpoint cluster region-abelson kinase including the T315I mutant. J Med Chem. 2013;56(9):3531–3545. doi: 10.1021/jm301891t. [DOI] [PubMed] [Google Scholar]
- 40.Sutherlin D.P., Sampath D., Berry M., et al. Discovery of (thienopyrimidin-2-yl)aminopyrimidines as potent, selective, and orally available pan-pi3-kinase and dual pan-PI3-kinase/mTOR inhibitors for the treatment of cancer. J Med Chem. 2010;53(3):1086–1097. doi: 10.1021/jm901284w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.